
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Substitution lability of the perfluorinated Cp* ligand in Rh(I) complexes†
Joshua
Parche
,
Susanne M.
Rupf
 ,
Robin
Sievers
and
Moritz
Malischewski
*
,
Robin
Sievers
and
Moritz
Malischewski
*
Freie Universität Berlin, Institute of Inorganic Chemistry, Fabeckstr. 34-36, 14195 Berlin, Germany. E-mail: moritz.malischewski@fu-berlin.de
First published on 28th March 2023
Abstract
Several cationic rhodium(I) complexes [Rh(COD)L2][C5(CF3)5] have been synthesized through substitution of the weakly bound [C5(CF3)5]− ligand from [Rh(COD)(C5(CF3)5)], further emphasizing its unique reactivity. Besides acetonitrile, pyridine derivatives with varying degrees of fluorination have been employed as ligands in order to investigate the influence of fluorination upon the binding affinity towards the resulting [Rh(COD)]+ fragment and the limit as to which the [C5(CF3)5]− ligand can be displaced. Furthermore, the newly synthesized compounds represent rare examples of rhodium complexes containing fluorinated pyridines as ligands.
Introduction
Fluorinated ligands, in particular F- or CF3-substituted aromatics, have been gaining increasing attention in organometallic chemistry over the last years. The electron withdrawing effect of fluorine may lead to a decreased bonding strength of fluorinated ligands towards metal centers in comparison to their electron rich counterparts. For example, η6-fluorobenzenes coordinated at rhodium(I) centers have been shown to have lower binding affinity with increasing degree of fluorination.1 These comparably weak binding interactions can be of great advantage in catalysis, for systems proceeding via an inner-sphere mechanism that requires vacant coordination sites for substrate binding. Due to the high reactivity and inherent instability of coordinatively unsaturated transition metal complexes, their vacant sites can be masked by labile ligands, which stabilize the respective complex while being easily displaced and not interfering with the catalytic process.1,2 Furthermore, the strong carbon–fluorine bond is relatively inert towards potential side-reactions such as oxidative addition, while also decreasing the probability of coordination through the electron-withdrawing group itself (e.g. cyano groups).3,4 In this sense, Weller et al. have demonstrated the synthesis of catalytically active rhodium(I) complexes containing a η6-monofluorobenzene ligand, prepared by using a simple one-pot procedure starting from [Rh(COD)2][BArF4] (COD = cycloocta-1,5-diene, ArF = C6H3-(3,5-CF3)2).5 The respective bench-stable complexes [Rh(C6H5F)(R2PCH2PR′2)][BArF4] (R,R′ = tBu or Cy) have been proven as efficient precatalysts for various reactions, including intermolecular hydroacylations and dehydrogenative couplings of substituted amine–boranes.5–10 In addition, similar rhodium(I) complexes containing weaker coordinating 1,2-difluorobenzene or 1,2,3-trifluorobenzene ligands allowed the isolation and structural characterization of previously inaccessible amine–borane σ-complexes, via displacement of the weakly coordinating ligands.11,12 Interestingly, the further reduced coordinating ability of 1,2,3,4-tetra- and pentafluorobenzene led to the π-complexation of the weakly coordinating anion (WCA) [BArF4]− in solution, forming zwitterionic complexes [Rh(R2PCH2PR′2)2(η6-(3,5-(CF3)2C6H3)BArF3)] (Fig. 1, top).12 In order to access these complexes containing highly fluorinated benzenes, the use of [Al(ORF)4]−, popularized by the group of Krossing, proved to be crucial to avoid displacement of the respective fluorobenzene.13,14 Complexes containing highly fluorinated benzenes were shown to effectively catalyze the Tishchenko reaction of cyclohexanecarboxaldehyde (C6H11CHO), while leading to shorter reaction times for ligands with higher degrees of fluorination.14 Although examples of η6-coordination towards rhodium(I) centers have been described for fluorobenzenes carrying one to five fluorine substituents, no respective coordination compounds of hexafluorobenzene have been structurally characterized. Instead, a η2-coordination was revealed after irradiation of rhodium alkene- or rhodium dihydride complexes in C6F6, leading to complexes with the general structure [Rh(C5H5)(PPh3)(η2-C6F6)] (Fig. 1, bottom).15–17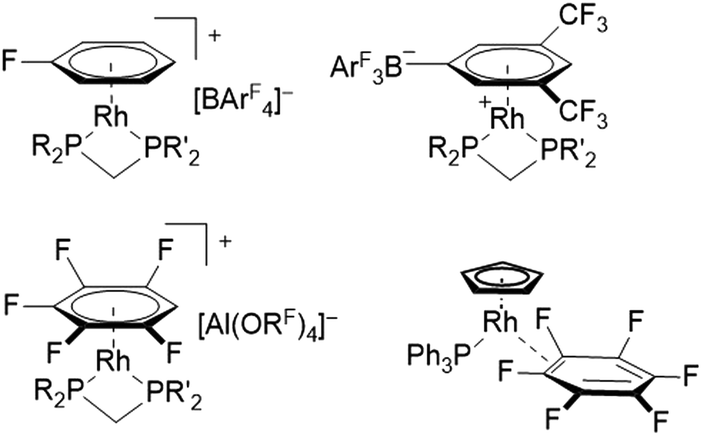 | ||
| Fig. 1 Examples of rhodium(I) complexes with fluorinated aromatic ligands. | ||
