
A biomimetic anti-biofouling coating in nanofluidic channels†
Sumire
Fukuda‡
ab and
Yan
Xu
 *acd
*acd
aDepartment of Chemical Engineering, Graduate School of Engineering, Osaka Prefecture University, 1-2, Gakuen-cho, Naka-ku, Sakai, Osaka 599-8570, Japan. E-mail: xu@chemeng.osakafu-u.ac.jp
bDepartment of Chemistry, Graduate School of Science, Osaka Prefecture University, 1-1, Gakuen-cho, Naka-ku, Sakai, Osaka 599-8531, Japan
cJapan Science and Technology Agency (JST), PRESTO, 4-1-8 Honcho, Kawaguchi, Saitama 332-0012, Japan
dNanoSquare Research Institute, Research Center for the 21st Century, Organization for Research Promotion, Osaka Prefecture University, 1-2, Gakuen-cho, Naka-ku, Sakai, Osaka 599-8570, Japan
First published on 17th January 2022
Abstract
Non-specific protein adsorption (NPA) is ubiquitous and generally considered a trigger for various biofoulings, adversely affecting diverse fields. Despite many approaches (generally polymer-based) available to combat NPA in a wide range of length scales, suppressing NPA in closed nanoscale spaces of emerging nanodevices such as nanofluidic devices remains a challenge due to the lack of suitable material and methodology to satisfy their ultra-small and closed features. In this study, a biomimetic, hydrosilane-functionalized 2-methacryloyloxyethyl phosphorylcholine (MPC) monomer material, which has tailored molecular characteristics and well-defined surface properties to overcome the challenges in the nanospaces, is elaborately designed and synthesized to form self-assembled, nanoscale, biomimetic coatings, enabling the efficient suppression of NPA in femtoliter-order, closed nanofluidic channels. The approach opens up a new path for exploring a strategic change of anti-biofouling coating from traditional polymer-based methodologies to a monomer-based methodology to overcome the challenge of suppressing NPA in closed nanospaces.
1. Introduction
Biofouling is the accumulation of biological entities on surfaces. Because it causes structural deficiencies or functional abnormalities of surfaces, biofouling is a general and critical issue that affects fields as diverse as mechanics, chemistry, biology, materials science, energy, electronics, drug discovery, clinical medicine, civil engineering, and environmental sciences. Since non-specific protein adsorption (NPA) is generally considered the first stage of various biofouling processes, suppressing NPA is an effective strategy to combat biofouling, in both the macroscopic and microscopic worlds.1 Many sophisticated coating methodologies based on various advanced materials have been developed for suppressing NPA in a wide range of length scales, from macroscopic and micrometer scales to recently open surfaces with nanometer scale structures.2 However, suppressing NPA in closed nanoscale spaces of emerging advanced nanodevices, such as chip-based nanofluidic devices,3,4 remains a challenge due to their ultra-small and closed features. Herein, we present a self-assembled, homogenous, density-controllable, biomimetic coating that enables efficient suppression of the NPA on the wall surface of nanofluidic channels. Our approach involves a well-tailored phosphorylcholine (PC)-containing monomer material, which is designed and synthesized to possess appropriate well-defined characteristics to satisfy the stringent features of tiny closed nanofluidic channels. We anticipate our approach to be a starting point for exploring a strategic change in the anti-biofouling coating from traditional polymer-based methodologies to a monomer-based coating methodology to overcome the challenges of suppressing NPA in closed nanoscale spaces.Chip-based nanofluidic devices (hereafter referred to as “nanofluidic devices”), which are planar, transparent devices with lithography-fabricated nanochannels, offer well-defined, closed, nanoscale spaces (i.e., nanofluidic channels), allowing mimicking nanoconfined fluidic conditions of intercellular and intracellular environments.5 Such characteristics of nanofluidic channels will allow us to obtain new insight into biological phenomena and processes and develop innovative applications. For example, nanofluidic channels are considered promising tools for transporting, separating, isolating, sorting, detecting, imaging, manipulating, and assembling proteins and other biomacromolecules with super-high precision, accuracy, and temporal-spatial resolution at a single-molecule level. However, NPA has been a critical issue, significantly impeding the exploration of these promising potentials.
While NPA is more prominent in nanofluidic channels because of their ultra-large surface-area-to-volume ratios and surface charge effects, most conventional coating materials and methodologies of suppressing NPA, such as surfactants,6,7 protein-based blocking agents,8,9 and polymer-based coatings,10,11 are difficult to be directly applied to nanofluidic channels owing to various reasons as follows. The use of surfactants is prone to generate bubbles in nanofluidic channels, resulting in significant difficulties in handling fluid in nanofluidic channels. For protein-based blocking agents and polymer-based coatings, the clogging of tiny nanofluidic channels is a significant problem because the length scales of proteins and polymers are at the same scale as that of the nanofluidic channels. Recently, surface passivation using lipid vesicles has shown potential for inhabiting NPA in nanochannels.12 However, the highly sophisticated and complicated process for handling lipid vesicles in nanoscale spaces and the unstable coating property resulting from either the physical adsorption coating mechanism or the mobility of the lipid membrane greatly limit its wide applications.
Polymers have been widely used among these conventional coating materials and have become primary materials, especially in coating small devices, such as microfluidic devices,13,14 the widely successful “brother” of nanofluidic devices. This widespread application is owing to the high degree of freedom in the design of monomers and chain structures of polymers, allowing the production of precise functions to meet the requirements of small devices. Among various polymers for anti-biofouling coatings, a polymer family containing 2-methacryloyloxyethyl phosphorylcholine (MPC) is one of the most attractive and widely used polymers.15–17 MPC is a phospholipid-like monomer with a zwitterionic PC moiety, which is bioinspired from the cell membrane structure and has been proven to be attributed to the high performance of MPC polymers in suppressing NPA.18 Despite their excellent performance in many devices and systems,19–21 including microfluidic devices,22–25 when extending their application to nanofluidic devices, the MPC polymers encounter the same difficult issue of the high risk of clogging the nanofluidic channels because the dimensions of the polymer molecules approach the length scales of the nanofluidic channels.
