
Strategies for the discovery of potential anticancer agents from plants collected from Southeast Asian tropical rainforests as a case study†
Esperanza J.
Carcache de Blanco
 a,
Ermias Mekuria
Addo
a,
H. Liva
Rakotondraibe
a,
Djaja D.
Soejarto
bc and
A. Douglas
Kinghorn
*a
a,
Ermias Mekuria
Addo
a,
H. Liva
Rakotondraibe
a,
Djaja D.
Soejarto
bc and
A. Douglas
Kinghorn
*a
aDivision of Medicinal Chemistry and Pharmacognosy, College of Pharmacy, The Ohio State University, Columbus, Ohio 43210, USA. E-mail: kinghorn.4@osu.edu
bDepartment of Pharmaceutical Sciences, College of Pharmacy, University of Illinois at Chicago, Chicago, IL 60612, USA
cScience and Education, Field Museum, Chicago, IL 60605, USA
First published on 17th May 2023
Abstract
Covering up to early 2023
The present review summarizes recent accomplishments made as part of a multidisciplinary, multi-institutional anticancer drug discovery project, wherein samples comprising higher plants were collected primarily from Southeast Asia, and also from Central America, and the West Indies. In the introductory paragraphs, a short perspective is provided on the current importance of plants in the discovery of cancer therapeutic agents, and the contributions of other groups working towards this objective are mentioned. For our own investigations, following their collection, tropical plants have been subjected to solvent extraction and biological evaluation for their antitumor potential. Several examples of purified plant lead bioactive compounds were obtained and characterized, and found to exhibit diverse structures, including those of the alkaloid, cardiac glycoside, coumarin, cucurbitacin, cyclobenzofuran (rocaglate), flavonoid, lignan, and terpenoid types. In order to maximize the efficiency of work on drug discovery from tropical plant species, strategies to optimize various research components have been developed, including those for the plant collections and taxonomic identification, in accordance with the requirements of contemporary international treaties and with a focus on species conservation. A major component of this aspect of the work is the development of collaborative research agreements with representatives of the source countries of tropical rainforest plants. The phytochemical aspects have included the preparation of plant extracts for initial screening and the selection of promising extracts for activity-guided fractionation. In an attempt to facilitate this process, a TOCSY-based NMR procedure has been applied for the determination of bioactive rocaglate derivatives in samples of Aglaia species (Meliaceae) collected for the project. Preliminary in vitro and in vivo mechanistic studies carried out by the authors are described for two tropical plant-derived bioactive lead compounds, corchorusoside C and (+)-betulin, including work conducted with a zebrafish (Danio rerio) model. In the concluding remarks, a number of lessons are summarized that our group has learned as a result of working on anticancer drug discovery using tropical plants, which we hope will be of interest to future workers.
 Esperanza J. Carcache de Blanco | Esperanza J. Carcache de Blanco is an Associate Professor, Division of Medicinal Chemistry and Pharmacognosy, College of Pharmacy, The Ohio State University. She holds a Pharmacy Licensure from the National Autonomous University of Nicaragua and was awarded a M.S. degree as a Fulbright Scholar from the College of Pharmacy, University of Illinois at Chicago (UIC), Chicago, Illinois. In 2003, she also gained her Ph.D. degree from this same institution. To date, she has published 95 peer-reviewed research and review articles, covering phytochemical and biological studies related to cancer and diabetes drug discovery as well as botanical dietary supplements. |
 Ermias Mekuria Addo | Ermias Mekuria Addo obtained his Bachelor of Pharmacy (2010) and Master of Science (2015) degrees from the School of Pharmacy, Addis Ababa University, Addis Ababa, Ethiopia. He then joined the graduate program in Pharmaceutical Sciences in 2016 in the Division of Medicinal Chemistry and Pharmacognosy, College of Pharmacy, The Ohio State University, and gained his M.S. and Ph.D. degrees in 2020 and 2021, respectively. He is currently a postdoctoral researcher and is interested in secondary metabolites from higher plants, mosses, lichens and marine organisms with activity against chronic diseases such as cancer. |
 H. Liva Rakotondraibe | Harinantenaina Liva R. Rakotondraibe is an Associate Professor, College of Pharmacy, The Ohio State University. He obtained a Diplome d'Etude Approfondie (DEA) in Organic Chemistry (University of Antananarivo, Madagascar, 1998) and a Ph.D degree in Pharmaceutical Sciences (Hiroshima University, Japan, 2003). In 2009, he moved to the U.S.A. as a Research Scientist with Prof. David Kingston at the Department of Chemistry, Virginia Tech, and worked on the International Cooperative Biodiversity Groups (ICBG/Madagascar) project. In 2013, he was appointed to the faculty of his present Institution. He has published more than 100 peer-reviewed papers on natural products research and related fields. |
 Djaja D. Soejarto | Djaja Doel Soejarto is Professor Emeritus at the Pharmacognosy Institute, College of Pharmacy, University of Illinois at Chicago (UIC). He earned his Ph.D. degree (Biology-Botany; 1969) from Harvard University. He served as Principal Investigator of an NCI contract on Plant Collection (Southeast Asia) with UIC (1986–2004) and was Principal Investigator of the Vietnam-Laos ICBG (1998–2010). He received the Distinguished Economic Botanist Award from the Society for Economic Botany (2011) and the Varro E. Tyler Prize in Botanicals from the American Society of Pharmacognosy (2022). Prof. Soejarto is also an Adjunct Curator (Honorary) in Science and Education, The Field Museum, Chicago. |
 A. Douglas Kinghorn | A. Douglas Kinghorn is Jack L. Beal Chair and Distinguished University Professor, at the College of Pharmacy, The Ohio State University (OSU). He received the 2010 Norman R. Farnsworth Research Achievement Award of the American Society of Pharmacognosy and the 2020 Egon Stahl Award in Gold from GA (Society for Medicinal Plant and Natural Product Research), both for lifetime contributions to natural products research. He is the former Editor-in-Chief (1994–2019) of the Journal of Natural Products and is a Series Editor of the book series “Progress in the Chemistry of Organic Natural Products (Springer Nature) (2008-present). |
1 Introduction
Of all the diseases that affect human health, cancer continues to pose a significant burden both globally and nationally. For example, the International Agency for Research on Cancer (IARC) of the World Health Organization (WHO) has estimated that for 2020 the incidence and death rate from cancer worldwide were, in turn, 19.3 and 10 million persons.1 In both incidence and mortality, Asia now leads, followed by Europe, the Americas, Africa and Oceania.1 In the U.S., the epidemiology established for cancer parallels the global pattern, and, despite a mortality rate decline of 32% from 1991 to 2019, this is still the second leading cause of death, and is only superseded by diseases of the heart and cardiovascular system.2 Furthermore, according to an American Cancer Society estimation, there were close to 2 million new cancer cases and more than 600![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cancer-related deaths in 2022.2 Based on these and other recent trends, Bray et al.3 have predicted that cancer will become the major cause of premature death before the end of this present century in most countries around the world. This is in spite of the availability and enhancement of effective treatment modalities such as targeted therapy, radiotherapy and surgery. Hence, a continued search for alternative cancer chemotherapeutic drugs with better safety margins and improved efficacy is still needed.
000 cancer-related deaths in 2022.2 Based on these and other recent trends, Bray et al.3 have predicted that cancer will become the major cause of premature death before the end of this present century in most countries around the world. This is in spite of the availability and enhancement of effective treatment modalities such as targeted therapy, radiotherapy and surgery. Hence, a continued search for alternative cancer chemotherapeutic drugs with better safety margins and improved efficacy is still needed.
Natural products have been at the forefront in the fight against many diseases and also represent a critical part of the current armamentarium against cancer.4 For example, in their analysis of the sources of approved cancer chemotherapeutic agents introduced into western medicine from 1946 to 2019, Newman and Cragg found more than 38% to be natural products or their derivatives.5 While both terrestrial and marine organisms have yielded useful oncolytic drugs, higher plants have been a main source of anticancer agents.4–6 The first plant-derived anticancer drugs introduced into the clinic were the dimeric indole alkaloids vincristine (1963) and vinblastine (1965), from the Madagascar periwinkle [Catharanthus roseus (L.) G. Don (Apocynaceae)].4–6 These were followed by the introduction of the two semi-synthetic derivatives, etoposide and teniposide, which are analogues of the aryltetralin lignan, podophyllotoxin, first obtained from the rhizomes of Podophyllum peltatum L. (Berberidaceae).4–6
As an excellent example of the continued role of a plant-derived constituent in anticancer drug discovery, the continued development as a lead antitumor compound of the plant-derived monoterpene indole alkaloid camptothecin, since its original discovery in 1966, is notable. Initially, when camptothecin, as obtained from the stem wood of Camptotheca acuminata Decne. (Nyssaceae), was evaluated as a sodium salt, it exhibited a lack of effectiveness, limited water solubility, and evidence of toxicity.7 However, following the discovery of its unusual mechanism of cellular action through the inhibition of topoisomerase I, synthetic studies aimed at optimizing this lead compound were conducted in a successful manner. This led to the clinical utilization of the camptothecin analogues topotecan, irinotecan (CPT-11), and belotecan.4,7 The first two of these compounds were approved initially for cancer chemotherapy by the U.S. FDA in 1993 and 1996, respectively, while belotecan was approved in South Korea in 2004.5–7 Moreover, continued progress on antibody–drug conjugates (ADCs) has led to the further utilization of camptothecin derivatives for therapeutic purposes. ADCs are prodrugs composed of an antibody connected by a suitable linker to another drug, called a “payload” or “warhead”, which is usually a highly cytotoxic small organic molecule.6,8 These drugs are based on the “magic bullet” concept of the 1908 Nobel Laureate Paul Ehrlich for targeted delivery in order to reduce off-target effects.9 Thus, a synthetic derivative of camptothecin, DXd, has been incorporated in the ADC trastuzumab deruxtecan (also known as DS-8201a; Enhertu®),10 which was designated initially with accelerated approval by the FDA in the U.S. in December 2019 against unresectable or metastatic HER2-positive breast cancer,11 based on the satisfactory results of a clinical trial in 184 patients.12 Next, it was then given regular approval for the same condition in May 2022,13 and, subsequently, has also been accorded accelerated approval, alone or in combination, such as for gastric cancer (January 2021)14 and non-small cell lung cancer (August 2022).15 Another derivative of camptothecin, SN-38, an active metabolite of irinotecan, has been conjugated with anti-Trop-2 monoclonal antibody to provide the ADC sacituzumab govitecan (also called IMMU-132; Trodelvy®),16 and was approved for metastatic triple-negative breast cancer in April 2020.17
The U.S. National Cancer Institute (NCI), National Institutes of Health (NIH), greatly facilitated the therapeutic development of the further plant-derived anticancer drug, paclitaxel, a nitrogen-containing diterpenoid purified initially from the bark of Taxus brevifolia Nutt. (Taxaceae).4,6,18 Currently, the Natural Products Branch of NCI maintains a worldwide collection of extracts and fractions derived from various plant, marine organism and microbial sources in its Natural Product Repository.19 Recently, several extracts in this repository have been pre-fractionated, including those from traditional medicinal plants collected in China, in an effort to make them amenable to high-throughput screening (HTS).19,20 Proof-of-concept studies on these pre-fractionated libraries along with additional HPLC-based fractionation have been successful in the dereplication, isolation and characterization of several cytotoxic secondary metabolites against the NCI-60 human tumor cell line panel.19–21
The search for antineoplastic agents from medicinal and other plants remains an active research area in various academic institutions internationally, and recently several potently cytotoxic compounds against various human-derived cancer cell lines have been described. Among these, examples of promising compounds include the taccalononide microtubule stabilizers from Tacca species (Dioscoreaceae),22 cyclotides from Hybanthus enneaspermus,23 withanolides from Physalis acutifolia (Solanaceae),24 brevipolide A from Hyptis brevipes (Lamiaceae) and 6-methoxytricin from Artemisia argyi (Asteraceae),25 and dichapetalin triterpenoids from Dichapetalum longipetalum (Dichapetalaceae).26
It should be pointed out that the main focus of the plant material acquisition described in this review is Southeast Asia, specifically Laos, Vietnam, and Indonesia, which collectively serves as a case study that is generally applicable to tropical countries in other continents.
2 Selected lead bioactive compounds obtained
For some 15 years, our collaborative team has been funded via a multidisciplinary program project (P01) award by the U.S. National Cancer Institute, titled “Discovery of Anticancer Agents of Diverse Natural Origin” (P01CA125066). Research teams at three universities [the Ohio State University (OSU), the University of Illinois at Chicago (UIC), and the University of North Carolina at Greensboro (UNCG)] and a fungus research company (Mycosynthetix, Inc., Hillsborough, NC) are included in the current project team. In addition to focusing on obtaining lead bioactive compounds from tropical plants, as summarized in the present review, compound leads from U.S. lichens and their mycobionts, aquatic and freshwater cyanobacteria, and filamentous fungi also are sought in this project. The organism collection and isolation chemistry aspects of the project are supported by three core components, dealing with, in turn, medicinal chemistry and pharmacokinetics, in vitro and in vivo biological evaluation, and biostatistics and overall administrative aspects. Since 2009, three comprehensive reviews have been published on the program project organization and have summarized the overall technical progress made.27–29 Also, recent reviews on facets of the higher plant-focused portion of the program have appeared.30–32In Fig. 1, eleven selected higher plant-derived lead compounds of interest (1–11) are shown that have been isolated by bioactivity-guided chromatographic fractionation in our program project work from tropical rainforest plants over the last decade. None of these compounds was featured previously in the two most recent comprehensive overview articles on the program project published in 2016 and 2022.28,29 These compounds exhibit a range of chemical structures and different types of biological activity germane to cancer. As wide a range of biological testing as possible on compounds 1–11 was conducted, by making a concerted effort to establish additional research collaborations. Some of the compounds featured had already been characterized structurally at the time of their isolation in our project, but they were found to exhibit biological activity of interest. The compounds in Fig. 1 are described in chronological order of their isolation.
