
A highly anisotropic family of hexagonal bipyramidal Dy(III) unsaturated 18-crown-6 complexes exceeding the blockade barrier over 2700 K: a computational exploration†
Shruti Moorthy ,
Ibtesham Tarannum‡
,
Kusum Kumari‡ and
Saurabh Kumar Singh*
,
Ibtesham Tarannum‡
,
Kusum Kumari‡ and
Saurabh Kumar Singh*
Department of Chemistry, Indian Institute of Technology, Hyderabad, Kandi, Sangareddy, Telangana 502284, India. E-mail: sksingh@chy.iith.ac.in
First published on 15th May 2024
Abstract
In the present work, we have explored a series of unsaturated hexa-18-crown-6 (U18C6) ligands towards designing highly anisotropic Dy(III) based single-ion magnets (SIMs) with the general formula [Dy(U18C6)X2]+ (where U18C6 = [C12H12O6] (1), [C12H12S6] (2), [C12H12Se6] (3), [C12H12O4S2] (4), [C12H12O4Se2] (5) and X = F, Cl, Br, I, OtBu and OSiPh3). By analysing the electronic structure, bonding and magnetic properties, we find that the U18C6 ligands prefer stabilising the highly symmetric eight-coordinated hexagonal bipyramidal geometry (HBPY-8), which is the source of the near-Ising type anisotropy in all the [Dy(U18C6)X2]+ complexes. Moreover, the ability of sulfur/selenium substituted U18C6 ligands to stabilize the highly anisotropic HBPY-8 geometry makes them more promising towards engineering the equatorial ligand field compared to substituted saturated 18C6 ligands where the exodentate arrangement of the S lone pairs results in low symmetry. Magnetic relaxation analysis predicts a record barrier height over 2700 K for [Dy(C12H12O6)F2]+ and [Dy(C12H12S6)X2]+ (where X = F, OtBu and OSiPh3) complexes, nearly 23% higher than those of the top performing Dy(III) based SIMs in the literature.
Introduction
Considerable attention has been directed towards designing lanthanide-based SIMs after the {TbPc2}− report by Ishikawa et al. showing exceptionally high barrier height for magnetic relaxation.1 The high stability of the trivalent oxidation state, simpler structures, and convenient functionalization to fine-tune the magnetic anisotropy make them the most appealing candidates for designing highly anisotropic SIMs.2–5 Among the studied lanthanide family of complexes, the Dy(III) ion is the best performing one for the isolation of highly anisotropic SIMs, including the [Dy(Cp*)(CpiPr5)][B(C6F5)4] (Cp* = pentamethyl-cyclopentadienyl, CpiPr5 = penta-iso-propylcyclopentadienyl) complex showing a record blocking temperature (TB) of ∼80 K.6 The giant first-order spin–orbit coupling (J = L + S) produces a 6H15/2 ground state, while the Kramer ion two-fold degeneracy guarantees magnetic bistability at zero-field, thus stabilizing the anisotropic f-electron density of the highest mJ |±15/2〉 as the ground state which is the key for the success of the oblate Dy(III) ion in highly anisotropic SIMs.7,8 Several strategies have been proposed, including designing two-coordinate linear complexes, shortening the Dy–L axial bonds compared to equatorial bonds, and achieving a higher-order axial symmetry around the Dy(III) ion to maximize the barrier height.5,8–25 Some notable works include the reports of organometallic sandwiched Dy(III) complexes with no equatorial ligands showing giant barrier heights between 1500 and 2200 K.6,24,26–28 In general, these complexes are highly air-sensitive in nature, and a recent theoretical study predicted the maximum barrier height to be 2200 K in the {DyCp2}+ family, which is a stumbling block for their further application.27In general, stabilizing air-stable highly anisotropic Dy(III) complexes requires a large coordination number (CN > 7) and higher-order axial symmetry such as square antiprismatic/axially compressed octahedral (D4d/D4h), pentagonal bipyramidal (D5h) and hexagonal bipyramidal (D6h) environments with short Dy–Lax bonds.14–22,29–34 Some notable work includes the report of [Dy(OtBu)2(L)4]+ with a barrier height exceeding 2000 K in an axially compressed octahedral (D4h) environment,17 [Dy(OtBu)2(py)5]+ with a pentagonal bipyramidal (PBP) geometry (D5h)35 and R/S-[Dy(LN6)(OSiPh3)2] in a D6h environment showing barrier heights more than 1800 K.21 Except for a few, in most of these complexes, the equatorial positions are occupied by anionic polydentate ligands, offering an undesirable equatorial ligand field for oblate type Dy(III) ions.18,19,36–38 Alternatively, the neutral crown ether macrocyclic ligands are relatively weak-field ligands compared to anionic polydentate ligands and hence offer a way to engineer the equatorial ligand field by modulating the donor atoms and ring size (see Fig. 1). Numerous Dy(III) complexes with various 12-crown-4 ether, 15-crown-5 ether, and 18-crown-6 ether ligands are reported in the literature and among all these, the cavity of saturated 18-crown-6 ether (18C6) is found to be suitable for stabilizing highly anisotropic [Dy(18C6)X2]+ (where X is the axial ligand) complexes.39–43 Surprisingly, most of the reported [Dy(18C6)X2]+ complexes show only moderate barrier heights (∼100 cm−1) due to the stabilization of the low-symmetry environment around Dy(III) ions resulting from the high conformational flexibility (C–C bond rotations) in the 18C6 ligands except for a [Dy(OtBu)Cl(18C6)][BPh4] complex showing a barrier height of ∼1000 K.22,25,39–43 In contrast, the unsaturated hexa-18-crown-6 (U18C6) ligands with C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C linkages offer conformation strain and prefer to stabilize the hexagonal bipyramidal environment around metal ions.44–46 Inspired by the exceptionally high coordinating capability of U18C6 towards the isolation of D6h geometry (preferable to generate an axial ligand field) and the suitable pore size of 18-crown-6 ligands to stabilize Ln(III) ions, here we investigated the electronic structure and magnetic properties in eleven Dy(III) complexes with the general formula [Dy(U18C6)X2]+ (where U18C6 = [C12H12O6] (1), [C12H12S6] (2), [C12H12Se6] (3), [C12H12O4S2] (4), [C12H12O4Se2] (5) and X = F, Cl, Br, I, OtBu and OSiPh3) (see Fig. 1) towards the search of new generation SIMs. All these complexes are named 1X–5X (where X = axial ligands, see Table S1†).
