
Ionizing radiation induces cross-linking of two noncovalently bound collagen mimetic peptide triple helices in the absence of a molecular environment
Marwa
Abdelmouleh
a,
Muhamed
Amin
bc,
Mathieu
Lalande
a,
Thomas
Schlathölter
 bd and
Jean-Christophe
Poully
*a
bd and
Jean-Christophe
Poully
*a
aCIMAP UMR 6252 CEA/CNRS/ENSICAEN/Université de Caen Normandie, Bd Becquerel 14070 Caen, France. E-mail: poully@ganil.fr
bUniversity College Groningen, Hoendiep 23/24, 9718BG Groningen, The Netherlands
cLaboratory of Computational Biology, National Heart, Lung and Blood Institute, National Institutes of Health, Bethesda, Maryland 20892, USA
dZernike Institute for Advanced Materials, University of Groningen, Nijenborgh 4, 9747AG Groningen, The Netherlands
First published on 6th October 2023
Abstract
Cross-linking is a fundamental molecular process that is highly important for many applications, in particular, to tune the properties of collagen-based biomaterials. Chemical reagents, the action of enzymes or physical factors such as heat or radiation can facilitate collagen cross-linking. Ionizing radiation has the advantages of being fast, efficient and free from potentially toxic reagents. Collagen cross-linking by ionizing radiation is thought to occur via a water-mediated pathway. In the past, synthesized collagen mimetic peptides have proven to be of great value for understanding the influence of the amino acid sequence on the stability of tertiary (fibrous) as well as secondary (triple helical) structures of collagen. Cross-linking of synthetic collagen mimetic peptides is often used for modifying the properties of biomaterials. In this work, for the first time, we apply radiation-induced cross-linking to synthetic collagen mimetic peptides and present an experimental and theoretical study of peptide hexamers consisting of two noncovalently bound triple helices in the absence of a molecular environment, i.e. in the gas phase. Our results show that X-ray photoabsorption of the hydroxylated hexamer leads to ionization and cross-linking of the two triple helices: thus, we found evidence that cross-linking can be achieved by ionizing radiation, without the presence of any reagent or water. We propose a cross-linking mechanism involving the creation of two radicals on hydroxyproline side-chains and their recombination, ultimately leading to covalent bond formation between the triple helices.
Introduction
The creation of bonds is at the heart of chemistry. The phenomenon of cross-linkage is due to covalent bond formation between atoms in a molecular system. These new bonds can lead to a modification of material properties. Cross-linking is therefore a key process in many fundamental domains such as polymer chemistry, materials science, biochemistry, etc. This is particularly true for collagen-based biomaterials that are currently widely used in medical applications such as prostheses, wound healing, cartilage renewal and more.1 Collagen is the most abundant protein in mammals, and the main constituent of the extracellular matrix of connective tissues such as skin, nails, cartilage, bones, etc. The advantages of collagen for designing biomaterials are thus biocompatibility, biodegradability, availability and versatility. Furthermore, collagen is naturally cross-linked in tissues, which is crucial for their mechanical properties. Artificial cross-linking of collagen proteins for biomaterials can be achieved by chemical reagents such as glutaraldehyde, genipin or chitosan, by physical factors such as heat or UV light, or via biological processes, involving for instance the microbial transglutaminase enzyme.2 Ionizing radiation also has the ability to cross-link collagen, as demonstrated by Hu et al.,3 for irradiation of collagen thin films with X-rays, gamma-rays and ions, and by Zhang et al.,4 who irradiated collagen hydrogels with gamma-rays. This technique has not become widespread yet, despite being fast, efficient and exempt from potentially toxic reagents. The mechanism proposed for radiation-induced cross-linking is indirect: radiation-induced water radiolysis, which is followed by hydroxyl radical attack and H abstraction from either glycine or tyrosine side-chains. The two radical sites then recombine to create a covalent bond. The same mechanism accounts for dityrosine formation by UV or gamma photon irradiation.5 It is important to note that in the latter case, the first step is the dehydrogenation of tyrosine hydroxyl groups, and the covalent bond is formed between two carbon atoms of the phenyl rings. Dityrosine cross-linking in collagen has recently been employed for engineering of a biomaterial for cell encapsulation.6 Interestingly, peptide (and also lipid) cross linking does not necessarily have to proceed via water hydrolysis, as shown recently in experimental UV absorption studies in the gas phase.7,8Another interesting outcome of cross-linking is the control and stabilization of the triple helical geometrical structure of collagen, in particular using collagen mimetic peptides.9 The latter are much shorter than native collagen, thus allowing for custom peptide synthesis of a controlled amino acid sequence and crystallization to investigate the influence of mutations on the structure and stability of the triple helix.10 Assemblies of triple helices of collagen mimetic peptides are also valuable for studying collagen fibrils that constitute collagen fibers, and last but not least, they are interesting building blocks for the design of biomaterials.11,12 However, to our knowledge, no previous studies have addressed cross-linking of collagen mimetic peptides by ionizing radiation.
In a previous work, we have experimentally shown that the triple helix structures of assemblies of (PPG)10 and (POG)10 (P, O and G being proline, hydroxyproline and glycine, respectively) collagen mimetic peptides can survive in the absence of a molecular environment (in the gas phase), and that the stability of this triple helix is an intrinsic property.13 In particular, the gas phase structure of the [((PPG)10)6+9H]9+ hexamer was found to be a noncovalent assembly of two triple helices, bound by dipole–dipole interactions but without H bonds, as in a crystal (see Fig. 1). Moreover, we have thoroughly studied the ionization and fragmentation of (PPG)10 and (POG)10 peptide monomers and trimers by VUV and X-ray photons as well as by carbon ion beams. We found that fragmentation proceeds in the electronic ground state of the ionized species, after internal conversion of initial electronic excitation into vibrational modes.14,15 Usually, in this process, noncovalent bonds are cleaved before covalent ones. Consistently, the ionization of noncovalent complexes by X-ray photoabsorption leads to their extensive dissociation.15–17 To date, the irradiation of higher-order assemblies of collagen mimetic peptides has not been investigated in the gas phase.
