
A new MOF@bioactive glass composite reinforced with silver nanoparticles – a new approach to designing antibacterial biomaterials†
Marzena
Fandzloch
 *a,
Adam W.
Augustyniak
b,
Joanna
Trzcińska-Wencel
c,
Patrycja
Golińska
c and
Katarzyna
Roszek
c
*a,
Adam W.
Augustyniak
b,
Joanna
Trzcińska-Wencel
c,
Patrycja
Golińska
c and
Katarzyna
Roszek
c
aInstitute of Low Temperature and Structure Research, Polish Academy of Sciences, Okólna 2, 50-422, Wrocław, Poland. E-mail: m.fandzloch@intibs.pl
bFaculty of Chemistry, University of Wrocław, F. Joliot-Curie 14, 50-383, Wrocław, Poland
cFaculty of Biological and Veterinary Sciences, Nicolaus Copernicus University in Toruń, Lwowska 1, 87-100, Toruń, Poland
First published on 18th June 2024
Abstract
Multifunctional materials that combine antimicrobial properties with the ability to stimulate bone formation are needed to overcome the problem of infected bone defects. As a novel approach, a new composite based on bioactive glass nanoparticles in a simple system of SiO2–CaO (BG) coated with NH4[Cu3(μ3-OH)(μ3-4-carboxypyrazolato)3] (Cu-MOF) with additionally anchored silver nanoparticles (AgNPs) was proposed. Ag@Cu-MOF@BG obtained by the spin coating approach in the form of a disc was characterized using PXRD, ATR-FTIR, XPS, ICP-OES, and TEM. Importantly, the material retained its bioactivity, although ion exchange in the bioactive glass administered as a disc is limited. Hydroxyapatite (HA) formation was identified in TEM images after 7 days of immersion of the composite in a physiological-like buffer (pH 7.4, 37 °C). The Cu and Ag contents of Ag@Cu-MOF@BG were as low as 0.013 and 0.018 wt% respectively, but the slow release of the AgNPs ensured its antibacterial nature. Ag@Cu-MOF@BG exhibited antibacterial activity against all tested bacteria (E. coli, S. aureus, P. aeruginosa, and K. pneumoniae) with the diameter of the inhibition zones of their growth between 8 and 10 mm and the reduction index determined to be ≥3. Moreover, the biocompatibility of the new composite has been demonstrated, as shown by cell culture assays with human dermal fibroblasts (HDFs). The results from the migration test also proved that the HDF cell's phenotypic properties were not changed, and the cell adhesion and migration ability were the same as in control indirect assays.
Introduction
Infected bone defects are a major challenge in orthopedic treatment, as they are chronic conditions in which pathogenic infection severely compromises the regenerative capacity of local tissues and bone.1,2Infectious diseases can spread and develop through open fractured sites or blood transmission from adjacent sites or implants. Excessive inflammatory responses prevent bone tissue regeneration and significantly interfere with the bone healing process.3 In fact, bacteria can damage the extracellular matrix, inhibit osteogenic differentiation and function, stimulate the production of osteoclasts, cause excessive bone resorption and impaired osteogenic function, and even cause delayed healing and fracture failure.4
Traditional treatment strategies for infected bone defects include thoroughly clearing the wound, implanting antibiotic-impregnated cement spacers, and administering antibiotics.5 However, the efficacy of systemic antibiotic administration is significantly undermined by the infection-induced disruption of the local blood supply and the resulting onset of osteonecrosis. This severely limits the effective concentration of antibiotics at the site of infection. In addition, the prolonged and widespread use of antibiotics has led to the gradual evolution of bacterial resistance and the emergence of highly resistant strains of bacteria.5 Therefore, alternative materials with antibacterial properties are being investigated. Among them, metal–organic frameworks (MOFs) are becoming increasingly prominent in this aspect.6 These highly designable structure and functional materials, due to their high porosity (e.g. MIL-53(Fe), MOF-5, or ZIF-8) have been proposed as antibacterial drug delivery systems.7 On the other hand, MOFs can lead to the slow release of metal ions, giving rise to antibacterial behavior (e.g. Ti-MIL-125-NH2,8 Ag-MOFs,7 and Zn-containing MOFs7).
There is also an increasing number of Cu-MOFs being tested and used for their antimicrobial activity.9–11 A well-known representative is [Cu3(BTC)2]n (H3BTC = 1,3,5-benzenetricarboxylic acid), commonly known as Cu-HKUST-1 or MOF-199, which has remarkable antibacterial activity against S. aureus and E. coli.11–15 The progressive degradation of Cu-MOFs and the leaching of Cu2+ ions as well as the ligand, e.g.L-glutamic acid from [Cu(glum)]n,16 are likely to disrupt the bacterial cell membrane. In addition, highly synergistic antibacterial activity due to ROS generation and delayed Cu2+ release was reported for [Cu2(cit)(H2O)2]n nano-MOF (cit = citrate), which was effective against E. coli and B. subtilis.17 Interestingly, the contact of bacteria with the MOF surface without metal ion release is another possible mechanism for the antibacterial activity of Cu-MOFs, such as for [Cu2(Glu)2(μ-L)]·x(H2O) containing glutarates (Glu) and bipyridyl ligands (L).18 These MOFs exhibit strong antibacterial activities against both Gram-positive bacteria (S. aureus and MRSA) and Gram-negative bacteria (E. coli, K. pneumonia, and P. aeruginosa) at very low bactericidal concentrations. Meanwhile, Sheta et al. presented nanoparticles of the MOF [{Cu(SB)(H2O)2}(H2O)4]n, where H2SB = Schiff bases, with a size of 7–19 nm suitable to penetrate the bacterial cell through nanometric pores in the membrane by a passive diffusion mechanism.7,19
One of the strategies to further enhance the antimicrobial efficacy of Cu-MOFs could be the loading with AgNPs. To date, Cu-BTC20 or polyCu-MOF (a Cu-based polymer-metal–organic framework)21 have been efficiently used as carriers for silver nanoparticles. The controlled release of Ag+ and Cu2+ metal ions significantly affected their bactericidal performance against S. aureus and E. coli.