C linkages offer conformation strain and prefer to stabilize the hexagonal bipyramidal environment around metal ions.44–46 Inspired by the exceptionally high coordinating capability of U18C6 towards the isolation of D6h geometry (preferable to generate an axial ligand field) and the suitable pore size of 18-crown-6 ligands to stabilize Ln(III) ions, here we investigated the electronic structure and magnetic properties in eleven Dy(III) complexes with the general formula [Dy(U18C6)X2]+ (where U18C6 = [C12H12O6] (1), [C12H12S6] (2), [C12H12Se6] (3), [C12H12O4S2] (4), [C12H12O4Se2] (5) and X = F, Cl, Br, I, OtBu and OSiPh3) (see Fig. 1) towards the search of new generation SIMs. All these complexes are named 1X–5X (where X = axial ligands, see Table S1†).
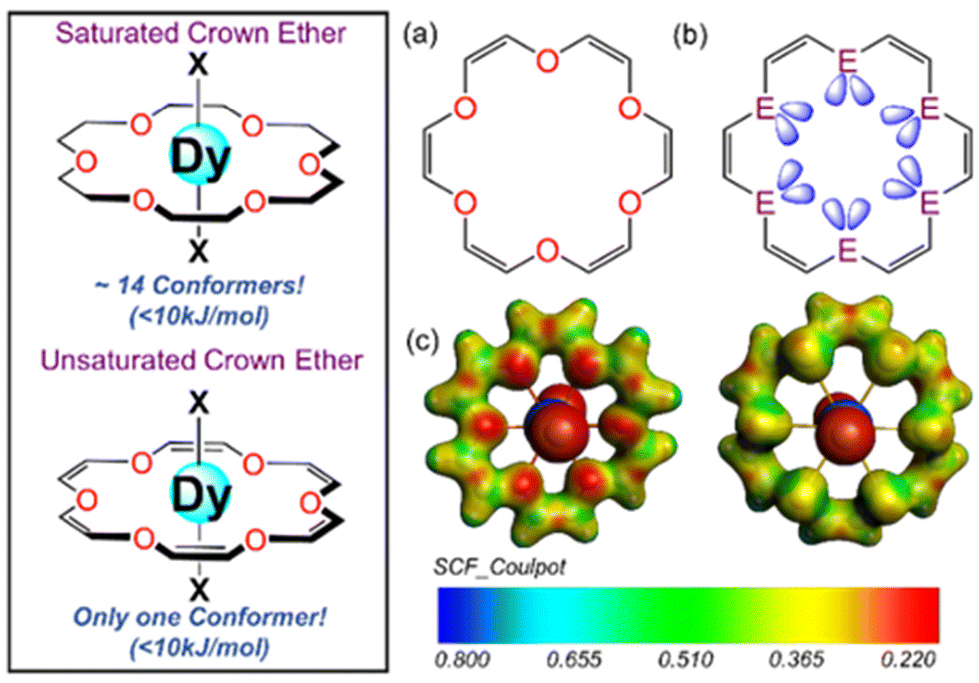 | ||
| Fig. 1 (a) Schematic representation of the unsaturated ring and (b) orientation of the lone pairs in the chalcogenide atoms (E = O/S/Se). (c) DFT-computed molecular electrostatic potential (MEP) maps for 1X and 2X. Red and blue represent the most electronegative and electropositive regions, respectively. | ||
Computational details
Gas phase geometry optimizations of all the complexes were carried out using ORCA 5.0.3 code47 at the BP86 level of theory.48,49 For the Dy atom, core electrons were replaced by the def2-ECP pseudopotential (for 28 core electrons, lmax = 5), while the triple ζ-quality def2-TZVP basis set was used to treat the valence electrons. For O, S, Se, F, Cl, and Br, we used the def2-TZVP basis set,50 while Sapporo-TZP basis sets were used for the I atom.51 The C and H atoms were treated using a def2-SVP basis set.50 The dispersion corrections were accounted for by using Grimme's dispersion with the Becke–Johnson (D3BJ) method as incorporated in the ORCA code.52 A very tight SCF (1 × 10−8 Eh) criterion was chosen for energy minimization. The “slowconv” and “KDIIS” criteria and large integration grid settings (GRID9 for Dy) were turned on throughout the calculations for smooth convergence. Vibrational frequency calculations show no negative frequency, thus confirming the stationary point as the local minimum. In addition, we tested our computational methodology by performing geometry optimization of twelve different reported mononuclear Dy(III) complexes (see Scheme S1†). DFT optimization nicely reproduces the X-ray crystal structures of all twelve complexes (see the ESI† for details), which provides confidence in applying our computational methodology to predict the novel geometry of [Dy(U18C6)X2]+ complexes.Conformational search analysis was carried out using xTB-CREST code to find all the conformers within an energy window of ∼10 kJ mol−1 (see the ESI† for details).53 All the obtained conformers were further optimised at the BP86 level of theory, and the lowest energy conformer was used for the bonding and magnetic property calculations. Single-point energy calculations were carried out using the hybrid PBE0 functional49 in ADF 2021 code to analyze the bonding interactions and energy decomposition analysis. Scalar relativistic effects were incorporated by using zeroth-order relativistic approximations (ZORA).54 The Slater-type all electron TZP basis set was used for the Dy atom and the DZP basis set for the remaining atoms, with “no frozen core” approximation (see the ESI† for details).
Complete-active space self-consistent field (CASSCF) calculations were performed on the DFT-optimized lowest energy structure to compute the magnetic properties.55 Here, we employed an all-electron SARC–DKH–TZVP basis set for the Dy(III) centre, the DKH-adapted version of def2–TZVP for O, S, Se, F, Cl, and Br atoms and Sapporo–DKH–TZP for the I atom.50,51,56 Using an active space of CAS(9,7), we computed 21 sextets and 224 quartets and performed the spin–orbit calculations using the spin–orbit mean field (SOMF-1X) operator. The computed spin-free energy and spin–orbit energies are provided in Tables S14–S19.† Ab initio ligand field theory (AILFT) calculations were performed at the CASSCF levels of theory to estimate the interelectronic repulsion in terms of Slater–Condon parameters and one electron energies to represent the f-orbital splitting.57 Next, we computed the effective demagnetisation barrier (Ueff) for the Orbach relaxation process as proposed earlier by Aravena et al.58 (see the ESI† for computational details).