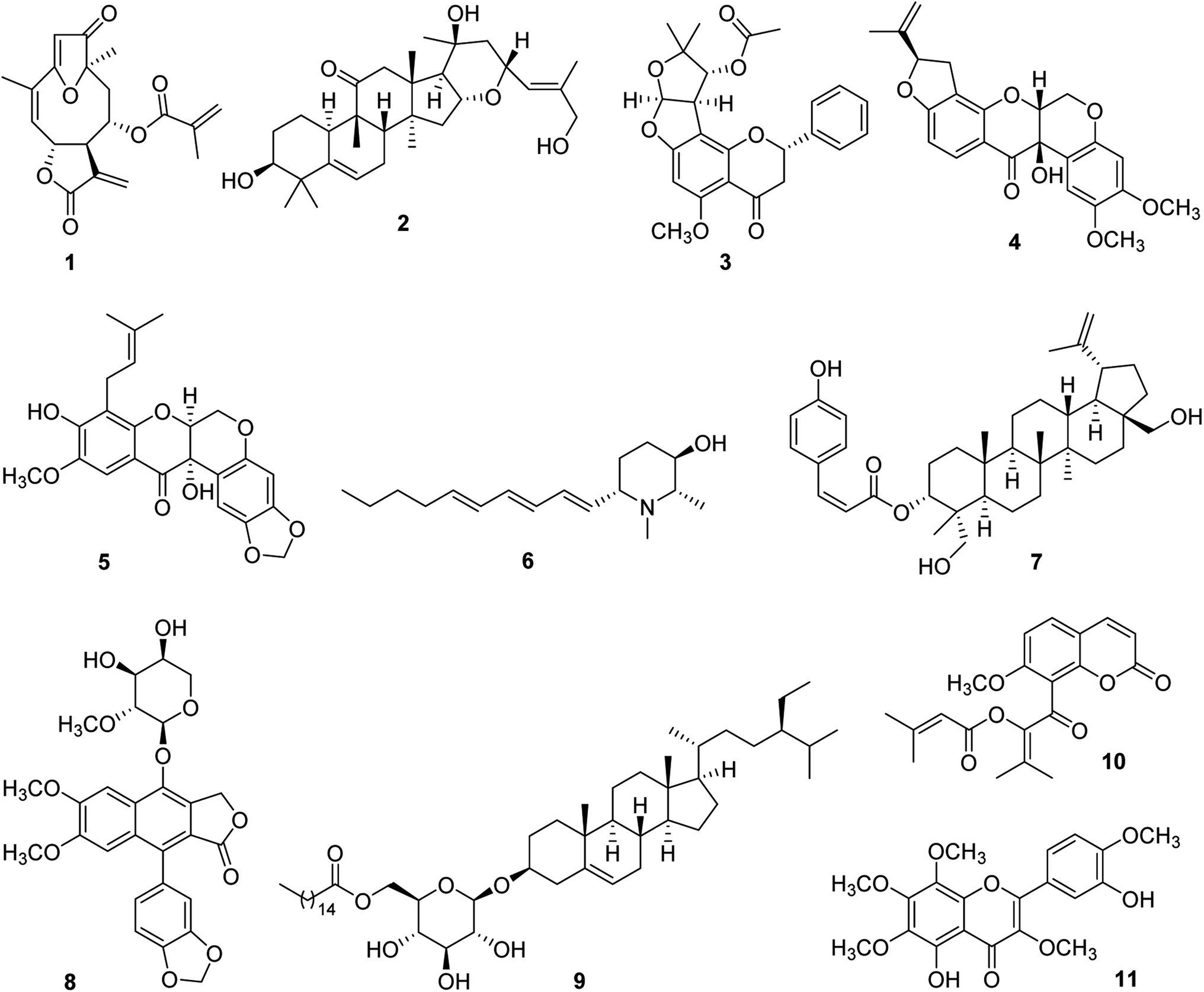 | ||
| Fig. 1 Structures of selected lead compounds isolated from tropical plants (1–11). | ||
Several germacranolide-type sesquiterpene lactones, phenylpropanol coumarates, and methylated flavonoids were purified and characterized from the leaves of Piptocoma rufescens Cass. (Asteraceae), collected in the Dominican Republic. Of these isolated compounds, the previously known sesquiterpene lactone, (−)-15-deoxygoyazensolide (1), was found to be cytotoxic for the HT-29 human cancer cell line (IC50 0.26 μM) and was inhibitory in a NF-κB p65 assay (IC50 3.2 μM).33 Since the activation of the cellular transcription factor nuclear factor kappa B (NF-κB) is associated with the induction of antiapoptotic proteins required by cancer cells, its inhibition by test compounds has been targeted.34 In a follow-up study, the molecular structure of compound 1 was confirmed by X-ray diffraction. Also, it showed inhibitory activity against the MOLM-13 and EOL-1 human acute myeloid leukemia cell lines, having IC50 values of 1.0 and 1.1 μM, respectively. This compound was found to be more potent for these two cell lines than a positive control sesquiterpene lactone, parthenolide.35 Synergistic cytotoxic effects were observed for compound 1, against both MOLM-13 and EOL-1 cells, when mixed with midostaurin,35 which is an approved drug for the treatment of advanced systemic mastocytosis.36
From separate samples of the fruits and stem bark of Elaeocarpus chinensis (Gardner & Champ.) Hook.f. ex Benth. (Elaeocarpaceae), collected in Vietnam, a number of cucurbitacin and 16,23-epoxycucurbitacin derivatives were generated in pure form. Of these, the new 16,23-epoxycucurbitacin, elaeocarpucin C (2), was isolated as a minor constituent (4 mg) from the fruits as a cytotoxic substance. The isolation procedure used was facilitated using a LC-MS dereplication procedure, coupled to a cytotoxicity assay, which suggested the probable presence of three cucurbitacins of known structure.37 When evaluated against the “gatekeeper” HT-29 human colon cancer cell line, compound 2 displayed an IC50 value of 0.41 μM.37 It was decided that this level of potency was sufficient to conduct a follow-up in vivo hollow fiber assay in mice on compound 2. This technique has been used in our research program as a secondary discriminator, in order to decide if lead compounds should be subjected to further in vivo evaluation in a mouse xenograft model,38 and originally was developed at the U.S. National Cancer Institute.39 For elaeocarpucin C (2), three human cancer cell lines were evaluated in the in vivo hollow fiber assay (HT-29 colon, MCF-7 breast, and MDA-MB-435 melanoma) by intraperitoneal (i.p.) injection. This work was conducted in the laboratory of Prof. Steven Swanson, who was then at the University of Illinois at Chicago. A sufficient amount (ca. 25 mg) of compound 2 was generated for testing in this assay using a derivative isolated in this same investigation (16α,23α-epoxy-3β,20β-dihydroxy-10αH,23βH-cucurbit-5,24-dien-11-one), by selectively oxidizing the C-27 allylic methyl group into a primary alcohol.37 However, no growth inhibitory activity was observed for elaeocarpucin C (2), using any of these cell types, when administered i.p. over a dose range of 0.5 to 10 mg kg−1. Accordingly, this compound was not selected for further biological evaluation.37
A further species collected in Vietnam was Indigofera spicata Forssk. (Fabaceae), also known by the common name of “false indigo”. From a chloroform-soluble partition of the combined flowers, fruits, leaves, and twigs of I. spicata were isolated four new flavanones, inclusive of (+)-5-methoxypurpurin (3), in addition to two additional flavanones, three rotenoids, and a chalcone, of which all were of previously known structure.40 The cytotoxicity of the isolated compounds was evaluated against three different cancer cell lines, namely, HT-29 human colon, 697 human acute lymphoblastic leukemia, and Raji human Burkett's lymphoma cells. The three known rotenoids that were isolated were deemed as being cytotoxic (IC50 < 10 μM) for one or more of these three cell lines, including cis-(6aβ,12aβ)-hydroxyrotenone (4), which was highly potent against the HT-29 cell line (IC50 0.1 μM). This compound showed a selectivity of over 1000 for this cancer cell line, when compared with its cytotoxicity for CCD-112CoN normal colon cells. It was selected therefore for evaluation in the murine in vivo hollow fiber assay, and was assessed after i.p. administration in terms of the growth inhibition of HT-29 cells, MCF-7 human breast cancer cells, and MDA-MD-435 human melanoma cells, over the dose range of 5–30 mg kg−1. Unfortunately, compound 4 was determined as toxic at this dose range, although it did show a 20% reduction in cell growth for MDA-MB-435 cells with a 2 mg kg−1 injection.40
In addition, the compounds purified from I. spicata were evaluated for their potential cancer chemopreventive activity, using a standard test system for this purpose, involving induction of the phase II detoxifying enzyme, quinone reductase (QR), using the Hepa 1c1c7 hepatoma cell line.41 Nine of the ten compounds isolated from I. spicata were assessed for their ability to induce quinone reductase, and eight were found to be active, each showing a CD value of <10 μM, namely, the concentration found to double the quinone reductase enzyme activity. However, the most promising activity in this assay system was exhibited by (+)-5-methoxypurpurin (3), which showed both a low CD value (0.2 ± 0.02 μM) and a high IC50 value (the concentration to inhibit 50% of the cell growth of the host cells). Thus, compound 3 gave a “chemopreventive index (CI)” (IC50/CD) of 376.7, and, in having selectivity in terms of inducing the enzyme QR over inhibiting the growth of the host cells, was considered to represent a promising lead as a cancer chemopreventive agent. In contrast, cis-(6aβ,12aβ)-hydroxyrotenone (4) gave a much lower CI value of 0.6 in this test system.40
Similar compounds to those obtained from I. spicata were isolated from another member of the plant family Fabaceae collected in Vietnam, namely, Milletia caerulea (Graham) Baker. In an initial investigation of the fruits of this species, three new rotenoids including caeruleanone C (5), were isolated, along with 11 rotenoids of known structure.42 Of these, caeruleanone C proved to be a potent inhibitor of mitochondrial transmembrane potential (MTP) (IC50 0.07 μM).42 The loss of MTP is regarded as an indication of apoptosis, and hence has been correlated with the anticancer potential of a given test compound.43 In addition, caeruleanone C (5) showed both good activity and relatively low cytotoxicity in the above-mentioned quinone reductase induction assay in Hepa 1c1c7 cells (CD 1.0 μM; IC50 for the host cells, 27.7 μM).42
Additional investigation of the fruits of M. caerulea afforded other examples of rotenoids as well as several isoflavonoids.44 Of these, the previously known rotenoid, (−)-3-hydroxyrotenone (4) [mentioned earlier from I. spicata as “cis-(6aβ,12aβ)-hydroxyrotenone”],40 was of the greatest interest biologically. This compound was the most highly cytotoxic of those tested for HT-29 human colon cells (and again gave an IC50 value of 0.1 μM), and also was inhibitory in a NF-κB p65 inhibition assay (IC50 5.3 μM).44 In a further in vitro bioassay used for the purified constituents of M. caerulea, compound 4 was active in a K-Ras inhibition assay, and exhibited an IC50 value of 3.1 μM.44 K-Ras is the protein product of the mutation gene KRAS, and has become an important target for the discovery of potential anticancer compounds.45
The plant Microcos paniculata L. (Malvaceae) was collected in Vietnam, and separate crude extracts were prepared from the branches, leaves, and stem bark. This led to the isolation of three new piperidine alkaloids, in addition to three other known compounds.46 Microgrewiapine A (6), one of the new compounds obtained, was purified in crystalline form from the stem bark as a minor constituent in only a 7 mg quantity. Compound 6 was the sole derivative isolated among the M. paniculata constituents obtained found to exhibit cytotoxicity against HT-29 human colon cells (IC50 6.8 μM).46 As a result of its lesser activity against CCD-112CoN normal colon cells (IC50 30.4 μM), compound 6 was observed to exhibit some cancer cell selectivity.46 Microgrewiapine A (6) was discerned as having several structural similarities, when compared with a positive control substance code-named KAB-18, a triphenylpiperidine ester and known ligand for neuronal nicotinic acetylcholine receptors (nAChRs).46,47 There is some evidence of the role in nAChRs in the development of cancer and in its progression.48 Therefore, it was decided to submit for testing compound 6 and several of the other alkaloids isolated from M. paniculata, to Prof. Dennis McKay, who was then at Ohio State University. These were evaluated in a functional calcium accumulation assay using HEKtsA201 cells stably expressing the nAChR of either human hα3β4 or hα4β2. At a concentration of 10 μM, compound 6 showed 58.2 ± 9.2% and 74.0 ± 14.2% inhibition for the hα3β4 and hα4β2 nAChR, respectively, and was thus somewhat comparable in its inhibitory potency with certain established nAChR antagonists.46
Partly as a result of its initially described biological activity, microgrewiapine A (6) has attracted subsequent interest from other investigators. This compound was isolated as a minor constituent of the leaves of M. paniculata by Wu et al., in a further phytochemical investigation, and its 2S,3R,6S absolute configuration was determined using a combination of X-ray crystallography and ECD calculations.49 In this same study, eight new 2,6-disubstituted piperidin-3-ol alkaloids were described from this same source plant and the absolute configurations of two further compounds isolated earlier in our own work (microgrewiapine B and microsamine A) were elucidated.46,49 Also, Wu et al. investigated the antiangiogenic activity of the alkaloids purified using human umbilical vein endothelial cells (HUVECs), and weak activity was found for microgrewiapine A (6) at a 20 μM concentration level.49 Two research groups have completed total synthesis procedures for microgrewiapine A (6). Consequently, it was pointed out that an error was made in our initial work in determining the optical rotation of this alkaloid.50–52 Thus, the value originally published for microgrewiapine A {[α]15D +15.4 (c 0.1, MeOH)}46 was corrected after chemical synthesis to {[α]20D −16.0 (c 0.8, MeOH)}.50 The availability of chemical synthesis routes should be useful in providing further quantities of microgrewiapine A (6) for more advanced biological studies than have been conducted to date.
