
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Direct and mild non-hydroxide activation of biomass to carbons with enhanced CO2 storage capacity†
Afnan
Altwala
ab and
Robert
Mokaya
 *a
*a
aSchool of Chemistry, University of Nottingham, University Park, Nottingham NG7 2RD, UK. E-mail: r.mokaya@nottingham.ac.uk
bDepartment of Chemistry, College of Science Al-Zulfi, Majmaah University, Al-Majmaah, 11952, Saudi Arabia
First published on 23rd March 2022
Abstract
Potassium oxalate (PO) was trialled as a non-corrosive and less toxic activating agent for the direct activation of biomass (sawdust, SD). The PO + SD mixtures were activated either in powder form or after compaction into pellets. The resulting activated carbons are highly microporous with surface area in the range of 550 to 2100 m2 g−1 and pore volume between 0.3 and 1.0 cm3 g−1. The porosity of the directly activated and compactivated carbons is similar to that of conventionally activated (via hydrothermal carbonisation) equivalents. In general, pelletized (i.e., compactivated) carbons achieved higher levels of porosity for any identical set (with respect to amount of PO and temperature) of preparation conditions. Unlike hydroxide activation, the amount of PO used, for PO/SD mass ratio between 2 and 4, does not have a significant effect on porosity. On the other hand, the activation temperature plays a critical role in determining the textural properties at any given PO/SD ratio. The porosity of the carbons is dominated by pores of size 6–8 Å, which are suitable for post-combustion (low pressure) CO2 storage. At 25 °C, the carbons capture up to 1.6 and 4.3 mmol g−1 of CO2 at 0.15 bar and 1 bar, respectively. Our findings show that the use of potassium oxalate as a mild activating agent via direct activation succeeds in addressing the need for non-corrosive and less toxic activators and also negates the need for hydrothermal treatment or pyrolysis of biomass prior to activation. The present carbons are attractive as sustainable energy materials especially for post-combustion CO2 capture and storage.
1. Introduction
Meeting the demand of low cost and environmentally friendly energy storage methods and avoiding the emission of environmental pollutants in mobile and stationary applications remains a challenge. To meet this challenge, new materials with properties targeted at specific applications are required. One such target is the storage of gases, such as H2, CO2 and CH4, which are relevant to energy production or environmental remediation. In this regard, porous solids are amongst the most studied materials for use in gas storage applications.1–4 One area of recent research on gas storage has focused on obtaining activated carbons from biomass, which is considered to be an environmentally sustainable source, is cheap and readily available.5–7 There are two main routes for generating activated carbon from biomass, namely physical or chemical activation.7 Physical activation is via high temperature thermal treatment of biomass in the presence of oxidising gases such as air, O2, CO2 and steam. Chemical activation, on the other hand, utilises activating agents such as KOH, H3PO4, ZnCl2 and NaOH as porogens for generating porosity in carbonaceous matter at medium to high temperature. KOH, via so-called hydroxide activation, is one of the most extensively explored activating agents for activated carbons. However, such hydroxide activation has disadvantages due to the toxicity and corrosive nature of KOH. Milder activating agents that offer carbons with similar or better textural qualities are therefore highly sought after. Therefore, beyond hydroxide activation, there is growing interest in the use of less corrosive and toxic activating agents.8–18 A number of studies have reported the use of potassium oxalate (PO) as activating agent for carbons with low to medium surface area (typically <1500 m2 g−1) for activation at 800 °C or below.8–18Another important consideration is that, for new activated carbons to be of interest, they should fall into one of the following categories: have new or improved properties, be easy/cheap to prepare, and be sustainable. Lower cost/easier to prepare materials typically result from a reduction in the number of fabrication steps required. Biomass is normally activated after the process of hydrothermal carbonisation (HTC) or pyrolysis, which converts the biomass into carbon-rich carbonaceous matter. Hydrothermal carbonisation involves heating (typically at 180–300 °C) in water under pressure to enable thermochemical decomposition of biomass to carbon-rich carbonaceous matter.19–22 The pyrolysis process, on the other hand, involves generation of carbonaceous matter via enrichment of carbon content during thermal treatment at temperatures of 600–900 °C under oxygen-free conditions.23–29 We have recently explored a direct and cheaper route to KOH activated biomass-derived carbons, which excluded the need for HTC or pyrolysis prior to activation.30 Recently, we have also reported on the process of compactivation, also known as mechanochemical activation, wherein mixtures of the precursor and KOH are compacted into pellets prior to activation.31 Given that the activation process is initially based on solid–solid interaction between the activating agent and the precursor, the compactivation method was developed with the aim of increasing proximity (solid–solid contacts) between the precursor and KOH.29,31 The desired outcome of compactivation is to improve the efficiency of the activating agent with respect to the porosity generated. It is now known that compactivation with KOH can generate carbons with higher (compared to conventional powder activation) surface area and pore volume.29,31 However, so far, compactivation has only been explored for KOH activation, and has not been performed for direct activation of raw biomass. It of interest to explore what impact the milder nature of PO has on the process and whether overall yields and porosity are affected by use of raw biomass rather than enriched carbonaceous matter derived from pyrolysis or HTC.
In this report, we explore the use of PO for the direct activation and compactivation of raw biomass (sawdust). This approach potentially offers several advantages that have, so far, not been probed in any one study, namely (i) use of a milder non-hydroxide activating agent, (ii) direct activation of biomass that negates the need for HTC or pyrolysis, and (iii) porosity modulation and optimisation (with respect to amount of activating agent) via the compactivation route. Overall, therefore, this approach offers a direct process that is simpler, cheaper, and more sustainable. Importantly, it is necessary that these advantages not compromise the porosity of the resulting carbons, i.e., the carbons should have similar properties to analogous carbons prepared via conventional methods. Based on the porosity of the resulting activated and compactivated carbons, we investigated their CO2 uptake and show that they offer very attractive trends especially for low-pressure (post-combustion) uptake.