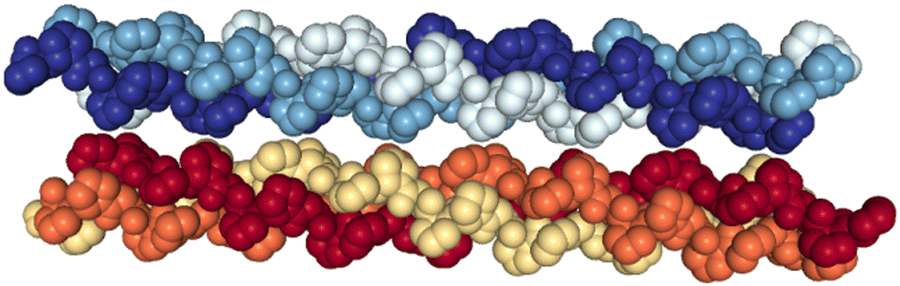 | ||
| Fig. 1 Crystal structure of the (PPG)10 collagen mimetic peptide hexamer (PDB 1K6F). Water molecules are not displayed. Heavy atoms of a given peptide are depicted with space filling and in the same color. | ||
Here, we report on the X-ray irradiation of [((PPG)10)6+9H]9+ and [((POG)10)6+9H]9+ hexamers in the gas phase. We employ mass spectrometry to analyze the cationic products of single photon absorption, in order to find evidence for photo-induced cross-linking of the two triple helices.
Experimental section
All experiments were performed using a home-made tandem mass spectrometer that has been described in detail elsewhere.18 Briefly, protonated molecular systems are brought in the gas phase with an electrospray ion source (ESI), and are focused and guided by RF electric fields through a funnel and an octopole. The molecular ion of interest is then selected by its mass over charge ratio (m/z) using a Quadrupole Mass Spectrometer (QMS), before being accumulated in a Paul trap and irradiated with the photon beam. All trapped cations are then extracted and analyzed using a time-of-flight (TOF) mass spectrometer before reaching a detector consisting of one microchannel plate (MCP), a scintillator and a photomultiplier (Bipolar TOF, Photonis). This allowed a post-acceleration voltage of −9.0 kV to be applied to the front side of the MCP, and thus increased detection efficiency compared to usual MCP detectors. For instance, around m/z = 2500, this efficiency is more than twice as compared to a post-acceleration voltage of −5.0 kV.X-Ray photon irradiation was carried out at the BESSY II synchrotron facility (U49/2-PGM1 beamline, HZB, Berlin, Germany), and a mechanical shutter was used to control the irradiation time in the 100–500 ms range.
The (PPG)10 and (POG)10 collagen mimetic peptides were purchased from Peptides International (>95% purity) and used without further purification. Powders were dissolved in equal proportions of water and methanol, with 1% of formic acid in volume. The final concentration was around 50 μmol L−1. For each molecular system studied, we recorded a reference mass spectrum without X-ray exposure (beam off), and two with the beam (beam on). We then subtracted the “beam off” spectrum from the average of the two “beam on” spectra. The result is a negative peak for the parent ion, and all positive peaks are product cations of the interaction involving a change in m/z. The resulting spectrum was smoothed by adjacent averaging over 20 points, and calibrated in m/z. Since the flux is about 1013 photons per s and the target density is low (107–108 molecules per cm3), photoabsorption events are independent. Therefore, a probability p for absorbing one photon gives the probability p2 for two photons. Since we tuned the irradiation time to induce less than 10% depletion of the precursor ion, it implies p2 + p < 0.1 (neglecting the absorption of more than two photons) and thus p < 0.09. At least 90% of the events that contribute to the observed mass spectra are thus due to single X-ray absorption events. We also irradiated the trap residual gas without the molecular ion beam (by turning off the electrospray needle voltage) to exclude the contribution of peaks due to residual gas contaminations.
Computational methods
All calculations were performed using the Gaussian 09 software.19 Geometry optimization was performed in the electronic ground singlet state of the neutral molecular species studied, with the default algorithm. Two levels of density functional theory were used: the B3LYP functional with the 6-31g(d) basis set and the wB97XD method that includes empirical dispersion as well as long-range corrections. The absence of negative frequencies was checked for molecular species with optimized geometry, to make sure that the structures were minima of the potential energy surface.Results and discussion
Non-hydroxylated hexamers