The above data indicate that Cu-MOFs, and in particular Ag@Cu-MOFs, can be effective antibacterial platforms and further extend the great potential of MOFs in the application of bone infections. The treatment of bone infections, however, is very demanding and requires multifunctional materials that combine therapy and regeneration.22 These properties, known as theragenerative or therepair, can be achieved by MOFs when combined with biomaterials.11,23,24 Among them, bioactive glasses seem to be very promising for orthopedic applications. Due to the formation of a bone-like hydroxyapatite layer on the surface, this type of material can form strong bonds with bone or soft tissue.25–28 In addition, specific concentrations of soluble Si and Ca ions released from bioactive glasses can stimulate the expression of several genes involved in osteogenesis and angiogenesis, resulting in rapid bone regeneration.29,30
To the best of our knowledge, two examples of composites based on bioactive glasses and MOFs have been developed to date to reduce bone infection.23,24 Both are based on zeolitic imidazolate framework-8 (ZIF-8) and ternary bioactive glass (SiO2–CaO–P2O5). One of these materials was obtained by deposition of vancomycin-loaded ZIF-8 (ZIF-8@VAN) on 3D-printed bioactive glass scaffolds. In addition to promoting the proliferation and osteogenic differentiation of rat bone marrow mesenchymal stem cells, this material has shown antibacterial activity against S. aureus.23 Meanwhile, Ahmed et al. proposed the use of a biocompatible composite based on a system of 60SiO2–10P2O5–(30−x)CaO–(x)ZIF-8 nanoparticles (x = 0 and 0.8 mol%) to solve the problem of infection related to orthopedic implant surgery.24
Recently, we demonstrated that the sol–gel-derived bioactive glasses in a simple binary system, depending on the size or amount of CaO, greatly enhanced the rate of hydroxyapatite formation.31 Based on bio- and hemocompatibility results, one of the nanoparticles of bioactive glasses (B_78S, herein referred to as BG) was found to be the most outstanding representative from a bioapplication viewpoint.
Taking into account this background and considering our previous results herein we present the use of bioactive glass nanoparticles (BG) as a component of a new MOF-based antibacterial composite. For the first time, we have used this bioactive glass in the structured form (discs), coated with a thin layer of a selected Cu-based MOF, namely NH4[Cu3(μ3-OH)(μ3-4-carboxypyrazolato)3] (denoted as Cu-MOF), with anchored silver nanoparticles to enhance the antibacterial effect. The physicochemical characterization of the new Ag@Cu-MOF@BG composite was extended to include bioactivity, in vitro biocompatibility, and antibacterial studies.
Experimental
Materials and physical measurements
All chemicals were commercially available and used without further purification. Calcium hydroxide (≥96%), TEOS (≥99%), copper(II) nitrate trihydrate (99–104%), and silver nitrate (≥99.0%) were purchased from Sigma Aldrich. Phosphate-buffered saline, Dulbecco's formula (DPBS, 10×), and polyethylene glycol 400 (PEG 400) were purchased from Alfa Aesar. Hydrochloric acid (35–38%), ethanol (96%), methanol, ammonium hydroxide solution (25%), and acetone were purchased from Avantor Performance Materials Poland S.A. 1H-Pyrazole-4-carboxylic acid (97%) was purchased from Thermo Fisher Scientific. Hydrofluoric acid (40%) and nitric acid (65%) were purchased from Chempur (Poland).Cu-MOF, Ag@Cu-MOF and BG were analyzed by powder X-ray diffraction (PXRD) using an X'Pert PRO diffractometer (PANalytical) with Cu Kα radiation (λ = 1.5418 Å). The mid-infrared spectra in the region of 4000–530 cm−1 were acquired in attenuated total reflectance (ATR) mode using a Thermo Scientific Nicolet Summit X FTIR spectrometer. In each case, the discs were ground in a mortar before the measurement. The specific surface area of Ag@Cu-MOF was determined based on N2 sorption measurements. The N2 adsorption–desorption isotherms were obtained at 77 K using a Micromeritics 3Flex surface characterization analyzer. Prior to isotherm acquisition, the materials were activated and outgassed (150 °C, 1.3 kPa) for 12 h. MicroActive software was used to determine specific surface area according to the Brunauer–Emmett–Teller (BET) method and total pore volume at a relative pressure (p/p0) = 0.9. Transmission electron microscopy (TEM) images were acquired using a FEI Tecnai G2 20 X-TWIN microscope. Silver particle size was calculated from the size of 100 particles determined using ES Vision software from TEM images. Scanning electron microscopy (SEM) images were recorded using a Hitachi S-3400N microscope. The composition of powders and filtrates was characterized using an iCAP™ 7400 ICP-OES analyzer. To quantify the content of elements (Ca, Si, P, Cu, Ag) by ICP-OES, 1–3 mg of powders were degraded in the mixture of HF/HCl (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, 6 mL) (for BG and Ag@Cu-MOF@BG) or 2 mL of HCl (for Cu-MOF and Ag@Cu-MOF). Afterward, the samples were diluted with distilled water. XPS data were collected using a Kratos X-ray photoelectron spectrometer. The binding energy was calibrated according to the C 1s peak of 285 eV. The Ag 3d spectral regions were peak-fitted through CasaXPS processing software. BG and Ag@Cu-MOF@BG discs (Fig. S1†) of 8 mg powder with a diameter of 5 mm were prepared using an evacuable Pellet Die (Specac) and hydraulic press (Sirio) with a pressure of 1 ton. For thin layer deposition, KLM Spin-Coater SCI-40 (Schaefer Tec) was used.
:1, 6 mL) (for BG and Ag@Cu-MOF@BG) or 2 mL of HCl (for Cu-MOF and Ag@Cu-MOF). Afterward, the samples were diluted with distilled water. XPS data were collected using a Kratos X-ray photoelectron spectrometer. The binding energy was calibrated according to the C 1s peak of 285 eV. The Ag 3d spectral regions were peak-fitted through CasaXPS processing software. BG and Ag@Cu-MOF@BG discs (Fig. S1†) of 8 mg powder with a diameter of 5 mm were prepared using an evacuable Pellet Die (Specac) and hydraulic press (Sirio) with a pressure of 1 ton. For thin layer deposition, KLM Spin-Coater SCI-40 (Schaefer Tec) was used.
Synthesis of materials
:15, 30 mL) and left for 3 days while dark blue crystals were formed. Afterward, these crystals were centrifuged, washed with distilled water (2 × 20 mL) and ethanol (96%) (1 × 20 mL), and dried for 24 h at 50 °C. ICP-OES: Cu = 22.45 ± 0.03 wt%.
:1, 20 mL). This suspension was then shaken on the platform at 600 rpm for 24 h at room temperature. Afterward, the precipitate was centrifuged, washed with methanol (1 × 20 mL), and dried at room temperature for 24 h. ICP-OES: Cu = 18.63 ± 0.17 wt%, Ag = 12.79 ± 1.05 wt%.