Thus, fluorinated benzenes were proved to be useful labile ligands which can mask reactive metal centers in order to facilitate catalytic reactions or to isolate reactive intermediates. In contrast, the use of weakly coordinated anionic aromatic ligands containing fluorine substituents is less explored.18
Undoubtedly the most prominent anionic aromatic ligand is the cyclopentadienyl ligand (Cp), which has gained enormous attention since its discovery in 1951, with known examples for nearly all metals.19,20 Consequently numerous Cp derivatives with different steric and electronic properties have been synthesized and coordinated. Most Cp ligands however are electron rich, thus acting as strong π-donors, e.g. the permethylated Cp (Cp*) being the best-known example. On the contrary, electron poor Cp derivatives are far less investigated, despite their potential to access electrophilic and oxidation resistant complexes with potential applications in catalysis.21–23
In this sense, an effective approach to access electron poor derivatives aims for perfunctionalization through the introduction of fluorine containing substituents. Nevertheless, perfluorinated Cp derivatives remain scarce due to their challenging synthesis and coordination.18 For example, the fully fluorinated Cp, [C5F5]−, has been first synthesized by Seppelt et al. in 1984. However, fast fluoride abstractions and further decomposition pathways prevented any coordination onto a metal center.24 Its first coordination was achieved in 1992 by Hughes et al. through an in-coordination-sphere generation of the [C5F5]− ligand by flash vacuum pyrolysis, forming the mixed ruthenocene [Ru(C5(CH3)5)(C5F5)].25 In 2015, Sünkel et al. further showed the generation of the [C5F5]− ligand by iterative deprotonations and electrophilic fluorinations of ferrocene (Fc), leading to [Fe(C5H5)(C5F5)].26 Interestingly, the corresponding diene HC5F5 proved to be only slightly more acidic (estimated pKa = 13–15) than HC5H5 (pKa = 15.5), due to the strong pronounced +M-effect of fluorine.24 In contrast, the introduction of perfluorinated alkyl groups, such as CF3, exhibit a stronger acidity, due to the absence of any conjugative donor effects. This effect is best visualized by the perfluorinated Cp* analog, [C5(CF3)5]−, which was first reported by Lemal et al. in 1980 with a synthetically challenging approach starting from a perfluorinated dewarthiophene derivative.27 In 1995, Chambers et al. simplified the synthetic access, by reacting hexachlorobuta-1,3-diene with KF, to obtain the non-isolatable K[C5(CF3)5], which was further reacted to form [NEt4][C5(CF3)5].28 In contrast to the HC5H5 and HC5F5, HC5(CF3)5 exhibits an extraordinary acidity (pKa = −2.2).27 The remarkable electron deficiency observed in [C5(CF3)5]− and the therefore expected weak bonding interactions to metal centers account for the difficulties concerning its coordination. Accordingly, more than four decades after its first synthesis the formation of metal complexes containing the [C5(CF3)5]− ligand remained elusive. In 2022 however, Malischewski et al. reported the first coordination of the perfluorinated Cp* analog, through a salt metathesis reaction of [Rh(COD)Cl]2 with AgBF4 in presence of [NEt4][C5(CF3)5], leading to the formation of the 18-electron complex [Rh(COD)(C5(CF3)5)].29 While ordinary electron rich Cp ligands mostly show an irreversible binding to their respective metal centers and are only substituted by arenes or olefins in presence of other reagents, an exceptional reactivity was observed for the [C5(CF3)5]− ligand.30,31 In presence of arenes and olefins, [C5(CF3)5]− was found to undergo an uncommon substitution, converting it into a WCA, further emphasizing the weak bonding interactions to the metal center. The displacement was shown by reacting [Rh(COD)(C5(CF3)5)] with toluene, leading to the quantitative formation of the isolatable cationic arene complex [Rh(COD)(PhMe)][C5(CF3)5]. Furthermore, the substitution of the Cp ligand was found to be fully reversible in weakly coordinating solvents such as CHCl3 or CH2Cl2, allowing it to switch between ligand and WCA depending on its environment (Scheme 1).29
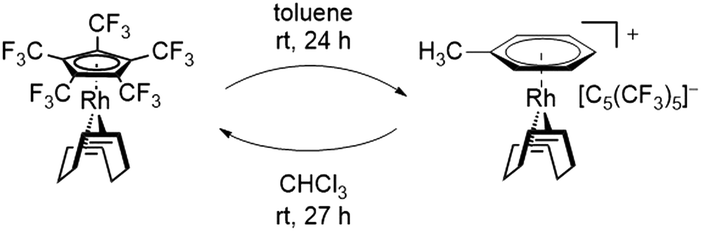 | ||
| Scheme 1 Quantitative substitution of [C5(CF3)5]− in [Rh(COD)(C5(CF3)5)] by toluene and fully reversible reaction in CHCl3. | ||
In this work we further describe the investigation of the substitution lability of [C5(CF3)5]−. In this context, several donor ligands, but especially fluorinated pyridines were applied in order to tune the basicity of the potential ligands and to explore the limits at which the [C5(CF3)5]− ligand can be displaced (Fig. 2).