Results and discussion
Initially, we carried out a conformational search analysis using the CREST code,53 which predicts the stabilization of only one stable eight-coordinated conformer within the 10 kJ mol−1 range for all complexes 1x–5x (X = F, Cl, Br and I) with the macrocyclic U18C6 ligand occupying the equatorial position around the central Dy(III) ion and the axial positions occupied by X ligands, resulting in the HBPY-8 geometry. Compared to the highly symmetric single conformer with U18C6 ligands, we observed 14 low-lying conformers within 10 kJ mol−1 with 18C6 ligands, significantly deviating from the HBPY-8 geometry (see Fig. 1). Next, we performed gas-phase geometry optimization of all the complexes at the BP86 level of theory (see Table S2†). The DFT optimized geometry of 1X–5X complexes shows that the Dy(III) ion lies in the plane formed by the six donor atoms of U18C6 while the two halide ions form a near linear ∠X–Dy–X bond angle ∼150°–180°. The average Dy–O bond length in 1X complexes ranges between 2.630 Å and 2.673 Å, which matches well with the previously reported Dy–O bond lengths with 18-crown-6 ligands (see Table S20†).39,40,42 In the 2X family of complexes, we noticed that the avg. Dy–S bond length ranges between 3.154 Å and 3.185 Å. Moreover, the computed Dy–X bond lengths in the 1X–5X family of complexes agree well with Dy–X bond lengths reported in the literature (see Table S20†), highlighting the robustness of our computational methodology in predicting X-ray geometry. The fluoride analogue has the shortest ∠X–Dy–X bond angle (150.2° for 1F and 150.6° for 2F), and the ∠X–Dy–X bond angle approaches linearity as we move towards heavier halides. Besides, in all the cases, the average axial to equatorial bond length ratio is always ≤1, indicating stabilization of the dominant axial ligand field around the Dy(III) ion in all the complexes.Next, we computed the binding energy between the {DyX2}+ fragment and {U18C6} fragments in complexes 1x–5x using an energy decomposition analysis (EDA) approach within the scalar relativistic density functional theory (SR-DFT). SR-DFT calculations predict stabilizing interactions between the fragments for all the complexes, with oxa crown complexes relatively more stabilized than thia and selena crown complexes (see the ESI† for details). The decomposition of total interaction energy (ΔEint) suggests that the electrostatic (ΔEelstat) and orbital interactions (ΔEorb) are the most dominant contributions (50–80%) to the ΔEint (see Fig. S2†). Tables S4 and S5† show that as we move from oxa to thia/selena crown complexes, the ΔEelstat interaction decreases significantly due to a decrease in the electronegativity as we move down from O to S/Se atom (see Fig. 1C and Fig. S1† for computed MEPs). In contrast, the strength of the ΔEorb value marginally increases as we move from O to S/Se, which is attributed to weak lanthanide-ligand covalency (see Tables S6 and S7†). In addition, we have also compared the EDA analysis results of 1Cl with those of U18C6 and 18C6 ligands, which show relatively higher binding energy with 18C6 ligands compared to U18C6 ligands (see Tables S4 and S5†).
Next, we performed complete active space self-consistent field (CASSCF) calculations on the DFT-optimized geometry to compute the relevant spin Hamiltonian (SH) parameters (see the ESI† for computational details). Before discussing the analysis of SH parameters and SIM behaviour in complexes 1X–5X, we first compared the magnetic properties of Dy(III) complexes with those of U18C6 and 18C6 ligands. Here, we have analyzed the magnetic anisotropy of 1Cl with that of saturated and unsaturated crown ethers (see Fig. 2 and S3†). From a structural point of view, the [Dy(U18C6)Cl2]+ complex is relatively more symmetric and closer to the HBPY-8 geometry (CShM = 0.756) compared to the [Dy(18C6)Cl2]+ complex (CShM = 1.592). The large CShM value arises from a distorted equatorial plane formed by the saturated crown ether ligand. A close inspection of the structural parameters reveals that the axial Dy–Cl bonds are relatively shorter compared to Dy–O bonds in [Dy(U18C6)Cl2]+ (avg. Dy–Cl![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :avg. Dy–O = 1:1.04), while an opposite trend is observed for the [Dy(18C6)Cl2]+ complex (avg. Dy–Cl:avg. Dy–O = 1:0.96). This indicates that the strength of the equatorial ligand field is expected to be more significant in saturated crown ether than in unsaturated crown ether, which is also evident from EDA analysis. CASSCF calculations evidence these minor structural differences and predict the wide energy span of the low-lying eight KDs in [Dy(U18C6)Cl2]+ (1132 cm−1) compared to the [Dy(18C6)Cl2]+ complex (740 cm−1). The ground state g-values are highly axial for both the complexes, while low-symmetry around the Dy(III) ion in the [Dy(18C6)Cl2]+ complex results in a non-negligible transverse component (gxx and gyy) in the ground state g-values. The analysis of mJ states and g-tensor orientation predicts magnetic blocking up to the 4th excited Kramer doublet (KD) for the [Dy(U18C6)Cl2]+ complex, while the [Dy(18C6)Cl2]+ complex relaxes via the 3rd excited KD. Ab initio computed magnetic relaxation shows extremely weak QTM (1.61 × 10−6μB) values for the [Dy(U18C6)Cl2]+ complex compared to that of the [Dy(18C6)Cl2]+ complex showing a nearly 1000 times higher QTM value (2.52 × 10−3μB). The U18C6 ligand has an advantage over 18C6 ligands as they offer a weak equatorial ligand field and higher-order symmetry around the Dy(III) ion, which helps in the complete quenching of the QTM in the [Dy(U18C6)Cl2]+ complex. The computed Ucal value of 1036 cm−1 for the [Dy(U18C6)Cl2]+ complex is ∼2× times higher than that of the [Dy(18C6)Cl2]+ complex (535 cm−1), indicating a promising behaviour of the U18C6 ligand towards designing highly anisotropic Dy(III) based SMMs. From these exciting results, we moved to the next section and analyzed the magnetic anisotropy and magnetic relaxation mechanism in complexes 1X–5X.
