
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThermal, photonic, and electrocatalysis in lignin depolymerization research
Wang Ziweiabc,
Shu Haoabc,
Chen Yizhenabc,
Liu Benabc,
Xu Yaoweiabc,
Wang Wanxiaabc,
Wang Kaiyuea,
Lei Menghengabc,
Guo Li *d and
Wang Lei*e
*d and
Wang Lei*e
aChina Tobacco Hubei Industrial Co., Ltd, Wuhan 430040, China
bHubei Xinye Reconstituted Tobacco Development Co., Ltd, Wuhan 430056, China
cApplied Technology Research of Reconstituted Tobacco Hubei Province Key Laboratory, Wuhan 430040, China
dKey Laboratory of Hubei Province for Coal Conversion and New Carbon Materials, School of Chemistry and Chemical Engineering, Wuhan University of Science and Technology, Heping Avenue 947, Wuhan 430081, China. E-mail: guoli@wust.edu.cn; Tel: +86-027-6886-2335
eHubei Provincial Key Laboratory of Green Materials for Light Industry, Hubei University of Technology, Wuhan 430068, China. E-mail: wanglei@hbut.edu.cn
First published on 6th November 2023
Abstract
In order to realize a sustainable bio-based future, it is essential to fully harness the potential of biomass, including lignin – a readily available biopolymer that ranks second in abundance and serves as a renewable source of aromatics. While lignin has traditionally been used for lower-value applications like fuel and power generation, unlocking its higher-value potential through diverse conversion and upgrading techniques is of paramount importance. This review focuses on the catalytic conversion of lignin, with a specific emphasis on selective depolymerization, a process that not only supports economically and environmentally sustainable biorefineries but also aligns with Green Chemistry principles, mitigating adverse environmental impacts. Furthermore, we provide a comprehensive discussion of reaction pathways and mechanisms, including C–O and C–C bond cleavage, among different catalysts. Lastly, we analyze and briefly discuss the prospects of rational catalyst design in biomass valorization.
1. Introduction
Petroleum has been the driving force behind human advancement ever since the industrial revolution, providing us with energy, fuels, and essential chemical feedstocks. However, the extensive global utilization of this fossil resource has not been without consequences, as highlighted by the findings of the Intergovernmental Panel on Climate Change (IPCC). Elevated concentrations of greenhouse gases (GHGs) in the atmosphere pose a growing threat, leading to alterations in climate and weather patterns.1 These changes manifest as global warming, rising sea levels, ocean acidification, and detrimental impacts on biodiversity and essential ecosystem services. The environmental harm has grown as a result of drilling and extraction activities expanding into increasingly fragile locations. Furthermore, our dependence on energy and the unpredictable access to oil supplies have been associated with global economic and political conflicts. Substituting finite, non-renewable fossil oil with biomass as the primary source of chemicals and fuels represents a substantial stride towards mitigating these issues. Biomass, created through the process of photosynthesis, assimilates carbon from the atmosphere (already part of the biospheric carbon cycle) and captures solar energy within chemical bonds. Consequently, utilizing fuels and chemicals derived from biomass essentially operates within a “closed circuit” of the biospheric carbon cycle, as opposed to introducing “open circuit” fossil carbon emissions that contribute to heightened climate change rates.2 Furthermore, as an additional advantage of this low-carbon “circular economy,” when practiced sustainably, plant biomass has the potential to not only capture but also partially sequester atmospheric CO2, thereby assisting in mitigating the environmental consequences of greenhouse gas emissions.At present, a variety of technologies are being utilized to convert biomass and waste streams into valuable products through thermal, biological, or chemical processes.3 Lignin, constituting the second most naturally abundant biopolymer in plant cell walls, ranks only behind cellulose in terms of abundance.4 Despite this, lignin, which is one of the three principal components of biomass, is typically either incinerated for process heat or integrated into animal feed, where it remains largely indigestible. Therefore, catalytic conversion to promote the valorization of lignin represents a viable approach for generating high-value chemicals. For example, lignin as well as its analogs/fragments also acts as antiviral compounds, showing inhibition against SARS-CoV-2 main protease.5 Besides, Lignin is a highly intricate macromolecule characterized by its extensive network of aromatic rings linked through both ether (C–O–C) and carbon–carbon (C–C) bonds. Typically, lignin is depolymerized to yield straightforward aromatic lignin monomers, which are compatible with existing industrial processes. Subsequently, these lignin monomers often undergo further refinement to transform them into high-value chemicals and fuels, suitable for a wide range of applications, including the pharmaceutical, chemical, and spice industries, among others (see Fig. 1).
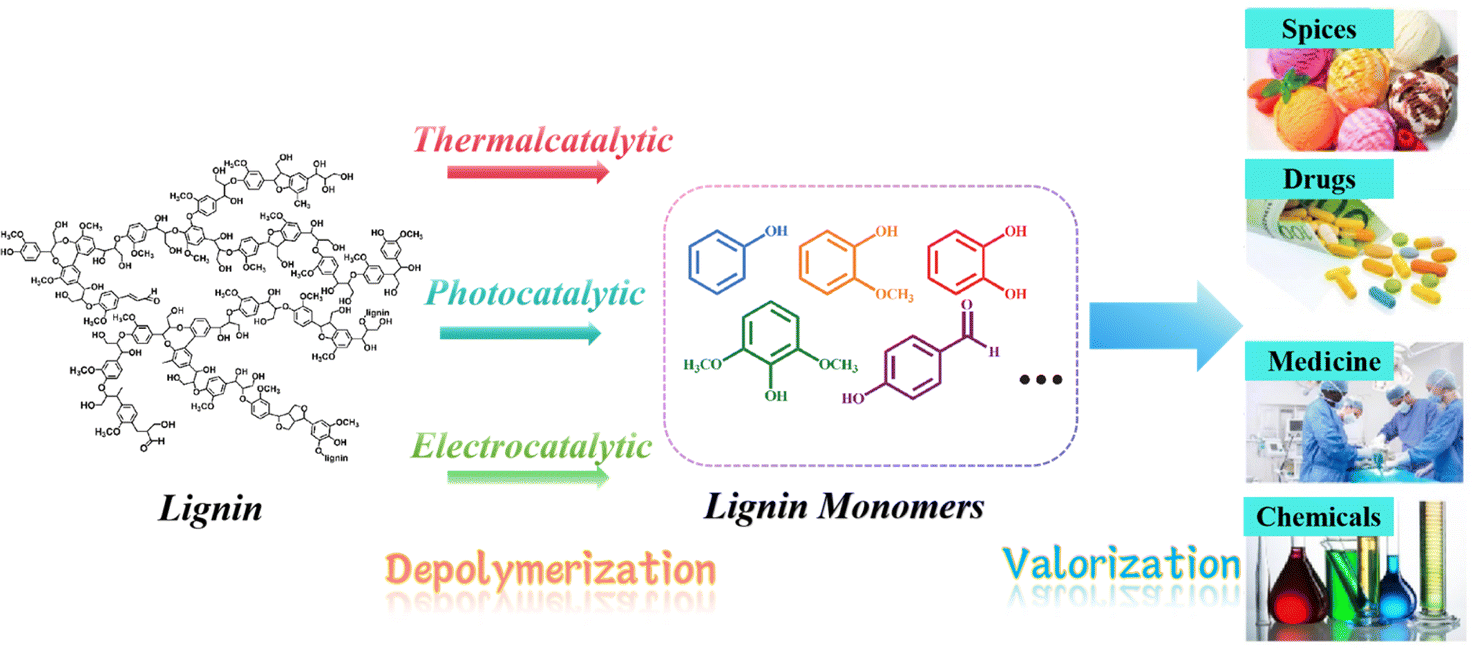 | ||
| Fig. 1 Catalytic lignin depolymerization pathways for utilization. | ||
In this context, we build upon our previous review and delve into model compound studies, which are often employed as tools to advance our comprehension of catalytic enhancements in the depolymerization of lignins. We begin by exploring the eco-friendly and sustainable aspects of catalysis, offering a concise overview of mechanisms involved in catalytic biomass transformation. Next, we summarize various upstream methods utilized for the depolymerization of lignin, including thermal, photonic, and electrical techniques (Fig. 2). Subsequently, we offer a critical assessment of the impact of catalyst structure on product distribution during pyrolysis and provide insight into the design of an efficient catalyst.
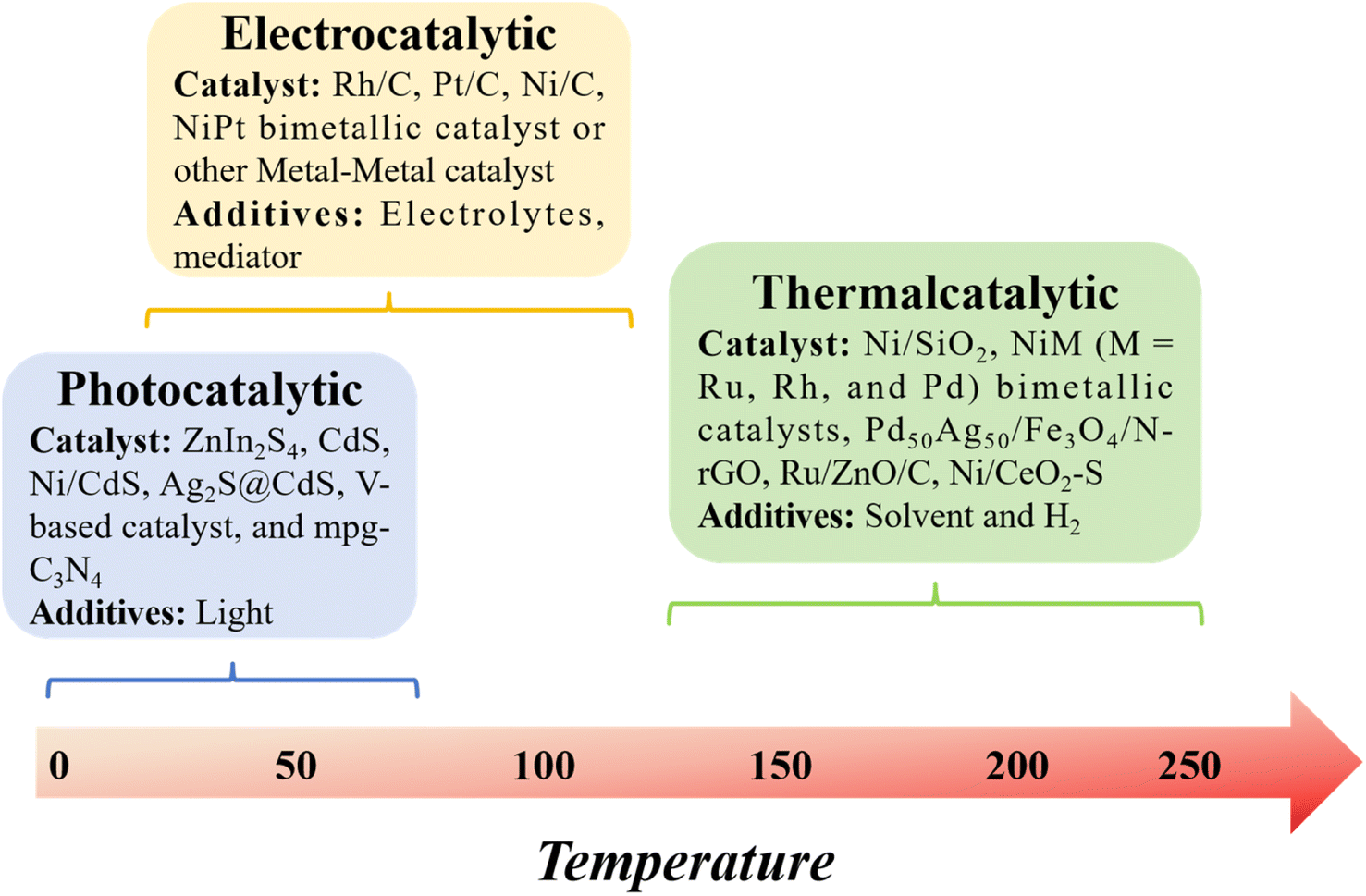 | ||
| Fig. 2 Comparison of thermal, photonic, and electrocatalysis in lignin depolymerization. | ||
2. Thermalcatalytic transformation of lignin