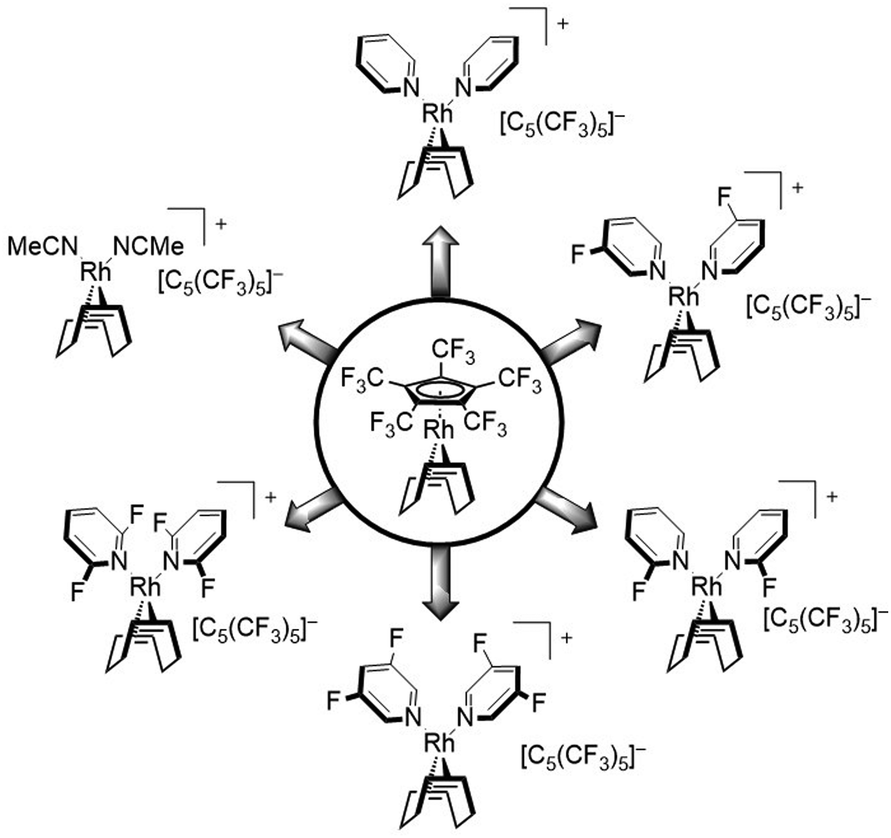 | ||
| Fig. 2 Investigated substitutions of [C5(CF3)5]− in [Rh(COD)(C5(CF3)5)] with different ligands. | ||
Results and discussion
The [C5(CF3)5]− ligand was synthesized as its tetraethylammonium salt according to the procedure by Chambers et al. with addition of 18-crown-6 to increase the solubility of KF, and subsequently coordinated to the rhodium metal center according to the procedure by Malischewski et al.28,29,32 The substitution reactions with the ligands shown in Fig. 2 could either be carried out in the respective ligand as solvent or in n-pentane with a moderate excess of the demanded ligand (except MeCN). When possible, it was preferred to perform the reaction in n-pentane as precipitation of the respective salts significantly reduced the reaction times. The reaction progress could be observed by 19F NMR spectroscopy, through comparison of the coordinated [C5(CF3)5]− and anionic [C5(CF3)5]− signals, in analogy to the substitution by toluene.29 The coordinated [C5(CF3)5]− ligand shows a singlet at δ = −51.1 ppm in CDCl3 (or δ = −51.46 ppm in CD2Cl2), while the uncoordinated [C5(CF3)5]− possesses a downfield shifted signal at δ = −50.18 ppm in CDCl3 (or δ = −50.52 ppm in CD2Cl2). Integration of the respective signals allows to observe the ratio of coordinated to non-coordinated [C5(CF3)5]−. Any unreacted [Rh(COD)(C5(CF3)5)] can be removed from cationic complexes by washing the respective mixture with n-pentane.First, the substitution of the perfluorinated Cp* from [Rh(COD)(C5(CF3)5)] in MeCN was investigated, in order to assess the displacement of [C5(CF3)5]− by two N-donor ligands. It was found that stirring the reaction for 1 h at room temperature, led to the complete substitution of the Cp ligand, turning it into a WCA and quantitatively forming the isolatable cationic 16-electron complex [Rh(COD)(MeCN)2][C5(CF3)5] (Fig. 3). The 19F NMR spectrum revealed a singlet at δ = −51.1 ppm in CDCl3, which is in agreement with the signals of [Rh(COD)(PhMe)][C5(CF3)5].29 In addition, a downfield shifted singlet was observed in the 1H NMR spectrum for the MeCN ligands (δ = 2.20 ppm in CD2Cl2) in comparison to free MeCN (δ = 1.97 ppm in CD2Cl2). Integration of the MeCN signals revealed a twofold coordination, when referencing to the COD signals.
 | ||
| Fig. 3 Substitution of [C5(CF3)5]− in [Rh(COD)(C5(CF3)5)] by MeCN and backreaction in CHCl3. 19F NMR spectrum (377 MHz, CDCl3, rt) measured 30 min after dissolving [Rh(COD)(MeCN)2][C5(CF3)5] in CDCl3, showing the presence of both MeCN substituted complex (left) and [Rh(COD)(C5(CF3)5)] (right). | ||
Furthermore, it was found that the backreaction to [Rh(COD)(C5(CF3)5)] proceeds rapidly, in comparison to the back reaction of [Rh(COD)(PhMe)][C5(CF3)5]. Within 30 min after dissolving [Rh(COD)(MeCN)2][C5(CF3)5] in CDCl3 (15 mg mL−1), both signals of substituted as well as coordinated [C5(CF3)5]− are observed with a ratio of 4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 respectively (Fig. 3). The equilibrium of the backreaction is strongly dependent on the concentration and is reached within 1 h. For a concentration of 2 mg mL−1 of [Rh(COD)(MeCN)2][C5(CF3)5] in CDCl3, an equilibrium of cationic to neutral complex of 1:2, respectively, is observed. Under strongly diluted conditions (0.5 mg mL−1) [Rh(COD)(MeCN)2][C5(CF3)5] can be fully reacted to [Rh(COD)(C5(CF3)5]. Dissolving the substituted complex in an increasingly polar solvent like CD2Cl2 led to a significantly less pronounced backreaction, reaching an equilibrium of 10:1 of cationic to neutral complex at a comparable concentration as in Fig. 3 (15 mg mL−1).
