
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
From pollution to energy storage: leveraging hydrogen sulfide with SU-101 cathodes in lithium–sulfur batteries†
Raul A. Marquez *a,
Juan L. Obeso‡
bc,
Rinish Reddy Vaidyula‡a,
Valeria B. López-Cervantes‡b,
Ricardo A. Peraltad,
Pablo Marín Rosasd,
José Antonio de los Reyese,
C. Buddie Mullins*afg and
Ilich A. Ibarra*bh
*a,
Juan L. Obeso‡
bc,
Rinish Reddy Vaidyula‡a,
Valeria B. López-Cervantes‡b,
Ricardo A. Peraltad,
Pablo Marín Rosasd,
José Antonio de los Reyese,
C. Buddie Mullins*afg and
Ilich A. Ibarra*bh
aDepartment of Chemistry, The University of Texas at Austin, Austin, Texas 78712, USA. E-mail: raul.marquez@utexas.edu
bLaboratorio de Fisicoquímica y Reactividad de Superficies (LaFReS), Instituto de Investigaciones en Materiales, Universidad Nacional Autónoma de México, Ciudad de México 04510, Mexico
cLaboratorio Nacional de Ciencia, Tecnología y Gestión Integrada del Agua (LNAgua), Instituto Politécnico Nacional, CICATA, U. Legaria, Ciudad de México 11500, Mexico
dDepartamento de Química, División de Ciencias Básicas e Ingeniería, Universidad Autónoma Metropolitana-Iztapalapa, Ciudad de México 09340, Mexico
eLaboratory of Environmental Catalysis, Universidad Autónoma Metropolitana-Iztapalapa, Ciudad de México 09340, Mexico
fMcKetta Department of Chemical Engineering, The University of Texas at Austin, Austin, Texas 78712, USA
gTexas Materials Institute, The University of Texas at Austin, Austin, Texas 78712, USA
hOn Sabbatical as “Catedra Dr. Douglas Hugh Everett” at Departamento de Química, Universidad Autónoma Metropolitana-Iztapalapa, Ciudad de México 09310, Mexico
First published on 14th June 2024
Abstract
Despite growing interest in developing metal–organic frameworks to capture toxic emissions, the potential for revalorizing these emissions has largely been overlooked. Captivated by the unique ability of SU-101 to transform H2S into polysulfides spontaneously, here we demonstrate how this remarkable capability can be leveraged to power lithium–sulfur batteries. Our proof-of-concept demonstrates how hydrogen sulfide emissions, efficiently captured by the SU-101 metal–organic framework, can be directly converted and utilized in a lithium–sulfur battery. Despite demonstrating a modest initial capacity of about 85 mA h g−1 and capacity retention of approximately 54%, analogous to other MOF-based Li–S batteries reported in the literature, the SU-101-Sat cathode delivered a durable and stable performance across 1000 cycles and maintained 99.87% coulombic efficiency. The stable response is credited to the controlled release of polysulfides generated from captured hydrogen sulfide. Such resilience is advantageous for developing compact batteries for extended use in special applications, including wearable technology, satellite components, and weather monitoring instruments. These findings open new pathways for waste valorization by employing metal–organic frameworks designed to spontaneously convert toxic gas emissions into valuable feedstocks, serving as potential candidates for more sustainable electrochemical energy conversion and storage devices.
Introduction
Air quality is an important environmental concern.1 In addition to greenhouse gases, carbon monoxide (CO), ozone (O3), ammonia (NH3), sulfur dioxide (SO2), volatile organic compounds (VOCs), particulate matter (PM), and hydrogen sulfide (H2S) represent some of the most predominant and hazardous air pollutants.2 H2S is a colorless, flammable gas, particularly corrosive (i.e., H2S is associated with acid rain), and is cataloged as a hazardous chemical. H2S is frequently found in biogas and natural gas and is produced by different chemical processes at industrial levels, such as in oil refineries.3H2S is highly toxic to humans since it can be rapidly absorbed through inhalation,4 causing illnesses of the respiratory, cardiovascular, and nervous systems, and specifically, it is related to laryngitis, pneumonia, bronchitis, and pulmonary edema.5 Even at relatively low concentrations, H2S is lethal, causing severe nervous system failure.6 Therefore, the effective sequestration of H2S is crucial for numerous industrial procedures before it is released into the atmosphere. Despite recent advances in H2S capture, current technologies (e.g., alkanolamines, ionic liquids, cryogenic sequential distillation, and physisorption in zeolites, activated carbons, and metal oxides) have encountered difficulties such as corrosion of pipelines, large amounts of wastewater, low capture and recovery of H2S, and high costs of re-use.7 Furthermore, there is a pressing need to develop further waste valorization strategies for exploiting these captured emissions.
Chemically-stable metal–organic frameworks (MOFs) have recently been investigated for the efficient capture of H2S, and promising results have been reported.8 However, once H2S is captured (by a reversible physisorption process) in MOFs, what steps are needed to convert H2S chemically to a different inert chemical molecule? Catalytic conversion of H2S within MOFs is a novel concept that was only recently demonstrated. Our research group reported the spontaneous chemical transformation of H2S inside the micropores of MOFs to generate polysulfides in situ at room temperature and atmospheric pressure.9 Based on this premise, we conceive a significant potential for the use of MOF adsorbents containing irreversibly chemisorbed sulfur species (polysulfides) as feedstocks for electrochemical energy conversion and storage technologies, such as sulfur-based batteries.10
Lithium–sulfur (Li–S) batteries are promising candidates for the next generation of energy storage systems due to their high theoretical capacity, low cost, and environmentally friendly fabrication.11 Li–S batteries show promise for overcoming dependence on fossil fuels, reducing exhaust emissions, and paving the way to produce batteries for the next generation of electric vehicles. Certainly, the commercial application of Li–S batteries faces many different challenges, such as (i) poor electrical conductivity of elemental sulfur and its final discharge product of Li2S; (ii) large volume expansion of sulfur during the lithiation process resulting in fast capacity decay; (iii) diffusion of soluble polysulfide intermediates affording to irreversible loss of active materials and corrosion of the lithium anode and (iv) the use of a metallic lithium anode can produce safety concerns due to the formation of uninhibited lithium dendrites.12 To solve these problems, different efforts have focused on designing cathode materials, improving novel electrolytes, adapting separators, and protecting the lithium anode.13
Previous studies have utilized MOFs as potential cathode materials for Li–S batteries, given their customizable pore and cage structures and pore dimensions that can effectively encapsulate sulfur.14 Typically, the MOF material is calcined at high temperatures (500–600 °C) and then subjected to sulfur impregnation. Some materials, such as MIL-101(Cr)@rGO/S (335 mA g h−1 after 50 cycles),15 HKUST-1 (240 mA g h−1 after 50 cycles),16 and Mn-MOF (190 mA g h−1 after 50 cycles),17 have demonstrated high reversible capacities. However, these approaches have reported low cyclability, raising concerns about the lifetime of sulfur-enriched MOF cathodes.
The decreased lifetime is often associated with the detrimental effects of polysulfide intermediates that shuttle between the electrodes and react with the Li anode, resulting in the depletion of active material and manifesting as a capacity decay over time.18,19 Only a few MOFs have demonstrated effective confinement of polysulfides through physical encapsulation and chemical adsorption.10 Therefore, identifying the optimal chemical compositions that can effectively suppress polysulfide release is crucial. Furthermore, the additional calcination and sulfur impregnation steps increase both the cost and energy demand of the production process, factors that become critical for commercialization and environmental valorization. To streamline synthesis and leverage the waste valorization potential of MOF adsorbents, the intrinsic formation of polysulfides following H2S adsorption presents new possibilities for enhancing Li–S battery technologies.
Herein, we report a proof-of-concept strategy that leverages MOF adsorbents designed to permanently capture toxic H2S, repurposing them as feedstocks for electrochemical energy storage applications. We show that cathodes containing SU-101 MOF (the polysulfide host) as the active material in a conventional Li–S battery deliver modest capacities, high coulombic efficiencies, and substantial cycling stability for 1000 cycles. These results open promising avenues for using MOFs in sustainable energy applications and waste valorization.