Given that the key component in the MPC polymers to suppress NPA is the PC moiety, we speculate that if we only assemble the PC moieties on the wall surface of nanofluidic channels, we might be able to achieve the corresponding NPA suppression effect in the closed tiny nanofluidic channels. Therefore, in this study, we have designed and synthesized a hydrosilane-functionalized MPC monomer (hereafter referred to as “MPC-Si”) and explored a new strategy to use the MPC-derived monomer rather than the MPC polymers to achieve stable, nanoscale, biomimetic anti-biofouling coating in nanofluidic channels fabricated in glass substrates, as conceptually depicted in Fig. 1. The significantly smaller sizes of the monomer molecule than those of polymer molecules can substantially decrease the risk of the clogging of nanofluidic channels. Although the new strategy is quite deviant from the traditional polymer-based approach, our study indicates that it would be a simple and effective way to overcome the challenges in suppressing NPA in closed nanoscale spaces.
 | ||
| Fig. 1 Conceptual drawing of the MPC-Si coating enabling suppression of non-specific protein adsorption in a nanofluidic channel. | ||
2. Results and discussion
2.1. Molecular design, synthesis, and identification of hydrosilane-functionalized MPC monomer (MPC-Si)
While several types of substrate materials can be used to fabricate microfluidic devices, currently, glass (commonly fused silica glass) is almost the only substrate that is ideal for developing nanofluidic devices. This is because glass meets strict nanofabrication requirements and possesses various excellent properties for chemical and biological applications. Hence, when designing the MPC-derived monomer, we focused on introducing a functional group to MPC to obtain stable coating on the glass surface via chemical bonding. Silanization is a fundamental process to functionalize the glass surface by forming self-assembled monolayers of silane compounds on the surface. Generally, silane compounds such as alkoxysilanes26,27 (Si–OR, where R is an alkyl group such as methyl and ethyl group) and hydrosilanes28 (Si–H) are used. Covalent siloxane (Si–O–Si) bonds can be formed between the silane compounds and silanol (Si–OH) of the glass surface. We consider that surface modification with alkoxysilanes is not favorable for nanofluidic channels because of the following reasons. First, the modification process requires the preparation of mixed solvents (usually need to prepare 95![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :5 mixtures of ethanol and water), pH adjustment (usually need to adjust to pH of 2 with concentrated acid), strict temperature control, and long reaction time (12 h). Such complicated multiple-step processes under strict conditions are difficult to be followed correctly in the tiny closed nanofluidic channels. Second, the Si–OR bond of alkoxysilanes is easily hydrolyzed, making it difficult to preserve and handle during silanization.26,27 By contrast, surface modification reaction between hydrosilanes and silanols of the glass surface can be easily performed in the presence of tris(penta fluorophenyl)borane (TPFB) as the catalyst in a short time at room temperature (25 °C). Moreover, the Si–H bond in hydrosilanes is very stable in the absence of the catalyst,28 quite favoring preservation and handling during silanization in the nanofluidic devices. Therefore, we focused on hydrosilane (Si–H) in this study.
:5 mixtures of ethanol and water), pH adjustment (usually need to adjust to pH of 2 with concentrated acid), strict temperature control, and long reaction time (12 h). Such complicated multiple-step processes under strict conditions are difficult to be followed correctly in the tiny closed nanofluidic channels. Second, the Si–OR bond of alkoxysilanes is easily hydrolyzed, making it difficult to preserve and handle during silanization.26,27 By contrast, surface modification reaction between hydrosilanes and silanols of the glass surface can be easily performed in the presence of tris(penta fluorophenyl)borane (TPFB) as the catalyst in a short time at room temperature (25 °C). Moreover, the Si–H bond in hydrosilanes is very stable in the absence of the catalyst,28 quite favoring preservation and handling during silanization in the nanofluidic devices. Therefore, we focused on hydrosilane (Si–H) in this study.
We designed a hydrosilane-functionalized MPC monomer (i.e., MPC-Si) by adding a hydrosilane group to the α,β-unsaturated carbonyl part in the methacrylate group of the MPC monomer (Fig. 2a). The addition reaction of α,β-unsaturated carbonyl compounds, 1,2-addition and 1,4-addition, also known as Michael addition, may occur. In 1,2-addition, the oxygen atom in the carbonyl group and the neighboring carbon atom will be attacked by the nucleophile, while for Michael addition (i.e., 1,4-addition), the oxygen atom and the third carbon atom next to it will be attacked. Owing to the influence of the oxygen atom, the neighboring carbon atom is positively polarized and easily attacked. Therefore, in terms of reaction kinetics, the 1,2-addition is more likely to occur. Nevertheless, when the nucleophile is bulky, the nucleophile will preferentially bind to the carbon far from the oxygen atoms to form a thermodynamically stable compound. In this study, a bulky nucleophile, 1,1,3,3-tetrametyldisilazane (TMD-Si), is used as a provider of hydrosilanes. Accordingly, the Michael addition will proceed preferentially (Fig. 2a). Since the Michael addition reactions usually proceed in the presence of a base29 or Lewis's acid catalyst,30 two typical catalysts, potassium carbonate (K2CO3; base) and lithium perchlorate (LiClO4; Lewis's acid), were chosen in this study. Hence, the synthesis of MPC-Si was performed according to the reaction as schemed in Fig. 2a.
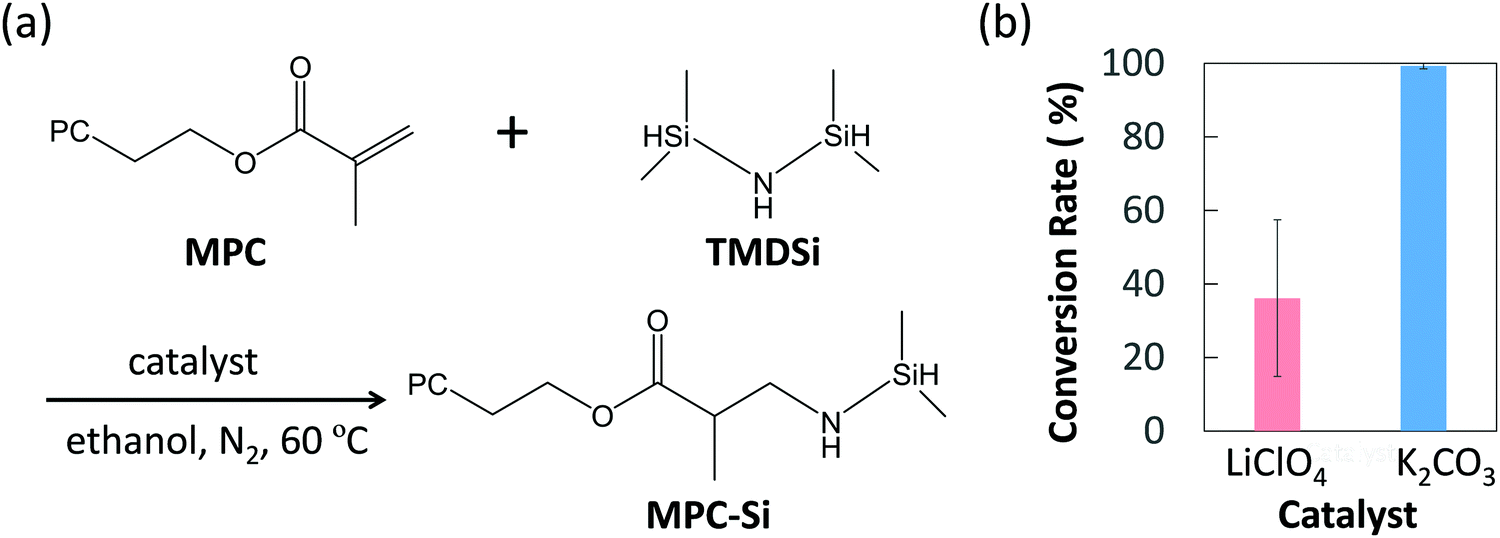 | ||
| Fig. 2 Synthesis of MPC-Si. (a) Synthesis scheme of MPC-Si via Michael addition, (b) the conversion rate depending on the type of the catalyst (Data are mean ± S.D, n = 3). | ||
The structures of purified products were identified by proton nuclear magnetic resonance (1H-NMR) spectra, and the results revealed that the MPC-Si was successfully synthesized as designed in both cases using different catalysts. To evaluate the efficiencies of two catalysts used, the conversion rates of the reactants were determined by comparing the peak intensities of proton of the methacrylate group from MPC (5.5–6.5 ppm) and the silane end group in MPC-Si (0–0.4 ppm) obtained from the 1H-NMR of the products. The conversion rate was 36.1 ± 21.3% for LiClO4, while it was 99.3 ± 0.8% for K2CO3 (Fig. 2b), revealing that the base catalyst K2CO3 was more efficient for synthesizing the MPC-Si. Furthermore, K2CO3 remained in a solid state in the ethanol solvent after the reaction, which ensured that it could be removed easily by simple filtration. Thus, we chose K2CO3 as the catalyst for synthesizing MPC-Si, used in the subsequent investigation.