Bioactivity test
Hydroxyapatite (HA) forming ability was determined in a physiological-like buffered (pH = 7.4) solution (DPBS). To maintain the previously established protocol,31 each disc was immersed in 5.333 mL DPBS in clean and sterile polyvinyl chloride bottles and placed in an incubator at a controlled temperature of 37 °C with continuous agitation (100 rpm) for 1, 4, 24, 72 h and 7 d. At each selected time, the disc was removed from DPBS, and changes in the pH of such filtrate were monitored. Subsequently, the disc was washed with distilled water (3 × 0.5 mL) and dried at 50 °C for 24 h. Each test was carried out in duplicate. The HA formation on the surface of Ag@Cu-MOF@BG and BG was determined by ATR-FTIR spectroscopy and TEM imaging. Changes in the composition of the powders after a 7-day incubation were monitored by ICP-OES.Microbial strains and bacterial inoculum preparation
The antimicrobial activity of tested samples was investigated towards Gram-negative: Escherichia coli ATCC 25922, Klebsiella pneumoniae ATCC 700603, Pseudomonas aeruginosa ATCC 10145, and Gram-positive: Staphylococcus aureus ATCC 25923 bacteria, and yeast Candida albicans ATCC 10231. All strains were purchased from the American Type Culture Collection (Manassas, VA, USA). The microbial suspensions with a density of 0.5 McFarland (1.5 × 108 CFU per mL) were prepared in sterile distilled water from cultures of bacterial strains and C. albicans grown in tryptic soy broth (TSB, Becton Dickinson) and Sabouraud dextrose broth (SDB, Becton Dickinson), respectively, for 24 h at 37 °C under shaking conditions (120 rpm).Determination of minimal inhibitory and minimal biocidal concentrations of Ag@Cu-MOF
The broth microdilution method was used to determine the minimal inhibitory concertation (MIC) of the test material in concentration ranges of 0.016–2048 mg mL−1 according to the Clinical Laboratory Standard Institute method.33 Ag@Cu-MOF was 2-fold diluted in the appropriate medium in sterile 96-well plates and inoculated with 1.5 μL of microbial suspension. The final volume of sample in each well was 150 μL, while the final concentration of microorganisms was equal to 1.5 × 106 CFU per mL. The cultures were incubated at 37 °C under shaking conditions (120 rpm) for 24 h. The MIC values were defined as the lowest concentration of Ag@Cu-MOF for which there was no visible microbial growth in the wells after incubation.To assess the minimal biocidal concentration (MBC) of Ag@Cu-MOF, 100 μL of the tested suspensions from wells with a concentration of ≥MIC were spread onto sterile tryptic soy agar (TSA, Becton Dickinson) or Sabouraud dextrose agar (SDA, Becton Dickinson) in Petri plates and incubated at 37 °C for 24 h. The lowest concentration of compounds that inhibited microbial growth ≥99.9% was determined as MBC values. All tests were performed in triplicate; the positive (inoculated medium) and negative (non-inoculated medium) controls were provided.34
Antimicrobial activity of Ag@Cu-MOF@BG
For evaluation of the antimicrobial efficiency of Ag@Cu-MOF@BG, the disc diffusion method was performed according to the protocol recommended by the Clinical and Laboratory Standards Institute.35 The Ag@Cu-MOF@BG discs were sterilized with a UVC lamp for 15 minutes on both sides and aseptically placed on the surface of TSA or SDA (for bacteria and yeast, respectively) inoculated with 100 μL of microbial inoculum (1.5 × 106 CFU per mL). Plates were incubated at 37 °C for 24 h. The diameter of the growth inhibition zones around discs was measured in mm.The biocidal activity of Ag@Cu-MOF@BG discs was determined by placing them in 1 mL of the microbial inoculum (approx. 1.5 × 106 CFU per mL) in 24-well plates for 24 h at 37 °C under shaking conditions (100 rpm). After incubation, the suspensions were collected from each well into sterile 2 mL Eppendorf tubes and serially 10-fold diluted. The aliquot (100 μL) of each sample dilution was spread aseptically on the surface of TSA or SDA medium in Petri plates and incubated for 24 h at 37 °C. The colonies were counted and the reduction factor (R) was calculated according to the formula:
| R = Ut − At |
The biocidal activity was determined if R ≥ 2 (99% reduction of microbial growth).
In vitro cytotoxicity assays
The cytotoxicity studies were assessed using human dermal fibroblasts (HDF, Biokom, Poland) grown in DMEM-LG (Dulbecco's modified Eagle's medium, low glucose) supplemented with 10% fetal bovine serum (FBS) and 1% antibiotics (penicillin, streptomycin).For the direct cytotoxicity test, 10 μL of suspension containing approximately 5 × 104 cells in culture media were seeded on each of the tested materials, i.e., different specimens in separate wells of a 12-well plate. The cells were left for 3 h for adhesion and then the culture medium was added and incubated for 24, 48, and 72 h. In an indirect test, the examined specimens were immersed in culture media for 72 h and then the suspension, as prepared, was added to a HDF cell culture seeded 24 hours prior to evaluation in a 12-well plate. The cell viability in both tests was assessed based on MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) (Sigma-Aldrich, Germany) assay, which shows the ability of the cells to reduce MTT. The absorbance of reduced formazan was measured at 570 nm using a Synergy HT Multi-detection reader (BioTek Instruments, Winooski, VT, USA).
In order to determine the rate of migration, HDF cells were seeded into 24-well plates and incubated for 24 h, then a scratch wound was created using a sterile pipette tip. Cells were washed once with the growth medium, and then the growth medium was added to the control sample, while the test samples contained medium obtained after 72 h-incubation with specimens (as in the indirect viability test). At time 0, after 6, 24, and 48 h, pictures were taken to measure the width of the wound using Motic Images Software. On the basis of the obtained results, the values were calculated for individual samples as (%) of the wound width relative to the control sample.