From the combined fruits, leaves, and twigs of Buxus cochinchinensis Pierre ex Gagnep. (Buxaceae), obtained from Vietnam, five new lupane-type triterpene coumaryl esters were characterized and were isolated together with four known compounds.53 One of the new esters was determined structurally as 3-O-(Z)-p-coumaroyl-23-hydroxy-3-epi-betulin (7), and it was found to be a cytotoxic constituent of its plant of origin when evaluated against HT-29 human colon cells, and exhibited an IC50 value of 3.3 μM. This triterpenoid was deemed as inactive in a follow-up NF-κB p65 ELISA assay (IC50 > 20 μM).53 In order to extend knowledge on their overall biological activity, arrangements were made for the purified constituents of B. cochinchinensis to be tested for their potential antimalarial activity by Dr Maria Cassera, then at the Virginia Tech Center for Drug Discovery. Using an in vitro assay on Plasmodium falciparum Dd2 parasite growth, the triterpenoid ester 7 was determined as being an active compound (IC50 1.02 ± 0.09 μM).53
The aerial parts of Phyllanthus songboiensis N.N. Thin (Phyllanthaceae), again collected in Vietnam, were subjected to activity-guided fractionation, leading to the purification of seven compounds including the known arylnaphthalene lignan, (+)-acutissimalignan A (8) and the known phytosterol, (−)-β-sitosterol-3-O-β-D-(6-O-palmitoyl)glucopyranoside (9). When evaluated against the HT-29 human colon cancer cell line, compound 8 was found to be the most active compound of those isolated, and it displayed considerable potency (IC50 19 nM).54 (+)-Acutissimalignan A (8) shows close structural similarities to the clinically used anticancer agent, etoposide, for which the enzyme DNA topoisomerase II (topo II) is a known molecular target. However, no activity was observed in an established assay for compound 8, for its ability to induce topo IIα-mediated cleavage of plasmid DNA, when evaluated by Prof. Jack Yalowich of Ohio State University at a dose of 100 μM.54 Accordingly, it was inferred that (+)-acutissimalignan A (8) has a different cellular mechanism of action than etoposide.54 The very high cytotoxic potency of 8 and its undetermined mode of action, were supportive of the synthetic structural optimization and more advanced biological evaluation of its biologically active lignan glycoside analogue, phyllanthusmin D, from Phyllanthus poilanei Beille.55,56 Also, using an assay to evaluate the stimulation of natural killer (NK) cells, the sterol long-chain ester 9 was determined as being the only active compound obtained from P. songboiensis, when tested at a dose of 10 μM in the presence of interleukin 12 (IL-12).54 Currently, there is considerable interest in discovering new lead compounds capable of stimulating NK cell activity.57
Work-up of the combined leaves, fruits, and stems of a final plant collected in Vietnam, Glycosmis ovoidea Pierre (Rutaceae), led to the purification of nine coumarins, including the previously described compound kimcoungin (10). As part of the study performed, the structure of compound 10 was confirmed by X-ray crystallography.58 In addition, a tenth constituent, 5,3′-dihydroxy-3,6,7,8,4′-pentamethoxyflavone (11), proved to be the only cytotoxic compound from G. ovoidea, as demonstrated by its evaluation against five human cancer cell lines.58 In a preliminary biological investigation, the mode of action of compound 11 was examined in some detail in terms of its ability to inhibit the growth of MCF-7 human breast cancer cells.59 Compounds 10 and 11 were purified from the same chromatographic fraction, and it was found experimentally that when they were co-treated as pure substances, this led to an approximately 100-fold potentiation of the MCF-7 cellular potency exhibited by compound 11 alone. In order to provide a mechanistic basis for this observation, a molecular docking study was conducted, indicating that 5,3′-dihydroxy-3,6,7,8,4′-pentamethoxyflavone (11) interacts with isoforms of the NF-κB complex. Using a confirmatory western blot experiment, kimcuongin (10) was determined to potentiate the effects of compound 11 through both the NF-κB and PARP-1 signal transduction pathways.58
3 Sourcing of tropical plant samples
Our plant-derived anticancer agent drug discovery program has consisted to date of two phases. The first phase (phase I) ran from 1990 to 2006 for the project “Novel Strategies for Plant-derived Anticancer Agents” (U19 CA52956), funded by the U.S. National Cancer Institute, National Institutes of Health (NCI, NIH), through an NCDDG (National Cooperative Natural Products Drug Discovery Groups) cooperative research program project award. During Phase I, plant materials were sourced primarily from Costa Rica, the Dominican Republic, Ecuador, Indonesia, Papua New Guinea, Peru, the Philippines, Thailand, the United States, and Zimbabwe.60,61 By 2005, our research agreements covering plants collected in Southeast Asia (Indonesia, Philippines, Thailand) had terminated. However, from 2005 to 2007, laboratory research continued on plant samples collected from the Dominican Republic, as inherited from phase I. The second phase (phase II) of our plant collection program has run from 2007 onwards with funding through the current program project (P01) grant mechanism, as referred to earlier in this review. The plant materials have been sourced for the P01 project primarily from Vietnam and Laos.28–31During both phases I and II of the anticancer project, the species obtained, constituting both primary screening plant samples and recollected plant samples in large quantities, were sourced through collaborative research agreements (memoranda of agreements; MOA). These were negotiated and consolidated between the University of Illinois at Chicago (UIC) and a collaborating host institution in the country from where the collections were made. Such agreements fulfill the requirements for access and benefit sharing (ABS) as set down in Article 1 of the UN Convention on Biological Diversity (CBD)62 and in Article 1 of the Nagoya Protocol,63 and in compliance with the access law of each source country. The collaborative research agreements (MOA) executed have the following statements and other components: (1) Introduction, (2) The purpose or scope of cooperation, (3) The objectives (areas of cooperation/goals), (4) The responsibilities of UIC, (5) The responsibilities of the partner, (6) The joint responsibilities, (7) IPR (Intellectual Property Rights) and benefit-sharing, (8) The transfer of biological materials, (9) Dispute resolution, (10) Renewal and amendment, (11) The term and termination, (12) Confidentiality and the binding nature of the agreement, and (13) The signature page.
The general description of the contractual contents of these MOA's consists of the following: (1) A research objective, (2) A research plan that defines the performance of each party, (3) Funding and budget, and (4) A record on biological material transfer and research plan.64
The framework of a recent MOA, between the University of Illinois at Chicago and the Institute of Ecology and Biological Resources of the Vietnam Academy of Science and Technology, Hanoi, Vietnam, is provided as ESI.† It must be noted that the processing of the negotiations and the drafting of a new MOA may take many months to accomplish, and several more months are required to review and renegotiate all points stipulated in the MOA to the satisfaction and acceptance of the parties concerned, leading toward its signing. For the renewal of an established MOA with a given source country, a renegotiation must be started several months before the end of the terms of the governing document, taking into consideration the performance and accomplishments of the collaborative research as stipulated in the MOA, and the prospect of research funding to support the continuing joint research.
All plant materials from tropical rainforest plants acquired for our anticancer projects were obtained following good collection practices. During phase I, botanist collaborators and staff botanists were requested not to obtain any species and genera that had previously been collected and studied in the project. Also, they avoided collecting plants in genera of limited interest, based on prior literature reviews on their phytochemistry and biology using the NAPRALERT database.65 In addition, a preference was expressed for the collection of taxa native to the particular geographic locality involved, especially those with ethnomedical information pertinent to cancer (unless the species was listed as being of no further interest). During phase II, the same collection guidance was followed, with one additional criterion added, namely, to “collect taxa that have traditional medicinal use in a broad sense (and not only cancer), based on previously available information from literature surveillance and from previous ethnomedical fieldwork”. Furthermore, during collection work, active consideration was given concerning the conservation status of locally occurring plants, to assure that the collections made of the available plant populations encountered would not endanger their continuing occurrence. Pertinent taxa included in the CITES Appendices66 or in the IUCN Red List67 were reviewed before each collection trip, and practices to help preserve the natural environment were observed.
Each plant sample collected (i.e., primary screening samples; recollected samples) was documented by a set of three or more voucher herbarium specimens as a basis for the taxonomic determination of its family, genus, and species and for future reference. Accurate field notes were compiled on the field characters of the plant, its geographic location, the GPS reading of the site, the collector's name and date of collection, and photographs of every plant collected. Three voucher specimens of each collection were prepared, complete with field data information, and one was deposited at a major herbarium institution of the collaborating host country, with a second set deposited at the herbarium of the Field Museum (John G. Searle Herbarium), Chicago, IL. The third voucher set was prepared in case it was needed for transmission to a specialist botanist, for assistance in taxonomic determination or confirmation of identification. Furthermore, all plant samples collected were fully dried and, for each primary screening and recollected sample, the collection identity, involving both the collection number of a given plant sample and the collection number of its voucher herbarium specimens, was kept and tracked very carefully. For the shipment of plant samples and their voucher herbarium specimens from a host country, where the collection was made, to the U.S., an export permit and a phytosanitary certificate accompanied all shipments. USDA plant import permits were obtained to facilitate the entry of species collected into the United States.68
During phase I of the project, 6128 primary screening plant samples, representing defined plant parts and usually in the range of 300–500 g of dried weight of a given acquisition, were acquired and tested. During phase II, 1199 primary screening samples were acquired from Vietnam (2007–2015), and 651 primary samples were acquired from Laos (2015–2018), with the collection sites shown in the map in Fig. 2. In the event of the activity of a promising species after initial biological testing, following satisfactory arrangements being made and with the required protocols followed, a larger quantity (up to 3–5 kg dry weight) was collected for activity-guided isolation and detailed biological evaluation. Altogether, 258 plant recollections were made in our joint Phase I and Phase II projects.