2.2. Surface modification and characterization
Owing to the extremely small and closed features of nanofluidic channels, it is difficult to directly characterize the details of surface modifications inside nanofluidic channels using surface characterization technologies such as X-ray photoelectron spectroscopy (XPS) analysis, contact angle analysis, and atomic force microscopy (AFM), which are widely used for characterization of open surfaces. Hence, glass substrates (i.e., fused silica glass substrates) were used to analyze optimal conditions of surface modification and characterize the properties of the modified surfaces. The surface modification can be easily achieved for glass substrates by a relatively simple dip-coating process and is detailed as follows. The cleaned glass substrates were dipped in an ethanol solution of MPC-Si of desired concentration containing TPFB (MPC-Si/TPFB = 100/1, mol/mol) as a catalyst for 5 min at room temperature (25 °C), where TPFB could promote the cleavage of Si–H bonding and the formation of Si–O–Si bonding (Fig. 3a). This simple process can be easily extended to the modification of nanofluidic channels, as described later. Experiments on the determination of the solubility of MPC-Si showed that the saturation concentration of MPC-Si in ethanol was 0.30 wt%. Thus, in this study, the upper limit of concentration was set to 0.30 wt%. Solutions of MPC-Si at concentrations of 0.10 wt% and 0.03 wt% were also prepared to determine the optimal concentration. In addition, uncoated glass substrates were used as a benchmark.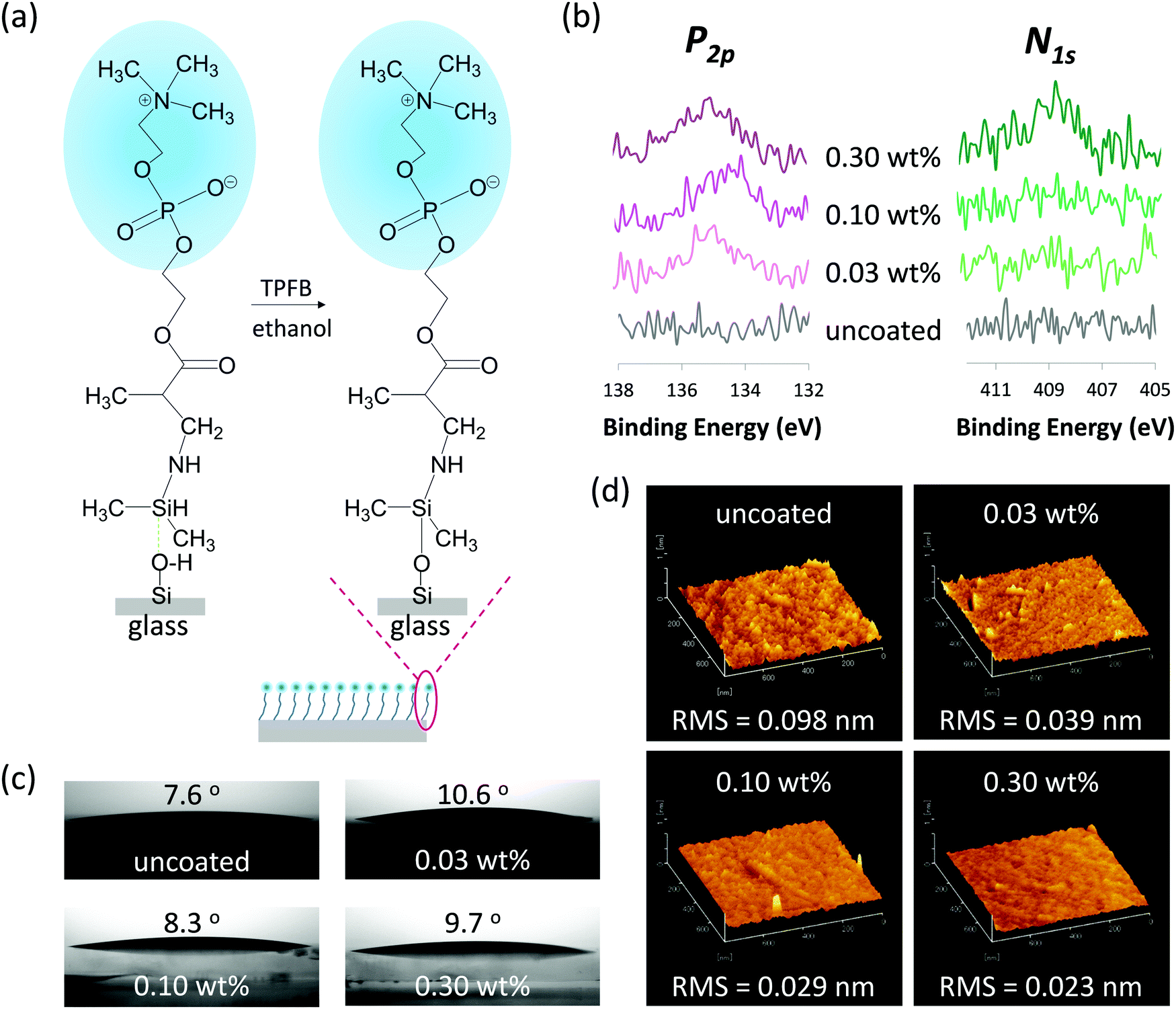 | ||
| Fig. 3 Formation and characterization of MPC-Si coatings on glass substrates. (a) Schematic drawing of self-assembly of MPC-Si molecules on the glass substrates via the silanization reaction to form MPC-Si coatings. (b) XPS spectra of phosphorus (P2p) and nitrogen (N1s), (c) water contact angle images, and (d) AFM images of the glass substrates coated with the 0.03 wt%, 0.10 wt%, and 0.30 wt% MPC-Si in comparison with those of uncoated glass substrates. | ||
To confirm whether MPC-Si was successfully coated on the surface, surface elemental analysis using XPS was performed. Fig. 3b shows the XPS spectra of phosphorus (P2p) and nitrogen (N1s), which are the characteristic elements in the PC head group of MPC-Si. The binding energy scales were corrected based on the binding energy of Au4f (84.0 eV) as an internal standard. As shown in Fig. 3b, while no signals were detected from the uncoated glass substrates, signals of both phosphorus (P2p, 134–135 eV) and nitrogen (N1s, 407–408 eV) were detected from the modified surface of the samples. This result reveals that MPC-Si was successfully coated on the glass substrate using the aforementioned simple coating process, independent of the concentrations used. In addition, the intensities of XPS peaks of both phosphorus (P2p) and nitrogen (N1s) increase as the concentration of MPC-Si coating increases (Fig. 3b), suggesting that the density of PC head groups on the glass surface increase as the concentration of MPC-Si increases, and hence the modification is controllable. Such a feature may provide us a simplistic measure to tune the density of PC head groups on the surface by only adjusting the concentration of MPC-Si coating, which is very favorable for developing a variety of potential applications in nanofluidic devices.