In Fig. 2, we show the mass spectra of the cationic products from single photon absorption by the [((PPG)10)6+9H]9+ hexamer. The most intense peak at m/z = 1266 can be attributed to the doubly charged peptide monomer cation (M2+) and/or to the sextuply charged trimer (T6+) that cannot be distinguished with the employed mass spectrometer. For the same reason, the weaker peak at m/z = 1519 can be assigned to non-dissociative ionization (NDI) of the precursor ion into [((PPG)10)6+9H]10+, also to the quintuply charged trimer cation (T5+). We have previously shown that collision-induced dissociation of the [((PPG)10)6+9H]9+ hexamer precursor ion leads to formation of the T5+ and T4+ trimers, consistent with the expected statistical distribution of protons in the precursor ion composed of two triple helical peptide trimers.13 As a consequence, single photoionization of one trimer in the precursor T5+/T4+ hexamer gives either T5+/T5+ or T6+/T4+ configurations, both with similar probabilities. Furthermore, inter-helix proton transfer can turn T6+/T4+ into T5+/T5+. Therefore, further dissociation into two trimers will mostly form the T5+ trimer (m/z = 1519). From Fig. 2, it is clear though that the corresponding peak is relatively weak, indicating a generally low yield of trimer photoproducts. It is therefore likely that the peak at m/z = 1266 is mostly due to M2+.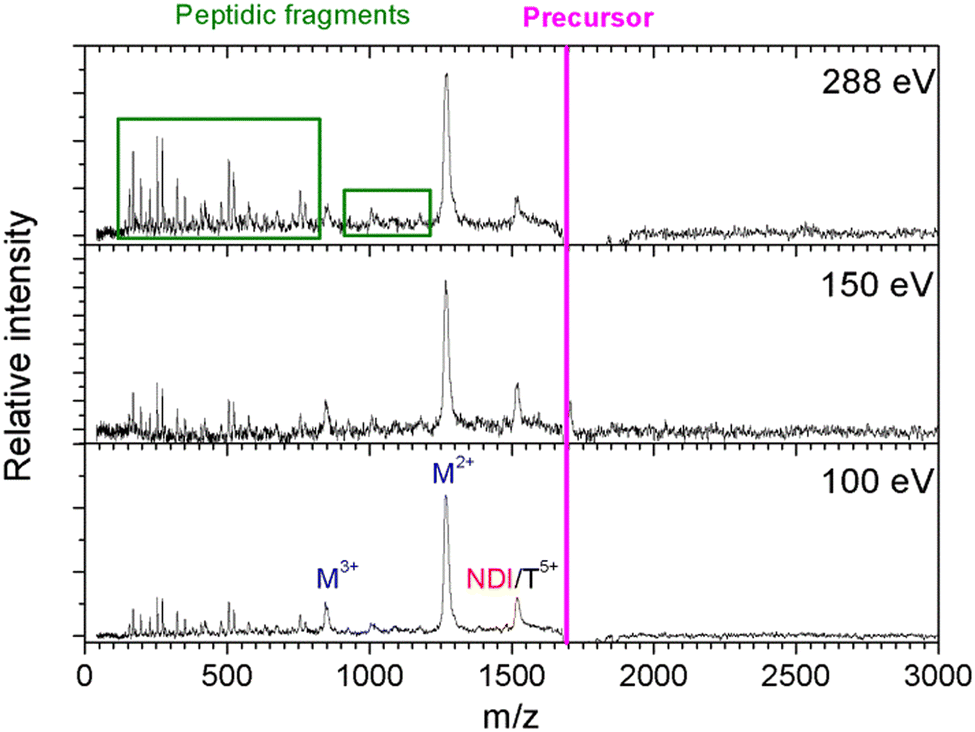 | ||
| Fig. 2 Mass spectra of single-photon absorption products of the collagen mimetic peptide hexamer [((PPG)10)6+9H]9+ (average m/z = 1688). NDI, M and T stand for the non-dissociative single ionization, monomer and trimer, respectively. | ||
The two trimers constituting the [((PPG)10)6+9H]9+ hexamer are bound via weak non-covalent dipole–dipole interactions; however, the total binding energy is about 2 eV.13 The relatively high binding energy reflects the high permanent dipole moment of the triple helix of about 65 D. The dipole is well aligned with the helix axis, which maximizes the binding energy between the two triple helices in the hexamer shown in Fig. 1. Creation of a vacancy in a carbon 1s orbital of biomolecular species after photoabsorption around 290 eV with subsequent Auger decay is known to deposit an average of about 20 eV of vibrational energy into the system,20 more than enough to detach both trimers from each other. The dominance of M2+ is evidence for a high probability of further dissociation of the triple helices into monomers after photoabsorption. We have already observed this process for photoabsorption of [((PPG)10)3+7H]7+ triple helices in the same photon energy range.14Scheme 1 illustrates this dissociation after single and double ionization of the [((PPG)10)6+9H]9+ precursor. We will show later that double ionization indeed occurs in this photon energy range for a very similar system. Moreover, previous studies have proven that soft X-ray single photon absorption induces non-dissociative multiple ionization of isolated protonated proteins.21Scheme 1 also indicates the statistical distribution of charges among monomers after dissociation: only M+ and M2+ are expected and the former is more than twice lower in abundance. This much lower abundance of M+ compared to that of M2+ partly explains the fact that we do not observe the singly charged monomer M+ peak at m/z = 2531. Another reason is the decrease of the detection efficiency with m/z by about a factor of 2 at 2531 compared to that at 1266. The absence of the M+ peak could also indicate that this monomer does not remain intact after photoionization and fragments into lower m/z ions, in particular for photon energies of 288 eV and above (see the next sub-section). A weak peak at m/z = 844.5 in the spectra from Fig. 2 can be assigned to the M3+ monomer trication. This observation is not accounted for by the dissociation mechanisms from Scheme 1, where a statistical distribution of charges is assumed following single or double ionization; thus, M3+ has to come from dissociation of the precursor hexamer into monomers before charge equilibrium.
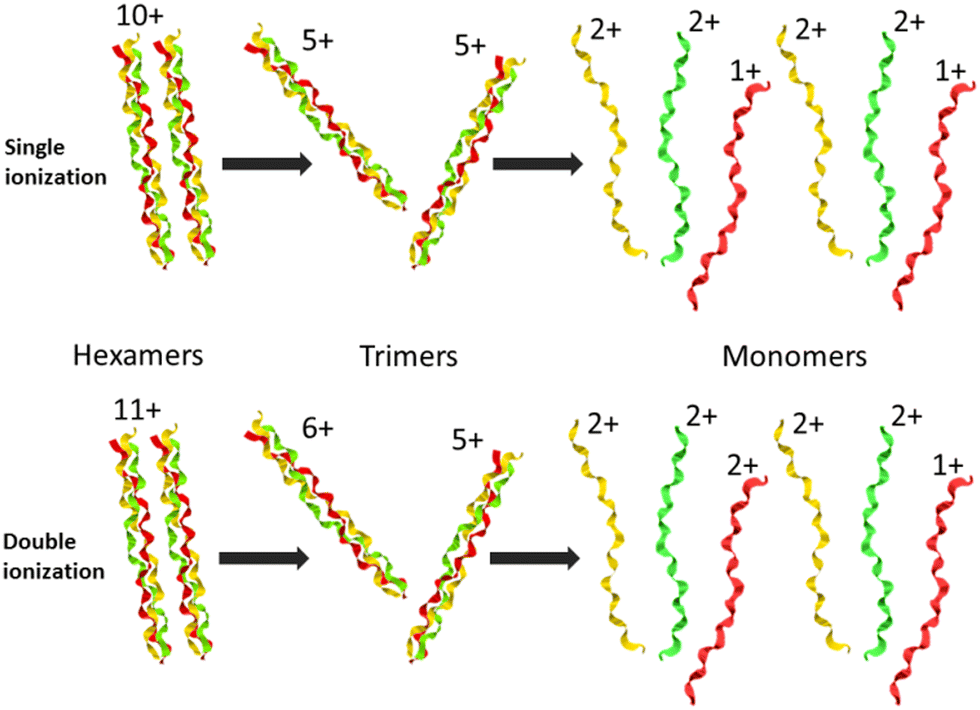 | ||
| Scheme 1 Dissociation of collagen peptide hexamers after X-ray photoabsorption, single or double ionization and vibrational energy redistribution. The charges of hexamers, trimers and monomers expected from a statistical distribution are indicated. | ||