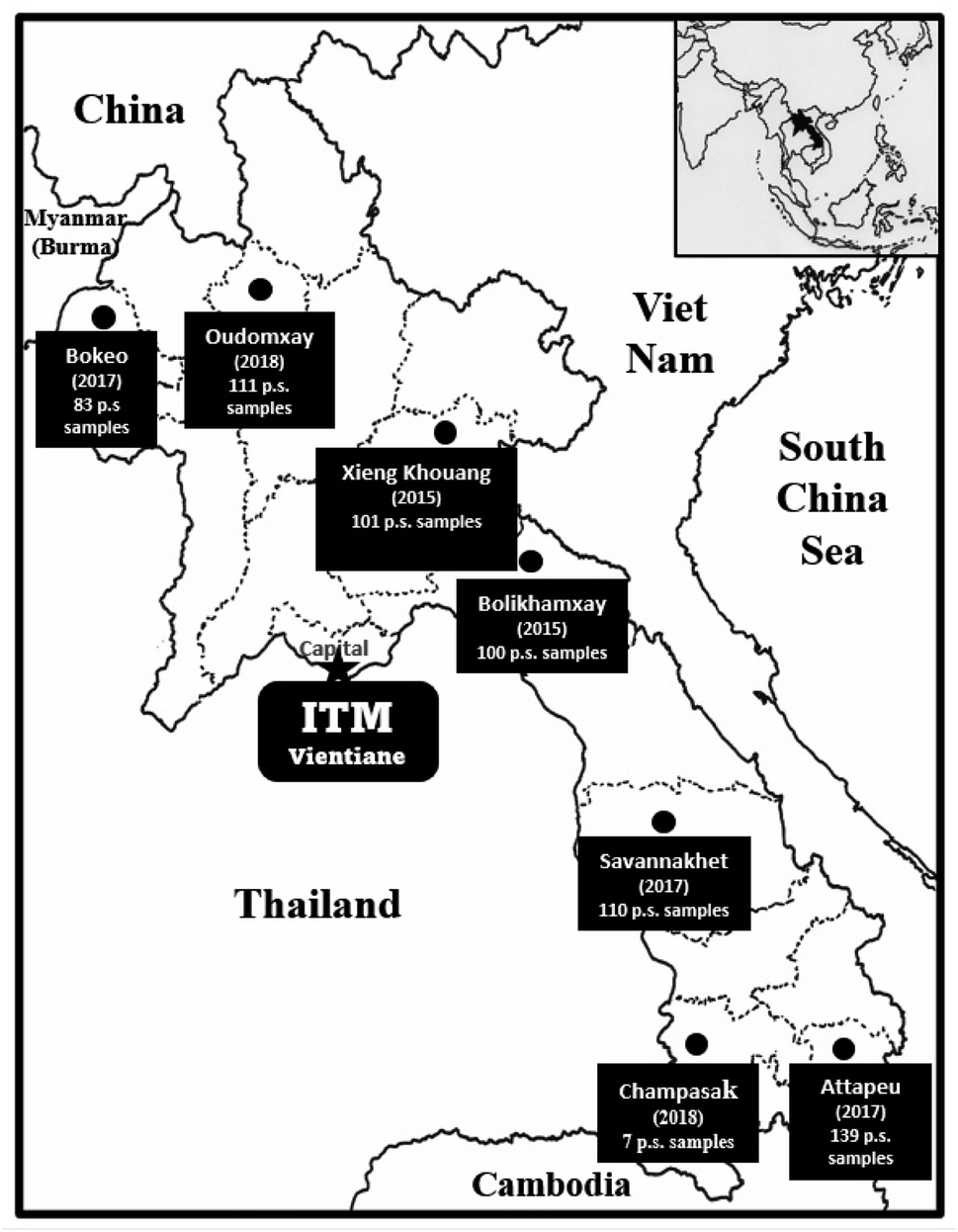 | ||
| Fig. 2 Plant collection expeditions to gather 651 primary screening (p.s.) samples in Laos over the period 2015 to 2018. | ||
4 General phytochemical considerations and initial biological testing of plant extracts and pure compounds
After their collection from tropical rainforest environments, in addition to shipping and initial processing, each plant sample has been received for preliminary laboratory investigation in our anticancer project in the form of a dried milled powder. It is necessary to extract a small quantity (20 g) of each of these plant samples with solvent in an efficient and reproducible manner, prior to preliminary in vitro biological screening. For over 25 years in our anticancer drug discovery project using tropical plant samples, we have used a simple extraction procedure to produce dried chloroform-soluble extracts, from which gallotannin and proanthocyanidin polyphenols are removed by treatment with 1% sodium chloride in water.27,28,60,64,69 This protocol for extraction was developed for our collaborative research project by the late Dr Monroe E. Wall, of Research Triangle Institute, Research Triangle Park, North Carolina.69 Both the rationale for removing the above-mentioned phenolic “interfering substances”, and a consideration of other possible small-scale extraction procedures, have been detailed previously.64For our present program project, initial in vitro testing is performed by Prof. Joanna Burdette at the University of Illinois at Chicago, using up to four main human cancer cell lines, namely, HT-29 colon, MDA-MD-231 breast, MDA-MD-435 melanoma, and OVCAR3 ovarian cells.70,71 Between 10–15% of all collected samples typically are found to be active (IC50 < 20 μg ml−1) against one or more of the present cell lines used. While a similar detailed study was not carried for phase II of our plant collections, a statistical correlation was made for 2628 plant samples and their demonstrated cytotoxicity (ED50 < 20 μg ml−1) against one or more cancer cell lines, for plants collected in phase I from tropical forests in five countries and from southern Florida in the United States. For these plant samples from the Dominican Republic, Ecuador, Indonesia, Peru, Thailand, and the United States, the range of cytotoxic plant extracts was between 15.8-29.6%, with the mean ED50 value being in the low micromolar range for the plants collected from all six of these locations.72
Pure compounds for our project are obtained from crude plant extracts deemed active in one of more initial cytotoxicity bioassays using “activity-guided fractionation”, as described earlier.27,64 Active compound purification studies are normally conducted on recollected plant samples. Efforts are being made to follow newer relevant technical developments, as exemplified by the use of safer, more environmentally acceptable solvents.73 In addition, members of our group have begun to use “molecular networking”, in which MS/MS data information is optimized on the basis of chemical similarity, in order to guide the purification process for biologically active compounds from tropical plants.74,75 Standard spectroscopic, spectrometric, and X-ray crystallographic procedures for compound structure elucidation are employed for the chemical characterization of the pure biologically active plant constituents that are obtained in our program. Recently, we have started to use electronic circular dichroism (ECD) calculations in support of compound structure elucidation, in collaboration with Prof. Xiaolin Cheng, of Ohio State University.71,76
The biological data obtained for a cytotoxic isolated compound may be supplemented by testing with in vitro target-based assays at Ohio State University, such as those involving activation of nuclear factor-κB (NF-κB),28,33,54 and the inhibition of K-ras.44 As mentioned earlier, the in vivo hollow fiber assay is used in our program project research as a secondary discriminator procedure, to prioritize promising in vitro-active isolated compounds for subsequent xenograft experiments using tumored murine models.28,37,38,40,55,70 Pure active compounds are evaluated also in more specialized oncology-related assays through additional collaborators. As an example, several of our plant-derived isolates have been evaluated against childhood sarcoma cell lines at Nationwide Children's Hospital, Columbus, OH, by Prof. Long-Sheng Chang.32,77
5 Dereplication of plant crude extracts and chromatographic fractions using a TOCSY NMR spectroscopic approach
Dereplication is a process to accelerate natural product drug discovery from solvent extracts prepared from organisms, by rapidly identifying already known compounds, to avoid the need to perform subsequent laborious isolation and structure elucidation procedures.74,75,78–81 Several different technical approaches toward dereplication are available. For example, mass spectrometry, typically combined with liquid chromatography, has been used widely in the dereplication of natural products, because of its high sensitivity and resolution as well as the development of efficient ionization techniques that allow the detection of many types of natural product compounds.78–81 Various dereplication approaches using mass spectrometry have been incorporated into our efforts to elucidate new anticancer agents from plants in the past.27,28,37,60,61,64 However, despite the many advantages of using mass spectrometry for natural products dereplication, not all of these compounds are ionizable, and also no single ionization technique can detect all compounds present in an extract. In addition, most natural product isomers cannot be differentiated by mass spectrometry alone.79,81Several strategies involving the use of NMR spectroscopic data to characterize specific features of natural products including their configuration have been used for both compound dereplication and structure identification purposes.82,83 One dereplication approach with NMR spectroscopy used by our group involves the one-dimensional Total Correlation SpectroscopY (TOCSY) experiment that enables the connection between all coupled protons in a given spin system.84 It has been found that a crowded 1H NMR spectrum can be simplified by isolating the individual spin networks constituting the spectrum.85 Several spin systems are contained in a given molecule, and, when taken together, these networks then form a diagnostic fingerprint that may be employed to differentiate and dereplicate known compounds. In the procedure used, the TOCSY NMR data is coupled with information about a given natural product metabolite obtained by mass spectrometry.84
For the present program project work, a combined TOCSY NMR spectroscopic experiment and mass spectrometry protocol has been applied to dereplicate the antiproliferative derivative silvestrol and certain of its structural analogues (Fig. 3), from extracts of Aglaia species (Meliaceae) collected in Vietnam and Laos. Initially, the cyclopenta[b]benzofuran (rocaglate) derivatives (−)-silvestrol (12) and (−)-5′′′-episilvestrol (13) were isolated and fully structurally characterized by our group using X-ray crystallography as the first rocaglate derivatives to contain a dioxanyl ring, and were purified from Aglaia foveolata Pannell collected from Central Kalimantan, Indonesia.86 In preliminary biological testing, silvestrol (12) was found to have low nanomolar inhibitory potency against several different cancer cell lines.86 Subsequently, it showed also activity in a number of in vivo test systems related to B-cell malignancies, when evaluated at the Ohio State University Comprehensive Cancer Center, as previously reviewed.27–29,32,87 In a key cellular mechanism of action study, performed by workers at McGill University in Montreal, silvestrol was found to interact with the RNA helicase eukaryotic initiation factor (eIF) 4A, and thus to inhibit protein translation.88 Since its initial discovery and complete structural characterization nearly 20 years ago, silvestrol has become available commercially through several suppliers as a standard laboratory eIF4A inhibitor for use in biomedical experiments.32 In addition, this compound and its structural analogue, rocaglamide, have become of interest as antiviral agents, due to additional work performed by other investigators.89 Unfortunately, during a preclinical investigation of this compound, through the auspices of the NExT program of the U.S. National Cancer Institute Division of Experimental Therapeutics, silvestrol was found to cause pulmonary toxicity in dogs.77 Accordingly, silvestrol (12) will not be further developed as a potential cancer chemotherapeutic drug.32,77 However, another rocaglate derivative, (−)-didesmethylrocaglamide (14), is still of interest for further investigation in this regard.32,77 The latter compound was isolated in our laboratory as a previously known rocaglate constituent from the combined fruits, leaves, and twigs of Aglaia perviridis Hiern collected in Vietnam,90 and has exhibited potent inhibitory activity against several childhood sarcoma cell lines, in collaborative work conducted at Nationwide Children's Hospital, Columbus, OH.77
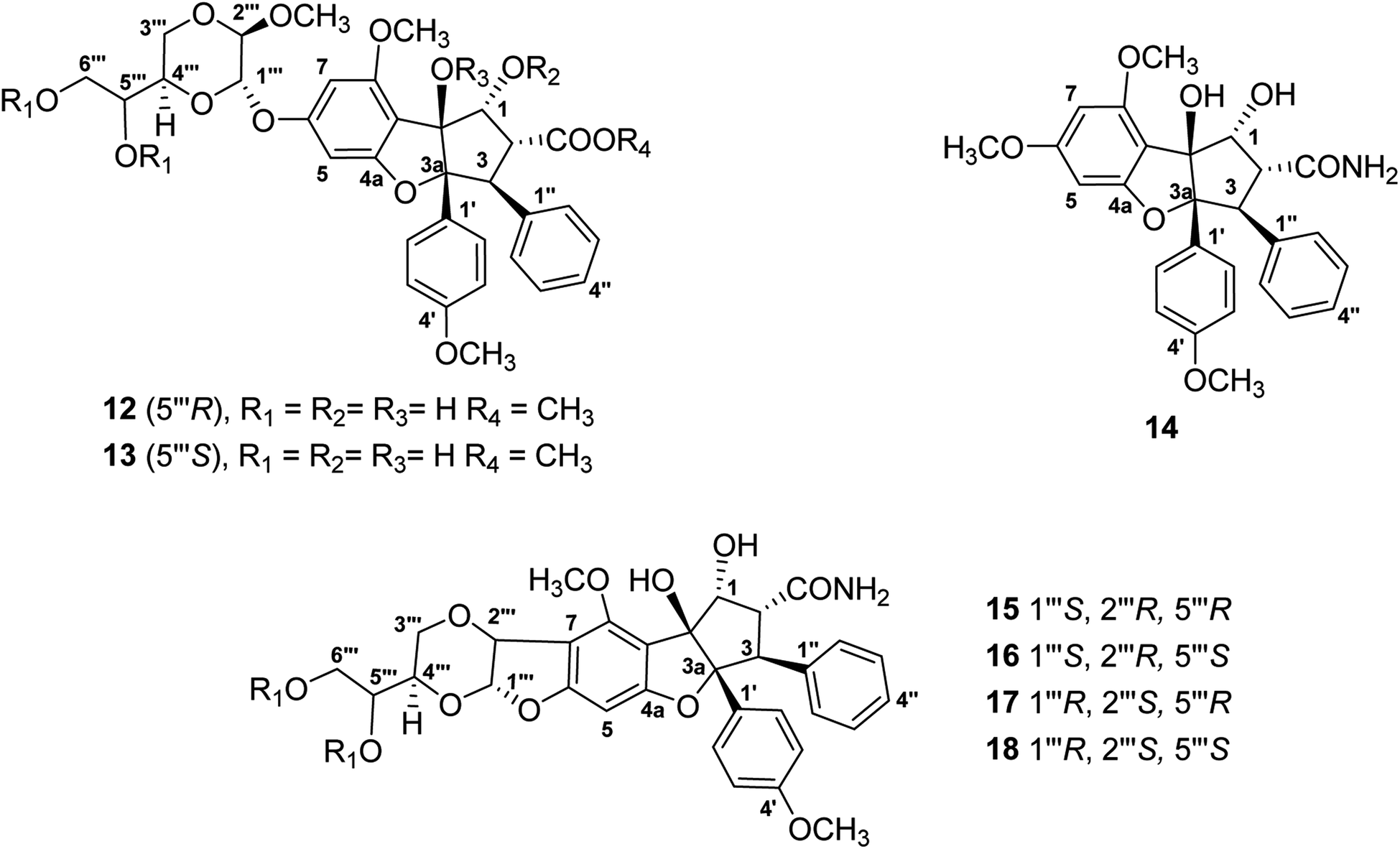 | ||
| Fig. 3 Structures of rocaglate derivatives (12–18) from Aglaia species subjected to dereplication studies. | ||
Thus far, compounds 12–14 have been found to be rare constituents of the genus Aglaia, which contains about 120 tropical species.32 Accordingly, to assist in rapidly identifying this group of compounds and related analogues from additional Aglaia species, construction of a spin network fingerprint (SNF) database has been conducted by our group. The 1H NMR spectrum of silvestrol (12) is characterized by resonances due to signals of monosubstituted (δH 7.05, 3H, m, H-3′′, H-4′′, and H-5′′, and δH 6.85, 2H, m, H-2′′ and H-6′′), para-disubstituted AA′BB′ (δH 7.11, 2H, J = 8.9 Hz, H-2′ and H-6′ and δH 6.68, 2H, J = 8.9 Hz, H-3′ and H-5′ and δH 6.42, 1H, J = 1.5 Hz, H-5) and tetrasubstituted rings, with the latter displaying two meta-coupled protons (δH 6.27, 1H, J = 1.5 Hz, H-7 and δH 6.42, 1H, J = 1.5 Hz, H-5). Methine signals arising from both the dioxanyl ring and furan ring of the rocaglate skeleton in compound 12 are also well resolved.86 A one-dimensional TOCSY NMR study of silvestrol (12) has identified five spin systems constituting the spin network fingerprint (SNF) of the molecule (Fig. 4). In this manner, silvestrol or a structurally closely related previously known compound of interest present in an Aglaia species extract or chromatographic fraction can be subjected to dereplication using 1D-TOCSY NMR by detecting and isolating its characteristic spin network fingerprint. The complete structure of the compound to be dereplicated can be deduced by connecting the identified spin networks using a HMBC experiment and a concomitant mass spectrometric analysis.