:avg. Dy–O = 1:1.04), while an opposite trend is observed for the [Dy(18C6)Cl2]+ complex (avg. Dy–Cl:avg. Dy–O = 1:0.96). This indicates that the strength of the equatorial ligand field is expected to be more significant in saturated crown ether than in unsaturated crown ether, which is also evident from EDA analysis. CASSCF calculations evidence these minor structural differences and predict the wide energy span of the low-lying eight KDs in [Dy(U18C6)Cl2]+ (1132 cm−1) compared to the [Dy(18C6)Cl2]+ complex (740 cm−1). The ground state g-values are highly axial for both the complexes, while low-symmetry around the Dy(III) ion in the [Dy(18C6)Cl2]+ complex results in a non-negligible transverse component (gxx and gyy) in the ground state g-values. The analysis of mJ states and g-tensor orientation predicts magnetic blocking up to the 4th excited Kramer doublet (KD) for the [Dy(U18C6)Cl2]+ complex, while the [Dy(18C6)Cl2]+ complex relaxes via the 3rd excited KD. Ab initio computed magnetic relaxation shows extremely weak QTM (1.61 × 10−6μB) values for the [Dy(U18C6)Cl2]+ complex compared to that of the [Dy(18C6)Cl2]+ complex showing a nearly 1000 times higher QTM value (2.52 × 10−3μB). The U18C6 ligand has an advantage over 18C6 ligands as they offer a weak equatorial ligand field and higher-order symmetry around the Dy(III) ion, which helps in the complete quenching of the QTM in the [Dy(U18C6)Cl2]+ complex. The computed Ucal value of 1036 cm−1 for the [Dy(U18C6)Cl2]+ complex is ∼2× times higher than that of the [Dy(18C6)Cl2]+ complex (535 cm−1), indicating a promising behaviour of the U18C6 ligand towards designing highly anisotropic Dy(III) based SMMs. From these exciting results, we moved to the next section and analyzed the magnetic anisotropy and magnetic relaxation mechanism in complexes 1X–5X.
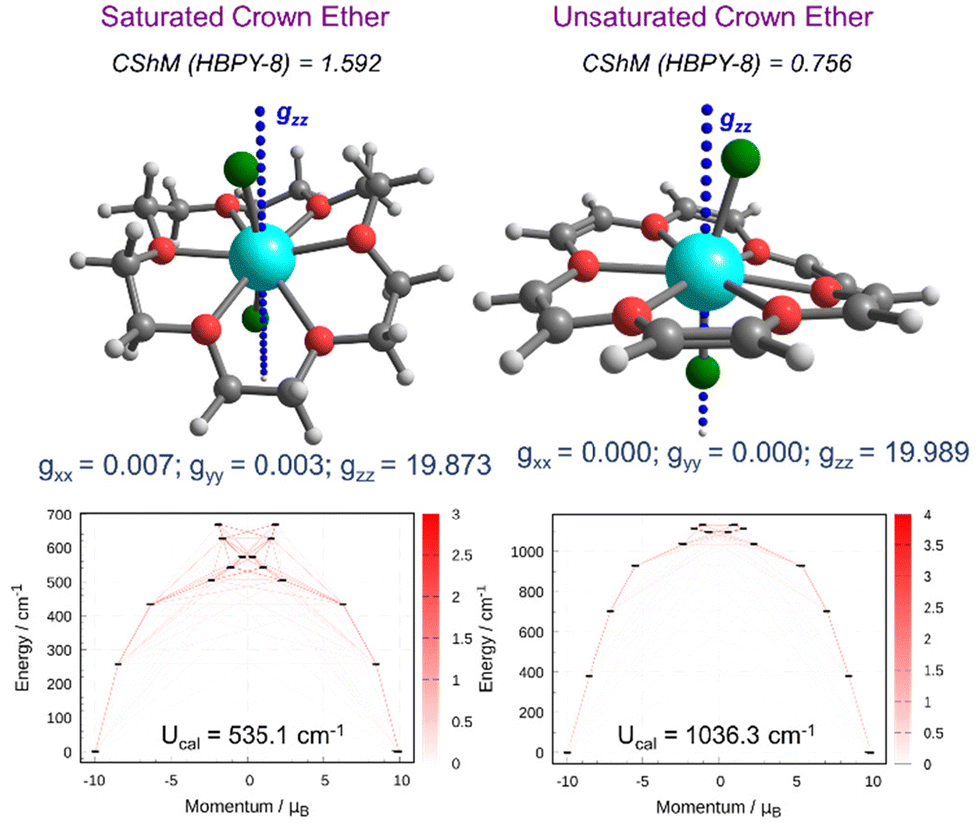 | ||
| Fig. 2 SINGLE_ANISO computed g-tensor orientation (top) and blockade barrier (bottom) for complexes [DyCl2(18C6)]+ (left) and [DyCl2(U18C6)]+ (right). Color code: Dy (cyan), O (red), Cl (green), C (grey) and H (white). | ||
Among all the complexes 1X–5X, we observed stabilization of mJ |±15/2〉 as the ground state, indicating the stabilization of the dominant axial ligand field. The computed ground state g-values (gzz ∼ 19.9 and gxx ∼ gyy ∼ 1 × 10−3) are highly axial and possess near-Ising type anisotropy for all the complexes. The computed gzz orientation of the ground state KD nearly passes through the X–Dy–X bond, indicating that the g-tensor orientation follows the highest order pseudo-C6 axis in all the complexes (see Fig. S5†). Among the studied 1X series, we observed that the first excited KD is 656.6, 381.3, 340.5 and 274.3 cm−1 above the ground state for 1F, 1Cl, 1Br and 1I, respectively (see Table S14†). The average axial to equatorial bond length ratios are 0.7 (1F), 0.9 (1Cl), 1.0 (1Br), and 1.1 (1I), indicating that lighter halides are structurally more favourable to generate an axial ligand field.59 In addition, we also observed an enormous negative charge on the F− ion compared to other halide ions (see Fig. S6†), which further leverages the axiality, resulting in the larger KD span for 1F. CASSCF computed ab initio ligand field theory (AILFT) predicts the following f-orbital manifold of 2815.3 (1F), 1626.9 (1Cl), 1315.3 (1Br), and 1035.2 (1I) cm−1 with giant f-orbital splitting for 1F, which corroborates the splitting of the KDs (see Fig. S7 and S8†). Calculations predict the magnetic relaxation via the 4th excited KD, i.e. mJ |±1/2〉 for all the complexes, setting the Ucal values of 1889, 1036.3, 844.6, and 644.8 cm−1 for 1F, 1Cl, 1Br and 1I, respectively (see Fig. S9†). Most importantly, the computed magnetic dipole matrix elements (kQTM) between ground state KDs are extremely weak (∼1 × 10−4μB), indicating quenching of quantum tunnelling of magnetization (QTM) in all the complexes. An extremely short Dy–F bond and sizeable negative charge on F− ions generate a more potent ligand field than heavier halides, resulting in a giant barrier height of 1889 cm−1 (2717 K) in 1F. Although complexes 1Br and 1I are not structurally favourable as the average axial/equatorial bond length ratio is ≥1, the isolation of the HBPY-8 geometry by neutral U18C6 ligands is the key for the highly anisotropic environment around the Dy(III) ion.
