
Alloying electrode coatings towards better magnesium batteries†
Clément
Pechberty
 ad,
Arthur
Hagopian
ad,
Jean-Bernard
Ledeuil
bd,
Dominique
Foix
bd,
Joachim
Allouche
bd,
Jean-Noël
Chotard
cd,
Olivera
Lužanin
ef,
Jan
Bitenc
e,
Robert
Dominko
efg,
Rémi
Dedryvère
bdg,
Jean-Sébastien
Filhol
ad,
Lorenzo
Stievano
adg and
Romain
Berthelot
*adg
ad,
Arthur
Hagopian
ad,
Jean-Bernard
Ledeuil
bd,
Dominique
Foix
bd,
Joachim
Allouche
bd,
Jean-Noël
Chotard
cd,
Olivera
Lužanin
ef,
Jan
Bitenc
e,
Robert
Dominko
efg,
Rémi
Dedryvère
bdg,
Jean-Sébastien
Filhol
ad,
Lorenzo
Stievano
adg and
Romain
Berthelot
*adg
aInstitut Charles Gerhardt de Montpellier (ICGM), Univ Montpellier, CNRS, ENSCM, Pôle Chimie Balard – 1919, Route de Mende, 34293 Montpellier Cedex 5, France. E-mail: romain.berthelot@umontpellier.fr
bIPREM, E2S-UPPA, CNRS, Univ. Pau & Pays Adour, Pau, France
cLRCS, CNRS UMR7314 Université de Picardie Jules Verne, HUB de l’Energie, 15 Rue Baudelocque, 80039 Amiens, France
dRéseau sur le Stockage Electrochimique de l’Energie (RS2E), CNRS FR3459, HUB de l’Energie, 15 Rue Baudelocque, 80039 Amiens, France
eDepartment of Materials Chemistry, National Institute of Chemistry, Hajdrihova 19, 1000 Ljubljana, Slovenia
fFaculty of Chemistry and Chemical Technology, University of Ljubljana, Večna Pot 113, 1000 Ljubljana, Slovenia
gAlistore-ERI, CNRS FR 3104, Hub de l’Energie, Rue Baudelocque, 80039 Amiens, France
First published on 3rd May 2022
Abstract
Mastering the metal–electrolyte interface is mandatory for the development of reliable rechargeable magnesium batteries. Nevertheless, most of the current electrolytes contain chloride species to bypass the surface passivation of magnesium, making them corrosive to other cell components and potentially irrelevant for industrial application. Here, we demonstrate a novel approach to bypass the use of such electrolytes via the mediation of an alloy-type interface prepared by coating the surface of a magnesium electrode with liquid gallium. Chemical alloying induces the formation of a surface layer, mainly composed of intermetallic Mg2Ga5, enabling significantly improved electrochemical performance with a simple chloride-free Mg(TFSI)2/DME electrolyte. Sensibly less-polarized and more stable plating/stripping is observed with symmetric cells at a current density of 0.1 mA cm−2, and longer cycle life is achieved in full cells with positive electrodes based on sulphur and organic composites. This proof-of-concept offers room for improvement in the coating protocol and could be tuned with other liquid metals. More importantly, it opens the door to electrolytes previously considered as non-compatible with magnesium metal, and consequently paves the way for the application of metal electrodes in practical magnesium batteries.
Introduction
Lithium-ion batteries have revolutionised the field of energy storage. At the root of the constant electrification of our societies by powering mobile electronics, now they equip new generations of hybrid and electric vehicles and are also considered for stationary storage of renewable energies. Consequently, the global battery production is significantly increasing and sustainability issues might arise even with implementing efficient battery recycling. In this regard, alternative electrochemical storage systems that offer high energy density together with reduced cost and low environmental footprint must be developed. Magnesium batteries, belonging to so-called post-lithium-ion systems, have attracted significant attention since the first rechargeable cell prototype was reported by Aurbach and co-workers.1 Indeed, using magnesium as the negative electrode theoretically enables high energy density batteries, thanks to the low density of magnesium, the low redox potential of the Mg2+/Mg couple and the double charge carried by Mg2+ cations.2–4 In addition, magnesium is abundant and cheap, safer than lithium, and already widely processed on an industrial scale. In spite of these promising features, magnesium batteries are still far from a realistic application.One of the reasons for such delayed commercialization is rooted in the passivation film created on the surface of the magnesium electrode in contact with common aprotic battery electrolyte solvents which, unlike lithium metal, inhibits the reversible plating/stripping process of magnesium.5–7 In the last 20 years, many electrolyte formulations have been proposed to bypass this major hurdle, from the early use of Grignard reagents to solutions employing borate-based magnesium salts.4,8,9 Unfortunately, despite significant improvements, no miracle formulation coupling good electrochemical performance with low-cost, easily scalable production and eco-friendly properties has been found until now. Only the use of chloride species enables breaking the passivation layer, however making the electrolytes corrosive and consequently not suitable for real applications.
Mastering the interface between magnesium and the electrolyte is then crucial. In 2018, Ban and co-workers designed a polymeric coating combining thermally-cyclized polyacrylonitrile and magnesium trifluoromethanesulfonate, and succeeded in further employing carbonate-based electrolytes.10 An organic coating made of reduced perylene diimide-ethylene diamine (rPDI) also enabled fast and reversible magnesium plating/stripping, however with the electrolyte Mg(TFSI)2-MgCl2/DME.11 Moving to inorganic coatings, the teams of Nazar and Archer almost simultaneously proposed the protection of lithium and sodium electrodes by using an alloy-type coating, created through the chemical reduction of a metallic salt in solution, followed by an alloying reaction with the alkali metal surface.12–15 Eventually, the passivating film formed in this way is in reality a composite layer, as insulating by-products are also present and offer a potential gradient to prevent plating onto the coating layer. The as-protected electrodes exhibit enhanced electrochemical performance, mainly induced by the minimised dendritic growth during plating.16 Although magnesium dendrites are not unexpected,17–19 similar protocols were thus applied to magnesium electrodes with the objective of using more conventional electrolytes. For example, in contact with a SnCl2 or BiCl3 solution, a composite layer containing the Mg2Sn or Mg3Bi2 alloy covers the magnesium electrode surface and enables fast ion transport.20,21 The creation of such a protective layer could be achieved by modifying the electrolyte formulation with the action of other salts (GeCl4, SiCl4, and Bi(OTf)3) in solution or as an electrolyte additive.22–25 Despite significant improvement in the electrochemical performance, using chloride species remains questionable with respect to possible corrosion side reactions with other components of the cell (Table S1†).