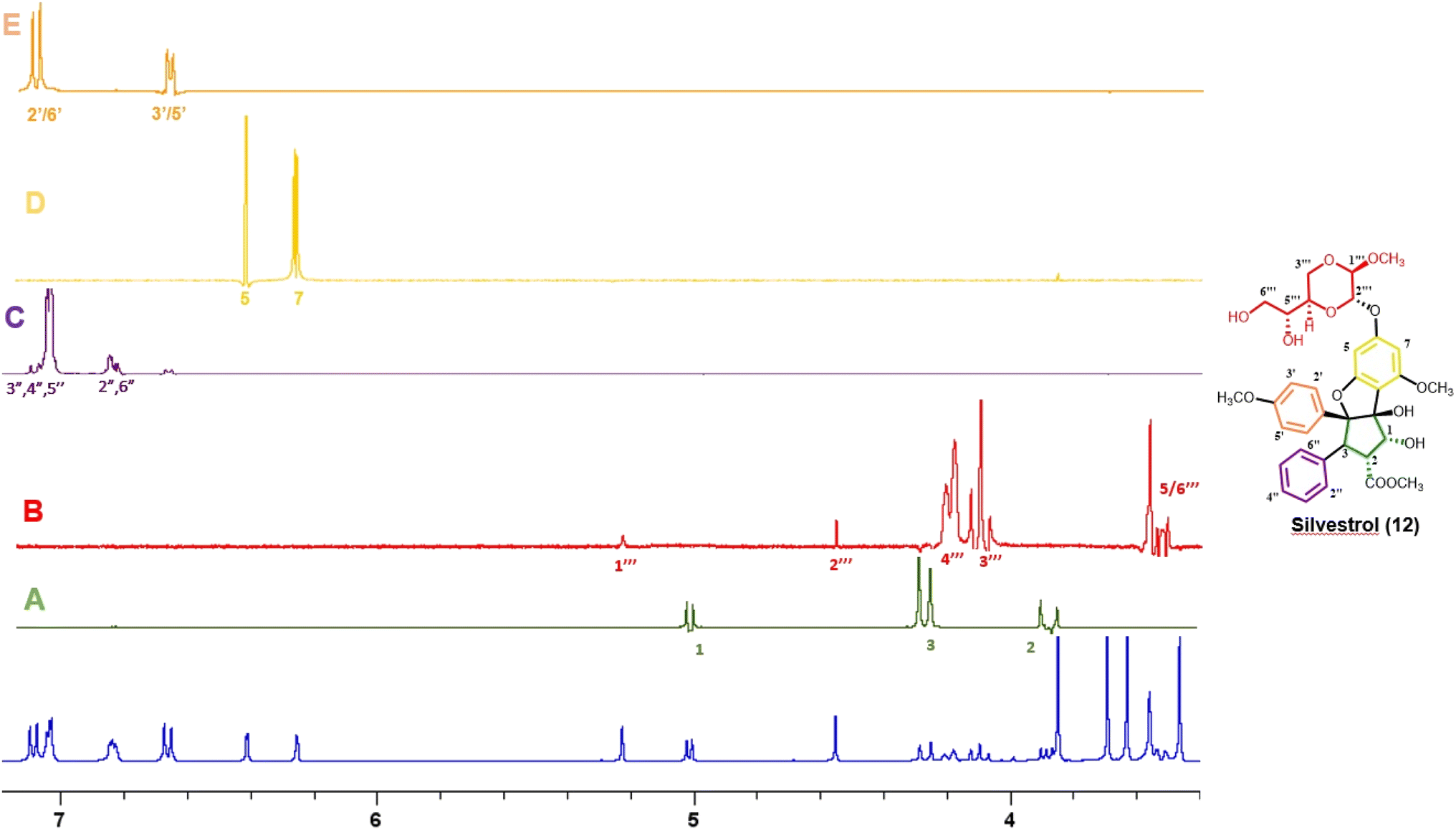 | ||
| Fig. 4 1H NMR spectra of silvestrol (blue) and spin network fingerprint (SNF) identified for this rocaglate derivative (A–E). | ||
The above method has been applied to detect four structurally modified cyclopenta[b]benzofuran derivatives each containing a dioxanyl ring (15–18), which were present in chromatographic fractions prepared from A. perviridis roots collected in Vietnam.76 Of these, compounds 15 and 16 could be characterized by the presence of doublet signals at δH 5.77, 1H, d, J = 4.0 Hz, H-1′′′ and δH 5.03, 1H, d, J = 4.0 Hz, H-2′′′ (for 15), and δH 5.66, 1H, d, J = 3.8 Hz, H-1′′′ and δH 5.05, 1H, d, J = 3.8 Hz, H-2′′′ (for 16), respectively.76 Although the characteristic 1H NMR signals of 15 and 16 were not apparent (due to their only low concentration levels present) in that of the mixture from which they were isolated, the two compounds could be dereplicated, using one-dimensional selective TOCSY irradiation of their characteristic 1H NMR resonances, and generated the desired coupled signals.76 In addition, efforts have been made to develop methodology to screen samples of several Aglaia species from Vietnam and Laos for the presence of didesmethylrocaglamide (14). This involved a preliminary screen using LC/MS analysis,91 followed by a 1D-TOCSY NMR spin network fingerprint analysis.92 The applicability of this combined procedure was validated by the detection of compound 14 in an extract prepared from A. perviridis.90–92
6 Follow up in vitro and in vivo preliminary mechanistic studies on selected bioactive compounds isolated
Corchorusoside C (19) (Fig. 5), a cardenolide with a five-membered lactone ring at C-17 and a disaccharide unit attached to C-3 of the steroidal skeleton, is considered below as the first of two recently isolated compounds for which more detailed biological testing and a preliminary mechanistic evaluation have been carried out at Ohio State University in our anticancer project. This cardiac glycoside was isolated from the stems of Streptocaulon juventas (Lour.) Merr. (Apocynaceae), which were collected in Vietnam for this study.93 Cardenolides have been found in our program project to exhibit in vitro cytotoxicity against several different human cancer cell lines.70,93 Also, some cardenolides have been found to activate or inhibit various cellular signal transduction mechanisms. For example, nerigoside from Nerium oleander induces inhibition of the ERK/GSK3β/β-catenin signaling pathway.94 In turn, oleandrin, also isolated from N. oleander, induces the blocking of nuclear factor kappa-light-chain B (NF-κB).95 Periplocin, biosynthesized by Periploca sepium, enhances DNA double-strand breaks and death-receptor mediated apoptosis.96 (+)-Strebloside, from Streblus asper, which represses p53 and inhibits NF-κB, has been determined to inhibit the Na+/K+ ATPase pump, which is considered the default mechanism of action for this compound class.70,93 Hence, inhibition of Na+/K+-ATPase activity stimulates an increase in intracellular Na+, triggering an increase in intracellular Ca2+, and thus causes a positive inotropic effect in cardiac muscle.97 Due to this mechanism, the cardenolide digoxin became a primary therapeutic agent for heart failure and for atrial tachyarrhythmias during the 20th century. Despite having a narrow therapeutic index, which is a cause for concern among this class of compounds, selected cardenolides still remain of interest for the potential treatment of other diseases, including cancer.98 | ||
| Fig. 5 Structures of two lead compounds (19 and 20) selected for mechanistic studies. | ||
Corchorusoside C (19), when evaluated in our research program, inhibited the proliferation of DU-145 prostate cancer cells with an IC50 value of 80 nM.93 Also, it showed a significantly reduced cytotoxicity against CCD-112CoN colon normal cells, with an IC50 value of 2.3 μM, when compared to HT-29 colon cancer cells (IC50 = 0.12 μM). Thus, it had a calculated selectivity index of 22.5-fold in favor of cancerous colon cells, suggesting a lower potential toxicity for this compound when compared to other cardenolides.93 Preliminary in vitro mechanistic studies revealed that compound 19 induces apoptosis through the NF-κB inflammatory pathway, and also activates the intrinsic apoptotic pathway, as confirmed by significant concentration-dependent decreases in the protein expression levels of NF-κB p65 and p50, IKKα and IKKβ, along with significant increases in expression levels of caspase 3, caspase 7, and PARP-1. NF-κB has been correlated with prostate cancer cellular survival, proliferation, and invasion,99 suggesting that corchoruscoside C has some potential in the treatment for prostate cancer. Also, the poly (ADP-ribose) polymerase (PARP) enzyme is an integral protein in the DNA damage-response mechanism,100 implying that the overexpression of PARP1 induced after treatment of DU-145 prostate cancer cells with compound 19 has a significant effect on DNA damage and mitochondrial outer membrane permeabilization.93 This modulation in protein expression levels of NF-κB and PARP-1 in vitro was confirmed in vivo by preliminary analysis by western blot using a zebrafish (Danio rerio) model.93 Due to the narrow therapeutic index for the cardenolide class of compounds, as mentioned above, a preliminary toxicity profile in zebrafish of compound 19 has been conducted. At a selected dose of 50 μM, this cardenolide was observed to have less developmental toxicity in zebrafish embryos 24 hours post fertilization (hpf) when compared to analogous treatments with digoxin or cycloheximide.93 This suggests not only a favorable toxicity profile of corchorusoside C (19) in vitro and in vivo as well as its potential for drug development by having potentially lower side effects and a wider therapeutic index than digoxin.93
A second isolated compound from our anticancer project has also been subjected to more detailed biological evaluation, namely, the pentacyclic triterpenoid, (+)-betulin [3-lup-20(29)-ene-3β,28-diol] (Fig. 5, 20).101 Betulin was isolated for this investigation from the bark of Cyrilla racemiflora L. (Cyrillaceae), collected in Dominica, and it was provided to our group for evaluation from an available extract repository, through an Administrative Supplement to our program project award.102 Further supplies of compound 20 were obtained from a commercial vendor for our biological work.101 (+)-Betulin, has been reported as exhibiting a broad range of biological effects, including antibacterial, antidiabetic, antifungal, antigastritis, anti-inflammatory, antiparasitic, and antiviral activities.101 The antiproliferative activity of compound 20 has been reported for human breast, central nervous system, cervical, hepatic, lung, ovarian, pancreatic, prostate, and stomach cancer, as well as leukemia cells.103–106 Also, betulin (20) is an inhibitor of the growth of animal cancer cells, such as the canine T-cell (CL-1), B-cell (CLBL-1) lymphoma, and canine osteosarcoma (D-17) cell lines.107 Additionally, (+)-betulin induces apoptosis in MCF-7 breast cancer cells, by regulating protein activation of the mitochondrial-intrinsic pathway.108 In our own work, compound 20 was found to inhibit the NF-κB complex both in vitro using in triple-negative MDA-MB-231 breast cancer cells, and in vivo, using zebrafish. Also, an in silico affinity comparison showed that the interaction with NF-κB p65 (1MY5) resulted in a Ki value of 0.596 μM for (+)-betulin. The binding pocket with NF-κB p65 included the residues Lys-221, Glu-222, Ile-224, Glu-225, Arg-236, Gly-237, Ser-238, Phe-239, and Gln-241.101 The transcription factor NF-κB is implicated in several cellular processes, such as inflammation, immune response, and tumorigenesis.109 NF-κB is found active constitutively in several cancer cells, and it has also been related to cell proliferation, apoptosis evasion, angiogenesis, and metastasis.34,110 Thus, inhibition of NF-κB by compound 20 leads to tumor regression and induces apoptosis, this supporting the NF-κB pathway as being a promising potential therapeutic target in cancer drug discovery.111
NF-κB family members form homo- or heterodimers by combinations of individual members such as p50, p52, p65 (RelA), RelB, and c-Rel. The most common heterodimer in cells is the p65/p50 complex.112 In our investigation, (+)-betulin (20) showed in TNBC MDA-MB-231 cells inhibition of activity and down-regulation of NF-κB p65 and p50,101 confirming a previous report using different non-cancerous cells, human cardiac AC16, where it reduced the active form of NF-κB p65, and also reduced the nucleus concentration and blocked the transcriptional activity.113 In addition, (+)-betulin has been found to inhibit the translocation of NF-κB p65 to the nucleus in non-human RAW 264.7 cells.114 The NF-κB signaling pathway elements may be activated via canonical and non-canonical routes. Ubiquitination of IκB by IKKα, IKKβ and IKKγ is required in the canonical pathway, with the involvement of p65/p50 and c-Rel/p50 dimers.109,110 In the non-canonical route, activation of NF-κB inducing kinase (NIK) triggers phosphorylation of the homodimer IKKα/IKKα, supporting ubiquitination of p100 to become p52.109,110 Hence, the IKK complex is important in NF-κB signaling. In work by our group, (+)-betulin (20) decreased in MDA-MB-231 cells the expression of the important IKKα and β proteins with particular emphasis on the down-regulation of IKKβ, indicating an apoptotic effect of this triterpenoid through the canonical NF-κB pathway.101 (+)-Betulin treatment also produced in MDA-MB-231 cells a reduction of the levels of intercellular adhesion molecule 1 (ICAM-1),101 an up-regulated protein in inflammatory conditions that is involved in adhesion of cancer cells.115 ICAM-1 is directly regulated by NF-κB and plays an important role in cancer metastasis, suggesting (+)-betulin (20) could have a potential in delaying metastatic breast cancer process.116
(+)-Betulin (20) has also been shown to induce the loss of mitochondrial transmembrane potential (MTP) in MDA-MB-231 cells.101 Mitochondrial apoptosis is prompted by mitochondrial outer membrane permeabilization and activation of caspases.117 Changes in MTP are regulated by the BCL-2 family that includes pro- or anti-apoptotic stimulators.118 Bcl-2 as an antiapoptotic protein inhibits the release of cytochrome c (Cyt c) from the mitochondria.118 Hence, the inhibitory activity of compound 20 against antiapoptotic BCL-2 members could make it a good candidate to treat triple-negative breast cancer (TNBC). Additionally, NF-κB has been correlated with the physiology of the mitochondria and up-regulates many antiapoptotic proteins, including the X chromosome-linked IAP (XIAP), and some members of the BCL-2 family such as Bcl-2, Bcl-XL and A1/Bfl-1.119,120 (+)-Betulin has been shown in our recent work to down-regulate Bcl-2 in MDA-MB-231 TNBC cells,101 which confirmed the findings of a previous study in HeLa cells wherein it triggered the release of mitochondrial cytochrome c and translocation to the mitochondrial membrane of Bax and Bak, proapoptotic members of the BCL-2 family.108 (+)-Betulin (20) also enhanced the expression of BAX, NOXA, PUMA, and PERP in MDA-MB-231 cells.121 Another study using HBL-100 cells has indicated that this triterpenoid can induce the down-regulation of Bcl-X protein as well as the up-regulation of BAX, cleaved caspase-3, and PARP. The regulation of these genes has been linked to up-regulation of the transcription factor TAp63 that belongs to the p53 family.122 Hence, inhibition of the NF-κB complex could be triggered by both intrinsic and extrinsic apoptosis that may involve the p53 pathway.