2. Experimental section
2.1 Material synthesis
Raw sawdust was sieved using 212 MC sieves to obtain homogenous powder. Potassium oxalate (PO) was ground into a fine powder and thoroughly mixed with the sawdust (SD) at PO/SD mass ratio of 2 or 4. Half of the PO/SD mixture was compacted for 10 min at a load of 10 tonnes in a 1.3 cm diameter die (equivalent to a compression pressure of 740 MPa) prior to activation. The light brown PO/SD mixtures (powders or pellets) were loaded on alumina boats and then placed in a furnace and heated, at ramp rate of 5 °C min−1, to 600, 700 or 800 °C under a flow of nitrogen. Samples were held at the target temperature for 1 h before being cooled to room temperature, whilst still under a flow of nitrogen. The resulting black matter was washed with 20 wt% HCl at ambient temperature to remove any inorganic species. The mixtures were then washed with deionised water until neutral pH was achieved for the filtrate, and dried in an oven. The activated carbons were denoted as DSDxT or DSDxTP where x is the PO/SD ratio (2 or 4) and T is the activation temperature (600, 700 or 800 °C), and P denotes compaction (i.e., palletisation) prior to activation. Thus, a powder sample activated at a PO/SD ratio of 4 and 700 °C is designated as DSD4700, while a compactivated sample prepared at a PO/SD ratio of 4 and 700 °C is designated as DSD4700P.2.2 Characterization methods
Thermogravimetric analysis (TGA) was performed using a TA Instruments Discovery analyser or TA Instruments SDT Q600 analyser under flowing air conditions (100 mL min−1). A PANalytical X’Pert PRO diffractometer was used to perform powder XRD analysis using Cu-Kα radiation (40 kV, 40 mA) with step size of 0.02° and 50 s time step. Elemental, CHN, analysis was performed on an Exeter Analytical CE-440 Elemental Analyser. Nitrogen sorption (at –196 °C) with a Micromeritics 3FLEX sorptometer was used for porosity analysis and to determine textural properties. Prior to analysis, the carbon samples were degassed under vacuum at 200 °C for 12 h. Surface area was calculated using the Brunauer–Emmett–Teller (BET) method applied to adsorption data according to Rouquerol rules, in the relative pressure (P/Po) range of 0.02–0.22, and pore volume was estimated from the total nitrogen uptake at close to saturation pressure (P/Po ≈ 0.99). The micropore surface area and micropore volume were determined via t-plot analysis. The pore size distribution was determined using Non-Local Density Functional Theory (NL-DFT) applied to nitrogen adsorption data. Scanning electron microscopy (SEM) images were recorded using an FEI Quanta200 microscope, operating at a 5 kV accelerating voltage.2.3 CO2 uptake measurements
CO2 uptake was determined using a Hiden Isochema XEMIS instrument at 25 °C and pressure of up to 20 bar. The carbons were outgassed for 3 h under vacuum at 240 °C prior to performing the CO2 uptake measurements.3. Results and discussion
3.1 Yield, nature and elemental composition of activated and compactivated carbons
This study explored the direct activation of biomass using a mild non-hydroxide activating agent with the aim of simplifying the activation process and making it more sustainable by moving away from some of the challenges associated with the corrosive nature of KOH activation. This work is the first time that direct activation of biomass, i.e., without the need for HTC or pyrolysis has been attempted with a mild activating agent. Furthermore, compactivated carbons were also prepared to explore any benefits of compaction of the PO/SD mixture prior to thermal activation. The elemental composition of the sawdust and the activated carbon yields, and the yields of the direct activation or direct compactivation are given in Table 1. The yield of activated (i.e., powder) samples ranges from 22% to 28% while for compactivated (i.e. pelletised) samples it is between 20% and 34%. It is important to note that the reported yields are with respect to the raw sawdust. This differs from most activated carbon yields in the literature that are typically reported with respect to carbonised matter derived from biomass via HTC or pyrolysis. This is an important distinction as conversion of raw biomass to carbonaceous matter (via HTC or pyrolysis) has a typical yield of between 30% and 45%. Previous studies have shown that the activated carbon yield from PO activation of sawdust hydrochar (i.e., after HTC) varied as follows; 35% (800 °C), 40% (700 °C) and 44% (600 °C).8 This means that the yield of activated carbon with respect to the raw sawdust was at most (assuming 45% yield for HTC) 16% (800 °C), 18% (700 °C) and 20% (600 °C).8 Thus, the data in Table 1 indicates that at any given temperature, the activated carbon yields for the direct PO activation of sawdust is higher than what is achieved via HTC. This greater yield, which is higher by nearly 50% at 600 °C, appears to be a general advantage of direct activation and has also been previously observed for KOH activated carbons.30| Sample | Yield [wt%] | C [%] | H[%] | N[%] | O[%] |
|---|---|---|---|---|---|
| Sawdust | 47.0 | 5.6 | 0.4 | 47.0 | |
| DSD2600 | 28 | 71.3 | 1.0 | 0.5 | 27.2 |
| DSD2700 | 28 | 65.6 | 1.1 | 0.2 | 33.1 |
| DSD2800 | 22 | 65.5 | 0 | 0 | 34.5 |
| DSD4600 | 28 | 65.5 | 0.5 | 0.3 | 33.7 |
| DSD4700 | 26 | 71.5 | 0.7 | 0.2 | 27.6 |
| DSD4800 | 22 | 72.5 | 0.1 | 0 | 27.4 |
| DSD2600P | 34 | 75.0 | 1.2 | 0.6 | 23.2 |
| DSD2700P | 20 | 73.0 | 0.8 | 0.2 | 26.0 |
| DSD2800P | 20 | 80.0 | 0.5 | 0.0 | 19.5 |
| DSD4600P | 30 | 72.0 | 0.5 | 0.5 | 27.0 |
| DSD4700P | 26 | 76.5 | 1.3 | 0.7 | 21.5 |
| DSD4800P | 20 | 86.0 | 0.1 | 0.1 | 13.8 |
For the present direct activation process, it is noteworthy that compaction of the PO/SD mixtures before activation (i.e., activation of pelletized mixtures) generates generally similar yields to activation of powder mixtures. Compaction of the PO/SD mixture is expected to engender closer contact between the PO and SD particles and under any given activation conditions (i.e., amount of PO and temperature) should lead to greater levels of activation and therefore lower activated carbon yields. Such a trend is now well established for KOH activation; in effect, compactions acts to improve the efficient use of KOH.29,31,32 The fact that the trend is not observed here for PO activation suggests that, unlike for hydroxide activation where the amount of KOH is critical, the PO/SD ratio is not a critical factor in determining the level or extent of activation. It is clear from the data in Table 1 that, in general, only the activation temperature determines the carbon yield; change of PO/SD ratio from 2 to 4 at any given temperature does not appear to have any significant effect on the carbon yield. On the other hand, higher activation temperatures lead to a lowering of carbon yield.