In addition to the cleavage of the noncovalent bonds between the two triple helices as well as between peptides within triple helices, we also observed the breakage of covalent bonds within the monomers. The latter process gives rise to the peaks in the low m/z range of the mass spectra of Fig. 2, assigned to backbone fragments that we have already observed after photoabsorption of (PPG)10 monomers and trimers in the VUV and soft X-ray ranges.14 These fragments imply that after photoabsorption, monomers can have sufficient vibrational energy to dissociate, even after the loss of all noncovalent interactions. However, the relative abundance of these fragments is lower than that in the case of trimers, at a given photon energy.15 This is due to the size of the precursor ion: a hexamer is twice as large as a trimer, and the excess vibrational energy is thus distributed over twice the degrees of freedom.20 Interestingly, the relative abundance of non-dissociative single ionization (leading to [((PPG)10)6+9H]10+ of m/z = 1519) is lower for the hexamer than that for the [((PPG)10)3+7H]7+ trimer ionized after photoabsorption at the same photon energy. This can be explained by the lower binding energy of [((PPG)10)6+9H]9+ as compared to [((PPG)10)3+7H]7+. The two triple helices in [((PPG)10)6+9H]9+ are presumably bound by dipole–dipole interactions while the three peptides in [((PPG)10)3+7H]7+ are linked by strong H bonds.13 All these findings are in line with photoabsorption followed by internal conversion to the electronic ground state, followed by inter- and intramolecular vibrational energy redistribution and eventually fragmentation, as previously found for other protonated molecular systems, in particular collagen peptide trimers.14,15
Hydroxylated hexamers
As a next step, we investigated the influence of proline hydroxylation on photoabsorption-induced processes. To this end, ten proline residues within the (PPG)10 collagen mimetic peptide were replaced by hydroxyprolines. In Fig. 3, we show the mass spectra of the [((POG)10)6+9H]9+ hexamer after photoabsorption in the 100–531 eV photon energy range. In the following, we will assume that in the gas-phase, the geometry of [((POG)10)6+9H]9+ is similar to the crystalline form (see Fig. 4), as we did for the case of [((PPG)10)6+9H]9+. For all photon energies between 100 and 288 eV, up to m/z = 1400, the mass spectrum is very similar to the one observed for [((PPG)10)6+9H]9+ (see Fig. 2), featuring peaks assigned to the same peptidic backbone fragments and peptide monomers with similar relative intensities. In sharp contrast, the peak at m/z = 1615.5 corresponding to NDI and/or the T5+ trimer has a much higher relative intensity for [((POG)10)6+9H]9+. This peak even dominates at 100 and 150 eV. The peak at m/z = 1469, observed for photon energies up to 300 eV, can unambiguously be attributed to non-dissociative double ionization (NDDI) of the precursor ion, forming [((POG)10)6+9H]11+. The equivalent peak at m/z = 1381 is absent from the spectra of [((PPG)10)6+9H]9+. To get more information, it is useful to look at the evolution of the relative intensity of the peaks at m/z = 1615.5 and 1469 from 288 to 300 eV, around the carbon K-edge energy. Previous studies on the protonated cytochrome C protein have shown that photoabsorption at 288 or 300 eV triggers C 1s excitation or ionization, followed by Auger decay, i.e. a total of one or two electrons are removed, respectively (see Scheme 2).21 Cytochrome C is a 12 kDa protein and for proteins of this size, backbone fragmentation is usually not observed after soft X-ray single photon absorption. Due to the large number of vibrational degrees of freedom, the vibrational energy of the photoionized system is insufficient for backbone fragmentation to occur.20 As a consequence, for cytochrome C NDI dominates over NDDI at 288 eV, whereas the opposite is observed at 300 eV. In Fig. 3, a similar transition from NDI to NDDI is observed for [((POG)10)6+9H]9+ if we assume the peak at m/z = 1615.5 to be dominated by NDI, rather than by T5+. This assumption is supported by the fact that the peak almost vanishes at 300 eV, even though the vibrational excitation energy should be very similar for both 1s excitation (288 eV) and 1s ionization (300 eV).20 Negligible T5+ at 300 eV thus implies negligible T5+ at 288 eV. Therefore, we found that non-dissociative single and double ionization is a major channel after photoionization of [((POG)10)6+9H]9+ up to 288 eV photon energy, and their relative probabilities behave like a covalent system of similar size around the carbon K-edge (see Fig. 3), which is not expected for a noncovalent complex: we will come back to this key point later.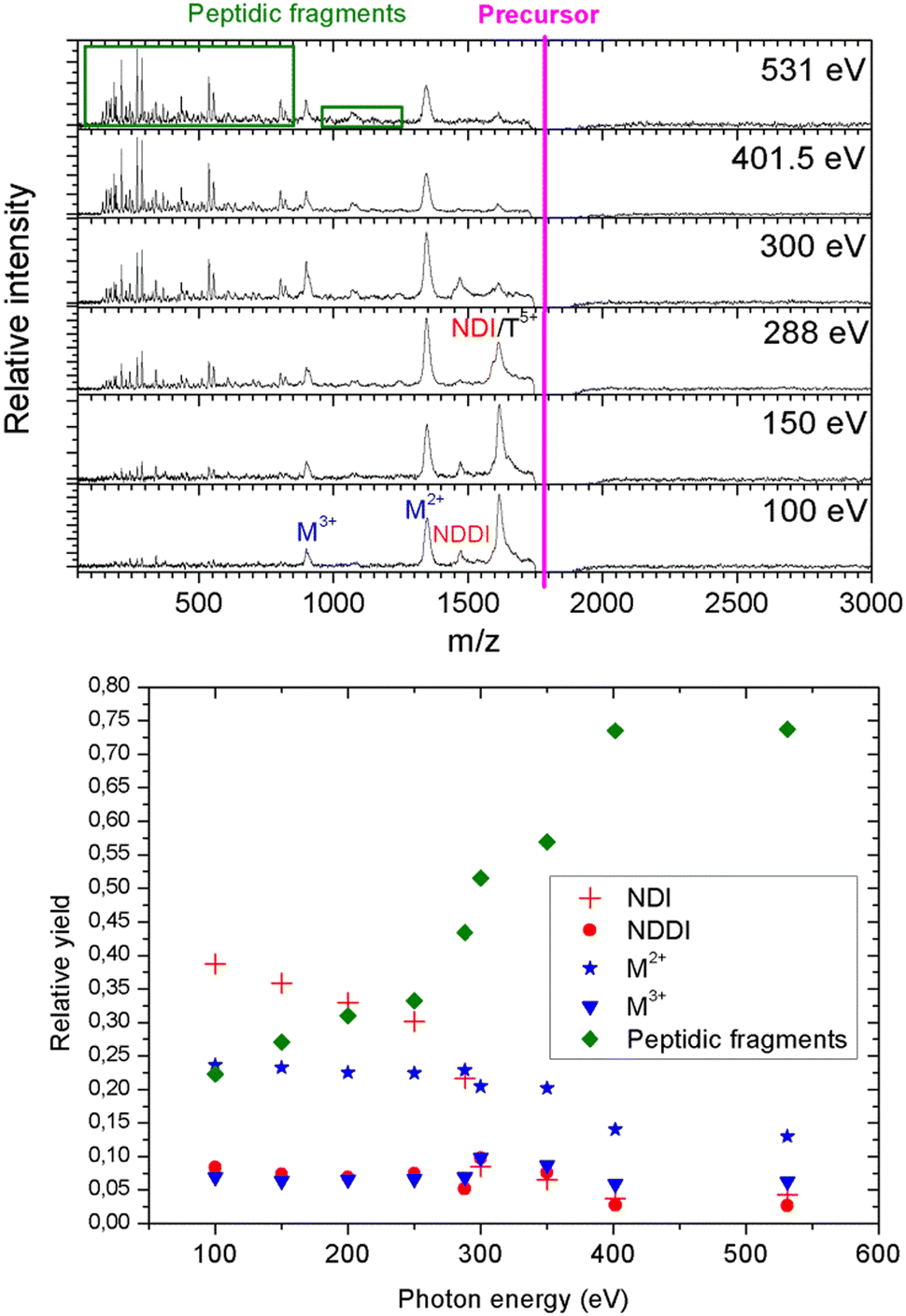 | ||