:1 respectively (Fig. 3). The equilibrium of the backreaction is strongly dependent on the concentration and is reached within 1 h. For a concentration of 2 mg mL−1 of [Rh(COD)(MeCN)2][C5(CF3)5] in CDCl3, an equilibrium of cationic to neutral complex of 1:2, respectively, is observed. Under strongly diluted conditions (0.5 mg mL−1) [Rh(COD)(MeCN)2][C5(CF3)5] can be fully reacted to [Rh(COD)(C5(CF3)5]. Dissolving the substituted complex in an increasingly polar solvent like CD2Cl2 led to a significantly less pronounced backreaction, reaching an equilibrium of 10:1 of cationic to neutral complex at a comparable concentration as in Fig. 3 (15 mg mL−1).
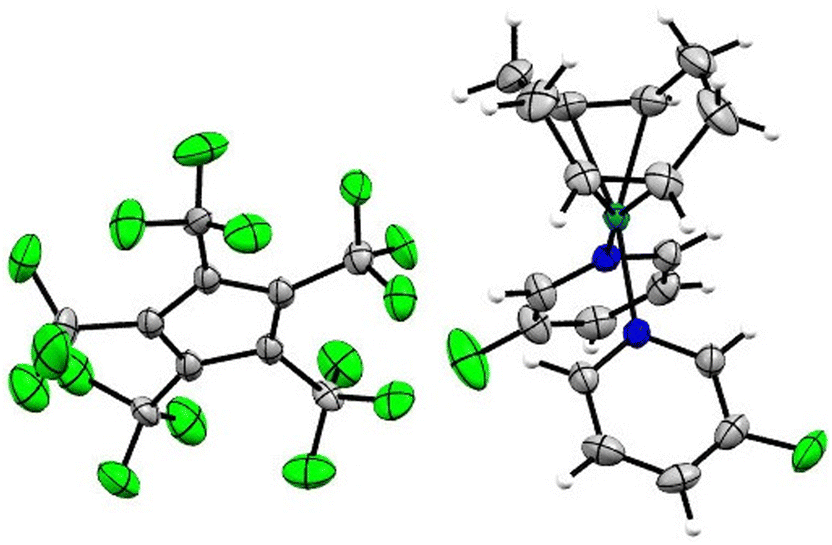
Single crystals of [Rh(COD)(MeCN)2][C5(CF3)5] in the monoclinic P21/n space group were obtained from solvent mixtures of CH2Cl2 and n-pentane by slowly cooling to −75 °C (Fig. 4). The crystal structure revealed an averaged Rh–N bond length of 2.057(7) Å and an average Rh–C distance to the COD ligand of 2.121(7) Å, comparable to the bond lengths of [Rh(COD)(MeCN)2][BF4], with average distances of 2.080(9) Å and 2.125(11) Å respectively.33
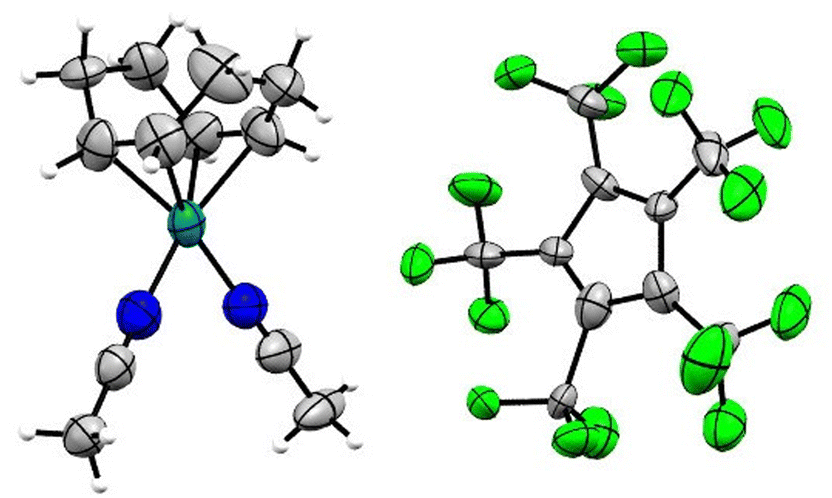 | ||
| Fig. 4 Molecular structure in solid state of [Rh(COD)(MeCN)2][C5(CF3)5]. Disorders are omitted for clarity. Ellipsoids are depicted with 50% probability level. Color code: white-hydrogen; grey-carbon; blue-nitrogen; green-fluorine; light-blue-rhodium. | ||
The substitution of [C5(CF3)5]− was further carried out with pyridine and several fluorine substituted derivatives (Scheme 2). Hereby, pyridine derivatives were employed with further decreasing basicity, depending on the position of the fluorine substituent as well as the degree of fluorination, in order to discover the substitution limit of [C5(CF3)5]−. The reactions could be performed by addition of the respective pyridine into a solution of [Rh(COD)(C5(CF3)5)] in n-pentane, which led to a rapid precipitation of a colorless solid. For all substitutions shown in Scheme 2 (except 2,6-difluoropyridine), 1H NMR studies revealed a twofold coordination of the respective pyridine ligand, similar to the substitution with MeCN. However, no noteworthy shift of the pyridine signals was observed. 19F NMR spectra clearly confirmed the conversion of [C5(CF3)5]− into a WCA and furthermore showed a downfield shifted signal for the respective fluorine substituent(s), e.g. δ = −68.3 ppm for uncoordinated 2-fluoropyridine vs. δ = −61.2 ppm for 2-fluoropyridine coordinated complex [Rh(COD)(2-C5H4FN)2][C5(CF3)5], in CD2Cl2.
