
Recent developments on 1,2-difunctionalization and hydrofunctionalization of unactivated alkenes and alkynes involving C–S bond formation
Bo
Dong
 a,
Jian
Shen
*ab and
Lan-Gui
Xie
*a
a,
Jian
Shen
*ab and
Lan-Gui
Xie
*a
aNational and Local Joint Engineering Research Center of Biomedical Functional Materials, School of Chemistry and Materials Science, Nanjing Normal University, Nanjing 210023, China. E-mail: jshen@njnu.edu.cn; xielg@njnu.edu.cn
bJiangsu Engineering Research Center of Interfacial Chemistry, Nanjing University, Nanjing 210023, China
First published on 20th January 2023
Abstract
Alkenes and alkynes are feedstock compounds and key units in many natural products, pharmaceuticals, agrochemicals, and organic functional materials. Hydrofunctionalization and 1,2-difunctionalization of alkenes and alkynes are of the most frequently used tools in organic synthesis for adding complexity to molecules. In this aspect, the construction of carbon–sulfur bonds through the functionalization of unactivated multiple carbon–carbon bonds represents a versatile way for the synthesis of sulfur-containing compounds. This review outlines recent progress on this topic, classified according to the activation of alkenes/alkynes following radical, electrophilic, and transition-metal catalyzed processes.
 Bo Dong | Bo Dong was born in Anhui Province, China. He obtained his Bachelor's degree from Fuyang Normal University in 2020. Currently, he is pursuing his doctorate under the supervision of Professor Lan-Gui Xie and Professor Jian Shen at Nanjing Normal University. He is engaged in the development of new synthetic methodologies toward the synthesis of bioactive sulfur-containing compounds. |
 Jian Shen | Jian Shen received his Bachelor's degree at Nanjing University in 1982, Master's degree at Nanjing University in 1991, and Doctorate at Nanjing University of Science and Technology in 2005. He has been a professor at Nanjing University since 1996, a professor at Nanjing Normal University since 2002 and the director of the National and Local Joint Engineering Research Center of Biomedical Functional Materials (Nanjing Normal University) and the Jiangsu Interface Chemical Engineering Technology Research Center (Nanjing University). Prof. Jian Shen has long been committed to the research on interface chemistry and biomaterials. |
 Lan-Gui Xie | Lan-Gui Xie received his BS from Anhui University (China, 2007) and PhD degree from University of Science and Technology of China (China, 2012). After his postdoctoral training at the Max-Planck-Institut für Kohlenforschung (Germany, 2012–2013) and the University of Vienna (Austria, 2013–2015), he moved to the University of Oxford (UK) as a Marie Curie Individual Fellow in 2016. He has been a professor at Nanjing Normal University (China) since 2018. He is currently interested in the development of new methodologies for the functionalization of alkenes/alkynes and the synthesis of sulfur-containing compounds. |
1 Introduction
Alkenes and alkynes are present in numerous drugs, agrochemicals, natural products and organic functional molecules, and play a key role as intermediates in organic synthesis.1 By selectively introducing two functionalities to π bonds, molecular complexity and diversity of alkene and alkyne scaffolds can be readily achieved, representing one of the most effective and atom-economical routes to synthesize compounds that exhibit biological or pharmacological activity.2Motifs containing C–S bonds prevail in bioactive compounds and synthetic intermediates.3 In nature, more than 1000 sulfur-containing products have been separated and determined from terrestrial creatures and marine organisms. In life, sulfur compounds exist in every cell of the body, including S-adenosyl methionine, which plays a key role in the metabolic process of life. In pharmaceuticals, sulfur-containing compounds represent a landmark in the history of antibiotics and still play a significant role in the development of new drugs4 (Scheme 1A). In agriculture, more than 30% of agricultural chemicals contain at least one sulfur atom, including herbicides and pesticides.5 The introduction of a sulfur atom into a known active molecule is one of the most important tools by which to find new agricultural chemicals. In organic synthesis, sulfur commonly exhibits five oxidation states, enabling its diverse utility as a synthetic intermediate. Organic chemists have developed many important reactions and reagents on the basis of sulfur molecules,6 such as the Pummerer rearrangement, Swern oxidation, Julia olefination and Burgess reagent.7 Therefore, the construction of C–S bonds is a highly active area in organic synthesis.
 | ||
| Scheme 1 Examples of sulfur-containing natural products and drugs alongside representative strategies for the functionalization of alkenes and alkynes involving C–S bond formation. | ||
In recent years, with the continuous efforts being made in the synthesis of sulfur-containing compounds, tremendous breakthroughs have been achieved.8 It is no exaggeration that the sulfenofunctionalization and hydrosulfenylation of alkenes or alkynes have emerged as a highly efficient tool toward the construction of C–S building blocks.9 This brief review10,11 focuses on and highlights the recent progress made in the formation of C–S bonds on the basis of the 1,2-difunctionalization and hydrofunctionalization of unactivated alkenes and alkynes, which are herein classified by the type of active intermediate that initiates the transformations: radical, electrophilic sulfenofunctionalization, and transition-metal catalyzed reactions (Scheme 1B).
2 Radical processes
2.1 Electrochemical systems
Electrochemistry has emerged as an attractive synthetic tool in organic synthesis with the capability of avoiding the use of toxic and highly-polluting oxidants or reductants, and satisfactory atom economy of the overall transformation in chemical reactions.12 Recently, strategies with significance for the difunctionalization of alkenes or alkynes involving carbon–sulfur bonds formation in electrochemical systems have been presented.13 In 2018, the electrochemical sulfenofunctionalization of styrenes (7) was achieved14 by Lei et al. employing thiophenols/thiols (8) as the thiolating agents (Scheme 2). Under mild conditions, a series of β-functionalized sulfides (10) were produced in good to excellent yields using a carbon anode and a platinum cathode in an undivided cell. Notably, various functional substrates were well-tolerated under the conditions, such as α- or β-alkyl styrenes, electron-rich and electron-deficient thiophenols, and thiols. However, the use of alkyl alkenes, such as cyclohexene and 2-ethyl-1-butene, resulted in lower yields. Alcohols, acetic acid and H2O were suitable O-nucleophiles. The authors also presented the sulfenoamination of styenes under similar conditions. Both alkyl amines and anilines were demonstrated to undergo C–N bond formation. A plausible mechanism was proposed on the basis of experimental observations. The key step in this process is the addition of a thiyl radical (12) to alkenes to generate a carbon-centered radical (13), which can be further oxidized to the benzylic cationic intermediate (14) at the anode.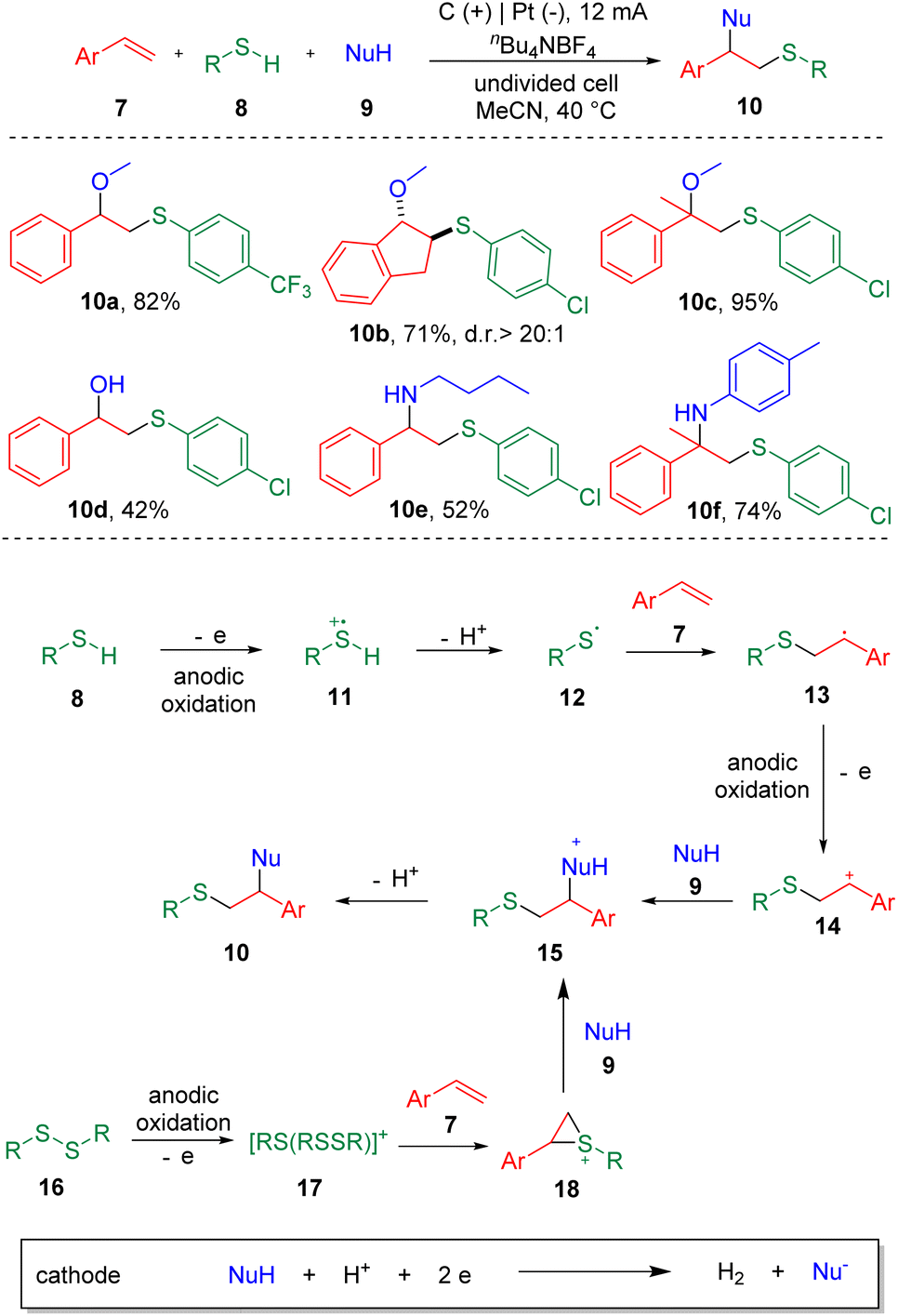 | ||
| Scheme 2 Sulfenoalkoxylation and sulfenoamination of alkenes. | ||
In the same year, the Lei et al. also reported15 the synthesis of β-alkoxysulfones (22) via the electrochemical oxidative alkoxysulfonylation of styrenes (19), using sulfonyl hydrazines (20) and alcohols (21) as the reagents (Scheme 3) to prepare a wide range of β-alkoxysulfones. Aryl sulfonyl hydrazines and 2-thiophenyl sulfonyl hydrazine (22a) underwent this transformation smoothly. Both electron-donating and electron-withdrawing functionalities could be tolerated on the phenyl ring of the styrenes. α-Methyl and α-phenyl styrenes were amenable. Both internal and terminal alkyl alkenes were unable to deliver the alkoxysulfonylation products. The reaction was conducted under chemical oxidant free conditions with molecular nitrogen and hydrogen as the byproducts. The results of cyclic voltammetry (CV) experiments showed that sulfonyl hydrazine (20) was oxidized preferentially at the anode to generate a sulfonyl radical (24), with the release of N2. A carbon-centered radical (25) was then produced by the addition of sulfonyl radical (24) to the C–C double bond, which then was oxidized to the corresponding carbon cation (26), delivering the product (22) after coupling with the alcohol nucleophile (21).
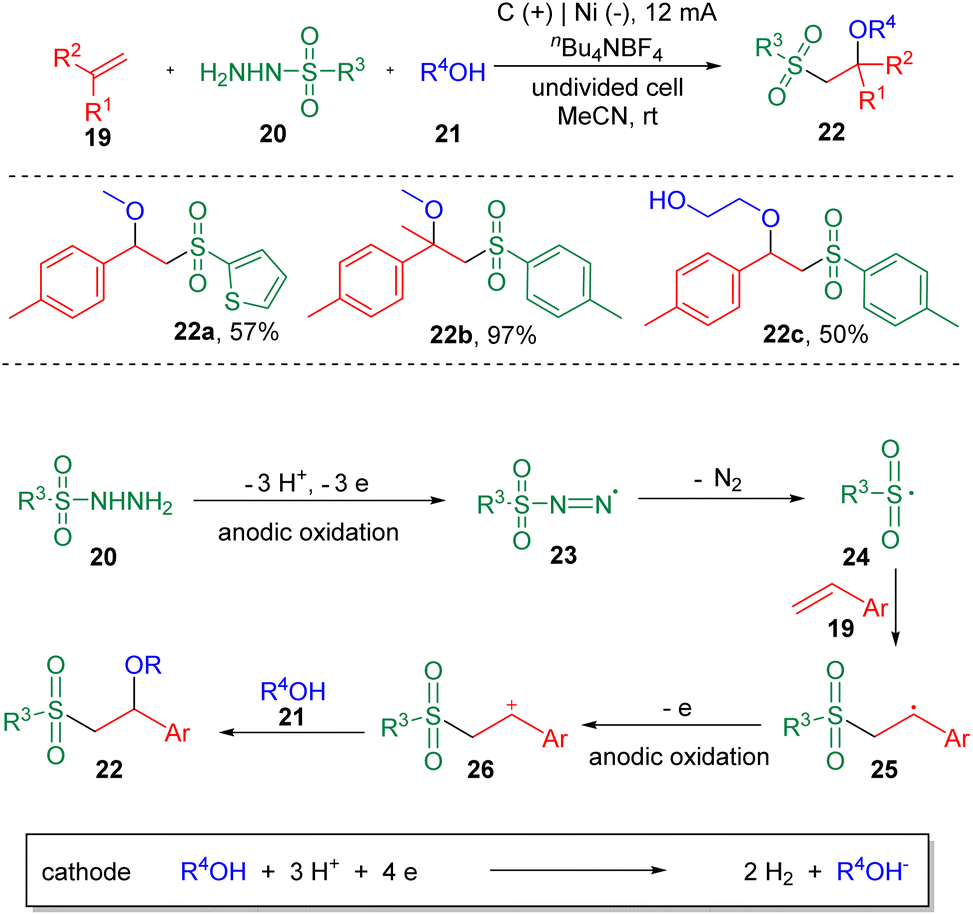 | ||
| Scheme 3 Alkoxysulfonylation of alkenes. | ||
In 2019, Sun's group16 described the electrochemical synthesis of a range of β-hydroxy- and β-alkoxy-sulfones (29) via the vicinal difunctionalization of styrenes (28) with aryl sulfinic acids (27) (Scheme 4). In this procedure, both water and alcohol were demonstrated to be applicable. The success of the installation of internal oxygen nucleophiles in the α-substitutes of the styrenes led to the synthesis of β-sulfonyllactone products (29d–29f).
 | ||
| Scheme 4 Oxysulfonylation of alkenes. | ||
Recently, a new protocol that enables the synthesis of tertiary β-hydroxysulfones (32) was developed by Lei et al.17via the electrochemical three-component reaction of α-trifluoromethyl styrenes (30), sodium sulfonates (31) and H2O (Scheme 5). A range of β-trifluoromethylsulfones were assembled by varying styrenes and sodium sulfonates. The authors claimed that this electrolysis process was sensitive to the electron effect and steric hindrance of the styrene partner, and proved that electron-deficient and ortho-substituted aryl α-CF3 alkenes were unable to undergo transformation. A labelling reaction using H218O was carried out, and the detection of the 18O-labelling product indicated that the OH group in the target product was derived from water. On the basis of CV experiments, the authors proposed that the sodium sulfonates (31) were oxidized prior to the α-CF3 styrenes (30) at the anode and produced the corresponding sulfonyl radicals, which then added to the carbon–carbon double bonds, delivering corresponding tertiary carbon radicals. Further oxidation led to the formation of the cation intermediates, which then underwent hydroxylation to produce the α-CF3-substituted tertiary alcohols.
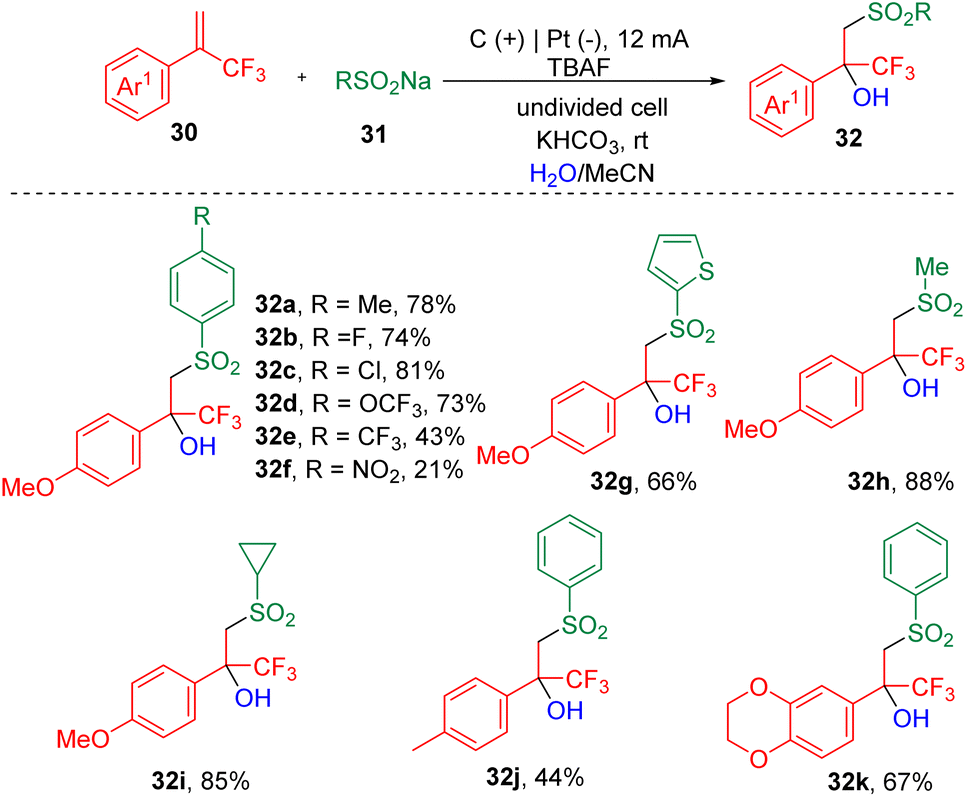 | ||
| Scheme 5 Hydroxysulfonylation of α-CF3 styrenes. | ||
The fluorosulfonylation of alkenes under electrochemical conditions has been reported by Ye and co-authors18 using sulfonylhydrazides (33) as the sulfonyl surrogates (Scheme 6). Trimethylamine trihydrofluoride (Et3N·3HF, 35) was used as the fluoride source and as the electrolyte, taking advantage of its ionic properties. In the optimization of conditions, the authors found the addition of phosphate dibasic salts enhanced the yield of the fluorosulfonylation product and attributed that to the possible increased concentration of hydrofluoride promoted by HPO42−via H-bonding. Regarding the scope of substrates, both aryl and aliphatic sulfonyl hydrazides proved to be applicable in delivering the target products in moderate to good yields. Heteroaryl hydrazide, represented by 2-thiophenyl sulfonyl hydrazine, enabled the difunctionalization of para-methylstyrene in 63% yield (36b). In the reaction scope, with respect to alkenes, both α- and β-substituted styrenes underwent fluorosulfonylation smoothly under standard conditions.
 | ||
| Scheme 6 Fluorosulfonylation of alkenes. | ||
The sulfonyl hydrazide (33) was proposed to endure consecutive anodic oxidations, leading to the formation of a sulfonyl radical (37), which then added to the styrene (34) and delivered a benzyl radical intermediate (38). Further anodic oxidation of the radical generated the benzyl carbocation (39), which was ready to undergo C–F bond formation in the presence of triethylamine trihydrofluoride (35), and eventually furnished the fluorosulfonylation product (36).