Thermal catalytic depolymerization, as we are aware, presents a compelling challenge due to the structural complexity and recalcitrance of this aromatic biopolymer. Lignin is intricately bound together by robust C–C and C–O bonds.6 The most prevalent linkages are illustrated in Fig. 3. Extensive efforts have been directed towards selectively cleaving the C–O bonds within the β-O-4 ether bond, constituting approximately 50% of all linkages. The cleavage of this particular linkage requires energy ranging from 68.2 to 71.8 kcal mol−1, contingent upon the substitution pattern. Additionally, competing ring hydrogenation reactions may reduce selectivity during lignin depolymerization. Consequently, the design of efficient and highly selective catalyst systems poses a substantial challenge.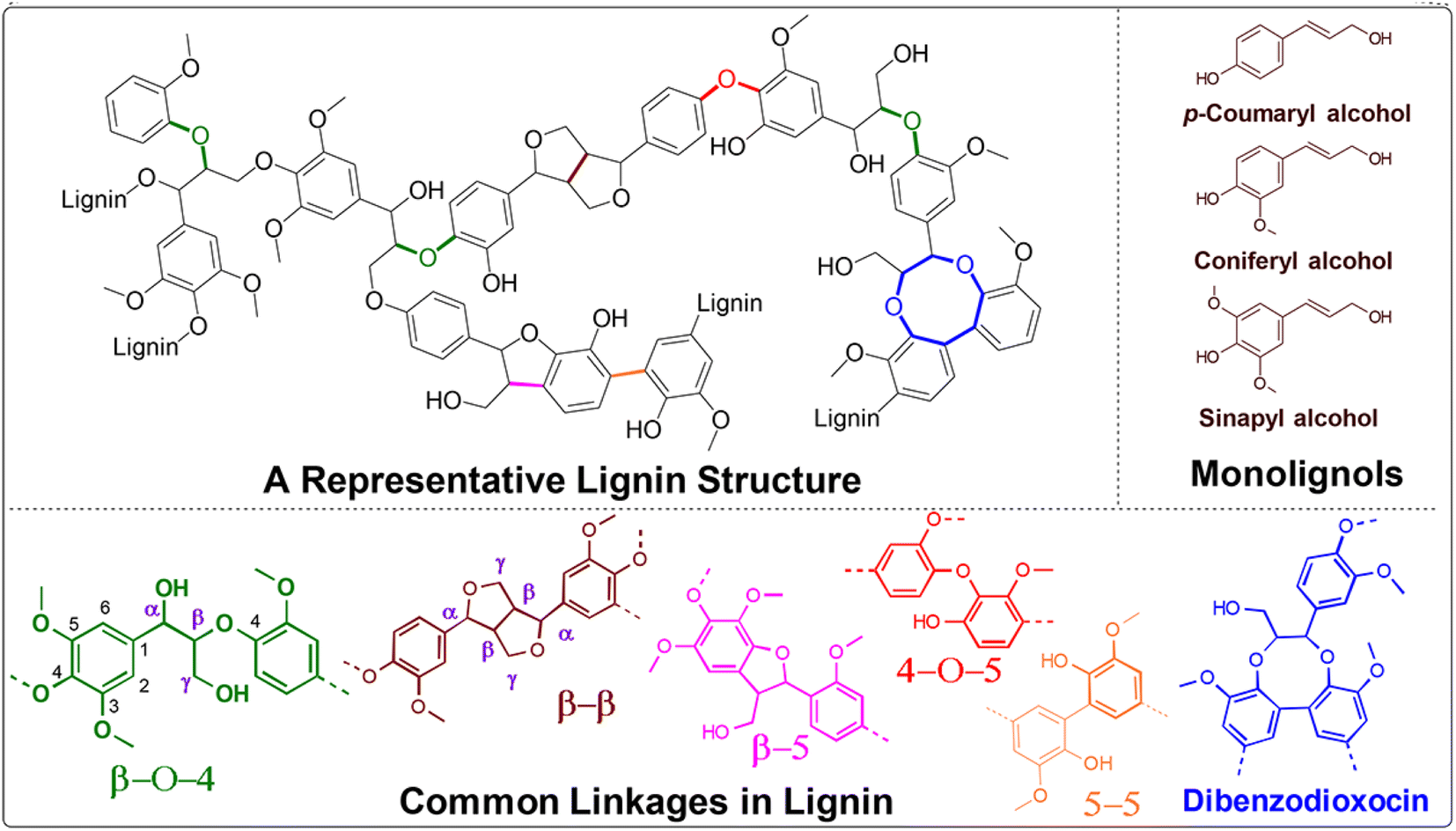 | ||
| Fig. 3 A representative lignin structure displaying typical lignin subunits and linkages encountered. | ||
Single atoms and metal nanoparticles exhibit exceptional hydrogenation or hydrogenolysis capabilities, making them promising catalysts for lignin depolymerization.7 Lercher et al. conducted a study where they synthesized a series of supported Ni/SiO2 catalysts using the deposition precipitation method.8 The rate of C–O bond cleavage increased as the size of the Ni particles increased within the range of 4.5–5.9 nm. However, when the particle size was further increased to 8.0 nm, the reaction rate decreased. Kinetic studies on the conversion of 2-phenylethyl phenyl ether (β-O-4) demonstrated the selective cleavage of the C–O–C bond occurred at the aliphatic carbon position, resulting in the production of phenol and ethylbenzene (see Fig. 4). The reaction of benzyl phenyl ether (α-O-4) on Ni/SiO2 indicated hydrogenolysis of benzyl phenyl ether to toluene and phenol. However, the cleavage of diphenyl ether (4-O-5) exhibited a different reaction mechanism compared to α-O-4 and β-O-4, with hydrogenolysis and hydrogenation occurring sequentially. Apart from the size of the metal nanoparticles, the properties of the support material also play a significant role in catalytic activity. Esposito et al. reported a TiN–Ni nanocomposite catalyst for C–O bond cleavage under relatively mild conditions in a fixed-bed reactor.9 To balance Fermi levels, electrons flow from Ni to the TiN side, resulting in electron-deficient Ni. This promotes the coordination of oxygen from ether linkages and accelerates the hydride transfer to the arene during reductive elimination. Moreover, comparing Ni/SiO2 and TiN–Ni, it was found that using TiN, there is a strong interaction between Ni species and the TiN support, which benefits the hydrogenolysis process of lignin.8,9
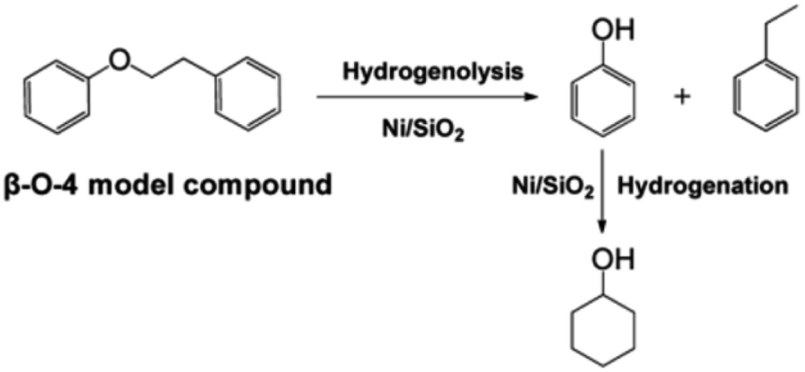 | ||
| Fig. 4 Reaction pathway for the cleavage of 2-phenylethyl phenyl ether (the model compound for the β-O-4 linkage in lignin) over Ni/SiO2 in the aqueous phase. Adapted from ref. 8 with permission. Copyright 2012 American Chemical Society. | ||
In the catalytic hydrolysis of lignin, there is a potential for a synergistic effect between different metals to improve the conversion of lignin into monomeric aromatic chemicals by enhancing H2 and substrate activation.10 Yan et al. conducted research involving a series of NiM (M = Ru, Rh, and Pd) bimetallic catalysts for the hydrogenolysis of lignin under mild conditions (130 °C, 10 bar H2). Their study proposed that the incorporation of Ru facilitated the reduction of Ni, leading to the formation of ultrasmall bimetallic catalyst particles and enhancing the C–O hydrogenolysis process. Besides, an elevated proportion of fraction of surface atoms (compared with Ni), which heightened activation of H2 and substrates. Meanwhile, Ru in the NiRu bimetallic catalyst may inhibit benzene ring hydrogenation.
In addition to the aforementioned Ni-based supported catalysts, catalysts have garnered significant attention in the development of lignin depolymerization methods, particularly in the reductive cleavage of C–O bonds. For example, Samec's group reported an efficient, robust, and environmentally friendly redox-neutral cleavage of β-O-4′ ether linkages in 2-aryloxy-1-arylethanol ethers using recyclable and commercially available Pd/C catalyst, resulting in high to excellent yields of aryl ketones and phenols.11 The proposed reaction mechanism suggests that both the hydrogens at the α and β positions of β-O-4′ ethanolaryl ethers participate in the hydrogenolysis of 2-aryloxy-1-arylethanols. The reaction initiates with a reversible dehydrogenation of 1, producing 4 and chemically adsorbed hydrogen on Pd/C (see Fig. 5). The basic medium may facilitate ketoenol tautomerization, thus promoting the adsorption of 4 onto the palladium surface to form C. Following a Horiuti–Polanyi-type mechanism, this methodology proceeds through a unique low-energy-barrier pathway (12 kcal mol−1) for the cleavage of the lignin C–O bond via an initial dehydrogenation step.
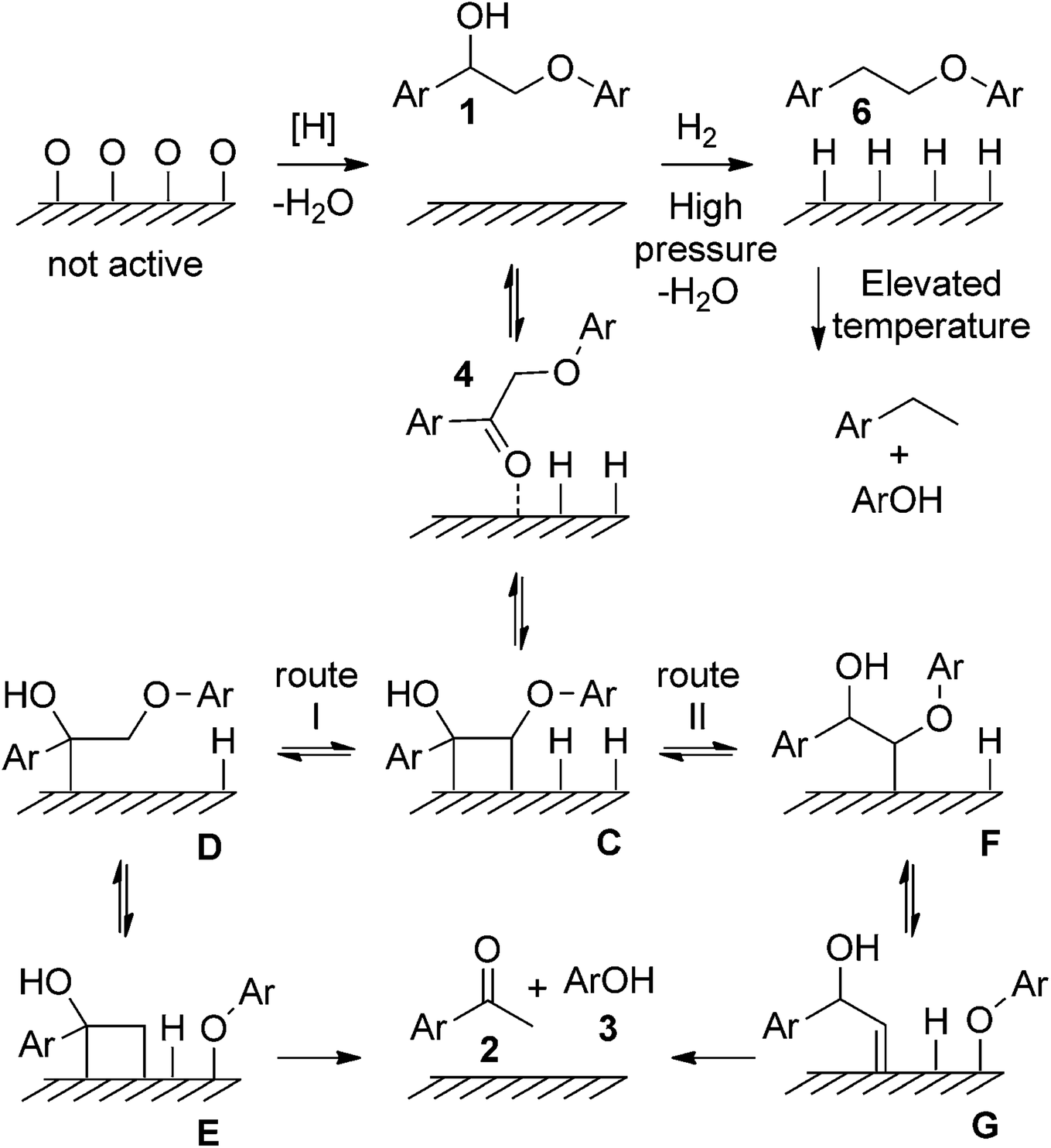 | ||
| Fig. 5 Proposed reaction mechanisms for the redox neutral transfer hydrogenolysis of 1. Metal atoms are depicted by surface for clarity. Adapted from ref. 11 with permission. Copyright 2015 Wiley-VCH. | ||
To effectively depolymerize and convert lignin model compounds, Kim et al. reported bimetallic PdAg catalyst supported on Fe3O4/nitrogen-doped reduced graphene oxide (N-rGO) and applied in the hydrogenolysis of β-O-4 and α-O-4 C–O model compounds.12 It was noted in this regard that Pd50Ag50/Fe3O4/N-rGO catalyst could accelerate the selective hydrogenolysis of β-O-4 and α-O-4 C–O model compounds via the H2 produced in situ from formic acid with high selectivity at 120 °C (Fig. 6). Bifunctional catalysts exhibit a synergistic influence on catalytic processes, simultaneously mitigating decarbonylation through steric hindrance and electronic effects while effectively reducing condensation reactions in the depolymerization process.13 Some bimetallic surfaces composed of 3d transition metals (like Fe, Ni, Cu, and Zn) in conjunction with noble metals (such as Ru, Pt, and Pd) exhibit a noteworthy shift of the D-band center towards the Fermi level, highlighting enhanced activation capabilities of these bifunctional catalysts.14 Recently, Li and co-works reported a bimetallic Ru/Zn@Beta catalyst for efficiently depolymerizing Kraft lignin via the Lewis-acid (Zn) and the hydrogen evolution site (Ru). In particular, as-fabricated 5% Ru/Zn(1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2)@Beta catalyst exhibited the excellent catalytic performance, an optimum yield of 96% ethyl acetate-soluble product was achieved after reacting at 290 °C for 9 h.
