
MIL-101(Cr)/aminoclay nanocomposites for conversion of CO2 into cyclic carbonates†
Jyoti a,
Sarita Kumaria,
Samiran Chakrabortya,
Prakash Kanooab,
Vinod Kumara and
Anindita Chakraborty*a
a,
Sarita Kumaria,
Samiran Chakrabortya,
Prakash Kanooab,
Vinod Kumara and
Anindita Chakraborty*a
aDepartment of Chemistry, School of Basic Sciences, Central University of Haryana, Mahendergarh 123031, Haryana, India. E-mail: achakraborty@cuh.ac.in
bSpecial Centre for Nano Sciences, Jawaharlal Nehru University, New Mehrauli Road, New Delhi, Delhi 110067, India
First published on 14th May 2024
Abstract
We present the use of an amine functionalized two-dimensional clay i.e., aminoclay (AC), in the chemistry of a three-dimensional metal–organic framework (MOF) i.e., MIL-101(Cr), to prepare MIL-101(Cr)/AC composites, which are exploited as catalysts for efficient conversion of CO2 gas into cyclic carbonates under ambient reaction conditions. Three different MOF nanocomposites, denoted as MIL-101(Cr)/AC-1, MIL-101(Cr)/AC-2, and MIL-101(Cr)/AC-3, were synthesized by an in situ process by adding different amounts of AC to the precursor solutions of the MIL-101(Cr). The composites were characterized by various techniques such as FT-IR, PXRD, FESEM, EDX, TGA, N2 adsorption, as well as CO2 and NH3-TPD measurements. The composites were exploited as heterogeneous catalysts for CO2 cycloaddition reactions with different epoxides and the catalytic activity was investigated at atmospheric pressure under solvent-free conditions. Among all the materials, MIL-101(Cr)/AC-2 shows the best catalytic efficiency under the optimized conditions and exhibits enhanced efficacy compared to various MIL-101(Cr)-based MOF catalysts, which typically need either high temperature and pressure or a longer reaction time or a combination of all the parameters. The present protocol using MIL-101(Cr)/AC-2 as the heterogeneous catalyst gives 99.9% conversion for all the substrates into the products at atmospheric pressure.
Introduction
CO2 capture and its subsequent conversion into valuable products offers a unique opportunity to address the serious environmental threats caused by the continuous increase in atmospheric CO2 level.1,2 This approach mitigates the adverse greenhouse effect and creates a circular and sustainable economy by producing important chemicals and fuels.3 In this regard, catalytic CO2 organic transformation reactions have attracted significant research interest.4,5 In particular, the CO2 cycloaddition reactions with epoxides to produce cyclic carbonates have widely been studied.6 The CO2 cycloaddition reactions typically need a synergy of a Lewis acid catalyst and a Lewis base co-catalyst. Traditional catalysts include metal oxides, metal–porphyrin complexes, lignin-based catalysts and metal–salen complexes.6–10 However, most of these catalysts work at elevated temperatures and pressures, and the purification and recycling process is also complicated for these homogeneous catalysts.Over the last few decades, metal–organic frameworks (MOFs) have evolved as novel inorganic–organic hybrid materials with a unique structure and diverse applications.11–16 The porous structure, periodicity, highly modular nature and the heterogeneous nature give MOFs an edge over the conventional homogenous catalysts.17–23 The chromium-terephthalate MOF MIL-101(Cr) is a prototype MOF which is well known for its high surface area, hydrothermal and chemical stability and diverse applications.24–26 The large mesopores, high thermal stability, good CO2 adsorption efficacy and the presence of Lewis acidic unsaturated metal sites (UMSs) available for epoxide binding should make it an ideal catalyst for the cycloaddition reaction of CO2 with epoxides. However, the crystal field effect makes the Cr(III) centres inert in the octahedral environment, which affects the catalytic efficacy.20 MIL-101(Cr) has been tested as a heterogeneous catalyst for the cycloaddition reactions of CO2 with epoxides by different research groups and successful reactions mostly require high pressure CO2 (8–100 bar) in the temperature range 298–393 K, with a reaction completion time of 24–48 h.27–30 To use the full potential of the MIL-101(Cr) framework as the desired catalyst, such drastic reaction conditions must be replaced with more ambient conditions, such as room temperature and atmospheric pressure. Recently, MIL-101 composites with polyoxometalates, ionic liquids and ionic polymers have shown enhanced catalytic properties;31–47 however, with the synergy of an easy synthetic method, efficient catalytic conversion at ambient temperature and atmospheric pressure in a quick time is yet to be achieved. We aimed to obtain enhanced catalytic CO2 conversions under ambient conditions using new composites based on the robust and porous structure of MIL-101(Cr). Novel MOF composites mitigate the typical shortcoming of MOFs like moisture sensitivity, poor water dispersibility, and poor thermal and chemical stability and exhibit enhanced/new properties based on judicious blending of MOFs with other functional components.48,49 We envisage that a synergistic combination of 2D layered clay, aminoclay (AC)50 with MIL-101(Cr) could result in such composites. Aminopropyl functionalized magnesium phyllosilicate (Scheme 1), popularly known as aminoclay (AC), has recently shown its potential to form novel MOF nanocomposites having enhanced CO2 adsorption, separation and catalytic conversion properties.51–53 In the present work, we report the synthesis of new MIL-101(Cr)/AC nanocomposites and their efficacy for CO2-epoxide cycloaddition reactions (Scheme 1). Three different composites were synthesized under solvothermal conditions by adding different amounts of AC to the precursor solution, MIL-101(Cr). The composite MIL-101(Cr)/AC-2 exhibits the best performance showing 99.9% conversion in the cycloaddition reaction of epichlorohydrin (EPH) with CO2 in 6 h at atmospheric pressure and 30 °C, while the pristine MOF shows a conversion of only 30% under the same reaction conditions. MIL-101(Cr)/AC-2 has also been tested as a heterogeneous catalyst using several other epoxide substrates and 99.9% conversion could be achieved with all the substrates under ambient reaction conditions.