 | ||
| Scheme 2 Substitution of [C5(CF3)5]− in [Rh(COD)(C5(CF3)5)] by different (fluorinated) pyridines with their corresponding proton affinities (B3LYP-D3/def2tzvp). The substitution by 2,6-difluoropyridine was carried out overnight. | ||
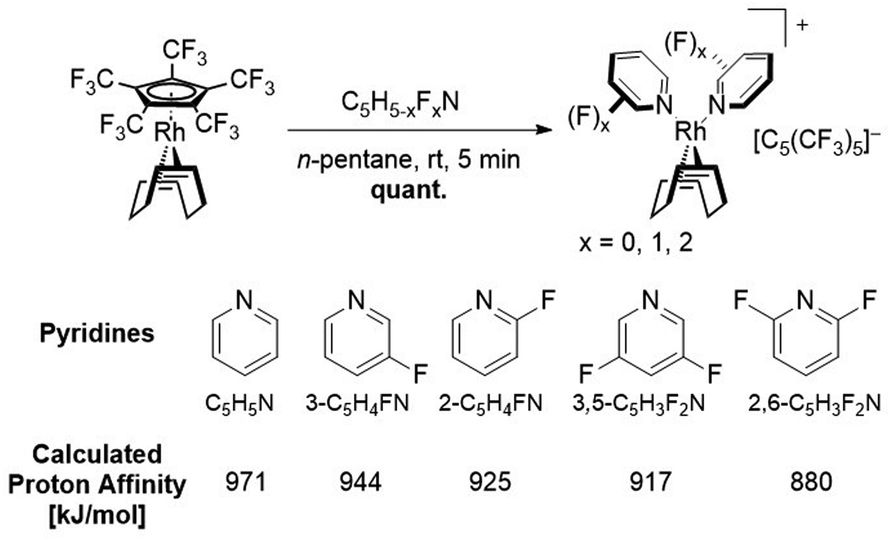
Single crystals of [Rh(COD)(3-C5H4FN)2][C5(CF3)5] were obtained from solvent mixtures of CH2Cl2 and n-pentane by slowly cooling to −75 °C. The compound crystallizes in the triclinic space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) . The asymmetric unit contains two [Rh(COD)(3-C5H4FN)2][C5(CF3)5] fragments, further confirming the twofold coordination of the pyridines (Fig. 5). Additionally, the solid-state structure shows an average Rh–N bond length of 2.111(3) Å and an average of 2.133(5) Å for the Rh–C distance to the COD ligand. A database survey in the Cambridge Crystallographic Database (CSD) revealed no results for rhodium complexes with fluorinated pyridine ligands, thus [Rh(COD)(3-C5H4FN)2][C5(CF3)5] represents a rare example thereof.
. The asymmetric unit contains two [Rh(COD)(3-C5H4FN)2][C5(CF3)5] fragments, further confirming the twofold coordination of the pyridines (Fig. 5). Additionally, the solid-state structure shows an average Rh–N bond length of 2.111(3) Å and an average of 2.133(5) Å for the Rh–C distance to the COD ligand. A database survey in the Cambridge Crystallographic Database (CSD) revealed no results for rhodium complexes with fluorinated pyridine ligands, thus [Rh(COD)(3-C5H4FN)2][C5(CF3)5] represents a rare example thereof.
 | ||
| Fig. 5 Molecular structure in solid state of [Rh(COD)(3-C5H4FN)2][C5(CF3)5]. Disorders are omitted for clarity. Ellipsoids are depicted with 50% probability level. Color code: white-hydrogen; grey-carbon; blue-nitrogen; green-fluorine; light-blue-rhodium. | ||
Surprisingly in contrast to the MeCN and toluene substitutions, no backreaction was observed for the pyridine substituted complexes in solution with CD2Cl2, except for the 2,6-difluoropyridine substituted complex. When dissolving [Rh(COD)(C5H5N)2][C5(CF3)5] in CHCl3, no backreaction was observed either, despite prolonged reaction times (up to 4 d). Dissolving [Rh(COD)(C5H5N)2][C5(CF3)5] in coordinating solvents such as MeCN did not lead to a substitution of the pyridine ligands, which was confirmed by 1H NMR. The backreaction of [Rh(COD)(3-C5H4FN)2][C5(CF3)5] to [Rh(COD)(C5(CF3)5)] proved to be minimal in CHCl3, despite low concentrations and prolonged reaction times (Fig. 6). On the other hand, substitution of complexes [Rh(COD)(2-C5H4FN)2][C5(CF3)5] and [Rh(COD)(3,5-C5H3F2N)2][C5(CF3)5] showed to be reversible in CDCl3, so they could be fully converted back into [Rh(COD)(C5(CF3)5)].