Results and discussion
Synthesis and structural characterization
Bioactive glass with a composition of 72SiO2–28CaO wt% was prepared by the sol–gel method using calcium hydroxide as the calcium source. As we have confirmed in previous studies, this network modifier, used instead of traditional calcium nitrate tetrahydrate, enables the reduction of the deviation between the nominal and experimental glass composition.36 This is noteworthy because the appropriate content of CaO is primarily responsible for the bioactivity of glasses and favors the nucleation of apatite onto the silica gel layer initially formed at the glass surface.37 The inherent amorphous structure of the obtained BG was evaluated by PXRD analysis (Fig. 1a). Meanwhile, the nanometric size of the spherical particles, typically less than 70 nm, was confirmed by TEM imaging (Fig. 1b).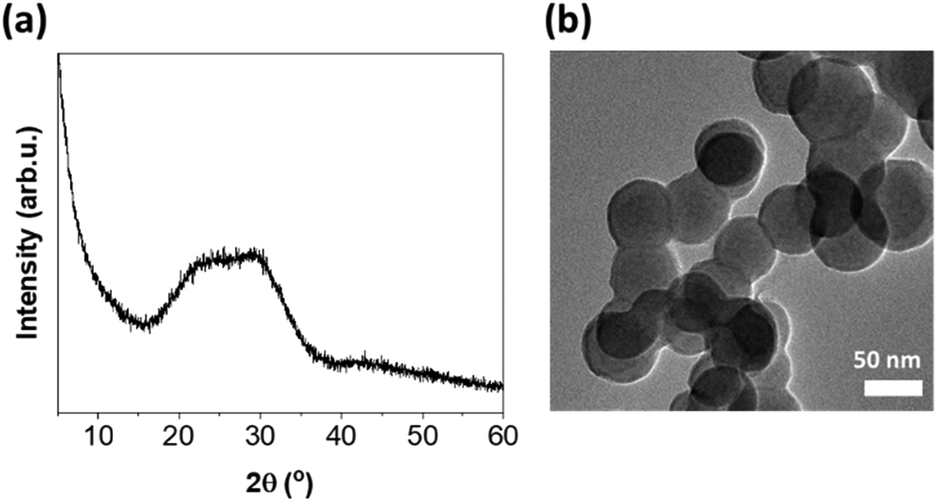 | ||
| Fig. 1 (a) PXRD and (b) TEM image of BG. | ||
The Cu-based MOF chosen as a component of the new composite is an anionic 3D porous framework composed of trinuclear Cu3(μ3-OH) clusters linked to six others by μ3-4-carboxypyrazolato bridges.32 This forms tetrahedral cages containing two extra-framework NH4+ cations and crystallization water molecules. Interestingly, the ion-exchange processes of the extra-framework cations can modulate the textural properties and thus influence the adsorption selectivity of various separation processes of gases and vapors or open up new application possibilities.32,38 The synthesized NH4[Cu3(μ3-OH)(μ3-4-carboxypyrazolato)3] MOF was systematically characterized by PXRD and ATR-FTIR, confirming the purity of the isolated material (Fig. S2†). The treatment of this Cu-MOF with silver nitrate in a 3-fold molar excess in a mixture of methanol and water resulted in the formation of silver nanoparticles (AgNPs) anchored to the Cu-MOF. They were identified by the inter-planar spacing d = 0.232 nm corresponding to the (111) plane of the fcc silver crystals (Fig. S3†). Notably, AgNPs (10 ± 3 nm in size) were fairly dispersed on the MOF surface. In addition, its homogeneous distribution was confirmed by EDS mapping (Fig. S4†). The Ag content determined on the basis of ICP-OES was almost 13 wt%. The IR spectrum registered for Ag@Cu-MOF indicated that the in situ formation of AgNPs did not affect the Cu-MOF structure (Fig. S5a†). Further characterization of Ag@Cu-MOF by PXRD did not reveal any characteristic diffraction peaks of AgNPs, probably due to poor crystallinity or small particle size (Fig. S5b†).
On the other hand, the presence of AgNPs had a significant effect on changes in the textural properties of Ag@Cu-MOF compared to pristine Cu-MOF. The N2 adsorption–desorption isotherm recorded for Ag@Cu-MOF (Fig. S6†) was a combination of IUPAC's type I and IV isotherms. The presence of micro- and mesopores was also typical of pristine Cu-MOF.38 However, it can be noted that the anchored AgNPs markedly modulated the porosity of the Cu-MOF. The BET surface area and pore volume decreased twice from 476 m2 g−1 and 0.22 cm3 g−1 for Cu-MOF38 to 230 m2 g−1 and 0.10 cm3 g−1 for Ag@Cu-MOF, respectively.
The resulting Ag@Cu-MOF was deposited onto a disc of BG using a spin coater (Scheme 1 and Fig. S1†). TEM imaging and FFT analysis clearly confirmed the presence of a Cu-MOF layer with embedded Ag nanoparticles on spherical BG nanoparticles (Fig. 2a–c). AgNPs were identified again by the inter-planar spacing d = 0.234 nm. Furthermore, the EDS mapping showed a uniform distribution of Ag and Cu along with Ca and Si throughout the measured area (Fig. S7†). The surface state of Ag@Cu-MOF@BG was also investigated using XPS analysis. The survey spectrum (Fig. S8a†) indicated the presence of Ca, Si, C, O, Cu, and Ag in the composite. Two peaks at 368.7 eV (Ag 3d5/2) and 374.6 eV (Ag 3d3/2) were observed in the Ag 3d spectrum (Fig. 2d), corresponding to metallic Ag0.39 Subsequently, the Cu 2p spectrum showed a peak at 933.7 eV corresponding to Cu2+ sites of the MOF (Fig. 2e). Besides, peaks of Ca and Si due to BG with binding energies of 351.1 eV (Ca 2p1/2), 347.6 eV (Ca 2p3/2), and 103.3 eV (Si 2p3/2) were observed (Fig. S8b and c†). In contrast, the IR spectrum of Ag@Cu-MOF@BG only confirmed the characteristic bands for bioactive glass, which are related to stretching vibrations of Si–O–Si40 (at 1028 cm−1 and 793 cm−1) (Fig. S9†). The absence of bands from the MOF is due to the thin Ag@Cu-MOF layer deposited, as the Cu and Ag contents are limited to 0.015–0.018 wt%.