Building on the findings of the fluorosulfonylation of styrenes, the Ye group19 then developed the controllable fluorosulfenylation and fluorosulfoxidation of styrenes (41) by using thiols (40) as the sulfur sources (Scheme 7). The selectivity of oxidation states of sulfur in the target products was enabled by varying the applied cell potential. These studies conceptionally indicated the possibility of using electrosynthesis to control the diversity of reactions via “dialed-in” potentials. Mechanistic insights (Scheme 8) suggested that a radical-polar crossover process with the episulfonium ion (46) as the key intermediate was involved. Very recently, the chlorosulfoxidation of alkenes with thiols and hydrochloride was also realized by the group.20 The authors managed to extend the scope using terminal and internal alkyl alkenes. The chloride was proposed to play the role of a redox mediator, presenting as either a chlorine radical or chlorine to oxidize β-chlorosulfide to the sulfur radical cation precursor, which was then converted to the chlorosulfoxide analogue by reacting with water.
 | ||
| Scheme 7 Controllable electrochemical fluorosulfenylation and fluorosulfoxidation of styrenes. | ||
 | ||
| Scheme 8 Proposed mechanism of electrochemical fluorosulfenylation and fluorosulfoxidation. | ||
In 2021, Lei and co-workers21 established the chlorosulfonylation of terminal alkenes and alkynes (47) by merging electrochemistry and metal catalysis (Scheme 9). The reaction proceeded through the atom-transfer radical addition (ATRA) reaction via convergent paired electrolysis. The ATRA reaction has long been recognized as a general and facile protocol to realize the 1,2-functionalization of alkenes and alkynes. By using manganese chloride as the chlorine atom-transfer catalyst and redox mediator, various sulfonyl chlorides (48), terminal aryl alkenes and alkynes (47) could be engaged and delivered structurally diverse β-chloride vinyl sulfones (49) in a highly regioselective manner. The CV experiments suggested that the oxidation of Mn(II) to Mn(III) was easier than the oxidation of Cl− to a chlorine radical. According to the mechanistic insights, the authors proposed that sulfonyl chloride (48) was firstly reduced at the cathode, to generate a sulfonyl radical (50) and chloride ion. The radical addition to styrene/alkyne (47) then led to the formation of a carbon radical intermediate (51). A chlorine radical, which was produced by the oxidation at the anode via the form of Mn(III)Cl3, integrated with the carbon radical (51) to deliver the vicinal chlorosulfonylation product (49).
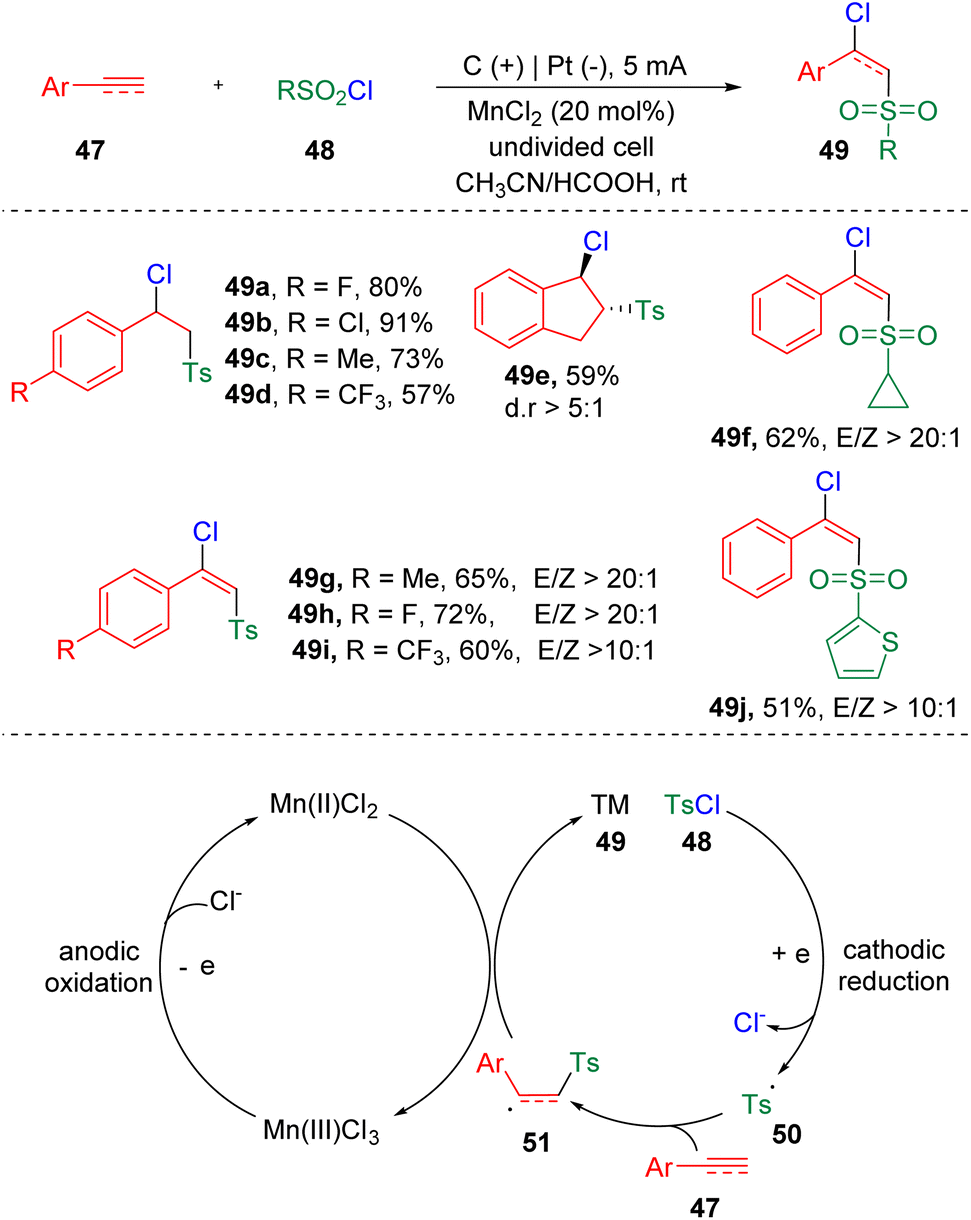 | ||
| Scheme 9 Electrochemical manganese-catalyzed chlorosulfonylation of styrenes and alkynes. | ||
2.2 Photochemical systems
Due to its inherent advantages of sustainability, photochemical synthesis has been considered as one of the mostly environmentally benign tools in the context of green chemistry and has attracted widespread attention in organic chemistry.22In 2018, the Glorius group23 developed a disulfide–ene reaction initiated by a triplet–triplet energy transfer (TTEnT) strategy with a photocatalyst (54) under visible-light conditions (Scheme 10). The protocol allowed the hydrothiolation of carbon–carbon double bonds under mild conditions in excellent chemoselectivity, and showed great tolerability to a large scope of functional groups. It was proposed that thiyl radicals (56) formed from disulfanes (53) could add to alkenes (52) with excellent regioselectivities in an anti-Markovnikov fashion. Benefiting from the adjustable photocatalyst redox potentials, the authors managed to apply the hydromethylthiolation method to alkenes bearing oxidation- or reduction-sensitive functionalities, such as amino acids, saccharides, nucleosides, single-stranded DNA, RNA (short RNA and total RNA) and human cell lysate containing various endogenous biomolecules. Phosphorescence lifetime quenching and radical scrambling experiments were carried out, and a mechanism was proposed. Following the excitation of photocatalyst [Ir–F] (54), the TTEnT activation of disulfanes (53) occurred and led to the homolytic cleavage of the S–S bond, delivering the corresponding thiyl radicals (56), which then added to a carbon–carbon double bond to initiate the entire transformation.
 | ||
| Scheme 10 Disulfide–ene reaction initiated by a triplet–triplet energy transfer (TTEnT) strategy. | ||
Recently, Liu's group24 combined styrenes (59) and (hetero)aryl thiols (60) to synthesize β-hydroxysulfides (61) via a visible-light-driven, EDA complex-promoted hydroxysulfenylation reaction (Scheme 11). Substrate screening showed that slightly electron-withdrawing (halides) and electron-donating (methyl or thiophenyl) functionalities were tolerated in this reaction. Interestingly, oxygen from the air was found to play both the role of oxidant and hydroxyl source. The authors proposed that a thiol anion (62), generated by the deprotonation of a thiol (60) with the inorganic base K2CO3, first interacted with the styrene to form an EDA complex (63), which was transferred to an excited state (64) under the irradiation of visible light, and underwent a single electron transfer (SET) process to deliver a thiyl radical (65) in the presence of O2. The cross-coupling of a thiyl radical (60), peroxide radical, and alkene (59) furnished the β-hydroperoxy sulfide (67), which were then cleaved to produce the target scaffold (61).
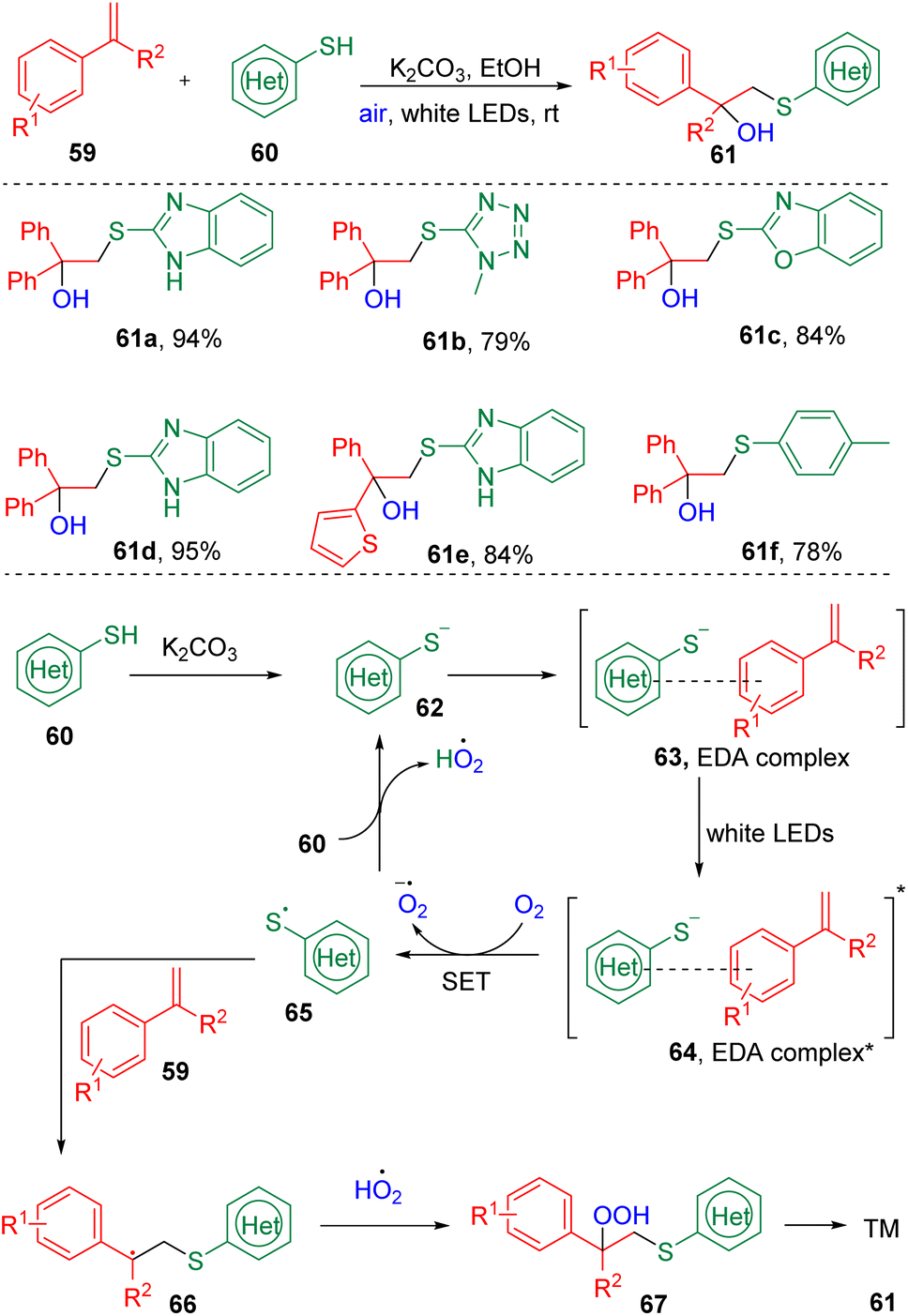 | ||
| Scheme 11 Light-induced EDA complex promoted hydroxysulfenylation. | ||
In 2021, Li and co-workers25 reported the visible-light-induced surfactant-promoted sulfonyl iodination of alkenes and alkynes in water (Scheme 12). Mono-substituted terminal alkenes and 1,2-dialkyl alkenes were proved suitable to provide anticipated products (69) in good to excellent yields with acetic acid as the additive. While, by the addition of sodium carbonate, α-alkylated styrenes were prone to deliver the eliminated alkenyl sulfones (ex. 69d). Furthermore, light-driven conditions were found also to be applicable to the iodosulfonylation of terminal and internal alkynes. Mechanistically, the key to this reaction was the aggregate formed by the CTAB (cetyltrimethylammonium bromide) surfactant, which acted as a reaction medium in water, stabilizing the transition state and reducing the activation energy of the reaction. While, the integration of iodide ion and sulfonyl chlorides performed as EDA complexes (70) through anion–π interactions, which triggered the visible-light-induced SET process to generate sulfonyl radicals (50) and an iodine radical, which eventually added to the carbon–carbon double/triple bonds.
 | ||
| Scheme 12 Light-induced surfactant-promoted iodosulfonylation. | ||
A photo-driven three-component reaction toward the synthesis of phenanthridine derived vinyl sulfones (Scheme 13) was showcased by Wang, Miao and co-workers.26 The authors proposed that the photochemical activation of the EDA complex (74), formed from 2-arylphenylisocyanide (72) and sulfinic acid (27), generated a sulfonyl radical (76), which then triggered the radical cascade process. Both E and Z target products were proved able to be selectively synthesized by controlling the irradiation conditions, in which 450 nm was found to preferably generate the E products, while 365 nm generated the Z olefins (major).
 | ||
| Scheme 13 Photo-driven selective synthesis of phenanthridine-derived vinyl sulfones. | ||
The group of Liao reported27 a photocatalysis-initiated strategy of fluorosulfonylation toward the construction of β-chloro alkenylsulfonyl fluorides (BCASF) (Scheme 14). The tremendous synthetic potential of the new class of sulfonyl fluorides (84) have been unveiled by numerous C–C and C–X bonds formation reactions, either via Suzuki and Sonogashira couplings, or direct nucleophilic substitutions with hetero-atom nucleophiles at the β-chloride position. Mechanistically, the authors proposed that a SET occurred between the excited photocatalyst and fluorosulfonyl chloride to generate an FSO2 radical (85), which then added to alkyne (83a), delivering the alkenyl radical 86a. The alkenyl radical abstracted a chloride from fluorosulfonyl chloride to form the target product (84a) and regenerated the FSO2 radical for the chain reaction.