Proteins are prone to bind to hydrophobic surfaces; that is, although the mechanism of NPA to a surface is complicated and associated with multiple comprehensive aspects of the surface properties, hydrophobic surfaces are usually unfavorable for suppressing the NPA.31,32 This phenomenon occurs because the hydrophobic surface may dehydrate protein structure to form hydrophobic interactions with the hydrophobic surface.32 Accordingly, the surface wettability of all samples was characterized by measuring the water contact angle on the sample surface. As shown in Fig. 3c, the modified surface of the glass substrates exhibited very small contact angles similar to that of uncoated glass, indicating that the surfaces were highly hydrophilic after modification. Considering the molecular structure of MPC-Si comprises a hydrophilic PC head group and a hydrophobic silane end group, the above result may imply that in the aqueous condition, the MPC-Si molecules can self-assemble on the glass surface with the PC head groups facing outward of the coating layer, as illustrated in Fig. 3a. This characteristic would be favorable for the MPC-Si to suppress NPA on the surface actively.
Owing to the ultrahigh surface-to-volume ratios of nanofluidic channels, the surface morphology of the channel wall influences or dominates a variety of nanofluidic phenomena, including NPA in the nanofluidic channels. A rough surface may bring risks in NPA;31,33 therefore, a smooth channel wall is usually favored and desired for handling proteins in the nanofluidic channels. Accordingly, AFM analysis was conducted to evaluate the surface morphologies of the samples. As revealed by the AFM images (Fig. 3d), all samples exhibited homogeneous surfaces. Further, a quantitative evaluation of these AFM images using the root mean square (RMS) of the roughness revealed that the surface of the glass substrate was much smoother after the MPC-Si coating (Fig. 3d). In addition, although all MPC-Si modified surfaces displayed low RMS values, these values gradually decreased as the concentration of MPC-Si increased (Fig. 3d), revealing that much more homogenous surface coating could be achieved by increasing the concentration of MPC-Si.
2.3. Evaluation of capability of suppressing NPA on modified glass substrates
The capability of MPC-Si coatings to suppress NPA was quantitively evaluated according to a Micro BCA protocol, which is a widely used standard method to measure small amounts of protein in a sample.34,35 Three typical types of proteins with different isoelectric point (pI) and different molecular weight (Mw), i.e., bovine serum albumin (BSA; pI = 4.7, Mw = 66.0 kDa), hemoglobin (Hb; pI = 6.8 to 7.0, Mw = 64.5 kDa), and cytochrome c (Cyt. C; pI = 10.0 to 10.5, Mw = 12.4 kDa), were chosen as representative proteins in terms of the charge condition and the size, which are considered important molecular factors of the protein associated with NPA. The pI of a protein is commonly defined as the pH at which the protein has no net charge. Hence, proteins with different pI values are considered to have different net charges on their surfaces at a neutral pH. In addition, the charge condition of a protein may induce electrostatic interactions with a solid surface, thereby unfortunately increasing NPA.31 Meanwhile, the sizes of proteins are usually proportional to their molecular weights. In some cases, the size of a protein may influence NPA on a surface through the steric effect.36Phosphate-buffered saline (PBS, pH 7.4) was used as a buffer for preparing protein solutions (0.32 g L−1). Hence, BSA, Hb, and Cyt. C were anionic, neutral, and cationic, respectively, under our experimental condition. As shown in Fig. 4a–c, all three types of proteins were heavily absorbed on the uncoated substrates with 0.83 μg cm−2, 1.67 μg cm−2, and 1.08 μg cm−2 of NPA for BSA, Hb, and Cyt. C, respectively. By contrast, the amounts of NPA for all three types of representative proteins with a wide range of pI values reduced on all MPC-Si modified surfaces (Fig. 4a–c). These results indicate that the MPC-Si coatings can suppress non-specific adsorption of anionic, neutral, and cationic proteins. Such characteristics, along with the insights from the result of the contact angle measurement (Fig. 3a and c), indicate that the zwitterionic PC head groups of the MPC-Si molecules assembled on the glass surface can be well oriented outward of the coating layer and play an essential role to suppress NPA as those in an MPC polymer.18,19,21,37