| Fig. 3 Top: Mass spectra of single-photon absorption products of the collagen mimetic peptide hexamer [((POG)10)6+9H]9+ (average m/z = 1795). Bottom: Relative yield of these products (area under the corresponding peaks normalized to the total yield of product ions) as a function of photon energy. NDI = non-dissociative single ionization, NDDI = non-dissociative double ionization, M2+ and M3+ are peptide monomers, and “peptidic fragments” is the total fragmentation yield of monomers. | ||
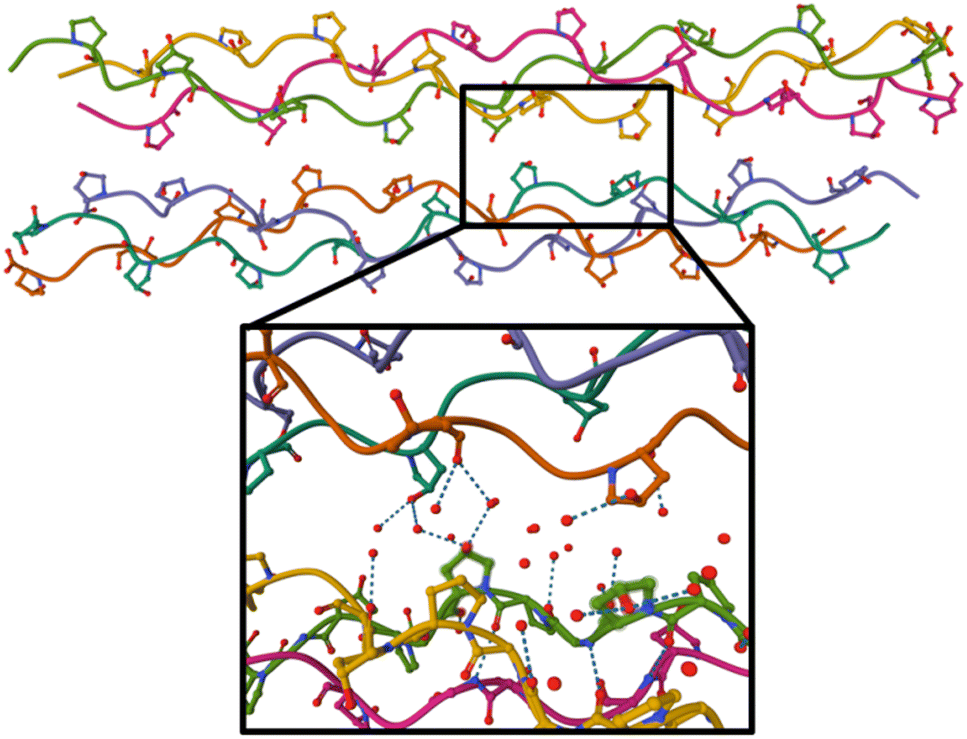 | ||
| Fig. 4 Crystal structure of the (GPO)9 collagen mimetic peptide hexamer (PDB 3B0S). Peptide backbones are depicted as lines, and only carbon, oxygen (in red) and nitrogen atoms (in blue) of hydroxyproline side-chains are shown. Water molecules are not displayed on the top figure, and on the bottom, only oxygen atoms of the water molecules within 5 Å around green highlighted hydroxyprolines are shown. H bonds are indicated by dashes. | ||
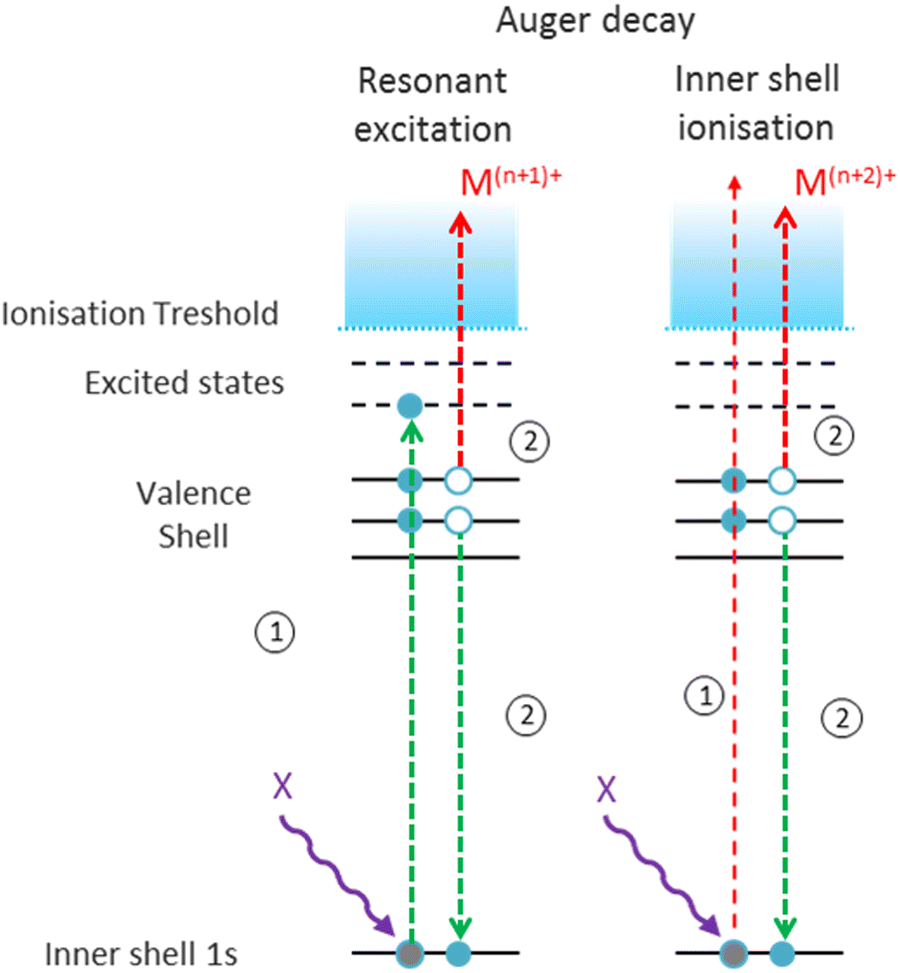 | ||
| Scheme 2 Electronic processes occurring within a molecular cation Mn+ after X-ray photoabsorption around atomic K-edges of heavy atoms. Green arrows illustrate resonant transitions leading to excitation or deexcitation, and red arrows depict ejection to the continuum. | ||
At 300 eV, it is also interesting to notice that the M3+ peak at around m/z = 898 is much more intense than at 288 eV for [((POG)10)6+9H]9+ (see Fig. 3). This confirms the hypothesis that we invoked in the previous paragraph to explain M3+ formation from the [((PPG)10)6+9H]9+ hexamer: double ionization of the precursor ion. Besides, the NDDI peak is absent from the [((PPG)10)6+9H]9+ spectra, presumably because this hexamer breaks upon double ionization, due to its lower binding energy. When photon energy increases from 100 to 531 eV, the peptide backbone fragments become much more abundant and the peaks assigned to NDI, NDDI, trimers and monomers decrease in intensity (see Fig. 3), indicating that the amount of vibrational energy in the system progressively increases with photon energy. Such an observation has already been made for other protonated systems, in particular the [((PPG)10)3+7H]7+ triple helix.15 The internal energy of the system after photoabsorption increases when electrons of deeper orbitals, for instance the 1s atomic orbitals of carbon at 285–300 eV, nitrogen around 400 eV and oxygen at 530–540 eV become accessible. Thus, our results are consistent with this increasing electronic energy being converted into vibrational energy in the electronic ground state of the system, provoking more fragmentation as photon energy rises. The present data confirm the general nature of this mechanism for single X-ray photoabsorption of molecular systems in the gas phase.
Observing NDI and NDDI as major processes after soft X-ray photoionization of a noncovalently bound system such as [((POG)10)6+9H]9+ is highly unusual, since weak intermolecular bonds are usually broken by the vibrational energy deposited by photoabsorption followed by internal conversion into the ground state (see the previous discussion about photoabsorption by [((PPG)10)6+9H]9+). This shows that hydroxyprolines strengthen the peptide hexamer enough to prevent the separation of the two triple helices upon soft X-ray photoabsorption. In the following, we explore the possible causes for this behavior.
Discussion