An alternative coating strategy, recently proposed for the alkali metals, is the direct reaction with a liquid metal. Protective layers of alkali amalgams were thus obtained on the surface of lithium, sodium and potassium, respectively, by the reaction with mercury drops. The as-protected electrodes exhibit an improved electrochemical behaviour such as less severe dendritic growth and improved stability towards moisture.26–28 However, the well-known toxicity of mercury precludes any chance of realistic applications. Furthermore, the very high density of the obtained amalgams would dramatically lower the theoretical specific energy density of possible full cells.
Searching for other lighter and environmentally-friendly liquid metals, we demonstrate in this work the feasibility of protecting the surface of a magnesium electrode with a gallium-based coating. Gallium has a relatively low abundance on the Earth's crust, but is very widely spread as a trace element and is generally obtained from processing to produce aluminium or zinc, and good recyclability enables its wide use in the electronics industry.29–31 Moreover gallium has been shown to be compatible with magnesium electrochemistry. Indeed, magnesium electrochemically alloys with gallium to form Mg2Ga5. Like other alloy-type electrodes,32 the reaction occurs at low potential (∼200 mV) and offers high specific capacity (∼300 mA h g−1). In addition, a self-healing behaviour is observed due to the near-room temperature solid–liquid phase change between solid Mg2Ga5 and liquid gallium.33
As shown hereafter, the chemical reaction between liquid gallium and magnesium forms an alloy layer mainly composed of Mg2Ga5 at the surface of the electrode. With the chloride-free electrolyte Mg(TFSI)2/DME, while the strong surface passivation of uncoated magnesium electrodes significantly alters the electrochemical activity, the as-protected magnesium electrodes exhibit enhanced performance. Consequently, extended cycling is achievable with full cells with various positive electrode composites. This proof-of-concept encourages similar coating strategies through the surface reaction with other liquid metals and may open the door to realistic application of magnesium batteries.
Surface alloying of the magnesium anode
The low melting point of gallium (29.8 °C) enables its easy melting before spreading drops on the surface of a magnesium electrode in an argon-filled glovebox. The metallic shiny aspect of magnesium rapidly evolves and turns grey after few hours (Fig. 1a). Multiple surface layers could be evidenced in cross-sectional scanning electron microscopy (SEM) images. Starting from the surface, we can distinguish the first thick layer of around 10–30 μm, then a very thin oxygen-rich layer (200–300 nm) and lastly an irregular micrometric sublayer (1–2 μm) observed at the interface with the magnesium bulk electrode (Fig. 1b, e and f). Elemental cross-sectional mapping by scanning Auger microscopy (SAM) shows distribution of gallium and magnesium through the interfaces. Semi-quantitative analysis based on the relative intensities of the Auger spectra of selected areas and their derivative curves, taking into account the relative sensitivity factors of Mg KLL and Ga LMM lines, unambiguously reveals a richer gallium content in the thick top layer than in the sublayer (Fig. 1e–g). The oxygen-rich thin interlayer most probably originates from the native oxide/carbonate surface layer that cannot be fully removed even when operating in a glovebox, as shown later by XPS, and possibly from contamination during sample preparation.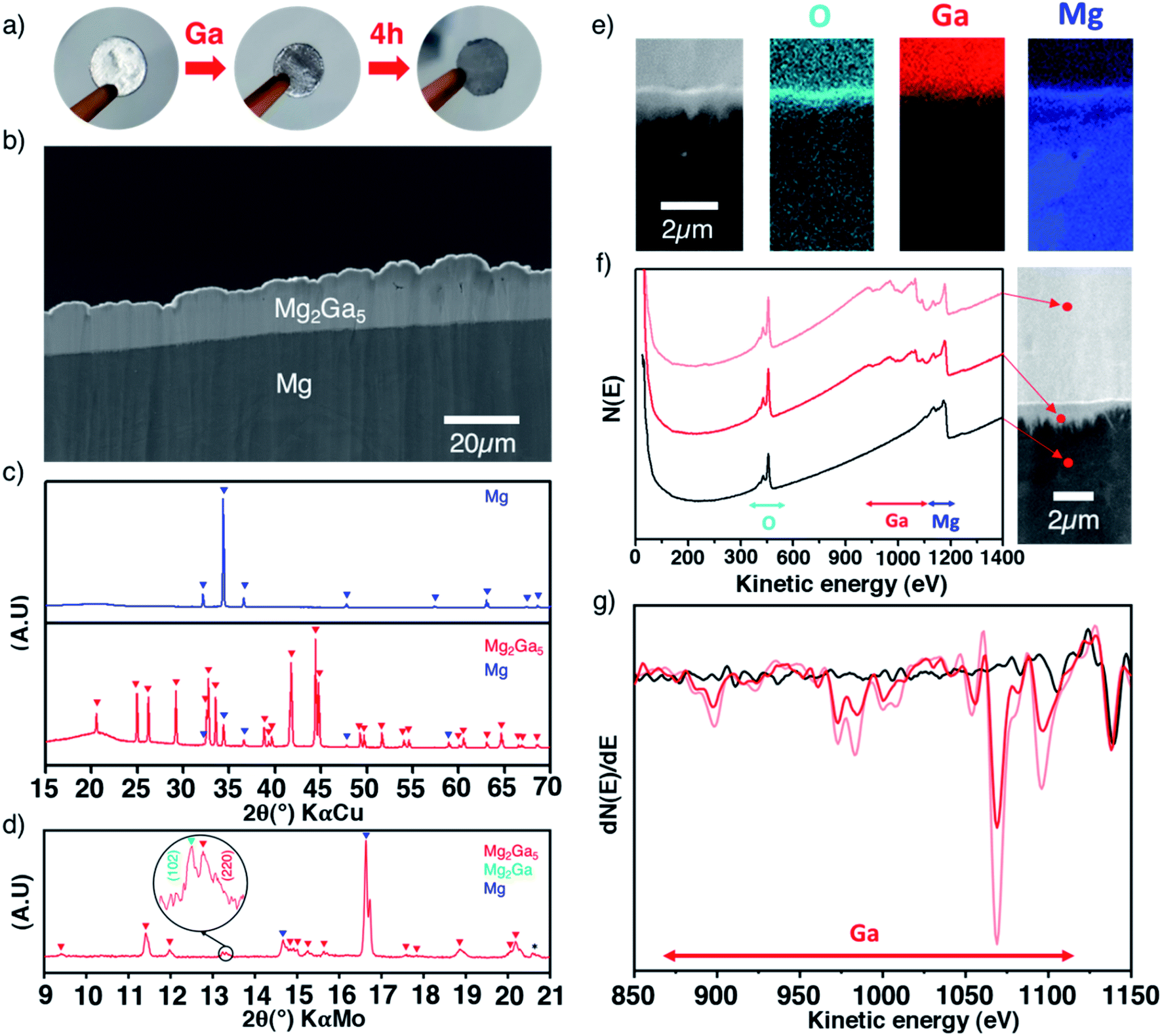 | ||