As expected, following activation, the carbon content (given as wt%) increases from 47% for the sawdust to a high of 86% for the sample compactivated at 800 °C (DSD4800P). The effect of activation temperature is such that carbons activated at the highest temperature (800 °C) have the most elemental C content (72.5% and 86% for powder and compacted samples, respectively). In general, compactivated carbons have higher C content compared to powder samples under any given activation conditions. The content of N, H and O decreases at higher activation temperature, which is the expected trend at higher levels of activation and is consistent with previous reports.23,24,29–32 Thermogravimetric analysis of the activated carbons, under flowing air conditions, was performed in order to assess the carbon purity (i.e., lack of inorganic matter) and thermal stability. The TGA curves (Fig. S1, ESI†) show a large mass loss between 450 and 650 °C due to carbon combustion, with an initial mass loss below 120 °C due to water removal. The TGA curves show that the residual mass, after heating in air at 800 °C, is typically lower than 5 wt%, which indicates that the activated carbons are generally free of inorganic residues. Samples activated at 600 °C have maximum burn-off at 500 °C compared to up to 650 °C for carbons activated at 800 °C, which exemplifies the effect of activation temperature on thermal stability. Higher activation temperatures results in greater thermal stability in the activated carbons, which is consistent with previous observations.33–39
The powder XRD patterns of the activated carbons (Fig. S2, ESI†) show that the amount of PO does not have any significant effect on the nature of the carbons – the XRD patterns of representative samples suggest a comparable level of graphitic ordering at any given activation temperature. The XRD patterns have very broad peaks at 2θ = 26° and 43°, which correspond to a very weakly graphitic/turbostratic nature in the carbons. The XRD patterns of carbons activated at higher temperature show a very weak peak, which indicates greater disruption of any graphitic ordering. Given that higher activation temperature leads to more thermally stable activated carbons, the ‘disruption’ of graphitisation as evidenced by the XRD patterns may be more to do with decrease in size of ‘graphitic’ domains rather than actual overall lowering of the level of graphitisation. The XRD patterns of some of the activated carbons exhibit sharp peaks, which could indicate the presence of inorganic residues left over by the activating agent. However, the TGA curves suggest that the presence of any inorganic impurities occurs only at a minor level.18–21
Scanning electron microscopy was used to probe the morphology of the carbons. The SEM images of the raw sawdust and activated carbons DSD4800 and DSD4800P are shown in Fig. 1. The morphology of the raw sawdust is comprised of extended fibrous structures typical for woody matter (Fig. S3, ESI†). After activation, the morphology shows some retention of fibrous and honeycomb-like structures. The morphology of the activated carbons includes smooth surfaces characterised by large conchoidal cavities, which is similar to what has previously been reported for carbons generated via direct or flash carbonisation activation routes.22,30,34 The aforementioned retention of some woody morphology is consistent with the milder nature of PO activation compared to KOH where the morphology is completely altered (compared to that of the biomass) especially at high levels of activation.35–37
 | ||
| Fig. 1 SEM images of raw sawdust and selected activated and compactivated carbons. | ||
3.2 Porosity of activated carbons and compactivated carbons