In order to fine-tune the magnetic anisotropy through ligand field modification, we have examined the complexes of sulfur and selenium-substituted crown ethers. For comparison, we have first examined the magnetic anisotropy of 2Cl and its saturated thia crown analogue. Unlike 2Cl where the endodentate arrangement of the S lone pairs forces the Dy(III) ion to sit in the middle of the ring, the exodentate nature of the S lone pair in saturated thia crown ligands throws the {DyCl2}+ fragment away from the center of the ring, stabilizing the low-symmetry environment (see Fig. 3). For complex 2Cl, we observed a near Ising-type ground state (gxx = gyy = 1 × 10−4 and gzz = 19.914) with magnetic blockade up to the 4th KD, resulting in a record Ucal of 1140 cm−1, a nearly 10% increase compared to that of 1Cl. On the other hand, a smaller Ucal value of 420.4 cm−1 is observed for the saturated analogue of 2Cl, which is nearly 3× times smaller than that of 2Cl. Moreover, the computed kQTM values are two orders of magnitude larger in the saturated analogue of 2Cl, suggesting strong QTM within the ground state. Comparative analysis indicates that ligand functionalization is not feasible in the saturated crown-ether based ligands as the exodentate arrangement of the S lone pair destroys the HBPY-8 arrangement, hence limiting the application of these ligands to engineer the equatorial ligand field.
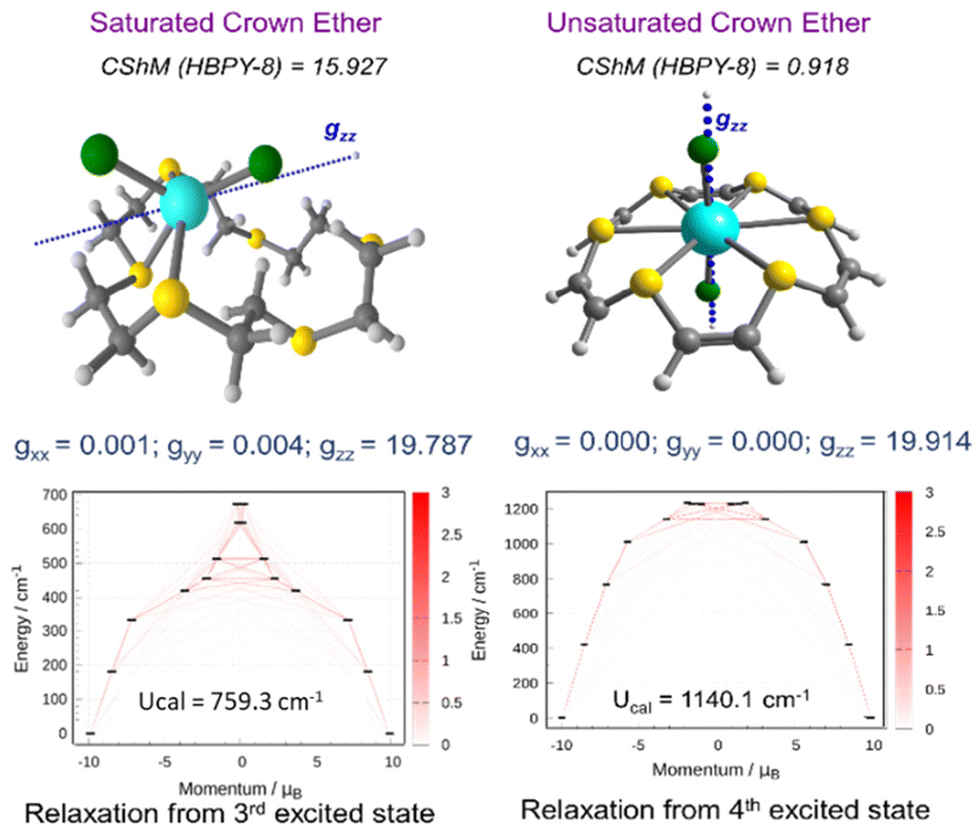 | ||
| Fig. 3 SINGLE_ANISO computed g-tensor orientation (top) and blockade barrier (bottom) for the saturated analogue of 2Cl (left) and 2Cl (right). Color code: Dy (cyan), S (yellow), Cl (green), C (grey) and H (white). | ||
Compared to the 1x series, we observed that complexes 2F–2I are highly symmetric, where six endodentate S atoms are arranged in a highly symmetric hexagonal manner. The computed CShM values are much smaller and closer to the ideal HBPY-8 geometry (see Table S2†). As a result, we observed a higher degree of axiality in the g-values and much smaller kQTM values compared to those of the 1X series (see Table 1). For 2X, we observed the following Ucal values: 1972.2 (2F), 1140.1 (2Cl), 934.6 (2Br), and 725.9 cm−1 (2I); these values are reasonably higher than those of the 1X series (see Table 1). The computed g-values and magnetic relaxation pattern for all 2F–2I are depicted in Fig. S9.† We noticed an increase of ∼100 cm−1 in the Ucal values as we moved from oxa crowns to thia crown complexes. Surprisingly, the increase in the barrier height is not colossal as the Dy-ligand covalency increases as we move from O to S, which counteracts the electrostatic effect, resulting in a modest increase in the Ucal value.