:2)@Beta catalyst exhibited the excellent catalytic performance, an optimum yield of 96% ethyl acetate-soluble product was achieved after reacting at 290 °C for 9 h.
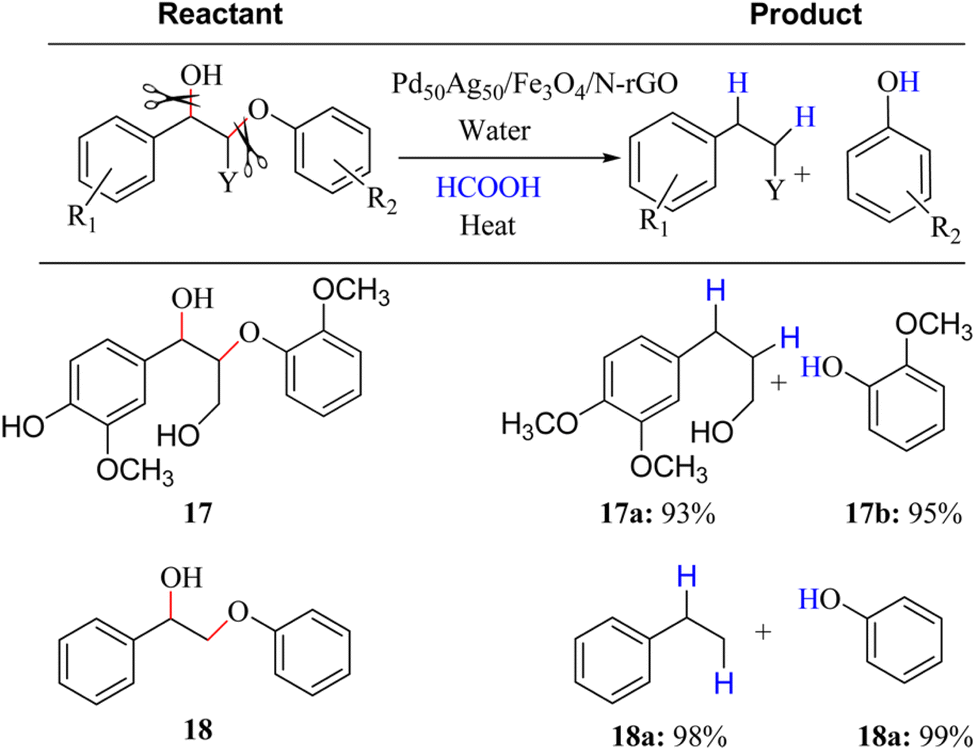 | ||
| Fig. 6 Selective hydrogenolysis reactions of β-O-4 linkage model aryl C–O group with formic acid over Pd50Ag50/Fe3O4/N-rGO catalyst. Reactant (0.1 mmol), formic acid (3.5 equiv.), catalyst (50 mg), time 3 h, temperature 120 °C; conversion and yield are based on NMR analysis using decane (0.1 mmol) as an internal standard. At least two measurements were taken for an average yield. In all cases complete conversion of β-O-4 linkage aryl C–O group was achieved. Adapted from ref. 12 with permission. Copyright 2015 American Chemical Society. | ||
The atomic dispersion of metal sites represents a breakthrough in catalyst design, enabling enhanced atomic utilization and catalytic activity.15 Song et al. reported an atomically dispersed Ru/ZnO/C catalyst, which exhibited excellent catalytic activity in the hydrogenolysis of C-lignin by cleaving C–O bonds within benzodioxane linkages.16 Analysis using HAADF-STEM and XAFS techniques revealed that the Ru species were distributed either in clusters or as individual atoms. This catalyst efficiently cleaved all C–O bonds within benzodioxane structures while selectively avoiding the reduction of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bonds, thus preferentially depolymerizing C-lignin biopolymer into propenylcatechol. A recent advancement in synthetic chemistry involves the creation of isolated single-atom sites (ISAS) dispersed on a variety of support materials. Li et al. found that Ni nanoparticles (NiNPs) deposited on CeO2 with oxygen vacancies (Ov) exhibit remarkable activity and selectivity in the hydrogenolysis of C–O bonds in diphenyl ether (DPE),17 a lignin model compound. In contrast, single-site Ni (SSNi) displays negligible reactivity in the same reaction (Fig. 7). The Ni/CeO2 catalyst containing NiNPs demonstrates an impressive turnover frequency (TOF) of up to 3486 h−1, particularly when using a 1.5-Ni/CeO2 ratio. The overall activity of Ni/CeO2 increases as the proportion of NiNPs rises, as they play a pivotal role in the adsorption and activation of both H2 and DPE. Notably, the adsorption of DPE on NiNPs is more influential for DPE hydrogenolysis compared to H2 adsorption. Most importantly, the activity for DPE hydrogenolysis experiences a significant enhancement with increasing oxygen vacancy (Ov) levels in Ni/CeO2.
C bonds, thus preferentially depolymerizing C-lignin biopolymer into propenylcatechol. A recent advancement in synthetic chemistry involves the creation of isolated single-atom sites (ISAS) dispersed on a variety of support materials. Li et al. found that Ni nanoparticles (NiNPs) deposited on CeO2 with oxygen vacancies (Ov) exhibit remarkable activity and selectivity in the hydrogenolysis of C–O bonds in diphenyl ether (DPE),17 a lignin model compound. In contrast, single-site Ni (SSNi) displays negligible reactivity in the same reaction (Fig. 7). The Ni/CeO2 catalyst containing NiNPs demonstrates an impressive turnover frequency (TOF) of up to 3486 h−1, particularly when using a 1.5-Ni/CeO2 ratio. The overall activity of Ni/CeO2 increases as the proportion of NiNPs rises, as they play a pivotal role in the adsorption and activation of both H2 and DPE. Notably, the adsorption of DPE on NiNPs is more influential for DPE hydrogenolysis compared to H2 adsorption. Most importantly, the activity for DPE hydrogenolysis experiences a significant enhancement with increasing oxygen vacancy (Ov) levels in Ni/CeO2.
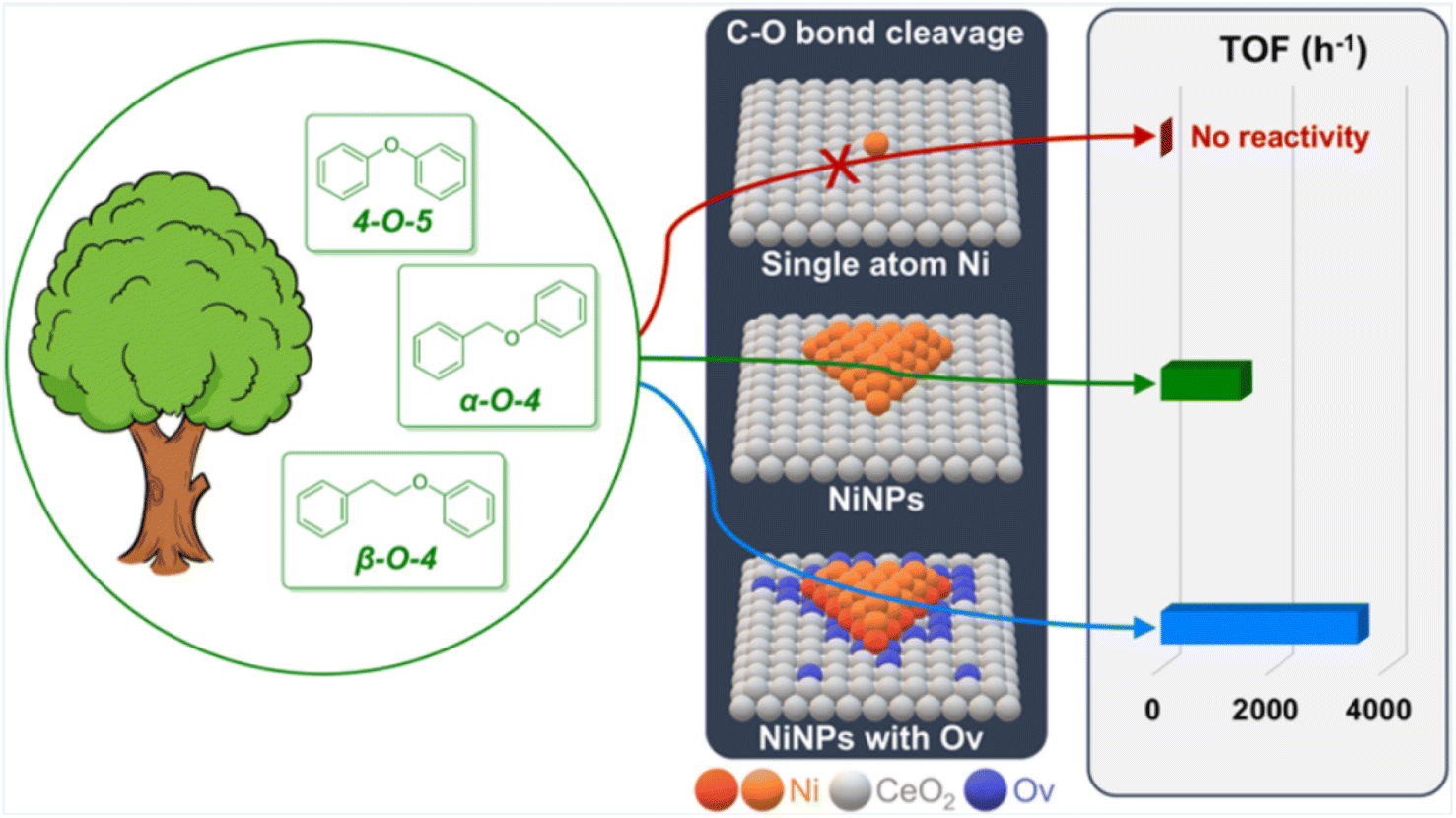 | ||
| Fig. 7 Comparison of catalyst activity of different sizes. Adapted from ref. 17 with permission. Copyright 2023 American Chemical Society. | ||
3. Photocatalytic transformation of lignin
Solar energy stands out as the most abundant natural energy source, with the potential to provide over a thousand times the total global energy consumption. However, its diffuse and intermittent nature presents challenges, making direct utilization a formidable task. Hence, there's an increasing need to develop technologies that efficiently convert solar energy into chemically stored and readily useable energy forms. In the past decade, emerging photocatalytic technology has evolved into a potent tool for converting biomass and its platform molecules into valuable products under relatively mild conditions.18Photocatalytic transfer hydrogenolysis was initially developed to cleave the β-O-4 linkage. This process involves the abstraction of a hydrogen atom from the benzylic alcohol, which is then transferred to break the aliphatic ether bond. Wang et al. introduced a novel self-hydrogen transfer hydrogenolysis method using a heterogeneous ZnIn2S4 photocatalyst to directly convert both model and organosolv lignin into valuable phenolic products (see Fig. 8).19 Under visible light exposure, an initial dehydrogenation process occurs, converting interlinking CαH–OH groups to CαO, creating what is referred to as a “hydrogen pool” on the ZnIn2S4 photocatalyst. Subsequently, the adsorbed hydrogen within this “hydrogen pool” is transferred to the (OCα)Cβ–O bond, leading to the cleavage of ether bonds. Lignin itself serves as a hydrogen source for this process, and while additional alcohols can act as hydrogen donors and enhance the reaction, they are not strictly necessary. With this groundbreaking approach, impressive yields ranging from 71% to 91% have been achieved for phenols and acetophenones derived from lignin β-O-4 models. Moreover, a substantial mass yield of 10% was obtained for p-hydroxyl acetophenone derivatives from dioxanesolv poplar lignin. This photocatalytic CTH (catalytic transfer hydrogenolysis) reaction deviates from the thermal catalytic transfer hydrogenolysis process, where the Hδ+ of α-OH typically transfers to phenol, maintaining its polarity if the CTH follows a monohydride mechanism. However, in this specific instance, the Hδ+ of α-OH ultimately transfers to acetophenone with an Hδ− form. This phenomenon suggests that during photocatalysis, there may be a reversal in the polarity of Hδ+ from α-OH to Hδ−. Consequently, the Hδ− is transferred to acetophenone instead of phenol, likely due to the smaller electronegativity of carbon in comparison to oxygen within the Cβ–O bond. A similar reversal in polarity of Hδ− for α-CH also occurs during the photocatalytic reaction, indicating that this photocatalytic transfer hydrogenolysis process may involve a polarity reversal in the abstracted hydrogen species.