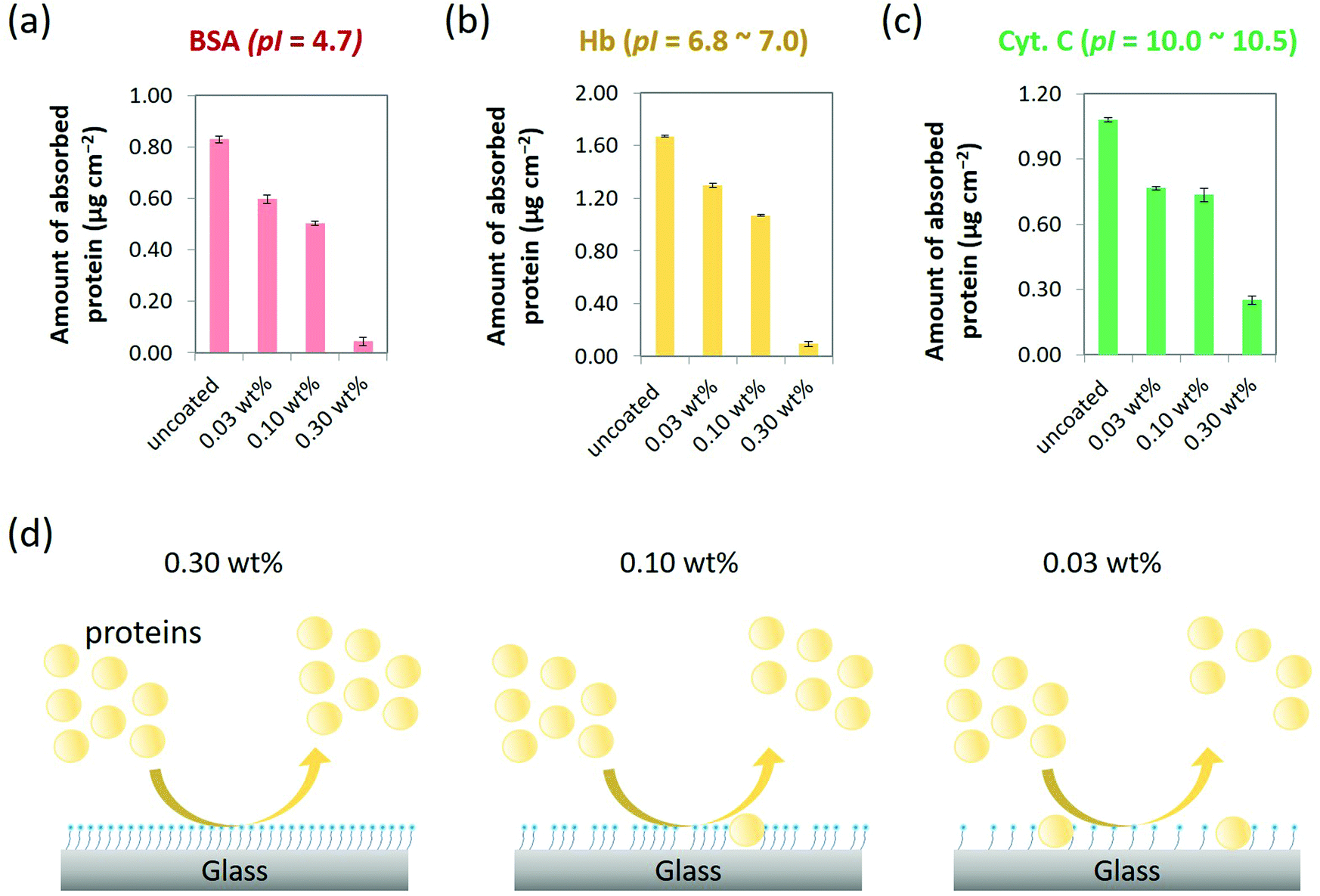 | ||
| Fig. 4 Amounts of typical (a) anionic (BSA), (b) neutral (Hb), and (c) cationic (Cyt. c) proteins adsorbed on the glass substrates coated with the 0.03 wt%, 0.10 wt%, and 0.30 wt% MPC-Si in comparison with those of uncoated glass substrates. (d) Different NPA suppressing capabilities exhibited by MPC-Si coatings of different concentrations, which may be ascribed to the difference in the density of the MPC-Si molecules on the surface. | ||
To further detail the differences in the performance of these coatings to suppress NPA, we calculated the reduction rate of the amount of NPA, which is defined as the percentage of [amount of adsorbed protein on the uncoated substrate – amount of adsorbed protein on the MPC-Si coated substrate]/amount of adsorbed protein on the uncoated substrate. For all three types of representative proteins, the reduction rate increased as the concentration of MPC-Si for coating increased. Such reduction trend is possibly ascribed to the fact that the density of the PC head groups on the coating surface increases as the concentration of MPC-Si for coating increases (Fig. 4d); similar insights were derived from the XPS analysis (Fig. 3b). Particularly, in comparison with the 0.03 wt% and 0.10 wt% MPC-Si coatings, the 0.30 wt% MPC-Si coating exhibited significantly higher performance in suppressing NPA of all three types of representative proteins (Fig. 4a–c), with remarkably high reduction rates of 95.2%, 94.6%, and 76.9% for BSA, Hb, and Cyt. C, respectively. As schematically illustrated in Fig. 4d, this result may suggest that while varying degrees of uncoated areas existed on the substrates at two lower concentrations (i.e., 0.03 wt% and 0.10 wt%), the substrate was coated fully and homogeneously covered by 0.30 wt% MPC-Si solution. This feature is consistent with the insights gained from the results of the AFM characterization (Fig. 3d). Accordingly, the 0.30 wt% MPC-Si solution was used for coating the nanofluidic channels in the following experiments.
2.4. Evaluation of NPA suppression capability in nanofluidic channels
Nanofluidic devices fabricated on glass substrates (40 mm × 30 mm × 0.7 mm) were used to evaluate the capability of MPC-Si to suppress NPA in nanofluidic channels (Fig. 5a). Each nanofluidic device comprises 240 parallel-arrayed straight nanofluidic channels (800 nm wide, 300 nm deep, 400 μm long, at intervals of 2 μm) in the center, bridging two arc-shaped microfluidic channels (500 μm wide and 2.8 μm deep) on the two sides (Fig. 5b). Such micro-/nanofluidic channel hybrid is a standard nanofluidic device structure widely used in the field of nanofluidics. With the hybrid structure, liquid can be easily introduced into the central nanofluidic channels from the adjacent microfluidic channel (Fig. 5c). Each nanofluidic channel has an ultra-small volume of 96 femtoliters (fL = 10−15 L); thus, the total volume of the nanofluidic channel array is approximately 23 picoliters (pL = 10−12 L), which is of the same order as that of a single mammalian cell. Wide intervals (2 μm) between the nanofluidic channels were deliberately designed to facilitate easy observation of the details of each nanofluidic channel.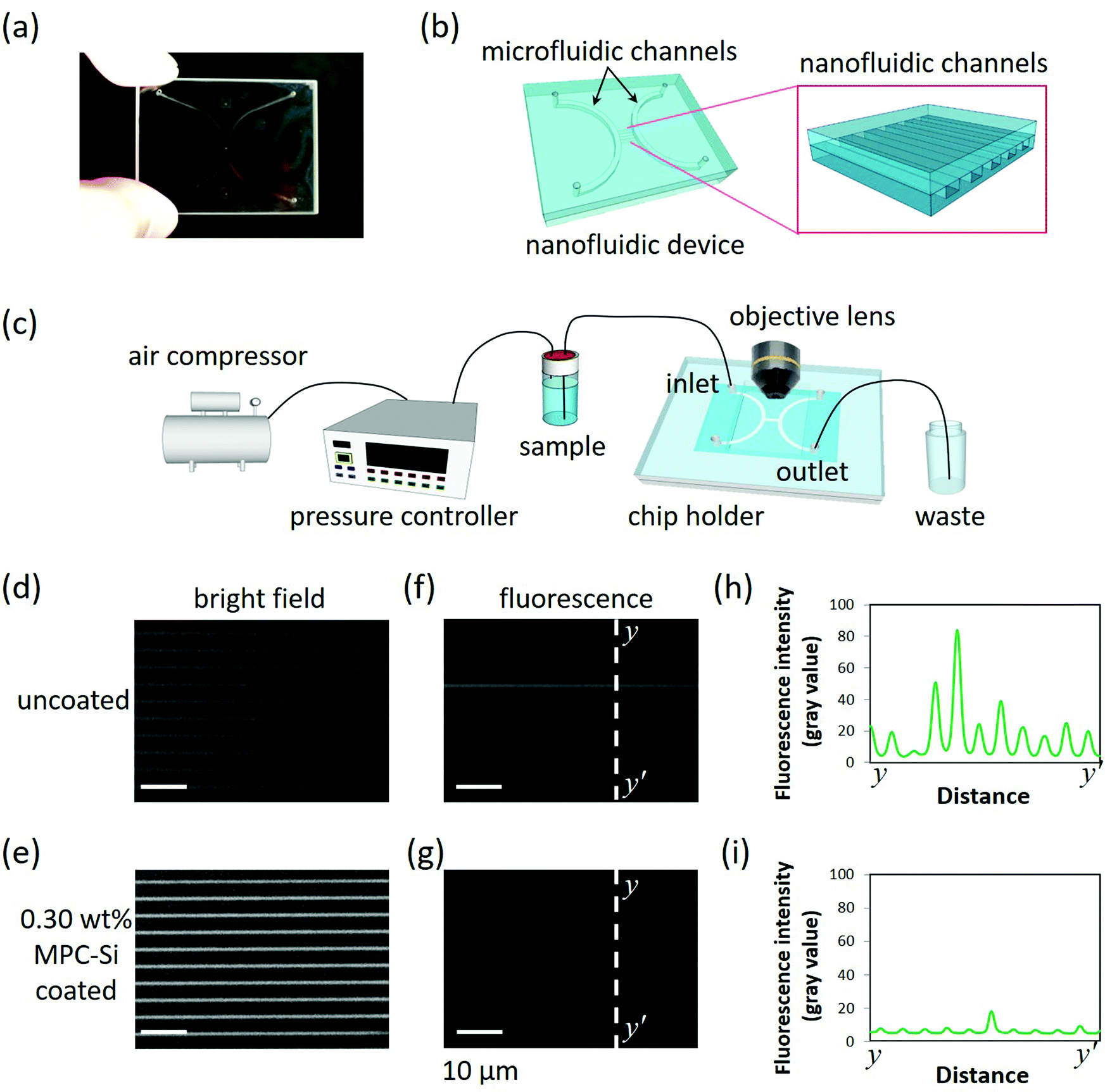 | ||
| Fig. 5 Evaluation of NPA suppression capability in (a) a nanofluidic device with (b) a standard micro-/nanofluidic channel hybrid. (c) Schematic drawing of the experimental setup for liquid introduction; bright-field microscopy images of (d) uncoated and (e) the 0.30 wt% MPC-Si coated nanofluidic channels before introducing the protein solution; fluorescence microscopy images of (f) uncoated and (g) the 0.30 wt% MPC-Si coated nanofluidic channels after protein adsorption experiments with BSA-FITC. (h and i) Corresponding line profiles of the fluorescence intensities of y–y′ lines on (f and g), respectively. | ||