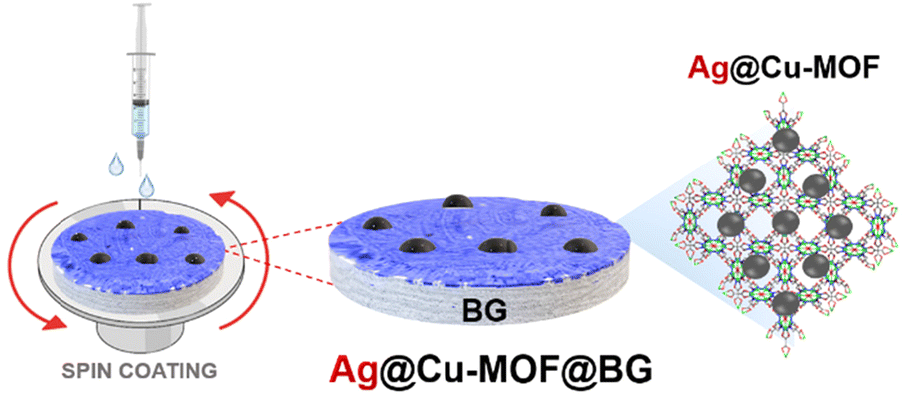 | ||
| Scheme 1 Preparation of the Ag@Cu-MOF@BG composite. | ||
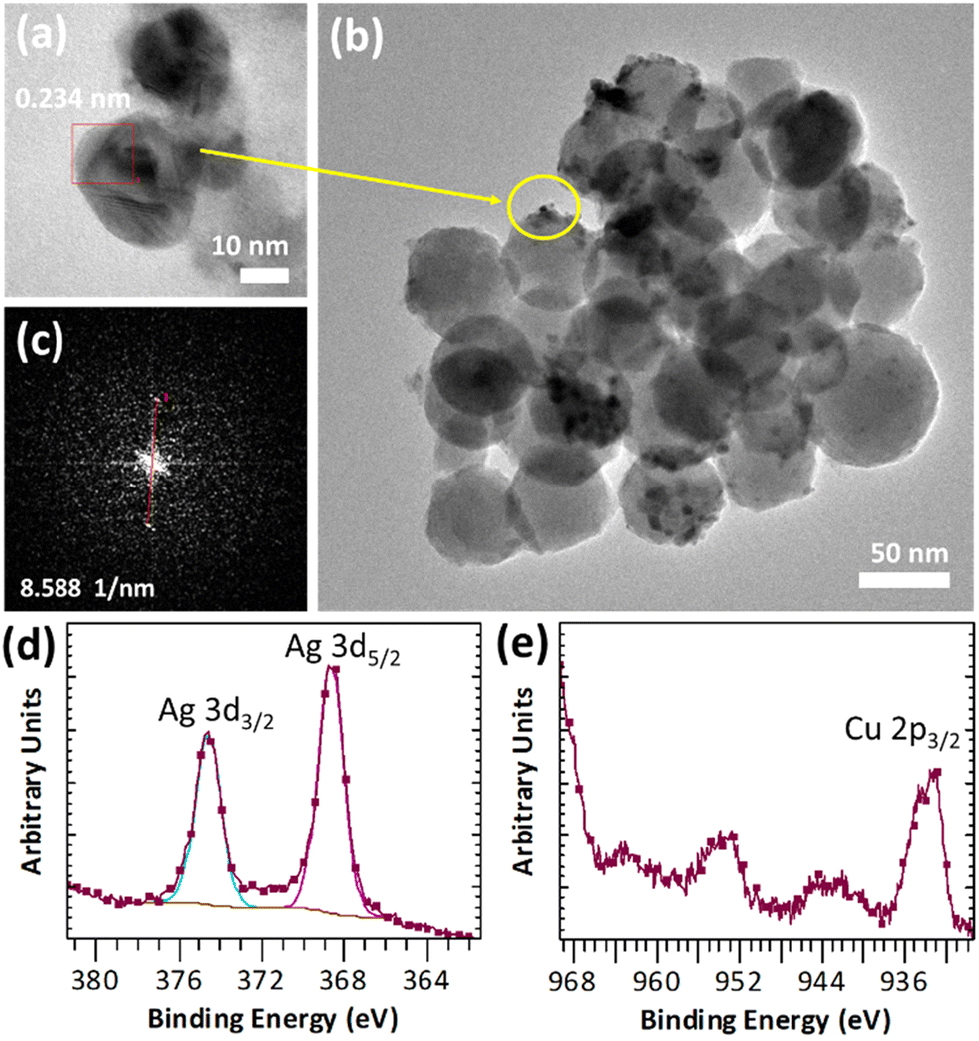 | ||
| Fig. 2 (a–c) TEM image with FFT and (d and e) high-resolution XPS spectra of Ag@Cu-MOF@BG. | ||
Bioactivity test
The in vitro bioactivity of the new composite was measured in a physiological-like buffer (DPBS) at 37 °C over 7 days. The formation of a hydroxyapatite (HA) layer on the surface, which is considered to indicate bioactivity, was also investigated for BG as a reference. At first, IR spectra showed that after 72 h of incubation of Ag@Cu-MOF@BG in the buffer, two bands characteristic of HA were observed, corresponding to P–O vibrations νPO43− at 600 and 562 cm−1 (Fig. 3a). For BG, these low intensity bands were visible at an earlier stage, after 4 h (Fig. S10†). Further ICP-OES analysis of the composition of the powders after different test durations showed that the P content, which is a source of HA, is already found after the first hour with values of 0.5 and 0.3 wt% for Ag@Cu-MOF@BG and BG, respectively. Within 7 days, this P content doubled for the composite (to 1 wt%) and increased fivefold for BG (to 1.5 wt%) (Fig. 3b and Fig. S11†). Meanwhile, the Si content decreased from 20–21 wt% to 18 wt% for both materials. In the case of BG, the gradual decrease in the Ca content and the increase at the end of the 7-day test correspond well with the changes in the pH value (Fig. S12a†). For Ag@Cu-MOF@AG, pH of the same value (7.7) was observed after 7 days. However, the rate of increase was different, with the main increase occurring after 24 h (Fig. S12b†). This is important because increasing the pH of the buffer solution is associated with the initial stages of hydroxycarbonate apatite (HCA) layer formation after immersing bioactive glasses in body fluid in vivo or simulated body fluid in vitro.41 In fact, the exchange of Ca2+ for H+ from the solution is responsible for the formation of silanol bonds (Si–OH) on the glass surface. According to the mechanism of glass bioactivity, high local pH leads to an attack of the silica glass network by OH−, breaking the Si–O–Si bonds. Finally, the soluble silica is lost to the solution as Si(OH)4, leaving more Si–OH (silanols) at the glass–solution interface. And it is a condensation of these Si–OH groups near the glass surface that forms the silica-rich layer, on which an amorphous CaO–P2O5-rich layer is subsequently formed.42,43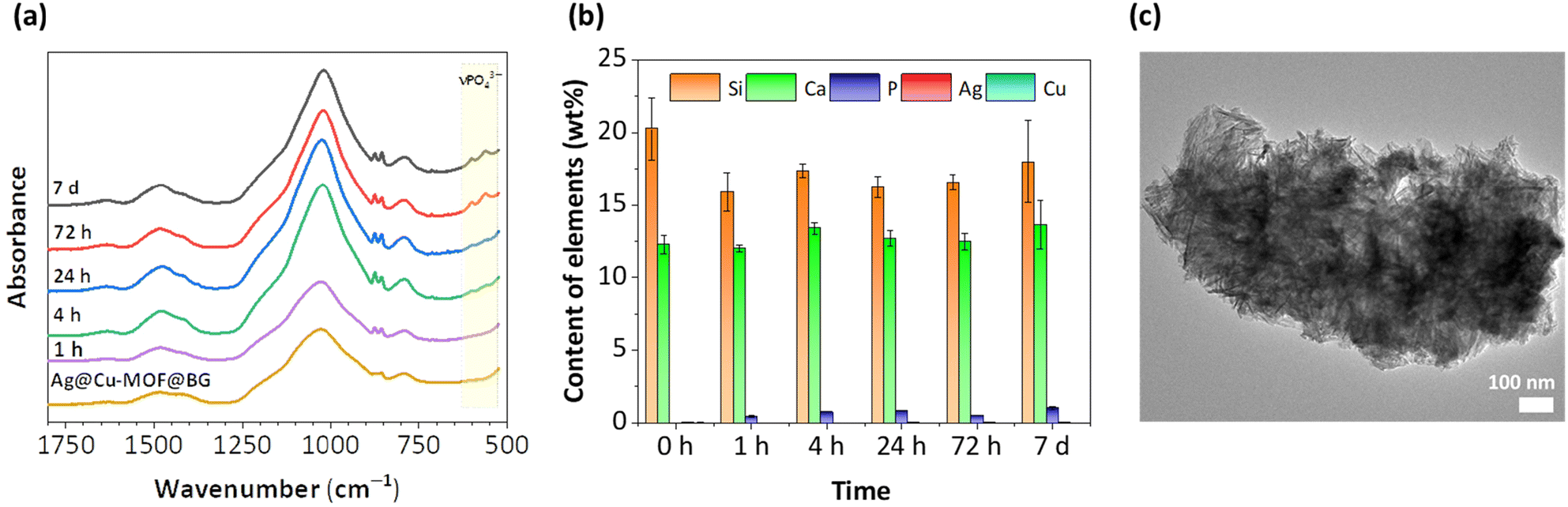 | ||
| Fig. 3 (a) ATR-FIR spectra of Ag@Cu-MOF@BG before and after immersion in DPBS (pH 7.4, 37 °C, 1 hour–7 days). (b) Analysis of the composition monitored by ICP-OES. (c) TEM image of Ag@Cu-MOF@BG after the 7th day of the bioactivity test. | ||
The bioactive glass used for the synthesis of Ag@Cu-MOF@BG, previously reported as B_78S, is well known for its excellent bioactivity.31 For this nanometric glass with a simple binary system of 78SiO2–22CaO (wt%), a CaP-rich layer was observed already after 2 h of immersion under physiological conditions. The change in the composition of B_78S was rapid, and after 4 hours of incubation the P content was equal to 18 at%, while the Si content decreased from 74 to 56 at%. However, to date, the bioactivity of this BG in a structured form (disc) has not been determined. A comparison of the bioactivity results of this bioactive glass tested in powder and disc form shows that their conversion rates to HA are significantly different. The more difficult exchange of ions in a compressed form is responsible for the slow changes in the composition of the BG disc. Nevertheless, as shown by TEM images, HA formation in the form of needle-like nanocrystallites was observed after 7 d of incubation for both BG and Ag@Cu-MOF@BG (Fig. 3c and Fig. S13†). Importantly, the presence of a thin Cu-MOF layer with anchored AgNPs did not hinder the formation of HA on the surface of the biomaterial.