| Fig. 1 Multi-technique characterization of the Mg–Ga alloy coating. Pictures of magnesium discs showing the colour changes after the reaction with liquid gallium (a), SEM cross-sectional image of a Ga-treated magnesium disc (b), XRD patterns of bare and treated discs in reflection and transmission modes ((c) and (d), respectively) with peak identification and a special enlarged view to detect the (102) peak of Mg2Ga, SAM mapping images of O, Ga and Mg elements at the interface (e), and Auger electron spectra of selected areas in direct mode “N(E)” and derivative mode “dN(E)/dE” displaying the chemical composition changes ((f) and (g)). | ||
X-ray diffraction was used to get more insights into the coating. The formation of highly crystalline Mg2Ga5 is evidenced in reflection geometry (Fig. 1c), with refined cell parameters in line with the literature (I4/mmm, a = 8.646(1) Å and c = 7.126(1) Å, Fig. S1†) while second acquisition performed on a high brilliance diffractometer (Mo-Kα rotating anode source) in transmission geometry enables the detection of an additional contribution (shown in Fig. 1d) that could be reasonably attributed to the (102) peak of Mg2Ga, a Ga-rich composition of the Mg–Ga phase diagram (Fig. S2†). Overall, in agreement with the gallium concentration gradient evidenced by Auger spectroscopy, the thickest top layer and the sublayer below can be reasonably assigned to Mg2Ga5 and Mg2Ga, respectively, as the latter is only detected in transmission geometry.
On the practical side, while controlling the weight of the gallium drops is not straightforward, the thickness of the deposit can be manually tuned during the coating process by adjusting the amount of gallium drops and/or by subsequently wiping out some gallium. Following this procedure, the electrode mass increase after gallium addition experimentally ranges from 18 to 56%. By simply considering the formation of only Mg2Ga5, this mass uptake corresponds to an average layer thickness of around 15 to 60 μm (Fig. S3†), in good agreement with our SEM observation. Importantly, owing to the molecular weight and density of magnesium, the coating formation does not consume an important volume of the magnesium electrode.
Benefits on the electrochemical behaviour
Unlike alkali metals, magnesium is not ductile and cannot be pressed and flattened on the surface of a current collector. Consequently, it appears necessary to cover both sides of each magnesium disc electrode before performing any electrochemical tests. For a better understanding of the magnesium plating and stripping, galvanostatic polarization measurements were first carried out in symmetric cells at a current density of 0.1 mA cm−2 for a better comparison with the existing literature (Table S1†) with a conventional electrolyte Mg(TFSI)2/DME at 0.8 M. Note that this relatively high concentration was chosen with the initial purpose of mitigating the diffusion of polysulphide species in the electrolyte usually observed with a carbon–sulphur positive electrode.With uncoated magnesium electrodes, after an initial peak, the overpotential is rapidly stabilized around 0.6 V during the first polarization sweep. However, in the subsequent ones the galvanostatic profile is strongly modified and the overpotential remains around 2 V, indicating a strong impedance of the plating/stripping process due to the surface passivation and consequent cycling failure generally after 100 hours (Fig. 2a).