A possible cause for observing non-dissociative single and double ionization of [((POG)10)6+9H]9+ and not [((PPG)10)6+9H]9+ following X-ray photoabsorption would be a much higher number of degrees of freedom and thus heat capacity for the hydroxylated hexamer, quenching fragmentation. However, this is not the case: hydroxyprolines bring only 3% more degrees of freedom. The only alternative explanation is a large increase of the dissociation energy of the hexamer upon hydroxylation. Previously, we have shown by the collision-induced dissociation of collagen mimetic peptide triple helices that the collision energy to induce 50% dissociation of [((POG)10)3+7H]7+ is 14% higher than that of [((PPG)10)3+7H]7+.13 We have attributed this difference to intra-helix stereoelectronic effects induced by the hydroxyl group of hydroxyprolines. Such effects are expected to play a role in the [((POG)10)6+9H]9+ hexamer as well (since we assume that it contains two triple helices) but only in dissociation into monomers, not in the separation of the two triple helices.13 Thus, stereoelectronic effects cannot explain the high abundance of NDI and NDDI for [((POG)10)6+9H]9+. Let us look at the crystal structure of (GPO)9 available in the Protein Data Bank (cf.Fig. 4):22 one notices that the two triple helices do not share any direct H bond; only water-mediated H-bonds exist between some hydroxyproline OH groups that are close to each other. In the gas phase, these water molecules are not present, which means that the corresponding H bonds do not exist. Even if several new direct OH⋯OH H bonds might form between hydroxyl groups after desolvation, these bonds would only slightly increase the binding energy of the hexamer since typical binding energies for such H bonds are about 0.14 eV (the binding energy of the methanol dimer held together by such a bond).23We propose that the process accounting for the large increase of the stability of the hydroxylated (POG)10 hexamer upon X-ray photoionization is the creation of a covalent bond between the two triple helices (cross-linking) as a result of photoionization. Since no such process is observed for the non-hydroxylated (PPG)10 hexamer, the hydroxyl group of hydroxyprolines must play a key role in cross-linking. By drawing a parallel to the mechanism of dityrosine formation, we can assume that the first step is the same, namely H abstraction and/or proton transfer from the hydroxyl groups of two hydroxyproline side-chains to create two radicals. If the latter are close enough, they might recombine to form an O–O bond: in Fig. 4, we can see that the closest hydroxyproline side-chain OH groups are only 5 Å apart. Moreover, they belong to different triple helices; this O–O bond would thus eventually cross-link the two triple helices. Alternatively, after H abstraction and/or proton transfer, the radicals might migrate to the carbon atoms linked to the oxygens of the two hydroxyprolines, to form two stable tertiary radicals. Recombination would create a covalent C–C bond, which has a typical bond dissociation energy (BDE) of 3.6–3.9 eV in alkanes,24 while peroxides are less stable (BDE ≈ 1.7 eV for the O–O bond of di-tert-butyl peroxide).25 This cross-linking would thus in any case significantly increase the binding energy of the hexamer and account for the presence of intense NDI and NDDI in the mass spectra of [((POG)10)6+9H]9+. This mechanism is supported by our previous studies on protonated collagen peptides containing hydroxyprolines: we found that hydroxyproline side-chains undergo a radical reaction upon photoionization in the same photon energy range than the one explored here.26 It should also be noted that a similar mechanism has been proposed for the cross-linking of protonated triacylglycerol lipid dimers upon UV photoabsorption by their noncovalent complexes with diiodoaniline in the gas phase.8 This mechanism starts with the abstraction of two I atoms from diiodoaniline, and the formation of this diradical is followed by H abstraction from a carbon atom of each of triacylglycerols, which finally induces their cross-linking via recombination of the two radicals. Our experimental results indicate that using ionizing photons, radical molecular ions of noncovalent complexes are created and can undergo recombination and cross-linking without the need of an external reagent such as diiodoaniline. In the next sub-section, we show that quantum-chemical calculations support this conclusion.