 | ||
| Fig. 6 19F NMR spectrum (377 MHz, CDCl3, rt) of [Rh(COD)(3-C5H4FN)2][C5(CF3)5] after 3 d in CDCl3 at rt. | ||
In strong contrast, the substitution with 2,6-difluoropyridine behaved differently in comparison to the other conducted pyridine substitutions. Performing the reaction in n-pentane did not lead to a fast precipitation of a solid, instead a yellow emulsion was formed after stirring the reaction overnight, discoloring the yellow n-pentane solution. After removing the remaining solvent the residue was washed with n-pentane, revealing an insoluble solid, strongly indicating the formation of a cationic complex under the given conditions. While 1H NMR spectra revealed the presence of two equivalents of 2,6-difluoropyridine when referenced to the COD signals, no signals of substituted [C5(CF3)5]− could be observed in the 19F NMR spectrum in either CD2Cl2 or CDCl3, indicating a rapid backreaction to [Rh(COD)(C5(CF3)5)] (see ESI, Fig. 20 and 21†). Elemental analysis studies however strongly indicate that the substitution with 2,6-difluoropyridine did proceed, as well as IR spectra of the newly formed species, which show new bands with strong similarities compared to IR spectra of the other (fluoro)pyridine substituted complexes (see Experimental section). It is therefore likely, that 2,6-difluoropyridine is able to substitute [C5(CF3)5]−, however the resulting complex [Rh(COD)(2,6-C5H3F2N)2][C5(CF3)5] is rather unstable, due to the backreaction to [Rh(COD)(C5(CF3)5)] in solution.
Conclusion
In conclusion, substitution of the [C5(CF3)5]− ligand of [Rh(COD)(C5(CF3)5)] was investigated with different N-donor ligands, emphasizing its extraordinary electron deficiency and the weak bonding interactions towards the metal center. Hereby the displacement of the [C5(CF3)5]− ligand was first investigated with MeCN, leading to the formation of isolatable 16-electron complex [Rh(COD)(MeCN)2][C5(CF3)5]. Different pyridines with decreasing basicities were subsequently employed, to explore the limits of the substitution for the [C5(CF3)5]− ligand. While all pyridines were shown to quantitatively substitute the perfluorinated Cp* ligand, the stability of the resulting cationic complexes was found to be strongly dependent on the basicity of the competing ligand. Cationic complexes containing relatively basic ligands, such as unsubstituted pyridine, were found to be stable in weakly coordinating solvents (e.g., CHCl3 or CH2Cl2), whereas for pyridines with weaker basicities, e.g. 2,6-difluoropyridine, the substitution proved to be fully reversible in solution, favouring the reformation of [Rh(COD)(C5(CF3)5)]. In general, the ionic products are less stable in CHCl3 than in CH2Cl2. Furthermore, newly synthesized complexes [Rh(COD)(C5H5−xFxN)2][C5(CF3)5] represent rare examples of rhodium(I) complexes containing (fluorinated) pyridine ligands.Experimental
General procedure
The substitutions with different pyridine derivatives were carried out by dissolving [Rh(COD)(C5(CF3)5)] (15.0 mg, 24.0 μmol, 1.0 eq.) with anhydrous n-pentane (1 mL) in a 10 mL Schlenk tube, followed by addition of the respective pyridine (0.2 mL, excess). The solution was then stirred at rt for 5 min, after which a colorless precipitate formed. The solvent was removed under high vacuum and the residue was washed with n-pentane (2 × 1 mL). The remaining solvent was removed under high vacuum to afford the respective cationic complex.Synthetic procedures
1 H NMR (600 MHz, CD2Cl2) δ [ppm]: 4.43 (s, 4H), 2.45 (m, 4H), 2.20 (s, 6H), 1.94 (q, 3J = 8.1 Hz, 4H). 13C{1H} NMR (151 MHz, CD2Cl2) δ [ppm]: 125.1 (s), 123.3 (s), 122.2 (s), 85.8 (d, 1JRh = 12.5 Hz), 30.9 (s). 19F NMR (377 MHz, CD2Cl2) δ [ppm]: −50.5 (s, 15F). FT-IR (ATR) ν [cm−1]: 2963 (w), 2322 (w), 2293 (w), 1648 (w), 1497 (m), 1205 (s), 1108 (s), 972 (m), 875 (w), 802 (m), 633 (m). HRMS (ESI-TOF, positive) m/z for [C10H15NRh]+ calculated: 252.0260; measured: 252.0260. HRMS (ESI-TOF, negative) m/z for [C10F15]− calculated: 404.9766; measured: 404.9486. EA (C22H18F15N2Rh) calculated: C: 37.84%, H: 2.60%, N: 4.01%; measured: C: 37.86%, H: 2.60%, N: 4.02%.
1 H NMR (600 MHz, CD2Cl2) δ [ppm]: 8.62–8.56 (m, 4H), 7.74 (tt, 3J = 7.7 Hz, 4J = 1.6 Hz, 2H), 7.37–7.31 (m, 4H), 4.09 (s, 4H), 2.67–2.57 (m, 4H), 2.04 (q, 3J = 7.9 Hz, 4H). 13C{1H} NMR (151 MHz, CD2Cl2) δ [ppm]: 149.9 (s), 138.9 (s), 126.2 (s), 124.7 (s), 122.9 (s), 85.7 (d, 1JRh = 12.1 Hz), 30.5 (s). 19F NMR (565 MHz, CD2Cl2) δ [ppm]: −50.5 (s, 15F). FT-IR (ATR) ν [cm−1]: 3021 (w), 2965 (w), 2925 (w), 2894 (w), 2877 (w), 2844 (w), 2360 (w), 1600 (w), 1496 (m), 1444 (m), 1296 (w), 1201 (vs), 1111 (vs), 997 (m), 971 (m), 874 (w), 836 (w), 802 (w), 760 (s), 698 (s), 632 (s). HRMS (ESI-TOF, negative) m/z for [C10F15]− calculated: 404.9766; measured: 404.9686. EA (C28H22F15N2Rh) calculated: C: 43.43%, H: 2.86%, N: 3.62%; measured: C: 44.60%, H: 3.13%, N: 3.57%.