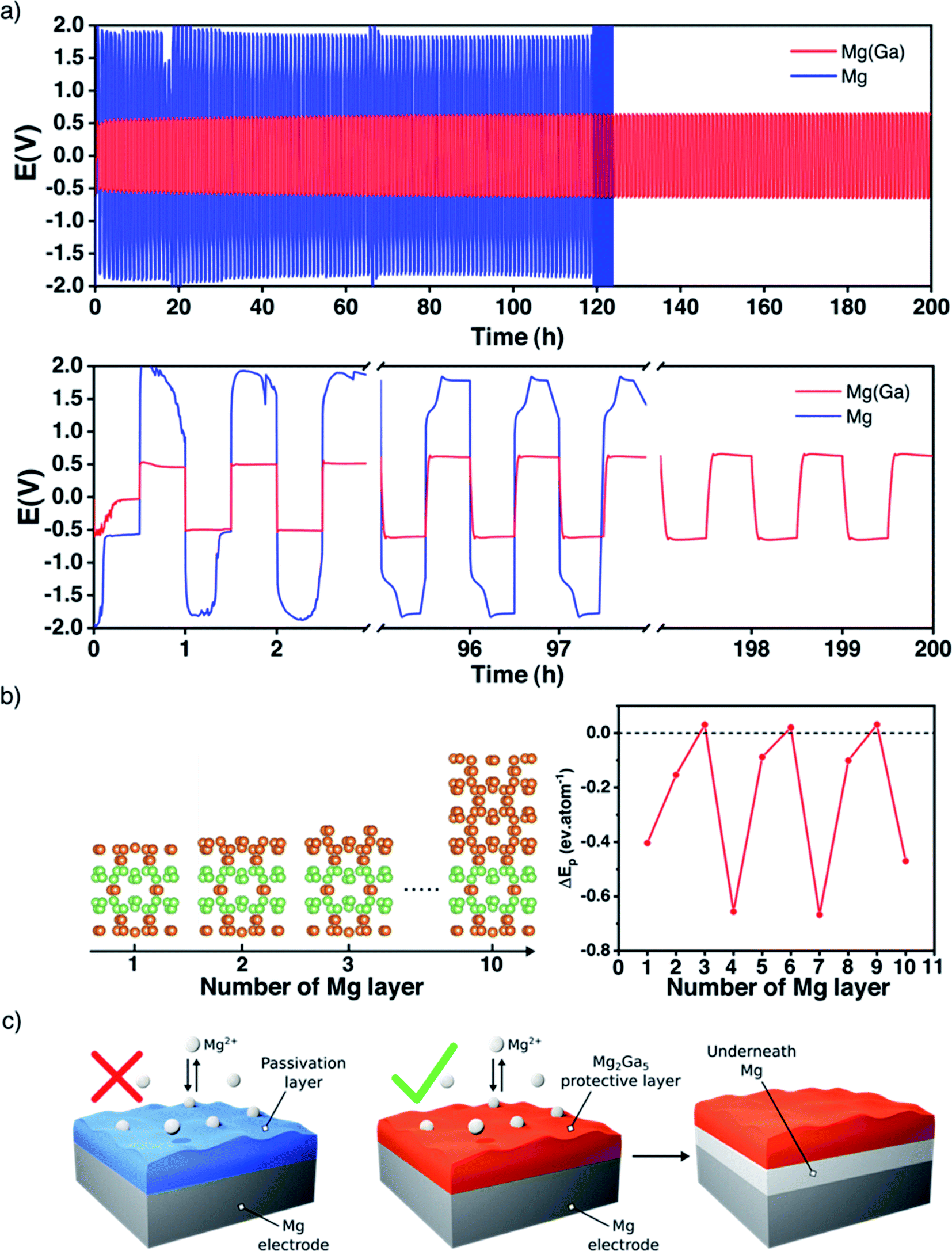 | ||
| Fig. 2 Enhanced magnesium plating/stripping. Evolution of the overpotential during subsequent magnesium plating/stripping processes in symmetrical cells with bare and Ga-protected magnesium electrodes (blue and red lines, respectively) in 0.8 M Mg(TFSI)2/DME electrolyte and at 40 °C, with sweeps of 30 min at a current density of 0.1 mA cm−2 (a); structures of the magnesium epitaxial surface on Mg2Ga5 with increasing number of layers and the corresponding DFT-calculated plating energy (b) and schematic representation of plating underneath (c). | ||
The picture is significantly different with gallium-protected magnesium electrodes. The overall overpotential remains around 0.5 V, and the profile is stable for several hundred cycles. In detail, each sweep exhibits a weak nucleation peak followed by a flat plateau throughout the whole electrochemical process. It is worth pointing out that a highly concentrated electrolyte was employed, and that the metastability of Mg(TFSI)2 is known to strongly participate in the surface passivation of magnesium.34,35 Despite this, the value of the overall potential is in line with previous studies where less concentrated electrolytes are used (Table S1†). Given that a current density of 0.1 mA cm−2 is far from realistic applications, additional tests were performed at higher current densities; under these conditions, however, the plating/stripping behavior of the Ga-coated electrode was not significantly improved in comparison with that of bare magnesium.
In the literature, the charge transfer and the plating mechanism processes occurring on the surface of these coated electrodes are not yet fully understood.16 The presence of insulating by-products (mainly chlorides) in the composite layers was proposed to create a potential gradient, further enabling the ionic diffusion across the coating and leading to plating occurring below the coating layer, further denoted as plating underneath.12 Moreover, the existence of chloride species dissolved in the electrolyte has been suggested to prevent further passivation through the formation of surface adsorbed species.36 With our coating approach, the protective surface layer is mainly composed of the alloys Mg2Ga5 and Mg2Ga. Density of states obtained from density functional theory (DFT) calculations confirms the metallic state of the alloys (Fig. S4†). The very thin oxygen-rich interlayer might be insulating and its influence on the properties of the coating is still unclear. Given its narrow thickness and its ubiquitous presence in all studied coatings, it cannot be considered as a major contributor to the observed improvement of the electrochemical properties.
As previously done for lithium systems,37,38 DFT calculations were carried out to rationalize the magnesium plating onto our Ga-coated electrode. Here, the stability of the different Mg2Ga5 surfaces was first analyzed as a function of the surface orientation and the applied potential. As a result, the (001) surface is found to be the most stable one in the potential range of interest (Fig. S5†). Then, the plating energy ΔEp, i.e., the energy difference between a magnesium layer epitaxially plated on Mg2Ga5 and the same amount of Mg atoms in their bulk environment, was computed for increasing number of adsorbed layers in order to qualitatively link the plating mechanism with the current rate, as the greater the current density, the larger the number of Mg atoms accumulated on the coating surface (Fig. 2b, see details in the Methods section and ESI†).