Experimental
Synthesis of SU-101 and H2S uptake
SU-101 was synthesized following a previously reported procedure.20 A total of 15 mg of ellagic acid and 38 mg of Bi(CH3CO2)3 were dissolved in 30 mL of water and acetic acid (6% vol. acetic acid). First, the solution was stirred strongly at room temperature for 48 hours. Later, the recovered powder was washed three times with water and ethanol. Subsequently, it was dried overnight at 60 °C. H2S saturation experiments were conducted using a custom-designed saturation chamber (Fig. S1†). A detailed description of this procedure is available in the ESI.†Material characterization
H2S adsorption measurements were conducted using an HP 5890 gas chromatograph. X-ray diffraction (XRD) patterns were recorded using a Rigaku MiniFlex 600 diffractometer equipped with a Cu Kα radiation source. Scanning electron microscopy (SEM) images and energy-dispersive X-ray spectroscopy (EDX) elemental maps were captured using a Thermo Scientific Apreo 2 microscope at a 10 kV accelerating voltage. Elemental mass fractions were quantified through total-reflectance X-ray fluorescence (TXRF) using an S2 PICOFOX spectrometer. XPS characterization was carried out with a PHI VersaProbe 4 instrument using a nonmonochromatic Al Kα source (1486.6 eV) using the charge neutralizer. The base pressure of the instrument was ∼10–9 torr. High-resolution spectra were collected over an analysis area of ∼250 × 250 μm2 using a pass energy of 10 eV. Binding energy calibration was carried out using the C 1s peak for adventitious hydrocarbons at 284.8 eV. Data analysis was performed using CasaXPS software. The spectral fitting parameters for the C 1s, O 1s, Bi 4f, and S 2p peaks were adopted from previous studies.21–23 Fitting components were modeled using a combination of Gaussian (70%) and Lorentzian (30%) profiles, denoted as GL(30) in CasaXPS, and a standard Shirley-type baseline.Electrode preparation and cell assembly
Battery performance tests were performed using LIR2032 coin-type cells following previous cathode and electrolyte compositions.14,19 For cathode preparation, slurries were prepared by mixing 40 wt% of the MOF powder (∼50 mg) with 50 wt% of carbon black (Super P, TIMCAL) and 10 wt% of polyvinylidene fluoride (PVDF) binder, for a total batch size of 125 mg. The mixture was carefully dissolved in ∼955 μL of N-methyl-2-pyrrolidone (NMP, density: 1.03 g cm−3) and stirred over 80 minutes using a small magnetic stir bar within 10 mL glass vials. The slurry was then spread onto aluminum foil current collectors using a compact film coater (Xiamen Tmax Battery Equipments Ltd, Model TMAX-TMH). Cathodes were cut into discs with a diameter of 13 mm. The thickness of the film was measured using a digital thickness gauge (Mitutoyo 543–400). A typical cathode contained approximately 0.6 mg of active material with a thickness of 8 μm, excluding the current collector. Coin cells were assembled using the MOF-containing cathode as the working electrode, lithium metal discs as the counter electrode, and a polypropylene microporous separator (Celgard 2400, thickness: 25 μm). The electrolyte (∼300 μL) consisted of a mixture of 1,3-dioxolane (DOL) and dimethoxyethane (DME) in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 volume ratio, 1 M lithium bis(trifluoromethane)sulfonimide (LiTFSI), and 0.2 M LiNO3. For carbonate-based electrolyte tests, the electrolyte consisted of a mixture of ethylene carbonate (EC) and diethyl carbonate (DEC) in a 1:1 volume ratio, 1 M LiPF6 as the supporting electrolyte, and 10 vol% of fluoroethylene carbonate (FEC) as an additive. Cell assembly was conducted inside an Ar-filled glovebox (MBRAUN UNIlab 2000), maintaining O2 and H2O contents below 0.1 ppm.
:1 volume ratio, 1 M lithium bis(trifluoromethane)sulfonimide (LiTFSI), and 0.2 M LiNO3. For carbonate-based electrolyte tests, the electrolyte consisted of a mixture of ethylene carbonate (EC) and diethyl carbonate (DEC) in a 1:1 volume ratio, 1 M LiPF6 as the supporting electrolyte, and 10 vol% of fluoroethylene carbonate (FEC) as an additive. Cell assembly was conducted inside an Ar-filled glovebox (MBRAUN UNIlab 2000), maintaining O2 and H2O contents below 0.1 ppm.
Electrochemical measurements
Tests were conducted using a Gamry Reference 620 potentiostat/galvanostat. The open-circuit potential (OCP) was measured 30 minutes before and after electrochemical measurements. Cyclic voltammetry (CV) scans were conducted within a potential window of 1.5 to 3.0 V vs. Li/Li+ at a scan rate of 0.1 mV s−1. Electrochemical impedance spectroscopy (EIS) measurements were conducted at stable OCP values at an AC amplitude of 10 mV over a frequency range from 100 kHz to 0.5 Hz. Galvanostatic charge–discharge (GCD) cycling was conducted by charging and discharging the cell at a constant current of 0.18 mA (equivalent to a C/2 rate or 0.5 A g−1) between 1.5 and 3.0 V vs. Li/Li+.Results and discussion
Our previous study demonstrated that the SU-101(Bi) MOF achieves a significant uptake of H2S (15.95 mmol g−1) from an N2 mixture.20 This uptake exceeds that of other MOF adsorbents such as MIL-47(V) (14.6 mmol g−1)24 and Ni-CPO (12.0 mmol g−1),25 and is comparable to other MOFs examined by our group, including MIL-53(Al)-TDC (18.1 mmol g−1),26 and MFM-300(Sc) (16.5 mmol g−1).9 The high H2S uptake has previously been attributed to the strong chemisorption of H2S, which results in the formation of low-order polysulfides.9,20 Building upon these reports, we synthesized samples of SU-101, saturated them with H2S gas, and used them as the active cathode material in Li–S batteries.The powder X-ray diffraction (PXRD) pattern of the pristine SU-101 MOF corroborates its phase purity (Fig. S2†). Next, a SU-101 sample was activated at 423 K for 10 h under vacuum (1.4 × 10−3 torr), and the BET surface area was calculated at 414 m2 g−1. These parameters are in good correlation with previously reported characterization.20 Two freshly synthesized samples of SU-101 were exposed to concentrated H2S (approximately 1500 ppm) in our custom in situ adsorption system (Fig. S1†) at different times. The first SU-101 sample was activated at 423 K for 10 h under vacuum and exposed to H2S for 4 h (labeled as SU-101-4h). H2S adsorption measurements reveal a complete uptake of about 4.3 mL after 13 minutes (Fig. S3†).
PXRD measurements were carried out to examine the integrity of the crystalline structure after exposure to a diluted H2S atmosphere (4.3 vol% H2S with 95.7 vol% of N2, approximately 50000 ppm). The crystalline structure of SU-101 remains largely unchanged after 4 h of exposure (Fig. S4†), in line with our previous findings.20 Another SU-101 sample was activated (vide supra) and exposed to H2S for 12 h (labeled as SU-101-Sat). Interestingly, the PXRD pattern indicates a significant change in the crystalline structure, likely due to the amorphization of the SU-101-Sat sample (Fig. S5†). However, the SU-101-Sat sample exhibits distinct diffraction peaks corresponding to the (220), (111), (060), and (002) planes of the simulated SU-101 structure and peak splitting at 28.7°.27,28 This amorphization phenomenon has been attributed to the de-coordination of the Bi–O bonds, as previously observed for other MOF materials.8 Therefore, two different SU-101 samples were tested to compare the Li–S battery performance: crystalline SU-101 (SU-101-4h) and partially amorphous SU-101 (SU-101-Sat).