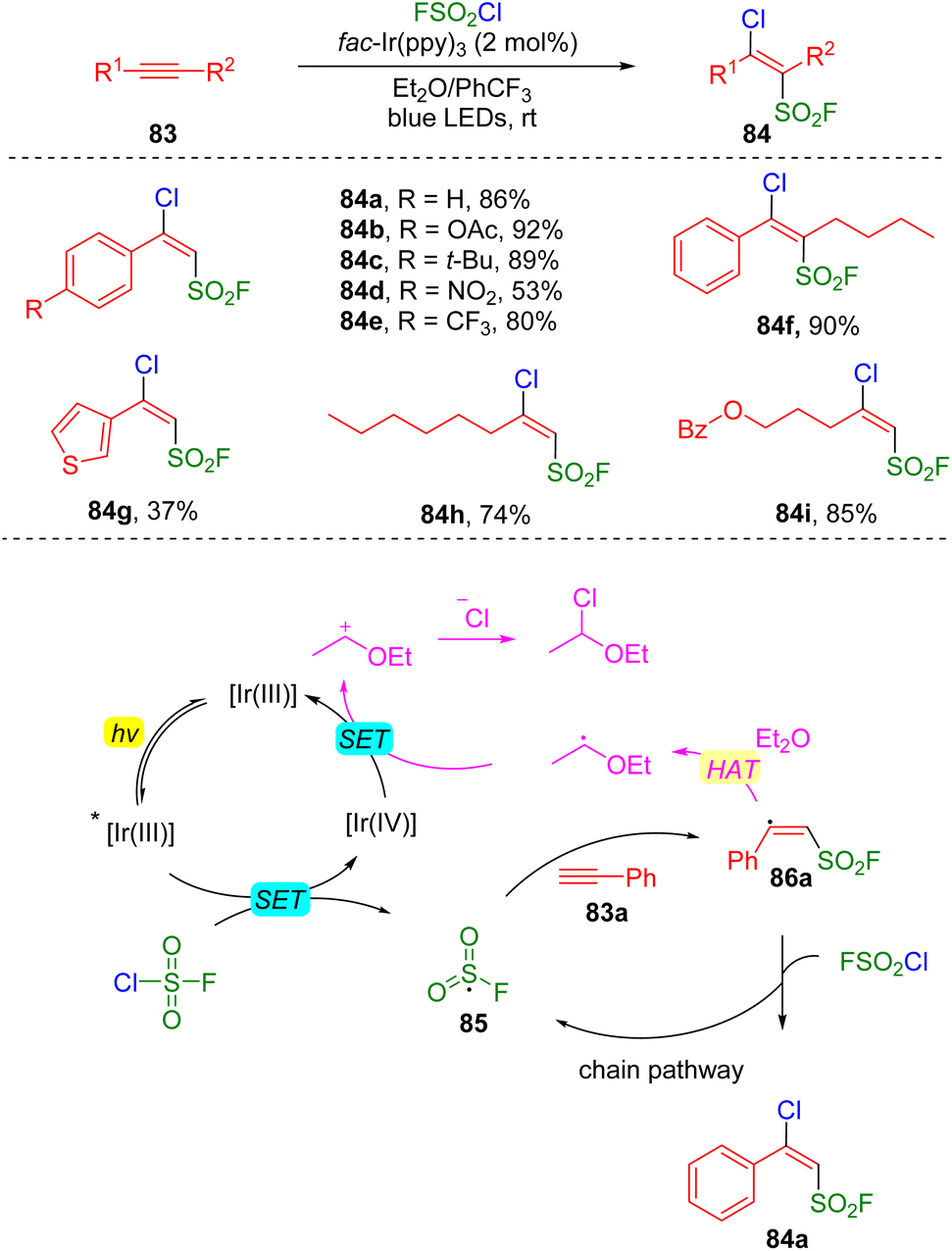 | ||
| Scheme 14 Chloro-fluorosulfonylation of alkynes. | ||
Very recently, Liao and Song further achieved28 the hydro-fluorosulfonylation of unactivated alkenes using oxygen-doped anthanthrene (ODA 89, E1/2(ODA*/ODA˙+) = −1.76 V vs. SCE) as the photocatalyst. The use of 1-fluorosulfonyl 2-aryl benzoimidazolium (FABI 88, E1/2 = −1.07 V vs. SCE) fulfilled the requirements of the acceptance of an electron from the exited photocatalyst and generation of an FSO2 radical (85) to trigger the addition to the C–C double bond (Scheme 15) and successfully avoided the chloride abstraction that occurred in their previous work, as discussed above. 1,4-Cyclohexadiene (94) was employed as the hydrogen donor via a hydrogen atom transfer process to deliver the cyclohexadiene radical (95), which then played underwent reductive quenching of the ODA˙+ species. The system was also proved to be applicable to the hydro-fluorosulfonylation of alkynes. A large scope of aliphatic sulfonyl fluorides that can be further applied to the late-stage modification and alkenylsulfonyl fluorides with high Z-selectivity were assembled under these conditions.
 | ||
| Scheme 15 Hydro-fluorosulfonylation of alkenes and alkynes. | ||
In 2019, by merging the photoredox and transition-metal catalysis, Rueping and co-workers29 achieved a three-component reaction of the difunctionalization of acetylenes (98) in the presence of aryl halides (97) and sodium sulfonates (31), leading to the facile synthesis of various trisubstituted alkenes (99) (Scheme 16). The stereoselectivities of this transformation (Z/E) were determined by the triplet state energy of the photocatalyst and were found to be switchable by varying the photocatalyst employed. An ET (triplet–triplet energy transfer) process was proposed to be responsible for the geometrical selectivities of the product. Employment of a photocatalyst with a triplet energy lower than the excited state of the product with anti geometry would furnish the anti product prominently. While, if the triplet energy of the photocatalyst was higher than that of both isomers, the syn product would be favored. A large scope of terminal aryl alkynes (98) were engaged in the difunctionalization. A few examples of alkenyl alkyne and internal alkynes were also proved to be applicable under these conditions. Detailed investigations revealed that the photocatalyzed reaction would be slightly hampered by the increased steric hindrance presented either in the aryl halide or in the acetylene partner. Stern–Volmer quenching experiments indicated that a sulfonyl radical intermediate was involved in the cross-coupling reaction, which was also supported by calculations based on density functional theory (DFT).
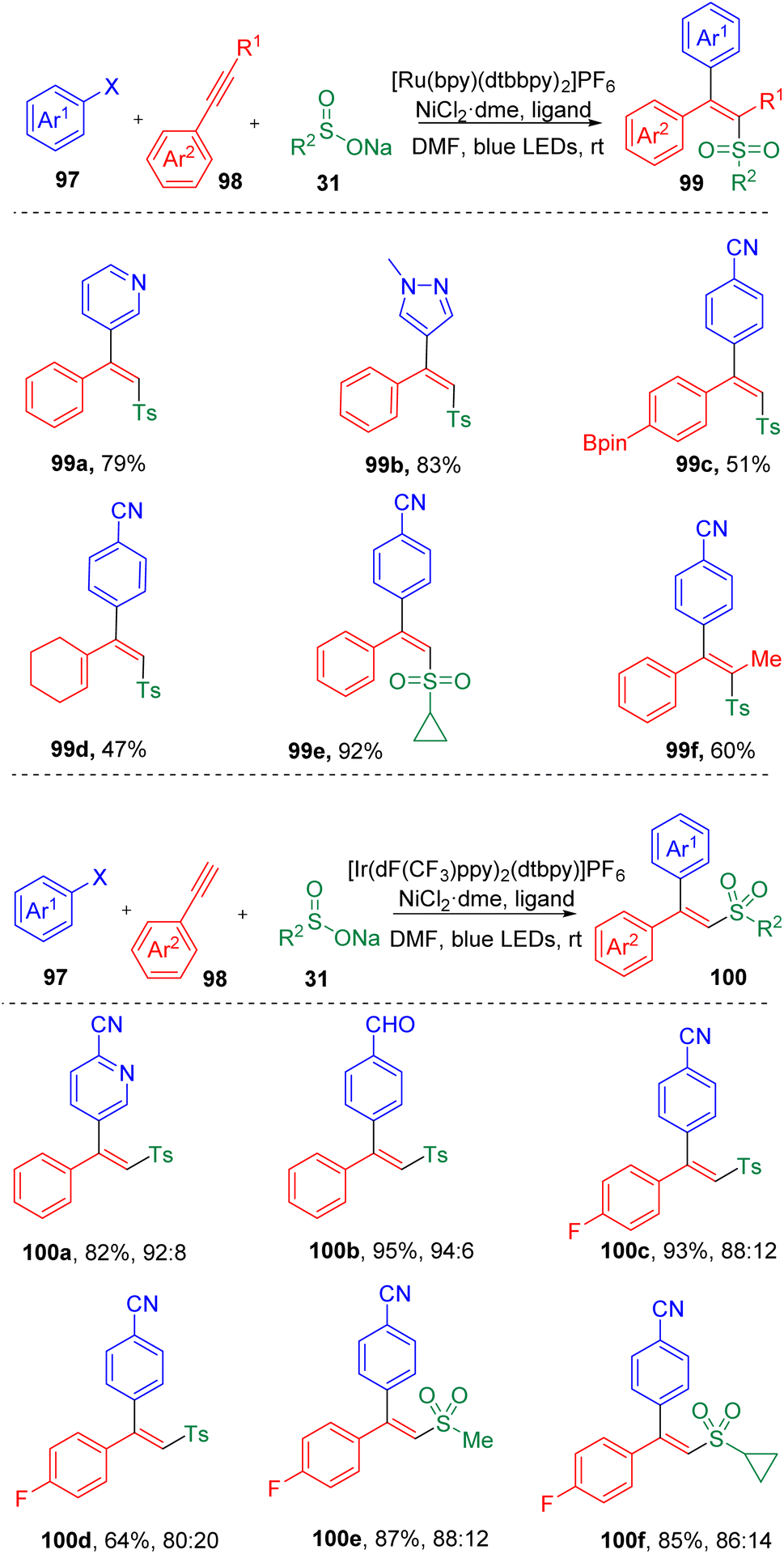 | ||
| Scheme 16 The photoredox/nickel dual-catalyzed sulfonylarylation of acetylene. | ||
2.3 Oxidative systems
The redox reaction is one of the mostly used reactions both in industry and in laboratory synthesis.30 The development of new methods through the use of green and inexpensive oxidants has positive and far-reaching implications in synthetic chemistry.Mal and co-workers31 systematically investigated the interesting aerial dioxygen activation and selective thiol–ene click (TEC) reactions with styrenes (Scheme 17). The authors found that carrying out the reactions neat led to the isolation of anti-Markovnikov products, while exclusively Markovnikov selectivity in the TEC reaction was observed using AgOTf as the additive. To rationalize the regioselectivity, the authors proposed that the anti-Markovnikov selectivity could possibly be attributed to the S–H⋯π (C–C double bond) and π–π (106) interactions between the arenes of the styrene (101) and thiophenol (102). While, in the presence of the thiophilic promoter AgOTf, the S–H⋯π interaction was inhibited and replaced by the styrene-Ag+ cation–π interaction (107), and thus proceeded in the manner of a normal Markovnikov click reaction. The authors also demonstrated that the synthesis of β-hydroxysulfides (104) via aerial dioxygen activation could be accomplished. Control experiments suggested the formation of the relatively stable benzyl radical (110), which was generated by the addition of a thiyl radical (109) to the carbon–carbon double bond. The formation of the thiyl radical (109) was proposed to proceed through SET initiated by tBuOLi or the oxidation of thiophenol by dioxygen.
 | ||
| Scheme 17 Selective thiol–ene click (TEC) reactions. | ||
Lei and colleagues32 disclosed an interesting radical redox reaction (Scheme 18). The process was proved to be switchable in synthesizing sulfoxides (114) or sulfides (115). The key reactive intermediate was proposed to be the β-hydroperoxy sulfide (119), which was formed via the addition of a thiyl radical (116) to the C–C double bond and oxygen oxidation (118) successively. The active β-hydroperoxy sulfide (119) underwent internal oxygen transfer to deliver the β-hydroxysulfoxide product (114). While, in DMSO medium, the presence of a triphenylphosphine additive furnished the β-hydroxysulfide (115) via intermolecular oxygen transfer.
 | ||
| Scheme 18 Controllable synthesis of β-hydroxy sulfoxides and sulfides. | ||
In 2017, Han's group33 designed a new copper-mediated aerobic oxidation strategy (Scheme 19) for the synthesis of thio-substituted lactones (121) via the difunctionalization of α-substituted styrenes (120). Both five- and six-membered lactones were accessible. 1,1-Dialkyl alkenes failed to undergo this transformation. Thiophenols with electron-donating substituents were found to have an enhanced reactivity in the reaction. Regarding the mechanistic proposal, the reaction was initiated by the formation of a CuOO˙ species from the oxidation of a Cu catalyst with O2. Sequential radical addition then occurred to produce the intermediate hydroxysulfide carboxylic acid (124). The authors claimed that the in situ generated sulfonic acid by-product from the oxidation of thiophenol promoted lactone formation.
 | ||
| Scheme 19 Copper-mediated sulfenyl lactonization. | ||
In 2018, the group of Zhu34 reported the radical-type cyanotrifluoromethylthiolation of unactivated alkynes through a well-designed strategy of internal nitrile migration (Scheme 20). The reaction was initiated by the addition of CF3S˙, generated by the combination of AgSCF3 and K2S2O8, to γ-cyanohydroxy alkynes (125). The formed highly active vinyl radical (127 and 127′) then induced the intramolecular migration of nitrile to deliver the energetically lower ketone products (126 and 126′). According to the observed results, the route (a) addition and the five-membered cyclic transition state for the migration was preferred rather than pathway (b), which produces the vinyl product with reversed regioselectivity.