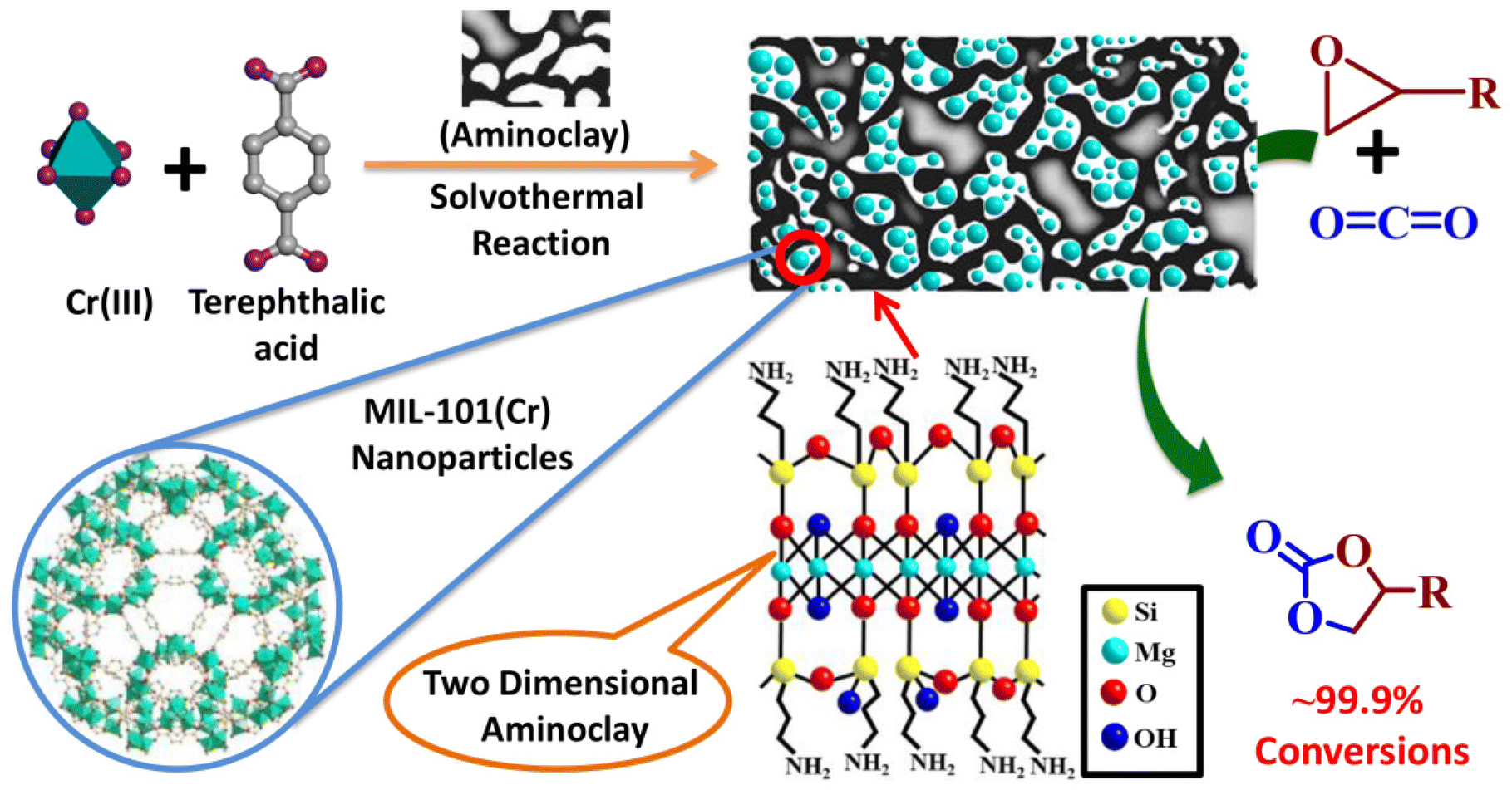 | ||
| Scheme 1 Synthesis of the self-assembled MIL-101(Cr)/AC composites, where the nanoparticles (NPs) of MIL-101(Cr) are assembled through the aminoclay (AC) layers. The composites are exploited as catalysts for the CO2 cycloaddition reaction with epoxides to produce cyclic carbonates. The structures of MIL-101(Cr) and AC are also shown in the scheme. | ||
Results and discussion
Synthesis and phase-purity of the composites
Pristine MIL-101(Cr) (PMIL) and aminoclay (AC) were synthesized by following the literature reported procedures.54,55 To synthesize the composites, a simple and facile in situ procedure was adopted where AC was dispersed in water first followed by the addition of the precursors of PMIL, which were then reacted under solvothermal conditions (see the Synthesis section for details). Three different composites named MIL-101(Cr)/AC-1, MIL-101(Cr)/AC-2 and MIL-101(Cr)/AC-3 were synthesized by successively increasing the relative amount of AC in the reaction mixtures. The well-correspondence between the simulated and experimental PXRD patterns (Fig. 1) of PMIL suggests high purity of the obtained sample. All the three composites exhibit PXRD patterns similar to that of PMIL (Fig. 1), indicating the formation of a pure phase of PMIL in the composites.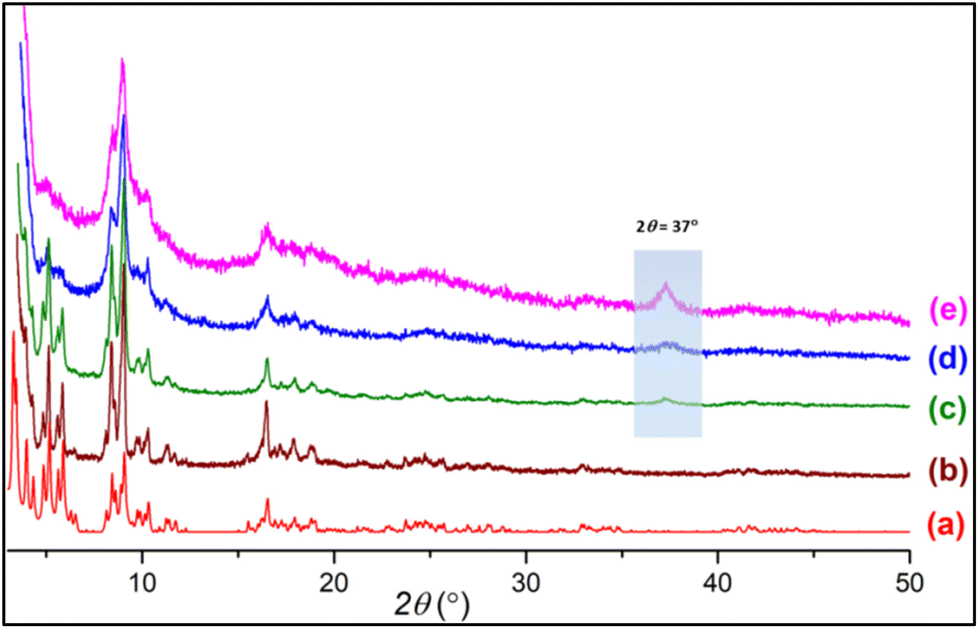 | ||
| Fig. 1 PXRD patterns of (a) simulated PMIL, (b) as-synthesized PMIL, (c) MIL-101(Cr)/AC-1, (d) MIL-101(Cr)/AC-2 and (e) MIL-101(Cr)/AC-3. | ||
The presence of the AC in the composites is difficult to interpret from the PXRD patterns as AC is weakly diffracting compared to the highly crystalline MOF and also the minor phase in the composites. However, with the increase in AC content, we observed the appearance of a broad reflection at 2θ = 37° in the composites with a higher clay content (particularly in MIL-101(Cr)/AC-3), which is attributed to the presence of the AC phase in the composite. This 2θ = 37° reflection is also observed in the PXRD pattern of the AC (Fig. S1†), which is ascribed to the d130,200 = 0.238 nm reflection originating from the 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 trioctahedral Mg-phyllosilicate clay with a talc-like structure of AC.50
:1 trioctahedral Mg-phyllosilicate clay with a talc-like structure of AC.50
IR spectra and thermal stabilities of the composites
To ascertain the functional groups of the samples, FT-IR spectra were recorded (Fig. 2 and S2†). The spectrum of PMIL shows the characteristic peaks of MIL-101(Cr) as reported in the literature.48,56 The IR spectrum of AC suggests its characteristic organoclay structure (Si–C 1116 cm−1, Si–O–Si 1014 cm−1)50,51 with the presence of a broad N–H stretching frequency band (∼3200–3700 cm−1) (Fig. S2†). The IR spectra for all the composites are very similar to that of the PMIL, with intense bands appearing in the range of ∼1400–1625 cm−1, which are corresponding to the stretching frequency of the carboxylate group of the 1,4-benzenedicarboxylate (H2bdc) ligand, originating from the PMIL. Selected FT-IR peaks (in cm−1) of MIL-101(Cr)AC-1: 1625 s, 1509 m, 1398 s, 1090 w, 1050 m, 1016 m, 883 m, 831 w, 745 s (Fig. S2†). Distinct bands around 1090 cm−1 and 1050 cm−1 appeared for the composite (which are absent in PMIL) and are ascribed to the Si–C and Si–O–Si stretching, respectively, and these bands originate from the AC phase (Fig. S2†). The shift in the above two vibrations in the composite compared to that in the pristine AC may be attributed to the plausible interaction of the NH2 groups of AC with the Cr(III) centres of the MOF. We envisage that in the composite, the nitrogen atoms of NH2 groups interact with Cr(III) centres and pull the alkyl chain towards itself leading to the weakening of the Si–C bond, which is reflected in the lower wavenumber (1090 cm−1) in the composite, compared to that of AC (1014 cm−1). On the other hand, the drift of the electron density of the alkyl chain towards Cr(III) makes Si atoms relatively electronegative which consequently pulls the oxygen atoms towards itself making the Si–O bond stronger, shifting it to a higher wavenumber (1050 cm−1 in the composite vs. 1014 cm−1 in AC). The IR spectra of other composites are similar to that of MIL-101(Cr)AC-1 and suggest the presence of both PMIL and the AC phase.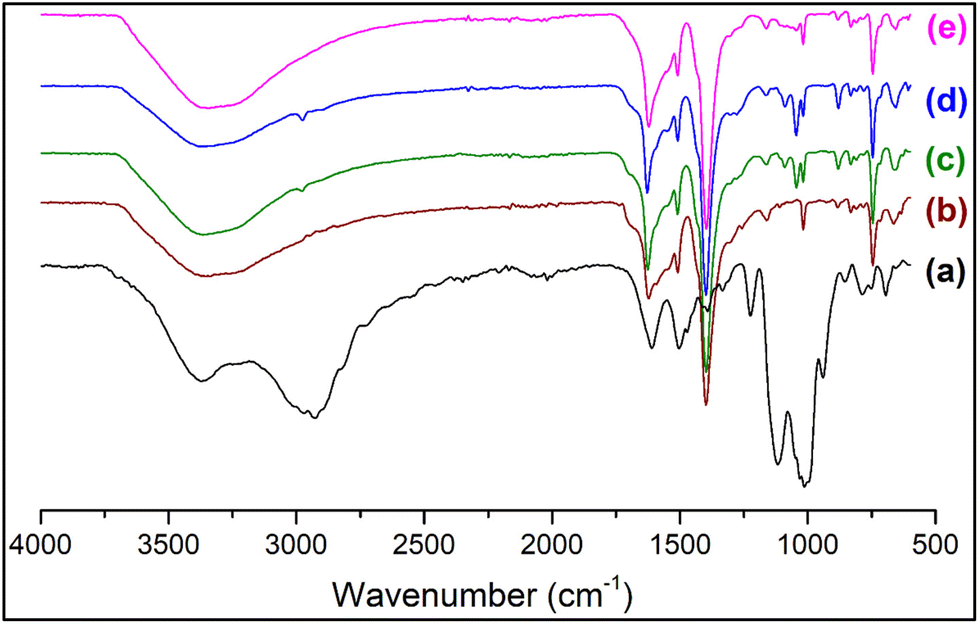 | ||
| Fig. 2 FTIR spectra of (a) aminoclay, (b) as-synthesized PMIL, (c) MIL-101(Cr)/AC-1, (d) MIL-101(Cr)/AC-2 and (e) MIL-101(Cr)/AC-3. | ||
To investigate the thermal stabilities of the samples, thermogravimetric analyses (TGA) (Fig. S3†) of the as-synthesized PMIL sample and all the composites were carried out. PMIL exhibits an initial weight loss of about 41% up to 100 °C, which matches well with the literature value and corresponds to a loss of ∼25 solvent (H2O) molecules from the framework.32,54 The dehydrated framework shows a plateau up to 350 °C temperature, beyond which framework decomposition occurs. All the composites exhibit similar profiles for thermal stability to that of PMIL and are stable up to the temperature range of 350–360 °C, which suggests that the composite formation does not affect the thermal stability of the framework structure. However, the initial weight loss percentages (Fig. S3†) for the composites are different compared to that of PMIL. The lesser initial loss may be attributed to the presence of a relatively small amount of the highly porous and solvated PMIL phase in the composites, where the AC content increases from MIL-101(Cr)/AC-1 to MIL-101(Cr)/AC-3, which is consistent with the synthetic conditions where an increasing amount of AC was added in the reaction mixtures for the latter composites.