For the composite, an additional analysis of the Ag content after immersion in DPBS was carried out, which showed the highest leaching of Ag during the first 4 h of incubation (Fig. S14†). At this time, the Ag content was reduced from 0.018 to 0.015 wt%, resulting in Ag leaching efficiency of approximately 15% given the initial content. The release of AgNPs in the initial stage will be important for the antibacterial effect of this material. After 24 h, the Ag content had increased to the initial level, suggesting that the released particles could be adsorbed back onto the BG surface.
Antimicrobial activity
The first step in exploring the therapeutic properties of the new materials was to evaluate the antimicrobial properties of Ag@Cu-MOF. For the antimicrobial activity against bacteria and yeast, the minimum inhibitory concentration (MIC) and the biocidal concentration (MBC) were determined, as shown in Table 1. A comparison of the results for Ag@Cu-MOF with those previously obtained for pristine Cu-MOF (known as Cu-capz)11 is indicative of the beneficial effect of the anchored AgNPs. Significantly higher susceptibility to the Ag@Cu-MOF was observed for all microorganisms tested (Table 1). Overall, Gram-negative bacteria showed significantly higher susceptibility to Ag@Cu-MOF than Gram-positive ones and yeasts. The MIC and MBC values of Ag@Cu-MOF against E. coli ATCC 25922 and K. pneumoniae ATCC 700603 were determined at a concentration of 62.5 μg mL−1 (MIC = MBC) while those against P. aeruginosa ATCC 10145 at a concentration of 31.125 μg mL−1 and 125 μg mL−1, respectively. The MIC values against S. aureus ATCC 25923 and C. albicans ATCC 10231 were determined at a concentration of 125 μg mL−1, while the MBC values against these microorganisms were determined at concentrations of 125 and 250 μg mL−1, respectively.| Microorganisms | Cu-MOFa | Ag@Cu-MOF | ||
|---|---|---|---|---|
| MIC | MBC | MIC | MBC | |
| a Published as Cu-capz;11 ND not determined. | ||||
| Escherichia coli ATCC 25922 | 2 | 5 | 62.5* | 62.5* |
| Klebsiella pneumoniae ATCC 700603 | ND | ND | 62.5* | 62.5* |
| Pseudomonas aeruginosa ATCC 10145 | 5 | 6 | 31.125* | 125* |
| Staphylococcus aureus ATCC 25923 | 2 | 3 | 125* | 125* |
| Candida albicans ATCC 10231 | ND | ND | 125* | 250* |
Finally, the effect of a thin layer of Ag@Cu-MOF on the antimicrobial properties of the coated bioactive glass was tested. The results of the evaluation of antimicrobial activity based on the growth inhibition zone from agar diffusion assays and the determination of the reduction index are shown in Table 2 and Fig. 4. The antimicrobial activity of Ag@Cu-MOF@BG was favorable against all bacteria tested. The diameter of the inhibition zones of growth of test microorganisms was found to be between 8 and 10 mm, while the reduction index was determined to be ≥3. The lower sensitivity to Ag@Cu-MOF@BG discs was recorded for C. albicans, where the zone of inhibition of growth was determined with a diameter of 7 mm, and the reduction index was <1, showing a lack of biocidal activity against yeasts.
 | ||
| Fig. 4 Antimicrobial activity of Ag@Cu-MOF@BG against selected microorganisms. | ||
| Microorganisms | Inhibition zone [mm] | Reduction index |
|---|---|---|
| Escherichia coli ATCC 25922 | 9 | ≥3 |
| Klebsiella pneumoniae ATCC 700603 | 9 | ≥3 |
| Pseudomonas aeruginosa ATCC 10145 | 10 | ≥3 |
| Staphylococcus aureus ATCC 25923 | 8 | ≥3 |
| Candida albicans ATCC 10231 | 7 | <1 |
MOFs can act as antimicrobial carriers by several routes. This can be mediated by their components or by agents loaded into the MOF and gradually released by diffusion or degradation of the MOF structure.44 Our results show that the antimicrobial activity of Cu-based MOFs is enhanced by the presence of small and uniformly distributed AgNPs synthesized in situ in the matrix. This is demonstrated by the significantly lower MIC and MBC values of Ag@Cu-MOF compared to Cu-MOF against all microorganisms tested. To date, several reports have revealed pleiotropic antimicrobial mechanisms of AgNPs related to the disruption of the ionic balance, interference with the cell membrane, impairment and damage to proteins or DNA, resulting in disruption of cell function and metabolism, leakage of cytoplasm, and cell death.34,45–47
The increase in the antimicrobial efficacy of Cu-based MOFs and AgNPs as a result of their incorporation has already been reported by other authors. For example, the synergistic effect of Cu-BTC and AgNPs against B. subtilis has been reported.48 Similarly, results presented by Soomro and coworkers revealed that copper(II) and guanosine 5′-monophosphate-based MOF (Cu/GMP) showed higher antimicrobial activity after impregnation with AgNPs (AgNPs@Cu/GMP).49 The inhibition zones for Cu/GMP and AgNPs@Cu/GMP were determined at 12 and 17 mm against S. aureus and 9 and 10 mm against E. coli, respectively. The MIC values for AgNPs@Cu/GMP were 18.75 μg mL−1 against S. aureus and 25 μg mL−1 against E. coli. In addition, ICP results suggested that antimicrobial activity of AgNPs@Cu/GMP was related to the pH-dependent release of metal ions from the composite. In another study, Guo et al. showed the biocidal activity of polyCu-MOF loaded with AgNPs (polyCu-MOF@AgNPs) against S. aureus and E. coli.21 This was due to the induction of damage to cell integrity through the generation of reactive oxygen species (ROS) and the disruption of bacterial metabolism.