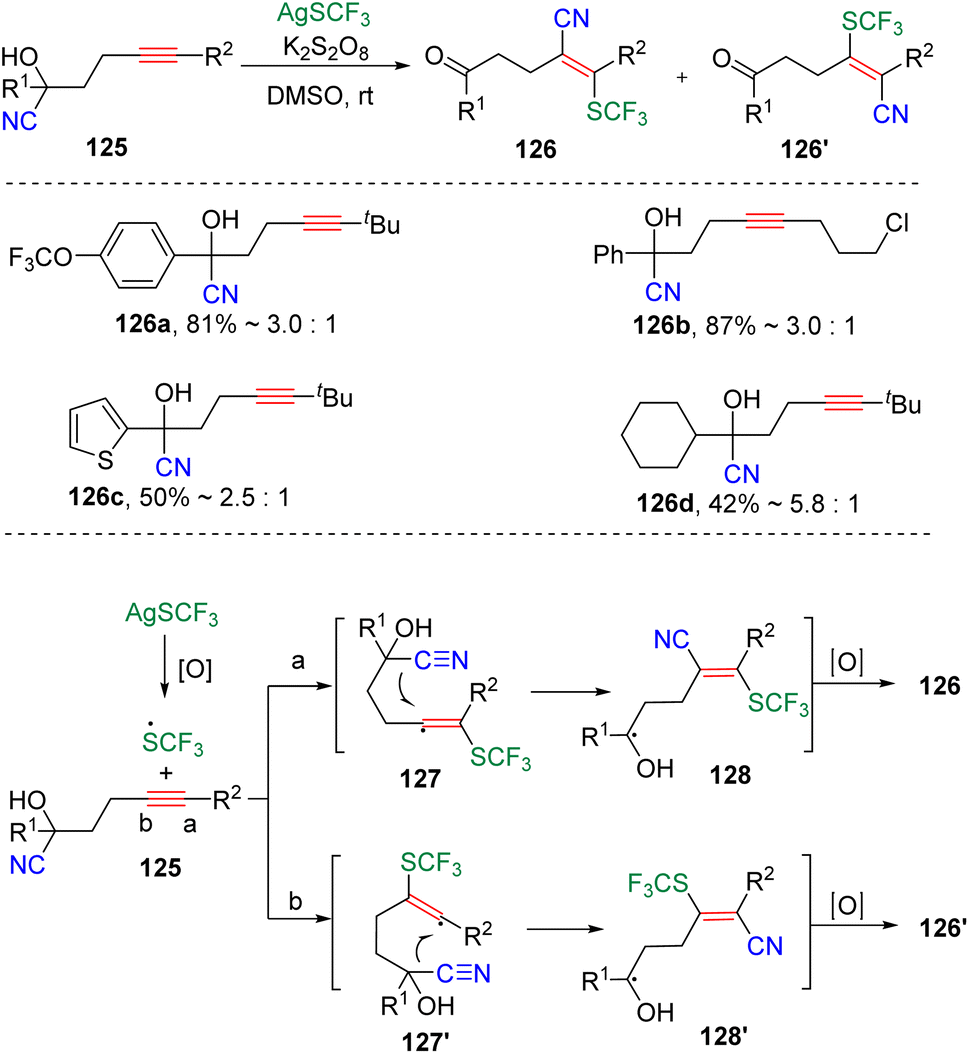 | ||
| Scheme 20 Cyanotrifluoromethylthiolation of alkynes. | ||
2.4 Transition-metal catalyzed system
In 2021, Lian, Zhang and co-authors35 investigated the copper-catalyzed chlorosulfonylation of styrenes (129) using exogenously generated sulfur dioxide (from SOgen), aryldiazonium tetrafluoroborates (130) and lithium chloride (Scheme 21). The versatility of this protocol was demonstrated by the tolerance of the functional groups in styrene derivatives and aryldiazonium tetrafluoroborates (130). Mechanistic studies revealed that the aryl radical (133) generated from the aryldiazonium tetrafluoroborate (130) was captured by sulfur dioxide to deliver the arylsulfonyl radical (134), which then added to the alkene to form the benzyl radical (137). The authors then proposed two plausible pathways for the formation of the target β-chloro alkylsulfones (132), (a) the benzyl radical (137) directly captures chloride from the Cu(II) specie (136), (b) the Cu(II) species (136) combine with the C-centered radical (137) and generate a Cu(III) species (138), which eventually provides the β-chlorosulfone products through reductive elimination.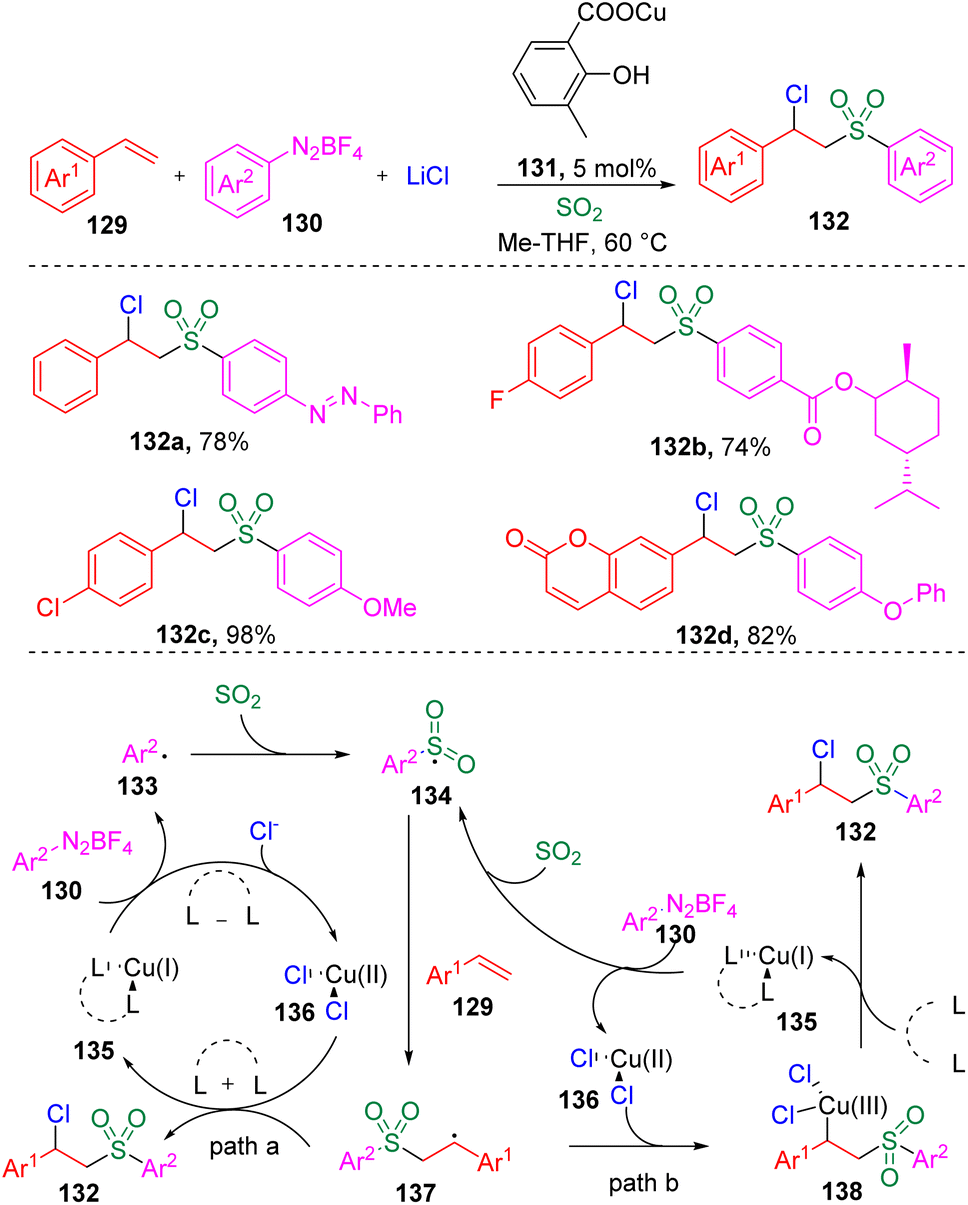 | ||
| Scheme 21 The chlorosulfonylation of styrenes via copper-catalyzed atom transfer radical addition. | ||
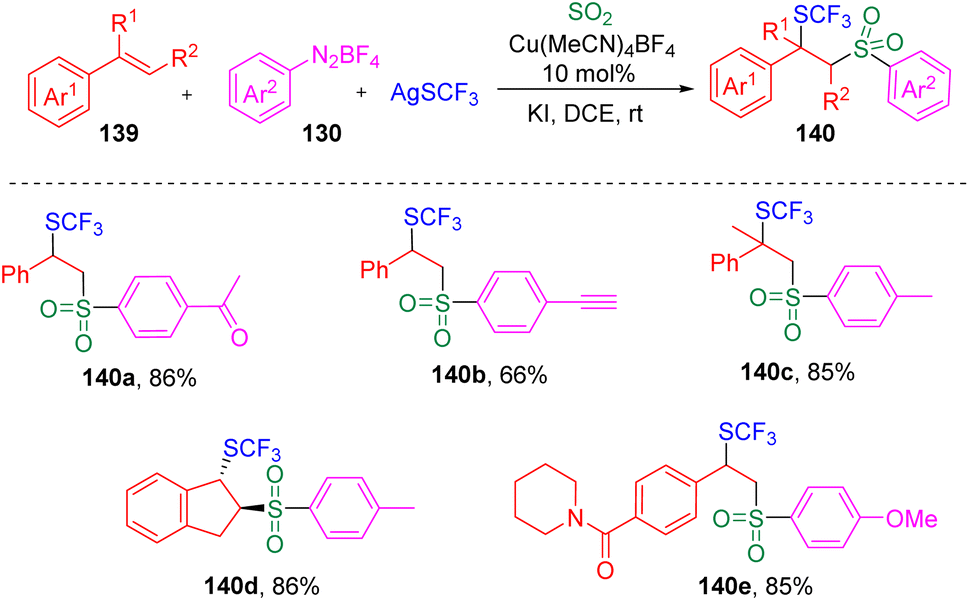
Recently, the same group36 further developed the copper-catalyzed multicomponent reaction to trifluoromethylthio-sulfonylation using AgSCF3 as a reaction partner (Scheme 22). The potential utility of this reaction in medicinal chemistry was demonstrated by the successful late-stage modification and further transformation of drug-based molecules.
 | ||
| Scheme 22 Copper-catalyzed trifluoromethylthio-sulfonylation. | ||
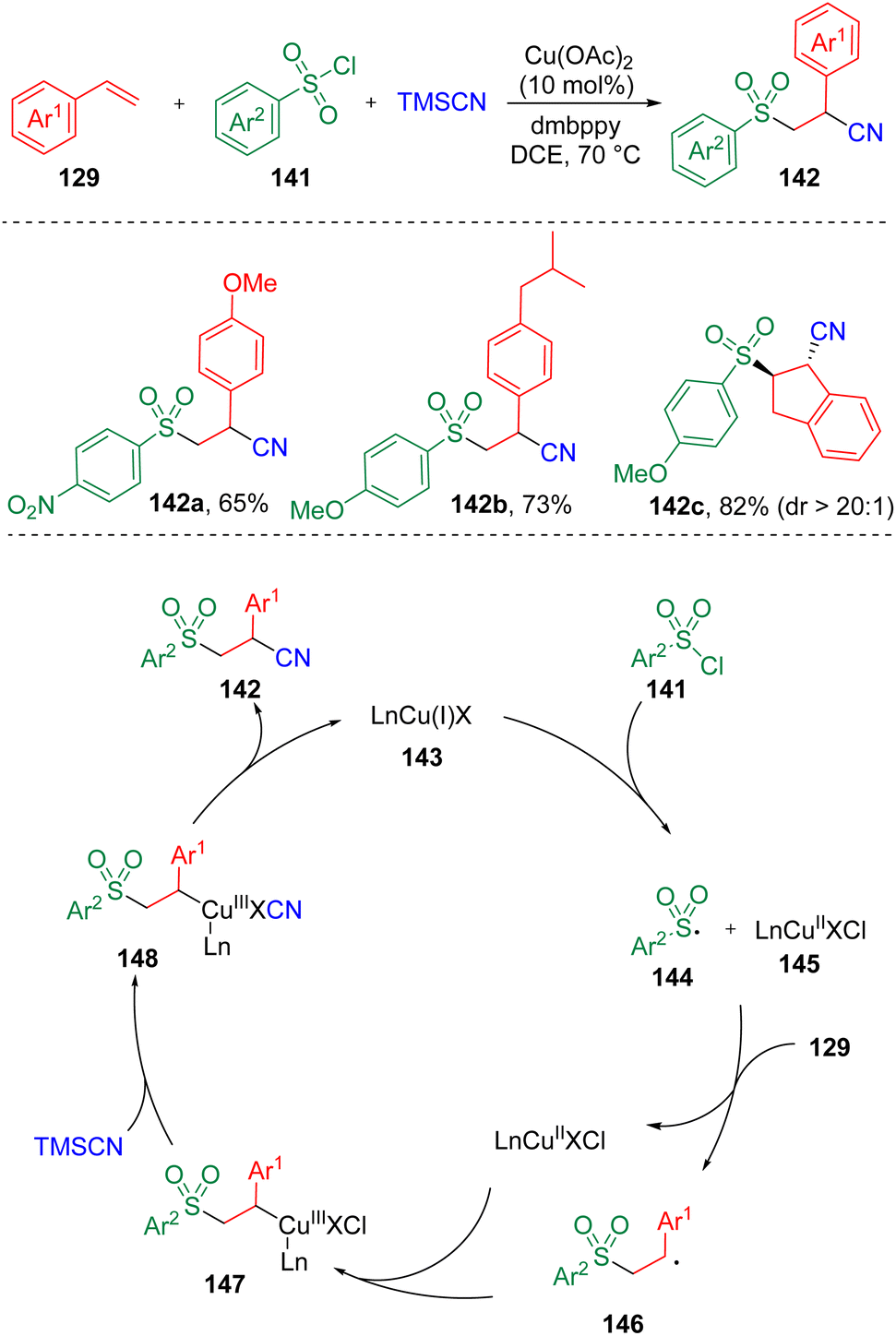
In 2020, Li and co-workers37 reported the synthesis of β-sulfonyl nitriles (142) via a copper-catalyzed regioselective sulfonylcyanation using TMSCN, sulfonyl chlorides (141), and styrenes (129) as substrates (Scheme 23). It was found that both electron-withdrawing and electron-donating substitutes on either aryl sulfonyl chlorides or styrenes could be well tolerated. Based on experimental observations in the presence of radical scavengers and the radical-clock experiment, a mechanistic process was proposed. The sulfonyl radical (144) generated by the SET process between sulfonyl chloride (141) and LnCu(I)X (143) underwent addition to styrene (129) and delivered a benzylic radical intermediate (146), which then reacted with LnCu(II)X (145). A Cl/CN exchange subsequently occurred in the presence of TMSCN to produce an organocopper(III) cyanide species (148). Reductive elimination then occurred to furnish the β-sulfonyl nitrile product (142).
 | ||
| Scheme 23 Copper-catalyzed sulfonylcyanation. | ||
Jia and collaborators38 described a copper-catalyzed three-component coupling reaction of styrenes (149), thiosulfonates (150) and arylboronic acids (151) (Scheme 24). Heterocyclic styrene derivatives, heterocyclic boronic acids and heteroaryl thiosulfonates exhibited impressive reactivities, delivering β-aryl sulfone products smoothly. The reaction system required N,N-dimethylformamide (DMF) as a co-solvent, which was proposed to be involved in the catalytic cycle. The key to the success of this copper-catalyzed intermolecular difunctionalization of styrene is the use of methylthiosulfonate (150) as the source of sulfonyl radical (50), which effectively inhibited the direct arylation of the sulfonyl radical by arylboronic acid.
 | ||
| Scheme 24 Copper-catalyzed three-component coupling reaction. | ||
3 Electrophilic sulfenofunctionalization of alkenes
The electrophilic sulfenofunctionalization of alkenes has been established as the classical method for the construction of carbon–sulfur bonds, which can be traced back to the report of sulfenochlorination by Lecher and Stöcklin in 1925.39 Mechanistic proposals and the following studies led to the finding of the three-membered cyclic sulfonium (thiiranium or episulfonium) ion as the key intermediate40 in the process of the electrophilic difunctionalization of alkene, therefore serving as a milestone in the understanding of the fundamental chemistry of the electrophilic activation of alkene.41 A significant difference of this chemistry with the previously discussed conceptions in this collection is the fixed stereoselectivity (formal trans difunctionalization) determined by the episulfonium intermediate, as the following nucleophilic attack generally occurs from the anti-side of the sulfur atom. Though complex interconversion could possibly happen in the reaction system, this process allows the prosperity of well-designed strategies toward the selective synthesis of various functionalized sulfur-containing compounds.3.1 Direct sulfonium transfer-promoted difunctionalization
The traditional procedure to obtain the episulfonium ion as the intermediate requires the use of sulfenyl chloride, and the successive addition of a Lewis acid in the following dechlorination stage.42 The instability and handling difficulty of the reagents largely limit the application of this conception in synthesis.Dimethyl(methylthio)sulfonium tetrafluoroborate (DMTSF) and dimethyl(methylthio)sulfonium trifluoromethanesulfonate (DMTSM) are commercially available and crystallizable electrophilic sulfenylating reagents.43 Their significant advantage over other easy-handling sulfenylating agents is the capability of the direct activation of alkenes without the assistance of acids or metal catalysts.44 Due to this, one can obtain the thiiranium ion intermediate in a much simple reaction system. In the 1980s, the groups of Trost and others45 systematically studied the application of DMTSF and DMTSM in the sulfenylation of alkenes and subsequent nucleophilic ring-opening additions. Therefore, various hetero-nucleophiles, such as amines, azide, acetate, phosphines, fluoride, have been employed in the sulfenofunctionalization of alkenes.46
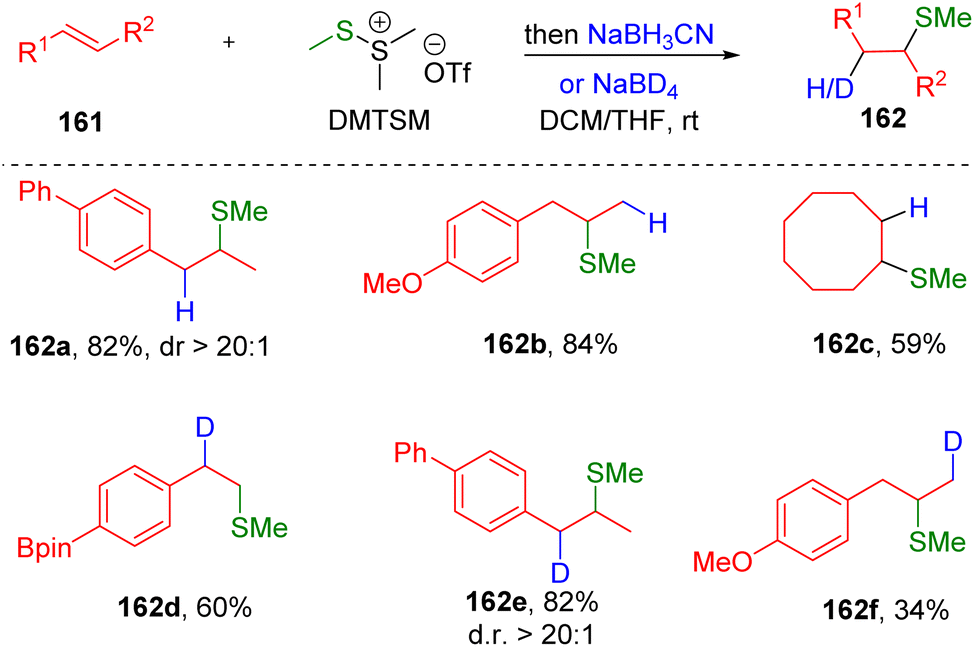
Encouraged by early studies, the group of Xie47 recently developed the hydromethylthiolation of alkenes with DMTSM as the electrophilic stimulator and sodium cyanoborohydride as the hydrogen source (Scheme 25). The transformation represents a transition metal-free procedure by which to synthesize hydromethylthioethers (162) from unactivated alkenes (161), successfully avoiding the use of volatile, corrosive, and flammable methanethiol. In addition, by employing the readily available deuteride source NaBD4 as a reductive agent, various β-deuterated methylthioethers (162) also proved to be accessible using this procedure. Scope screening indicated that the regioselectivities were different between asymmetrical styrenes (162a, 162d and 162e) and alkyl olefins (162b and 162f). The authors noted that the episulfonium ion intermediate formed by the reaction of DMTSM with a terminal styrene substrate might be able to convert to the corresponding carbocation, because of the effect of conjugation from the phenyl ring. Additionally, the benzyl carbocation is in fact the species that undergoes hydrogenation, while the nucleophilic ring-opening addition of hydride to the episulfonium ion occurs in the case of alkyl alkenes.
 | ||
| Scheme 25 Hydro/deuteromethylthiolation of unactivated alkenes. | ||
Very recently, direct conjugate addition of DMTSM (MeS- and TfO-) to terminal alkyl alkynes has been disclosed by Xie, Huang and co-authors,48 providing simple protocol regio- and stereo-selectively access to β-methylthio vinyl triflates (165) under neutral conditions (Scheme 26). The thiirenium ion (164) formed by the mixing of alkyne (163) and DMTSM was proposed as the key intermediate, which undergoes ring-opening under the nucleophilic attack of triflate. The reactivity of the thiirenium ion (164) is defined by the trans addition process in the vinyl triflate (165) synthesis, while an electronically biased site-selectivity was used to rationalize the addition of the triflate to the more steric carbon atom. Functionalities such as nitriles, esters, protected alcohols, chloride and OTs (p-toluenesulfonate) were tolerated under the conditions. Vinyl triflation of bioactive molecules derived alkynes were realized, such as febuxostat, α-D-glucofuranose, ciprofibrate and fenofibric acid. The authors also presented the reactivity of haloalkynes, including aryl/alkyl bromoalkynes and chloroalkynes (165d–165f), in this transformation. However, other internal alkynes, such as 4-octyne and diphenylacetylene, did not deliver the corresponding vinyl triflate target products.
 | ||
| Scheme 26 Conjugate addition of DMTSM to alkynes. | ||
Mechanistic investigations (Scheme 27) suggested that by the transfer of sulfonium from DMTSM/DMTSF to alkenes (161), an equilibrium of two intermediates, the active episulfonium ion 166 and the sluggish thiosulfonium ion 167, would be delivered. The conversion of thiosulfonium ion 167 to its episulfonium analog 166 to undergo nucleophilic attack ensured the overall efficiency of the transformation in the presence of heteronucleophiles and cyanide. However, the use of a typical carbon nucleophile (generally carbon metal reagents), which are more alkaline, led to the deprotonation and decomposition of intermediate 167. Consequently, the intermolecular electrophilic carbosulfenylation of alkenes via episulfonium intermediates proved to be problematic. The Trost group49 published the leading example using the low basic alkynylalaneate (172) as a suitable sp carbon nucleophile.