Textural properties of the composites
The as-synthesized PMIL particles are known to be octahedral and nanometer-sized, as reported in the literature.32 We recorded the FESEM images for our as-synthesized PMIL, which reveal that faceted octahedral-like particles are present having sizes in the range of 100–150 nm (Fig. S4†). A literature survey suggests that the FESEM images of AC exhibit a typical layered structure attributed to the 2D sheet-like organoclay structure.50 To get an idea about the morphology of the composites, FESEM images were recorded for all the composites, which show a distinct difference compared to both PMIL and AC. For MIL-101(Cr)/AC-1, an assembly of fused particles is observed, where the MOF nanoparticles (NPs) appeared to be coated with AC layers (Fig. 3a, b and S5†). Such a morphology is in clear contrast to the faceted particles of the pristine framework. The size histogram plot (Fig. S6†) shows that the particles have a maximum distribution in the size range of 70–90 nm. The assembly of particles is more evident in the case of MIL-101(Cr)/AC-2, where distinct faces of the PMIL particles disappear and the particles gradually become more spherical-like particles, with an increase in the AC content in the composite (Fig. 3c, d and S7†). The AC layer coating PMIL is visible in the case of MIL-101(Cr)/AC-2 in the presence of layered AC junctions between the PMIL NPs (Fig. 3d). We envisage that the assembly of the PMIL particles through AC in the composites occurs due to the specific interaction between unsaturated surface functionalities (e.g., Cr3+ sites) in the PMIL NPs and the amine-functionalized clay, which results in a self-assembled MOF–clay nanocomposite. To the best of our knowledge, only one report is known so far to document such self-assembled nanocomposites, where the MOF (ZIF-8) particles were ‘glued’ by clay (AC) driven by the interaction between the Zn2+ sites of ZIF-8 and the NH2 groups of AC.51 The size histogram plot reveals that the maximum particles of MIL-101(Cr)/AC-2 have sizes in the range of 50–70 nm (Fig. S8†). It is noteworthy that the average sizes of particles decrease in the order: PMIL < MIL-101(Cr)/AC-1 < MIL-101(Cr)/AC-2. This may be attributed to the downsizing efficacy of AC through coordination modulation via the NH2 group, as literature reports indicate that AC can also act as a functional template to assist growth of MOF NPs.52 The FESEM images of MIL-101(Cr)/AC-3 exhibit that NPs are also fused similar to the first two composites, but with an increased amount of AC coating (Fig. S9†). This is attributed to the use of excessive AC in the reaction mixture, which results in an inhomogeneous composite having too much AC content. It's difficult to comment on the size of the particles in MIL-101(Cr)/AC-3, as the PMIL particles are highly coated and deeply buried in the increased number of AC layers. | ||
| Fig. 3 (a) FESEM images of composite MIL-101(Cr)/AC-1, (b) magnified view of a portion from MIL-101(Cr)/AC-1 showing the AC coated fused particles, (c) FESEM images of composite MIL-101(Cr)/AC-2 and (d) magnified view of a portion from MIL-101(Cr)/AC-2 showing the presence of layered AC junctions between the PMIL NPs. | ||
To understand the element content and their distribution, we performed an energy dispersive X-ray (EDX) study and element analysis for one of the composites, MIL-101(Cr)/AC-2. The EDX spectrum (Fig. S10†) clearly shows the presence of Mg and Si elements coming from AC and Cr from PMIL and thus confirms the presence of both the parent materials (PMIL and AC) in the composite. Furthermore, elemental mapping indicates that the different elements are distributed uniformly throughout the sample, suggesting the formation of a homogeneous composite (Fig. 4). The homogeneous nature of MIL-101(Cr)/AC-2 prompted us to check its solution processability, as enhanced dispersibility can be expected from such a homogeneous composite. When the same amount of PMIL and MIL-101(Cr)/AC-2 was dispersed in methanol (5 mg in 2 ml), PMIL settled down within 15 min, but the dispersion of MIL-101(Cr)/AC-2 remained stable even after 4 days (Fig. S11†). This result clearly demonstrates the high solution processability of the composite.
 | ||
| Fig. 4 Elemental mapping of MIL-101(Cr)/AC-2 indicating uniform distribution of various elements (Cr, Mg, Si, N, O) throughout the material. | ||
Adsorption and TPD studies
In order to investigate the porous nature and to know the surface area of the samples, N2 adsorption isotherms at 77 K of the activated PMIL and the composites were recorded (Fig. 5). The as-synthesized PMIL sample shows high uptake of N2 gas and a typical type-1 profile with a BET surface area of 2300 m2 g−1. The value of the surface area is consistent with the literature reported values5,25 of MIl-101(Cr). On the other hand, the densely packed 2D layers of AC are known to inhibit any N2 uptake at 77 K (ref. 51–53) and exhibit negligible N2 uptake (Fig. 5). MIL-101(Cr)/AC-1 exhibits a type-I profile, but the total uptake (∼800 cc g−1) at P/P0 = 1 and the BET surface area (1434 m2 g−1) are much lower than those of PMIL. Such a decrease in the surface area is attributed to the presence of the non-porous AC phase in the composite. Similarly, the other two composites also exhibit type-I profiles with lower N2 uptake (Fig. 5), and the uptake gradually decreases with an increase in AC content from MIL-101(Cr)/AC-1 to MIL-101(Cr)/AC-3. The BET surface areas are found to be 1332 m2 g−1 and 921 m2 g−1 for MIL-101(Cr)/AC-2 and MIL-101(Cr)/AC-3, respectively. Furthermore, the pore size distribution plot (Fig. S12†) of the PMIL and all the MIL-101(Cr)/AC composites shows the presence of characteristics pores of the MIL-101(Cr) framework (∼29 Å and ∼34 Å) which matches well with the literature.24 In the case of MIL-101(Cr)/AC-2, we observed a broad distribution centered near ∼45 Å and we attribute this to the formation of some random large pores at the MOF–clay interface, which may also enhance the substrate diffusion and enhance the catalytic efficacy.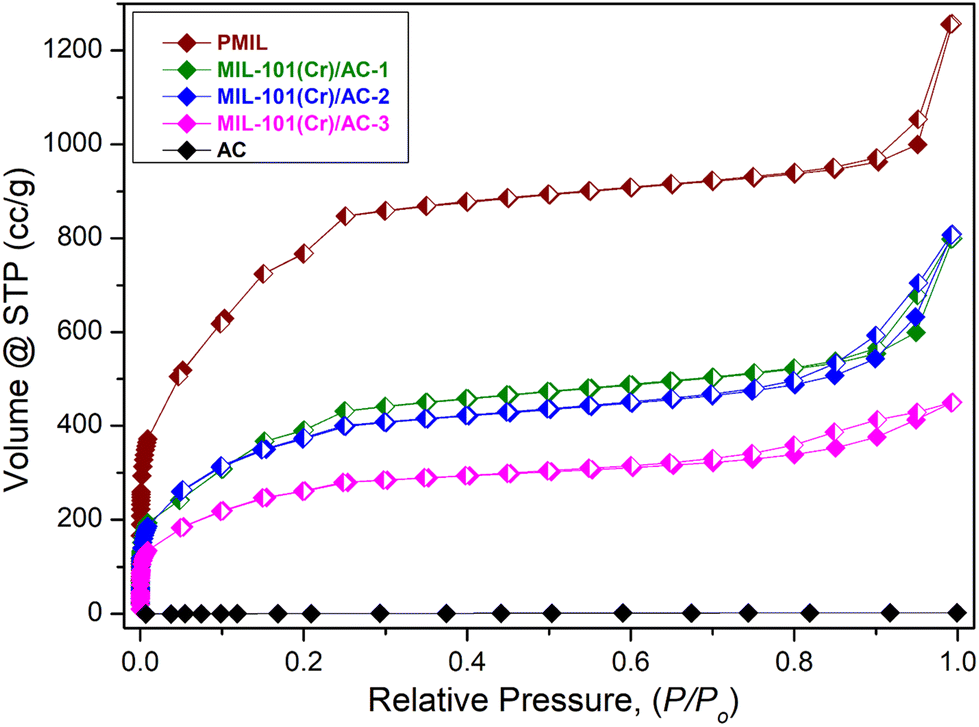 | ||
| Fig. 5 N2 Adsorption isotherms of PMIL, MIL-101(Cr)/AC-1, MIL-101(Cr)/AC-2, MIL-101(Cr)/AC-3 and AC at 77 K. Open symbol denotes adsorption and closed symbol denotes desorption of gas molecules. | ||
To investigate the presence of any Lewis acidic and basic sites in MIL-101(Cr)/AC-2, we performed TPD experiments with NH3 and CO2 (Fig. S13†). The acidity and basicity were determined from the peak area of the desorbed NH3 and CO2. The NH3-TPD profile shows a desorption peak in the temperature range of 30–350 °C with 1.629 mmol g−1 of desorption capacity, suggesting moderate acidic sites, which may be attributed to the Cr(III) UMSs. The CO2-TPD profile shows a desorption peak in the temperature range of 200–350 °C with 1.125 mmol g−1 of desorption capacity, suggesting the presence of moderate basic sites in the composites, which may be attributed to the NH2 groups at the MOF–clay interface.