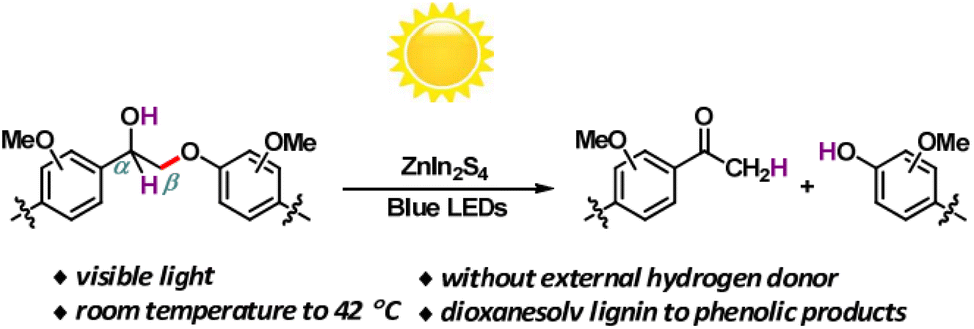 | ||
| Fig. 8 Visible-light-driven fragmentation of lignin β-O-4 models and dioxanesolv lignin. Adapted from ref. 19 with permission. Copyright 2017 American Chemical Society. | ||
The concept of “lignin-first,” which involves the initial catalytic conversion of native lignin present in biomass, offers an exciting opportunity to utilize complete lignocellulosic biomass more efficiently. This approach has recently gained significant interest and attention. Wang et al. have developed an efficient solar energy-driven lignin-first approach for the fractionation and valorization of lignocellulosic biomass catalyzed by CdS quantum dots (QDs). These CdS QDs cleave the β-O-4 bond at room temperature under visible light, following an electron–hole coupled (EHCO) mechanism via a Cα radical intermediate.20 As depicted in Fig. 9, the Cα–H bond cleavage has the least positive oxidative potential (1.1 V versus SHE), indicating that the Cα–H bond is the first to break upon oxidation by holes. Furthermore, the valence band maximum (VBM) of CdS (1.7 V versus SHE) is more positive than the oxidative potential of the Cα–H bond. Therefore, the photoexcited hole can effectively oxidize PP-ol into the Cα radical. Computational results confirm the experimental findings, highlighting the crucial role played by the Cα hydrogen in the reaction. The colloidal nature of quantum dots (QDs) promotes close contact with the solid biomass substrate. This enhanced interaction improves the accessibility of β-O-4 linkages in biomass to the catalyst, thereby enabling the efficient conversion of native lignin into aromatic monomers under mild reaction conditions.
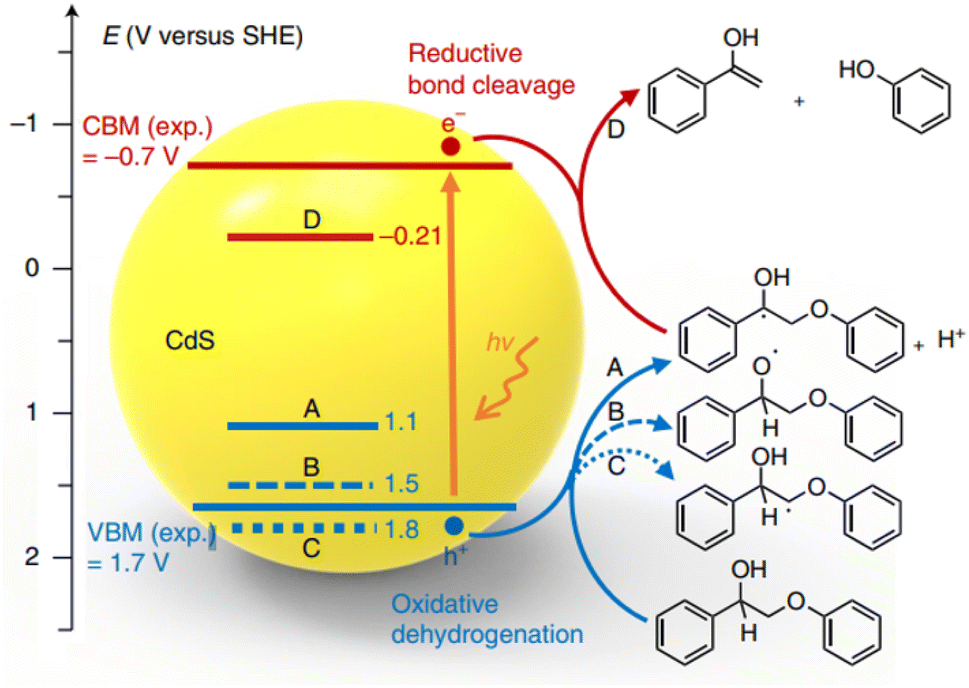 | ||
| Fig. 9 Potentials of oxidative dehydrogenation of PP-ol via three different paths (A, B, C) and potential of reductive cleavage of β-O-4 bond in the Cα radical intermediate (D) against the positions of VBM and CBM of CdS. Experimental values of VBM and CBM were obtained through UV-visible and electrochemical measurements of bulk CdS. Adapted from ref. 20 with permission. Copyright 2018 Springer Nature. | ||
Sun et al. employed ultrathin 2D cocatalysts (Fe, Co, Ni, or Cu) supported on CdS nanosheets for both alcohol oxidation and H2 evolution under visible light irradiation.21 Solar-driven hydrogen (H2) production using semiconductor/co-catalyst systems is widely considered a promising approach for transforming and storing intermittent solar energy as H2. As a result, they combined oxidative biomass valorization with solar-driven H2 production using semiconductor/co-catalyst systems, achieving the oxidative photocleavage of lignin model compounds in highly selective ways. Mechanistic studies (see Fig. 10) revealed that the excited holes generated in Ni/CdS through visible light irradiation were capable of oxidizing the Cα–OH group in compound 5, leading to the formation of the corresponding ketone product 6. It's important to note that the excited electrons on Ni/CdS promote the generation of adsorbed hydrogen species on the surface of Ni/CdS. These hydrogen species preferentially transfer to the Cβ–O bond, resulting in the cleavage of the ether bond and the formation of acetophenone (7) and phenol (8).
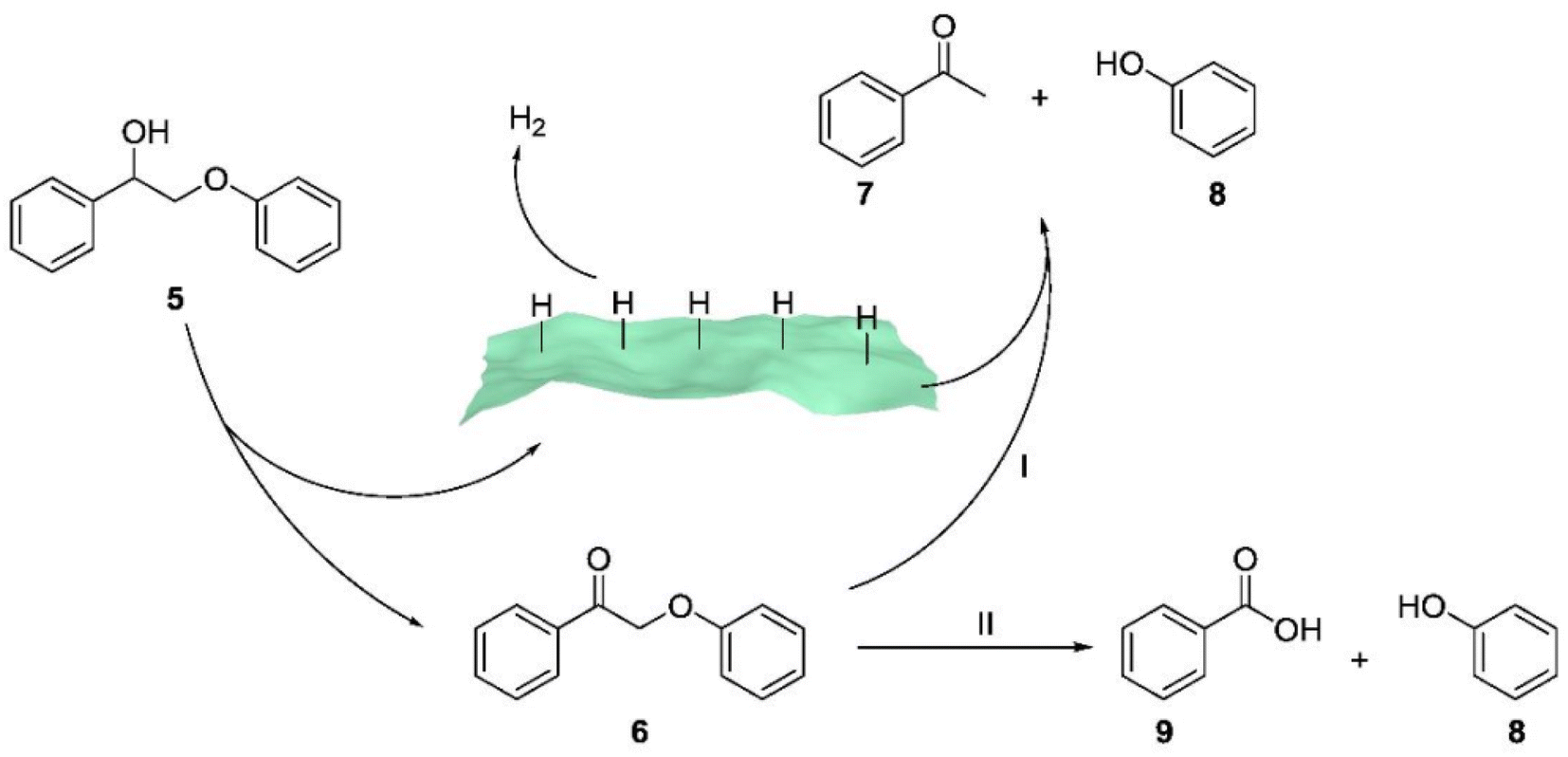 | ||
| Fig. 10 Proposed mechanistic steps for the two pathways of the photocatalytic oxidation of 2-phenoxy-1-phenylethanol (5) on Ni/CdS to produce different products with high selectivity. 2-Phenoxy-1-phenylethanone (6) is yielded as the sole product in CH3CN, which will be fully transformed to acetophenone (7) and phenol (8) in CH3CN/0.1 M KOH (v/v = 2/8) (pathway I) or benzoic acid (9) and phenol through a chemical oxidation (pathway II). Adapted from ref. 21 with permission. Copyright 2019 American Chemical Society. | ||
The photogenerated electron and hole can effectively and simultaneously react with reductants and oxidants to enable one-step redox-neutral reactions are required. An Ag+-exchanged CdS photocatalyst (denoted as Ag2S@CdS) is employed as an active visible-light-driven photocatalyst for lignin fragmentation. When Ag+ is substituted for Cd2+ via low Ag+ exchange, trace amounts of Ag+ produce small Ag2S domains on the surface of the CdS particles, which can induce electronic structure change. Therefore, the enhanced photocatalytic performance of Ag2S@CdS with small Ag2S domains the surface of the CdS particles is explored for the visible-light-driven fragmentation of lignin into the desired aromatic monomers.22 Typical n-type semiconductor characteristics i.e., reduction by an electron in the conduction band (CB) is slower than oxidation by a hole, leading to the accumulation of electrons can account for the poor performance of the photocatalytic redox conversion of pristine CdS (Fig. 11). The Fermi level is rather close to the CB minimum in CdS, due to its characteristics of an n-type semiconductor. Upon an increase in the amount of exchanged Ag+, the Fermi level was gradually shifted downward toward the VB maximum. Under visible light irradiation, electrons in the VB are excited to the CB in CdS, occurring with simultaneous hole generation in the VB. Consequently, the photoexcited electrons could effectively transfer from CdS to Ag2S@CdS induced by the potential energy due to a downward shift in the Fermi level. This phenomenon accelerates the transfer of photoexcited electrons in the CB to the oxidized lignin model compounds for reductive cleavage. The increasing H+ affinity can have a great effect on this process by assisting the electron transfer in the CB. Consequently, the photogenerated electrons and holes in Ag2S@CdS would be efficiently separated, allowing them to reach the surface and simultaneously trigger the photocatalytic oxidation reaction at the VB and the reductive cleavage reaction at the CB.