Overall, (+)-betulin (20) has exhibited cellular properties of potential interest for further development in cancer therapy. Studies have revealed that compound 20 induced apoptosis by inhibition of the NF-κB pathway when using MDA-MB-231 triple-negative breast cancer (TNBC) cells. Many NF-κB proteins have been shown to be down-regulated by (+)-betulin. Studies using in silico methods also reported interactions of compound 20 with NF-κB pathway elements. Although it showed some cancer cell line cytotoxicity, (+)-betulin did not show toxicity in a zebrafish model at the concentration used.101 The latter observation is supportive of (+)-betulin (20) through its polyvalent activity in being a feasible lead compound for TNBC.
7 Conclusions
In this short review describing recent progress made in a project oriented towards the discovery of potential anticancer agents of natural origin, a number of biologically active compounds were obtained from selected tropical rainforest higher plants. These plants were collected in a responsible and sustainable manner according to international treaties. The plant specialized metabolites of interest as described represent the cardiac glycoside, coumarin, cucurbitacin, cyclopenta[b]benzofuran (rocaglate), flavanone, flavone, lignan, piperidine alkaloid, rotenoid, sesquiterpene lactone, and triterpenoid structural types. Methodology has been developed for the detection and dereplication of rocaglate derivatives in certain Aglaia species using a 1D-TOCSY NMR procedure. The initial biological testing performed on compounds has involved evaluation against small panels of human cancer cells, and additional evaluations using target-based assays and in vivo testing also have been carried out. Promising compounds have been subjected to preliminary mechanism of action testing, requiring the study of a wider range of biological effects.As a result of working on the phytochemistry and bioactivity evaluation of secondary metabolites of tropical rainforest species for several years, a number of lessons have been learned by members of our group in order to perform this type of research at an optimum level. From the point-of-view of obtaining plant collections from a given source country, it has been found that formulating and finalizing a collaborative research agreement (MOA) will be time-consuming to conduct. Once a mutually signed agreement is available, if possible primary plant samples for initial extraction and biological screening should be obtained from different geographical locations within a given country, and for each of these species several different plant parts should be obtained. It has been our experience that the taxonomic knowledge of tropical rainforest plants found in inaccessible locations may not be fully developed, and so, on occasion it might be necessary to seek input from a specialist taxonomist for the plant genus and family concerned. When a new bioactive compound is isolated from a tropical plant, it is advisable to try to obtain a suitable crystal in order to confirm the structure and absolute configuration by X-ray diffraction. We have attempted to keep current with enhancements made in the use of NMR spectroscopic methodology for natural product structure elucidation, and we have also incorporated ECD spectroscopic calculations for confirmation of the proposed structures for selected new compounds, as mentioned earlier in this review. Furthermore, to enhance the prospects of discovering new bioactive compounds, dereplication methods are valuable in order to potentially save time when well-known common cytotoxic compounds are suspected to be present. Isolated compounds with potential cancer chemotherapeutic effects should be evaluated in as many pertinent in vitro bioassays as are available, including those that assess their toxicity potential, so that a decision can be made in each case as to whether it should be produced in greater quantity by either scale-up isolation or synthesis for in vivo biological testing. It is desirable to learn as much as possible of the mechanisms of cellular activity of lead antitumor compounds. Synthetic chemistry work on a plant lead compound can generate also more potent biologically active analogues. For compounds that might have clinical potential and require preclinical work, the resources of outside collaborators or government organizations will need to be used. Even if a lead compound with cytotoxic activity is not isolated in sufficient quantity for subsequent in vivo testing, it is of interest when it is assessed in additional in vitro assays related to disease states other than cancer, as has been exemplified in this review. Technical progress on this project has been enhanced by the multidisciplinary nature of our senior investigator group, and the frequent interactions between the personnel concerned.
The requirements for a bioactive plant natural product of interest to be selected for formal anticancer agent preclinical development are rather stringent. Hence, despite showing in vitro activity of some interest in each case, it does not seem likely that any of compounds 1–11 (Fig. 1) will be developed further towards potential clinical use, mainly due to their general lack of demonstrated in vivo activity. In contrast, the previously mentioned silvestrol (12) (Fig. 3) was found to possess several favorable chemical and biological attributes before being selected for further evaluation. Thus, in addition to being highly cytotoxic at the low nM level, it was found as the prototype member of a new rocaglate derivative subclass containing a dioxanyl ring, and was amenable to total chemical synthesis.32,86 Silvestrol proved to be inhibitory against several different patient-derived leukemia and lymphoma in vivo models, and acts at the cellular level in an unusual manner as a protein translation inhibitor by targeting eukaryotic factor 4A.32,86 While it is unfortunate that pulmonary toxicity was observed for silvestrol that prevented its further investigation as a potential oncolytic agent, other rocaglate derivatives based on plant lead compounds are still of considerable interest in this regard.86
8 Conflicts of interest
There are no conflicts to declare.9 Acknowledgements
We wish to thank many current and former faculty and staff colleagues, postdoctoral associates, and graduate and undergraduate students who have participated in this multidisciplinary project, and whose names are included in the biography below. We are grateful to our taxonomic collaborators in Central America and the Caribbean and in Southeast Asia for their kind cooperation concerning the plant collections. The laboratory work covered in this review was supported by program project grant P01 CA125066 and the administrative supplements 3P01CA125066-10S1 and 3P01CA125066-12S1, funded by the National Cancer Institute, NIH, Bethesda, MD, USA. Also funding through the supplement 3P01CA125066-05S2, from the National Center for Complementary and Integrative Health, NIH, Bethesda, MD, USA, is gratefully acknowledged.10 References
- H. Sung, J. Ferlay, R. L. Siegel, M. Laversanne, I. Soerjomataram, A. Jemal and F. Bray, Ca-Cancer J. Clin., 2021, 71, 209–249 CrossRef PubMed.
- R. L. Siegel, K. D. Miller, H. E. Fuchs and A. Jemal, Ca-Cancer J. Clin., 2022, 72, 7–33 CrossRef PubMed.
- F. Bray, M. Laversanne, E. Weiderpass and I. Soerjomataram, Cancer, 2021, 127, 3029–3030 CrossRef PubMed.
- G. M. Cragg and D. J. Newman, Biochim. Biophys. Acta, 2013, 1830, 3670–3695 CrossRef CAS PubMed.
- D. J. Newman and G. M. Cragg, J. Nat. Prod., 2020, 83, 770–803 CrossRef CAS PubMed.
- G. Agarwal, P. J. Blanco Carcache, E. Mekuria Addo and A. D. Kinghorn, Biotechnol. Adv., 2020, 38, 107337 CrossRef CAS PubMed.
- J. F. Pizzolato and L. B. Saltz, Lancet, 2003, 361, 2235–2242 CrossRef CAS PubMed.
- Y. Jin, M. A. Schladesch, X. Huang, M. J. Balunas and A. J. Weimer, Pharmacol. Ther., 2022, 229, 107917 CrossRef CAS PubMed.
- K. C. Nicolaou and S. Rigol, Angew. Chem., Int. Ed. Engl., 2019, 58, 11206–11241 CrossRef CAS PubMed.
- Y. Ogitani, T. Aida, K. Hagihara, J. Yamaguchi, C. Ishii, N. Harada, M. Soma, H. Okamoto, M. Oitate, S. Arakawa, T. Hirai, R. Atsumi, T. Nakada, I. Hayakawa, Y. Abe and T. Agatsuma, Clin. Cancer Res., 2016, 22, 5097–5108 CrossRef CAS PubMed.
- U.S. Food and Drug Administration, FDA approves fam-trastuzumab deruxtecan-nxki for unresectable or metastatic HER2-positive breast cancer, https://www.fda.gov/drugs/resources-information-approved-drugs/fda-approves-fam-trastuzumab-deruxtecan-nxki-unresectable-or-metastatic-her2-positive-breast-cancer, accessed October 14, 2022 Search PubMed.
- S. Modi, C. Saura, T. Yamashita, Y. H. Park, S. B. Kim, K. Tamura, F. Andre, H. Iwata, Y. Ito, J. Tsurutani, J. Sohn, N. Denduluri, C. Perrin, K. Aogi, E. Tokunaga, S. A. Im, K. S. Lee, S. A. Hurvitz, J. Cortes, C. Lee, S. Chen, L. Zhang, J. Shahidi, A. Yver and I. Krop, N. Engl. J. Med., 2020, 382, 610–621 CrossRef CAS PubMed.
- U.S. Food and Drug Administration, FDA grants regular approval to fam-trastuzumab deruxtecan-nxki for breast cancer, https://www.fda.gov/drugs/resources-information-approved-drugs/fda-grants-regular-approval-fam-trastuzumab-deruxtecan-nxki-breast-cancer, accessed October 14, 2022 Search PubMed.
- U.S. Food and Drug Administration, FDA approves fam-trastuzumab deruxtecan-nxki for HER2-positive gastric adenocarcinomas, https://www.fda.gov/drugs/resources-information-approved-drugs/fda-approves-fam-trastuzumab-deruxtecan-nxki-her2-positive-gastric-adenocarcinomas, accessed October 14, 2022 Search PubMed.
- U.S. Food and Drug Administration FDA grants accelerated approval to fam-trastuzumab deruxtecan-nxki for HER2-mutant non-small cell lung cancer, https://www.fda.gov/drugs/resources-information-approved-drugs/fda-grants-accelerated-approval-fam-trastuzumab-deruxtecan-nxki-her2-mutant-non-small-cell-lung, accessed October 14, 2022.
- T. M. Cardillo, S. V. Govindan, R. M. Sharkey, P. Trisal, R. Arrojo, D. Liu, E. A. Rossi, C. H. Chang and D. M. Goldenberg, Bioconjugate Chem., 2015, 26, 919–931 CrossRef CAS PubMed.
- U.S. Food and Drug Administration, FDA grants accelerated approval to sacituzumab govitecan-hziy for metastatic triple negative breast cancer, https://www.fda.gov/drugs/resources-information-approved-drugs/fda-grants-accelerated-approval-sacituzumab-govitecan-hziy-metastatic-triple-negative-breast-cancer, accessed October 14, 2022.
- A. Chabner and T. G. Roberts Jr, Nat. Rev. Cancer, 2005, 5, 65–72 CrossRef PubMed.