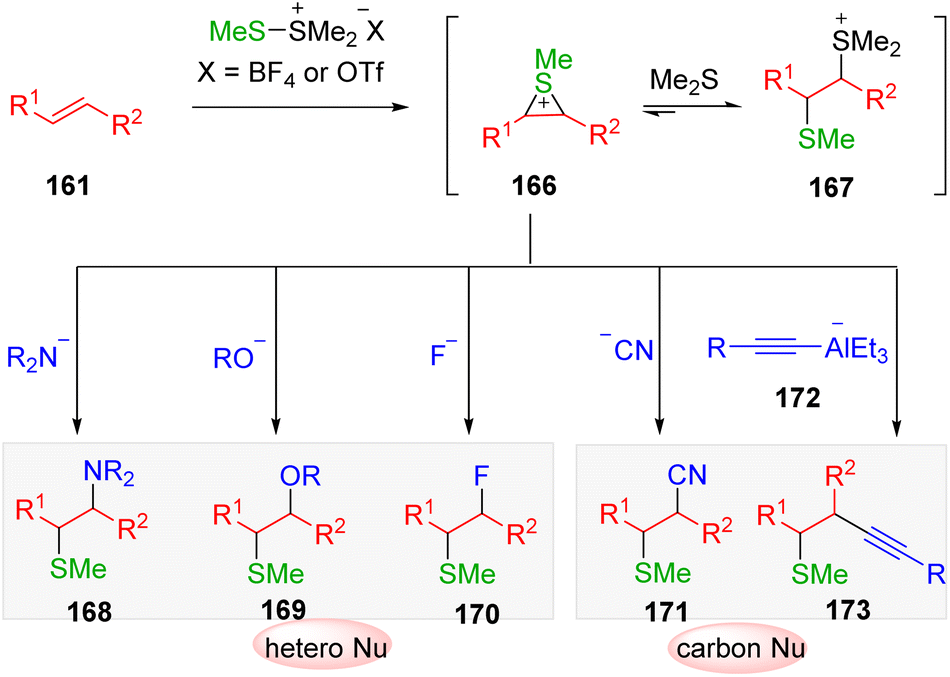 | ||
| Scheme 27 Intermediates and reactions of sulfonium transfer from DMTSM or DMTSF. | ||
Inspired by the work on alkynyl sulfenylation, Xie and colleagues50 have succeeded in the intermolecular arylsulfenylation of alkenes (161) via an episulfonium ion (176) in the presence of DMTSM employing organozinc reagents (174) as the carbon nucleophiles (Scheme 28). The key to this transformation was attributed to the relatively covalent C–Zn bond, which determined the low basicity of these nucleophilic reaction partners. The authors also found that the magnesium salts generated from the preparation of the zinc reagents were crucial to the carbon–carbon bond formation. It was proposed that magnesium salts could perform as Lewis acids to coordinate with dimethylthioether via the formation of the complex 178, driving the conversion of thiosulfonium ions (177) to episulfonium ions (176) and thereby facilitating carbon–carbon bond formation upon the addition of organozinc reagents.
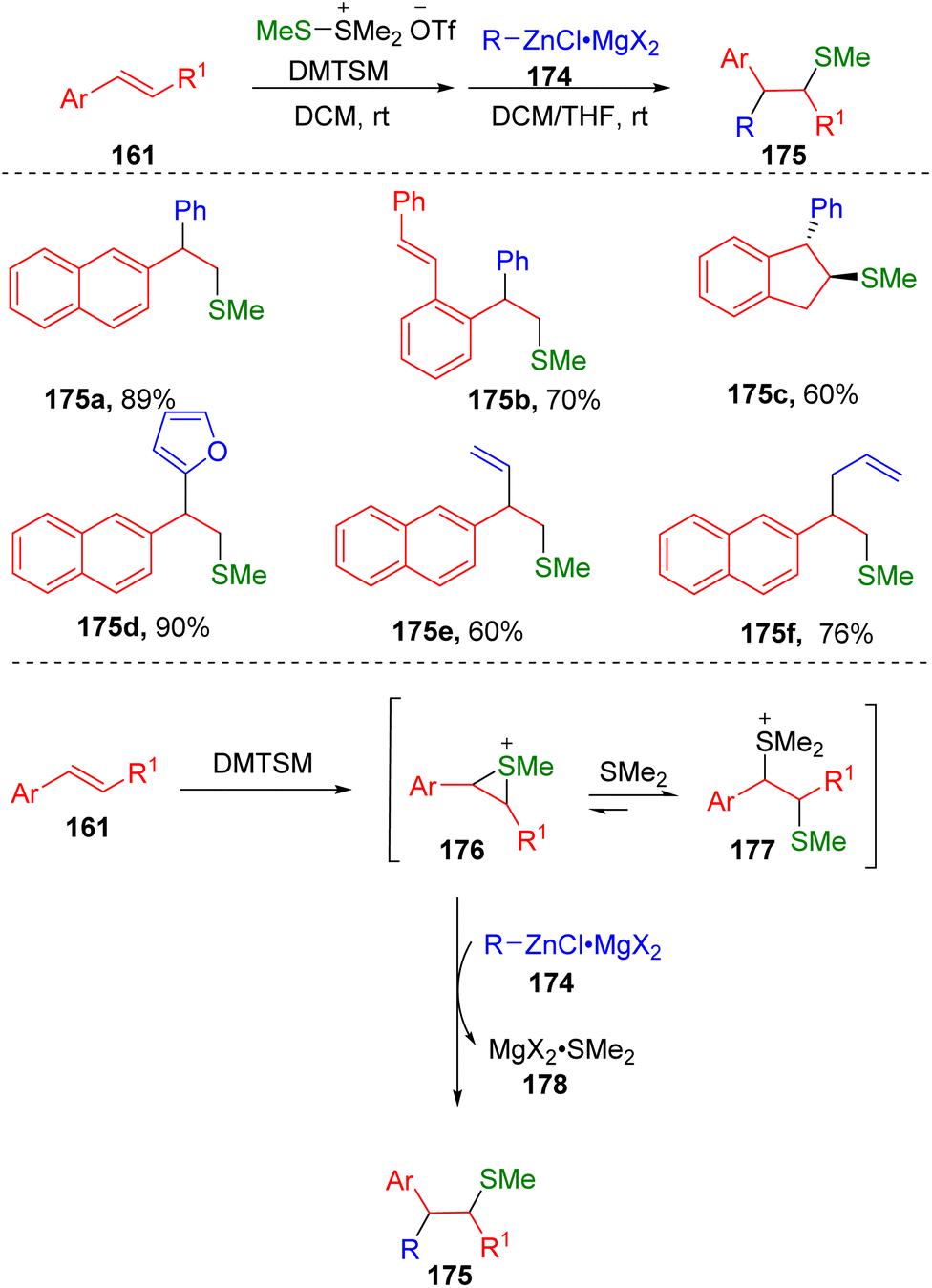 | ||
| Scheme 28 Intermolecular carbosulfenylation of alkenes with organozincs. | ||
In 2017, Snyder and co-workers51 synthesized isolable alkyldisulfanium salts (180) via the reaction of dithioethers with chlorine and SbCl5 (Scheme 29). The following tests showed that these new types of electrophilic thiolating agents are capable of initiating polyene cyclization with the installation of MeS-, EtS- or CF3CH2CH2S-functionalities. The group52 also designed and synthesized chiral sulfanium salts (183) to promote cyclization, which showed low enantioselectivities.
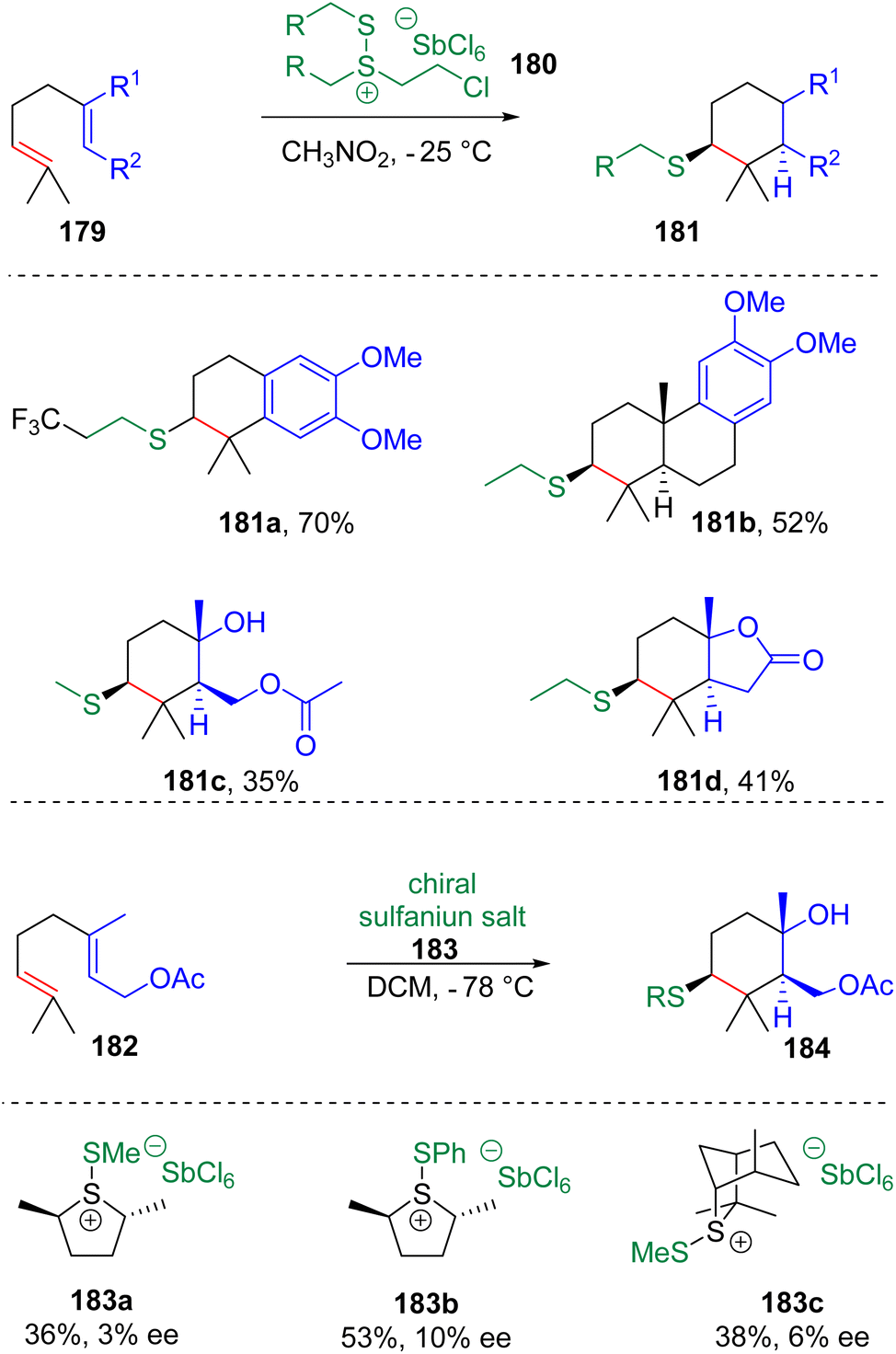 | ||
| Scheme 29 Alkyldisulfanium salts promoted cyclization. | ||
3.2 Lewis base catalyzed sulfenylfunctionalization
In 2009, the group of Denmark53a initially observed the transfer of sulfeniums between thiiranium ions and alkenes, and explored the synthesis and reactivity of the enantiomerically enriched thiiranium ion in the presence of various nucleophiles.53b On the basis of this work, the group applied the concept of a Lewis base activated Lewis acid to the enantioselective sulfenyl functionalization of alkenes in the presence of a BINAM-based phosphoramide catalyst (186 in Scheme 30). Thus, highly impressive intramolecular cyclizations with oxygen54a–d and nitrogen nucleophiles54e–g, as well as electron-rich arenes,54h–j were achieved. Systematic mechanistic studies then provided in-depth understanding to the factors that promoted the stereo-control of the reactions.55 | ||
| Scheme 30 Lewis base catalyzed enantioselective sulfenocyclization. | ||
In 2018, Denmark and colleagues56a using homogeranylarenes and ortho-geranylphenols as substrates, elaborated on the catalytic intramolecular sulfenocyclization of polyenes (185) in a diastereoselective and enantioselective manner (Scheme 30). Studies demonstrated that the use of hexafluoroisopropyl alcohol (HFIP) as a solvent in the reaction was crucial. It was proposed that the solvophobic interaction in the highly polar reaction medium forced the polyene substrates to endure conformation with a minimized surface area, therefore making the terminal C–C double bond more reactive toward the sulfenylating agent, and determining the selectivity of the cascade transformation.56b The authors managed to apply this sulfenyl polyene cyclization in the enantioselective synthesis of (+)-ferruginol and (+)-hinokiol.
A well-designed enantioselective carbosulfenylation was also presented by Denmark and co-workers (Scheme 31).57 On the basis of the positive effect of HFIP on Lewis base-catalyzed sulfenocyclization, an alkenylboronate was first reacted with phenyllithium to form a boronate complex (198), and then subjected to chiral selenophosphoramide (186) catalyzed sulfenylating conditions. Thiiranium ion formation and the following 1,2-migration of the zwitterionic thiiranium–boronate complex (199) eventually delivered carbosulfenylation products (197) with exclusive stereoselectivity and excellent enantioselectivity.
 | ||
| Scheme 31 Enantioselective carbosulfenylation of alkenylboronates. | ||
On the basis of the Lewis base activated Lewis acid concept, the group of Zhao58 initially designed bifunctional catalysts by combining a chalcogenide as the activator of sulfenylating agents and an NH group as the hydrogen-bond donor to bind with nucleophiles and offer assistance in regio- and stereo-selectivities in the ring-opening process of sulfonium intermediates. Therefore, Zhao and co-authors synthesized a series of indene-based sulfide/selenide chiral catalysts (ex. 202), and had great success in applying this concept to enantioselective trifluoromethylsulfenylating intramolecular cyclization reactions, including lactonization,59a lactamization59b and oxazoline formation,59c as well as the desymmetrization process to access various trifluoromethylsulfenyl tetrahydronaphthalenes.59d Additionally, more importantly, the group further established the intermolecular version of the enatioselective trifluoromethylsulfenylating functionalization of alkenes with the catalytic system they developed.59e,f
In 2020, Zhao and co-authors60 published an enantioselective, intramolecular carbosulfenylation of alkynes 200 in the presence of a chiral bifunctional sulfide based catalyst (202) (Scheme 32). Scope screening showed both arylsulfenylating and trifluoromethylsulfenylating agents (201) were applicable. The reaction provided a reliable strategy to construct axially chiral vinyl arenes (203) bearing amine and sulfide functionalities. The products were modifiable to be converted to various axially chiral biaryl amino sulfides, which could possibly serve as precursors of potential (S, N)-based ligands and catalysts.
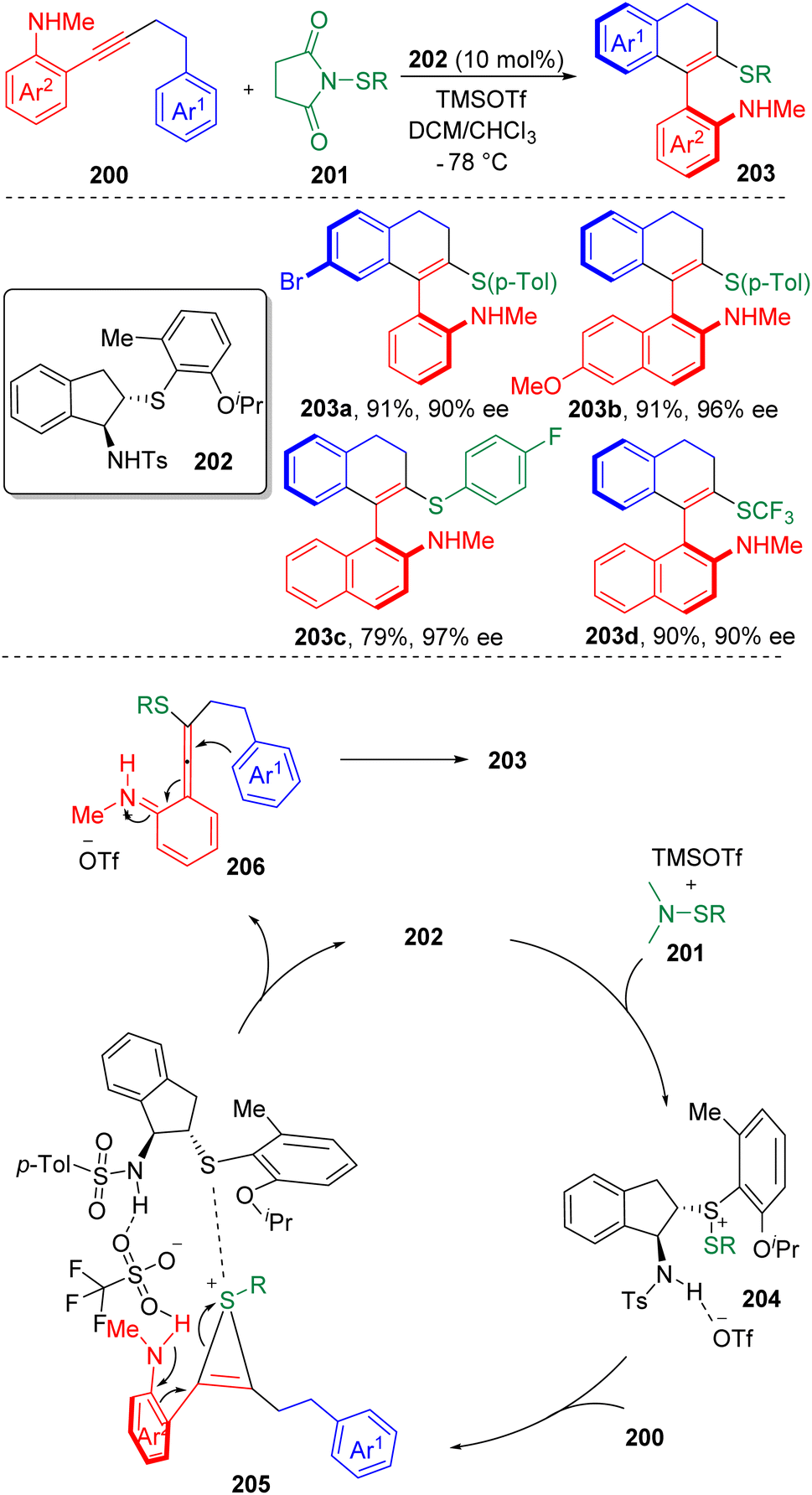 | ||
| Scheme 32 Enantioselective cyclization of alkynes. | ||
The Denmark group61 successfully transplanted their HFIP assisted Lewis base catalysis into the intermolecular, enantioselective sulfenoamination of olefins (Scheme 33). The authors proposed that the protonation of the sulfenylating agent (195) with HFIP promoted the transfer of sulfur to the selenophosphoramide catalyst (186) to form cationic species (191). Enantioenriched and configurationally stable thiiranium ions (209) were formed by the transfer of sulfenyl groups from the cationic species (191) to alkenes (161), and were then captured by intermolecular nucleophiles to provide products. Benzylamines, anilines and alkenes with various functional groups were evaluated, delivering 1,2-sulfenoaminated products (208) in good to excellent yields.