The nitrogen sorption isotherms and the corresponding pore size distribution (PSD) curves for carbons directly activated or compactivated at PO/SD ratio of 2 are shown in Fig. 2. All samples exhibit type I isotherms typical of microporous materials. Due to micropore filling, a significant proportion of nitrogen sorption takes place at a relative pressure (P/Po) below 0.1, followed by a sharp knee and plateau with no significant adsorption occurring at P/Po greater than 0.1. For samples activated at 800 °C, the isotherms show a slight broadening of the adsorption ‘knee’, which suggests limited widening in the pore size distribution. The amount of nitrogen adsorbed appears to be correlated with the temperature of activation, and increases at higher activation temperature. The isotherms of the compactivated carbons (Fig. 2C) are similar to those of powder samples (Fig. 2A), except for higher amounts of nitrogen adsorbed. This is the first indication that direct compactivation with PO generates higher levels of porosity compared to direct activation. The pore size distribution curves of powder samples (Fig. 2B) show no pores of width greater than 20 Å with most pores being of size less than 10 Å. A similar trend is observed for the compactivated samples except that they have a slightly higher proportion of pores in the 10–20 Å range. Regarding pore maxima, increasing the activation temperature from 600 to 800 °C leads to an increase such that while samples prepared at 600 °C or 700 °C show an average pore width of 6–8 Å, this rises to 6–12 Å at 800 °C. Compactivation does not significantly alter the pore size distribution (Fig. 2D) except for a slight shift to larger micropores especially for sample DSD2800P. Thus, despite the increase in overall porosity occasioned by compactivation (as per the amount of nitrogen adsorbed – Fig. 2A and C), the pore size remains largely unchanged (Fig. 2B and D).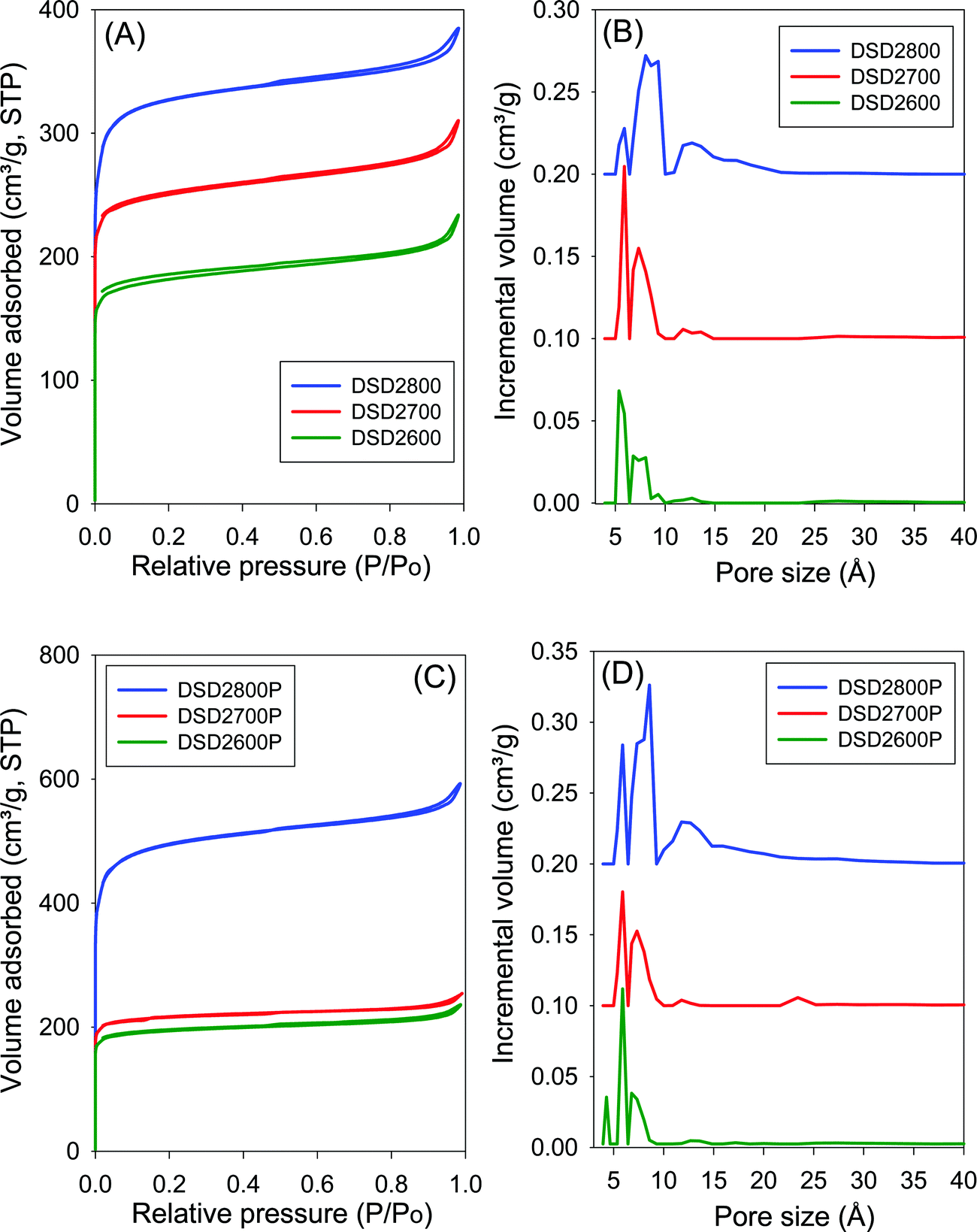 | ||
| Fig. 2 Nitrogen sorption isotherms (A and C) and pore size distributions curves (B and D) of carbons directly activated (A and B) or compactivated (C and D) from sawdust (SD) at 600, 700 or 800 °C, and PO/SD ratio of 2. The PSD curves are offset (y-axis) by 0.1 (DSD2700 and DSD2700P) or 0.2 (DSD2800 ad DSD2800P). | ||
Fig. 3 shows the nitrogen sorption isotherms and corresponding pore size distribution curves for carbons prepared at PO/SD ratio of 4. The isotherms are typically type I and very similar to those in Fig. 2 both in terms of shape and amount of nitrogen adsorbed. Clearly, increasing the amount of PO does not appear to have any significant effect on the porosity, which is consistent with the results of XRD analysis as described above. The PSD curves in Fig. 3 are comparable to those of in Fig. 2, which means that increase in the PO/SD ratio does not lead to pore size expansion. This means that control of the porosity of the carbons is best achieved by choice of activation temperature rather than the amount of PO or a combination of both. This is a departure from what is observed for direct activation of sawdust with KOH wherein increase in the KOH/SD ratio from 2 to 4 generated carbons with larger pores and also included the presence of small mesopores.30 According to Fig. 3 (B and D), direct activation at PO/SD ratio of 4 results in the formation of larger micropores with hardly any mesopores even for samples activated at 800 °C. The presence of supermicropores for samples activated at 800 °C could be related to CO2 and CO gases released during the activation at high temperature.8 Generally, though, the compactivated samples show higher nitrogen sorption compared to equivalent powder samples, which is in agreement with previous studies using KOH as activating agent.31,32 The increase in porosity induced by compactivation is, however, not as high as that observed for KOH activation. This is consistent with the fact that the amount of PO is not a critical factor in determining the porosity. It is likely, therefore, that PO exists in an excess amount, which means that the close contact with sawdust particles engendered by the compaction does not have any significant effect of the overall efficiency of the activator.
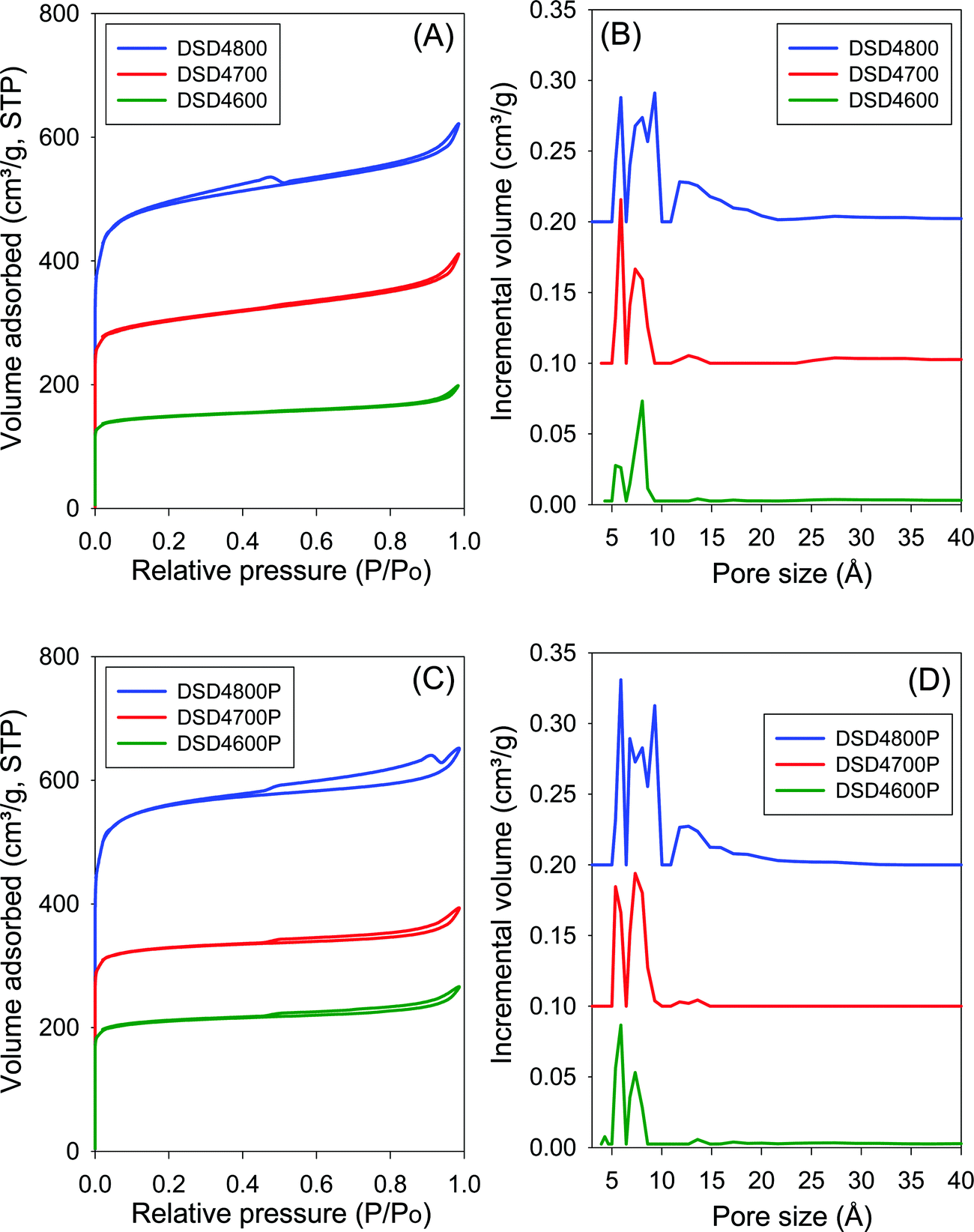 | ||
| Fig. 3 Nitrogen sorption isotherms (A and C) and pore size distributions curves (B and D) of carbons directly activated (A and B) or compactivated (C and D) from sawdust (SD) at 600, 700 or 800 °C, and PO/SD ratio of 4. The PSD curves are offset (y-axis) by 0.1 (DSD4700 and DSD4700P) or 0.2 (DSD4800 and DSD4800P). | ||