- C. C. Thornburg, J. R. Britt, J. R. Evans, R. K. Akee, J. A. Whitt, S. K. Trinh, M. J. Harris, J. R. Thompson, T. L. Ewing, S. M. Shipley, P. G. Grothaus, D. J. Newman, J. P. Schneider, T. Grkovic and B. R. O'Keefe, ACS Chem. Biol., 2018, 13, 2484–2497 CrossRef CAS PubMed.
- M. He, T. Grkovic, J. R. Evans, C. C. Thornburg, R. K. Akee, J. R. Thompson, J. A. Whitt, M. J. Harris, J. A. Loyal, J. R. Britt, L. Jia, J. D. White, D. J. Newman and B. R. O'Keefe, Fitoterapia, 2019, 137, 104285 CrossRef CAS PubMed.
- T. Grkovic, R. K. Akee, C. C. Thornburg, S. K. Trinh, J. R. Britt, M. J. Harris, J. R. Evans, U. Kang, S. Ensel, C. J. Henrich, K. R. Gustafson, J. P. Schneider and B. R. O'Keefe, ACS Chem. Biol., 2020, 15, 1104–1114 CrossRef CAS PubMed.
- J. Li, A. L. Risinger and S. L. Mooberry, Bioorg. Med. Chem., 2014, 22, 5091–5096 CrossRef CAS PubMed.
- Q. Du, L. Y. Chan, E. K. Gilding, S. T. Henriques, N. D. Condon, A. S. Ravipati, Q. Kaas, Y.-H. Huang and D. J. Craik, J. Biol. Chem., 2020, 295, 10911–10925 CrossRef CAS PubMed.
- G.-B. Xu, Y.-M. Xu, E. M. K. Wijeratne, F. Ranjbar, M. X. Liu and A. A. L. Gunatilaka, J. Nat. Prod., 2021, 84, 187–194 CrossRef CAS PubMed.
- T. Hell, A. Rutz, L. Dürr, M. Dobrzyński, J. K. Reinhardt, T. Lehner, M. Keller, A. John, M. Gupta, O. Pertz, M. Hamburger, J. L. Wolfender and E. Garo, J. Nat. Prod., 2022, 85, 1540–1554 CrossRef CAS PubMed.
- B. Zhou, X.-N. Gao, M.-M. Zhang, H.-C. Liu and J.-M. Yue, J. Nat. Prod., 2023, 86, 209–221 CrossRef CAS PubMed.
- A. D. Kinghorn, E. J. Carcache-Blanco, H.-B. Chai, J. Orjala, N. R. Farnsworth, D. D. Soejarto, N. H. Oberlies, M. C. Wani, D. J. Kroll, C. J. Pearce, S. M. Swanson, R. A. Kramer, W. C. Rose, C. R. Fairchild, G. D. Vite, S. Emanuel, D. Jarjoura and F. O. Cope, Pure Appl. Chem., 2009, 81, 1051–1063 CrossRef CAS PubMed.
- A. D. Kinghorn, E. J. Carcache de Blanco, D. M. Lucas, H. L. Rakotondraibe, J. Orjala, D. D. Soejarto, N. H. Oberlies, C. J. Pearce, M. C. Wani, B. R. Stockwell, J. E. Burdette, S. M. Swanson, J. R. Fuchs, M. A. Phelps, L.-H. Xu, X. Zhang and Y. Y. Shen, Anticancer Res., 2016, 36, 5623–5638 CrossRef CAS PubMed.
- L. N. Aldrich, J. E. Burdette, E. J. Carcache de Blanco, C. C. Coss, A. S. Eustaquio, J. R. Fuchs, A. D. Kinghorn, A. MacFarlane, B. K. Mize, N. H. Oberlies, J. Orjala, C. J. Pearce, M. A. Phelps, H. L. Rakotondraibe, Y. Ren, D. D. Soejarto, B. R. Stockwell, J. C. Yalowich and X. Zhang, J. Nat. Prod., 2022, 85, 702–719 CrossRef CAS PubMed.
- L. Bueno Pérez, P. C. Still, C. B. Naman, Y. Ren, L. Pan, H.-B. Chai, E. J. Carcache de Blanco, T. N. Ninh, B. V. Thanh, S. M. Swanson, D. D. Soejarto and A. D. Kinghorn, Phytochem. Rev., 2014, 13, 727–739 CrossRef PubMed.
- Y. Ren, E. J. Carcache de Blanco, J. R. Fuchs, D. D. Soejarto, J. E. Burdette, S. M. Swanson and A. D. Kinghorn, J. Nat. Prod., 2019, 82, 657–679 CrossRef CAS PubMed.
- G. Agarwal, L.-S. Chang, D. D. Soejarto and A. D. Kinghorn, Planta Med., 2021, 87, 937–948 CrossRef CAS PubMed.
- Y. Ren, U. Muñoz-Acuña, F. Jiménez, R. García, M. Mejía, H. Chai, J. C. Gallucci, N. R. Farnsworth, D. D. Soejarto, E. J. Carcache de Blanco and A. D. Kinghorn, Tetrahedron, 2012, 68, 2671–2678 CrossRef CAS PubMed.
- A. Garg and B. B. Aggarwal, Leukemia, 2002, 16, 1053–1068 CrossRef CAS PubMed.
- Y. Ren, J. C. Gallucci, X. Li, L. Chen, J. Yu and A. D. Kinghorn, J. Nat. Prod., 2018, 81, 554–561 CrossRef CAS PubMed.
- Y. L. Kasamon, C. W. Ko, S. Subramaniam, L. Ma, Y. Yang, L. Nie, S. Shord, D. Przepiorka, A. T. Farrell, A. E. McKee and R. Pazdur, Oncologist, 2018, 23, 1511–1519 CrossRef CAS PubMed.
- L. Pan, Y. Yong, Y. Deng, D. D. Lantvit, T. N. Ninh, H. Chai, E. J. Carcache de Blanco, D. D. Soejarto, S. M. Swanson and A. D. Kinghorn, J. Nat. Prod., 2012, 75, 444–452 CrossRef CAS PubMed.
- Q. Mi, J. M. Pezzuto, N. R. Farnsworth, M. C. Wani, A. D. Kinghorn and S. M. Swanson, J. Nat. Prod., 2009, 72, 573–580 CrossRef CAS PubMed.
- J. J. Casciari, M. G. Hollingshead, M. C. Alley, J. G. Mayo, L. Malspeis, S. Miyauchi, M. R. Grever and J. N. Weinstein, J. Natl. Cancer Inst., 1994, 86, 1846–1852 CrossRef CAS PubMed.
- L. Bueno Pérez, J. Li, D. D. Lanvit, L. Pan, T. N. Ninh, H.-B. Chai, D. D. Soejarto, S. M. Swanson, D. M. Lucas and A. D. Kinghorn, J. Nat. Prod., 2013, 76, 1498–1504 CrossRef PubMed.
- M. Cuendet, C. P. Oldham, R. C. Moon and J. M. Pezzuto, J. Nat. Prod., 2006, 69, 460–463 CrossRef CAS PubMed.
- L. Bueno Pérez, L. Pan, U. Muñoz Acuña, J. Li, H.-B. Chai, J. C. Galucci, T. N. Ninh, E. J. Carcache de Blanco, D. D. Soejarto and A. D. Kinghorn, Org. Lett., 2014, 16, 1462–1465 CrossRef PubMed.
- S. Salvioli, A. Ardizzoni, C. Franceschi and A. Cossarizza, FEBS Lett., 1997, 411, 77–82 CrossRef CAS PubMed.
- Y. Ren, P. A. Benatrehina, U. Muñoz Acuña, C. Yuan, H.-B. Chai, T. N. Ninh, E. J. Carcache de Blanco, D. D. Soejarto and A. D. Kinghorn, Planta Med., 2016, 82, 1096–1104 CrossRef CAS PubMed.
- A. T. Baines, D. Xu and C. J. Der, Future Med. Chem., 2011, 3, 1787–1808 CrossRef CAS PubMed.
- P. C. Still, B. Yi, T. Gonzalez-Cestari, L. Pan, R. E. Pavlovicz, H.-B. Chai, T. N. Ninh, C. Li, D. D. Soejarto, D. B. McKay and A. D. Kinghorn, J. Nat. Prod., 2013, 76, 243–249 CrossRef CAS PubMed.
- R. E. Pavlovicz, B. J. Henderson, A. B. Bonnell, R. T. Boyd, D. B. McKay and C. Li, PLoS One, 2011, 6, e24949 CrossRef CAS PubMed.
- S. Singh, S. Pillai and S. Chellappan, J. Oncol., 2011, 456743 Search PubMed.
- Z.-L. Wu, W.-Y. Zhang, J.-C. Zhong, Z.-J. Huang, W. Xu, M.-F. Chen, S.-Q. Weng, D.-M. Zhang, C.-T. Che, W.-C. Ye and Y. Wang, J. Nat. Prod., 2022, 85, 375–383 CrossRef CAS PubMed.
- L. Macha and H.-J. Ha, J. Org. Chem., 2019, 84, 94–103 CrossRef CAS PubMed.
- S. C. Davies, A. M. Fletcher, P. M. Roberts, C. E. Taylor and J. E. Thomson, Tetrahedron, 2021, 89, 13208 CrossRef.
- S. C. Davies, A. M. Fletcher, P. M. Roberts, C. E. Taylor and J. E. Thomson, J. Nat. Prod., 2022, 85, 306–312 CrossRef CAS PubMed.
- L. Pan, U. Muñoz Acuña, H. Chai, H.-Y. Park, T. N. Ninh, B. V. Thanh, E. F. Merino, M. B. Cassera, H. L. Rakotondraibe, E. J. Carcache de Blanco, D. D. Soejarto and A. D. Kinghorn, Planta Med., 2015, 81, 1133–1140 CrossRef CAS PubMed.
- Y. Ren, C. Yuan, Y. Deng, R. Kanagasabai, T. N. Ninh, V. T. Tu, H.-B. Chai, D. D. Soejarto, J. R. Fuchs, J. C. Yalowich, J. Yu and A. D. Kinghorn, Phytochemistry, 2015, 111, 132–140 CrossRef CAS PubMed.
- Y. Ren, D. D. Lantvit, Y. Deng, R. Kanagasabai, J. C. Gallucci, T. N. Ninh, H.-B. Chai, D. D. Soejarto, J. R. Fuchs, J. C. Yalowich, J. Yu, S. M. Swanson and A. D. Kinghorn, J. Nat. Prod., 2014, 77, 1494–1504 CrossRef CAS PubMed.
- J. L. Woodard, A. C. Huntsman, P. A. Patel, H.-B. Chai, R. Kanagasabai, S. Karmahapatra, A. N. Young, Y. Ren, J. C. Yalowich, A. D. Kinghorn, J. E. Burdette and J. R. Fuchs, Bioorg. Med. Chem., 2018, 26, 2354–2364 CrossRef CAS PubMed.
- S.-Y. Wu, T. Fu, Y.-Z. Jiang and Z.-M. Shao, Mol. Cancer, 2020, 19, 120 CrossRef CAS PubMed.
- P. J. Blanco Carcache, G. D. Anaya-Eugenio, Y. N. Ninh, C. E. Moore, J. Rivera-Chávez, Y. Ren, D. D. Soejarto and A. D. Kinghorn, Fitoterapia, 2022, 162, 105265 CrossRef CAS PubMed.
- G. D. Anaya-Eugenio, P. J. Blanco-Carcache, T. N. Ninh, Y. Ren, D. D. Soejarto and A. D. Kinghorn, Phytother. Res., 2021, 35, 1634–1645 CrossRef CAS PubMed.
- A. D. Kinghorn, N. R. Farnsworth, D. D. Soejarto, G. A. Cordell, J. M. Pezzuto, G. O. Udeani, M. C. Wani, M. E. Wall, H. A. Navarro, R. A. Kramer, A. T. Menendez, C. R. Fairchild, K. E. Lane, S. Forenza, D. M. Vyas, K. S. Lam and Y.-Z. Shu, Pure Appl. Chem., 1999, 71, 1611–1618 CrossRef CAS.
- A. D. Kinghorn, N. R. Farnsworth, D. D. Soejarto, G. A. Cordell, S. M. Swanson, J. M. Pezzuto, M. C. Wani, M. E. Wall, N. C. Oberlies, D. J. Kroll, R. A. Kramer, W. C. Rose, G. D. Vite, C. R. Fairchild, R. W. Peterson and R. Wild, Pharm. Biol., 2003, 41(Suppl.), 53–67 CrossRef.
- Convention on Biological Diversity, text of the Convention, https://www.cbd.int/convention/articles/?a=cbd-01, accessed October 14, 2022 Search PubMed.
- Convention on Biological Diversity, text of the Nagoya Protocol, https://www.cbd.int/abs/text/articles/?sec=abs-0, accessed October 14, 2022 Search PubMed.
- J. M. Henkin, Y. Ren, D. D. Soejarto and A. D. Kinghorn, in Progress in the Chemistry of Organic Natural Products, ed. A. D. Kinghorn, H. Falk, S. Gibbons, J. Kobayashi, Y. Asakawa and J.-K. Liu, Springer, Cham, Switzerland, 2018, 107, pp. 1–94 Search PubMed.