Elemental analysis conducted via TXRF revealed sulfur mass fractions of 6.6 and 11.3% for the SU-101-4h and SU-101-Sat samples, respectively (Fig. S6 and Table S1†). SEM images of the Bi(III)-based MOF particles reveal agglomerates of approximately 15 μm in diameter (Fig. 1a). A closer inspection of these particles reveals a spike-like morphology (Fig. 1b), while EDX elemental mappings reveal uniform bismuth, oxygen, and sulfur distribution across the particles (Fig. 1c). Notably, the EDX analysis confirms the presence of sulfur in the MOF particles. Furthermore, the formation of polysulfides was confirmed by Raman spectroscopy (Fig. S7†), with bands at 243 and 445 cm−1 assigned to Sn2− and S42− species, respectively.20
 | ||
| Fig. 1 SEM images of the as-synthesized SU-101-Sat MOF at (a) low and (b) high magnifications. (c) EDX mapping for Bi, O, and S elements in the SU-101-Sat sample. | ||
X-ray photoelectron spectroscopy (XPS) characterization was conducted to examine the chemical composition and bonding of the SU-101-Sat MOF. As shown in Fig. 2a, the XPS survey reveals signals for C, S, O, and Bi, which align with the EDX analysis. The C 1s, O 1s, Bi 4f, and S 2p regions were inspected closely, and components were fit accordingly. The C 1s peak exhibits clear components for C–C, C–H, C–OH, and O–C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O functional groups (Fig. 2b), which are present in the building unit of SU-101, ellagic acid.20 Furthermore, a small component originating from the (CO)–O–Bi interaction in the SU-101 MOF can be seen at ∼291 eV.21 The O 1s region also shows components characteristic of ellagic acid and its interaction with the metallic Bi center (Fig. 2c). The main two signals are caused by oxygen atoms in the carboxylic group interacting with the Bi center, namely (CO)–
O functional groups (Fig. 2b), which are present in the building unit of SU-101, ellagic acid.20 Furthermore, a small component originating from the (CO)–O–Bi interaction in the SU-101 MOF can be seen at ∼291 eV.21 The O 1s region also shows components characteristic of ellagic acid and its interaction with the metallic Bi center (Fig. 2c). The main two signals are caused by oxygen atoms in the carboxylic group interacting with the Bi center, namely (CO)–![[O with combining low line]](https://www.rsc.org/images/entities/char_004f_0332.gif) –Bi and (C)–O–Bi at around 533.4 and 531.8 eV, respectively. Moreover, a contribution from O–Bi–O bonds can be seen at approximately 530.3 eV.21 There are no signals ascribable to adsorbed O, confirming the absence of water molecules in the MOF.22
–Bi and (C)–O–Bi at around 533.4 and 531.8 eV, respectively. Moreover, a contribution from O–Bi–O bonds can be seen at approximately 530.3 eV.21 There are no signals ascribable to adsorbed O, confirming the absence of water molecules in the MOF.22
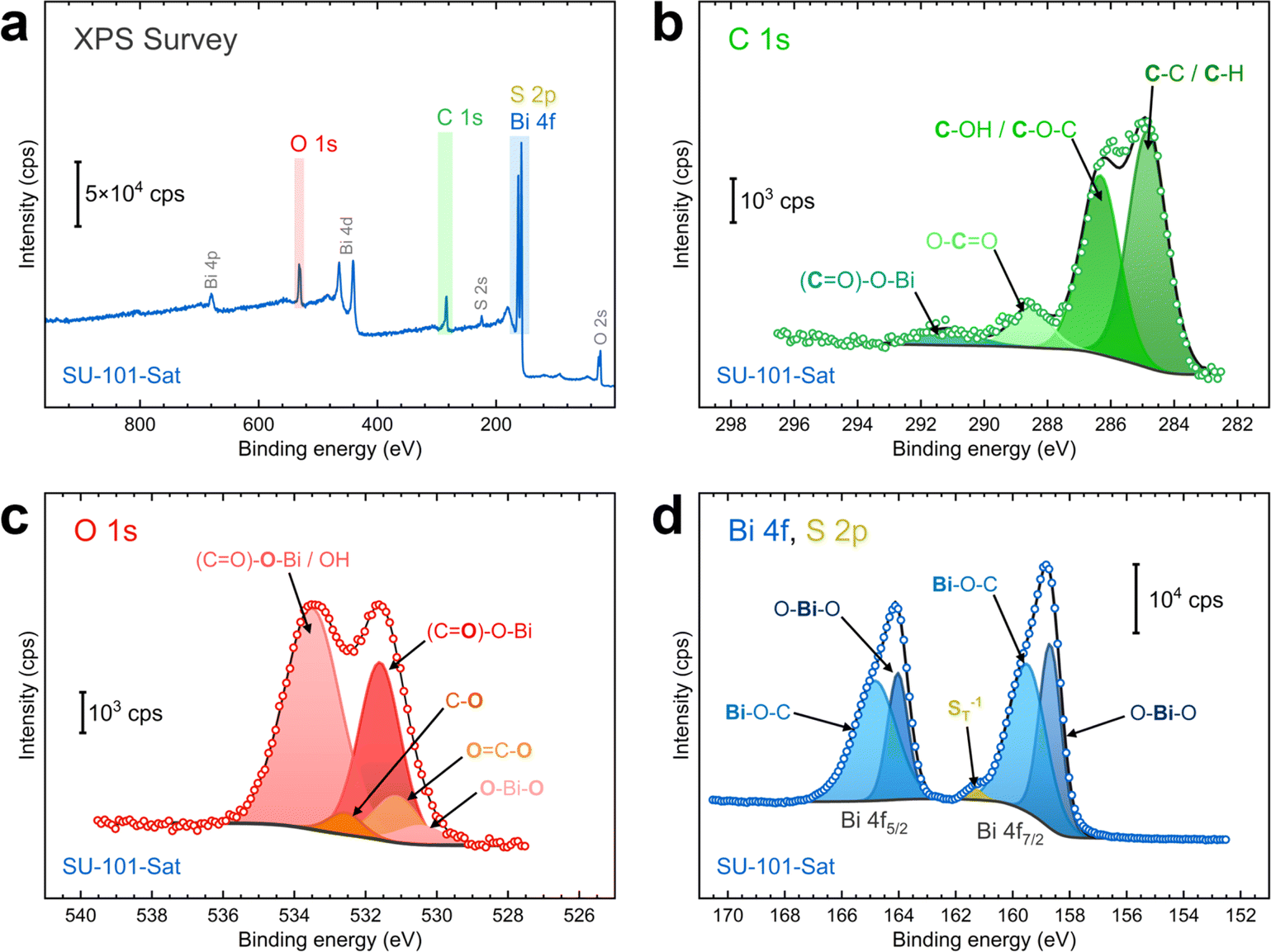 | ||
| Fig. 2 XPS characterization of the SU-101-Sat sample: (a) XPS survey, (b) C 1s region, (c) O 1s region, and (d) Bi 4f/S 2p regions. The labels indicate the specific bonds of the functional groups related to each component fitted to the spectra. Green, red, yellow, and blue tones refer to carbon-, oxygen-, sulfur-, and bismuth-based groups. | ||
As shown in Fig. 2d, the Bi 4f region exhibits two asymmetric peaks at 159.3 and 164.6 eV, corresponding to the Bi 4f7/2 and 4f5/2 peaks of Bi3+ species with a spin–orbit splitting of 5.3 eV.21,22 In contrast to the typical symmetric peaks of Bi2O3, the presence of a shoulder at higher binding energies is attributed to the Bi–O–C interaction, typically seen in carboxylic groups coordinating Bi3+.21 Furthermore, a small component was added at approximately 161.7 eV, attributed to terminal sulfur atoms (n-ST−1) in polysulfide species.23,29,30 Note, however, that there are no additional peaks at higher binding energies, which are typically ascribed to bridging S0 species (–S0B–, ∼163.9 eV), and oxidized products such as thiosulfate (167.2 eV), polythionate (168.2 eV), and sulfate (169.5 eV).23,29–32 This result suggests that sulfur is present as a short-chain polysulfide or sulfide species in the SU-101-Sat MOF material after saturation.
Additional XPS analyses were performed on the pristine SU-101 and SU-101-4h samples to examine the effects of H2S saturation. The pristine sample exhibits a composition featuring oxygen- and carbon-based functional groups similar to those observed in the SU-101-Sat sample. Bismuth is predominantly involved in Bi–O–C interactions and has no components related to sulfur species (Fig. S8†). In contrast, the SU-101-4h sample reveals a small contribution from an additional component attributed to terminal sulfur (Fig. S9†). Furthermore, the O–Bi–O component, absent in the pristine sample, becomes more pronounced with increasing saturation times (see Fig. 2d, S8d, and S9d†). In contrast, the same component in the O 1s region decreases in intensity (see Fig. 2c, S8c, and S9c†). These results indicate that the Bi–O interaction is likely affected by the incorporation of sulfur. It is also conceivable that the O–Bi–O component in the Bi 4f region might represent a combination of Bi–O and Bi–S interactions. However, this is challenging to confirm due to the complex overlap of Bi 4f and S 2p signals.