Most of the magnesium adsorbed layer configurations lead to a strongly negative ΔEp, suggesting an plating underneath favorably independent of the current rate. Note that only the 3-, 6- and 9- layer configurations possess a slightly positive plating energy, well below room temperature kBT. Therefore, for such configurations, the surface plating is either unstable or metastable at low temperature. At room temperature, because of the extremely weak surface stabilization, even the metastable Mg layers should dissolve in the Mg2Ga5 coating because of both entropic (defect creation) and enthalpic (increase of the number of the strong Mg–Ga bonds relative to the weaker Ga–Ga ones) effects. To summarise, from a thermodynamic point of view, a magnesium layer epitaxially plated above the Mg2Ga5 coating is expected to migrate underneath it to form a magnesium bulk layer on the electrode surface. The physical reason for such a behaviour can be revealed through the competition between the adsorption energy and the epitaxial stress. The chemical bond between the adsorbed Mg-layer and the Mg2Ga5 substrate appears weak, with an adsorption energy of only −0.12 eV per atom, and cannot compensate the epitaxial stress of the Mg-layer which is constrained to the Mg2Ga5 crystallographic environment (+0.22 eV per atom).
To confirm the plating underneath the coating suggested by the electrochemical results and the DFT calculations, the evolution of the Ga-coated electrode during repeated plating and stripping cycles was followed by in situ XRD in transmission mode. Indeed, the Mg2Ga5 layer is highly crystalline and any structural evolution should be detectable in this way. The use of a rotating Mo anode X-ray source enables collection of high-quality diffraction patterns with a relatively short acquisition time (3 minutes), allowing us to follow precisely any changes occurring through the whole electrode (bulk magnesium, alloy coating and the interface with the liquid electrolyte).
At first glance, the pristine highly crystalline Mg2Ga5 and the secondary diffraction peaks assigned to Mg2Ga remain unmodified all along the electrochemical process (Fig. 3a). Only tiny peak broadening is observed, which can be linked to a decrease of crystallinity and/or increase of the lattice strain of the Mg2Ga5 crystallites (Fig. S7†).
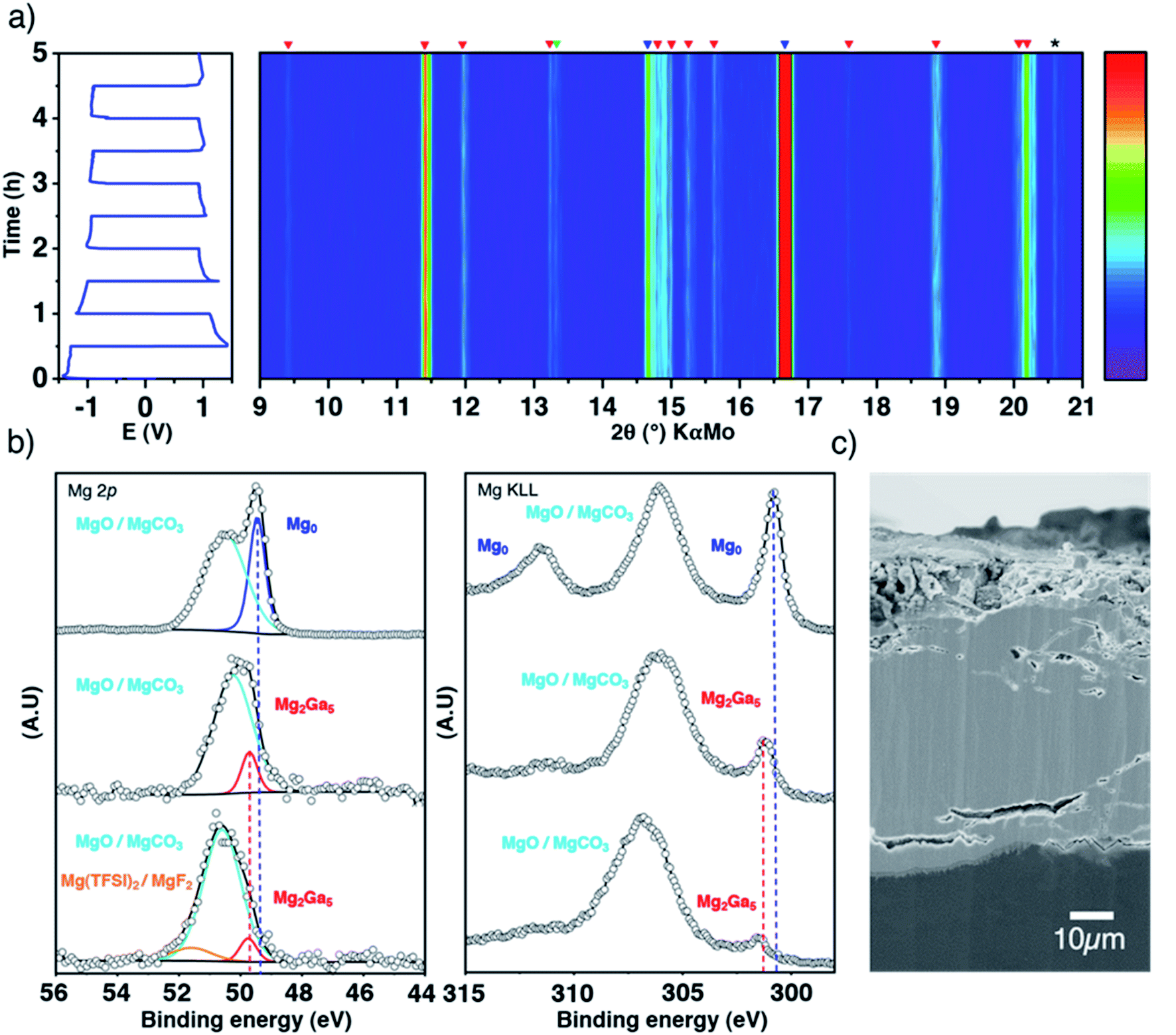 | ||
| Fig. 3 Inside the coating evolution. Top-view representation of XRD patterns collected in situ in transmission mode during galvanostatic polarization measurements of a symmetric cell at 0.1 mA cm−2, with blue, red and green markers for Mg, Mg2Ga5 and Mg2Ga, respectively (a); XPS spectra (Mg 2p and Auger Mg KLL) of pristine Mg, Ga-coated Mg before and after 50 hours of plating/stripping, normalized by the most intense contribution (b); SEM cross-sectional image of the coated electrode after plating/stripping (c). Note that XRD acquisitions were performed at room temperature as the in situ cell does not allow working at 40 °C, even if the coating efficiency is not optimized at such temperature (Fig. S6†). | ||