Results of theoretical calculations
To investigate the cross-linking process between the two triple helices of the hydroxylated peptide hexamer in the gas phase, we performed quantum-chemical calculations at the density functional theory (DFT) level. All water molecules in the crystal structure of the (GPO)9 collagen mimetic peptide hexamer (PDB 3B0S) were removed. Since considering the whole hexamer is computationally unaffordable, we had to choose smaller systems. As a first step, only one peptide from each helix was kept, and the residues far away from the closest hydroxyproline side chains were removed: this is called system A. In addition, the H atoms of both hydroxyl groups were removed to create two radicals, and system A was subject to geometry optimization using several levels of DFT (see the “Computational methods” section). To probe the effect of the other peptides of the hexamer, system B was created by including the residues of the other peptides that share H bonds with system A: since system B is much larger than system A (244 vs. 98 atoms), only the wb97xd level was used. In all cases, during geometry optimization, an O–O covalent bond is formed by radical recombination. The same calculations were repeated for the two radicals located at the carbon atoms linked to hydroxyproline oxygens: a C–C covalent bond is now formed. The resulting structures for system A are visualized in Fig. 5. Much more thorough theoretical investigations would be needed to firmly establish the mechanism responsible for ionization-induced cross-linking between the hexamers, but the present results are the first supporting the existence of such a process in collagen mimetic peptides.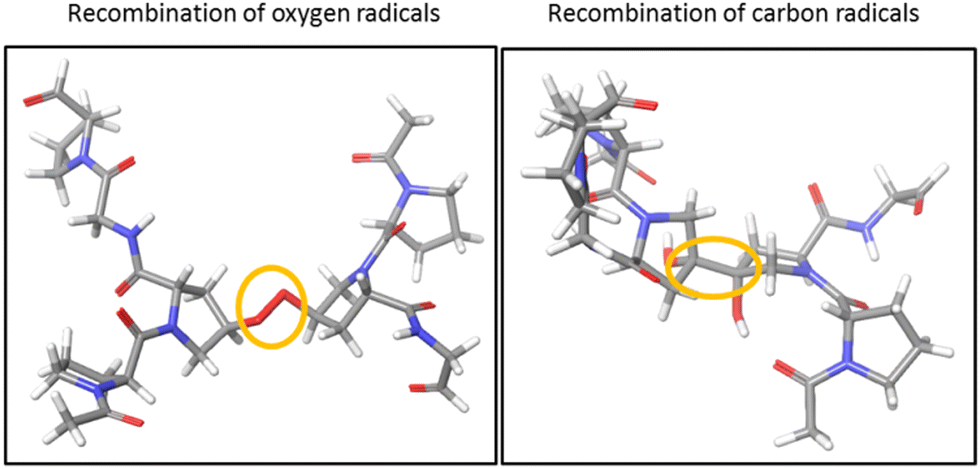 | ||
| Fig. 5 Optimized structures of system A for radicals initially located at oxygens (left) and carbons (right) of the hydroxyproline side chains. The O–O and C–C bonds formed between the hydroxyprolines are encircled. Carbons are depicted in light grey, oxygens in red, nitrogens in blue, and hydrogens in white. | ||
Conclusions
X-Ray photoionization of dimers of collagen mimetic peptide triple helices in the gas phase is in most cases followed by fragmentation due to excess vibrational energy. However, we also observe pronounced non-dissociative single and double ionization for peptides containing hydroxyprolines. We attribute this behavior to the creation of a covalent bond between the two triple helices, after the formation of two radicals and their recombination. Quantum-chemical calculations support this conclusion. However, the exact mechanism is yet to be identified with the help of theoretical calculations and also experiments such as multi-stage collision-induced dissociation tandem high-resolution mass spectrometry of the product ions of single or double ionization. Our findings show that cross-linking of molecular systems can be achieved by ionizing radiation, without the presence of any reagent or water, and also extend the possibilities of designing biomaterials by exposure of collagen mimetic peptides to ionizing radiation. It would be valuable to change the nature of the ionizing radiation, and for instance irradiate triple helix assemblies with electrons or ion beams since we expect more multiple ejections of valence electrons, and thus potentially more covalent bonds to be formed. Emission of several valence electrons from multiply deprotonated DNA oligonucleotides also occurs after X-ray photoabsorption or carbon ion beam irradiation;27 another perspective is thus to seek for cross-linking of oligonucleotide dimers upon ionizing radiation.Author contributions
Marwa Abdelmouleh: investigation, Muhamed Amin: investigation and writing – review and editing. Mathieu Lalande: investigation and writing – review and editing. Thomas Schlathölter: conceptualization, funding acquisition, project administration, and writing – review and editing. Jean-Christophe Poully: investigation, formal analysis, funding acquisition, project administration, supervision, and writing – original draft.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank HZB for the allocation of synchrotron radiation beam time and local contacts for their support during experiments. The French “Conseil Régional de Normandie” is acknowledged for a PhD funding (#15P01339). This project has received funding from the European Union's Horizon 2020 research and innovation program under grant agreement no. 730872.References
- X. Yu, C. Tang, S. Xiong, Q. Yuan, Z. Gu, Z. Li and Y. Hu, Modification of Collagen for Biomedical Applications: A Review of Physical and Chemical Methods, Curr. Org. Chem., 2016, 20, 1797–1812 CrossRef CAS.
- K. Adamiak and A. Sionkowska, Current methods of collagen cross-linking: Review, Int. J. Biol. Macromol., 2020, 161, 550–560 CrossRef CAS PubMed.
- X. Hu, W. K. Raja, B. An, O. Tokareva, P. Cebe and D. L. Kaplan, Stability of Silk and Collagen Protein Materials in Space, Sci. Rep., 2013, 3, 3428 CrossRef PubMed.
- X. Zhang, L. Xu, X. Huang, S. Wei and M. Zhai, Structural study and preliminary biological evaluation on the collagen hydrogel crosslinked by gamma-irradiation, J. Biomed. Mater. Res., Part A, 2012, 100A, 2960–2969 CrossRef CAS.