Catalytic study
We envisage that the composites, particularly MIL-101(Cr)/AC-2, can be efficient as a catalyst for the CO2 conversion, as the catalytic reaction can occur at pores as well as at the surface of the composite and at the MOF–clay interface where the basic NH2 groups are present. The synergy of Lewis acidic Cr(III) UMS and Lewis basic NH2 sites was expected to enhance the catalytic activity.For a comparative study, we studied the catalytic performance of all the composites, along with the pristine MOF (PMIL) and AC in the CO2 cycloaddition reactions with epoxides, using tetrabutylammonium bromide (TBA+Br−) as a co-catalyst under solvent-free conditions and conversion (%) for the product formation was calculated using the 1H NMR spectrum (see the Experimental section for details). To start with, EPH as the model epoxide substrate (Table 1) was selected and catalytic reactions were performed with different catalysts under the identical optimized reaction conditions. AC shows only 10% catalytic activity (entry 1, Table 1), while a Cr(III) salt exhibits only 27% conversion (entry 2, Table 1) up to 6 h, and the catalyst is homogeneous. The heterogenous catalyst PMIL exhibits only a slight increase in the conversion (entry 3, Table 1) compared to that of the homogeneous Cr(III) salt, and the poor conversion (30%) can be attributed to the inertness of the Cr(III) centres of PMIL, which typically needs high CO2 pressure for the cycloaddition reaction.32,35,51 To our delight, under the same reaction conditions, the composites exhibit an enhanced performance compared to the PMIL. MIL-101(Cr)/AC-1, MIL-101(Cr)/AC-2 and MIL-101(Cr)/AC-3 show conversions of 44, 99.9 and 42%, respectively (entries 4–6, Table 1) in 6 h and the conversion (%) was calculated using 1H NMR spectra (Fig. S14†). We also recorded the FT-IR spectra (Fig. S15†) of the model substrate i.e., EPH and the reaction mixture of the product in the cycloaddition reaction of EPH with CO2 using the MIL-101(Cr)/AC-2 catalyst. The IR spectra (Fig. S15†) clearly suggest the conversion of the EPH to the cyclic carbonate product exhibiting the characteristic carbonyl stretching frequency at 1783 cm−1 arising from the carbonate group of the product. To confirm that the enhanced catalytic efficacy is indeed the effect of the composite formation, an experiment under the same reaction conditions was carried out, using a physical mixture of PMIL and AC as the catalyst (entry 7, Table 1). This physical mixture exhibits a negligible conversion (5%) of the substrate. This observation clearly indicates that a physical mixture does not have the synergistic effect as in the case of a composite. Such negligible conversion may be attributed to the densely packed non-porous AC layers in the mixture which inhibit accessibility to the catalytic sites. The catalytic reaction without any catalyst and only using the co-catalyst, TBA+Br−, was also performed at 30 °C and 40 °C (entries 8 and 9, Table 1), which shows only 20% and 30% conversion within 6 h, respectively. Reactions were also performed using the catalyst (MIL-101(Cr)/AC-2) only at 30 °C and we got negligible conversion (entry 29, Table S1†), suggesting that the presence of the co-catalyst is essential for effective reaction. Apart from TBAB as a co-catalyst, we also tried catalytic reactions using other co-catalysts such as tetrabutylammonium chloride (TBAC) and tetrabutylammonium iodide (TBAI) to compare the effectiveness of the co-catalyst on the catalytic activity of the reaction. An ideal co-catalyst for catalytic reaction should have suitable nucleophilicity as well as better leaving group ability. Upon co-catalyzing the CO2/EPH cycloaddition with TBAC, we observed a conversion of approximately 50% within 6 h and 80% within 24 h. However, when employing TBAI, the conversion was only around 5% within 6 h, with a subsequent increase to 50% after 24 h of reaction, while the reaction is completing within 6 h using TBAB as a co-catalyst. From our observations the enhanced efficiency of TBAB over TBAI may be attributed to its smaller molecular size, which enables facile diffusion within the confined channels of the MOF structure. Additionally, the observed superiority of TBAB compared to TBAC could be attributed to the better leaving group ability of bromide ions in TBAB, which facilitates the ring-opening reaction and subsequent carbonate formation more effectively. The efficient catalytic activity of TBAB as a co-catalyst may be attributed to the less energy barrier for opening of the epoxide ring of the substrate for the addition of CO2. So, the intricate interaction among molecular properties emphasizes the impact of co-catalysts on determining the effectiveness of the cycloaddition reaction.57–60 We have also conducted an additional experiment to evaluate the catalytic performance of MIL-101(Cr)/AC-2 with EPH at an elevated temperature (at 70 °C). As a result of our analysis, we observed that the CO2 cycloaddition reaction is accomplished in 5 h at 70 °C. This indicates that the temperature has minimal impact on the catalytic reaction under the optimized conditions, as it also reaches completion within 6 h at 30 °C.
|
|||
|---|---|---|---|
| Entry | Catalysts | Time (h) | Conversion (%) |
| a Reaction conditions: 9.2 mmol (851 mg) EPH, 30 mg activated catalyst and 0.92 mmol (296 mg) TBA+Br− under 1 bar CO2 and 30 °C.b Physical mixture.c Without the catalyst at 30 °C.d Without the catalyst at 40 °C. Conversion (%) were calculated from the 1H NMR spectra by integration of epoxide peaks versus cyclic carbonate peaks. | |||
| 1 | Aminoclay | 6 | 10 |
| 2 | Cr(NO3)3·9H2O | 6 | 27 |
| 3 | PMIL | 6 | 30 |
| 4 | MIL-101(Cr)/AC-1 | 6 | 44 |
| 5 | MIL-101(Cr)/AC-2 | 6 | 99.9 |
| 6 | MIL-101(Cr)/AC-3 | 6 | 42 |
| 7 | PMIL + ACb | 6 | 5 |
| 8 | TBABc | 6 | 20 |
| 9 | TBABd | 6 | 30 |
Among all the composites, MIL-101(Cr)/AC-2 shows the best performance owing to its homogeneous structure having a uniform distribution of the different elements, as revealed by the FESEM images. Furthermore, this composite also exhibits the smallest size particles (50–70 nm) among all the composites, and the network structure of the fused small NPs is expected to enhance the substrate diffusion to the catalytic sites, as the catalytic efficacy is found to be high for well-dispersed MOF NPs.51,61,62 Finally, the synergy of the Lewis acidic Cr(III) UMSs and the Lewis basic NH2 groups, as revealed by the TPD experiments, is expected to enhance the catalytic efficacy in this composite.
A plausible mechanism for CO2-epoxide cycloaddition using the MIL-101(Cr)/AC composites as catalysts is presented in Scheme 2. This involves coordination of an epoxide oxygen atom with a Lewis acidic site (LAS), which is the Cr(III) UMS in the composite structure. This results in polarization of the C–O bond and subsequently Br− of the co-catalyst (TBA+Br−) attacks the less sterically hindered carbon atom of the epoxide nucleophilically, causing a heterolytic cleavage. Subsequently, the CO2 carbon atom is activated by the Lewis basic site (LBS), which we assign to the NH2 groups in the composite structure, which are possibly present at the MOF–clay interface. This facilitates the nucleophilic attack on the CO2 carbon atom by the ring-opened alkoxide intermediate. This process allows carbon dioxide to enter and intramolecular ring-closure takes place to produce cyclic carbonate, regenerating the catalyst and co-catalyst.