1 H NMR (600 MHz, CD2Cl2) δ [ppm]: 8.54 (t, 3J = 2.5 Hz, 2H), 8.46 (dd, 3J = 5.4 Hz, 2H), 7.57–7.51 (m, 2H), 7.39 (dt, 3J = 8.6, 3J = 5.3 Hz, 2H), 4.12 (s, 4H), 2.68–2.59 (m, 4H), 2.06 (q, 3J = 7.7 Hz, 4H). 13C{1H} NMR (151 MHz, CD2Cl2) δ [ppm]: 161.2 (s), 159.5 (s), 146.7 (d, J = 4.4 Hz), 139.3 (d, J = 29.6 Hz), 127.7 (d, J = 6.2 Hz), 126.8 (d, J = 17.5 Hz), 86.9 (d, 1JRh = 12.0 Hz), 30.9 (s). 19F NMR (565 MHz, CD2Cl2) δ [ppm]: −50.5 (s, 15F), −119.4 (s, 2F). FT-IR (ATR) ν [cm−1]: 3011 (w), 2957 (w), 2894 (w), 2847 (w), 1586 (w), 1481 (m), 1435 (m), 1258 (m), 1208 (vs), 1117 (vs), 976 (w), 845 (m), 799 (s), 696 (s), 633 (s), 537 (m). HRMS (ESI-TOF, negative) m/z for [C10F15]− calculated: 404.9766; measured: 404.9486. EA (C28H20F17N2Rh) calculated: C: 41.50%, H: 2.49%, N: 3.46%; measured: C: 41.88%, H: 2.71%, N: 3.59%.
1 H NMR (600 MHz, CD2Cl2) δ [ppm]: 8.49 (s, 2H), 7.85 (q, 3J = 7.6 Hz, 2H), 7.29 (s, 2H), 7.06–6.98 (d, 3J = 8.3 Hz, 2H), 4.19 (s, 4H), 2.69–2.60 (m, 4H), 2.02 (q, 3J = 7.8 Hz, 4H). 13C{1H} NMR (151 MHz, CD2Cl2) δ [ppm]: 148.3 (d, J = 6.5 Hz), 144.4 (s), 125.0 (s), 123.7 (s), 123.2 (s), 112.4 (s), 85.1 (d, 1JRh = 12.5 Hz), 30.9 (s). 19F NMR (565 MHz, CD2Cl2) δ [ppm]: −50.5 (s, 15F), −61.2 (s, 2F). FT-IR (ATR) ν [cm−1]: 3012 (w), 2962 (w), 2922 (w), 2894 (w), 2845 (w), 1615 (m), 1576 (w), 1533 (m), 1479 (s), 1446 (s), 1337 (m), 1260 (m), 1205 (vs), 1100 (vs), 865 (s), 800 (s), 771 (vs), 734 (m), 696 (m), 633 (vs), 560 (m). HRMS (ESI-TOF, negative) m/z for [C10F15]− calculated: 404.9766; measured: 404.9668. EA (C28H20F17N2Rh) calculated: C: 41.50%, H: 2.49%, N: 3.46%; measured: C: 41.78%, H: 2.93%, N: 3.54%.
1 H NMR (600 MHz, CD2Cl2) δ [ppm]: 8.46 (d, 3J = 2.3 Hz, 4H), 7.41 (tt, 3J = 7.9 Hz, 4J = 2.3 Hz, 2H), 4.13 (s, 4H), 2.68–2.60 (m, 4H), 2.06 (q, 3J = 7.6 Hz, 4H). 13C{1H} NMR (151 MHz, CD2Cl2) δ [ppm]: 161.3 (s), 136.0 (d, J = 27.3 Hz), 125.1 (s), 115.5 (s), 87.5 (s), 30.8 (s). 19F NMR (565 MHz, CD2Cl2) δ [ppm]: −50.6 (s, 15F), −116.3 (s, 4F). FT-IR (ATR) ν [cm−1]: 3088 (w), 3013 (w), 2961 (w), 2925 (w), 2897 (w), 2361 (w), 1599 (m), 1494 (m), 1438 (m), 1317 (m), 1210 (vs), 1116 (vs), 977 (m), 871 (m), 801 (w), 688 (m), 633 (m), 533 (m). HRMS (ESI-TOF, negative) m/z for [C10F15]− calculated: 404.9766; measured: 404.9684. EA (C28H18F19N2Rh) calculated: C: 39.74%, H: 2.14%, N: 3.31%; measured: C: 37.68%, H: 3.15%, N: 3.44%.
FT-IR (ATR) ν [cm−1]: 2961 (w), 2927 (w), 2893 (w), 2844 (w), 1629 (s), 1567 (w), 1529 (m), 1493 (s), 1467 (w), 1329 (w), 1207 (vs), 1112 (vs), 1007 (s), 876 (w), 792 (vs), 741 (m), 697 (w), 632 (s), 569 (m). EA (C28H18F19N2Rh) calculated: C: 39.74%, H: 2.14%, N: 3.31%; measured: C: 39.76%, H: 2.18%, N: 3.32%.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
Gefördert durch die Deutsche Forschungsgemeinschaft (DFG)—Projektnummer 387284271—SFB 1349. Computing time was made available by High-Performance Computing ZEDAT/FU Berlin. The authors acknowledge the assistance of the Core Facility BioSupraMol supported by the DFG. The authors acknowledge generous support by the Fonds of the Chemical Industry (Kekulé fellowship for Robin Sievers).References
- S. D. Pike, I. Pernik, R. Theron, J. S. McIndoe and A. S. Weller, J. Organomet. Chem., 2015, 784, 75–83 CrossRef CAS.