| gzz | kQTM | Ucal | Ueff | TB | |
|---|---|---|---|---|---|
| 1F | 19.908 | 3.04 × 10−5 | 1889.0 | 1710 | 67.5 |
| 1Cl | 19.989 | 1.61 × 10−6 | 1036.3 | 1087 | 37.0 |
| 1Br | 19.906 | 1.43 × 10−5 | 844.6 | 890 | 30.2 |
| 1I | 19.904 | 4.11 × 10−5 | 644.8 | 686 | 23.0 |
| 2F | 19.913 | 1.31 × 10−5 | 1972.2 | 1863 | 70.4 |
| 2Cl | 19.914 | 1.32 × 10−6 | 1140.1 | 1196 | 40.7 |
| 2Br | 19.913 | 8.96 × 10−7 | 934.6 | 1008 | 33.4 |
| 2I | 19.912 | 1.02 × 10−5 | 725.9 | 797 | 25.9 |
| 3Cl | 19.915 | 3.63 × 10−6 | 1133.3 | 1201 | 40.5 |
| 4Cl | 19.907 | 1.72 × 10−4 | 942.9 | 960 | 33.7 |
| 5Cl | 19.909 | 1.16 × 10−4 | 1043.0 | 1039 | 37.3 |
| 2OSiPh3 | 19.905 | 3.60 × 10−5 | 1903.3 | 1623 | 67.9 |
| 2OtBu | 19.913 | 2.86 × 10−6 | 2397.2 | 1886 | 85.6 |
Next, we investigated the selena crown complex (3Cl), where DFT predicts a substantially more bent ∠Cl–Dy–Cl bond angle than those for 2Cl and 1Cl, owing to the huge cavity provided by selena crowns (see Table S3†). As a result, we do not see any improvement in the g-values and magnetic relaxation pattern for 3Cl compared to those of 2Cl. Next, we studied magnetic anisotropy in 4Cl and 5Cl complexes arising from the oxa-thia (O4S2) and oxa-selena (O4Se2) ligands, and we observed stabilization of the mJ |±15/2〉 and reasonably large Ucal values of 942.9 cm−1 (1319 K) and 1043 cm−1 (1460 K) for complexes 4Cl and 5Cl, respectively. Among all the studied complexes, we observed that U18C6 prefers to stabilize the HBPY-8 environment around the Dy(III) ion, resulting in the magnetization blockade 4th KD and generating giant Ucal values in the range of ∼650–1900 cm−1 (∼900–2800 K) for these complexes. Next, we studied two model complexes of the best-performing 2X series, where the bulky OtBu and OSiPh3 ligands replace the halides at axial positions. The synthesis and isolation of these complexes often require a bulky group at the axial position to prevent multiple coordination and to maintain the axiality.32,61,62 DFT optimization predicts the stabilization of the HBPY-8 geometry around both complexes. Magnetic anisotropy and magnetic relaxation analyses indicate a giant barrier height of ∼2600 K and 3450 K for 2OSiPh3 and 2OtBu complexes, respectively, further highlighting the possibility of designing U18C6 ligand-based air-stable Dy(III) complexes (see Table S19 and Fig. S13†).
Next, we computed the effective demagnetization barrier (Ueff) using the ab initio computed KD's energy and transition dipole moments to analyze the Orbach magnetic relaxation pathways.58 For all the complexes, the temperature-dependent effective demagnetization barrier (Ueff) and relative contribution from each KD are provided in Table S12, Fig. S11 and S12,† and Fig. 4. A close inspection of the relaxation pattern suggests that at low temperatures (0–100 K), the Ueff values nearly remain zero, indicating all the population to be in the mJ |±15/2〉 ground state, which is blocked due to the minimal kQTM value (∼1 × 10−4μB). As the temperature increases beyond 100 K, the Ueff value increases by climbing to the higher excited KD with sequential absorption of the thermally available phonons, while the saturation in the Ueff plot at room temperature indicates rapid relaxation by spontaneously emitting phonons. For 2F, we noticed saturation in the Ueff with maximum contributions from the following three KDs : KD6 24% + KD5 18% + KD8 16% (see Table S12†). Most importantly, the Ueff values are nearly similar to the ab initio computed Ucal values for all the studied complexes, further confirming giant barrier height in these complexes (see Table 1 and Fig. 4). The computed Ueff value of ∼2700 K for 2F and 2OtBu is nearly ∼23% higher than the best reported Ucal value for the {DyCp2}+ family in the literature.
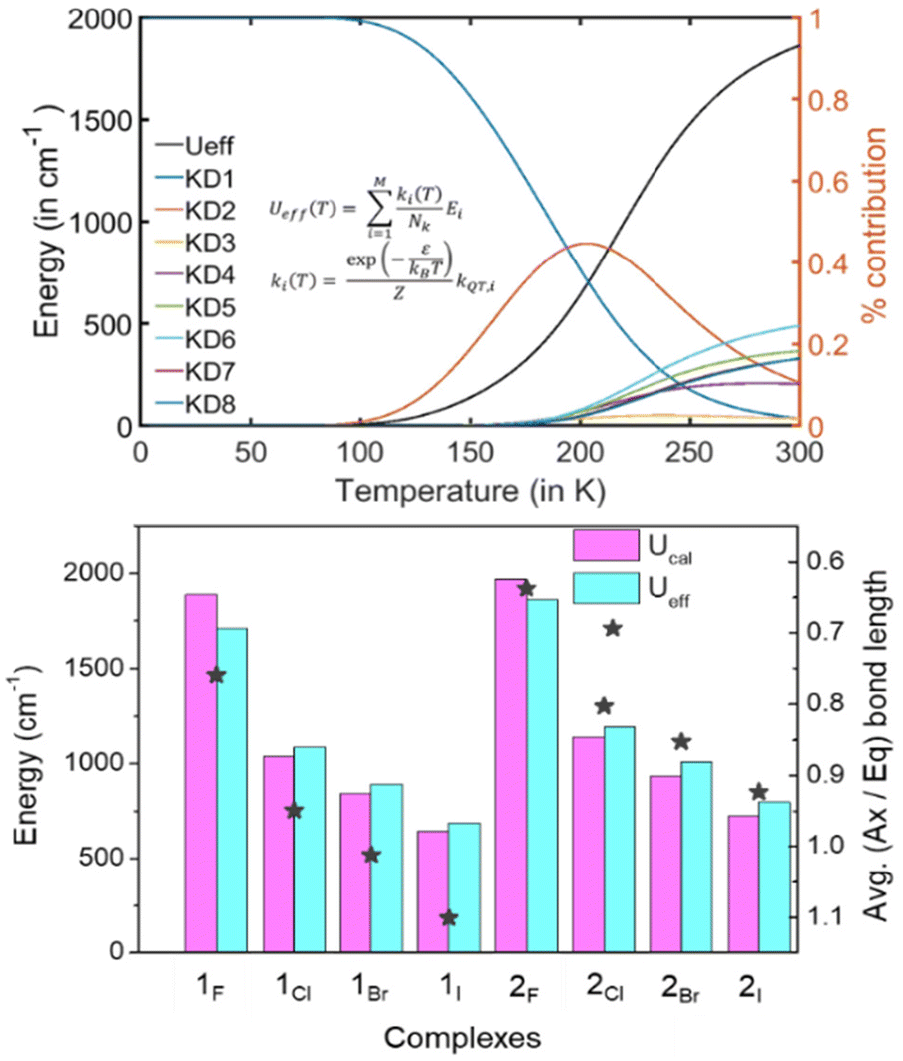 | ||
| Fig. 4 Temperature dependence of calculated Ueff along with the relative contributions from the KDs for 2F (top) and CASSCF computed Ucal and Ueff values along with the ratio of axial and equatorial bond distances for 1X–2X complexes (bottom). | ||
Conclusion
Our comprehensive computational study indicates that, in contrast to 18C6 ligands, U18C6 ligands are highly selective towards stabilizing the hexagonal bipyramidal geometry (D6h), which is the key for generating near-Ising type anisotropy. Magnetic anisotropy calculations predict mJ |±15/2〉 as the ground state for all the complexes 1X–5X, which quenches the ground state QTM. By modulating the donor atoms, we predicted that the endodentate arrangement of S atoms in the thia crown complexes stabilizes higher-order symmetry and offers a recipe to achieve giant barrier height in the 2X series. Our calculations predict a giant barrier height of ∼2700 K in 1F and 2F complexes, which surpasses the record barrier height reported for the family of {DyCp2}+ complexes. Overall, our finding indicates that the U18C6 ligand framework holds great potential for the development of high-temperature Dy(III) SMMs, which can outperform well-known organometallic sandwiched Dy(III) complexes.Conflicts of interest
There are no conflicts to declare.Acknowledgements
SKS acknowledges the Science and Engineering Research Board (CRG/2023/002936) and IIT Hyderabad for generous funding. SM and KK acknowledge PMRF. IT is thankful for the UGC-NFOBC fellowship. The support and resources provided by PARAM Seva at IITH are acknowledged. This work is dedicated to Professor R. Murugavel on the occasion of his 60th Birthday.References
- N. Ishikawa, M. Sugita, T. Ishikawa, S. Koshihara and Y. Kaizu, J. Am. Chem. Soc., 2003, 125, 8694–8695 CrossRef CAS PubMed.