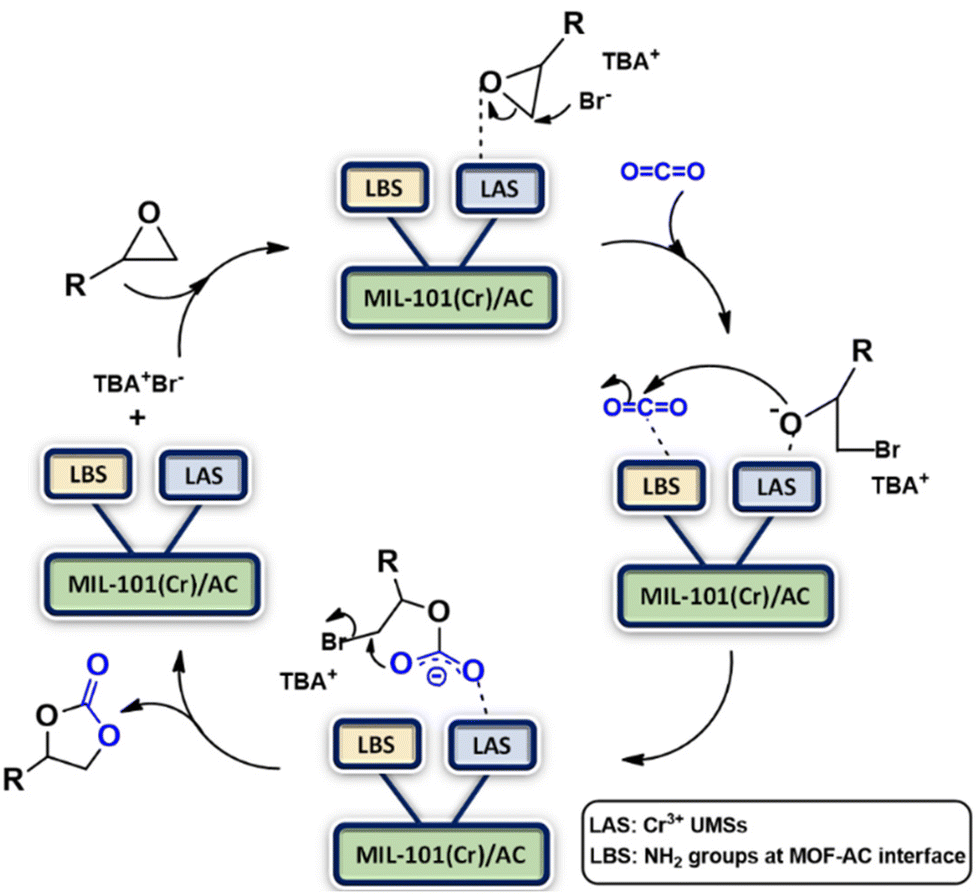 | ||
| Scheme 2 Proposed mechanism for cycloaddition of CO2 with epoxides catalyzed by MIL-101(Cr)/AC-2 involving LASs, LBSs and TBA+Br−. | ||
The conversion decreases from MIL-101(Cr)/AC-2 to MIL-101(Cr)/AC-3, which is attributed to the inhomogeneous nature of MIL-101(Cr)/AC-3, however it still shows a considerable conversion (comparable to that of MIL-101(Cr)/AC-1). To study the reaction completion time using each of the composites as a catalyst, we continued the reactions for a longer time and the conversions were recorded at regular time intervals (entries 15 and 25, Table S1, Fig. S16 and S17†). We also checked the heterogeneity of one of the composites, MIL-101(Cr)/AC-2. To confirm the heterogeneity for the catalytic process of the cycloaddition reaction of EPH, filtration tests were carried out. After 1 h of catalytic reaction at 1 bar of CO2 and at temperature 30 °C, the solid catalyst was removed, and a conversion of 33% was obtained. The catalytic reaction was further continued with the filtrate and it was observed that after removal of the solid catalyst, the conversions were 36, 37, 38, 39 and 40% at 2, 3, 4, 5 and 6 h, respectively, which clearly indicates that there is no significant conversion after the filtration (Fig. 6). On the other hand, the undisturbed catalyst shows conversions of 43, 63, 69, 92 and 99.9% at 2, 3, 4, 5 and 6 h, respectively (Fig. 6). These observations further confirm the heterogeneity of the catalyst. The recyclability test was also performed with the recycled MIL-101(Cr)/AC-2 catalyst in the cycloaddition reaction of EPH with CO2 and no significant decrease in the catalytic conversion was observed up to five cycles (88% conversion after the 5th cycle) (Fig. 7). Following each catalytic run, the heterogeneous catalyst can be effectively recovered from the reaction mixture by filtration and can be reused without losing its active properties. When comparing with the recovered catalyst's PXRD and FTIR data (Fig. S18 and S19†) with that of the MIL-101(Cr)/AC-2, no significant changes were observed, implying that the catalyst's structural integrity has not been compromised.
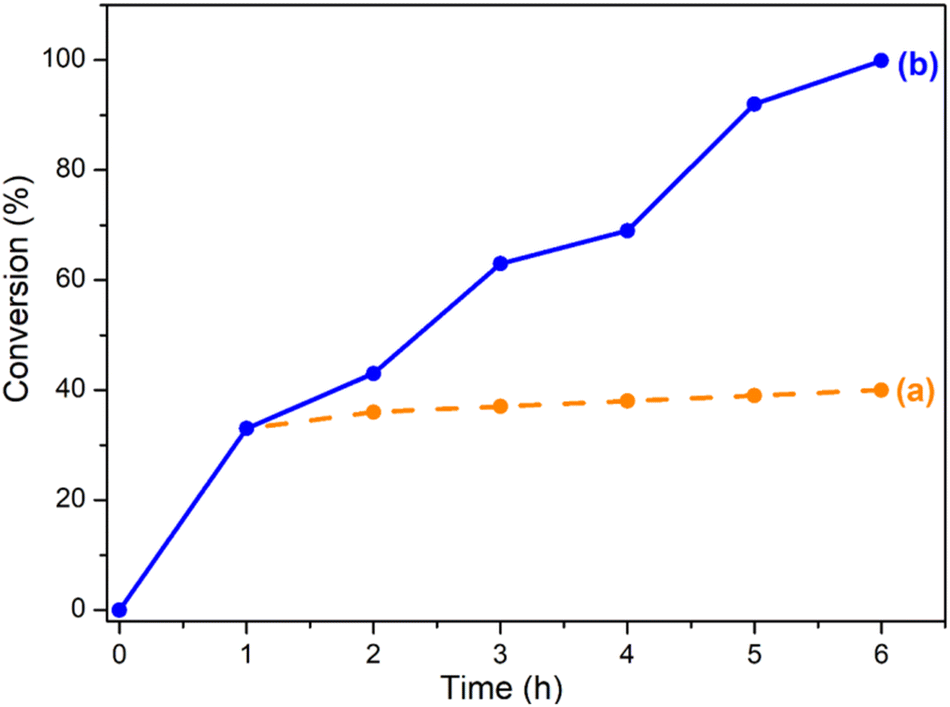 | ||
| Fig. 6 CO2 conversion % with EPH (a) catalyst MIL-101(Cr)/AC-2 removed after 1 h of catalytic reaction and (b) reaction continued undisturbed with catalyst MIL-101(Cr)/AC-2 up to 6 h. | ||
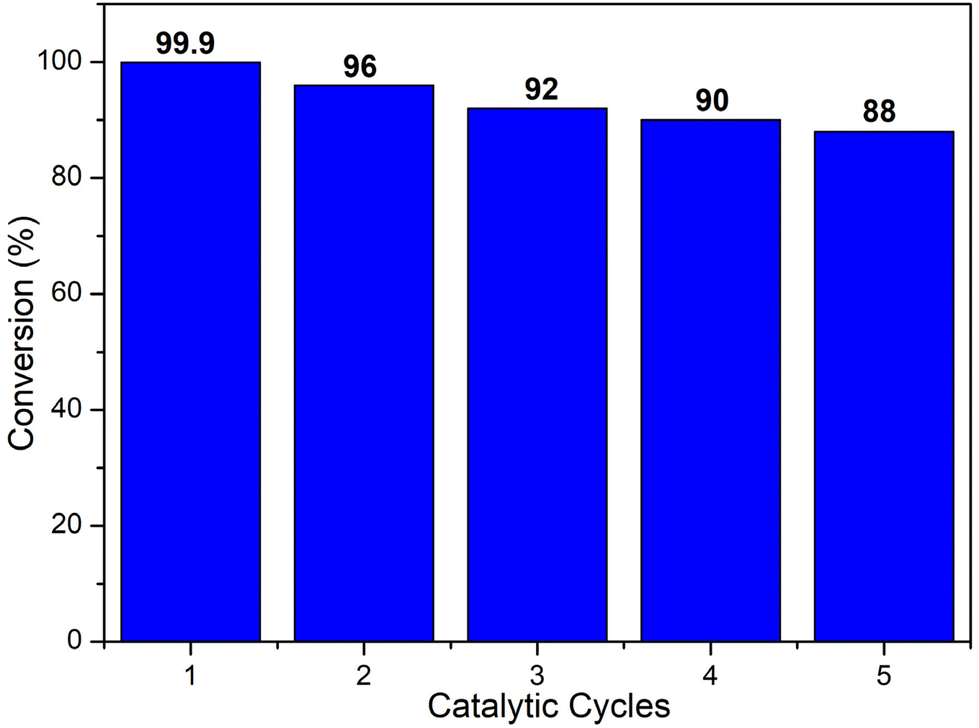 | ||
| Fig. 7 Recyclability test using MIL-101(Cr)/AC-2 as a catalyst for the cycloaddition reaction of CO2 with EPH. Identical reaction conditions to those of Table 1 were employed. | ||
The superior catalytic performance of MIL-101(Cr)/AC-2 inspired us to further expand the application of this novel composite as a catalyst in the cycloaddition of CO2 with other epoxides. After the catalytic reaction with EPH, we carried out a catalytic reaction with styrene oxide (SO) which is a larger rigid epoxide with a bulky phenyl ring attached to it. Under the optimized reaction conditions for EPH, there was no significant conversion with both the PMIL and the composite while SO was used as the substrate. Therefore, we carried out this reaction at a little elevated temperature (i.e., 40 °C) which resulted in good CO2 conversion efficiency for MIL-101(Cr)/AC-2. CO2 conversion using PMIL was also carried out at 40 °C with SO keeping all other conditions the same. In the first few hours (i.e., up to 12 h), PMIL does not show any conversion, but the composite shows significant conversion (60% at 12 h). As the reaction continued, PMIL shows a little conversion (5, 12, and 15% at 24, 42 and 48 h, respectively), whereas MIL-101(Cr)/AC-2 shows conversion of 71.4, 92.5 and 99.9% at 24, 42 and 48 h, respectively (Fig. 8 and Table S2†).