Using the optimal coating conditions obtained from the dip-coating procedure (Fig. 3 and 4), a simple one-step filling coating process was adopted to modify the nanofluidic channels using a home-built pressure-driven flow system (Fig. 5c). The 0.30 wt% MPC-Si solution with TPFB catalyst (MPC-Si/TPFB = 100/1, mol/mol) was introduced into the nanofluidic channels from the left microfluidic channel. During the process, the solution of MPC-Si continuously flowed through the tiny nanofluidic channels, and no clogging was observed. This fact indicates that despite the high saturation concentration (0.30 wt%), MPC-Si did not induce clogging of the nanofluidic channels, consistent with our previous observations. After the channels were filled with the solution, the flow was stopped by regulating the air pressure, and the inlets and outlets were closed. Further, these channels were kept at room temperature for 1 h to achieve a uniform coating. After that, the coated nanofluidic channels were subjected to continuous strong fluidic rinsing by flowing absolute ethanol under high pressure at 430 kPa for overnight to remove the residuals. Finally, the coating process was completed by drying the channels under a nitrogen gas stream. As described later, the coating kept a high performance although experiencing such high-pressure nanofluidic operation, suggesting that the MPC-Si coating was very stable. As a benchmark, a nanofluidic device without MPC-Si coating was also used in this investigation. Similar to the uncoated nanofluidic device (Fig. 5d), the entire passage of the coated nanofluidic device could be clearly visible from the bright-field image (Fig. 5e) obtained using an upright optical microscope (BX53, Olympus). This comparison further confirms that MPC-Si coating did not induce clogging in the nanofluidic channels. In addition, the channel areas of the coated nanofluidic channels in the bright-field image (Fig. 5e) are brighter than those of the uncoated nanofluidic channels (Fig. 5d). Such a difference may be attributed to a small change in the refractive index of the channel surface after a thin coating layer is formed, implying that the nanofluidic channels were successfully coated by MPC-Si. In addition, the quite uniform brightness in the channel shown in Fig. 5e may also suggest that the MPC-Si coating in the nanofluidic channels was homogeneous.
BSA conjugated with fluorescein isothiocyanate (BSA-FITC) was used to evaluate the capability of the MPC-Si coating to suppress NPA. BSA is a protein well known for its easy adsorption to various surfaces (as shown in Fig. 4); therefore, it is generally used as a model protein to study NPA and evaluate the performance of anti-NPA coatings. In addition, BSA-FITC has been widely used in fluorescence imaging of NPA at the micro-/nanoscales in some small devices owing to the excellent fluorescence properties of FITC. A BSA-FITC (PBS as a buffer) solution at a high concentration of 0.32 g L−1 was used, a same level as those of intracellular proteins.38 After all channels of the nanofluidic device were filled completely with the protein solution using the liquid input system (Fig. 5c), the device was kept at 4 °C for 12 h to guarantee sufficient interactions between the protein molecules and the wall surface of the nanofluidic channels. Subsequently, the protein solution in the channels was replaced and rinsed with PBS to remove the unabsorbed protein molecules. Finally, the protein adsorption in the channels was evaluated by fluorescence imaging using the microscope with an electron multiplying charge-coupled device (EM-CCD) camera (iXon Ultra 897, Andor), whose ultimate sensitivity allows the detection of fluorescence signals even at the ultraweak levels. In contrast to the uncoated nanofluidic channels, where apparent strong fluorescence ascribing to adsorbed BSA-FITC molecules was observed (Fig. 5f), almost no fluorescence was detected in the MPC-Si coated nanofluidic channels (Fig. 5g). As shown in Fig. 5h and i, fluorescence intensity profiles corresponding to the dotted lines y–y′ in both fluorescence images (Fig. 5f and g) further support the observation quantitatively. These results indicate that while a significant amount of BSA-FITC was adsorbed in the uncoated nanofluidic channels, almost no BSA-FITC was absorbed in the MPC-Si coated nanofluidic channels. Therefore, the newly developed MPC-Si coating solution possesses a high capability to modify tiny nanofluidic channels and effectively suppress NPA in nanofluidic channels.