One of the key aims of this study was to explore the impact of a simpler and more direct activation route on the porosity of activated carbons. This is an important consideration given the mild nature of PO as an activating agent. Table 2 summarizes the textural properties of the directly activated and compactivated carbons. The surface area of activated carbons is in the range of 550–1860 m2 g−1 with pore volume of between 0.3 and 0.96 cm3 g−1. The surface area and pore volume are mainly determined by the activation temperature. Both textural parameters increase as activation temperature rises from 600 to 800 °C. The amount of PO (given as PO/SD ratio of 2 or 4) does not appear to have any effect of the surface area and pore volume especially for activation at 600 and 700 °C. However, for activation at 800 °C, a higher amount of PO generates greater surface area and pore volume. Thus the surface area and pore volume of DSD4800 (1859 m2 g−1 and 0.96 cm3 g−1, respectively) is higher than that of DSD2800 (1238 m2 g−1 and 0.59 cm3 g−1, respectively). The activated carbons exhibit high to very high microporosity as indicated by the magnitude and proportion of surface area and pore volume arising from micropores (Table 2). The proportion of surface area arising from micropores is between 80 and 86% while for pore volume it is 59 to 71%. The activation temperature has no effect on the proportion of microporosity. However, a higher amount of PO appears to very slightly reduce the microporosity. It is also clear from the textural data in Table 2 that compactivation generates carbons with higher surface area and pore volume compared to equivalent activated samples and that, interestingly, this increase in overall prosity does not compromise the microporosity. The microporosity of the compactivated carbons is either similar of slightly higher than that of equivalent activated carbons.
| Sample | Surface area (m2 g−1) | Micropore surface areaa (m2 g−1) | Pore volume (cm3 g−1) | Micropore volumeb (cm3 g−1) | CO2 uptake. (mmol g−1) | ||
|---|---|---|---|---|---|---|---|
| 0.15 bar | 1 bar | 20 bar | |||||
| a The values in the parenthesis refer to % micropore surface area. b The values in the parenthesis refer to % micropore volume. | |||||||
| DSD2600 | 682 | 574 (84) | 0.36 | 0.23 (64) | 1.0 | 2.7 | 4.9 |
| DSD2700 | 945 | 813 (86) | 0.48 | 0.33 (69) | 1.2 | 4.1 | 8.5 |
| DSD2800 | 1238 | 1053 (85) | 0.59 | 0.42 (71) | 0.9 | 3.4 | 12.3 |
| DSD4600 | 556 | 465 (84) | 0.30 | 0.18 (60) | 1.0 | 2.5 | 4.8 |
| DSD4700 | 1131 | 906 (80) | 0.63 | 0.37 (59) | 0.9 | 3.0 | 6.8 |
| DSD4800 | 1859 | 1497 (81) | 0.96 | 0.60 (63) | 0.8 | 3.2 | 11.5 |
| DSD2600P | 730 | 638 (87) | 0.36 | 0.26 (72) | 1.2 | 2.9 | 5.1 |
| DSD2700P | 822 | 699 (85) | 0.39 | 0.28 (72) | 1.0 | 3.6 | 7.7 |
| DSD2800P | 1893 | 1545 (82) | 0.92 | 0.61 (66) | 1.0 | 4.0 | 13.0 |
| DSD4600P | 793 | 693 (87) | 0.41 | 0.28 (68) | 1.2 | 2.9 | 5.2 |
| DSD4700P | 1242 | 1123 (91) | 0.61 | 0.45 (74) | 1.6 | 3.8 | 7.8 |
| DSD4800P | 2121 | 1816 (86) | 1.01 | 0.73 (72) | 1.1 | 4.3 | 12.2 |
It is interesting to note that the porosity of the directly activated carbons is comparable (Table S1, ESI†) to that of equivalent (in terms of amount of PO and activation temperature) conventionally activated samples prepared via HTC prior to activation of the hydrochar. The microporosity of the two sets of samples is also similar especially for carbons prepared at PO/SD or PO/hydrochar ratio of 2. At PO/SD or PO/hydrochar ratio of 4, the directly activated carbons have very slightly lower levels of microporosity. The overall picture that emerges is that direct activation or compactivation of biomass with PO does not compromise the porosity of the resulting carbons. This is similar to what has previously been observed when the harsher activating agent, KOH, is used.30 Thus direct activation or compactivation of biomass with PO offers simplicity but without introducing any disadvantages with respect to achievable porosity.