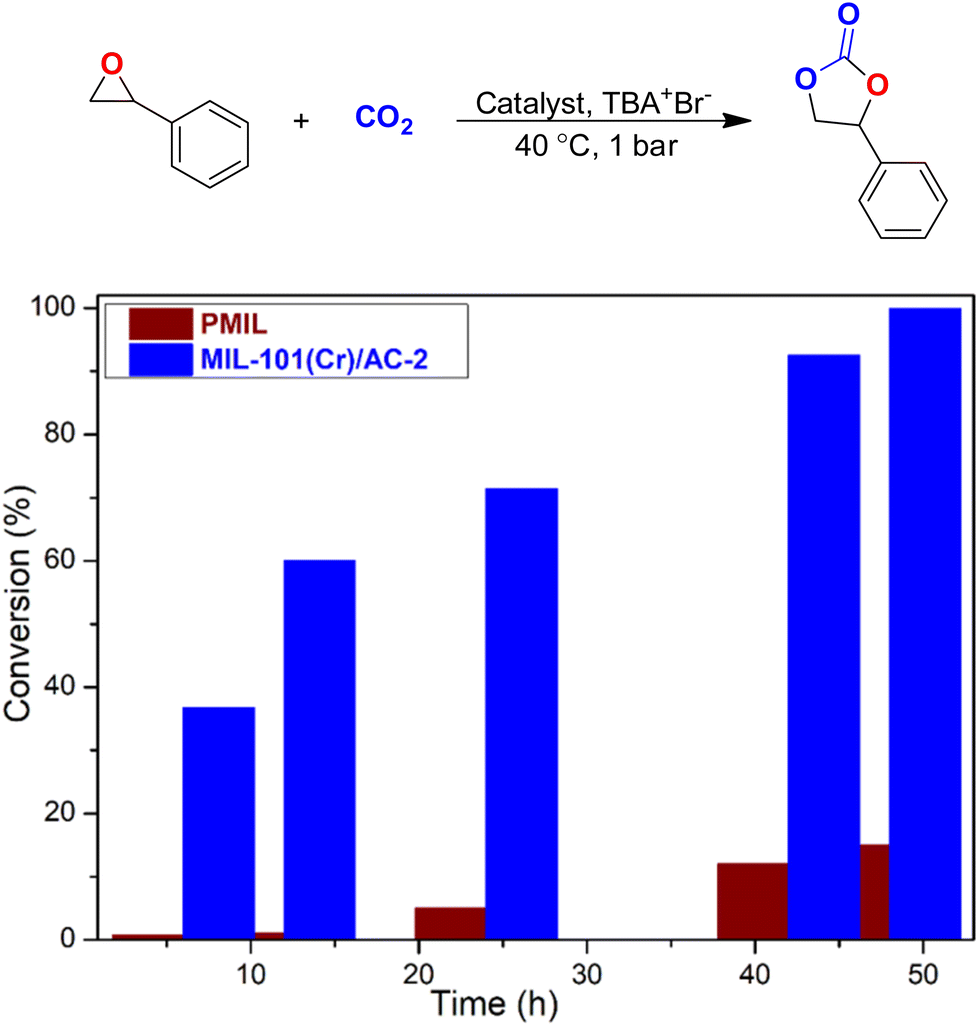 | ||
| Fig. 8 Conversion as a function of time using PMIL and MIL-101(Cr)/AC-2 as catalysts for the cycloaddition reaction of CO2 with SO. Conversion (%) were calculated from the 1H NMR spectra by integration of epoxide versus cyclic carbonate peaks. | ||
These results also indicate that the catalytic activity is significantly enhanced for the composite when a bulkier substrate is taken. The optimization of reaction conditions with various other substrates was also performed and we observed that, some of the substrates were not showing efficient catalytic activity at 30 °C and required a little elevated temperature (40 °C), which may be attributed to the different sizes (Table S3†) and reactivities of the substrates. The catalytic activity of the MIL-101(Cr)/AC-2 catalyst was examined (Table 2). It was observed that the small epoxides [propylene oxide (PO) and epoxy butane (EB)] undergo full conversion (99.9%) within 6 h (entries 2 and 3, Table 2), whereas for larger or bulkier epoxides [allyl glycidyl ether (AGE), butyl glycidyl ether (BGE)] (Table S3†), the reactions are relatively slow, but also shows full CO2 conversion when the reaction time is extended up to 12 h for AGE and 15 h for BGE at 40 °C (entries 4 and 5, Table 2).
|
|||||
|---|---|---|---|---|---|
| Entry | Substrates | Products | Temperature (°C) | Time (h) | Conversion (%) |
| a Reaction conditions: 9.2 mmol respective epoxide substrates, 30 mg activated catalyst and 0.92 mmol (296 mg) TBA+Br− under 1.0 bar CO2 and at the optimized temperature (30/40 °C). Conversion (%) were calculated from the 1H NMR spectra by integration of epoxide peaks (a) versus cyclic carbonate peaks (a′) (Fig. S20–S24†). | |||||
| 1 | 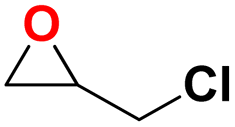 |
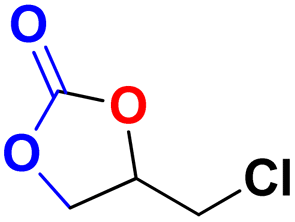 |
30 | 6 | 99.9 |
| 2 | 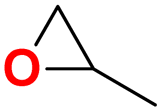 |
 |
30 | 6 | 99.9 |
| 3 |  |
 |
40 | 6 | 99.9 |
| 4 |  |
 |
40 | 12 | 99.9 |
| 5 |  |
 |
40 | 15 | 99.9 |
| 6 |  |
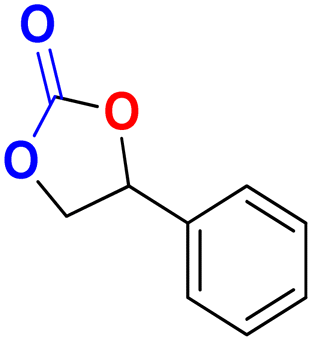 |
40 | 48 | 99.9 |
In general, the percentage conversion decreases as the size of the substrate molecule (Table S3†) increases. It becomes difficult for bigger molecules to interact with Cr(III) because of the steric hindrance and can be considered as a key factor of barrier for the interaction of the epoxide with the chromium center in the catalyst.63 The results indicate that efficient conversion of the bulky substrates can be achieved using elevated temperatures and an extended reaction time.
From the above results, the features of MIL-101(Cr)/AC-2 as a heterogeneous catalyst for the CO2 conversion reaction can be summarized as below:
(a) Excellent conversion at ambient temperature and atmospheric pressure while the parent MOF catalyst typically requires high temperature and pressure.
(b) Complete conversion in a shorter reaction time.
(c) Enhanced solution dispersibility and processibility (compared to the parent MOF).
(d) Easy recyclability as compared to other traditional organic homogeneous catalysts.
The above advantages make our composite indeed a better catalyst than the pristine MOF. In order to compare the efficiency of MIL-101(Cr)/AC-2 as a catalyst towards the CO2 conversion reactions with respect to the reported MIL-101(Cr) and MIl-101(Cr)-based MOF catalysts, a literature survey was done and the results are summarized in Table S4.† Table S4† indicates that most of the pristine MOF catalysts need relatively higher temperatures (30–100 °C) and high pressure (5–20 bar) for the CO2-epoxide cycloaddition reactions. The catalyst MIL-101(Cr)/AC-2 presents almost complete conversion comparatively in less time and under ambient reaction conditions. Thus, our composite catalyst would be the one of the best candidates among the MIL-101(Cr) based materials.