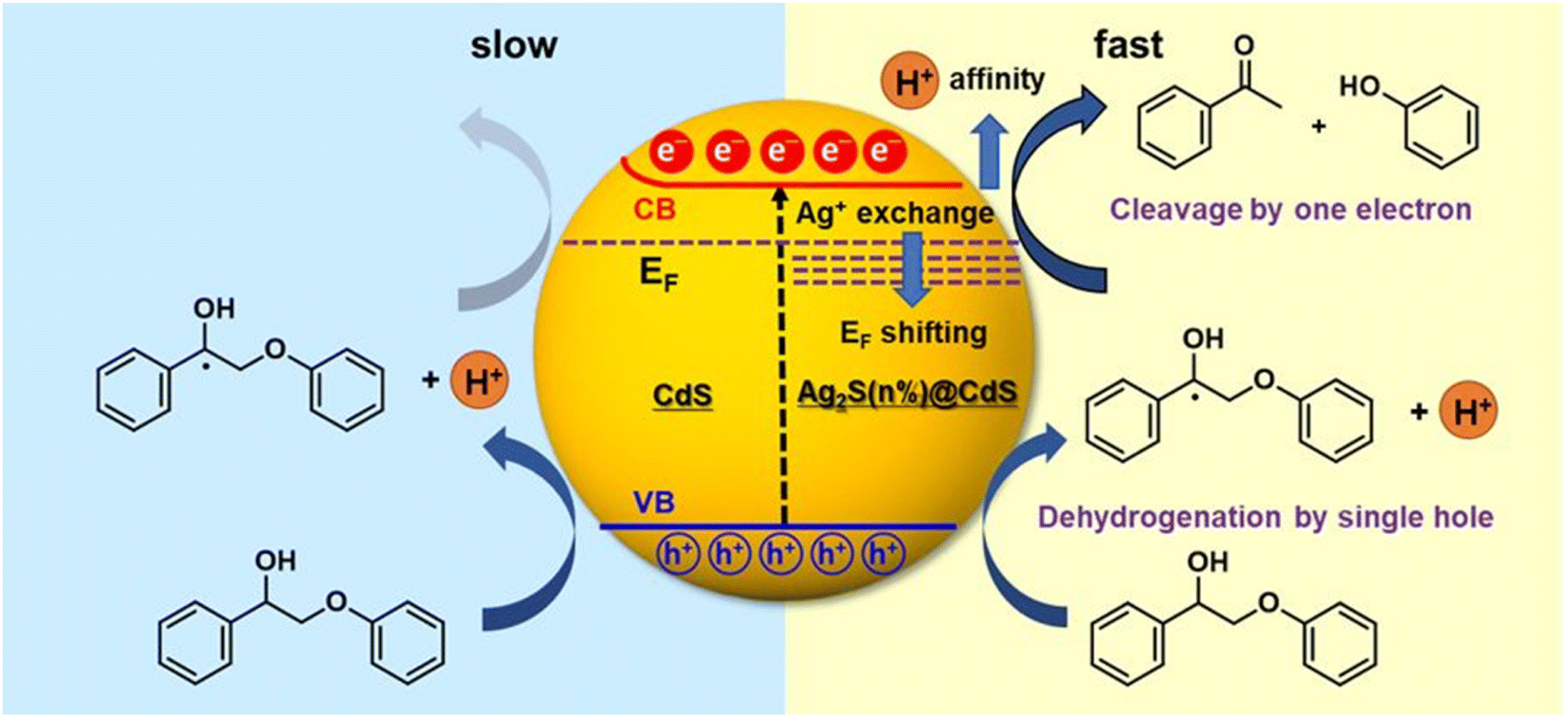 | ||
| Fig. 11 Schematic representation comparing the simultaneous oxidative dehydrogenation and reductive cleavage of the β-O-4 bond in the Cα radical intermediate in Ag2S@CdS (right) with the oxidative dehydrogenation and pinned Fermi level of CdS (left). Adapted from ref. 22 with permission. Copyright 2020 American Chemical Society. | ||
Furthermore, hydrogen abstraction from the C(sp3)–H bond at the benzylic Cα atom to cleave the β-O-4 linkage has also been successfully achieved. Density functional theory (DFT) calculations have shown that the bond dissociation energy (BDE) of the benzylic Cα–H bond is notably high, at 470 kJ mol−1. In contrast, the BDEs of the Cα–H and Cβ–H bonds in the transient negative ion (TNI) [C14H14O]– are considerably lower, at 250 kJ mol−1 and 329 kJ mol−1, respectively.2c,23 Theoretical and experimental investigations have demonstrated that the intense electromagnetic near-field generated at illuminated plasmonic nanoparticles (NPs) promotes the chemisorption of the β-O-4 compound. Additionally, the transfer of photo-generated hot electrons from the NPs to the adsorbed molecules leads to hydrogen abstraction and the direct cleavage of the unreactive ether Cβ–O bond under moderate reaction conditions. This hydrogen abstraction significantly weakens the Cβ–O bond, facilitating its homolytic cleavage under mild conditions. Polar Hδ− species are extracted from alcohols on illuminated silver NPs. The relatively modest hydrogenation ability of these species enables the reductive cleavage of alkyl–aryl ether bonds, selectively generating unsaturated products like styrene, which is challenging to obtain through thermal catalytic reactions.
To achieve hydrogenolysis of aryl ethers under mild conditions without the need for H2 gas, Li et al. developed a synergistic approach that combines photocatalytic hydrogen transfer with acid catalysis. This method enables the hydrogenolysis of diphenyl ether (DPE) using alcohol as a hydrogen donor at room temperature.24 In this process, weakly coordinated Pt0 atoms and positively charged Pt species on the catalyst surface, when exposed to light irradiation, enhance surface electron availability and store more hydrogen species. This unique property is pivotal for efficient photocatalytic hydrogen transfer.
The reaction proceeds through the cleavage of the aryl C–O bond prior to the saturation of the aryl ring, not only facilitating the reduction of diphenyl ether (DPE) but also enabling the reduction of phenols and aryl alkyl ethers. The presence of acid is crucial to ensuring the highly selective cleavage of the aryl C–O bond. Density functional theory (DFT) calculations have revealed that the most stable adsorption modes of DPE and protonated DPE are co-planar adsorptions. The adsorption energy of DPE is slightly more negative than that of protonated DPE, suggesting that protonation weakens the adsorption of DPE slightly. The cleavage of the C–O bond in protonated DPE has a lower activation barrier (0.81 eV) compared to DPE (1.48 eV), and the formation of cleaved products from protonated DPE is more exothermic than that from DPE. Consequently, C–O bond cleavage is kinetically easier and more thermodynamically favorable after protonation. The activation of the C–O bond through protonation is further supported by the larger C–O bond length in the adsorbed state (1.46 Å) and transition state of protonated DPE (1.77 Å). These calculations align well with the experimental findings.
While the cleavage of C–O linkages in lignin can yield monomeric aromatics, it typically achieves a theoretical yield of no more than 50%. To achieve higher yields of monomeric products from lignin depolymerization, it's necessary to also cleave the interlinking C–C bonds. However, C–C bond cleavage is more challenging due to its non-polar and robust nature. Additionally, C–C bond cleavage may produce aldehydes, which can react with phenolic aromatics, leading to re-condensation of products or intermediates and the formation of more recalcitrant structures under reaction conditions. Wang et al. employed vanadium catalysts to cleave the C–C bonds in β-1 and β-O-4 interlinkages present in lignin models under visible light irradiation.25 The initiation of light irradiation triggers electron transfer from the substrate molecules to the vanadium center, leading to the cleavage of the Cα–Cβ bond and the formation of radical intermediates. Both non-phenolic and phenolic β-1 model compounds exhibit high conversion and selectivity in Cα–Cβ bond cleavage when using the VO(Oi Pr)3 catalyst (see Fig. 12).
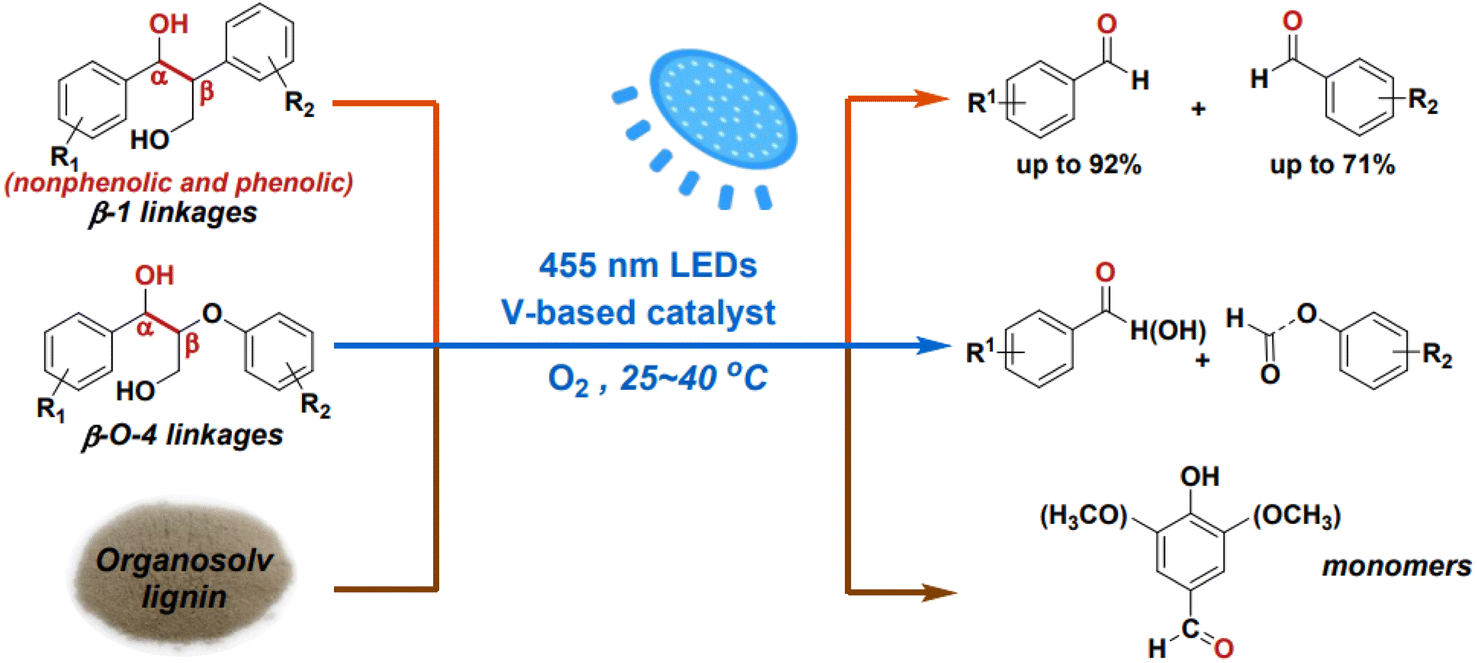 | ||
| Fig. 12 Photocatalytic aerobic Cα–Cβ bond cleavage of lignin linkages. Adapted from ref. 25 with permission. Copyright 2020 American Chemical Society. | ||
Heterogeneous photocatalysts have been developed to facilitate the cleavage of the Cα–Cβ bond in β-O-4 and β-1 linkages. A visible-light responsive mesoporous graphitic carbon nitride (mpg-C3N4) semiconductor was found to catalyze the fragmentation of both β-O-4 and β-1 lignin models by cleaving the Cα–Cβ bond under blue LED irradiation (λ = 455 nm) in the presence of O2.26 A proposed mechanism for the photodriven transformation of lignin molecule 1 using mpg-C3N4 as the catalyst is illustrated in Fig. 13.
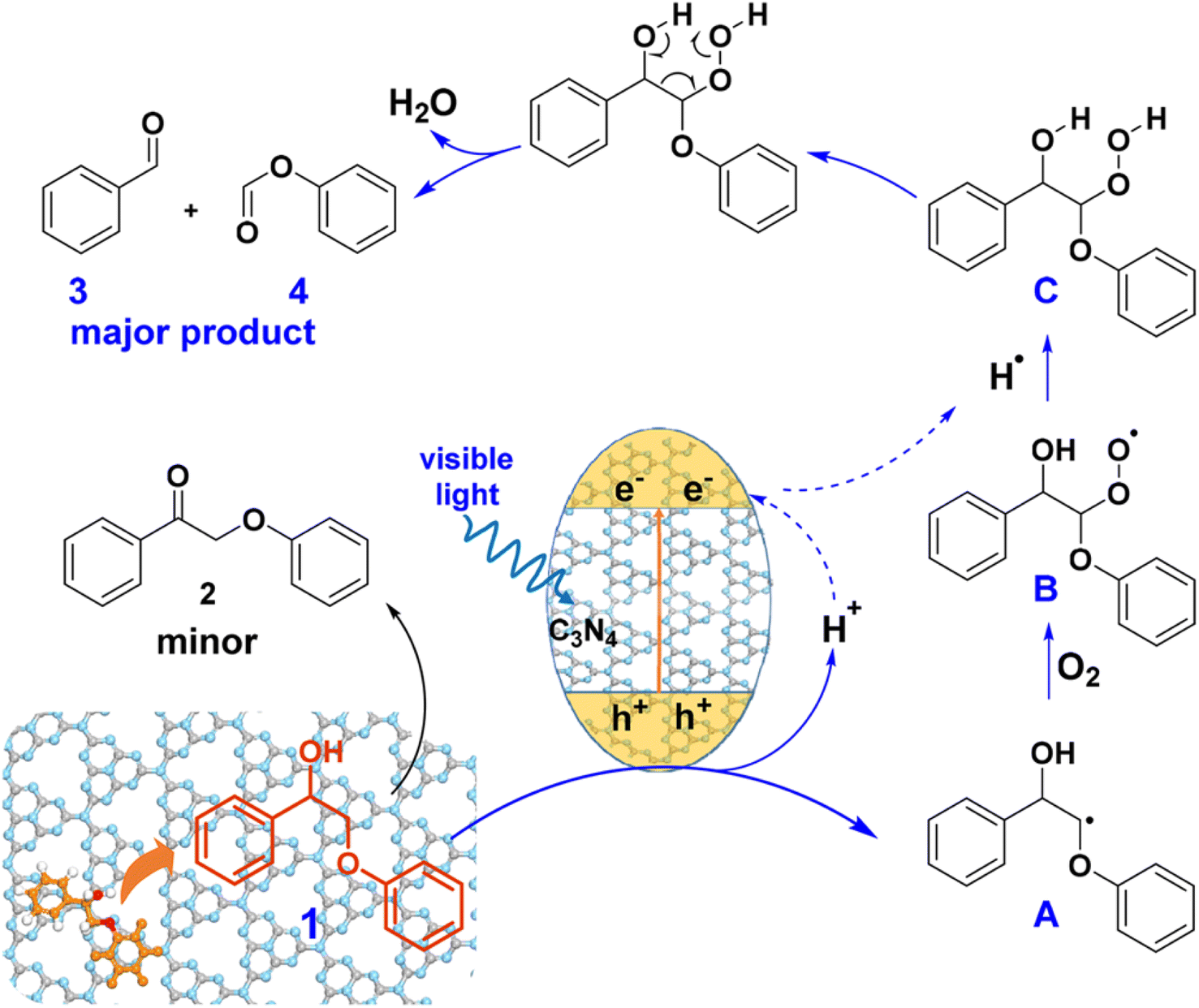 | ||
| Fig. 13 Proposed mechanism of mpg-C3N4 catalyzed transformation of molecule 1. Adapted from ref. 26 with permission. Copyright 2018 American Chemical Society. | ||
When exposed to visible light irradiation, mpg-C3N4 becomes excited, generating holes and electrons separately in the valence and conduction bands. For the reactant molecule interacting with the mpg-C3N4 surface, a hydrogen atom from Cβ is abstracted due to the action of photo-generated holes and surface basic sites on mpg-C3N4, resulting in the formation of a Cβ-centered radical (A).27 This radical further combines with O2 and hydrogen to produce a peroxide intermediate C. Subsequent electron transfer in C, facilitated through a six-membered ring transition state, induces the cleavage of both the Cα–Cβ and O–O bonds, resulting in the formation of aromatic aldehyde and phenyl formate as the major products. Some of the aldehyde product can undergo further oxidation to form benzoic acid. On the other side, photogenerated electrons can reduce O2 molecules to form the superoxide radical anion O2˙−, which can deprotonate benzylic alcohol to produce the alkoxide anion and ultimately form byproduct 2.28 Therefore, the construction of a novel and efficient multiphase photocatalytic system offers a new strategy for the photocatalytic cleavage of lignin models under visible light.