3.3 CO2 uptake of activated and compactivated carbons
Given the high microporosity of the present carbons, we investigated their CO2 uptake at 25 °C with particular focus on low pressure (>1 bar) storage capacity. Fig. 4 shows the CO2 uptake isotherms for activated (i.e., powder) samples, and the storage capacity at 0.15 bar, 1 bar and 20 bar is summarised in Table 2. The CO2 uptake at 1 bar, which is often used as a measure of performance for post-combustion capture from flue gas streams from fossil fuel power stations, is in the range of 2.5 to 4.1 mmol g−1. The highest CO2 uptake at 1 bar is for sample DSD2700, which also has the highest proportion of micropore surface area (Table 2). The CO2 uptake rises significantly from ca. 2.5 mmol g−1 (activation at 600 °C) to a high of 4.1 mmol g−1 (700 °C) and then reduces to ca. 3.4 mmol g−1 for samples activated 800 °C.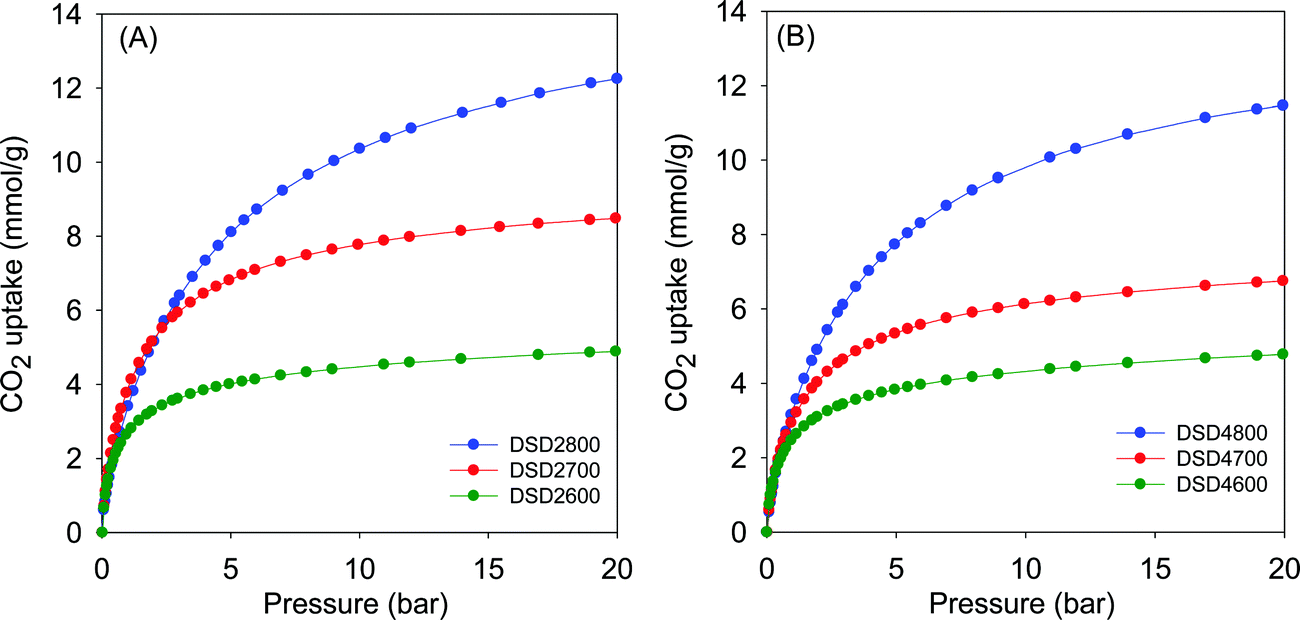 | ||
| Fig. 4 CO2 uptake isotherms of carbons directly activated from sawdust (SD) at 600, 700 or 800 °C, and PO/SD ratio of 2 (A) or 4 (B). | ||
Fig. 5 shows the CO2 uptake isotherms for compactivated samples, and Table 2 summarises the uptake at 0.15 bar, 1 bar and 20 bar. For compactivated carbons, the CO2 uptake at 1 bar is in the range 2.9–4.3 mmol g−1, and the highest uptake is for sample DSD4800P. For compacted samples, the rise is from 2.9 mmol g−1 (600 °C) to ca. 3.8 mmol g−1 (700 °C) and up to 4.3 mmol g−1 for the sample compactivated at 800 °C (DSD4800P). The uptake data, therefoe, suggest that at 25 °C and 1 bar, the total surface area does not determine CO2 uptake capacity but rather samples with the largest proportion of micropore surface area have the better performance. This is consistent with previous studies in which the preponderance of small pores (6–8 Å) has been noted to determine low pressure CO2 uptake. It is noteworthy that the highest CO2 uptake at 1 bar of 4.3 mmol g−1 for the present sawdust-derived direct PO activated carbons is comparable to that previously reported for conventionally (via HTC) PO activated sawdust derived carbons8 and also for directly activated sawdust-derived carbons with KOH as activating agent.30 A benefit of the current PO activation route is that it is milder and more environmentally friendly and appears to offer a simpler optimisation of pore size by choice of activation temperature. Thus despite being prepared via a simpler, milder, potentially cheaper and more direct route, the directly activated and compactivated carbons show attractive performance for gravimetric CO2 uptake, which is comparable or better than for current benchmark porous carbons (Table S2, ESI†).40–52
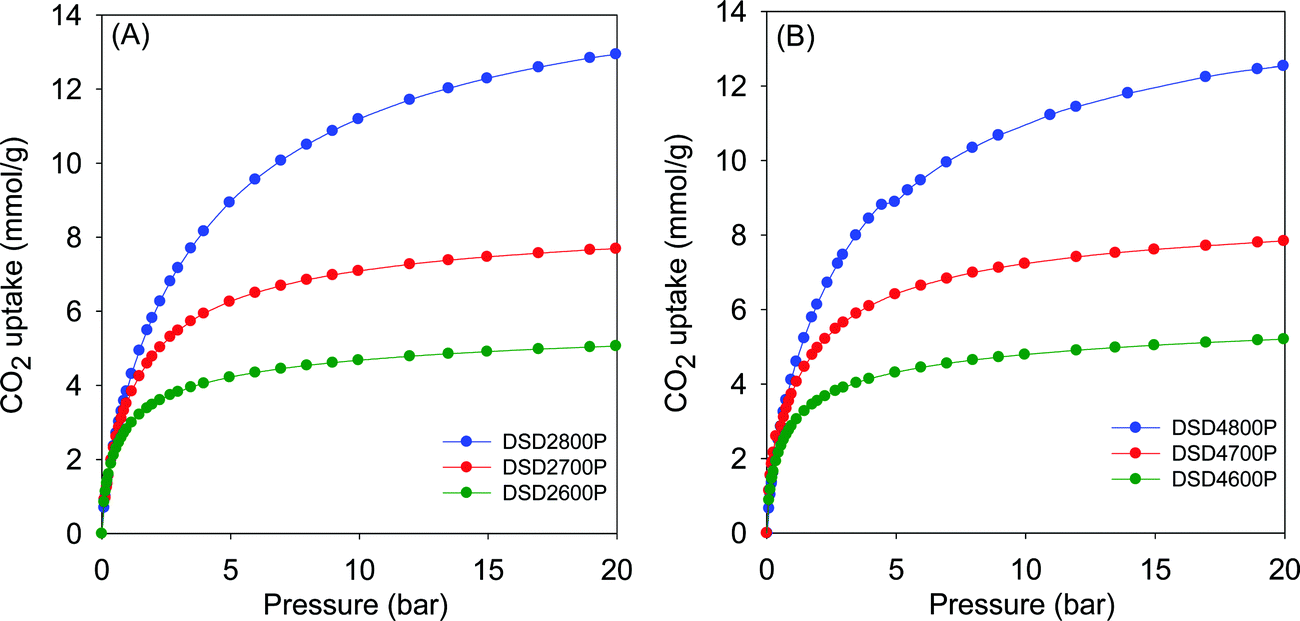 | ||
| Fig. 5 CO2 uptake isotherms of carbons directly compactivated from sawdust (SD) at 600, 700 or 800 °C, and PO/SD ratio of 2 (A) or 4 (B). | ||
The CO2 uptake at 0.15 bar is considered to be a close mimic of capture performance from post-combustion flue gas streams emanating from power stations, which typically contain of ca. 15–20% CO2 with the remainder being mainly N2 (70–75%), and water (ca.%).53,54 For activated (powder) carbons, the uptake at 0.15 bar is in the narrow range of 0.8 and 1.1 mmol g−1 with samples activated at 800 °C having the lowest storage capacity. For compactivated carbons, the uptake is higher at between 1.0 and 1.6 mmol g−1. The trend generally matches that of uptake at 1 bar and is clearly related to the microporosity of the carbons. It is noteworthy that sample DSD4700P achieves a very high uptake at 0.15 bar of 1.6 mmol g−1, which is one of the highest reported for porous carbons (Table S2, ESI†).40–52
The trend is reversed for the CO2 uptake at the higher pressures of 20 bar. The highest uptakes are achieved for samples activated at 800 °C, and the lowest values are achieved for samples activated at 600 °C. This trend mirrors the variation in surface area and confirmed the fact that the CO2 uptake at moderate to high pressure is dependent on the total surface area rather than pore size, whereas at lower pressure the level of microporosity plays an important role in determining the uptake capacity. Additionally, the CO2 uptake at 20 bar is not affected by the amount of PO, which is a consequence of the fact that changing the PO/SD ratio from 2 to 4 does not alter the porosity in any significant way. This confirms that in optimising or tailoring the textural properties of the present carbons for targeted CO2 uptake applications, the activation temperature is more effective than the PO/SD ratio. The possibility of using low amounts of PO whilst still achieving the full range of porosity is also a positive outcome of the present synthesis approach.