Conclusions
In summary, this work showcases the synthesis of new MIL-101(Cr)-Aminoclay composites and their utilization as heterogeneous catalysts for efficient conversion of CO2 gas into valuable cyclic carbonates. The highly porous and robust framework of MIL-101 is a well-studied prototype MOF containing Lewis acidic Cr(III) sites, but when employed as a heterogeneous catalyst for the cycloaddition reaction of CO2 with various epoxides, it mostly exhibits a poor conversion and requires drastic reaction conditions such as high temperature and pressure for a good conversion. On the other hand, the synergy of the two-dimensional clay, AC with the MIL-101(Cr) yielded novel nanocomposites which act as efficient catalysts under ambient conditions. One of the composites exhibits excellent catalytic efficiency showing 99.9% conversion with a variety of substrates at atmospheric pressure. This composite acts as a heterogeneous catalyst with recycling efficiency and also exhibits highly enhanced solution processability compared to the pristine MOF. It is believed that the present work would open a new route towards the fabrication of MOF–clay composites for promising energy and environmental applications like CO2 conversion and enhance the scope of novel MOF composites with enhanced functions.Experimental
Materials
No further purification was done to any of the commercially available reagents or solvents that were used. Terephthalic acid (C8H6O4), chromium nitrate nonahydrate (Cr(NO3)3·9H2O), magnesium chloride hexahydrate (MgCl2.6H2O), 3-aminopropyltrimethoxysilane (APTMS), ammonium fluoride (NH4F), tetrabutylammonium bromide (Bu4NBr), epichlorohydrin (EPH), styrene oxide (SO), propylene oxide (PO), allyl glycidyl ether (AGE), butyl glycidyl ether (BGE), and epoxy butane (EB) were purchased from Sigma-Aldrich Chemical Company, tetrabutylammonium chloride (TBAC) was purchased from TCI Chemicals (India) Pvt. Ltd. and tetrabutylammonium iodide (TBAI) was purchased from CDH Chemicals.Physical measurements
Powder X-ray diffraction (PXRD) patterns were recorded on a Rigaku Smart Lab SE instrument using Cu-Kα radiation (λ = 1.5406 Å) in the range of 2–50°. Fourier transform infrared spectroscopy (FTIR), recorded on a Bruker IFS 66v/S spectrophotometer in the area 4000–600 cm−1, was used to characterize the synthesized MOF and its composites. The Mettler Toledo-TGA 850 instrument was used to measure the thermal stability in a N2 atmosphere within the temperature range 30–800 °C at a heating rate of 5 °C min−1. Morphological studies have been carried out using a Zeiss GeminiSEM 500 Field Emission Electron Microscope (FESEM) by placing samples on a silicon wafer under high vacuum with an accelerating voltage of 20 kV. Energy dispersive spectroscopy (EDS) analysis was performed with an EDAX genesis instrument attached to the FESEM column. Adsorption isotherms of N2 (at 77 K) were recorded with the desolvated samples using the AUTOSORB iQ2 instrument. To prepare the desolvated samples, PMIL and the composites were degassed at 120 °C under 10–1 pa vacuum for 12 h before measurements. The surface areas of the samples were calculated from N2 adsorption data using the ASiQwin software. The temperature-programmed desorption of carbon dioxide (CO2-TPD) and ammonia (NH3-TPD) was performed using a BELCAT II instrument. Prior to each measurement i.e., CO2-TPD and NH3-TPD, 50 mg of sample were placed in a quartz reactor heated (10 °C min−1) to 120 °C in a flow of helium (50 ml min−1) for 12 h. After heating the sample was cooled to 30 °C and saturated with CO2 or NH3, using a flow of a gas of CO2 or NH3 in helium (50 ml min−1) for 30 min. Then, the sample was purged with helium for 30 min to remove the physically absorbed CO2 or NH3. Finally, the TPD experiment was carried out with a linear heating rate of 10 °C min−1 in a flow of He (50 ml min−1). The desorbed products were analyzed using a mass spectrometer. 1H NMR spectra were recorded in CDCl3 solvent using Magritek, Spinsolve Benchtop NMR.Catalytic study
Prior to catalytic studies, all the catalysts were activated at 120 °C under vacuum for 12 h. The performance of the different catalysts in the CO2 fixation experiments was checked under different reaction conditions varying the reaction time and temperature and the optimized reaction conditions were used for further catalytic reactions. All the catalytic reactions were performed by taking 30 mg of the respective activated catalyst (as entered in Table 1), 0.92 mmol (296 mg) of the co-catalyst tetrabutylammonium bromide (TBA+Br−) (10 mol% with respect to substrate), and 9.2 mmol of the substrate in a 30 ml Schlenk tube with CO2 purged at 1 bar (using CO2 balloon). For the catalytic reaction using a physical mixture of PMIL and AC, 95 mg of PMIL and 5 mg of AC (5 weight% w.r.t. total amount) were taken. Catalytic reactions were also performed without any catalyst and only using the co-catalyst TBA+Br−, at 30 °C and 40 °C (Table 1). Every cycloaddition reaction was carried out under solvent-free conditions in the Schlenk-tube with magnetic stirring. After the reaction, the solid heterogenous catalyst was removed by centrifugation and the product was purified by separation using ethyl acetate and water. The conversion (%) was calculated based on 1H NMR analysis by integration of epoxide peaks (a) versus cyclic carbonate peaks (a′) that are circled in the structures (refer to the 1H NMR spectra in the ESI†). For the recyclability tests, the recovered catalyst was washed with fresh CHCl3 thoroughly, air-dried and finally was activated at 120 °C under vacuum for 12 h perform further catalytic reactions under identical conditions.Synthesis
Conflicts of interest
There are no conflicts to declare.Acknowledgements
A. C. sincerely acknowledges the Department of Science and Technology (DST), New Delhi, India for an INSPIRE Faculty Fellowship (DST/INSPIRE/04/2020/001603). The authors are grateful to the Central University of Haryana, Mahendergarh for infrastructural support and characterization facilities. Jyoti acknowledges CUH for providing a research fellowship during the initial two years of research and UGC, New Delhi for the current Junior Research Fellowship (NTA Ref. No.: 221610140712). S. K. acknowledges CSIR, New Delhi for Senior Research Fellowship (09/1152(0021)/2020-EMR-I). Jyoti also acknowledges Ms. Ekta and Mr. Aman Kumar for their valuable contribution in analysing some NMR data. The authors gratefully acknowledge Prof. T. K. Maji of JNCASR, Bangalore and Prof. Richard Layfield and Dr Siddhartha De of University of Sussex, UK for some of the measurements.References
- Carbon dioxide: projected emissions and concentrations, https://ipcc-data.org/observ/ddc_co2.html.
- E. S. S. Pérez, C. R. Murdock, S. A. Didas and C. W. Jones, Chem. Rev., 2016, 116, 11840 CrossRef PubMed.
- A. Kätelhön, R. Meys, S. Deutz, S. Suh and A. Bardow, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 11187 CrossRef PubMed.
- Q. Liu, L. Wu, R. Jackstell and M. Beller, Nat. Commun., 2015, 6, 5933 CrossRef PubMed.
- M. Ding, R. W. Flaig, H.-L. Jiang and O. M. Yaghi, Chem. Soc. Rev., 2019, 48, 2783 RSC.
- C. Martín, G. Fiorani and A. W. Kleij, ACS Catal., 2015, 5, 1353 CrossRef.
- J. Chun, S. Kang, N. Kang, S. M. Lee, H. J. Kimb and S. U. Son, J. Mater. Chem. A, 2013, 1, 5517 RSC.
- L. Guo, R. Dou, Y. Wu, R. Zhang, L. Wang, Y. Wang, Z. Gong, J. Chen and X. Wu, ACS Sustainable Chem. Eng., 2019, 7, 16585 CrossRef CAS.
- S. Saengsaen, S. D. Gobbo and V. D'Elia, Chem. Eng. Res. Des., 2023, 191, 630 CrossRef CAS.
- Y. Chen, R. Luo, Q. Xu, J. Jiang, X. Zhou and H. Ji, ChemSusChem, 2017, 10, 2534 CrossRef CAS PubMed.
- H.-C. Zhou, J. R. Long and O. M. Yaghi, Chem. Rev., 2012, 112, 673 CrossRef CAS PubMed.
- T. Islamoglu, Z. Chen, M. C. Wasson, C. T. Buru, K. O. Kirlikovali, U. Afrin, M. R. Mian and O. K. Farha, Chem. Rev., 2020, 120, 8130 CrossRef CAS PubMed.
- S. Roy, A. Chakraborty and T. K. Maji, Coord. Chem. Rev., 2014, 273, 139 CrossRef.
- P. Yadav, P. Bhardwaj, M. Maruthi, A. Chakraborty and P. Kanoo, Dalton Trans., 2023, 52, 11725 RSC.
- P. Yadav, S. Kumari, A. Yadav, P. Bhardwaj, M. Maruthi, A. Chakraborty and P. Kanoo, ACS Omega, 2023, 8, 28367 CrossRef CAS PubMed.
- A. Yadav, S. Kumari, P. Yadav, A. Hazra, A. Chakraborty and P. Kanoo, Dalton Trans., 2022, 51, 15496 RSC.
- A. Yadav and P. Kanoo, Chem. – Asian J., 2019, 14, 3531 CrossRef CAS PubMed.