The interference between Bi and S species complicates the analysis of sulfur interactions in the S 2p region. Therefore, we also probed the S 2s region to determine the composition of sulfur in the SU-101 MOFs. While the pristine SU-101 samples exhibit no peaks in the S 2s region, both the SU-101-4h and SU-101-Sat samples display clear signals, with the signal for the SU-101-Sat sample being more intense (Fig. S10†). The peaks were fitted into two components at approximately 225.8 and 227.8 eV. Given that the S 2p and S 2s peaks are separated by about 64.0 ± 0.2 eV,33 the sulfur species data in the S 2p region (SO42−: 169.5 eV; SO32−: 166.8 eV; Sn: 164.0 eV; –S0B–: ∼163.9 eV; S22−: 162.9 eV; S2−: 161.9 eV; n-ST−1: ∼161.7 eV) enable us to assign the observed components in the S 2s region.23,29,30,33 More reduced species are present at lower binding energies, while more oxidized species appear at higher ones. Consequently, the stronger component at the lower binding energy (∼225.8 eV) can be attributed to either terminal sulfur (n-ST−1, ∼225.7 eV) or S2− (225.9 eV), while the less intense peak at higher binding energy may correspond to either bridging sulfur (–S0B–, ∼227.9 eV) or elemental sulfur (Sn, ∼228.0 eV). Consistent with previous studies,33,34 these results suggest that sulfur within the MOF is predominantly reduced, likely as short-chain polysulfides, with only a small fraction of sulfur existing as Sn species.
Building on previous studies of SU-101,9,10,20,35 and based on the physical and chemical characterization results from this study, we propose a reaction mechanism for the formation of polysulfides (illustrated in Fig. S11†). First, the SU-101 MOF is activated by the removal of water molecules. Subsequently, H2S molecules adsorb onto exposed Bi3+ sites. These coordinated H2S molecules then undergo a dissociation step where protons are removed. Given the proximity (3.78 Å) between adjacent Bi3+ sites, the sulfur atoms are oxidized, forming an S–S bond and the concurrent release of H2. Finally, the resultant sulfur species are released, completing the reaction cycle.
To evaluate the battery performance, we prepared cathodes from SU-101 slurries (Fig. S12a†) and paired them with Li metal anodes to assemble coin cells (Fig. S12b†). A detailed battery composition checklist is shown in Table S2.† Cyclic voltammetry tests were conducted to characterize the redox processes of these MOF-containing Li–S batteries. The batteries comprised an MOF-containing cathode encapsulated with a separator and a Li metal anode in a commercial coin cell setup (Fig. 3a). As shown in Fig. 3b, the cathodes containing the SU-101 MOFs show clear reduction and oxidation signals. During the reduction scan, sulfur species within SU-101-Sat reduce to Li2S sequentially, transitioning through various polysulfide intermediates. In the case of the SU-101-Sat, the cathode exhibits multiple subtle peaks and plateaus between 2.5 and 1.9 V vs. Li/Li+, indicative of the transformation from S8 to longer-chain intermediates (Li2Sx where x ≥ 4) at relatively higher potentials and to shorter-chain intermediates (Li2Sx where x < 4) at lower potentials.18,36,37 These transformations align with the XPS results (Fig. S10†), suggesting that the initial fraction of Sn species is converted into intermediates during cycling. A pronounced peak appears around 1.7 V, associated with the strong confinement of sulfur within the partially amorphous MOF (SU-101-Sat). The sharpness of this peak has been attributed to strong chemical adsorption and rapid conversion.14
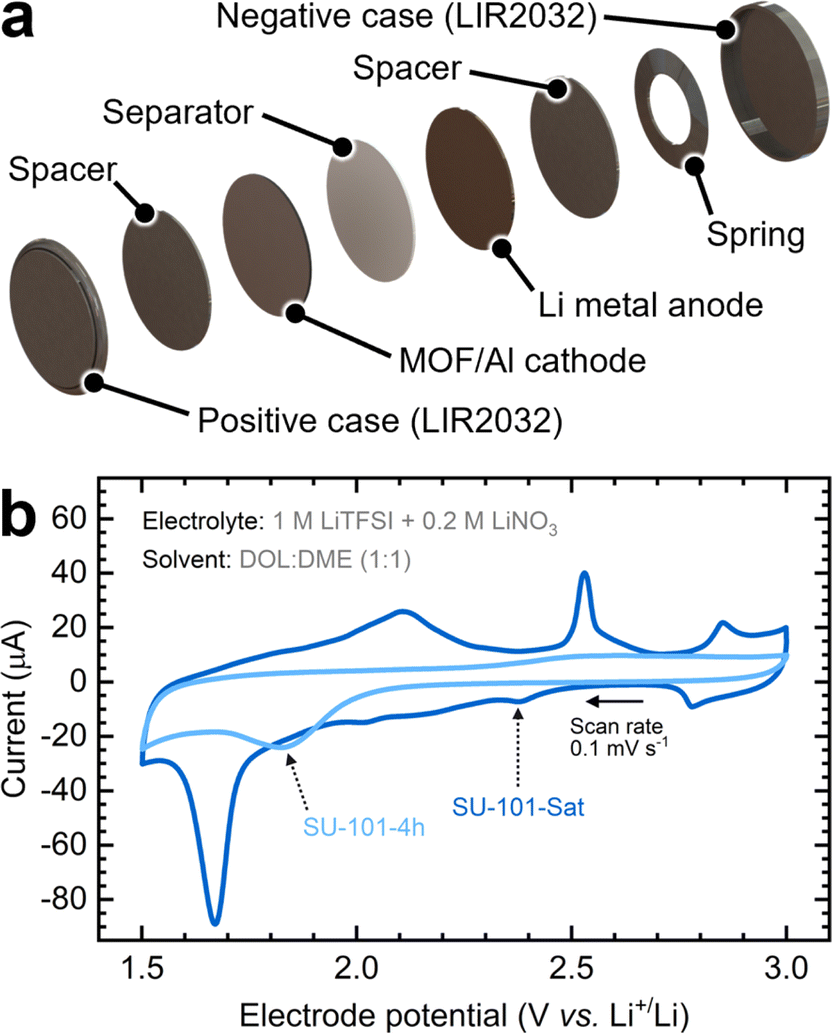 | ||
| Fig. 3 (a) Exploded-view 3D rendering of the Li–S battery coin cell and its components. (b) Cyclic voltammograms of SU-101-4h and SU-101-Sat cathodes assembled into coin cells. The second CV scan is shown for each cathode. The arrow indicates the scan direction at the specified scan rate. | ||
During the oxidation scan, two peaks emerge at approximately 2.1 V and 2.5 V, corresponding to the gradual oxidation of short-chain polysulfides back to longer chains and ultimately to S8.14 A pair of redox peaks, observed at around 2.8 V and 2.9 V, exhibit some degree of reversibility and persistence over successive cycles. These peaks, previously unreported in the literature on MOF-based Li–S batteries,14,19,36 can be attributed to the high potential polarization of polysulfide peaks influenced by the interaction of the metal center (Bi(III)) with polysulfides of short length as also demonstrated by XPS.37–39
Interestingly, the SU-101-4h cathode displays more subdued redox signals, suggesting that the polysulfide intermediates undergo gradual reactions, resulting in broader redox peaks and extended plateaus.18 This diminished intensity might also stem from less sulfur participating in the reactions, unlike the SU-101-Sat cathode material, which was saturated with H2S for an extended period, and the crystalline structure of SU-101 was partially lost (Fig. S5†).