3. Conclusions
In this study, we present a simple, self-assembled, homogenous, density-controllable, biomimetic coating enabling the efficient suppression of NPA in nanofluidic channels. The delicate coating was based on a well-tailored, hydrosilane-functionalized MPC monomer material (i.e., MPC-Si), especially designed and synthesized to have well-defined molecular characteristics and excellent anti-biofouling surface properties to meet the stringent features of tiny and closed nanofluidic channels. The use of the new monomer material allowed us to develop the first proof of concept to demonstrate that, similar to the MPC polymers, the MPC monomer can possess high NPA suppressing capability if high-density biomimetic PC head groups can be assembled on the surface. We believe that our approach provides a general and straightforward solution to the critical issue of NPA, which has impeded the application of nanofluidics to a wide range of fields associated with proteins and other biomacromolecules. Further, it offers a starting point for exploring a strategic change in the anti-biofouling coating from traditional polymer-based methodologies to a monomer-based methodology to overcome the challenges of suppressing NPA in closed nanoscale spaces. Last but not least, in the future the further elucidation of the mechanism of MPC-Si to suppress NPA by quantitative characterization of the detailed assembly structure, orientation, and density of MPC-Si molecules on the glass surface using both experimental and molecular simulation techniques39 would contribute to the improvement of the capability and applications of MPC-Si.4. Experimental
4.1. Materials and reagents
The MPC monomer was kindly provided by Prof. Kazuhiko Ishihara, University of Tokyo. TMD-Si was purchased from Tokyo Kasei Kogyo Co. (Tokyo, Japan). Ethanol (99.9%, solvent for Michael addition), BSA, BSA-FITC, Hb, PBS were purchased from Merck KgaA (Darmstadt, Germany). K2CO3, TPFB, Ethanol-d6 (CD3CD2OD, 99.5% for NMR spectroscopy), and Cyt. C from horse heart were purchased from Wako Pure Chemical (Tokyo, Japan). Fused silica glass substrates used for surface modification (10 mm × 10 mm × 0.7 mm) and fabricated nanofluidic device (30 mm × 40 mm × 0.7 mm) were purchased from Sendai Quartz (Sendai, Japan).4.2. Synthesis of MPC-Si
The Michael addition reaction of MPC monomer and TMD-Si was conducted in a flowing nitrogen atmosphere and water-cooling reflux. First, ethanol (40 mL) was added as the solvent into a rounded-bottom flask (100 mL, TOP, Tokyo, Japan). Then, the MPC monomer (1 mmol) and TMD-Si (5 mmol) were added as reactants, and LiClO4 or K2CO3 (2 mmol) was added as catalyst. The reaction was performed at 60 °C for 12 h. For the synthesis using the catalyst K2CO3, after the reaction, K2CO3 was filtered, and the solvent was evaporated with a rotary evaporator (N-1110, EYELA, Tokyo, Japan). For the synthesis using the catalyst LiClO4, the product was further purified using the silica gel column chromatography and MPC-Si could be obtained. Then, MPC-Si was dried in vacuo. The structure of MPC-Si was identified by 1H-NMR spectra (JEOL, JMN-ECS400, Tokyo, Japan) (CD3CD2OD, (δ): 0.03–0.11 (CH3Si–), 1.07–1.11 (CH3–CH–), 3.21 (–CH2–NH–Si–), 3.24 (CH3–N–), 3.39–3.49 (–CH–CO–), 3.62–3.76 (–CH2–CH2–N–), 3.85–3.92 (–CH2–O–CO), 4.10–4.13 (H–Si–), 4.21–4.30 (–CH2–OP), as shown in Fig. S1, ESI.†4.3. Surface coating and characterization
First, the glass substrates were perfectly cleaned with the piranha solution for 10 min. Then, the cleaned glass substrates were dip-coated in MPC-Si solutions with the desired concentration containing TPFB for 5 min. Finally, the coated substrates were rinsed with ethanol to remove the residuals and dried under nitrogen gas flow. The formation of MPC-Si coatings on the surfaces was confirmed by surface elemental analysis using XPS (ESCA-3200, Shimadzu, Kyoto, Japan). The wettability of the sample surfaces was characterized by measuring the water contact angle using a contact angle meter (DM 500, Kyowa Interface Science, Saitama, Japan). The surface morphologies and roughness of the sample surfaces were characterized by AFM (SPA-400, Hitachi High-Tech, Tokyo, Japan).4.4. Quantitative evaluation of NPA
The amount of non-specifically adsorbed proteins on the sample surfaces was quantitatively evaluated according to a standard protocol reported elsewhere.34,35 A brief description of the procedure is as follows. First, solutions of BSA, Hb, and Cyt. C (0.32 g L−1; PBS as a buffer) were prepared. Further, for each protein adsorption experiment, uncoated and the MPS-Si coated substrates were immersed in the protein solution at 37 °C for 2 h to sufficiently absorb the protein. Then, the substrates were rinsed twice under a stirring condition at 300 rpm in PBS for 5 min to wash off the unabsorbed proteins. Subsequently, to detach and collect total non-specifically adsorbed proteins on the substrates, each substrate was ultrasonically rinsed in 2.0 mL of sodium dodecyl sulfate (SDS) solution in a small sealed case for 10 min. Finally, the amount of total protein collected in the SDS solution from each substrate was measured using the Micro BCA Protein Assay Kit (Pierce Biotechnology, Rockford, IL, USA).4.5. Fabrication of nanofluidic devices
The nanofluidic devices were fabricated according to previously reported processes.40–42 A brief description of the process is as follows. Nanochannels were fabricated on a glass substrate by electron-beam (EB) lithography using an EB system (ELS-7500; Elionix, Tokyo, Japan), followed by dry etching using a high-density inductively coupled plasma etching system (NE-550; Ulvac, Kanagawa, Japan). Microchannels with inlets and outlets were fabricated on another glass substrate by a conventional photolithography process followed by plasma dry etching and through-hole punching. Subsequently, the substrate with nanochannels and the substrate with microchannels were bonded using previously reported bonding methods43–45 form the nanofluidic device finally. The devices can be reused after regeneration according to a protocol previously reported by us.46Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was partially supported by JSPS KAKENHI (Grant No. JP26706010, JP18H01848, JP21H04640, JP19KK0129, and JP20H00497), JST, PRESTO (Grant No. JPMJPR18H5), MEXT KAKENHI (Grant No. JP26107714, JP19H04678, and JP21H05231), the Asahi Glass Foundation, and the National Natural Science Foundation of China (NSFC) (Grant No. 21628501). The authors thank Prof. Kazuhiko Ishihara at the University of Tokyo for kindly providing MPC monomers, Prof. Hiroaki Kawata and Prof. Yoshihiko Hirai at Osaka Prefecture University for their assistance in plasma dry etching of glass nanofluidic channels, and Prof. Hiroshi Inoue and Prof. Eiji Higuchi at Osaka Prefecture University for their assistance in XPS determination.References
- V. B. Damodaran and N. S. Murthy, Biomater. Res., 2016, 20, 18 CrossRef PubMed.
- I. Banerjee, R. C. Pangule and R. S. Kane, Adv. Mater., 2011, 23, 690–718 CrossRef CAS PubMed.
- Y. Xu, Adv. Mater., 2018, 30, 1702419 CrossRef PubMed.
- D. G. Haywood, A. Saha-Shah, L. A. Baker and S. C. Jacobson, Anal. Chem., 2015, 87, 172–187 CrossRef CAS PubMed.
- J. Yang and Y. Xu, Chin. Chem. Lett., 2021 DOI:10.1016/J.CCLET.2021.09.066.
- L. Shen, A. Guo and X. Zhu, Surf. Sci., 2011, 605, 494–499 CrossRef CAS.
- N. B. Holland, Y. Qiu, M. Ruegsegger and R. E. Marchant, Nature, 1998, 392, 799–801 CrossRef CAS PubMed.