 | ||
| Scheme 33 Enantioselective sulfenoamination of alkenes. | ||
In 2019, Zhao and co-workers62 achieved the intermolecular azidothiolation and oxythiolation of alkenes in the presence of an indene-based chiral selenide catalyst (212) and Tf2NH additive (Scheme 34). The efficiency of azidothiolation was illustrated by the high yields and high enantio-/diastereo-selectivities. Both alkyl and aryl thiolating agents (201) bearing halogens, double bonds and nitrile substitutes were suitable to initiate the azidothiolation. Different oxygen nucleophiles, including water, proved to be applicable to the oxythiolation reaction. The key to the control of selectivity was attributed to the proton on the nitrogen neighbouring the α carbon of the carbon–carbon double bond in 210. According to the principles of the Lewis base activation of Lewis acid, in the presence of Tf2NH, the selenide atom of the chiral catalyst (212) first coordinated with the electrophilic thiolating reagent (201), which then transferred a sulfonium to the olefin (210). The authors claimed that in the thiiranium ion intermediate (214), a hydrogen bonding interaction existed between the proton on the sulfonamide and the Tf2NH, which assisted the determination of the diastereo- and enantioselectivities.
 | ||
| Scheme 34 Enantioselective azidosulfenylation and oxysulfenylation of alkenes. | ||
Recently, the Zhao group63 further developed the concept, and achieved the first enantioselective, intermolecular carbosulfenolation of N-allyl sulfonamides (210) using electron-rich arenes (215) as the carbon nucleophiles (Scheme 35). Various β-sulfide alkyl phenols (216) with alkyl or halide functionalities were synthesized with high regio-, enantio-, and diastereoselectivities. Electrophilic arylthiolating agents (201) proved to be variable, with the toleration of alkyl (Me- and Et-) and halide groups (Br- and Cl-). Control experiments ruled out the process of the O-alkylation of phenol and the following rearrangement, and indicated the possibility of direct C-type attack by a phenol to the sulfonium intermediate.
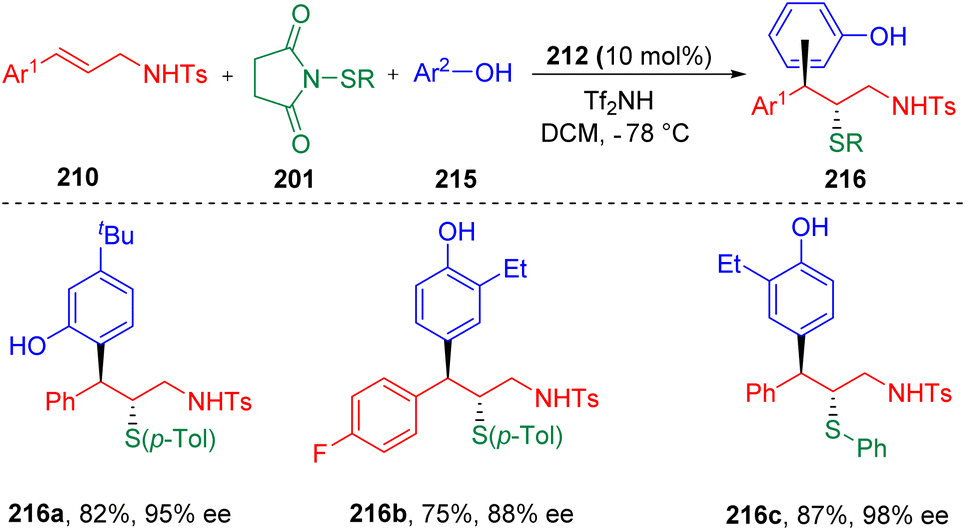 | ||
| Scheme 35 Enantioselective intermolecular thioarylation of N-allyl sulfonamides. | ||
The Chen group applied64 this concept to the enatioselective sulfenylation and cyclization of unsaturated carboxylic acids (217), providing an efficient method to access chiral sulfenylated lactones (219, Scheme 36). By comparing the experimental results, the authors found that electron-withdrawing groups reduced the yields of the reactions, while large steric hindrance led to lower ee values. The biologically active natural products (−)-nicotlactone B and (−)-galbacin were formally synthesized using this procedure to reveal the utility of this protocol.
 | ||
| Scheme 36 Enantioselective sulfenyl lactonization of unsaturated carboxylic acids. | ||
Shortly after this work, the group reported65 the divergent 6-endo and 5-exo lactonization of homoallylic acids, on the basis of Lewis base/Brønsted acid co-catalyzed thiolation. The authors found that using catalytic amount of acid (10 mol% EtSO3H), the kinetically favored 6-endo product was mainly formed, while the thermodynamically more stable 5-exo product was predominately generated under stoichiometric acid conditions.
In 2019, Chen and co-authors synthesized66N-thiocyano dibenzenesulfonimide (222) and demonstrated its capability as a new electrophilic thiocyanation reagent to achieve various thiocyano oxyfunctionalizations of alkenes (Scheme 37). The transformation was not limited to intramolecular cyclizations, but also amenable to the intermolecular oxyfunctionalization of alkenes when alcohol was used as a solvent.
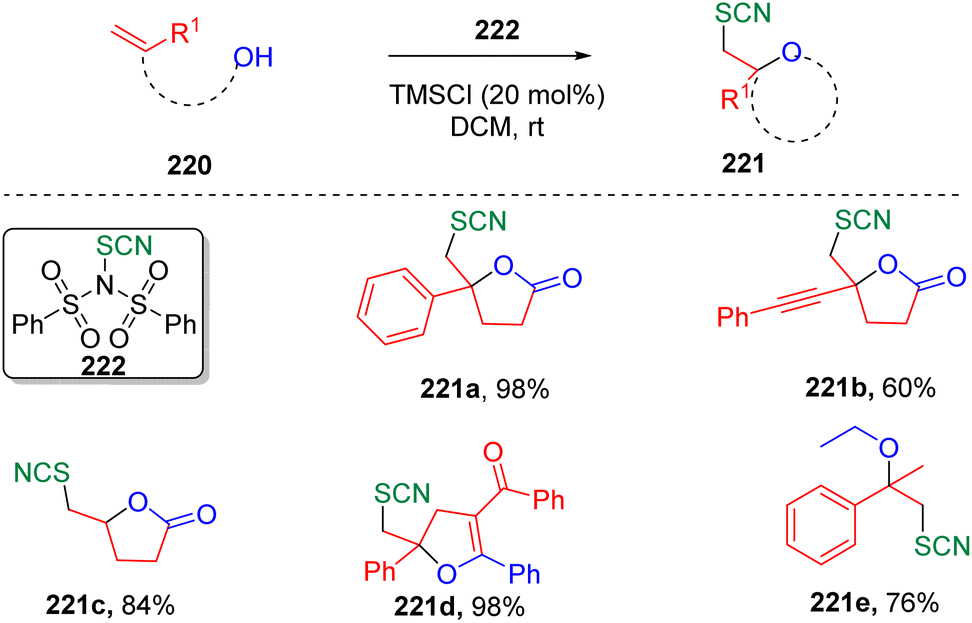 | ||
| Scheme 37 TMSCl-catalyzed electrophilic thiocyano oxyfunctionalization. | ||
On the basis of their previous findings,67 Chen, Tu, Bao and co-authors68 further established the enantioselective sulfenylation/semipinacol rearrangement of di- or tri-substituted allylic alcohols (Scheme 38), in the presence of a BINAM-based phosphoramide catalyst (186 in Scheme 30), in the aim of producing chiral all-carbon quaternary centers. In the reaction discovery, the authors found that using methanesulfonic acid and ethanesulfonic acid as the cocatalyst only afforded the product in moderate yields and moderate enantioselectivities. Improved results were obtained using 10 mol% chiral BINOL-derived phosphoric acid (CPA, 224) to combine with the chiral Lewis base catalyst (186). The active species was determined by 31P NMR spectroscopy. Based on computational studies (DFT calculations), the sulfenyl transfer was proposed to occur via SN2 electrophilic attack of the active species (226) at the terminal alkenyl carbon of 223. Steric hindrance and relative bond lengths of Se⋯S and C⋯S bonds determined the optimal transition state and enantioselectivity of the transformation after the subsequent ring opening of the episulfonium ion (227) and 1,2-carbon migration.
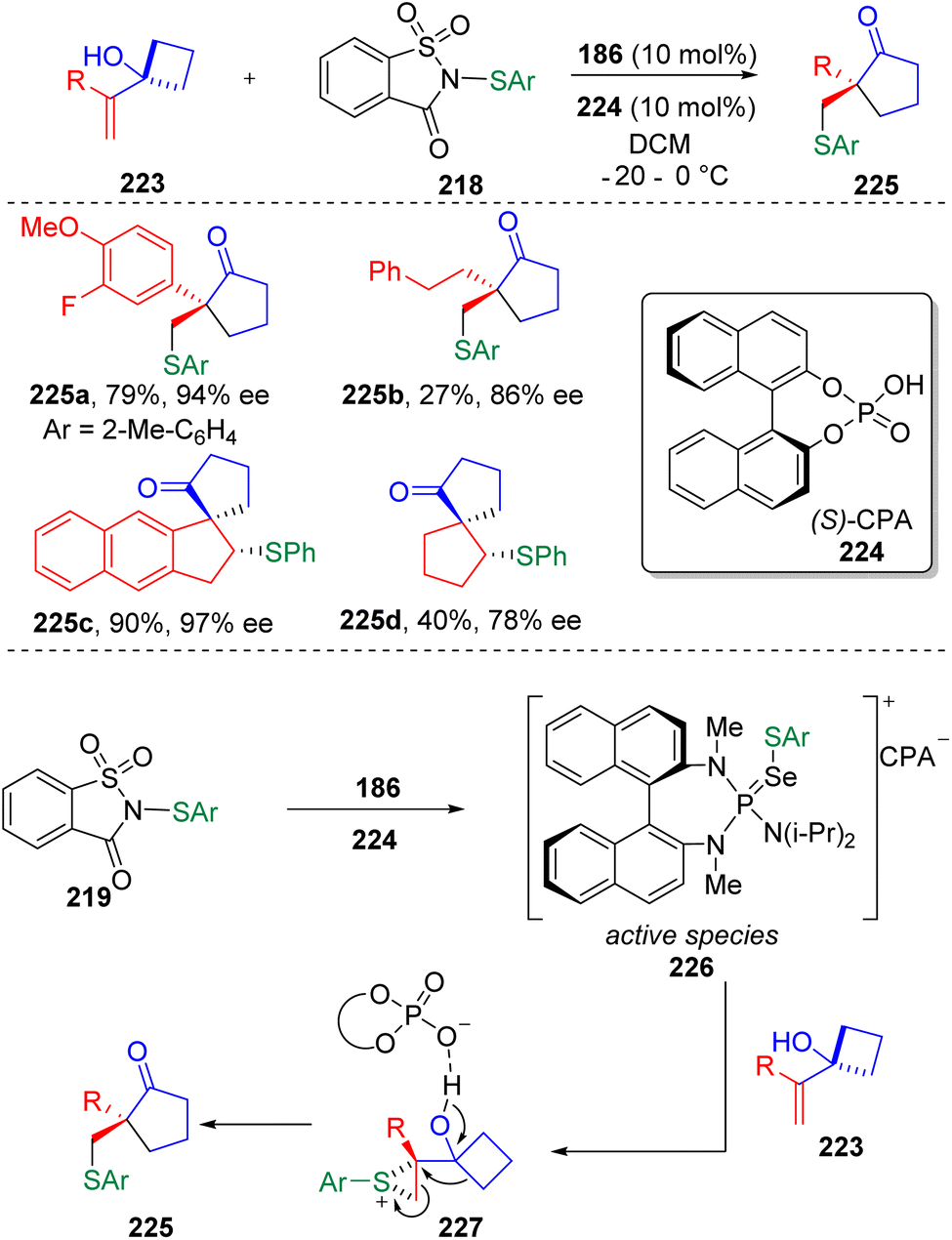 | ||
| Scheme 38 Enantioselective sulfenylation/semipinacol rearrangement. | ||
In 2020, Xu and co-workers69 presented an interesting fluorosulfenylation of alkenes with N-thiosuccinimides (201) and pyridine hydrofluoride (229) (Scheme 39). The authors conceptionally assumed that the in situ generated sulfenyl fluorides converted the alkenes (228) into episulfonium ion intermediates (232) and delivered the vicinal fluorosulfenyl products (230) after the nucleophilic ring-opening addition. Other fluoride sources did not give the desired products, even Et3N-HF and DMPU-HF. Non-polar solvents such as dichloroethane and dichloromethane were preferred by the transformation. The reaction proceeded well with terminal alkyl alkenes and symmetrical internal alkenes, with the tolerance of functionalities of sulfonyl esters (OTs and OMs), thiophene, furan, azides, etc. Regarding the thiolating partner, both alkyl thiols and thiophenol-derived N-thiosuccinimides were applicable and underwent fluorosulfenylation smoothly. Furthermore, taking advantage of the leaving ability of bromide at the benzyl position, the authors found that difluorosulfenylated products could be obtained by subjecting the α-bromostyrenes to the standard conditions, with β-bromo-β-fluoroalkyl phenyl thioethers as the intermediates of the step-wise protocol.
 | ||
| Scheme 39 Fluorosulfenylation of alkenes using N-thiosuccinimides and pyridine hydrofluoride. | ||
The Zhao group70 also applied the Lewis base catalyzed activation of sulfenylating agents (235) to the intermolecular difunctionalization of alkynes (234) (Scheme 40). Thus, the method of the trifluoromethylthiolative triflation of alkynes was developed to access the regio- and stereoselective synthesis of trifluoromethylthiolated tetrasubstituted alkenes (237). Based on experimental evidence, the authors proposed a possible pathway for the transformation. The catalytic diaryl selenide (236) performed as a Lewis base to interact with the thiolating agent (235), and promoted the transfer of trifluoromethylthio sulfonium to alkyne substrates (234). The formed thiirenium ion intermediate (239) then underwent nucleophilic addition in the presence of TfO− to give the target product (237) and release the selenide catalyst (236) for the next cycle of the catalysis.
 | ||
| Scheme 40 Lewis base catalyzed trifluoromethylthiolative triflation of alkynes. | ||
4 Transition-metal catalyzed carbosulfenylation
In 2022, Engle and co-workers71 introduced a novel directing Ni-catalyzed syn-carbosulfenylation reaction of unactivated alkenes (240) with organoboron nucleophiles (241) (Scheme 41). The key to the process is the use of N-alkyl-N-(arylsulfinyl)arylsulfonamides (242) as electrophilic sulfur sources, which provide the opportunity to control the efficiency and selectivity through the tunable electronic and steric properties of the sulfonamide fragment. On this basis, several sulfenylating reagents were employed and it was determined that N-methyl-N-para-methoxysulfonamide derived arylsulfenyl agents (242) were reactive enough to engage ArS to the alkylnickel(I) intermediate (247) before the β-hydride elimination occurred, while sluggish in the direct sulfenylation of alkene (240) or the arylnickel(I) intermediate (245). DFT parameterization of the lowest unoccupied molecular orbital (LUMO) energy and bond dissociation energy (BDE) of the N–S bond in 242 against the yields was conducted to rationalize this phenomenon. The catalytic cycle of the three-component coupling reaction first involved the transmetalation of an aryl boronic reagent to Ni(I) species 244. The formed carbon-nickel intermediate 245 coordinated with alkene substrates (240) and activated the carbon–carbon double bond. An internal migratory insertion of the aryl group led to the generation of metallacycle species 247, which was then oxidized by the sulfenylating reagent (242) to deliver the key Ni(III) intermediate (248). C–S bond formation via the reductive elimination regenerated the active Ni(I) catalyst (244). Soon after that, the Engle group72 further expanded the scope of the nucleophile to C(sp3) partners and developed the 1,2-alkylsulfenylation of unactivated alkenes with alkylzinc reagents and the sulfonamide sulfenyl electrophiles. To surrogate the above mentioned undesired process and the β-hydride elimination of alkyl nucleophile, a stronger bidentate directing auxiliary (8-aminoquinoline, AQ) was employed. | ||
| Scheme 41 Ni-catalyzed syn-carbosulfenylation reaction. | ||
Besides sulfonamide sulfenylating agents, the Wang group73 found that simple aryl disulfides (250, Scheme 42) were also competent to oxidize Ni(I) (254) to the sulfenyl Ni(III) intermediate (255) and furnish the 1,2-arylsulfenylation products (251) chemo- and regioselectively from unactivated alkenes (248) and arylboronic acids (249). In this process, the assistance of the intramolecular bidentate coordination of PA auxiliary (picolinamide) was proposed to stabilize the active nickel intermediates.