The cycling performance of the MOF-containing cathodes was further examined through galvanostatic charge/discharge tests at a C/2 rate using an identical coin cell setup. Typical potential versus time curves depicting the charge and discharge profiles at a constant current were used to extract the capacity for each cycle (Fig. S13†). The SU-101-Sat cathode exhibits distinct plateaus aligning with the redox signals identified in CV scans. The charge profile is characterized by a notable slope change at approximately 2.1 V and a more pronounced plateau at ∼2.7 V, while the discharge profile presents a sharp slope transition at ∼2.7 V and a prolonged plateau below 1.9 V, indicative of the sequential reduction of short-chain polysulfides to insoluble Li2S.18 As shown in Fig. 4a, this pattern is also discernible when examining the charge/discharge profiles based on the capacity after 100 cycles. The SU-101-Sat cathode achieves the highest capacity at ∼66 mA h g−1, followed closely by the SU-101-4h cathode at ∼48 mA h g−1. As shown in Fig. 4b, the cycling performance tests indicate an initial capacity reduction for the SU-101-Sat cathode, which stabilizes after 80 cycles. In contrast, the SU-101-4h cathode demonstrates a more consistent cycling performance. These observations suggest that while the SU-101-Sat possesses a more abundant sulfur content available for cycling, a portion is depleted during the cycles until a stable composition is attained. On the other hand, the SU-101-4h cathode contains a lower amount of sulfur available, slightly below the stable composition threshold. The decreased sulfur content accounts for the ∼22% capacity reduction compared to the SU-101-Sat cathode. Capacity measurements over cycling show that both SU-101 compositions (SU-101-4h and SU-101-Sat) maintain average coulombic efficiencies of 99.88% and 99.76%, respectively, confirming the reversibility of the Li–S battery (Fig. S14†).
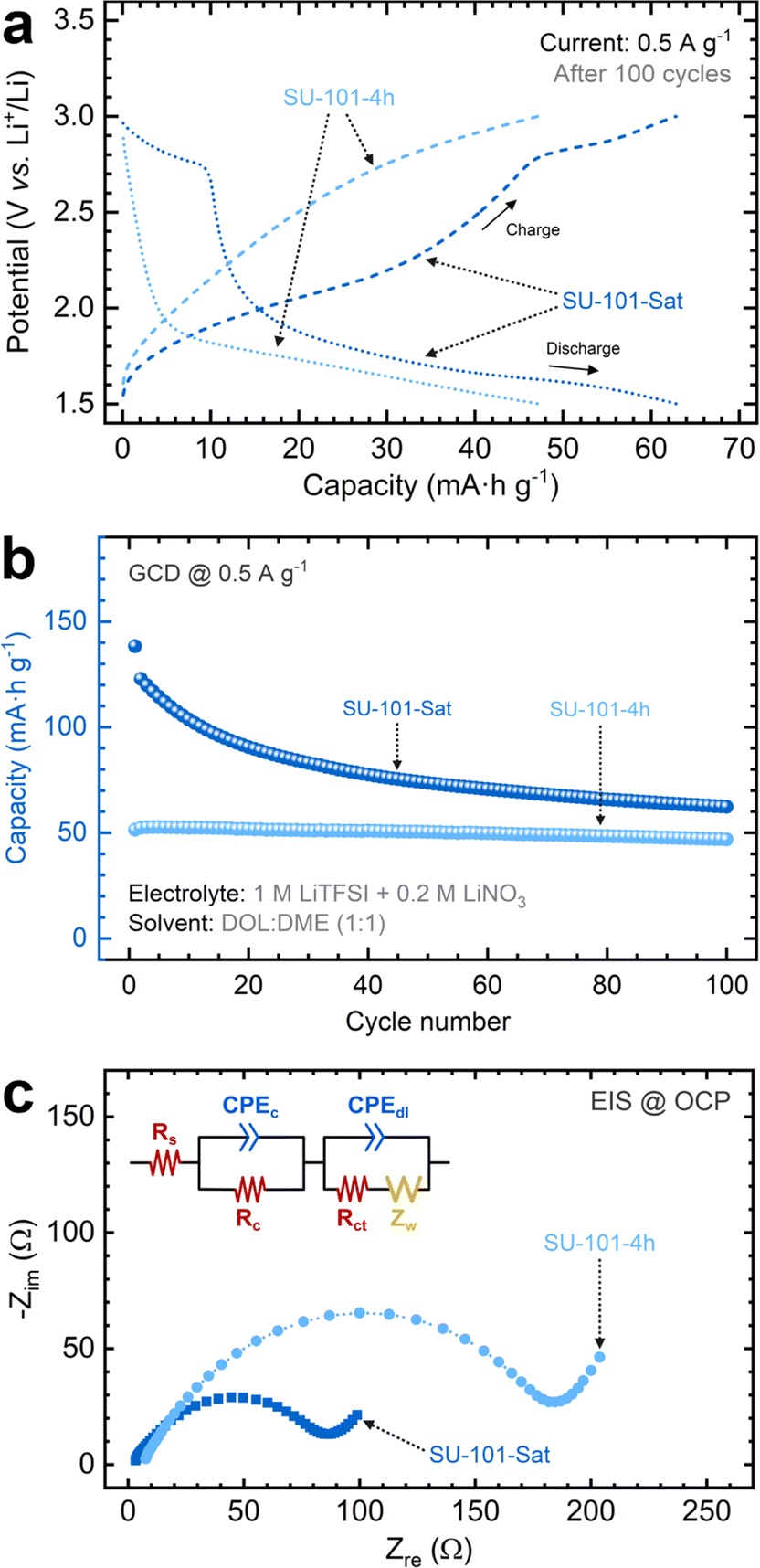 | ||
| Fig. 4 (a) Galvanostatic charge–discharge curves of the SU-101-4h and SU-101-Sat cathodes after cycling at a C/2 rate (0.5 A g−1) between 1.5 and 3.0 V vs. Li/Li+. Charge profiles are depicted with dashed lines, while discharge profiles are depicted with dotted lines. Arrows indicate the charge and discharge directions. (b) Capacity decay tests of SU-101-4h and SU-101-Sat cathodes for 100 cycles. Each sphere represents the capacity measured from one charge–discharge cycle. (c) Nyquist plots after cycling performance tests measured at the open circuit potential (OCP). The inset depicts the equivalent circuit model used to fit Nyquist plots. | ||
EIS analysis was conducted to study the influence of the electrochemical processes on the ionic and electronic resistances of the cells. Fig. 4c shows Nyquist plots for SU-101 cathodes (SU-101-4h and SU-101-Sat), revealing distinct characteristics. A modified equivalent circuit model was applied to derive parameters such as the ohmic resistance (Rs), charge-transfer resistance (Rct), double-layer capacitance (Cdl), and the Warburg impedance (Zw) from the Nyquist plots (Table S3†). The Rs values for the SU-101-Sat and SU-101-4h cathodes after cycling are ∼2.6 and ∼6.8 Ω, respectively, aligning with expectations for this electrolyte composition.37–40 A similar pattern emerges for the Rct values, with the SU-101-Sat cathode demonstrating the lowest resistance at 74.9 Ω. Note, however, that the Rct values increase slightly after cycling, which can be attributed to the formation of Li2S2 and Li2S films on the cathode surfaces. These films act as barriers, being ionically and electronically insulating.18 The intrinsic low electrical conductivity of SU-101-4h and SU-101-Sat, coupled with other polysulfide species dissolved in the electrolyte, further explains this increase in Rct.18,41 However, this increase does not lead to complete battery failure after 100 cycles. Furthermore, the data in Table S3† reveals that both SU-101-4h and SU-101-Sat exhibit comparably similar Cdl values, suggesting similar surface areas, independently of the crystallinity grade of SU-101 (i.e., crystalline vs. amorphous). Finally, both cathodes experience increased Zw values after cycling, indicating higher resistance towards Li ion diffusion and aligning with the Rct trend explained by the surface passivation of insoluble, short-chain polysulfides. However, the SU-101-Sat cathode exhibits a lower Zw value, indicating decreased lithium ion diffusion resistance compared to SU-101-4h.38
Given the superior performance of the SU-101-Sat cathode compared to SU-101-4h, we extended the cycling performance tests to 1000 cycles. CV scans illustrate a marked decrease in intensity for the reduction and oxidation peaks with prolonged cycling, as depicted in Fig. 5a. The peak at ∼1.7 V attributed to confined sulfur decreases after cycling, indicating the consumption of some initially retained sulfur within the amorphous SU-101. Nonetheless, persistent redox signals between 2.3 and 2.0 V denote the ongoing and reversible presence of intermediate polysulfide species during cycling. Fig. 5b demonstrates that prolonged cycling of the SU-101-Sat cathode results in a capacity reduction of approximately 52%. However, charge and discharge plateaus remain distinctly visible. The cycling performance curves shown in Fig. 5c reveal that most capacity decay occurs in the initial 400 cycles without losing coulombic efficiency until the coin cell achieves a stable response (capacity: 34 mA h g−1). Further EIS analysis (Table S4†) reveals a slight increase in the Rct and the Zw values, aligning with the same trend observed after 100 cycles to the formation of insulating films during cycling.