4. Conclusion
We have shown that potassium oxalate (PO) is an effective activation agent for the formation of activated carbons from biomass via a direct and simpler synthesis approach. This is notable, as potassium oxalate is milder than the typically used KOH, which is more corrosive/toxic. Carbons were prepared directly via activation with PO as powders or by including a compaction step (compactivation), in which the mixture of PO and sawdust are compacted together prior to thermal activation. Highly microporous activated and compactivated carbon materials were generated from sawdust. Direct activation of sawdust, without recourse to hydrothermal carbonisation or pyrolysis, in combination with the use of milder PO as activating agent, offers significant advantages over previous approaches. Despite the clear attractions of the new synthesis approach in terms of reduced steps and a milder activating agent, the properties of the generated carbons were comparable to those prepared via routes that are more complicated. Furthermore, the new approach enabled higher yields of activated carbons. The carbons have surface area in the range 550 to 2100 m2 g−1 and pore volume of between 0.3 and 1.0 cm3 g−1. For both powder and compacted samples, surface area increased with increasing activation temperature, but with the amount of PO having little effect thus enabling simpler control of the porosity via choice of activating temperature. The carbons, due to the highly microporous nature, were found to be excellent for the uptake of CO2 at 25 °C, with storage capacity of up to 1.6 and 4.3 mmol g−1 at 0.15 bar and 1 bar, respectively. Our findings show that the use of potassium oxalate as a mild activating agent via direct activation succeeds in addressing the need for non-corrosive and less toxic activators and also negates the need for hydrothermal treatment or pyrolysis of biomass prior to activation. The resulting activated carbons have attractive performance as sustainable energy materials especially for post-combustion CO2 capture and storage.Author contributions
Afnan Altwala: investigation and writing – original draft; Robert Mokaya: funding, conceptualization, supervision, and writing – review and editing.Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
We are thankful to the Nanoscale and Microscale Research Centre (nmRC) at the University of Nottingham for assistance with SEM analysis. We thank Majmaah University, the Kingdom of Saudi Arabia, for funding a PhD studentship for Afnan Altwala. RM thanks the Royal Society for a Research Grant, and for a Royal Society Wolfson Research Merit Award.References
- K. V. Kumar, K. Preuss, M. M. Titirici and F. Rodriguez-Reinoso, Chem. Rev., 2017, 117, 1796 CrossRef CAS PubMed.
- B. Li, H.-M. Wen, W. Zhou, J. Q. Xu and B. Chen, Chem, 2016, 1, 557 CAS.
- Y. He, W. Zhou, G. Qian and B. Chen, Chem. Soc. Rev., 2014, 43, 5657 RSC.
- T. A. Makal, J. R. Li, W. Lu and H. C. Zhou, Chem. Soc. Rev., 2012, 41, 7761 RSC.
- G. Singh, K. S. Lakhi, S. Sil, S. V. Bhosale, I. Kim, K. Albahily and A. Vinu, Carbon, 2019, 148, 164 CrossRef CAS.
- P. González-García, Renewable Sustainable Energy Rev., 2018, 82, 1393 CrossRef.
- M. Sevilla and R. Mokaya, Energy Environ. Sci., 2014, 7, 1250 RSC.
- A. M. Aljumialy and R. Mokaya, Mater. Adv., 2020, 1, 3267 RSC.
- J. Li, Q. Jiang, L. Wei, L. Zhong and X. Wang, J. Mater. Chem. A, 2020, 8, 1469 RSC.
- M. Sevilla, N. Alam and R. Mokaya, J. Phys. Chem. C, 2010, 114, 11314 CrossRef CAS.
- Q. Jiang, Y. Wang, Y. Gao and Y. Zhang, Environ. Sci. Pollut. Res., 2019, 26, 30268 CrossRef CAS PubMed.
- M. Sevilla, A. M. Al-Jumialy, A. B. Fuertes and R. Mokaya, ACS Appl. Mater. Interfaces, 2018, 10, 1623 CrossRef CAS PubMed.
- J. Wei, S. Wan, P. Zhang, H. Ding, X. Chen, H. M. Xiong, S. Gao and X. Wei, New J. Chem., 2018, 42, 6763 RSC.