- W. D. Loub, N. R. Farnsworth, D. D. Soejarto and M. L. Quinn, J. Chem. Inf. Comput. Sci., 1985, 25, 99–103 CrossRef CAS PubMed . Also: Pharmacognosy Institute, University of Illinois at Chicago, The NAPRALERT Database of Natural Products, Ethnomedicine, Pharmacology, and Botany, 2022, https://pharmacognosy.pharmacy.uic.edu/napralert/, accessed October 14, 2022 Search PubMed.
- The CITES Appendices, https://cites.org/eng/app/index.php, accessed October 14, 2022 Search PubMed.
- IUCN Red List of Threatened Species, https://www.iucnredlist.org/, accessed October 14, 2022 Search PubMed.
- USDA APHIS, Plants and Plant Products Permits, https://www.aphis.usda.gov/aphis/ourfocus/planthealth/import-information/permits/plants-and-plant-products-permits, accessed October 14, 2022 Search PubMed.
- M. E. Wall, M. C. Wani, D. M. Brown, F. Fullas, J. B. Oswald, F. F. Josephson, N. M. Thornton, J. M. Pezzuto, C. W. W. Beecher, N. R. Farnsworth, G. A. Cordell and A. D. Kinghorn, Phytomedicine, 1996, 3, 281–285 CrossRef CAS PubMed.
- W.-L. Chen, Y. Ren, J. Ren, C. Erxleben, M. E. Johnson, S. Gentile, A. D. Kinghorn, S. M. Swanson and J. E. Burdette, J. Nat. Prod., 2017, 80, 659–669 CrossRef CAS PubMed.
- Y. Ren, Q. Tan, K. Heath, S. Wu, J. R. Wilson, J. Ren, P. Shriwas, C. Yuan, T. N. Ninh, H.-B. Chai, X. Chen, D. D. Soejarto, M. E. Johnson, X. Cheng, J. B. Burdette and A. D. Kinghorn, Bioorg. Med. Chem., 2020, 28, 115301 CrossRef CAS PubMed.
- M. J. Balunas, W. P. Jones, Y.-W. Chin, Q. Mi, N. R. Farnsworth, D. D. Soejarto, G. A. Cordell, S. M. Swanson, J. M. Pezzuto, H.-B. Chai and A. D. Kinghorn, Chem. Biodiversity, 2006, 3, 897–915 CrossRef CAS PubMed.
- D. Prat, A. Wells, J. Hayler, H. Sneddon, C. R. McElroy, S. Abou-Shehada and P. J. Dunn, Green Chem., 2016, 18, 288–296 RSC.
- J. Y. Yang, L. M. Sanchez, C. M. Rath, X. Liu, P. D. Boudreau, N. Bruns, E. Glukhov, A. Wodtke, R. de Felicio, A. Fenner, W. R. Wong, R. G. Linington, L. Zhang, H. M. Debonsi, W. H. Gerwick and P. C. Dorrestein, J. Nat. Prod., 2013, 76, 1686–1699 CrossRef CAS PubMed.
- R. A. Quinn, L.-F. Nothias, O. Vining, M. Meehan, E. Esquenazi and P. C. Dorrestein, Trends Pharmacol. Sci., 2017, 38, 143–154 CrossRef CAS PubMed.
- G. Agarwal, J. R. Wilson, S. Kurina, G. D. Anaya-Eugenio, T. N. Ninh, J. E. Burdette, E. J. Carcache de Blanco, X. Cheng, D. D. Soejarto, H. L. Rakotondraibe and A. D. Kinghorn, J. Nat. Prod., 2019, 82, 2870–2877 CrossRef CAS PubMed.
- L.-S. Chang, J. L. Oblinger, S. S. Burns, J. Huang, L. W. Anderson, M. G. Hollingshead, R. Shen, L. Pan, G. Agarwal, Y. Ren, R. Roberts, B. R. O'Keefe, A. D. Kinghorn and J. M. Collins, Mol. Cancer Ther., 2020, 19, 731–741 CrossRef CAS PubMed.
- S. P. Gaudêncio and F. Pereira, Nat. Prod. Rep., 2015, 32, 779–810 RSC.
- C. L. Zani and A. R. Carroll, J. Nat. Prod., 2017, 80, 1758–1766 CrossRef CAS PubMed.
- J.-L. Wolfender, J.-M. Nuzillard, J. J. J. van der Hooft, J. H. Renault and S. Bertrand, Anal. Chem., 2019, 91, 704–742 CrossRef CAS PubMed.
- J. M. Egan, J. A. van Santen, D. Y. Liu and R. G. Linington, J. Nat. Prod., 2021, 84, 1044–1055 CrossRef CAS PubMed.
- R. B. Williams, M. O'Neil-Johnson, A. J. Williams, P. Wheeler, R. Pol and A. Moser, Org. Biomol. Chem., 2015, 13, 9957–9962 RSC.
- L. Buedenbender, L. J. Habener, T. Grkovic, D. I. Kurtboke, S. Duffy, V. M. Avery and A. R. Carroll, J. Nat. Prod., 2018, 81, 957–965 CrossRef CAS PubMed.
- C. Diaz-Allen, R. W. Spjut, A. D. Kinghorn and H. L. Rakotondraibe, Trends Org. Chem., 2021, 22, 99–114 Search PubMed.
- P. Charisiadis, C. G. Tsiafoulis, V. Exarchou, A. G. Tzakos and I. P. Gerothanassis, J. Agric. Food Chem., 2012, 60, 4508–4513 CrossRef CAS PubMed.
- B. Y. Hwang, B.-N. Su, H. Chai, Q. Mi, L. B. S. Kardono, J. J. Afriastini, S. Riswan, B. D. Santarsiero, A. D. Mesecar, C. R. Fairchild, R. Wild, G. D. Vite, W. C. Rose, N. R. Farnsworth, G. A. Cordell, J. M. Pezzuto, S. M. Swanson and A. D. Kinghorn, J. Org. Chem., 2004, 69, 3350–3358 CrossRef CAS PubMed; B. Y. Hwang, B.-N. Su, H. Chai, Q. Mi, L. B. S. Kardono, J. J. Afriastini, S. Riswan, B. D. Santarsiero, A. D. Mesecar, C. R. Fairchild, R. Wild, G. D. Vite, W. C. Rose, N. R. Farnsworth, G. A. Cordell, J. M. Pezzuto, S. M. Swanson and A. D. Kinghorn, J. Org. Chem., 2004, 69, 6156 CrossRef.
- L. Pan, J. L. Woodard, D. M. Lucas, J. R. Fuchs and A. D. Kinghorn, Nat. Prod. Rep., 2014, 31, 924–939 RSC.
- R. Cencic, M. Carrier, G. Galicia-Vazquez, M.-E. Bordeleau, R. Sukarieh, A. Bourdeau, B. Brem, J. G. Teodoro, H. Greger, M. L. Tremblay, J. A. Porco Jr and J. Pelletier, PLoS One, 2009, 4, e5223 CrossRef PubMed.
- G. Schulz, C. Victoria, A. Kirschning and E. Steinmann, Nat. Prod. Rep., 2021, 39, 18–23 RSC.
- L. Pan, U. Muñoz Acuña, J. Li, N. Jena, T. N. Ninh, C. M. Pannell, H.-B. Chai, J. R. Fuchs, E. J. Carcache de Blanco, D. D. Soejarto and A. D. Kinghorn, J. Nat. Prod., 2013, 76, 394–404 CrossRef CAS PubMed.
- G. Agarwal, Phytochemical Study of Flavaglines from Aglaia perviridis and Their Rapid Dereplication in Selected Tropical Plant Samples, Ph.D. dissertation, The Ohio State University, 2019.
- C. Diaz-Allen, Developing a 1D-TOCSY NMR-based Dereplication Technique to Facilitate the Isolation of New Cytotoxic Compounds from Natural Products, Ph.D. dissertation, The Ohio State University, 2022.
- G. D. Anaya-Eugenio, E. Mekuria Addo, N. Ezzone, J. M. Henkin, T. N. Ninh, Y. Ren, D. D. Soejarto, A. D. Kinghorn and E. J. Carcache de Blanco, J. Nat. Prod., 2019, 82, 1645–1655 CrossRef CAS PubMed.
- S.-Y. Wen, Y.-Y. Chen, C.-M. Deng, C.-Q. Zhang and M.-M. Jiang, Phytomedicine, 2019, 57, 352–363 CrossRef CAS PubMed.
- S. K. Manna, N. K. Sah, R. A. Newman, A. Cisneros and B. B. Aggarwal, Cancer Res., 2000, 60, 3838–3847 CAS.
- B. Lohberger, S. Wagner, J. Wohlmuther, H. Kaltenegger, N. Stuendl, A. Leithner, B. Rinner, O. Kunert, R. Bauer and N. Kretschmer, Phytomedicine, 2018, 51, 162–170 CrossRef CAS PubMed.
- W. Schoner and G. Scheiner-Bobis, Am. J. Cardiovasc. Drugs, 2007, 7, 173–189 CrossRef CAS PubMed.
- N. R. Z. Schneider, C. Cerella, C. M. O. Simðes and M. Diederich, Molecules, 2017, 22, 1932 CrossRef CAS PubMed.
- J. Staal and R. Beyaert, Cells, 2018, 7, 122–141 CrossRef CAS PubMed.
- E. Nizialek and E. S. Antonarakis, Cancer Manage. Res., 2020, 12, 8105–8114 CrossRef CAS PubMed.
- G. D. Anaya-Eugenio, N. A. Eggers, Y. Ren, J. A. Rivera-Chávez, A. D. Kinghorn and E. J. Carcache de Blanco, Anticancer Res., 2020, 40, 6637–6647 CrossRef CAS PubMed.
- Y. Ren, A. VanSchoiack, H.-B. Chai, M. Goetz and A. D. Kinghorn, J. Nat. Prod., 2015, 78, 2440–2446 CrossRef CAS PubMed.
- S. K. Król, M. Kiełbus, A. Rivero-Müller and A. Stepulak, BioMed Res. Int., 2015, 2015, 584189 Search PubMed.
- A. Boparai, J. Niazi, N. Bajwa and P. Singh, Open Access J. Trans. Med. Res., 2017, 1, 53–59 Search PubMed.
- H. M. So, H. J. Eom, D. Lee, S. Kim, K. S. Kang, I. K. Lee, K. H. Baek, J. Y. Park and K. H. Kim, Arch. Pharmacal Res., 2018, 41, 815–822 CrossRef CAS PubMed.
- A. Hordyjewska, A. Ostapiuk and A. Horecka, J. Pre-Clin. Clin. Res., 2018, 12, 72–75 CrossRef.
- J. Zhao, R. Li, A. Pawlak, M. Henklewska, A. Sysak, L. Wen, J. E. Yi and B. Obmińska-Mrukowicz, In Vivo, 2018, 32, 1081–1088 CrossRef CAS PubMed.
- Y. Li, K. He, Y. Huang, D. Zheng, C. Gao, L. Cui and Y. H. Jin, Mol. Carcinog., 2010, 49, 630–640 CAS.
- A. Oeckinghaus and S. Ghosh, Cold Spring Harbor Perspect. Biol., 2009, 1, a000034 Search PubMed.
- B. B. Aggarwal, Cancer Cell, 2004, 6, 203–208 CrossRef CAS PubMed.
- Y. Xia, S. Shen and I. M. Verma, Cancer Immunol. Res., 2014, 2, 823–830 CrossRef CAS PubMed.
- B. Hoesel and J. A. Schmid, Mol. Cancer, 2013, 12, 1–15 CrossRef PubMed.
- S. Zhang, Q. Zhao, N. Fang and J. Yu, Eur. Rev. Med. Pharmacol. Sci., 2015, 19, 455–460 Search PubMed.
- Q. Wu, H. Li, J. Qiu and H. Feng, Microb. Pathog., 2014, 75, 21–28 CrossRef CAS PubMed.
- H. Kobayashi, K. C. Boelte and P. C. Lin, Curr. Med. Chem., 2007, 14, 377–386 CrossRef CAS PubMed.
- A. Benedicto, I. Romayor and B. Arteta, Oncol. Lett., 2017, 14, 3883–3892 CrossRef PubMed.
- H. Kalkavan and D. R. Green, Cell Death Differ., 2018, 25, 46–55 CrossRef CAS PubMed.
- J. T. Opferman and A. Kothari, Cell Death Differ., 2018, 25, 37–45 CrossRef CAS PubMed.
- B. C. Albensi, Front. Cell Dev. Biol., 2019, 7, 154 CrossRef PubMed.
- M. Paul, K. Kemparaju and K. S. Girish, Biochem. Biophys. Res. Commun., 2017, 493, 1471–1477 CrossRef CAS PubMed.
- R.-J. Hsu, Y.-C. Hsu, S.-P. Chen, C.-L. Fu, J.-C. Yu, F.-W. Chang, Y.-H. Chen, J.-M. Liu, J.-Y. Ho and C.-P. Yu, BMC Complementary Altern. Med., 2015, 15, 1–9 CrossRef CAS PubMed.
- E. Bębenek, M. Kadela-Tomanek, E. Chrobak, J. Wietrzyk, J. Sadowska and S. Boryczka, Med. Chem. Res., 2017, 26, 1–8 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2np00080f |
| This journal is © The Royal Society of Chemistry 2023 |