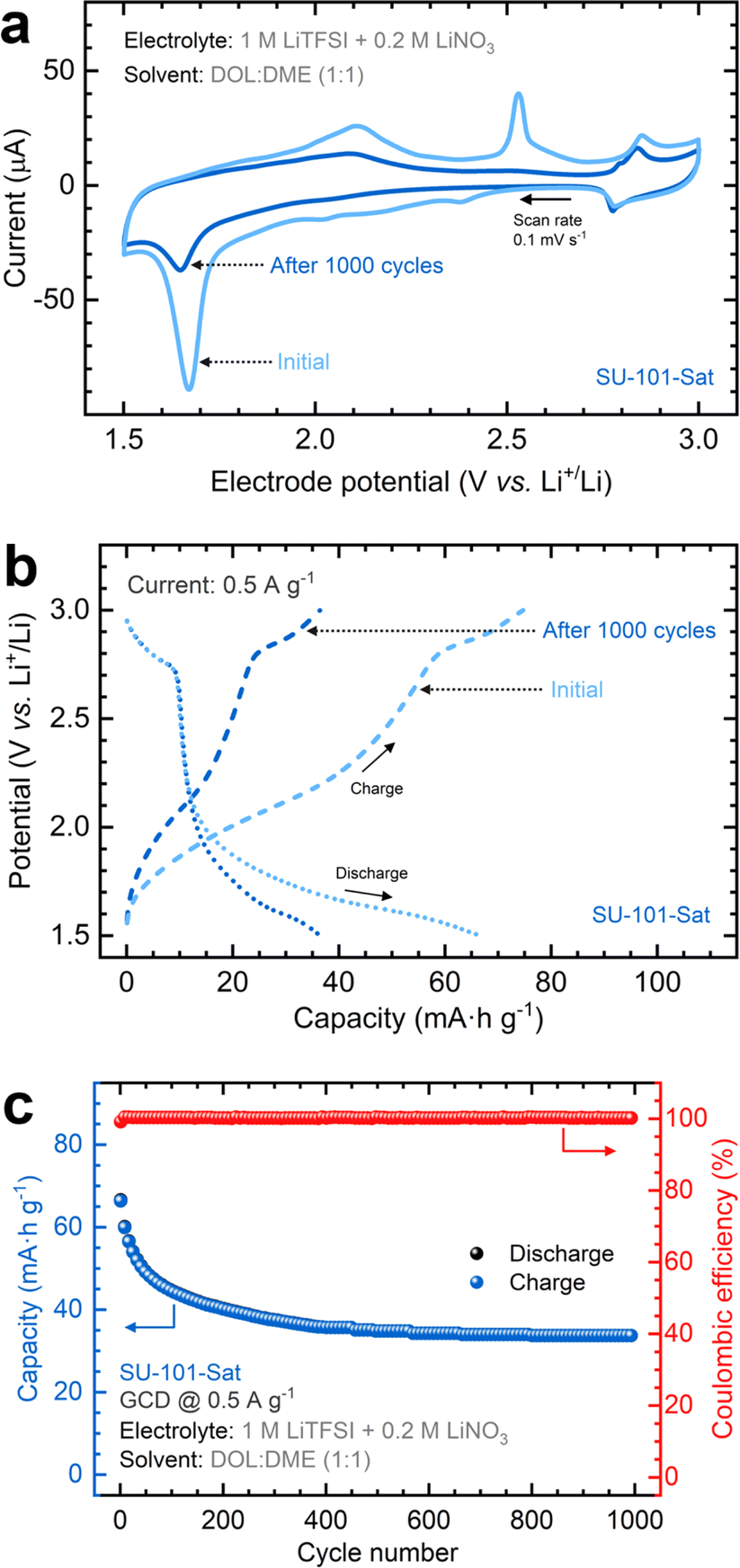 | ||
| Fig. 5 Extended cycling performance tests for the SU-101-Sat MOF cathode: (a) cyclic voltammograms and (b) galvanostatic charge–discharge profiles before and after 1000 cycles. (c) Cycling performance plot showing the capacity on the left axis and the coulombic efficiency on the right axis. Each sphere represents the capacity measured during charge (blue) and discharge (black). Discharge and charge profiles are plotted concurrently. Red spheres represent the coulombic efficiency measured for each charge–discharge cycle. Cycling was conducted at a rate of C/2 (0.5 A g−1) between 1.5 and 3.0 V vs. Li/Li+. Electrolyte: 1 M LiTFSI + 0.2 M LiNO3 in DME:DOL 1:1 solvent. | ||
To evaluate the performance variability of cathodes prepared in different batches, replicate experiments were conducted, with the results included in the ESI (Fig. S15).† Despite the variation in capacity values, particularly in the initial capacity (84.6 mA h g−1 on average), all three cells displayed similar decay patterns, resulting in an average final capacity of 45.6 mA h g−1 after 1000 cycles, corresponding to an average capacity retention of about 54%.
These findings suggest that an excess uptake of polysulfides is initially consumed during the early cycles until a stable solid-electrolyte interphase is formed. Although the SU-101-Sat cathode exhibited an initial capacity decay, it ultimately achieved the highest final capacity and the lowest charge-transfer and diffusion resistances compared to SU-101-4h. However, both materials demonstrated satisfactory reversibility, maintaining average coulombic efficiencies of about 99.8%. This finding is attributed to the control over and release of polysulfide species facilitated by the unique architecture and chemistry of SU-101, which preserved the crystalline structure, particularly achieving only partial amorphization. It is worth noting that, in contrast to conventional practices, the sulfur in the cathode is not deliberately introduced during synthesis. Instead, it is sequestered by SU-101 from the spontaneous transformation of H2S to polysulfides (at room temperature and atmospheric pressure) and subsequently serves as the electroactive element in the Li–S battery.
We also explored using carbonate-based electrolytes, which require specialized sulfur cathodes designed to limit polysulfide formation to short-chain species.42 Coin cells were assembled using the configuration detailed in Table S2,† substituting the ether-based electrolyte with a mixture of EC:DEC (1:1 in volume) in 1 M LiPF6 and FEC as an additive. Fig. S16† illustrates the battery performance results of the SU-101-Sat cathode operating with the carbonate-based electrolyte.
CV scans of the SU-101-Sat cathode in the carbonate electrolyte are quite similar to those in the ether-based electrolyte (Fig. S16a†), particularly with notable peaks between 2.5 and 1.9 V vs. Li/Li+ attributed to the formation of shorter-chain sulfur intermediates and the distinct peak at 1.7 V attributed to sulfur confinement. However, the absence of an oxidation peak at 2.5 V suggests that the gradual oxidation of polysulfides back into S8 was limited. Despite exhibiting similar ohmic resistance, capacitance, and Warburg impedance values to those in the ether electrolyte, EIS analysis reveals a significant increase in the charge-transfer resistance when operating in the carbonate electrolyte (Fig. S16b and Table S5†). Cycling performance tests for over 1000 cycles revealed a battery capacity nearly five times lower than that observed in the ether electrolyte. After an initially higher discharging step (Fig. S16c†), the capacity quickly stabilized around 7 mA h g−1 (Fig. S16d†). Thus, despite a lower experimental capacity, the SU-101-Sat cathode in the carbonate electrolyte displays stable performance over 1000 cycles, with a coulombic efficiency of 99.7%.
The abrupt drop in capacity and subsequent stabilization, along with a significantly lower initial coulombic efficiency (approximately 89.9%, see Fig. S16d†), suggest that the cathode material undergoes an initial chemical transformation that drastically reduces the capacity in carbonate electrolytes. In line with the observed increase in charge-transfer resistance and the absence of a strong oxidation peak at 2.5 V, it seems that not all sulfur intermediates participate in the reaction, and operation in carbonate electrolytes restricts the battery's reversibility. The chemistry and associated transformations in this environment are beyond the scope of our study, and we encourage future investigations to examine the interaction of carbonate electrolytes with MOF cathodes more thoroughly.
Finally, the capacity of the SU-101-Sat cathode (45.6 mA h g−1) is relatively modest compared to other examples of Li–S batteries in the literature (Table S6†). Although this capacity does not set new records, the high cycle life and coulombic efficiency over 1000 cycles offer a valuable opportunity for creating low-energy batteries with long cyclability. These batteries could be suitable for powering small portable devices, satellite components, and weather station sensors.43,44 Hence, this proof-of-concept Li–S battery not only showcases the potential of MOF materials in capturing toxic gas emissions but also demonstrates a successful example of sustainable waste valorization and its successful application in electrochemical energy technologies.