4. Electrocatalytic transformation of lignin
Lignin holds great promise as a valuable resource for producing renewable organic chemicals and fuels. In the context of a sustainable biorefinery, the valorization of lignin is crucial for improving the economic viability of overall biomass conversion processes.29 Electrocatalysis and electrochemical processes are expected to play a significant role in lignin valorization because they offer the possibility of utilizing renewable electricity sources to produce “green” hydrogen (H2) and other necessary reagents in situ for biomass conversion. Recent advancements in the electrocatalytic hydrogenation and hydrogenolysis of lignin derivatives, including oxygenated aromatic compounds, present promising pathways for the synthesis of industrially relevant chemicals. For example, ketonealcohol (KA) oil, a mixture of cyclohexanone and cyclohexanol, can be produced for the manufacturing of nylon polymers. Electrocatalytic hydrogenation (ECH) has been employed in the electrocatalytic valorization of lignin-derived substrates or lignin itself in recent studies.Electrocatalytic hydrogenation (ECH) is a process that operates with the influence of electricity at ambient temperature and atmospheric pressure, eliminating the need for hydrogen gas or organic hydrogen donors. This characteristic makes it a promising method for lignin conversion.30 However, the aqueous ECH process faces challenges related to competitive hydrogen evolution, which results in reduced faradaic efficiency. This issue significantly hampers the performance of ECH because the electrons are diverted from producing the desired target products to reducing proton ions, leading to the generation of hydrogen gas instead.
One of the reasons for this problem is the necessity for unsaturated substrates to travel long distances to reach the pores of the electrode where the noble metal catalyst is situated, and where the actual reaction occurs. Concurrently, active hydrogen species (H*) can easily combine to form H2 gas, which strongly inhibits the hydrogenation reactions. Despite the use of 3D porous electrodes with extensive surface areas in ECH, transport and kinetic diffusion limitations persistently hinder the reaction rate.31 Furthermore, conventional electrocatalytic techniques rely on conductive polymeric binders to attach active catalyst particles to the electrodes. This approach inherently leads to the loss of catalytic active sites and presents significant stability challenges for the adhered catalysts, especially when exposed to high operating current densities and the reaction solution. As a result of these factors, most ECH processes are known to operate at low current densities (around 40 mA cm−2) with limited faradaic efficiency, typically falling within the range of 20–60%.32
The ECH process offers several advantages, including: (i) greener hydrogen gas: it generates hydrogen gas internally and continuously from water splitting reactions, eliminating the need for an external hydrogen supply. (ii) Milder reaction conditions: ECH operates under lower temperature and pressure using aqueous electrolytes, eliminating the need for organic solvents and addressing issues related to catalyst deactivation due to coking. (iii) Cleaner process: it results in higher carbon recovery and produces useful by-products, such as hydrogen and oxygen gases. These by-products can be effectively utilized for fuel cell applications when implemented on a large scale. (iv) Simpler operation: ECH allows for the control of operating parameters, such as current/voltage input, making the process more controllable and adaptable to specific requirements.33
Electrocatalyst preparation and application techniques involve the design of electrocatalytic materials. Noble metal catalysts like Pt, Pd, Ru, and Rh are commonly used components of electrode materials. For instance, Amouzegar et al. reported a Pt-supported Vulcan XC-72R catalyst with Teflon as the binding material, which exhibited higher selectivity and current efficiency (85%).34 The superior performance in the electrocatalytic hydrogenation of phenol can be attributed to the enhanced adsorption of phenol on the catalyst surface.
Savadogo et al. discovered that the reaction is not sensitive to the Pt particle size, but the efficiency and selectivity are influenced by the electrode material, such as the presence of alloying components like Pt–Co/C.35 Pt/C electrode is significantly more active than platinized platinum (Pt/Pt) electrode due to increased Pt electron density in its interaction with the carbon support, leading to a metal–support interaction effect that weakens the Pt–H bond. Additionally, the study of the reaction mechanism revealed that the rate-determining step (RDS) for the phenol electrocatalytic hydrogenation to cyclohexanol is the surface reaction between the adsorbed species, not the adsorption of phenol.
Song et al. also found that the electrocatalytic hydrogenation proceeds via a Langmuir-type mechanism in which surface hydrogen is produced by the reduction of protons when the catalyst contacts the electrode, rather than H2 dissociation as in thermal hydrogenation (Fig. 14).31c In this mechanism, the electrocatalytic hydrogenation of phenol follows a Langmuir–Hinshelwood mechanism in which H+ ions and the substrate are adsorbed on the catalyst metal particles. Upon contact with the electrode, protons are reduced, generating adsorbed hydrogen radicals that hydrogenate the adsorbed hydrocarbons.
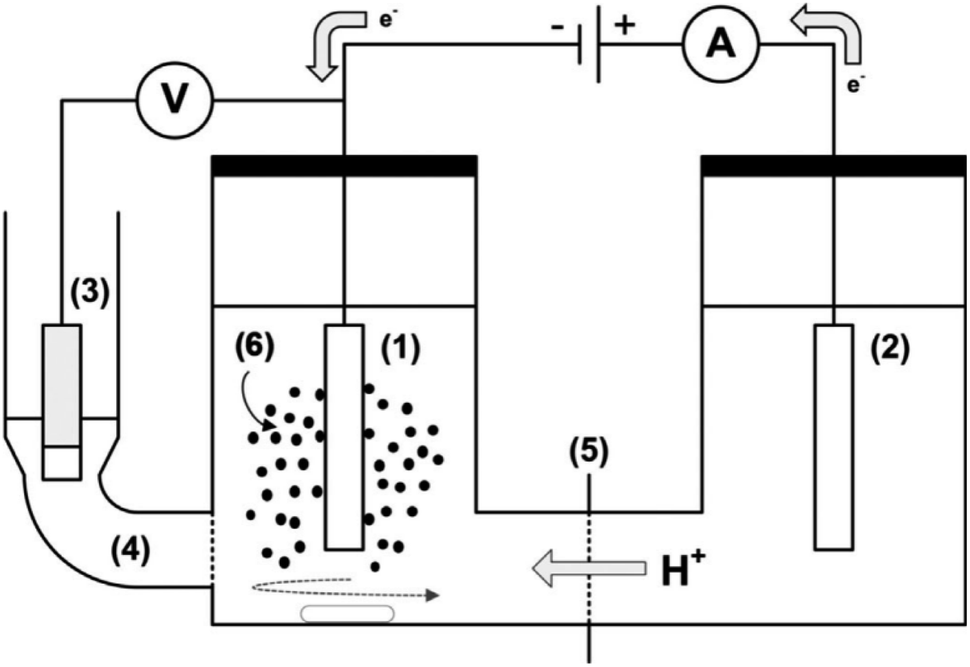 | ||
| Fig. 14 A sketch of electrochemical cell setup (H-cell) for ECH of guaiacol and phenol: (1) cathode, (2) anode, (3) reference electrode (Ag/AgCl/3 M KCl), (4) fritted side compartment (Luggin probe) for reference electrode filled with 3 M KCl, (5) cation exchange membrane, and (6) catalyst slurry. Adapted from ref. 33 with permission. Copyright 2019 Wiley-VCH. | ||
To improve the liquid–solid mass transfer in electrochemical reactions, a stirred slurry electrochemical reactor (SSER) was developed. It can be used as a fluidized bed or a moving bed electrocatalytic reactor.33 In the case of the neutral–acid pair, proton diffusion and migration through the membrane from the anolyte to the catholyte provide the protons required for electrocatalytic hydrogenation (ECH). Typically, the two major hydrogenation products are cyclohexanol and 2-methoxycyclohexanol. However, under constant cathode superficial current density (−182 mA cm−2) and higher temperature (e.g., 60 °C), ECH favors a pathway that predominantly produces cyclohexanone (Fig. 15). The conversion pathways of guaiacol are influenced by temperature and cathode potential-dependent surface coverage of adsorbed hydrogen radicals generated through the electroreduction of protons.
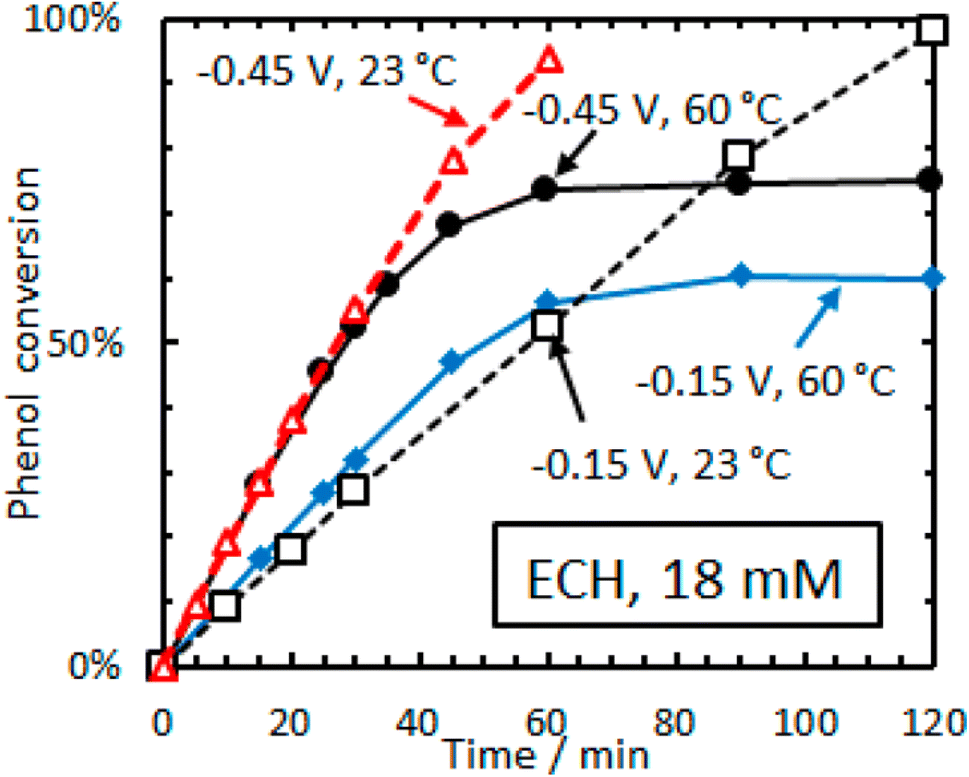 | ||
| Fig. 15 Conversion versus time for ECH of phenol (18 mM) on 5 wt% Rh/C in aqueous acetate buffer. Adapted from ref. 36 with permission. Copyright 2016 American Chemical Society. | ||
Ruthenium is also an important hydrogenation catalyst, demonstrating remarkable catalytic activity in hydrogenation and hydrolysis reactions. Singh et al. reported a counterintuitive temperature effect in the electrocatalytic hydrogenation (ECH) of phenol (18 mM) over rhodium on carbon (Rh/C) at low cathode potentials (−0.15 to −0.45 V vs. RHE).36 The phenol conversion reached a plateau (60–75%) at higher temperature (60 °C) but almost 100% at room temperature (23 °C), suggesting the possibility of thermal dehydrogenation of phenol species that could block the active sites. Additionally, lower Hads (adsorbed hydrogen) coverage was observed at elevated temperatures (Fig. 16). The reaction rate is highly dependent on PH2, indicating that the H* coverage is low and is determined by the steady-state balance between its adsorption rate to form H* and the desorption rate of H* to form H2. The observation that H* is primarily removed as H2, even in ECH, is consistent with the low faradaic efficiency reported. Similarly, the cathode potential significantly affects the Hads coverage, with ECH rates increasing faster than the rates of the hydrogen evolution reaction (HER) with increasingly negative potentials. This leads to higher cyclohexanol production at room temperature (23 °C) using low phenol concentrations (16 mM).37 At higher temperatures (30–50 °C) and higher phenol concentrations (105 mM) under potentiostatic control, cyclohexanol was also obtained as the most selective product in the ECH of phenol using H2SO4 (0.2 M) and 5 wt% Pt/C. Interestingly, the product distribution shifted to cyclohexanone under galvanostatic control (j = −109 mA cm−2) at a constant temperature (50 °C), demonstrating the influence of temperature-potential synergy on the Hads coverage on the catalyst surface.