- J. Luzon and R. Sessoli, Dalton Trans., 2012, 41, 13556–13567 RSC.
- D. N. Woodruff, R. E. P. Winpenny and R. A. Layfield, Chem. Rev., 2013, 113, 5110–5148 CrossRef CAS PubMed.
- S. G. McAdams, A.-M. Ariciu, A. K. Kostopoulos, J. P. S. Walsh and F. Tuna, Coord. Chem. Rev., 2017, 346, 216–239 CrossRef CAS.
- P. Zhang, L. Zhang and J. Tang, Dalton Trans., 2015, 44, 3923–3929 RSC.
- F.-S. Guo, B. M. Day, Y.-C. Chen, M.-L. Tong, A. Mansikkamäki and R. A. Layfield, Science, 2018, 362, 1400–1403 CrossRef CAS PubMed.
- N. F. Chilton, Annu. Rev. Mater. Res., 2022, 52, 79–101 CrossRef CAS.
- J. D. Rinehart and J. R. Long, Chem. Sci., 2011, 2, 2078–2085 RSC.
- A. Zabala-Lekuona, J. M. Seco and E. Colacio, Coord. Chem. Rev., 2021, 441, 213984 CrossRef CAS.
- B.-C. Liu, N. Ge, Y.-Q. Zhai, T. Zhang, Y.-S. Ding and Y.-Z. Zheng, Chem. Commun., 2019, 55, 9355–9358 RSC.
- N. F. Chilton, Inorg. Chem., 2015, 54, 2097–2099 CrossRef CAS PubMed.
- K. Kotrle, M. Atanasov, F. Neese and R. Herchel, Inorg. Chem., 2023, 62, 17499–17509 CrossRef CAS PubMed.
- I. Tarannum, S. Moorthy and S. K. Singh, Dalton Trans., 2023, 52, 15576–15589 RSC.
- S. Bala, G.-Z. Huang, Z.-Y. Ruan, S.-G. Wu, Y. Liu, L.-F. Wang, J.-L. Liu and M.-L. Tong, Chem. Commun., 2019, 55, 9939–9942 RSC.
- B. Zhang, X. Guo, P. Tan, W. Lv, X. Bai, Y. Zhou, A. Yuan, L. Chen, D. Liu, H.-H. Cui, R. Wang and X.-T. Chen, Inorg. Chem., 2022, 61, 19726–19734 CrossRef CAS PubMed.
- H. Wu, B. Guo, X. Han, M. Li, B. Yin, Y. Chen, Y. Bian, H. Ke and S. Chen, Cryst. Growth Des., 2023, 23, 309–319 CrossRef CAS.
- X.-L. Ding, Y.-Q. Zhai, T. Han, W.-P. Chen, Y.-S. Ding and Y.-Z. Zheng, Chem. – Eur. J., 2021, 27, 2623–2627 CrossRef CAS PubMed.
- P. Kalita, N. Ahmed, S. Moorthy, V. Béreau, A. K. Bar, P. Kumar, P. Nayak, J.-P. Sutter, S. K. Singh and V. Chandrasekhar, Dalton Trans., 2023, 52, 2804–2815 RSC.
- P. Kalita, N. Ahmed, A. K. Bar, S. Dey, A. Jana, G. Rajaraman, J.-P. Sutter and V. Chandrasekhar, Inorg. Chem., 2020, 59, 6603–6612 CrossRef CAS PubMed.
- A. B. Canaj, S. Dey, C. Wilson, O. Céspedes, G. Rajaraman and M. Murrie, Chem. Commun., 2020, 56, 12037–12040 RSC.
- C. Zhao, Z. Zhu, X.-L. Li and J. Tang, Inorg. Chem. Front., 2022, 9, 4049–4055 RSC.
- Y.-S. Ding, W. J. A. Blackmore, Y.-Q. Zhai, M. J. Giansiracusa, D. Reta, I. Vitorica-Yrezabal, R. E. P. Winpenny, N. F. Chilton and Y.-Z. Zheng, Inorg. Chem., 2022, 61, 227–235 CrossRef CAS PubMed.
- N. F. Chilton, S. K. Langley, B. Moubaraki, A. Soncini, S. R. Batten and K. S. Murray, Chem. Sci., 2013, 4, 1719–1730 RSC.
- A. H. Vincent, Y. L. Whyatt, N. F. Chilton and J. R. Long, J. Am. Chem. Soc., 2023, 145, 1572–1579 CrossRef CAS PubMed.
- K.-X. Yu, Y.-S. Ding, Y.-Q. Zhai, T. Han and Y.-Z. Zheng, Dalton Trans., 2020, 49, 3222–3227 RSC.
- P. Evans, D. Reta, G. F. S. Whitehead, N. F. Chilton and D. P. Mills, J. Am. Chem. Soc., 2019, 141, 19935–19940 CrossRef CAS PubMed.
- D. Reta, J. G. C. Kragskow and N. F. Chilton, J. Am. Chem. Soc., 2021, 143, 5943–5950 CrossRef CAS PubMed.