A major concern regarding the use of MOFs in energy technology applications is their economic viability and scalability. Although there is a clear need for more robust and specific techno-economic assessments in the future, using these specific MOF materials could prove competitive for industrial applications. For instance, a bio-derived MIL-160(Al) MOF costs about $55 per kg at a production scale of 100 tons per year, which could be further reduced to $29.5 per kg at 1 kton per year.45 The SU-101 MOF is synthesized from ellagic acid, a linker abundant in strawberries, raspberries, grapes, walnuts, pecans, and pomegranates.46,47 Thus, a bio-derived SU-101 material could potentially be synthesized at a large scale using a similar approach to MIL-160(Al).20 Given the biocompatibility of ellagic acid and the low toxicity of bismuth, the synthesis of bio-derived SU-101 offers a competitive and environmentally friendly approach for commercializing MOF-based technologies.
The waste valorization strategy proposed in our study offers critical environmental benefits. A significant challenge with adsorbent materials used for toxic gas capture is that they merely transfer pollutants into a solid residue, failing to remove them effectively from the environment.48 Our approach not only captures H2S but also repurposes the resulting solid waste into useful materials for energy technology applications, thereby fostering a circular economy where materials are reused rather than discarded, minimizing additional waste generation.49 Furthermore, life cycle assessment (LCA) studies have enhanced our understanding of the environmental impact of MOFs and Li–S battery technologies. LCA studies have identified solvent use during MOF synthesis as a critical hotspot for environmental impact,50 particularly with organic solvents like DMF and THF. In contrast, solvents like ethanol and methanol are considered more environmentally friendly.51,52 Our synthesis approach for SU-101, which occurs in aqueous media, supports the sustainability of our method.
Regarding the environmental impact of Li–S batteries, the electricity source, cycle life, and specific energy density are critical factors in the cradle-to-grave LCA impacts. The cyclability is particularly relevant, as longer lifetimes mean cells need replacing less frequently, which reduces environmental impacts across all categories. The LCA study by Wickerts and coworkers assumes a baseline of 1500 cycles, aligning closely with our reported lifespan of 1000 cycles, which is significantly higher than previous studies (<500 cycles, see Table S6†).53 Moreover, compared to traditional Li-ion batteries, which often contain toxic metals like nickel, cobalt, and manganese, Li–S batteries have a lower environmental footprint.54,55 Given the proven biocompatibility and lower toxicity of the Bi-based SU-101,20 our approach is also expected to have a lower environmental impact than other conventional battery technologies containing more toxic metals.
Following the demonstration of our proof-of-concept study, we encourage the community to explore further the potential of these promising materials in electrochemical energy storage and conversion. There is a substantial opportunity to enhance our understanding of battery performance, economic viability, and the environmental impacts of waste valorization strategies using toxic gas adsorbents and MOF-based cathode materials.
Conclusions
We present an experimental proof-of-concept SU-101-Sat MOF cathode exhibiting remarkable performance similar to standard Li–S batteries reported in the literature. Despite an initial capacity reduction, commonly observed in MOF-derived Li–S batteries during the initial conditioning phase, this cathode exhibited a stable performance and maintained nearly 99.8% coulombic efficiency across 1000 cycles. This resilience is attributed to the controlled release of polysulfides, a characteristic feature facilitated by the chemical composition of SU-101 (i.e., Bi(III) metal centers) and the unique chemical transformation of H2S to polysulfides at room temperature and atmospheric pressure by the SU-101 MOF. These findings highlight the potential of MOF materials to spontaneously transform toxic H2S emissions into polysulfides independently of fully maintaining the crystalline structure of the framework. This approach is a promising candidate for implementing toxic waste valorization strategies and subsequent applications in electrochemical energy storage applications.Author contributions
R.A. Marquez: conceptualization, investigation, methodology, formal analysis, visualization, writing – original draft, project administration. J. L. Obeso: investigation, methodology, formal analysis, visualization, writing – original draft. R. R. Vaidyula: investigation, methodology, writing – review & editing. V. B. López-Cervantes: investigation, methodology, writing – review & editing. R. A. Peralta: conceptualization. P. Marín Rosas: investigation. J. A. de los Reyes: resources, funding acquisition. C. B. Mullins: writing – review & editing, resources, funding acquisition, project administration. I. A. Ibarra: writing – original draft, resources, funding acquisition, project administration. All authors have approved the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge PAPIIT-UNAM (Grant IN202820), Mexico and the Welch Foundation (Grant F-1436), United States, for their generous financial support. R. A. M., V. B. L.-C., and J. L. O. acknowledge CONAHCYT for their PhD scholarship award (919871, 1005649, 1003953). Thanks to U. Winnberg (Pharma View Consulting SC) for scientific discussions and G. Ibarra-Winnberg for conceptualizing the design of this contribution.References
- A. Cincinelli and T. Martellini, Int. J. Environ. Res. Public Health, 2017, 14, 1286 CrossRef PubMed.
- Z. Jianwen, L. Da and F. Wenxing, J. Hazard. Mater., 2014, 264, 350–362 CrossRef PubMed.
- S. Çetinyokuş, Process Saf. Prog., 2023, 42, 469–480 CrossRef.
- S. L. Malone Rubright, L. L. Pearce and J. Peterson, Nitric Oxide, 2017, 71, 1–13 CrossRef CAS PubMed.
- D. H. Truong, M. A. Eghbal, W. Hindmarsh, S. H. Roth and P. J. O'Brien, Drug Metab. Rev., 2006, 38, 733–744 CrossRef CAS PubMed.
- A. A. Khan, M. M. Schuler, M. G. Prior, S. Yong, R. W. Coppock, L. Z. Florence and L. E. Lillie, Toxicol. Appl. Pharmacol., 1990, 103, 482–490 CrossRef CAS PubMed.
- M. S. Shah, M. Tsapatsis and J. I. Siepmann, Chem. Rev., 2017, 117, 9755–9803 CrossRef CAS PubMed.
- E. Martínez-Ahumada, A. López-Olvera, V. Jancik, J. E. Sánchez-Bautista, E. González-Zamora, V. Martis, D. R. Williams and I. A. Ibarra, Organometallics, 2020, 39, 883–915 CrossRef.
- J. G. Flores, J. A. Zárate-Colín, E. Sánchez-González, J. R. Valenzuela, A. Gutiérrez-Alejandre, J. Ramírez, V. Jancik, J. Aguilar-Pliego, M. C. Zorrilla, H. A. Lara-García, E. González-Zamora, G. Guzmán-González, I. González, G. Maurin and I. A. Ibarra, ACS Appl. Mater. Interfaces, 2020, 12, 18885–18892 CrossRef CAS.
- J. L. Obeso, D. R. Amaro, C. V. Flores, A. Gutiérrez-Alejandre, R. A. Peralta, C. Leyva and I. A. Ibarra, Coord. Chem. Rev., 2023, 485, 215135 CrossRef CAS.
- R. Fang, S. Zhao, Z. Sun, D. Wang, H. Cheng and F. Li, Adv. Mater., 2017, 29, 1606823 CrossRef PubMed.
- M. Zhao, B. Li, H. Peng, H. Yuan, J. Wei and J. Huang, Angew. Chem., Int. Ed., 2020, 59, 12636–12652 CrossRef CAS PubMed.
- S. Ponnada, M. S. Kiai, D. B. Gorle, S. Rajagopal, S. Andra, A. Nowduri and K. Muniasamy, Energy Fuels, 2021, 35, 11089–11117 CrossRef CAS.
- Z. Wang, X. Li, Y. Cui, Y. Yang, H. Pan, Z. Wang, C. Wu, B. Chen and G. Qian, Cryst. Growth Des., 2013, 13, 5116–5120 CrossRef CAS.
- W. Bao, Z. Zhang, Y. Qu, C. Zhou, X. Wang and J. Li, J. Alloys Compd., 2014, 582, 334–340 CrossRef CAS.
- Z. Zhang, Y. An, J. Feng, L. Ci, B. Duan, W. Huang, C. Dong and S. Xiong, Mater. Lett., 2016, 181, 340–344 CrossRef CAS.