Focusing now on the composition of the electrode surface, ex situ XPS analysis was performed on coated magnesium before and after 50 hours of successive plating and stripping in a symmetrical cell in order to approach what could occur at the electrode–electrolyte interface in full cells (Fig. 3b). The presence of the Mg2Ga5 alloy in Ga-coated magnesium is evidenced by a significant shift of the thin Auger Mg KLL metallic signature (301.8 eV compared to 301.2 eV for bare magnesium). Due to the greater amount of oxide and carbonate on the surface of Ga-coated Mg, the relative contribution of the alloy vs. MgO/MgCO3 decreases, which can be observed in the Mg 2p spectrum as well. After cycling, Mg2Ga5 remains present as no shift of the peak is observed. Magnesium metal is consequently not plated above the coating, in agreement with operando XRD and DFT calculations. Additionally, the presence of Mg(TFSI)2 salt and its degradation product MgF2 can also be observed by XPS (Mg 2p in Fig. 3, and F 1s and Ga 3d in Fig. S8†). Cross-sectional electron microscopy coupled with Auger electron spectroscopy reveals a global resistance of the coating in spite of some changes in the morphology, with the apparition of small cracks in some parts of the protective layer (Fig. S9†). After 50 hours of continuous plating, which could correspond to long unique discharge in a full cell, electrolyte decomposition localized on these cracks could be observed (Fig. S10†).
Towards better magnesium batteries
The gallium alloy coating was shown to enhance the electrochemical features and remain chemically constant during polarization measurements. The coated magnesium electrodes were then evaluated in a full cell configuration using the same Mg(TFSI)2-based electrolyte. We first tested the protected Ga-treated magnesium electrode with the Chevrel phase Mo6S8 prepared by the conventional procedure (Fig. S11†), which can be considered as the standard electrode material for magnesium-based batteries for which the electrochemical activity with magnesium is well established.1,39,40 However, Mo6S8 is characterised by a relatively low reversible capacity and by a low working voltage and therefore should not be considered for realistic applications. Galvanostatic profiles obtained with bare and protected magnesium electrodes appear quite similar in the first cycles with two typical plateaus assigned to distinct insertion sites of magnesium ions in the sulphide framework (Fig. 4a and b). Electrolyte decomposition parasite reactions could explain the exceeding initial discharge capacity and certainly induce increasing polarization which causes battery failure after a few cycles. Remarkably, the potential values of the plateaus are very close for both cells, either in charge or in discharge. This is another observation that supports the role of the alloy layer as a composition-invariant coating. Indeed, the potential is significantly lower with the negative electrode of bulk Mg2Ga5 prepared by mechanical alloying with a difference of 200 mV (i.e., approaching the potential of the alloying/dealloying process of gallium33) (Fig. S12 and S13†).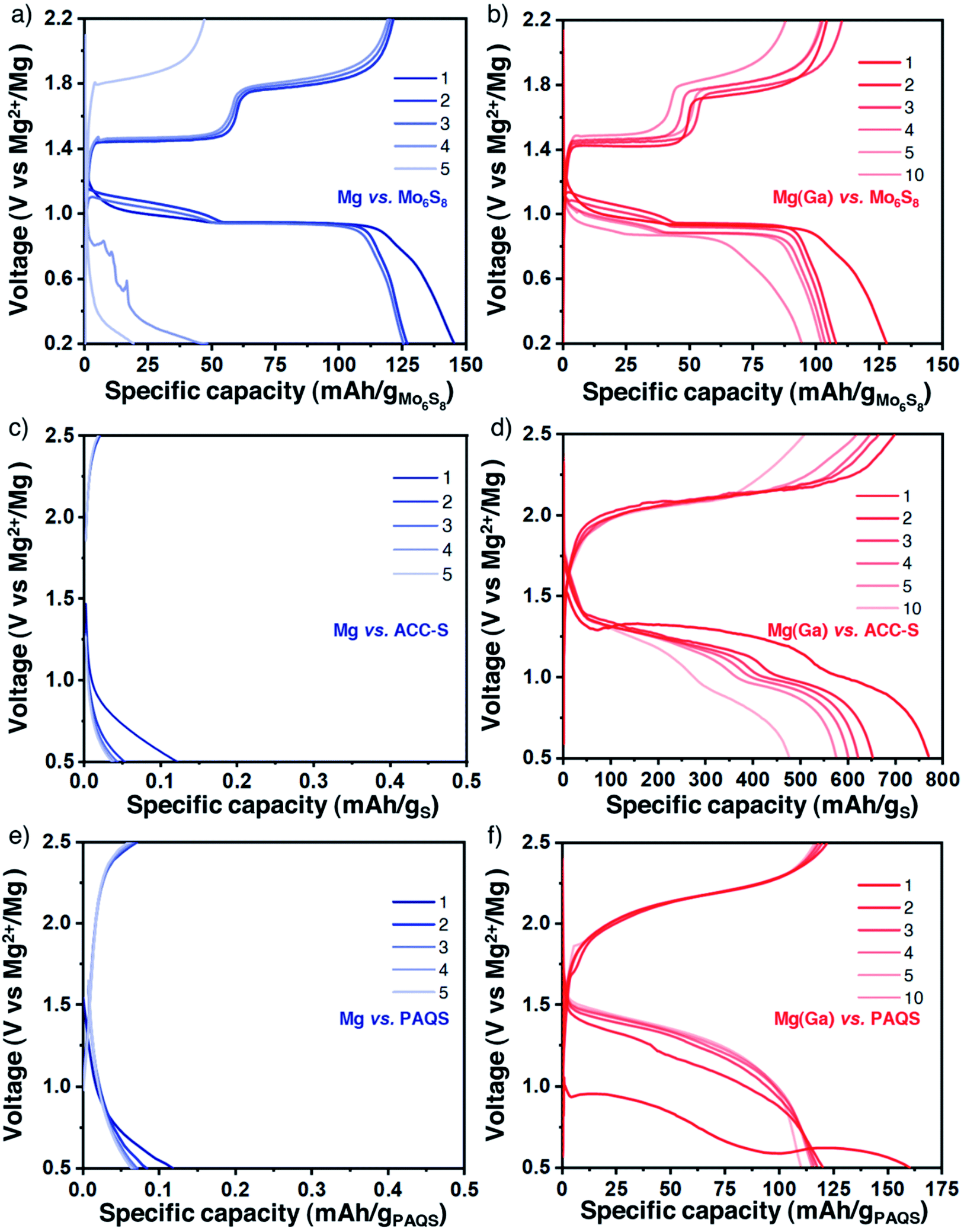 | ||