- A. Bavykina, N. Kolobov, I. S. Khan, J. A. Bau, A. Ramirez and J. Gascon, Chem. Rev., 2020, 120(16), 8468 CrossRef CAS PubMed.
- L. Zhu, X.-Q. Liu, H.-L. Jiang and L.-B. Sun, Chem. Rev., 2017, 117, 8129 CrossRef CAS PubMed.
- M. H. Beyzavi, C. J. Stephenson, Y. Liu, O. Karagiaridi, J. T. Hupp and O. K. Farha, Front. Energy Res., 2015, 2, 1 Search PubMed.
- C. Liu, L. Shi, J. Zhang and J. Sun, Chem. Eng. J., 2022, 427, 131633 CrossRef CAS.
- Z. Gao, L. Liang, X. Zhang, P. Xu and J. Sun, ACS Appl. Mater. Interfaces, 2021, 13, 61334 CrossRef CAS PubMed.
- S. Subramanian, J. Park, J. Byun, Y. Jung and T. C. Yavuz, ACS Appl. Mater. Interfaces, 2018, 10, 9478 CrossRef CAS PubMed.
- G. Férey, C. Serre, C. M. Draznieks, F. Millange, S. Surblé, J. Dutour and I. Margiolaki, Angew. Chem., Int. Ed., 2004, 43, 6296 CrossRef PubMed.
- S. Bhattacharjee, C. Chen and W.-S. Ahn, RSC Adv., 2014, 4, 52500 RSC.
- M. Y. Zorainy, M. G. Alalm, S. Kaliaguine and D. C. Boffito, J. Mater. Chem. A, 2021, 9, 22159 RSC.
- O. V. Zalomaeva, A. M. Chibiryaev, K. A. Kovalenko, O. A. Kholdeeva, B. S. Balzhinimaev and V. P. Fedin, J. Catal., 2013, 298, 179 CrossRef CAS.
- O. V. Zalomaeva, N. V. Maksimchuk, A. M. Chibiryaev, K. A. Kovalenko, V. P. Fedin and B. S. Balzhinimaev, J. Energy Chem., 2013, 22, 130 CrossRef CAS.
- J. Kim, S.-N. Kim, H.-G. Jang, G. Seo and W.-S. Ahn, Appl. Catal., A, 2013, 453, 175 CrossRef CAS.
- E. Akimana, J. Wang, N. V. Likhanova, S. Chaemchuen and F. Verpoort, Catalysts, 2020, 10, 453 CrossRef CAS.
- A. Marandi, M. Bahadori, S. Tangestaninejad, M. Moghadam, V. Mirkhani, I. Mohammadpoor-Baltork, R. Frohnhoven, S. Mathur, A. Sandleben and A. Klein, New J. Chem., 2019, 43, 15585 RSC.
- B. Aguila, Q. Sun, X. Wang, E. O'Rourke, A. M. Al-Enizi, A. Nafady and S. Ma, Angew. Chem., Int. Ed., 2018, 57, 1 CrossRef PubMed.
- M. Ding and H.-L. Jiang, ACS Catal., 2018, 8, 3194 CrossRef CAS.
- M. Bahadori, S. Tangestaninejad, M. Bertmer, M. Moghadam, V. Mirkhani, I. Mohammadpoor-Baltork, R. Kardanpour and F. Zadehahmadi, ACS Sustainable Chem. Eng., 2019, 7, 3962 CrossRef CAS.
- F. Li, Y. Chen, A. Gao, W. Tong, C. Ji, Y. Cheng and Y.-H. Zhou, New J. Chem., 2022, 46, 18418 RSC.
- Y. Liu, J. Li, Z. Zhang, Y. Hou, L. Wang and J. Zhang, Inorg. Chem., 2022, 61, 17438 CrossRef CAS PubMed.
- W. Dai, P. Mao, Y. Liu, S. Zhang, B. Li, L. Yang, X. Luo and J. Zou, J. CO2 Util., 2020, 36, 295 CrossRef CAS.
- Y. Sun, H. Huang, H. Vardhan, B. Aguila, C. Zhong, J. A. Perman, A. M. Al-Enizi, A. Nafady and S. Ma, ACS Appl. Mater. Interfaces, 2018, 10, 27124 CrossRef CAS PubMed.
- D. Liu, G. Li and H. Liu, Appl. Surf. Sci., 2018, 428, 218 CrossRef CAS.
- L.-F. Xiong, R. Bu, S.-L. Yang and E.-Q. Gao, Microporous Mesoporous Mater., 2022, 339, 111984 CrossRef CAS.
- L.-J. Zhou, W. Sun, N.-N. Yang, P. Li, T. Gong, W.-J. Sun, Q. Sui and E.-Q. Gao, ChemSusChem, 2019, 12, 2202 CrossRef CAS PubMed.
- W.-S. Liu, L.-J. Zhou, G. Li, S.-L. Yang and E.-Q. Gao, ACS Sustainable Chem. Eng., 2021, 9, 1880 CrossRef CAS.
- L.-F. Xiong, L.-J. Zhou, R. Bu, S.-L. Yang and E.-Q. Gao, Microporous Mesoporous Mater., 2022, 330, 111601 CrossRef CAS.
- J. Long, W. Dai, M. Zou, B. Li, S. Zhang, L. Yang, J. Mao, P. Mao, S. Luo and X. Luo, Microporous Mesoporous Mater., 2021, 318, 111027 CrossRef CAS.
- Y. Jiang, D. Li, Y. Zhao and J. Sun, J. Colloid Interface Sci., 2022, 618, 22 CrossRef CAS PubMed.
- D. Ma, B. Li, K. Liu, X. Zhang, W. Zou, Y. Yang, G. Li, Z. Shi and S. Feng, J. Mater. Chem. A, 2015, 3, 23136 RSC.
- Y. Jiang, Z. Wang, P. Xu and J. Sun, Cryst. Growth Des., 2021, 21, 3689 CrossRef CAS.
- Q.-L. Zhu and Q. Xu, Chem. Soc. Rev., 2014, 43, 5468 RSC.
- R. Lin, M. Chai, Y. Zhou, V. Chen, T. D. Bennett and J. Hou, Chem. Soc. Rev., 2023, 52, 4149 RSC.
- K. R. Datta, A. Achari and M. Eswaramoorthy, J. Mater. Chem. A, 2013, 1, 6707 RSC.
- A. Chakraborty, S. Laha, K. Kamali, C. Narayana, M. Eswaramoorthy and T. K. Maji, Inorg. Chem., 2017, 56, 9426 CrossRef CAS PubMed.
- A. Chakraborty, A. Achari, M. Eswaramoorthy and T. K. Maji, Chem. Commun., 2016, 52, 11378 RSC.
- A. Chakraborty, S. Roy, M. Eswaramoorthy and T. K. Maji, J. Mater. Chem. A, 2017, 5, 8423 RSC.
- D. Jiang, A. D. Burrows and K. J. Edler, CrystEngComm, 2011, 13, 6916 RSC.
- G. Johnsy, K. K. R. Datta, V. A. Sajeevkumar, S. N. Sabapathy, A. S. Bawa and M. Eswaramoorthy, ACS Appl. Mater. Interfaces, 2009, 1, 2796 CrossRef CAS PubMed.
- X. Quan, Z. Sun, H. Meng, Y. Han, J. Wu, J. Xu, Y. Xu and X. Zhang, Dalton Trans., 2019, 48, 5384 RSC.
- A. Rehman, F. Saleem, F. Javed, A. Ikhlaq, S. W. Ahmad and A. Harvey, J. Environ. Chem. Eng., 2021, 9, 105113 CrossRef CAS.
- J.-Q. Wang, D.-L. Kong, J.-Y. Chen, F. Cai and L.-N. He, J. Mol. Catal. A: Chem., 2006, 249, 143 CrossRef CAS.
- A. K. Guptaa, N. Guhab, S. Krishnanb, P. Mathura and D. K. Rai, J. CO2 Util., 2020, 39, 101173 CrossRef.
- B. Parmar, P. Patel, R. S. Pillai, R. K. Tak, R. I. Kureshy, N.-u. H. Khan and E. Suresh, Inorg. Chem., 2019, 58, 10084 CrossRef CAS PubMed.
- D. Tanaka, A. Henke, K. Albrecht, M. Moeller, K. Nakagawa, S. Kitagawa and J. Groll, Nat. Chem., 2010, 2, 410 CrossRef CAS PubMed.
- T. Kiyonaga, M. Higuchi, T. Kajiwara, Y. Takashima, J. Duan, K. Nagashima and S. Kitagawa, Chem. Commun., 2015, 51, 2728 RSC.
- R. Das, T. Ezhil, A. S. Palakkal, D. Muthukumar, R. S. Pillai and C. M. Nagaraja, J. Mater. Chem. A, 2021, 9, 23127 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: PXRD, IR spectra, TGA, FESEM images, size histogram plots, the EDX spectrum, pore size distribution, CO2-TPD and NH3-TPD, 1H NMR spectra, tables and reaction schemes. See DOI: https://doi.org/10.1039/d4dt00849a |
| This journal is © The Royal Society of Chemistry 2024 |