- S. T. H. Willems, P. H. M. Budzelaar, N. N. P. Moonen, R. de Gelder, J. M. M. Smits and A. W. Gal, Chem. – Eur. J., 2002, 8, 1310–1320 CrossRef CAS PubMed.
- V. V. Grushin and H. Alper, Chem. Rev., 1994, 94, 1047–1062 CrossRef CAS.
- R. J. Less, T. C. Wilson, M. McPartlin, P. T. Wood and D. S. Wright, Chem. Commun., 2011, 47, 10007–10009 RSC.
- A. B. Chaplin, J. F. Hooper, A. S. Weller and M. C. Willis, J. Am. Chem. Soc., 2012, 134, 4885–4897 CrossRef CAS PubMed.
- A. Prades, M. Fernández, S. D. Pike, M. C. Willis and A. S. Weller, Angew. Chem., Int. Ed., 2015, 54, 8520–8524 CrossRef CAS PubMed.
- I. Pernik, J. F. Hooper, A. B. Chaplin, A. S. Weller and M. C. Willis, ACS Catal., 2012, 2, 2779–2786 CrossRef CAS.
- T. M. Douglas, J. L. Nôtre, S. K. Brayshaw, C. G. Frost and A. S. Weller, Chem. Commun., 2006, 3408–3410 RSC.
- R. Dallanegra, A. P. M. Robertson, A. B. Chaplin, I. Manners and A. S. Weller, Chem. Commun., 2011, 47, 3763–3765 RSC.
- J. F. Hooper, R. D. Young, I. Pernik, A. S. Weller and M. C. Willis, Chem. Sci., 2013, 4, 1568–1572 RSC.
- T. M. Douglas, A. B. Chaplin, A. S. Weller, X. Yang and M. B. Hall, J. Am. Chem. Soc., 2009, 131, 15440–15456 CrossRef CAS PubMed.
- A. L. Colebatch, A. I. McKay, N. A. Beattie, S. A. Macgregor and A. S. Weller, Eur. J. Inorg. Chem., 2017, 2017, 4533–4540 CrossRef CAS.
- I. Krossing, Chem. – Eur. J., 2001, 7, 490–502 CrossRef CAS PubMed.
- A. I. McKay, J. Barwick-Silk, M. Savage, M. C. Willis and A. S. Weller, Chem. – Eur. J., 2020, 26, 2883–2889 CrossRef CAS PubMed.
- S. T. Belt, S. B. Duckett, M. Helliwell and R. N. Perutz, J. Chem. Soc., Chem. Commun., 1989, 928–930 RSC.
- C. L. Higgitt, A. H. Klahn, M. H. Moore, B. Oelckers, M. G. Partridge and R. N. Perutz, J. Chem. Soc., Dalton Trans., 1997, 1269–1280 RSC.
- L. Lefort, T. W. Crane, M. D. Farwell, D. M. Baruch, J. A. Kaeuper, R. J. Lachicotte and W. D. Jones, Organometallics, 1998, 17, 3889–3899 CrossRef CAS.
- R. Sievers, J. Parche, N. G. Kub and M. Malischewski, Synlett, 2023 DOI:10.1055/s-0042-1751426.
- T. J. Kealy and P. L. Pauson, Nature, 1951, 168, 1039–1040 CrossRef CAS.
- C. Janiak and H. Schumann, in Adv. Organomet. Chem, ed. F. G. A. Stone and R. West, Academic Press, 1991, vol. 33, pp. 291–393 Search PubMed.
- D. W. Macomber, W. P. Hart and M. D. Rausch, in Adv. Organomet. Chem, Academic Press, 1982, vol. 21, pp. 1–55 Search PubMed.
- S. Lauk and A. Schäfer, Eur. J. Inorg. Chem., 2021, 2021, 5026–5036 CrossRef CAS.
- F. Mazzotta, G. Zitzer, B. Speiser and D. Kunz, Chem. – Eur. J., 2020, 26, 16291–16305 CrossRef CAS PubMed.
- G. Paprott and K. Seppelt, J. Am. Chem. Soc., 1984, 106, 4060–4061 CrossRef CAS.
- O. J. Curnow and R. P. Hughes, J. Am. Chem. Soc., 1992, 114, 5895–5897 CrossRef CAS.
- K. Sünkel, S. Weigand, A. Hoffmann, S. Blomeyer, C. G. Reuter, Y. V. Vishnevskiy and N. W. Mitzel, J. Am. Chem. Soc., 2015, 137, 126–129 CrossRef PubMed.
- E. D. Laganis and D. M. Lemal, J. Am. Chem. Soc., 1980, 102, 6633–6634 CrossRef CAS.
- R. D. Chambers, S. J. Mullins, A. J. Roche and J. F. S. Vaughan, J. Chem. Soc., Chem. Commun., 1995, 841–842 RSC.
- R. Sievers, M. Sellin, S. M. Rupf, J. Parche and M. Malischewski, Angew. Chem., Int. Ed., 2022, e202211147 CAS.
- D. W. Slocum, T. R. Engelmann, R. L. Fellows, M. Moronski and S. Duraj, J. Organomet. Chem., 1984, 260, C21–C25 CrossRef CAS.
- S. G. Shore, W. L. Hsu, M. R. Churchill and C. Bueno, J. Am. Chem. Soc., 1983, 105, 655–656 CrossRef CAS.
- R. Alberto , personal communication.
- S. O. Kang, V. M. Lynch, V. W. Day and E. V. Anslyn, Organometallics, 2011, 30, 6233–6240 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: General conditions. Copies of NMR spectra. DFT calculations. CCDC 2240960 and 2240962. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d3dt00425b |
| This journal is © The Royal Society of Chemistry 2023 |