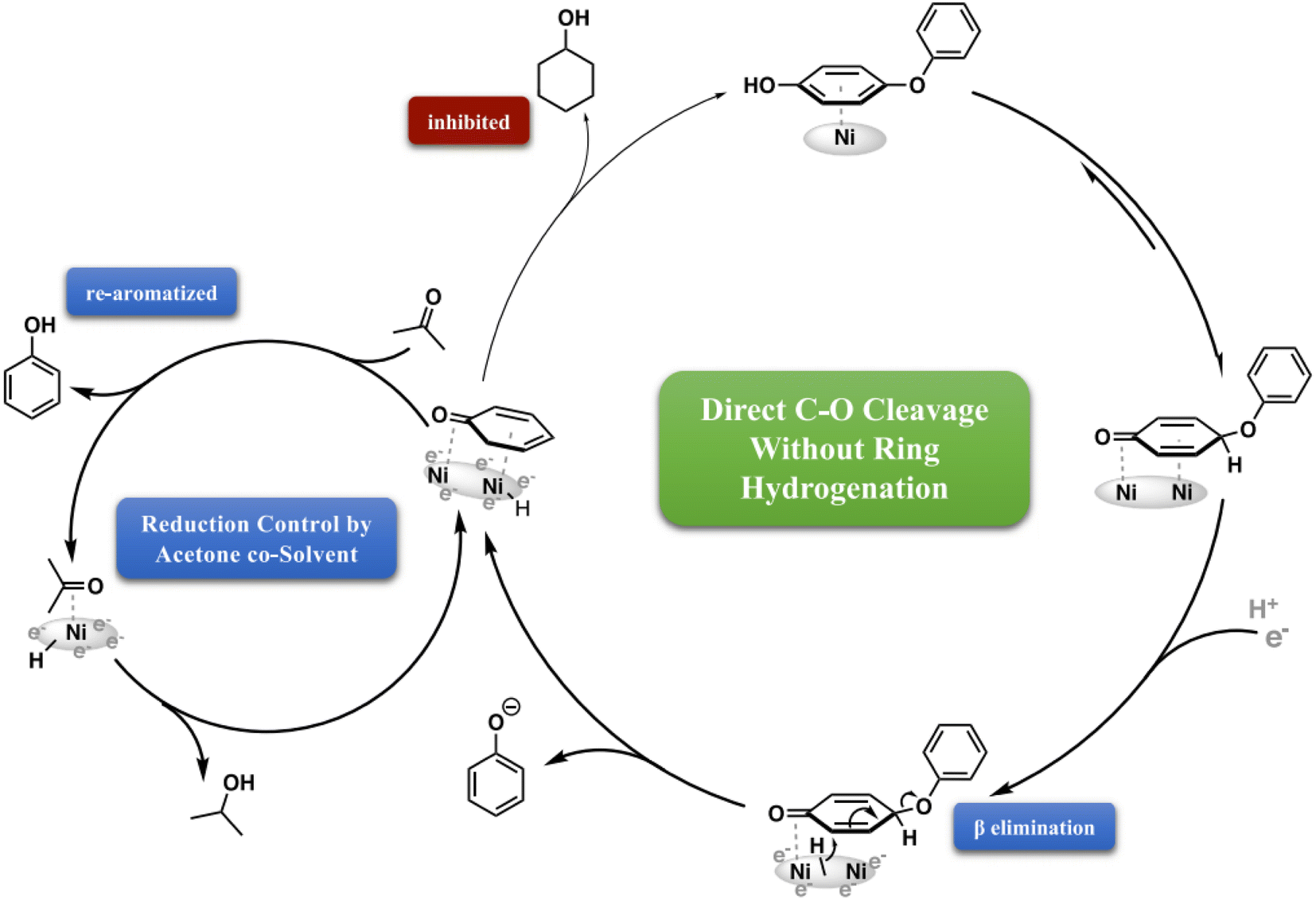 | ||
| Fig. 16 Selective inhibition of aromatic ring reduction by acetone co-solvent. Schematic illustration of product selectivity control by the inclusion of acetone as a co-solvent during ECH of 4-phenoxyphenol. Adapted from ref. 38 with permission. Copyright 2022 Springer Nature. | ||
In addition to noble metals, certain base metal catalysts such as Ni, Fe, or Co have also demonstrated high activity in electrocatalytic hydrogenation. Zhou et al. achieved direct C–O cleavage without initial benzene ring saturation by using electrocatalytic hydrogenation (ECH) over skeletal Ni cathodes in a mild, aqueous process.38 Two distinct cleavage mechanisms were identified: (a) dual-ring coordination and C–H activation: This mechanism involves the activation of two adjacent rings, leading to vicinal elimination to form phenol and a surface-bound aryne intermediate. The aryne intermediate is subsequently hydrogenated and released as the arene. (b) Surface binding in keto form: in this mechanism, the phenolic ring of hydroxy-substituted substrates binds to the catalyst surface in a keto form, followed by the direct displacement of the departing phenol.
As phenol is produced through the cleavage of phenoxyphenol, its reabsorption is outcompeted by acetone adsorption, which blocks the sites responsible for converting phenol to cyclohexanol. Acetone, present as a co-solvent throughout the reaction, slows down the cleavage of the ether C–O bond but does not completely inhibit it, as it does the phenol hydrogenation. A significant difference between these two processes lies in the energy barrier for the reaction. Cleavage of the phenoxide requires only one H2 equivalent and is favored by an increase in entropy. However, reducing the bound phenol necessitates the transfer of three H2 equivalents, breaking the aromaticity and strong surface binding of the phenol ring. As a result, acetone selectively inhibits phenol monomer reduction, allowing intentional control over the choice of saturated or aromatic monomers.38
Multimetallic catalysts are well-known for their ability to fine-tune catalytic performance due to their versatility in compositional, electronic, and geometric properties.39 Various metal elements have been extensively studied for their potential in constructing multimetallic catalysts, which find applications in both electrocatalytic and thermocatalytic reactions.
For instance, Wu et al. have developed an innovative integration approach using nitrogen-doped hierarchically porous carbon (NHPC) as a support material.40 They designed and synthesized NiPt and FeRu catalysts, which, when supported by NHPC, exhibit remarkable efficiency and selectivity as cathode and anode catalysts. These catalysts achieve selectivity rates of over 99.9% for both cyclohexanone and benzoquinone production. Experimental and theoretical studies suggest the following reaction mechanism: (1) phenol molecules are initially adsorbed onto the surface of the NiPt NPs in the catalytic system. (2) Under electrostatic attraction, the phenolic hydroxyl-group-derived proton transfers to the NiPt NPs. (3) The aromatic ring is activated by the proton dissociated from the HSO4− anion, leading to the fast tautomerization of the adsorbed phenol molecule into 2,4-cyclohexadienone. (4) Reduction is initiated by the attack of an electron on the Cδ+ site of the conjugated ethylene bonds in 2,4-cyclohexadienone. (5) Proton transfer occurs to the Cδ− site. (6) Charge redistribution takes place in the resulting species, where new Cδ+ and Cδ− sites accept the transfer of the second electron and second proton. (7) The ethylene bond in 3-cyclohexenone reacts with two electrons and two protons based on the above reduction pathway, ultimately yielding the cyclohexanone product. Compared to the commercial Pt/C catalyst, the alloyed NiPt NPs have a higher electron cloud density on active Pt sites due to the electron transfer from Ni to Pt. This prevents the adsorption of the carbonyl on the NiPt NPs and avoids the further reduction of the cyclohexanone product, resulting in enhanced catalytic performance.
5. Summary and outlook
Lignin, a natural resource rich in potential, holds promise for the production of valuable aromatic chemicals. Over the years, various catalytic systems, including thermal catalysts, photocatalysts, and electrocatalysts, have been explored for lignin deconstruction. However, the search for an optimal and technologically feasible method for lignin transformation continues.Among these catalytic systems, small organic molecule-catalyzed lignin oxidation has emerged as a promising candidate for further development. This preference is due to a deeper understanding of its intricate mechanism, distinguishing it from the complexities associated with enzymatic or surface catalysis. Recently, the challenging aspect of lignin depolymerization into monomers primarily stems from several factors. Firstly, the complexity of the process is a result of the intricate lignin structure, characterized by a variety of targeted linkages. Additionally, the different transformations of the lignin structure and the condensation modes of lignin fragments/monomers into stable linkages contribute to this complexity. Moreover, the task of separating a wide range of monomers with similar molecular structures poses a formidable challenge in lignin depolymerization, leading to increased cost disparities between the biomass-based and fossil-based routes in chemical production. Consequently, the primary hurdle in lignin depolymerization lies in the development of strategies employing selective and active catalysts capable of breaking the prevalent C–O/C–C linkage bonds while preserving the aromatic rings, and yielding final products with a narrow species distribution.
Moreover, the design of catalysts for lignin depolymerization faces several challenges, including the following aspects: (a) lignin is a highly diverse polymer with variations in structure due to its biological source and growth conditions. This diversity makes it difficult to develop universal catalysts, as different types of lignin require different catalysts for effective depolymerization; (b) lignin contains numerous C–O and C–C bonds, and selectively breaking these bonds to obtain desired monomers is a challenging task. Catalysts must be capable of selectively cleaving C–O/C–C bonds while preserving the integrity of other lignin structures; (c) during the lignin depolymerization process, some reaction products or impurities may adsorb onto the catalyst's surface, leading to catalyst poisoning and a reduction in its activity. Therefore, developing mild and efficient catalysts for lignin depolymerization is a significant challenge.
This review serves as a compilation of accumulated insights, offering a comprehensive overview of catalytic system design for lignin oxidation. It is our hope that this summary will inspire and motivate further research and innovation in this critical field, ultimately unlocking the full potential of lignin as a valuable resource.
Author contributions
Wang Ziwei: methodology, formal analysis, writing – review & editing. Shu Hao: investigation, methodology, visualization. Chen Yizhen: visualization, formal analysis, supervision, funding acquisition. Liu Ben: formal analysis, supervision. Xu Yaowei: visualization, formal analysis. Wang Wanxia: visualization, formal analysis. Wang Kaiyue: visualization, formal analysis, supervision, funding acquisition. Lei Mengheng: visualization, formal analysis. Guo Li: methodology, supervision, funding acquisition. Wang Lei: methodology, supervision, writing – review & editing. All authors have read and agreed to the published version of the manuscript.Conflicts of interest
The authors report no conflict of interest in any capacity, i.e., competing or financial.Acknowledgements
This work was supported by the National Natural Science Foundation of China (No. 21902053 and 22002114), the Research Project supported by Foundation of State Key Laboratory of High-efficiency Utilization of Coal and Green Chemical Engineering (Grant No. 2021-K73). This work was supported by scientific research project of the Education Department of Hubei Province (Q20201701). All authors are thankful to their representative universities/institutes for literature services.References
- S. Mohr, J. Wang, G. Ellem, J. Ward and D. Giurco, Projection of world fossil fuels by country, Fuel, 2015, 141, 120–135 CrossRef CAS.
- (a) A. Pineda and A. F. Lee, Heterogeneously catalyzed lignin depolymerization, Appl. Petrochem. Res., 2016, 6, 243–256 CrossRef CAS PubMed; (b) R. E. Key and J. J. Bozell, Progress toward lignin valorization via selective catalytic technologies and the tailoring of biosynthetic pathways, ACS Sustainable Chem. Eng., 2016, 4, 5123–5135 CrossRef CAS; (c) S.-H. Li, S. Liu, J. C. Colmenares and Y.-J. Xu, Progress toward lignin valorization via selective catalytic technologies and the tailoring of biosynthetic pathways, Green Chem., 2016, 18, 594–607 RSC; (d) W. Guan, C.-W. Tsang, C. S. K. Lin, C. Len, H. Hu and C. Liang, A review on high catalytic efficiency of solid acid catalysts for lignin valorization, Bioresour. Technol., 2020, 298, 122432 CrossRef CAS PubMed; (e) C. O. Tuck, E. Pérez, I. T. Horváth, R. A. Sheldon and M. Poliakoff, Valorization of biomass: deriving more value from waste, Science, 2012, 337, 695–699 CrossRef CAS PubMed.
- (a) T. Ren, S. You, Z. Zhang, Y. Wang, W. Qi, R. Su and Z. He, Highly selective reductive catalytic fractionation at atmospheric pressure without hydrogen, Green Chem., 2021, 23, 1648–1657 RSC; (b) S. S. Wong, R. Shu, J. Zhang, H. Liu and N. Yan, Downstream processing of lignin derived feedstock into end products, Chem. Soc. Rev., 2020, 49, 5510–5560 RSC.