- Z. Zhang, H. Yoshikawa and K. Awaga, Chem. Mater., 2016, 28, 1298–1303 CrossRef CAS.
- X. Huang, T. Qiu, X. Zhang, L. Wang, B. Luo and L. Wang, Mater. Chem. Front., 2020, 4, 2517–2547 RSC.
- X. Liu, S. Wang, A. Wang, Z. Wang, J. Chen, Q. Zeng, P. Chen, W. Liu, Z. Li and L. Zhang, J. Mater. Chem. A, 2019, 7, 24515–24523 RSC.
- E. S. Grape, J. G. Flores, T. Hidalgo, E. Martínez-Ahumada, A. Gutiérrez-Alejandre, A. Hautier, D. R. Williams, M. O'Keeffe, L. Öhrström, T. Willhammar, P. Horcajada, I. A. Ibarra and A. K. Inge, J. Am. Chem. Soc., 2020, 142, 16795–16804 CrossRef CAS PubMed.
- J. Rodriguez-Pereira, S. A. Rincón-Ortiz and R. Ospina, Surf. Sci. Spectra, 2020, 27, 024001 CrossRef CAS.
- V. H. Nguyen, T. D. Nguyen and T. Van Nguyen, Top. Catal., 2020, 63, 1109–1120 CrossRef CAS.
- M. Fantauzzi, B. Elsener, D. Atzei, A. Rigoldi and A. Rossi, RSC Adv., 2015, 5, 75953–75963 RSC.
- L. Hamon, C. Serre, T. Devic, T. Loiseau, F. Millange, G. Férey and G. D. Weireld, J. Am. Chem. Soc., 2009, 131, 8775–8777 CrossRef CAS PubMed.
- P. K. Allan, P. S. Wheatley, D. Aldous, M. I. Mohideen, C. Tang, J. A. Hriljac, I. L. Megson, K. W. Chapman, G. D. Weireld, S. Vaesen and R. E. Morris, Dalton Trans., 2012, 41, 4060–4066 RSC.
- J. A. Zárate, E. Sánchez-González, T. Jurado-Vázquez, A. Gutiérrez-Alejandre, E. González-Zamora, I. Castillo, G. Maurin and I. A. Ibarra, Chem. Commun., 2019, 55, 3049–3052 RSC.
- Y.-H. Zou, X. Wang, F. Ning, J. Yi and Y. Liu, Sep. Purif. Technol., 2023, 317, 123806 CrossRef CAS.
- Q. Zhang, Y. Liu, Z. Wang, P. Wang, Z. Zheng, H. Cheng, X. Qin, X. Zhang, Y. Dai and B. Huang, J. Colloid Interface Sci., 2022, 617, 578–584 CrossRef CAS PubMed.
- F. Liu, Q. Xiao, H. B. Wu, F. Sun, X. Liu, F. Li, Z. Le, L. Shen, G. Wang, M. Cai and Y. Lu, ACS Nano, 2017, 11, 2697–2705 CrossRef CAS.
- X. Liang, C. Hart, Q. Pang, A. Garsuch, T. Weiss and L. F. Nazar, Nat. Commun., 2015, 6, 5682 CrossRef PubMed.
- Z. Zhang, J.-N. Wang, A.-H. Shao, D.-G. Xiong, J.-W. Liu, C.-Y. Lao, K. Xi, S.-Y. Lu, Q. Jiang, J. Yu, H.-L. Li, Z.-Y. Yang and R. V. Kumar, Sci. China Mater., 2020, 63, 2443–2455 CrossRef CAS.
- Y. Wu, T. Momma, S. Ahn, T. Yokoshima, H. Nara and T. Osaka, J. Power Sources, 2017, 366, 65–71 CrossRef CAS.
- G. A. Bukhtiyarova, V. I. Bukhtiyarov, N. S. Sakaeva, V. V. Kaichev and B. P. Zolotovskii, J. Mol. Catal. A: Chem., 2000, 158, 251–255 CrossRef CAS.
- R. Malakooti, L. Cademartiri, Y. Akçakir, S. Petrov, A. Migliori and G. A. Ozin, Adv. Mater., 2006, 18, 2189–2194 CrossRef CAS.
- A. López-Olvera, J. G. Flores, J. Aguilar-Pliego, C. K. Brozek, A. Gutiérrez-Alejandre and I. A. Ibarra, Chem. Mater., 2021, 33, 6269–6276 CrossRef.
- R. Demir-Cakan, M. Morcrette, F. Nouar, C. Davoisne, T. Devic, D. Gonbeau, R. Dominko, C. Serre, G. Férey and J.-M. Tarascon, J. Am. Chem. Soc., 2011, 133, 16154–16160 CrossRef CAS PubMed.
- K. Zou, X. Chen, W. Jing, X. Dai, P. Wang, Y. Liu, R. Qiao, M. Shi, Y. Chen, J. Sun and Y. Liu, Energy Storage Mater., 2022, 48, 133–144 CrossRef.
- Z. Li, K. Hu, J. Guo, Y. Zhang, M. Zhang and J. Lian, J. Mater. Sci., 2023, 58, 2234–2248 CrossRef CAS.
- T. Li, C. Ma, Y. Li, F. Tu, C. Jiao, Z. Li and S. Yao, Ionics, 2022, 28, 1701–1711 CrossRef CAS.
- X. Zhou, X. Luo, H. Wang, J. Yang, H. Xu, M. Jia and J. Tang, J. Mater. Sci., 2019, 54, 9622–9631 CrossRef CAS.
- S. Suriyakumar, G. J. Rani and A. M. Stephan, Ionics, 2020, 26, 2201–2210 CrossRef CAS.
- X. Li, M. Banis, A. Lushington, X. Yang, Q. Sun, Y. Zhao, C. Liu, Q. Li, B. Wang, W. Xiao, C. Wang, M. Li, J. Liang, R. Li, Y. Hu, L. Goncharova, H. Zhang, T.-K. Sham and X. Sun, Nat. Commun., 2018, 9, 4509 CrossRef.
- S. Dörfler, S. Walus, J. Locke, A. Fotouhi, D. J. Auger, N. Shateri, T. Abendroth, P. Härtel, H. Althues and S. Kaskel, Energy Technol., 2021, 9, 2000694 CrossRef PubMed.
- A. Fotouhi, D. J. Auger, L. O'Neill, T. Cleaver and S. Walus, Energies, 2017, 10, 1937 CrossRef.
- M. I. Severino, E. Gkaniatsou, F. Nouar, M. L. Pinto and C. Serre, Faraday Discuss., 2021, 231, 326–341 RSC.
- K. R. Määttä-Riihinen, A. Kamal-Eldin and A. R. Törrönen, J. Agric. Food Chem., 2004, 52, 6178–6187 CrossRef PubMed.
- M. N. Clifford and A. Scalbert, J. Sci. Food Agric., 2000, 80, 1118–1125 CrossRef CAS.
- E.-S. M. El-Sayed and D. Yuan, Green Chem., 2020, 22, 4082–4104 RSC.
- S. Geisendorf and F. Pietrulla, Thunderbird Int. Bus. Rev., 2018, 60, 771–782 CrossRef.
- P. G. Jessop and A. R. MacDonald, Green Chem., 2023, 25, 9457–9462 RSC.
- C. A. Grande, R. Blom, A. Spjelkavik, V. Moreau and J. Payet, Sustainable Mater. Technol., 2017, 14, 11–18 CrossRef CAS.
- H. U. Escobar-Hernandez, Y. Quan, M. I. Papadaki and Q. Wang, ACS Sustainable Chem. Eng., 2023, 11, 4219–4225 CrossRef CAS.
- S. Wickerts, R. Arvidsson, A. Nordelöf, M. Svanström and P. Johansson, ACS Sustainable Chem. Eng., 2023, 11, 9553–9563 CrossRef CAS.
- G. Benveniste, A. Sánchez, H. Rallo, C. Corchero and B. Amante, Resour. Conserv. Recycl. Adv., 2022, 15, 200086 Search PubMed.
- Y. Deng, J. Li, T. Li, X. Gao and C. Yuan, J. Power Sources, 2017, 343, 284–295 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental details and procedures, supporting figures and tables, additional characterization data. See DOI: https://doi.org/10.1039/d4ta03620d |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2024 |