 | ||
| Scheme 42 Ni-catalyzed carbosulfenylation with disulfides. | ||
5. Conclusions
In summary, the recent progress in the field of C–S bond formation via the hydrofunctionalization and 1,2-difunctionalization of unactivated alkenes and alkynes was reviewed. Transformations were elucidated according to the types of reaction model, mainly divided as radical and thiiranium ion processes. The scopes, limitations and main mechanisms were discussed. Although significant accomplishments have been achieved in this topic over the past few years, a few challenges still remain for synthetic chemists; for example: the electrochemical difunctionalization of unactivated alkenes other than styrene substrates, as well as general intermolecular carbosulfenylation in electrophilic sulfenylative reaction and photochemical systems. We hope that this review will be beneficial for researchers who are working on related topics and encourage the development of more efficient and general reactions and methodologies in this field.Abbreviations
| TBAF | Tetrabutylammonium fluoride |
| EDA complex | Electron donor–acceptor complex |
| DMSO: | Dimethyl sulfoxide |
| DCE | 1,2-Dichloroethane |
| Dtbbpy | 4,4′-Di-tert-butyl-2,2′-bipyridine |
| BINAM | (S)-(−)-1,1′-Binaphthyl-2,2′-diamine |
| TMSOTf | Trimethylsilyl trifluoromethanesulfonate |
| Tf2NH | Bis(trifluoromethylsulfonyl)amine |
| Ni(cod)2 | Bis(1,5-cyclooctadiene)nickel(0) |
| DME | 1,2-Dimethoxyethane |
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are grateful to the National Key Research and Development Project of China (No. 2021YFC2100100), the National Natural Science Foundation of China (No. 21901123) and the Jiangsu Specially Appointed Professor Plan.References
- (a) G. Li, X. Huo, X. Jiang and W. Zhang, Asymmetric Synthesis of Allylic Compounds via Hydrofunctionalization and Difunctionalization of Dienes, Allenes, and Alkynes, Chem. Soc. Rev., 2020, 49, 2060 RSC; (b) K. Wu, Y. Liang and N. Jiao, Azidation in the Difunctionalization of Olefins, Molecules, 2016, 21, 352 CrossRef PubMed; (c) W. Liu and W. Kong, Ni-Catalyzed Stereoselective Difunctionalization of Alkynes, Org. Chem. Front., 2020, 7, 3941 RSC; (d) G. Zhang, Y. Liu, J. Zhao, Y. Li and Q. Zhang, Radical Cascade Reactions of Unsaturated C-C Bonds Involving Migration, Sci. China: Chem., 2019, 62, 1476 CrossRef CAS.
- (a) Z.-L. Li, G.-C. Fang, Q.-S. Gu and X.-Y. Liu, Recent Advances in Copper-Catalyzed Radical Involved Asymmetric 1,2-Difunctionalization of Alkenes, Chem. Soc. Rev., 2020, 49, 32 RSC; (b) H.-Y. Tu, S. Zhu, F.-L. Qing and L. Chu, Recent Advances in Nickel-Catalyzed Three-Component Difunctionalization of Unactivated Alkenes, Synthesis, 2020, 52, 1346 CrossRef CAS; (c) X. Chen, F. Xiao and W.-M. He, Recent Developments in the Difunctionalization of Alkenes with C–N Bond Formation, Org. Chem. Front., 2021, 8, 5206 RSC; (d) Y. Li, D. Wu, H.-G. Cheng and G. Yin, Difunctionalization of Alkenes Involving Metal Migration, Angew. Chem., Int. Ed., 2020, 59, 7990 CrossRef CAS PubMed.
- (a) C.-S. Jiang, W. E. G. Müller, H. C. Schröder and Y.-W. Guo, Disulfide- and Multisulfide-Containing Metabolites from Marine Organisms, Chem. Rev., 2012, 112, 2179 CrossRef CAS PubMed; (b) J. Zeng, Y. Liu, W. Chen, X. Zhao, L. Meng and Q. Wan, Glycosyl Sulfoxides in Glycosylation Reactions, Top. Curr. Chem., 2018, 376, 27 CrossRef PubMed; (c) N. Wang, P. Saidhareddya and X. Jiang, Nat. Prod. Rep., 2020, 37, 246 RSC.
- (a) Q. Wang, J. Guan, J. Wan and Z. Li, Disulfide Based Prodrugs for Cancer Therapy, RSC Adv., 2020, 10, 24397 RSC; (b) K. A. Scott and J. T. Njardarson, Analysis of US FDA-Approved Drugs Containing Sulfur Atoms, Top. Curr. Chem., 2018, 376, 5 CrossRef; (c) J. Zhao and X. Jiang, Chin. Chem. Lett., 2018, 29, 1079 CrossRef CAS.
- P. Devendar and G.-F. Yang, Sulfur-Containing Agrochemicals, Top. Curr. Chem., 2017, 375, 82 CrossRef.
- K. Dong, X.-S. Liu, X. Wei, Y. Zhao and L. Liu, Borane-Catalyzed S–H Insertion Reaction of Thiophenols and Thiols with α-Aryl-α-diazoesters, Green Synth. Catal., 2021, 2, 385 CrossRef.
- (a) L. Kurti and B. Czako, Strategic Applications of Named Reactions in Organic Synthesis, Elsevier Academic Press, 2005 Search PubMed; (b) Sulfur-Mediated Rearrangements I, ed. E. Schaumann, Springer, 2007 Search PubMed.
- (a) D. Kaiser, I. Klose, R. Oost, J. Neuhaus and N. Maulide, Bond-Forming and -Breaking Reactions at Sulfur(IV): Sulfoxides, Sulfonium Salts, Sulfur Ylides, and Sulfinate Salts, Chem. Rev., 2019, 119, 8701 CrossRef CAS PubMed; (b) L. Tang, Q. Hu, M. Elsaid, C. Liu, K. Yang and H. Ge, Recent Advances in Direct α-C(sp3)-H Bond Functionalization of Thioethers, Green Synth. Catal., 2022, 3, 203 CrossRef.
- B. Wang, Y. Zhou, X. Luo, W. Chen, S. Yang, S. Luo and Z. Wang, Research Progress in C-S Bond Formation Reaction of Olefins with Organic Sulfur Reagents under Photocatalyst-Free and Non-Electrochemical Conditions, Chin. J. Org. Chem., 2021, 41, 171 CrossRef.
- (a) W. Guo, K. Tao, W. Tan, M. Zhao, L. Zheng and X. Fan, Recent Advances in Photocatalytic C–S/P–S Bond Formation via the Generation of Sulfur Centered Radicals and Functionalization, Org. Chem. Front., 2019, 6, 2048 RSC; (b) F. Denes, M. Pichowicz, G. Povie and P. Renaud, Thiyl Radicals in Organic Synthesis, Chem. Rev., 2014, 114, 2587 CrossRef CAS PubMed.
- (a) L. Liao and X. Zhao, Indane-Based Chiral Aryl Chalcogenide Catalysts: Development and Applications in Asymmetric Electrophilic Reactions, Acc. Chem. Res., 2022, 55, 2439 CrossRef CAS PubMed; (b) A. Matviitsuk, J. L. Panger and S. E. Denmark, Catalytic, Enantioselective Sulfenofunctionalization of Alkenes: Development and Recent Advances, Angew. Chem., Int. Ed., 2020, 59, 19796 CrossRef CAS PubMed; (c) B. Majia, Stereoselective Haliranium, Thiiranium and Seleniranium Ion-Triggered Friedel–Crafts-Type Alkylations for Polyene Cyclizations, Adv. Synth. Catal., 2019, 361, 3453 CrossRef.
- (a) X. Cheng, A. Lei, T.-S. Mei, H.-C. Xu, K. Xu and C. Zeng, Recent Applications of Homogeneous Catalysis in Electrochemical Organic Synthesis, CCS Chem., 2022, 4, 1120 CrossRef CAS; (b) P. Wang, X. Gao, P. Huang and A. Lei, Recent Advances in Electrochemical Oxidative Cross-Coupling of Alkenes with H2 Evolution, ChemCatChem, 2020, 12, 27 CrossRef CAS; (c) M. Yan, Y. Kawamata and P. S. Baran, Synthetic Organic Electrochemistry: Calling All Engineers, Angew. Chem., Int. Ed., 2018, 57, 4149 CrossRef CAS PubMed.
- (a) C. Song, K. Liu, X. Dong, C.-W. Chiang and A. Lei, Recent Advances in Electrochemical Oxidative Cross-Coupling for the Construction of C–S Bonds, Synlett, 2019, 30, 1149 CrossRef CAS; (b) M. He, P. Zhong, H. Liu, C. Ou, Y. Pan and H. Tang, Electrochemically Mediated Three-Component Synthesis of Isothioureas Using Thiols as Sulfur Source, Green Synth. Catal., 2022 DOI:10.1016/j.gresc.2022.03.002.
- Y. Yuan, Y. Chen, S. Tang, Z. Huang and A. Lei, Electrochemical Oxidative Oxysulfenylation and Aminosulfenylation of Alkenes with Hydrogen Evolution, Sci. Adv., 2018, 4, eaat5312 CrossRef CAS PubMed.
- Y. Yuan, Y. Cao, Y. Lin, Y. Li, Z. Huang and A. Lei, Electrochemical Oxidative Alkoxysulfonylation of Alkenes Using Sulfonyl Hydrazines and Alcohols with Hydrogen Evolution, ACS Catal., 2018, 8, 10871 CrossRef CAS.
- Z. Zhang, J. Yan, D. Ma and J. Sun, Electrochemical Synthesis of β-Hydroxy-, β-Alkoxy-, and β-Carbonyloxy Sulfones by Vicinal Difunctionalization of Olefins, Chin. Chem. Lett., 2019, 30, 1509 CrossRef CAS.
- X. Luo, S. Wang and A. Lei, Electrochemical-Induced Hydroxysulfonylation of α-CF3 Alkenes to Access Tertiary β-Hydroxysulfones, Adv. Synth. Catal., 2022, 364, 1016 CrossRef CAS.
- Y.-M. Jiang, Y. Yu, S.-F. Wu, H. Yan, Y. Yuan and K.-Y. Ye, Electrochemical Fluorosulfonylation of Styrenes, Chem. Commun., 2021, 57, 11481 RSC.
- Y. Yu, Y. Jiang, S. Wu, Z. Shi, J. Wu, Y. Yuan and K. Ye, Electrochemistry Enabled Selective Vicinal Fluorosulfenylation and Fluorosulfoxidation of Alkenes, Chin. Chem. Lett., 2022, 33, 2009 CrossRef CAS.
- Y. Yu, Y.-M. Jiang, X.-B. Zhu, Y.-Y. Lin, Y. Yuan and K.-Y. Ye, Electrochemical β-Chlorosulfoxidation of Alkenes, Org. Chem. Front., 2022, 9, 5586 RSC.
- M. Yu, H. Wang, Y. Gao, F. Bu, H. Cong and A. Lei, Manganese-Catalyzed Chlorosulfonylation of Terminal Alkene and Alkyne via Convergent Paired Electrolysis, Cell Rep. Phys. Sci., 2021, 2, 100476 CrossRef CAS.
- (a) Y. Chen, L.-Q. Lu, D.-G. Yu, C.-J. Zhu and W.-J. Xiao, Visible Light-Driven Organic Photochemical Synthesis in China, Sci. China: Chem., 2019, 62, 24 CrossRef CAS; (b) A. Y. Chan, I. B. Perry, N. B. Bissonnette, B. F. Buksh, G. A. Edwards, L. I. Frye, O. L. Garry, M. N. Lavagnino, B. X. Li, Y. Liang, E. Mao, A. Millet, J. V. Oakley, N. L. Reed, H. A. Sakai, C. P. Seath and D. W. C. MacMillan, Metallaphotoredox: The Merger of Photoredox and Transition Metal Catalysis, Chem. Rev., 2022, 122, 1485 CrossRef CAS PubMed; (c) P. Wang, Q. Zhao, W. Xiao and J. Chen, Recent Advances in Visible-Light Photoredox-Catalyzed Nitrogen Radical Cyclization, Green Synth. Catal., 2020, 1, 42 CrossRef.
- A. Kahnt, A. Rentmeister, D. Guldi and F. Glorius, The Energy-Transfer-Enabled Biocompatible Disulfide–ene Reaction, M. Teders, C. Henkel, R. Kleinmans, Nat. Chem., 2018, 10, 981 CrossRef PubMed.
- Z. Chen, F. Xue, T. Liu, B. Wang, Y. Zhang, W. Jin, Y. Xia and C. Liu, Synthesis of β-Hydroxysulfides via Visible-Light-Driven and EDA Complex-Promoted Hydroxysulfenylation of Styrenes with Heterocyclic Thiols in EtOH under Photocatalyst-Free Conditions, Green Chem., 2022, 24, 3250 RSC.
- L. Lin, Z. Yang, J. Liu, J. Wang, J. Zheng, J.-L. Li, X. Zhang, X.-W. Liu, H. Jiang and J. Li, Visible-Light-Induced Surfactant-Promoted Sulfonylation of Alkenes and Alkynes with Sulfonyl Chloride by the Formation of an EDA-Complex with NaI in Water at Room Temperature, Green Chem., 2021, 23, 5467 RSC.
- Y. Li, T. Miao, P.-H. Li and L. Wang, Photo-Driven Synthesis of C6-Polyfunctionalized Phenanthridines from Three-Component Reactions of Isocyanides, Alkynes, and Sulfinic Acids by Electron Donor−Acceptor Complex, Org. Lett., 2018, 20, 1735 CrossRef CAS PubMed.
- X. Nie, T. Xu, Y. Hong, H. Zhang, C. Mao and S. Liao, Introducing A New Class of Sulfonyl Fluoride Hubs via Radical Chloro-Fluorosulfonylation of Alkynes, Angew. Chem., Int. Ed., 2021, 60, 22035 CrossRef CAS PubMed.
- P. Wang, H. Zhang, M. Zhao, S. Ji, L. Lin, N. Yang, X. Nie, J. Song and S. Liao, Radical Hydro-Fluorosulfonylation of Unactivated Alkenes and Alkynes, Angew. Chem., Int. Ed., 2022, 61, e202207684 CAS.
- C. Zhu, H. Yue, B. Maity, I. Atodiresei, L. Cavallo and M. Rueping, A Multicomponent Synthesis of Stereodefined Olefins via Nickel catalysis and Single Electron/triplet Energy Transfer, Nat. Catal., 2019, 2, 678 CrossRef CAS.
- (a) J. Xu and Q. Song, Radical Promoted Difunctionalization of Unsaturated Carbon-Carbon Bonds in the Presence of Dioxygen, Chin. J. Org. Chem., 2016, 36, 1151 CrossRef CAS; (b) G. Yin, X. Mu and G. Liu, Palladium(II)-Catalyzed Oxidative Difunctionalization of Alkenes: Bond Forming at a High-Valent Palladium Center, Acc. Chem. Res., 2016, 49, 2413 CrossRef CAS PubMed.
- K. Choudhuri, A. Mandal and P. Mal, Aerial Dioxygen Activation vs. Thiol–ene Click Reaction within a System, Chem. Commun., 2018, 54, 3759 RSC.
- H. Wang, Q. Lu, C. Qian, C. Liu, W. Liu, K. Chen and A. Lei, Solvent-Enabled Radical Selectivities: Controlled Syntheses of Sulfoxides and Sulfides, Angew. Chem., Int. Ed., 2016, 55, 1094 CrossRef CAS.
- B. Du, Y. Wang, H. Mei, J. Han and Y. Pan, Copper(II) Acetate-Catalyzed Hydroxysulfenylation-Initiated Lactonization of Unsaturated Carboxylic Acids with Oxygen as Oxidant and Oxygenation Reagent, Adv. Synth. Catal., 2017, 359, 1684 CrossRef CAS.
- M. Ji, J. Yu and C. Zhu, Cyanotrifluoromethylthiolation of Unactivated Dialkyl-Substituted Alkynes via Cyano Migration: Synthesis of Trifluoromethylthiolated Acrylonitriles, Chem. Commun., 2018, 54, 6812 RSC.
- Y. Li, L. Shen, M. Zhou, B. Xiong, X. Zhang and Z. Lian, Copper-Catalyzed Chloro-Arylsulfonylation of Styrene Derivatives via the Insertion of Sulfur Dioxide, Org. Lett., 2021, 23, 5880 CrossRef CAS PubMed.
- G. Chen, J. Xu, B. Xiong, H. Song, X. Zhang, X. Ma and Z. Lian, Copper-Catalyzed Trifluoromethylthio-arylsulfonylation of Styrene Derivatives via the Insertion of Sulfur Dioxide, Org. Lett., 2022, 24, 1207 CrossRef CAS PubMed.
- L. Liu, M. Si, S. Han, Y. Zhang and J. Li, Copper-Catalyzed Regioselective Sulfonylcyanations of Vinylarenes, Org. Chem. Front., 2020, 7, 2029 RSC.
- Q. Liang, P. J. Walsh and T. Jia, Copper-Catalyzed Intermolecular Difunctionalization of Styrenes with Thiosulfonates and Arylboronic Acids via a Radical Relay Pathway, ACS Catal., 2020, 10, 2633 CrossRef CAS.
- H. Lecher, F. Holschneider, K. Köberle, W. Speer and P. Stöcklin, Phenyl-schwefelchlorid (II), Chem. Ber., 1925, 58, 409 CrossRef.
- (a) N. Kharasch and C. M. Buess, Derivatives of Sulfenic Acids. II. Characterization of Olefins with 2,4-Dinitrobenzenesulfenyl Chloride, J. Am. Chem. Soc., 1949, 71, 2724 CrossRef CAS; (b) M. Z. Krimer, E. A. Vorob'ev and W. A. Smit, Generation and Chemical Reactions of Episolfonium Ions, Tetrahedron Lett., 1975, 16, 2451 CrossRef.