| Fig. 4 Enhanced cycling in full magnesium batteries. The galvanostatic profile of magnesium batteries with bare and coated magnesium electrodes (blue lines, left, and red lines, right, respectively) assembled successively with different positive electrodes: Mo6S8 (a and b, at 6 mA g−1 for Mo6S8), carbon–sulphur composite ACC/S (c and d, at 167 mA g−1 for sulphur, corresponding to 0.3 mA cm−2 for Mg) and carbon–organic composite PAQS-CNT (e and f, at 122 mA g−1 for PAQS, corresponding to 0.21 mA cm−2 for Mg). Battery tests performed at 40 °C. | ||
On the way towards realistic magnesium batteries, sulphur- and organic-based composites were then considered. Indeed, given the sluggish diffusion of Mg2+ ions in classical insertion hosts such as high-voltage layered oxides or polyanionic compounds, working with sulphur, a conversion-type electrode, is a promising pathway and Mg/S cells could potentially exhibit high energy density while meeting sustainability and durability criteria.41–43 Nevertheless, the well-known intrinsic drawbacks of sulphur-based electrodes, from the insulating character of sulphur to the well-known polysulphide shuttle effect, could lower these promising values and therefore still need to be overcome. Besides sulphur, organic compounds, mainly quinone derivatives involving a carbonyl group as the redox centre, are now regularly highlighted as possible cathode materials in divalent batteries. Despite the solubility issue, which can be mitigated with polymer synthesis, promising electrochemical cycling could be performed with appropriate electrolytes.44–51 We used sulphur-impregnated activated carbon clothes (ACC-S, Fig. S14†) which are commonly used as model electrodes in magnesium batteries despite their limited sulphur loading (10 wt% of sulphur, corresponding to a surface density of around 1 mg cm−2).52–54 In parallel, the popular redox polymer poly (anthraquinonyl sulphide) (PAQS, Fig. S15†) was chosen due to its reversible electrochemical mechanism with divalent ions with Mg(B(hfip)4)2 and Mg(TFSI)2-2MgCl2 electrolytes and combined with carbon nanotubes (CNT) for improved capacity utilization.
For both electrode composites, the comparison between bare and protected magnesium electrodes strikingly highlights the benefits of the alloy coating. The strong passivation of magnesium metal induces an immediate cell failure (Fig. 4c and d for ACC-S and Fig. 4e and f for PAQS-CNT). In contrast, despite a certain increase of overpotential, features of the plateaus are not strongly modified, indicating no change in the electrochemical mechanisms with coated magnesium electrodes for both ACC-S and PAQS-CNT composites.
With the ACC-S positive electrode, XPS analysis performed ex situ after 15 cycles confirms the chemical integrity of the coating and also highlights the important polysulphide species dissolution and their well-known shuttle to the negative electrode side that strongly participates in the important capacity fading (Fig. S16 and S17†). Concerning the PAQS-CNT positive electrode, the capacity decay here could also be mainly attributed to partial dissolution of the active material (Fig. S17 and S18†).