- F.-S. Guo, B. M. Day, Y.-C. Chen, M.-L. Tong, A. Mansikkamäki and R. A. Layfield, Angew. Chem., Int. Ed., 2017, 56, 11445–11449 CrossRef CAS PubMed.
- Y.-C. Chen, J.-L. Liu, L. Ungur, J. Liu, Q.-W. Li, L.-F. Wang, Z.-P. Ni, L. F. Chibotaru, X.-M. Chen and M.-L. Tong, J. Am. Chem. Soc., 2016, 138, 2829–2837 CrossRef CAS PubMed.
- Y. Chen, J. Liu, Y. Lan, Z. Zhong, A. Mansikkamäki, L. Ungur, Q. Li, J. Jia, L. F. Chibotaru, J. Han, W. Wernsdorfer, X. Chen and M. Tong, Chem. – Eur. J., 2017, 23, 5708–5715 CrossRef CAS PubMed.
- J. Liu, Y.-C. Chen, J.-L. Liu, V. Vieru, L. Ungur, J.-H. Jia, L. F. Chibotaru, Y. Lan, W. Wernsdorfer, S. Gao, X.-M. Chen and M.-L. Tong, J. Am. Chem. Soc., 2016, 138, 5441–5450 CrossRef CAS PubMed.
- A. B. Canaj, S. Dey, E. R. Martí, C. Wilson, G. Rajaraman and M. Murrie, Angew. Chem., Int. Ed., 2019, 58, 14146–14151 CrossRef CAS PubMed.
- S. Jia, X. Zhu, B. Yin, Y. Dong, A. Sun and D. Li, Cryst. Growth Des., 2023, 23, 6967–6973 CrossRef CAS.
- S. K. Gupta, T. Rajeshkumar, G. Rajaraman and R. Murugavel, Chem. Sci., 2016, 7, 5181–5191 RSC.
- Y. Ding, N. F. Chilton, R. E. P. Winpenny and Y. Zheng, Angew. Chem., Int. Ed., 2016, 55, 16071–16074 CrossRef CAS PubMed.
- J. Acharya, P. Kalita and V. Chandrasekhar, Magnetochemistry, 2020, 7, 1 CrossRef.
- Y. Gil, A. Castro-Alvarez, P. Fuentealba, E. Spodine and D. Aravena, Chem. – Eur. J., 2022, 28, e202200336 CrossRef CAS PubMed.
- V. Singh, D. Das, S. Anga, J.-P. Sutter, V. Chandrasekhar and A. K. Bar, ACS Omega, 2022, 7, 25881–25890 CrossRef CAS PubMed.
- Y.-S. Ding, T. Han, Y.-Q. Hu, M. Xu, S. Yang and Y.-Z. Zheng, Inorg. Chem. Front., 2016, 3, 798–807 RSC.
- A. Borah, S. Dey, S. K. Gupta, G. Rajaraman and R. Murugavel, Dalton Trans., 2023, 52, 8943–8955 RSC.
- J.-C. G. Bünzli and D. Wessner, Helv. Chim. Acta, 1981, 64, 582–598 CrossRef.
- L. Maxwell, M. Amoza and E. Ruiz, Inorg. Chem., 2018, 57, 13225–13234 CrossRef CAS PubMed.
- E. Rousset, M. Piccardo, M.-E. Boulon, R. W. Gable, A. Soncini, L. Sorace and C. Boskovic, Chem. – Eur. J., 2018, 24, 14768–14785 CrossRef CAS PubMed.
- M. G. Ferrier, C. A. Valdez, S. K. Singh, S. Hok, D. Ray, L. Gagliardi and J. D. Despotopulos, Inorg. Chem., 2022, 61, 807–817 CrossRef CAS PubMed.
- T. Shimizu, M. Kawaguchi, T. Tsuchiya, K. Hirabayashi and N. Kamigata, J. Org. Chem., 2005, 70, 5036–5044 CrossRef CAS PubMed.
- T. Tsuchiya, T. Shimizu, K. Hirabayashi and N. Kamigata, J. Org. Chem., 2002, 67, 6632–6637 CrossRef CAS PubMed.
- F. Neese, F. Wennmohs, U. Becker and C. Riplinger, J. Chem. Phys., 2020, 152, 224108 CrossRef CAS PubMed.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
- J. P. Perdew, J. Tao, V. N. Staroverov and G. E. Scuseria, J. Chem. Phys., 2004, 120, 6898–6911 CrossRef CAS PubMed.
- F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC.
- T. Noro, M. Sekiya and T. Koga, Theor. Chem. Acc., 2012, 131, 1124 Search PubMed.
- S. Grimme, S. Ehrlich and L. Goerigk, J. Comput. Chem., 2011, 32, 1456–1465 CrossRef CAS PubMed.
- P. Pracht, F. Bohle and S. Grimme, Phys. Chem. Chem. Phys., 2020, 22, 7169–7192 RSC.
- E. V. Lenthe, E. J. Baerends and J. G. Snijders, J. Chem. Phys., 1993, 99, 4597–4610 CrossRef.
- B. O. Roos, P. R. Taylor and P. E. M. Sigbahn, Chem. Phys., 1980, 48, 157–173 CrossRef CAS.
- D. A. Pantazis and F. Neese, J. Chem. Theory Comput., 2009, 5, 2229–2238 CrossRef CAS PubMed.
- M. Atanasov, D. Ganyushin, K. Sivalingam and F. Neese, in Molecular Electronic Structures of Transition Metal Complexes II, ed. D. M. P. Mingos, P. Day and J. P. Dahl, Springer, Berlin, Heidelberg, 2012, pp. 149–220 Search PubMed.
- D. Aravena, J. Phys. Chem. Lett., 2018, 9, 5327–5333 CrossRef CAS PubMed.
- E. Regincós Martí, A. B. Canaj, T. Sharma, A. Celmina, C. Wilson, G. Rajaraman and M. Murrie, Inorg. Chem., 2022, 61, 9906–9917 CrossRef PubMed.
- A. Castro-Alvarez, Y. Gil, L. Llanos and D. Aravena, Inorg. Chem. Front., 2020, 7, 2478–2486 RSC.
- V. S. Parmar, D. P. Mills and R. E. P. Winpenny, Chem. – Eur. J., 2021, 27, 7625–7645 CrossRef CAS PubMed.
- L. R. Thomas-Hargreaves, M. J. Giansiracusa, M. Gregson, E. Zanda, F. O'Donnell, A. J. Wooles, N. F. Chilton and S. T. Liddle, Chem. Sci., 2021, 12, 3911–3920 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4dt00632a |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2024 |