- C. Giulivi, N. J. Traaseth and K. J. A. Davies, Tyrosine oxidation products: analysis and biological relevance, Amino Acids, 2003, 25, 227–232 CrossRef CAS PubMed.
- B. Partlow, M. Applegate, F. Omenetto and D. Kaplan, Dityrosine Cross-Linking in Designing Biomaterials, ACS Biomater. Sci. Eng., 2016, 2, 2108–2121 CrossRef CAS PubMed.
- H. Nguyen, P. Andrikopoulos, L. Rulisek, C. Shaffer and F. Turecek, Photodissociative Cross-Linking of Non-covalent Peptide-Peptide Ion Complexes in the Gas Phase, J. Am. Soc. Mass Spectrom., 2018, 29, 1706–1720 CrossRef CAS PubMed.
- S. Nie, H. T. Pham, S. J. Blanksby and G. E. Reid, Photoinduced Intermolecular Cross-Linking of Gas Phase Triacylglycerol Lipid Ions, Eur. J. Mass Spectrom., 2015, 21, 287–296 CrossRef CAS PubMed.
- S. A. H. Hulgan and J. D. Hartgerink, Recent Advances in Collagen Mimetic Peptide Structure and Design, Biomacromolecules, 2022, 23, 1475–1489 CrossRef CAS PubMed.
- B. Brodsky, G. Thiagarajan, B. Madhan and K. Kar, Triple-helical peptides: An approach to collagen conformation, stability, and self-association, Biopolymers, 2008, 89, 345–353 CrossRef CAS PubMed.
- S. M. Yu, Y. Li and D. Kim, Collagen mimetic peptides: progress towards functional applications, Soft Matter, 2011, 7, 7927–7938 RSC.
- Y. Li and S. M. Yu, Targeting and mimicking collagens via triple helical peptide assembly, Curr. Opin. Chem. Biol., 2013, 17, 968–975 CrossRef CAS PubMed.
- M. Lalande, C. Comby-Zerbino, M. Bouakil, P. Dugourd, F. Chirot and J.-C. Poully, Isolated Collagen Mimetic Peptide Assemblies Have Stable Triple-Helix Structures, Chem. – Eur. J., 2018, 24, 13728–13733 CrossRef CAS PubMed.
- L. Schwob, M. Lalande, J. Rangama, D. Egorov, R. Hoekstra, R. Pandey, S. Eden, T. Schlathölter, V. Vizcaino and J.-C. Poully, Single-photon absorption of isolated collagen mimetic peptides and triple-helix models in the VUV-X energy range, Phys. Chem. Chem. Phys., 2017, 19, 18321–18329 RSC.
- M. Lalande, M. Abdelmouleh, M. Ryszka, V. Vizcaino, J. Rangama, A. Mery, F. Durantel, T. Schlathölter and J.-C. Poully, Irradiation of isolated collagen mimetic peptides by x rays and carbon ions at the Bragg-peak energy, Phys. Rev. A, 2018, 98, 062701 CrossRef CAS.
- M. Abdelmouleh, M. Lalande, J. El Feghaly, V. Vizcaino, A. Rebelo, S. Eden, T. Schlathölter and J.-C. Poully, Mass Spectral Signatures of Complex Post-Translational Modifications in Proteins: A Proof-of-Principle Based on X-ray Irradiated Vancomycin, J. Am. Soc. Mass Spectrom., 2020, 31, 1738–1743 CrossRef CAS PubMed.
- M. Abdelmouleh, A. Rodriguez, J. Leroux, P. Christodoulou, B. Bernay, T. Schlathölter and J. Poully, X-ray photoabsorption-induced processes within protonated rifamycin sodium salts in the gas phase, Eur. Phys. J. D, 2021, 75 DOI:10.1140/epjd/s10053-021-00092-w.
- S. Bari, R. Hoekstra and T. Schlathölter, Peptide fragmentation by keV ion-induced dissociation, Phys. Chem. Chem. Phys., 2010, 12, 3376–3383 RSC.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery, Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, T. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 09, Gaussian, Inc., Wallingford CT, 2016 Search PubMed.
- D. Egorov, L. Schwob, M. Lalande, R. Hoekstra and T. Schlathölter, Near edge X-ray absorption mass spectrometry of gas phase proteins: the influence of protein size, Phys. Chem. Chem. Phys., 2016, 18, 26213–26223 RSC.
- A. R. Milosavljevic, F. Canon, C. Nicolas, C. Miron, L. Nahon and A. Giuliani, Gas-Phase Protein Inner-Shell Spectroscopy by Coupling an Ion Trap with a Soft X-ray Beamline, J. Phys. Chem. Lett., 2012, 3, 1191–1196 CrossRef CAS PubMed.
- K. Okuyama, K. Miyama, K. Mizuno and H. P. Bächinger, Crystal structure of (Gly-Pro-Hyp)9: Implications for the collagen molecular model, Biopolymers, 2012, 97, 607–616 CrossRef CAS PubMed.
- F. C. Hagemeister, C. J. Gruenloh and T. S. Zwier, Density Functional Theory Calculations of the Structures, Binding Energies, and Infrared Spectra of Methanol Clusters, J. Phys. Chem. A, 1998, 102, 82–94 CrossRef CAS.
- S. J. Blanksby and G. B. Ellison, Bond Dissociation Energies of Organic Molecules, Acc. Chem. Res., 2003, 36, 255–263 CrossRef CAS PubMed.
- R. D. Bach and H. B. Schlegel, Bond Dissociation Energy of Peroxides Revisited, J. Phys. Chem. A, 2020, 124, 4742–4751 CrossRef CAS PubMed.
- L. Schwob, M. Lalande, D. Egorov, J. Rangama, R. Hoekstra, V. Vizcaino, T. Schlathölter and J.-C. Poully, Radical-driven processes within a peptidic sequence of type I collagen upon single-photon ionisation in the gas phase, Phys. Chem. Chem. Phys., 2017, 19, 22895–22904 RSC.
- W. Li, O. Kavatsyuk, W. Douma, X. Wang, R. Hoekstra, D. Mayer, M. S. Robinson, M. Gühr, M. Lalande, M. Abdelmouleh, M. Ryszka, J. C. Poully and T. Schlathölter, Multiple valence electron detachment following Auger decay of inner-shell vacancies in gas-phase DNA, Chem. Sci., 2021, 12, 13177–13186 RSC.
| This journal is © the Owner Societies 2023 |