- S. Van den Bosch, T. Renders, S. Kennis, S.-F. Koelewijn, G. Van den Bossche, T. Vangeel, A. Deneyer, D. Depuydt, C. Courtin and J. Thevelein, Integrating lignin valorization and bio-ethanol production: on the role of Ni-Al2O3 catalyst pellets during lignin-first fractionation, Green Chem., 2017, 19, 3313–3326 RSC.
- X. Li and Y. Song, Structure and function of SARS-CoV and SARS-CoV-2 main proteases and their inhibition: A comprehensive review, Eur. J. Med. Chem., 2023, 260, 115772 CrossRef CAS PubMed.
- C. Zhang, X. Shen, Y. Jin, J. Cheng, C. Cai and F. Wang, Catalytic strategies and mechanism analysis orbiting the center of critical intermediates in lignin depolymerization, Chem. Rev., 2023, 123, 4510–4601 CrossRef CAS PubMed.
- G. Meng, W. Lan, L. Zhang, S. Wang, T. Zhang, S. Zhang, M. Xu, Y. Wang, J. Zhang and F. Yue, Synergy of Single Atoms and Lewis Acid Sites for Efficient and Selective Lignin Disassembly into Monolignol Derivatives, J. Am. Chem. Soc., 2023, 145, 12884–12893 CrossRef CAS PubMed.
- J. He, C. Zhao and J. A. Lercher, Ni-catalyzed cleavage of aryl ethers in the aqueous phase, J. Am. Chem. Soc., 2012, 134, 20768–20775 CrossRef CAS PubMed.
- (a) V. Molinari, C. Giordano, M. Antonietti and D. Esposito, Titanium nitride-nickel nanocomposite as heterogeneous catalyst for the hydrogenolysis of aryl ethers, J. Am. Chem. Soc., 2014, 136, 1758–1761 CrossRef CAS PubMed; (b) V. Molinari, G. Clavel, M. Graglia, M. Antonietti and D. Esposito, Mild continuous hydrogenolysis of kraft lignin over titanium nitride-nickel catalyst, ACS Catal., 2016, 6, 1663–1670 CrossRef CAS.
- J. Zhang, J. Teo, X. Chen, H. Asakura, T. Tanaka, K. Teramura and N. Yan, A series of NiM (M= Ru, Rh, and Pd) bimetallic catalysts for effective lignin hydrogenolysis in water, ACS Catal., 2014, 4, 1574–1583 CrossRef CAS.
- M. V. Galkin, C. Dahlstrand and J. S. Samec, Mild and Robust Redox-Neutral Pd/C-Catalyzed Lignol β-O-4′ Bond Cleavage Through a Low-Energy-Barrier Pathway, ChemSusChem, 2015, 8, 2187–2192 CrossRef CAS PubMed.
- A. K. Singh, S. Jang, J. Y. Kim, S. Sharma, K. Basavaraju, M.-G. Kim, K.-R. Kim, J. S. Lee, H. H. Lee and D.-P. Kim, One-Pot Defunctionalization of Lignin-Derived Compounds by Dual-Functional Pd50Ag50/Fe3O4/N-rGO Catalyst, ACS Catal., 2015, 5, 6964–6972 CrossRef CAS.
- K. B. Jung, J. Lee, J.-M. Ha, H. Lee, D. J. Suh, C.-H. Jun and J. Jae, Effective hydrodeoxygenation of lignin-derived phenols using bimetallic RuRe catalysts: Effect of carbon supports, Catal. Today, 2018, 303, 191–199 CrossRef CAS.
- T. Liu, Z. Tian, W. Zhang, B. Luo, L. Lei, C. Wang, J. Liu, R. Shu and Y. Chen, Selective hydrodeoxygenation of lignin-derived phenols to alkyl cyclohexanols over highly dispersed RuFe bimetallic catalysts, Fuel, 2023, 339, 126916 CrossRef CAS.
- Z. Li, S. Ji, Y. Liu, X. Cao, S. Tian, Y. Chen, Z. Niu and Y. Li, Well-defined materials for heterogeneous catalysis: from nanoparticles to isolated single-atom sites, Chem. Rev., 2019, 120, 623–682 CrossRef PubMed.
- S. Wang, K. Zhang, H. Li, L.-P. Xiao and G. Song, Selective hydrogenolysis of catechyl lignin into propenylcatechol over an atomically dispersed ruthenium catalyst, Nat. Commun., 2021, 12, 416 CrossRef CAS PubMed.
- J. Xie, Y. Xi, W. Gao, H. Zhang, Y. Wu, R. Zhang, H. Yang, Y. Peng, F. Li, Z. Li and C. Li, Hydrogenolysis of lignin model compounds on Ni nanoparticles surrounding the oxygen vacancy of CeO2, ACS Catal., 2023, 13, 9577–9587 CrossRef CAS.
- X. Wu, N. Luo, S. Xie, H. Zhang, Q. Zhang, F. Wang and Y. Wang, Photocatalytic transformations of lignocellulosic biomass into chemicals, Chem. Soc. Rev., 2020, 49, 6198–6223 RSC.
- N. Luo, M. Wang, H. Li, J. Zhang, T. Hou, H. Chen, X. Zhang, J. Lu and F. Wang, Visible-light-driven self-hydrogen transfer hydrogenolysis of lignin models and extracts into phenolic products, ACS Catal., 2017, 7, 4571–4580 CrossRef CAS.
- X. Wu, X. Fan, S. Xie, J. Lin, J. Cheng, Q. Zhang, L. Chen and Y. Wang, Solar energy-driven lignin-first approach to full utilization of lignocellulosic biomass under mild conditions, Nat. Catal., 2018, 1, 772–780 CrossRef CAS.
- G. Han, T. Yan, W. Zhang, Y. C. Zhang, D. Y. Lee, Z. Cao and Y. Sun, Highly Selective Photocatalytic Valorization of Lignin Model Compounds Using Ultrathin Metal/CdS, ACS Catal., 2019, 9, 11341–11349 CrossRef CAS.
- H. Yoo, M.-W. Lee, S. Lee, J. Lee, S. Cho, H. Lee, H. G. Cha and H. S. Kim, Enhancing photocatalytic β-O-4 bond cleavage in lignin model compounds by silver-exchanged cadmium sulfide, ACS Catal., 2020, 10, 8465–8475 CrossRef CAS.
- (a) G. Magallanes, M. D. Karkas, I. Bosque, S. Lee, S. Maldonado and C. R. Stephenson, Lignin C–C bond cleavage induced by consecutive two-photon excitation of a metal-free photocatalyst, ACS Catal., 2019, 9, 2252–2260 CrossRef CAS; (b) N. Luo, M. Wang, H. Li, J. Zhang, H. Liu and F. Wang, Photocatalytic oxidation–hydrogenolysis of lignin β-O-4 models via a dual light wavelength switching strategy, ACS Catal., 2016, 6, 7716–7721 CrossRef CAS.
- Z. Peng, Z. Wu, X. Sun and H. Li, Photocatalytic transfer hydrogenolysis of aryl ethers, Green Chem., 2023, 25, 6869–6880 RSC.
- H. Liu, H. Li, N. Luo and F. Wang, Visible-light-induced oxidative lignin C-C bond cleavage to aldehydes using vanadium catalysts, ACS Catal., 2019, 10, 632–643 CrossRef.
- H. Liu, H. Li, J. Lu, S. Zeng, M. Wang, N. Luo, S. Xu and F. Wang, Photocatalytic cleavage of C-C bond in lignin models under visible light on mesoporous graphitic carbon nitride through π–π stacking interaction, ACS Catal., 2018, 8, 4761–4771 CrossRef CAS.
- T. Hou, N. Luo, H. Li, M. Heggen, J. Lu, Y. Wang and F. Wang, Yin and Yang Dual Characters of CuOx Clusters for C-C Bond Oxidation Driven by Visible Light, ACS Catal., 2017, 7, 3850–3859 CrossRef CAS.
- F. Su, S. C. Mathew, G. Lipner, X. Fu, M. Antonietti, S. Blechert and X. Wang, mpg-C3N4-Catalyzed Selective Oxidation of Alcohols Using O2 and Visible Light, J. Am. Chem. Soc., 2010, 132, 16299–16301 CrossRef CAS PubMed.
- Y. P. Wijaya, K. J. Smith, C. S. Kim and E. L. Gyenge, Electrocatalytic hydrogenation and depolymerization pathways for lignin valorization: toward mild synthesis of chemicals and fuels from biomass, Green Chem., 2020, 22, 7233–7264 RSC.
- (a) C. H. Lam, S. Das, N. C. Erickson, C. D. Hyzer, M. Garedew, J. E. Anderson, T. J. Wallington, M. A. Tamor, J. E. Jackson and C. M. Saffron, Towards sustainable hydrocarbon fuels with biomass fast pyrolysis oil and electrocatalytic upgrading, Sustainable Energy Fuels, 2017, 1, 258–266 RSC; (b) M. Garedew, D. Young-Farhat, J. E. Jackson and C. M. Saffron, Electrocatalytic upgrading of phenolic compounds observed after lignin pyrolysis, ACS Sustainable Chem. Eng., 2019, 7, 8375–8386 CrossRef CAS.
- (a) Z. Li, M. Garedew, C. H. Lam, J. E. Jackson, D. J. Miller and C. M. Saffron, Mild electrocatalytic hydrogenation and hydrodeoxygenation of bio-oil derived phenolic compounds using ruthenium supported on activated carbon cloth, Green Chem., 2012, 14, 2540–2549 RSC; (b) C. H. Lam, C. B. Lowe, Z. Li, K. N. Longe, J. T. Rayburn, M. A. Caldwell, C. E. Houdek, J. B. Maguire, C. M. Saffron and D. J. Miller, Electrocatalytic upgrading of model lignin monomers with earth abundant metal electrodes, Green Chem., 2015, 17, 601–609 RSC; (c) Y. Song, O. Y. Gutiérrez, J. Herranz and J. A. Lercher, Aqueous phase electrocatalysis and thermal catalysis for the hydrogenation of phenol at mild conditions, Appl. Catal., B, 2016, 182, 236–246 CrossRef CAS; (d) S. Jung and E. J. Biddinger, Electrocatalytic hydrogenation and hydrogenolysis of furfural and the impact of homogeneous side reactions of furanic compounds in acidic electrolytes, ACS Sustainable Chem. Eng., 2016, 4, 6500–6508 CrossRef CAS.
- Y. Wang, Y. Liu, J. He and Y. Zhang, CeCl3-Promoted Simultaneous Photocatalytic Cleavage and Amination of Cα-Cβ Bond in Lignin Model Compounds and Native Lignin, Sci. Bull., 2019, 64, 1658–1666 CrossRef CAS PubMed.
- Y. P. Wijaya, T. Grossmann-Neuhaeusler, R. D. Dhewangga Putra, K. J. Smith, C. S. Kim and E. L. Gyenge, Electrocatalytic hydrogenation of guaiacol in diverse electrolytes using a stirred slurry reactor, ChemSusChem, 2020, 13, 629–639 CrossRef CAS PubMed.
- K. Amouzegar and O. Savadogo, Electrocatalytic hydrogenation of phenol on highly dispersed Pt electrodes, Electrochim. Acta, 1994, 39, 557–559 CrossRef CAS.
- K. Amouzegar and O. Savadogo, Electrocatalytic hydrogenation of phenol on dispersed Pt: Effect of metal electrochemically active surface area and electrode material, J. Appl. Electrochem., 1997, 27, 539–542 CrossRef CAS.
- N. Singh, Y. Song, O. Y. Gutiérrez, D. M. Camaioni, C. T. Campbell and J. Lercher, Electrocatalytic hydrogenation of phenol over platinum and rhodium: unexpected temperature effects resolved, ACS Catal., 2016, 6, 7466–7470 CrossRef CAS.
- Y. Song, S. H. Chia, U. Sanyal, O. Y. Gutiérrez and J. A. Lercher, Integrated catalytic and electrocatalytic conversion of substituted phenols and diaryl ethers, J. Catal., 2016, 344, 263–272 CrossRef CAS.
- Y. Zhou, G. E. Klinger, E. L. Hegg, C. M. Saffron and J. E. Jackson, Skeletal Ni electrode-catalyzed CO cleavage of diaryl ethers entails direct elimination via benzyne intermediates, Nat. Commun., 2022, 13, 2050 CrossRef CAS PubMed.
- (a) K. Liu, R. Qin and N. Zheng, Insights into the interfacial effects in heterogeneous metal nanocatalysts toward selective hydrogenation, J. Am. Chem. Soc., 2021, 143, 4483–4499 CrossRef CAS PubMed; (b) C.-L. Yang, L.-N. Wang, P. Yin, J. Liu, M.-X. Chen, Q.-Q. Yan, Z.-S. Wang, S.-L. Xu, S.-Q. Chu and C. Cui, Insights into the interfacial effects in heterogeneous metal nanocatalysts toward selective hydrogenation, Science, 2021, 374, 459–464 CrossRef CAS PubMed.
- R. Wu, Q. Meng, J. Yan, H. Liu, Q. Zhu, L. Zheng, J. Zhang and B. Han, Electrochemical strategy for the simultaneous production of cyclohexanone and benzoquinone by the reaction of phenol and water, J. Am. Chem. Soc., 2020, 6, eabd1951 Search PubMed.
| This journal is © The Royal Society of Chemistry 2023 |