- M. Sevilla, G. A. Ferrero and A. B. Fuertes, Carbon, 2017, 114, 50 CrossRef CAS.
- M. Sevilla, G. A. Ferrero and A. B. Fuertes, Chem. Mater., 2017, 29, 6900 CrossRef CAS.
- H. Lin, C. Xu, Q. Wang, J. Wu, Y. Wang, Y. Zhang and G. Fan, Int. J. Hydrogen Energy, 2019, 44, 21527 CrossRef CAS.
- J. Ludwinowicz and M. Jaroniec, Carbon, 2015, 82, 297 CrossRef CAS.
- Y. Chen, W. Chen, B. Huang and M. Huang, Chem. Eng. Res. Des., 2013, 91, 1783 CrossRef CAS.
- T. Wang, Y. Zhai, Y. Zhu, C. Li and G. Zeng, Renewable Sustainable Energy Rev., 2018, 90, 223 CrossRef CAS.
- M. M. Titirici and M. Antonietti, Chem. Soc. Rev., 2010, 39, 103 RSC.
- M. M. Titirici, R. J. White, C. Falco and M. Sevilla, Energy Environ. Sci., 2012, 5, 6796 RSC.
- M. Sevilla, A. B. Fuertes and R. Mokaya, Energy Environ. Sci., 2011, 3, 1400 RSC.
- A. Altwala and R. Mokaya, Energy Environ. Sci., 2020, 13, 2967 RSC.
- N. Balahmar, A. C. Mitchell and R. Mokaya, Adv. Energy Mater., 2015, 5, 1 Search PubMed.
- C. Quan, A. Li and N. Gao, Procedia Environ. Sci., 2013, 18, 776 CrossRef CAS.
- N. Soltani, A. Bahrami, M. I. Pech-Canul and L. A. González, Chem. Eng. J., 2015, 264, 899 CrossRef CAS.
- C. R. Lohri, H. M. Rajabu, D. J. Sweeney and C. Zurbrügg, Renewable Sustainable Energy Rev., 2016, 59, 1514 CrossRef CAS.
- J. F. González, J. M. Encinar, C. M. González-García, E. Sabio, A. Ramiro, J. L. Canito and J. Gañán, Appl. Surf. Sci., 2006, 252, 5999 CrossRef.
- E. Haffner-Staton, N. Balahmar and R. Mokaya, J. Mater. Chem. A, 2016, 4, 13324 RSC.
- N. Balahmar, A. S. Al-Jumialy and R. Mokaya, J. Mater. Chem. A, 2017, 5, 12330 RSC.
- B. Adeniran and R. Mokaya, Nano Energy, 2015, 16, 173 CrossRef CAS.
- N. Balahmar, A. C. Mitchell and R. Mokaya, Adv. Energy Mater., 2015, 5, 1500867 CrossRef.
- M. Sevilla, W. Sangchoom, N. Balahmar, A. B. Fuertes and R. Mokaya, ACS Sustainable Chem. Eng., 2016, 4, 4710 CrossRef CAS.
- E. A. Hirst, A. Taylor and R. Mokaya, J. Mater. Chem. A, 2018, 6, 12393 RSC.
- N. Balahmar and R. Mokaya, J. Mater. Chem. A, 2019, 7, 17466 RSC.
- H. M. Coromina, D. A. Walsh and R. Mokaya, J. Mater. Chem. A, 2016, 4, 280 RSC.
- W. Sangchoom and R. Mokaya, ACS Sustainable Chem. Eng., 2015, 3, 1658 CrossRef CAS.
- B. Adeniran, E. Masika and R. Mokaya, J. Mater. Chem. A, 2014, 2, 14696 RSC.
- B. Adeniran and R. Mokaya, J. Mater. Chem. A, 2015, 3, 5148 RSC.
- Z. Zhang, J. Zhou, W. Xing, Q. Xue, Z. Yan, S. Zhuo and S. Z. Qiao, Phys. Chem. Chem. Phys., 2013, 15, 2523 RSC.
- A. Almasoudi and R. Mokaya, J. Mater. Chem. A, 2014, 2, 10960 RSC.
- N. P. Wickramaratne and M. Jaroniec, J. Mater. Chem. A, 2013, 1, 112 RSC.
- N. P. Wickramaratne and M. Jaroniec, ACS Appl. Mater. Interfaces, 2013, 5, 1849 CrossRef CAS PubMed.
- X. Fan, L. Zhang, G. Zhang, Z. Shu and J. Shi, Carbon, 2013, 61, 423 CrossRef CAS.
- B. Adeniran and R. Mokaya, Chem. Mater., 2016, 28, 994 CrossRef CAS.
- A. Almasoudi and R. Mokaya, J. Mater. Chem., 2012, 22, 146 RSC.
- M. Nandi, K. Okada, A. Dutta, A. Bhaumik, J. Maruyama, D. Derksa and H. Uyama, Chem. Commun., 2012, 48, 10283 RSC.
- Y. D. Xia, R. Mokaya, G. S. Walker and Y. Q. Zhu, Adv. Energy Mater., 2011, 1, 678 CrossRef CAS.
- A. Wahby, J. M. Ramos-Fernandez, M. Martnez-Escandell, A. Sepulveda-Escribano, J. Silvestre-Albero and F. Rodriguez-Reinoso, ChemSusChem, 2010, 3, 974 CrossRef CAS PubMed.
- D. Lee, C. Zhang, C. Wei, B. L. Ashfeld and H. Gao, J. Mater. Chem. A, 2013, 1, 14862 RSC.
- G. Srinivas, J. Burress and T. Yildirim, Energy Environ. Sci., 2012, 5, 6453 RSC.
- J. Silvestre-Albero, A. Wahby, A. Sepulveda-Escribano, M. Martinez-Escandell, K. Kaneko and F. Rodriguez-Reinoso, Chem. Commun., 2011, 47, 6840 RSC.
- Z. Zhang, K. Wang, J. D. Atkinson, X. Yan, X. Li, M. J. Rood and Z. Yan, J. Hazard. Mater., 2012, 229–230, 183 CrossRef CAS PubMed.
- J. D. Figueroa, T. Fout, S. Plasynski, H. McIlvried and R. D. Srivastava, Int. J. Greenhouse Gas Control, 2008, 2, 9 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Three additional figures; TGA curves, XRD patterns and SEM images along with two tables with comparative textural data and CO2 uptake. See DOI: 10.1039/d1ya00085c |
| This journal is © The Royal Society of Chemistry 2022 |