- W. A. Smit, R. Caple and I. P. Smoliakova, Stepwise Electrophilic Addition. Some Novel Synthetic Ramifications of an Old Concept, Chem. Rev., 1994, 94, 2359 CrossRef CAS.
- C. M. Rayner, Synthetic Transformations Involving Thiiranium Ion Intermediates, in Organosulfur Chemistry, Synthetic Aspects, ed. P. Page, Academic Press, 1995, ch. 3 Search PubMed.
- (a) H. Minato, T. Miura and M. Kobayashi, Reactions of Dimethylmethyl-thiosulfonium Fluoroborate with Some Nucleophiles, Chem. Lett., 1975, 4, 701 CrossRef; (b) K. Okuma, T. Koike, S. Yamamoto, K. Yonekura and H. Ohta, Reaction of Olefins with Dimethylthiomethylsulfonium Salts and Triphenylphosphine. A New Convenient Synthesis of Vinylphosphonium Salts, Chem. Lett., 1989, 18, 1953 CrossRef.
- (a) G. K. Helmkamp, B. A. Olsen and D. J. Pettitt, The Addition of Dialkylalkylthiosulfonium Salts to Alkenes, J. Org. Chem., 1965, 30, 676 CrossRef CAS PubMed; (b) S. A. Anderson, J. K. Kim and M. C. Caserio, Methylthiolation of 2,3-bis(methylthio)butanes, J. Org. Chem., 1978, 43, 4822 CrossRef CAS; (c) S. H. Smallcombe and M. C. Caserio, Kinetics of Sulfur-Sulfur Bond Cleavage in Methylated Methyl Disulfide by Nuclear Magnetic Resonance, J. Am. Chem. Soc., 1971, 93, 5826 CrossRef CAS; (d) K. Okuma, I. Takeshita, T. Yasuda and K. Shioji, Regioselective Synthesis of Benzazetines and Indoles from Alkenylanilides and Dimethyl(methylthio)sulfonium Trifluoromethanesulfonate, Chem. Lett., 2006, 35, 1122 CrossRef CAS; (e) K. Okuma, T. Yasuda, I. Takeshita, K. Shiojia and Y. Yokomori, Novel Formation of Indoles and 3,1-Benzoxazines from O-Alkenylanilides and Dimethyl(methylthio)sulfonium Trifluoromethanesulfonate, Tetrahedron, 2007, 63, 8250 CrossRef CAS; (f) F. Alonso, Y. Moglie, G. Radivoy and M. Yus, Alkenes as Azido Precursors for the One-Pot Synthesis of 1,2,3-Triazoles Catalyzed by Copper Nanoparticles on Activated Carbon, J. Org. Chem., 2013, 78, 5031 CrossRef CAS PubMed; (g) J. Wang, M. Tang, W. Gu, S. Huang and L.-G. Xie, Synthesis of Pyrrole via Formal Cycloaddition of Allyl Ketone and Amine under Metal-Free Conditions, J. Org. Chem., 2022, 87, 12482 CrossRef CAS PubMed.
- (a) B. M. Trost and E. Murayama, Dimethylmethylthiosulfonium Fluoroborate. A Chemoselective Initiator for Thionium Ion Induced Cyclizations, J. Am. Chem. Soc., 1981, 103, 6529 CrossRef CAS; (b) G. Capozzi, V. Lucchini, F. Marcuzzi and G. Modena, Intramolecular Cyclization Using Methyl(bismethylthio)sulfonium Salts. Part I. 2,3-Dihydrobenzofurans, J. Chem. Soc., Perkin Trans. 1, 1981, 3106 RSC; (c) B. M. Trost and T. Shibata, Nucleophilic Attack on Olefins Initiated by Dimethyl(methylthio)sulfonium Fluoroborate (DMTSF). Azasulfenylation, J. Am. Chem. Soc., 1982, 104, 3225 CrossRef CAS; (d) B. M. Trost, T. Shibata and S. J. Martin, Nucleophilic Attack on Ole-fins Initiated by Dimethyl(methylthio)sulfonium Fluoroborate (DMTSF). Cyanosulfenylation and Oxy- and Oxosulfenylation, J. Am. Chem. Soc., 1982, 104, 3228 CrossRef CAS; (e) G. J. O'Malley and M. P. Cava, Synthetic Applications of (Dimethylthio)sulfonium Fluoroborate: Sulfenyletherification and Sulfenyllactonization, Tetrahedron Lett., 1985, 26, 6159 CrossRef; (f) G. Haufe, Formal Addition of Methanesulfenyl Fluoride to Unsatureated Substrates, Tetrahedron Lett., 1988, 29, 2311 CrossRef CAS; (g) M. C. Caserio and J. K. Kim, Thioammonium Ions. Azasulfenylation Reactions, J. Am. Chem. Soc., 1982, 104, 3231 CrossRef CAS.
- E. J. Adams and S. Oscarson, Dimethyl(methylthio)sulfonium Tetrafluoroborate (DMTSF), in Encyclopedia of Reagents for Organic Synthesis, Wiley, 2005, DOI:10.1002/047084289X.rd349.pub2.
- S. Chen, J. Wang and L.-G. Xie, Transition Metal-Free Formal Hydro/deuteromethylthiolation of Unactivated Alkenes, Org. Biomol. Chem., 2021, 19, 4037 RSC.
- M. Tang, Y. Wei, S. Huang and L.-G. Xie, Regio- and Stereoselective Synthesis of β-Methylthio Vinyl Triflates, Org. Lett., 2022, 24, 7026 CrossRef CAS PubMed.
- B. M. Trost and S. J. Martin, Alkynyl Sulfenylation. A Direct Approach for Nucleophilic Addition and Substitution of Olefins by Carbanions, J. Am. Chem. Soc., 1984, 106, 4263 CrossRef CAS.
- M. Tang, S. Han, S. Huang, S. Huang and L.-G. Xie, Carbosulfenylation of Alkenes with Organozinc Reagents and Dimethyl(methylthio)sulfonium Trifluoromethanesulfonate, Org. Lett., 2020, 22, 9729 CrossRef CAS PubMed.
- F. T. Schevenels, M. Shen and S. A. Snyder, Alkyldisulfanium Salts: Isolable, Electrophilic Sulfur Reagents Competent for Polyene Cyclizations, Org. Lett., 2017, 19, 2 CrossRef CAS PubMed.
- C. J. F. Cole, H. M. Chi, K. C. DeBacker and S. A. Snyder, Synthesis of Enhanced, Isolable Disulfanium Salts and their Application to Thiiranium-Promoted Polyene Cyclizations, Synthesis, 2018, 50, 4351 CrossRef CAS.
- (a) S. E. Denmark, W. R. Collins and D. Matthew, Cullen, Observation of Direct Sulfenium and Selenenium Group Transfer from Thiiranium and Seleniranium Ions to Alkenes, J. Am. Chem. Soc., 2009, 131, 3490 CrossRef CAS PubMed; (b) S. E. Denmark and T. Vogler, Synthesis and Reactivity of Enantiomerically Enriched Thiiranium Ions, Chem. – Eur. J., 2009, 15, 11737 CrossRef CAS PubMed.
- (a) S. E. Denmark, J. P. Kornfilt and T. Vogler, Catalytic Asymmetric Thiofunctionalization of Unactivated Alkenes, J. Am. Chem. Soc., 2011, 133, 15308 CrossRef CAS PubMed; (b) S. E. Denmark and D. J. P. Kornfilt, Catalytic, Enantioselective, Intramolecular Sulfenofunctionalization of Alkenes with Phenols, J. Org. Chem., 2017, 82, 3192 CrossRef CAS PubMed; (c) A. Matviitsuk and S. E. Denmark, Enantio- and Diastereoselective, Lewis Base Catalyzed, Cascade Sulfenoacetalization of Alkenyl Aldehydes, Angew. Chem., Int. Ed., 2019, 58, 12486 CrossRef CAS PubMed; (d) K. M. Hilby and S. E. Denmark, Lewis Base Catalyzed, Sulfenium Ion Initiated Enantioselective, Spiroketalization Cascade, J. Org. Chem., 2021, 86, 14250 CrossRef CAS PubMed; (e) S. E. Denmark and H. M. Chi, Lewis Base Catalyzed, Enantioselective, Intramolecular Sulfenoamination of Olefins, J. Am. Chem. Soc., 2014, 136, 8915 CrossRef CAS PubMed; (f) S. E. Denmark and H. M. Chi, Catalytic, Enantioselective, Intramolecular Sulfenoamination of Alkenes with Anilines, J. Org. Chem., 2017, 82, 3826 CrossRef CAS PubMed; (g) J. L. Panger and S. E. Denmark, Enantioselective Synthesis of γ-Lactams by Lewis Base Catalyzed Sulfenoamidation of Alkenes, Org. Lett., 2020, 22, 2501 CrossRef CAS PubMed; (h) S. E. Denmark and A. Jaunet, Catalytic, Enantioselective, Intramolecular Carbosulfenylation of Olefins, J. Am. Chem. Soc., 2013, 135, 6419 CrossRef CAS PubMed; (i) S. E. Denmark and A. Jaunet, Catalytic, Enantioselective, Intramolecular Carbosulfenylation of Olefins. Preparative and Stereochemical Aspects, J. Org. Chem., 2014, 79, 140 CrossRef CAS PubMed; (j) T. Menard, A. Laverny and S. E. Denmark, Synthesis of Enantioenriched 3,4-Disubstituted Chromans through Lewis Base Catalyzed Carbosulfenylation, J. Org. Chem., 2021, 86, 14290 CrossRef CAS PubMed.
- (a) S. E. Denmark and H. M. Chi, Catalytic, Enantioselective, Intramolecular Carbosulfenylation of Olefins. Mechanistic Aspects: A Remarkable Case of Negative Catalysis, J. Am. Chem. Soc., 2014, 136, 3655 CrossRef CAS PubMed; (b) S. E. Denmark, E. Hartmann, D. J. P. Kornfilt and H. Wang, Mechanistic, Crystallographic, and Computational Studies on the Catalytic, Enantioselective Sulfenofunctionalization of Alkenes, Nat. Chem., 2014, 6, 1056 CrossRef CAS PubMed; (c) E. Hartmann and S. E. Denmark, Structural, Mechanistic, Spectroscopic, and Preparative Studies on the Lewis Base Catalyzed, Enantioselective Sulfenofunctionalization of Alkenes, Helv. Chim. Acta, 2017, 100, e1700158 CrossRef PubMed.
- (a) Z. Tao, K. A. Robb, K. Zhao and S. E. Denmark, Enantioselective, Lewis Base-Catalyzed Sulfenocyclization of Polyenes, J. Am. Chem. Soc., 2018, 140, 3569 CrossRef CAS PubMed; (b) K. A. Robb, S. V. Athavale and S. E. Denmark, Unusual Kinetic Profiles for Lewis Base-Catalyzed Sulfenocyclization of ortho-Geranylphenols in Hexafluoroisopropyl Alcohol, Synlett, 2019, 30, 1656 CrossRef CAS PubMed.
- Z. Tao, K. A. Robb, J. L. Panger and S. E. Denmark, Enantioselective, Lewis Base-Catalyzed Carbosulfenylation of Alkenylboronates by 1,2-Boronate Migration, J. Am. Chem. Soc., 2018, 140, 15621 CrossRef CAS PubMed.
- Q. Jiang and X. Zhao, Chiral Bifunctional Chalcogenide-Catalyzed Enantioselective Electrophilic Thiofunctionalization of Alkenes, Chin. J. Org. Chem., 2021, 41, 443 CrossRef.
- (a) X. Liu, R. An, X. Zhang, J. Luo and X. Zhao, Enantioselective Trifluoromethylthiolating Lactonization Catalyzed by an Indane-Based Chiral Sulfide, Angew. Chem., Int. Ed., 2016, 55, 5846 CrossRef CAS PubMed; (b) J. Luo, Y. Liu and X. Zhao, Chiral Selenide-Catalyzed Enantioselective Construction of Saturated Trifluoromethylthiolated Azaheterocycles, Org. Lett., 2017, 19, 3434 CrossRef CAS PubMed; (c) T. Qin, Q. Jiang, J. Ji, J. Luo and X. Zhao, Chiral Selenide-Catalyzed Enantioselective Synthesis of Trifluoromethylthiolated 2,5-Disubstituted Oxazolines, Org. Biomol. Chem., 2019, 17, 1763 RSC; (d) J. Luo, Q. Cao, X. Cao and X. Zhao, Selenide-Catalyzed Enantioselective Synthesis of Trifluoromethylthiolated Tetrahydronaphthalenes by Merging Desymmetrization and Trifluoromethylthiolation, Nat. Commun., 2018, 9, 527 CrossRef PubMed; (e) X. Liu, Y.-Y. Liang, J.-Y. Ji, J. Luo and X.-D. Zhao, Chiral Selenide-Catalyzed Enantioselective Allylic Reaction and Intermolecular Difunctionalization of Alkenes: Efficient Construction of C-SCF3 Stereogenic Molecules, J. Am. Chem. Soc., 2018, 140, 4782 CrossRef CAS PubMed; (f) J. Xu, Y. Zhang, T. Qin and X.-D. Zhao, Catalytic Regio- and Enantioselective Oxytrifluoromethylthiolation of Aliphatic Internal Alkenes by Neighboring Group Assistance, Org. Lett., 2018, 20, 6384 CrossRef CAS PubMed.
- Y. Liang, J. Ji, X. Zhang, Q. Jiang, J. Luo and X. Zhao, Enantioselective Construction of Axially Chiral Amino Sulfide Vinyl Arenes by Chiral Sulfide-Catalyzed Electrophilic Carbothiolation of Alkynes, Angew. Chem., Int. Ed., 2020, 59, 4959 CrossRef CAS PubMed.
- A. Roth and S. E. Denmark, Enantioselective, Lewis Base-Catalyzed, Intermolecular Sulfenoamination of Alkenes, J. Am. Chem. Soc., 2019, 141, 13767 CrossRef CAS PubMed.
- Y. Liang and X. Zhao, Enantioselective Construction of Chiral Sulfides via Catalytic Electrophilic Azidothiolation and Oxythiolation of N-Allyl Sulfonamides, ACS Catal., 2019, 9, 6896 CrossRef CAS.
- Y. Zhang, Y. Liang and X. Zhao, Chiral Selenide-Catalyzed, Highly Regio- and Enantioselective Intermolecular Thioarylation of Alkenes with Phenols, ACS Catal., 2021, 11, 3755 CrossRef CAS.
- H.-Y. Luo, Y.-Y. Xie, X.-F. Song, J.-W. Dong, D. Zhu and Z.-M. Chen, Lewis Base-Catalyzed Asymmetric Sulfenylation of Alkenes: Construction of Sulfenylated Lactones and Application to the Formal Syntheses of (-)-Nicotlactone B and (-)-Galbacin, Chem. Commun., 2019, 55, 9367 RSC.
- H.-Y. Luo, J.-W. Dong, Y.-Y. Xie, X.-F. Song, D. Zhu, T. Ding, Y. Liu and Z.-M. Chen, Lewis Base/Brønsted Acid Co-catalyzed Asymmetric Thiolation of Alkenes with Acid-Controlled Divergent Regioselectivity, Chem. – Eur. J., 2019, 25, 15411 CrossRef CAS PubMed.
- A.-H. Ye, Y. Zhang, Y.-Y. Xie, H.-Y. Luo, J.-W. Dong, X.-D. Liu, X.-F. Song, T. Ding and Z.-M. Chen, TMSCl-Catalyzed Electrophilic Thiocyano Oxyfunctionalization of Alkenes Using N-Thiocyano-dibenzenesulfonimide, Org. Lett., 2019, 21, 5106 CrossRef CAS PubMed.
- X.-F. Song, A.-H. Ye, Y.-Y. Xie, J.-W. Dong, C. Chen, Y. Zhang and Z.-M. Chen, Lewis-Acid-Mediated Thiocyano Semipinacol Rearrangement of Allylic Alcohols for Construction of α-Quaternary Center β-Thiocyano Carbonyls, Org. Lett., 2019, 21, 9550 CrossRef CAS PubMed.
- Y.-Y. Xie, Z.-M. Chen, H.-Y. Luo, H. Shao, Y.-Q. Tu, X. Bao, R.-F. Cao, S.-Y. Zhang and J.-M. Tian, Lewis Base/Brønsted Acid Co-catalyzed Enantioselective Sulfenylation/Semipinacol Rearrangement of Di- and Trisubstituted Allylic Alcohols, Angew. Chem., Int. Ed., 2019, 58, 12491 CrossRef CAS PubMed.
- S. Liu, X. Zeng and B. Xu, Practical Fluorothiolation and Difluorothiolation of Alkenes Using Pyridine-HF and N-Thiosuccinimides, Org. Chem. Front., 2020, 7, 119 RSC.
- J. Wu, J. Xu and X. Zhao, Selenide-Catalyzed Stereoselective Construction of Tetrasubstituted Trifluoromethylthiolated Alkenes with Alkynes, Chem. – Eur. J., 2016, 22, 15265 CrossRef PubMed.
- Z.-Q. Li, Y. Cao, T. Kang and K. M. Engle, Electrophilic Sulfur Reagent Design Enables Directed syn-Carbosulfenylation of Unactivated Alkene, J. Am. Chem. Soc., 2022, 144, 7189 CrossRef CAS PubMed.
- Z.-Q. Li, W.-J. He, H.-Q. Ni and K. M. Engle, Directed, Nickel-Catalyzed 1,2-Alkylsulfenylation of Alkenyl Carbonyl Compounds, Chem. Sci., 2022, 13, 6567 RSC.
- L. Zhu, X. Meng, L. Xie, Q. Shen, W. Li, L. Zhang and C. Wang, Regioselective 1,2-Carbosulfenylation of Unactivated Alkenes via Directed Nickel Catalysis, Org. Chem. Front., 2022, 9, 3068 RSC.
| This journal is © the Partner Organisations 2023 |