To the best of our knowledge, the Ag@Cu-MOF system has not been combined with biomaterials to date. The results presented show that the activity of the new composite was slightly lower in the eukaryotic C. albicans cells than in the prokaryotic bacterial cells when Ag@Cu-MOF was combined with bioactive glass nanoparticles.
However, the demonstration of antibacterial properties of Ag@Cu-MOF@BG indicates the potential use of such composites for pathogen control as well as various implementations in the pharmaceutical or biomedical sectors. This is also supported by previous results for the Zn-MOF@bioactive glass in a ternary system (SiO2–CaO–P2O5), where the MOF is ZIF-8. This type of composite exhibited the presence of an inhibition zone against E. coli, S. aureus, and C. albicans equal to 17.66, 13.33, and 16.33 mm, respectively.24 After loading with gentamicin, the diameter of the inhibition zone increased significantly (up to 37.33 mm) against all of the microorganisms tested. On the other hand, ZIF-8@VAN@BG scaffolds showed significant antibacterial activity against S. aureus due to the inhibitory effect of the released vancomycin. Furthermore, with increasing ZIF-8@VAN deposition on the scaffolds, the antibacterial activity of ZIF-8@VAN@BG increased from 62.5% to 93.5%.23
Biocompatibility studies
Finally, human dermal fibroblasts (HDF) were used as a cellular model to assess the biocompatibility of the new composite. In the direct viability test, HDF cells exhibited decreased (to approximately 14–20% of control) viability after 24, 48, and 72 h of growing on BG and Ag@Cu-MOF@BG (Fig. 5a). As the specimens were prepared in the form of discs, the HDF cells were seeded directly onto them and cell growth was strongly dependent on adhesion. Thus, the reduced adhesion underpinned the hindered proliferation and lower viability on both surfaces of pristine bioactive glass nanoparticles and after Ag@Cu-MOF coating. However, it can be assumed that by using these highly bioactive materials in the biological system, the hydroxyapatite layer is rapidly formed, making the surface more cell-friendly and suitable for adhesion. To confirm that the specimens did not release any toxic compounds into the cell culture medium, we performed the indirect assays. The obtained set of results confirmed that the discs neither release toxic agents nor adsorb any vital compounds from the media. Therefore, the viability of the HDF cells was maintained compared to the control (Fig. 5a). The results of the migration test also showed that the phenotypic characteristics of the HDF cells were not altered and that the ability of the cells to migrate was the same as in the control (Fig. 5b and c). | ||
| Fig. 5 (a) Comparison of BG and Ag@Cu-MOF@BG influence on HDF viability in direct and indirect assay. (b) Representative images of migration of HDF cells incubated with standard medium: A – time 0, C – after 24 h, and with medium pre-incubated with Ag@Cu-MOF@BG: B – time 0, D – after 24 h. (c) Quantitative results of measuring scratch width after 6, 24 and 48 h related to the initial scratch width. | ||
Conclusions
Herein, it was demonstrated that the integration of a MOF and bioactive glass may be a future direction for the development of theragenerative biomaterials. The new Ag@Cu-MOF@BG composite retained the benefits of the bioactive glass and acquired new antimicrobial properties. A selected Cu-MOF, namely NH4[Cu3(μ3-OH)(μ3-4-carboxypyrazolato)3], provides a suitable template for the in situ synthesis of well-dispersed silver nanoparticles. The presence of a very thin Ag@Cu-MOF layer on the bioactive glass surface was sufficient to show antibacterial activity against E. coli, S. aureus, P. aeruginosa and K. pneumoniae with the reduction index determined to be ≥3. In spite of all these properties, the Ag@Cu-MOF@BG composite was non-toxic to human dermal fibroblasts as it does not inhibit their adhesion, proliferation, and migration.Author contributions
The manuscript was written through the contributions of all authors. M. Fandzloch participated in the conceptualization, methodology, investigation, writing – original draft preparation, visualization, funding acquisition, resource procurement, supervision, and project administration. J. Trzcińska-Wencel and A. Augustyniak performed the investigation. P. Golińska, K. Roszek conducted the investigation and resource procurement. All authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support of the National Science Centre (NCN, Poland) with grant no. 2019/35/D/ST5/02243 is gratefully acknowledged.References
- H. Qian, T. Lei, L. Hua, Y. Zhang, D. Wang, J. Nan, W. Liu, Y. Sun, Y. Hu and P. Lei, Bioact. Mater., 2023, 24, 450 CAS
.
- A. Wubneh, E. K. Tsekoura, C. Ayranci and H. Uludağ, Acta Biomater., 2018, 80, 1 CrossRef CAS PubMed
- J. Chen, H. Zhou, Y. Fan, G. Gao, Y. Ying and J. Li, Chem. Eng. J., 2023, 471, 144537 CrossRef CAS
- Y. Cui, H. Liu, Y. Tian, Y. Fan, S. Li, G. Wang, Y. Wang, C. Peng and D. Wu, Mater. Today Bio, 2022, 16, 100409 CrossRef CAS PubMed
- W. Shuaishuai, Z. Tongtong, W. Dapeng, Z. Mingran, W. Xukai, Y. Yue, D. Hengliang, W. Guangzhi and Z. Minglei, Front. Bioeng. Biotechnol., 2023, 11, 1081446 CrossRef PubMed
- D. Han, X. Liu and S. Wu, Chem. Soc. Rev., 2022, 51, 7138 RSC
- C. Pettinari, R. Pettinari, C. Di Nicola, A. Tombesi, S. Scuri and F. Marchetti, Coord. Chem. Rev., 2021, 446, 214121 CrossRef CAS