- B. Sweryda-Krawiec, H. Devaraj, A. George Jacob and J. J. Hickman, Langmuir, 2004, 20, 2054–2056 CrossRef CAS PubMed.
- T. Bonnard, A. Jayapadman, J. A. Putri, J. Cui, Y. Ju, C. Carmichael, T. A. Angelovich, S. H. Cody, S. French, K. Pascaud, H. A. Pearce, S. Jagdale, F. Caruso and C. E. Hagemeyer, ACS Nano, 2018, 12, 6988–6996 CrossRef CAS PubMed.
- A. M. C. Maan, A. H. Hofman, W. M. Vos and M. Kamperman, Adv. Funct. Mater., 2020, 30, 2000936 CrossRef CAS.
- R. F. Landis, A. Gupta, Y.-W. Lee, L.-S. Wang, B. Golba, B. Couillaud, R. Ridolfo, R. Das and V. M. Rotello, ACS Nano, 2017, 11, 946–952 CrossRef CAS.
- F. Persson, J. Fritzsche, K. U. Mir, M. Modesti, F. Westerlund and J. O. Tegenfeldt, Nano Lett., 2012, 12, 2260–2265 CrossRef CAS PubMed.
- A. Shakeri, S. Khan and T. F. Didar, Lab Chip, 2021, 21, 3053–3075 RSC.
- A. Gokaltun, M. L. Yarmush, A. Asatekin and O. B. Usta, Technology, 2017, 5, 1–12 CrossRef PubMed.
- K. Ishihara, J. Biomed. Mater. Res., Part A, 2019, 107, 933–943 CrossRef CAS PubMed.
- S. Monge, B. Canniccioni, A. Graillot and J.-J. Robin, Biomacromolecules, 2011, 12, 1973–1982 CrossRef CAS PubMed.
- Y. Iwasaki and K. Ishihara, Sci. Technol. Adv. Mater., 2012, 13, 064101 CrossRef CAS PubMed.
- K. Ishihara, H. Nomura, T. Mihara, K. Kurita, Y. Iwasaki and N. Nakabayashi, J. Biomed. Mater. Res., 1998, 39, 323–330 CrossRef CAS PubMed.
- M. Chen, W. H. Briscoe, S. P. Armes and J. Klein, Science, 2009, 323, 1698–1701 CrossRef CAS PubMed.
- H. Ou, T. Cheng, Y. Zhang, J. Liu, Y. Ding, J. Zhen, W. Shen, Y. Xu, W. Yang, P. Niu, J. Liu, Y. An, Y. Liu and L. Shi, Acta Biomater., 2018, 65, 339–348 CrossRef CAS PubMed.
- T. Moro, Y. Takatori, K. Ishihara, T. Konno, Y. Takigawa, T. Matsushita, U. Chung, K. Nakamura and H. Kawaguchi, Nat. Mater., 2004, 3, 829–836 CrossRef CAS PubMed.
- Y. Xu, M. Takai, T. Konno and K. Ishihara, Lab Chip, 2007, 7, 199–206 RSC.
- J. Sibarani, M. Takai and K. Ishihara, Colloids Surf., B, 2007, 54, 88–93 CrossRef CAS PubMed.
- Y. Xu, K. Sato, K. Mawatari, T. Konno, K. Jang, K. Ishihara and T. Kitamori, Adv. Mater., 2010, 22, 3017–3021 CrossRef CAS PubMed.
- Y. Xu, K. Mawatari, T. Konno, T. Kitamori and K. Ishihara, ACS Appl. Mater. Interfaces, 2015, 7, 23089–23097 CrossRef CAS PubMed.
- G. L. Witucki, J. Coat. Technol. Res., 1993, 65, 822 Search PubMed.
- F. D. Osterholtz and E. R. Pohl, J. Adhes. Sci. Technol., 1992, 6, 127–149 CrossRef CAS.
- N. Moitra, S. Ichii, T. Kamei, K. Kanamori, Y. Zhu, K. Takeda, K. Nakanishi and T. Shimada, J. Am. Chem. Soc., 2014, 136, 11570–11573 CrossRef CAS.
- A. Michael, J. Prakt. Chem., 1887, 35, 349–356 CrossRef.
- S. Kobayashi and T. Mukaiyama, Chem. Lett., 1986, 221–224 CrossRef CAS.
- V. Hlady and J. Buijs, Curr. Opin. Biotechnol, 1996, 7, 72–77 CrossRef CAS.
- P. Roach, D. Farrar and C. C. Perry, J. Am. Chem. Soc., 2005, 127, 8168–8173 CrossRef CAS PubMed.
- K. Rechendorff, M. B. Hovgaard, M. Foss, V. P. Zhdanov and F. Besenbacher, Langmuir, 2006, 22, 10885–10888 CrossRef CAS PubMed.
- P. K. Smith, R. I. Krohn, G. T. Hermanson, A. K. Mallia, F. H. Gartner, M. D. Provenzano, E. K. Fujimoto, N. M. Goeke, B. J. Olson and D. C. Klenk, Anal. Biochem., 1985, 150, 76–85 CrossRef CAS.
- Y. Xu, M. Takai and K. Ishihara, Biomaterials, 2009, 30, 4930–4938 CrossRef CAS PubMed.
- E. A. Vogler, Biomaterials, 2012, 33, 1201–1237 CrossRef CAS.
- Y. Xu, M. Takai and K. Ishihara, Biomacromolecules, 2009, 10, 267–274 CrossRef CAS.
- N. J. Mudrak, P. S. Rana and M. A. Model, Cytometry, Part A, 2018, 93, 297–304 CrossRef PubMed.
- S. Chen, J. Zheng, L. Li and S. Jiang, J. Am. Chem. Soc., 2005, 127, 14473–14478 CrossRef CAS PubMed.
- Y. Xu, M. Shinomiya and A. Harada, Adv. Mater., 2016, 28, 2209–2216 CrossRef CAS PubMed.
- Y. Xu and B. Xu, Small, 2015, 11, 6165–6171 CrossRef CAS.
- H. Kawagishi, S. Kawamata and Y. Xu, Nano Lett., 2021, 21, 10555–10561 CrossRef CAS.
- Y. Xu, N. Matsumoto, Q. Wu, Y. Shimatani and H. Kawata, Lab Chip, 2015, 15, 1989–1993 RSC.
- H. Kamai and Y. Xu, Micromachines, 2021, 12, 775 CrossRef PubMed.
- Y. Xu, C. Wang, L. Li, N. Matsumoto, K. Jang, Y. Dong, K. Mawatari, T. Suga and T. Kitamori, Lab Chip, 2013, 13, 1048–1052 RSC.
- Y. Xu, Q. Wu, Y. Shimatani and K. Yamaguchi, Lab Chip, 2015, 15, 3856–3861 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1tb02627e |
| ‡ Current address: S. Fukuda (Former name: Qian Wu), School of Medicine, Kobe University, 7-5-1, Kusunoki-cho, Chuo-ku, Kobe, Hyogo 650-0017, Japan. |
| This journal is © The Royal Society of Chemistry 2022 |