- Z. A. Khan, E. S. Goda, A. ur Rehman and M. Sohail, Mater. Sci. Eng., B, 2021, 269, 115146 CrossRef CAS
- X. Xu, M. Ding, K. Liu, F. Lv, Y. Miao, Y. Liu, Y. Gong, Y. Huo and H. Li, Front. Chem., 2023, 11, 1124303 CrossRef CAS PubMed
- S. N. K. Lelouche, L. Albentosa-González, P. Clemente-Casares, C. Biglione, A. Rodríguez-Diéguez, J. Tolosa Barrilero, J. C. García-Martínez and P. Horcajada, Nanomaterials, 2023, 12, 2294 CrossRef PubMed
- M. Fandzloch, W. Bodylska, J. Trzcińska-Wencel, P. Golińska, K. Roszek, J. Wiśniewska, M. Bartmański, A. Lewińska and A. Jaromin, ACS Biomater. Sci. Eng., 2023, 9, 4646 CrossRef CAS PubMed
- S. Elmehrath, K. Ahsan, N. Munawar, A. Alzamly, H. L. Nguyen and Y. Greish, RSC Adv., 2024, 14, 15821 RSC
- S. Shams, W. Ahmad, A. Hussain Memon, S. Shams, Y. Wei, Q. Yuan and H. Liang, New J. Chem., 2020, 44, 17671 RSC
- R. Ballesteros-Garrido, R. Montagud-Martínez and G. Rodrigo, ACS Appl. Mater. Interfaces, 2019, 11, 19878 CrossRef CAS PubMed
- H. S. Rodríguez, J. P. Hinestroza, C. Ochoa-Puentes, C. A. Sierra and C. Y. Soto, J. Appl. Polym. Sci., 2014, 131, 40815 CrossRef
- M. Can, S. Demirci, A. K. Sunol and N. Sahiner, Microporous Mesoporous Mater., 2020, 309, 110533 CrossRef CAS
- A. Rauf, A. A. Khawaja, M. Javed, S. Mahmood, S. Iqbal, S. Nadeem, M. Jahangir, M. Ahmad, A. Bahadur and M. Alshalwi, Inorg. Chem. Commun., 2024, 159, 111802 CrossRef CAS
- J. H. Jo, H.-C. Kim, S. Huh and Y. Kim, Dalton Trans., 2019, 48, 8084 RSC
- S. M. Sheta, S. M. El-Sheikh and M. M. Abd-Elzaher, Dalton Trans., 2018, 47, 4847 RSC
- S. Soltani, K. Akhbari and A. Phuruangra, Appl. Organomet. Chem., 2022, 36, e6634 CrossRef CAS
- C. Guo, F. Cheng, G. Liang, S. Zhang, Q. Jia, L. He and M. Du, Chem. Eng. J., 2022, 435, 134915 CrossRef CAS
- B. Chen, H. Xiang, S. Pan, L. Yu, T. Xu and Yu Chen, Adv. Funct. Mater., 2020, 30, 2002621 CrossRef CAS
- L. Han, Z. Huang, M. Zhu, Y. Zhu and H. Li, Ceram. Int., 2022, 48, 6890 CrossRef CAS
- M. M. Ahmed, A. E. Omar, H. S. Zayed and M. Moaness, Appl. Phys. A, 2024, 130, 209 CrossRef CAS
- X. Kesse, C. Vichery and J.-M. Nedelec, ACS Omega, 2019, 4, 5768 CrossRef CAS PubMed
- A. Hoppe, N. S. Güldal and A. R. Boccaccini, Biomaterials, 2011, 32, 2757 CrossRef CAS PubMed
- J. R. Jones, D. S. Brauer, L. Hupa and D. C. Greenspan, Int. J. Appl. Glass Sci., 2016, 7, 423 CrossRef CAS
- L. L. Hench, Biomed. Glasses, 2015, 1, 1 Search PubMed
- G. Gao, A. F. Schilling, T. Yonezawa, J. Wang, G. Dai and X. Cui, Biotechnol. J., 2014, 9, 1304 CrossRef CAS PubMed
- S. Kargozar, M. Mozafari, S. J. Hashemian, P. B. Milan, S. Hamzehlou, M. Soleimani, M. T. Joghataei, M. Gholipourmalekabadi, A. Korourian, K. Mousavizadeh and A. M. Seifalian, J. Biomed. Mater. Res., 2018, 106, 61 CrossRef CAS PubMed
- M. Fandzloch, W. Bodylska, K. Roszek, D. Szymański, A. Jaromin and A. Lukowiak, Part. Part. Syst. Charact., 2023, 40, 2200184 CrossRef CAS
- E. Quartapelle Procopio, F. Linares, C. Montoro, V. Colombo, A. Maspero, E. Barea and J. R. Navarro, Angew. Chem., Int. Ed., 2010, 49, 7308 CrossRef CAS PubMed
- Clinical and Laboratory Standards Institute (CLSI) (2012). Methods for Dilution Antimicrobial Susceptibility Tests for Bacteria that Grow Aerobically; Approved Standard 9th. Document M07-A9, CLSI Wayne, USA.
- J. Trzcińska-Wencel, M. Wypij, M. Rai and P. Golińska, Front. Microbiol., 2023, 14, 1125685 CrossRef PubMed
- Clinical and Laboratory Standards Institute (CLSI) (2012). Performance Standards for Antimicrobial Disk Susceptibility Tests, Approved Standard, 7th ed., CLSI document M02-A11. Clinical and Laboratory Standards Institute, 950 West Valley Road, Suite 2500, Wayne, Pennsylvania 19087, USA.
- M. Fandzloch, W. Bodylska, K. Roszek, K. Halubek-Gluchowska, A. Jaromin, Y. Gerasymchuk and A. Lukowiak, Nanoscale, 2022, 14, 5514 RSC
- C. Ohtsuki, T. Kokubo and T. Yamamuro, J. Non-Cryst. Solids, 1992, 143, 84 CrossRef CAS
- W. Bodylska, M. Fandzloch, R. Szukiewicz and A. Lukowiak, Nanomaterials, 2022, 12, 4480 CrossRef CAS PubMed
- W. Zhang, L. Wang and J. Zhang, Res. Chem. Intermed., 2019, 45, 4801 CrossRef CAS
- J. C. Moses and B. B. Mandal, ACS Appl. Mater. Interfaces, 2022, 14, 14961 CrossRef CAS PubMed
- J. R. Jones, Acta Biomater., 2013, 9, 4457 CrossRef CAS PubMed
- A. E. Clark, C. G. Pantano and L. L. Hench, J. Am. Ceram. Soc., 1976, 59, 37 CrossRef CAS
- L. L. Hench, J. Am. Ceram. Soc., 1991, 74, 1487 CrossRef CAS
- T. C. Livesey, L. A. Mahmoud, M. G. Katsikogianni and S. Nayak, Pharmaceutics, 2023, 15, 274 CrossRef CAS PubMed
- M. A. Huq, Int. J. Mol. Sci., 2020, 21, 1510 CrossRef CAS PubMed
- S. Wahab, T. Khan, M. Adil and A. Khan, Heliyon, 2021, 7, e07448 CrossRef CAS PubMed
- A. M. Alotaibi, N. B. Alsaleh, A. T. Aljasham, E. A. Tawfik, M. M. Almutairi, M. A. Assiri and M. M. Almutairi, Antibiotics, 2022, 11, 1219 CrossRef CAS PubMed
- Y. Gao, H. Dou, Y. Ma, G. Tian, D. M. Weragoda, S. Li and B. Chen, J. Environ. Chem. Eng., 2024, 12, 112133 CrossRef CAS
- N. A. Soomro, S. A. Amur, Y. Wei, A. H. Shah, M. Jiao, H. Liang and Q. Yuan, J. Cluster Sci., 2021, 32, 1519 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available: PXRD, ATR-FTIR, TEM, N2 adsorption–desorption isotherm, SEM-EDS, XPS, ICP-OES, and changes in the pH value. See DOI: https://doi.org/10.1039/d4dt01190b |
| This journal is © The Royal Society of Chemistry 2024 |