Conclusions and perspectives
In this work, we propose an innovative protocol to create an alloy layer on the surface of magnesium to unlock the use of non-corrosive chloride-free electrolytes for magnesium batteries. From preliminary galvanostatic polarization measurements to full cells with various positive electrode materials, the Ga-coated magnesium electrodes exhibit enhanced electrochemical performance. The magnesium surface passivation, which usually precludes any electrochemical activity, does not occur, and reversible magnesium plating and stripping is demonstrated. For this proof-of-concept, liquid gallium was used to create a relatively thick alloy layer. Although gallium is relatively light, the alloy layer impacts the overall energy density (Fig. S19 and S20†). Therefore, the coating protocol must be further optimized to create a thinner and more robust gallium-based protective layer that resists higher current densities, together with a larger systematic benchmark of the electrolyte compositions. Designing a self-healing composite coating could also be achieved with an alloy coating based on liquid eutectic compositions.55 In addition, different types of magnesium electrolytes need to be tested, keeping in mind the final purpose of using carbonate-based electrolytes for benchmarking high-voltage positive electrode materials, together with a detailed understanding of the evolution of both the protective layer and the solid-electrolyte interphase during battery cycling. Overall, this study opens the door for the large-scale application of low-melting-point elements and compounds to protect sensitive metal electrodes such as magnesium and calcium.Methods
Ga-coated Mg electrodes
Mg–Ga electrodes were prepared in an argon-filled glovebox with <0.5 ppm oxygen and <0.5 ppm H2O at room temperature. Mg foil (Goodfellow, 99.9%, 250 μm) was first polished with a blade to remove the native oxide layer. Then, the coating was made by dropping liquid Ga (from molten ingots, Alfa-Aesar) droplets onto the surface of Mg and spreading over until a homogeneous liquid layer was formed. After few hours, the liquid layer reacted with Mg to form a solid alloy layer.Positive electrode materials
Chevrel phase Mo6S8, and ACC-S and PAQS-CNT composites were prepared following previous reports.44,52–54,56,57 Details on syntheses, characterization and electrode formulation are provided in the ESI.†Electrochemical tests
Electrodes were tested in coin-cells (2032, 316L stainless steel) assembled in an argon-filled glove-box, and one glass-fibre (Whatman, GF/A) surrounded by two polypropylene membranes (Celgard 2325) was used as a separator and wetted with 100 μL of electrolyte. Tests have been performed mainly in an oven set at 40 °C, and also at room temperature, using research-grade potentiostats (Neware and Bio-Logic).X-ray diffraction (XRD)
Samples were prepared in an Ar-filled glove box and then transferred under a protective airtight polymeric film to limit the moisture reaction. XRD measurements in reflection mode were performed with a Panalytical X'Pert Pro diffractometer operating with Cu Kα radiation. For the operando XRD measurement in transmission mode, a specially designed cell using two Be windows acting as both the X-ray transparent window and current collector was assembled in an Ar-filled glovebox. Transmission XRD acquisitions were carried out with a Bruker D8 Discover diffractometer equipped with a rotating anode (Mo-Kα radiation) and a Dectris EIGER2 R 500K detector. In parallel, electrochemical measurements were performed on a BCS potentiostat (Bio-Logic).Auger electron spectroscopy (AES) and scanning Auger microscopy (SAM)
Coated magnesium discs were mechanically cut and then polished by using an Ar+ ion beam in a cross-section polisher (model IB-09010CP, Jeol Ltd., Tokyo, Japan) operating at 4 keV for 2 hours (working pressure of 1 × 10−4 Pa). Polished discs were transferred without any air exposure into the Auger electron nano probe (JAMP 9500F, Jeol Ltd.) to perform SAM imaging and the AES analyses of the electrode cross-cut sections. The Auger analyses were carried out under UHV conditions (pressure < 2 × 10−7 Pa), using the following beam energy and current conditions: 10 keV and 8 nA, respectively.X-ray photoelectron spectroscopy (XPS)
Samples were transferred without any air exposure into an Ar-filled glove box (<1 ppm O2, <1 ppm H2O) directly connected to an XPS spectrometer (Thermofischer Scientific Escalab 250 Xi) using focused monochromatized Al-Kα radiation (hν = 1486.6 eV). No sputtering was used to clean the surface to analyze in order to preserve the surface chemistry. The analyzed surface area of the samples was a 650 μm diameter disk. The spectra were fit and analysed in CasaXPS software.DFT calculations
Periodic calculations were performed within the density functional theory (DFT) framework, using the Vienna ab initio simulation package (VASP) implemented with projector augmented wave (PAW) pseudopotentials.58,59 Exchange-correlation effects have been accounted for by generalized gradient approximation (GGA) using the functional of Perdew, Burke and Ernzerhof (PBE).60 Surface calculations were performed on Mg2Ga5 (100), (010) and (001) slabs. The surrounding environment was described with an implicit solvent using the polarizable continuum model (PCM) as implemented in VASPsol.61 To vary the electrode potential, the grand canonical density functional theory (GC-DFT) framework has been used in association with the homogeneous background method (HBM) as detailed in previous studies.62 More details can be found in the ESI† section.Author contributions
C. P. carried out the electrode coating and electrochemical characterization experiments, the operando XRD measurement (with J.-N. C.) and the surface and cross-sectional analysis (with D. F., J.-B. L., J. A. and R. D); A. H. and J.-S. F. performed and discussed the DFT calculations; O. L., J. B. and R. D. provided the PAQS-CNT composite and characterized its electrochemical behaviour; R. B. supervised the overall study; C. P. L. S. and R. B. wrote the manuscript and all authors discussed the experiments and final manuscript.Conflicts of interest
The authors declare no competing financial interests.Acknowledgements
The authors gratefully acknowledge financial support from the French National Research Agency (project MISTRALE, ANR-19-CE05-0013, and Labex STORE-EX, ANR-10-LABX-76-01), and Cellule Energie CNRS (project PEPS DIBAPA). O. L., J. B. and R. D. would like to thank the European Union's Horizon 2020 research and innovation program under the Marie Skłodowska-Curie grant agreement No. 860403. J. B. and R. D. would also like to acknowledge support of Slovenia Research Agency under research programs P2-0393 and P2-0423.References
- D. Aurbach, et al. Prototype systems for rechargeable magnesium batteries, Nature, 2000, 407, 724–727 CrossRef CAS PubMed.
- A. Ponrouch, et al. Multivalent rechargeable batteries, Energy Storage Mater., 2019, 20, 253–262 CrossRef.
- R. Dominko, et al. Magnesium batteries: Current picture and missing pieces of the puzzle, J. Power Sources, 2020, 478, 229027 CrossRef CAS.
- R. Mohtadi, O. Tutusaus, T. S. Arthur, Z. Zhao-Karger and M. Fichtner, The metamorphosis of rechargeable magnesium batteries, Joule, 2021, 5, 581–617 CrossRef CAS.
- J. D. Forero-Saboya, D. S. Tchitchekova, P. Johansson, M. R. Palacín and A. Ponrouch, Interfaces and Interphases in Ca and Mg Batteries, Adv. Mater. Interfaces, 2022, 9, 2101578 CrossRef CAS.
- A. Kopač Lautar, J. Bitenc, R. Dominko and J.-S. Filhol, Building Ab Initio Interface Pourbaix diagrams to Investigate Electrolyte Stability in the Electrochemical Double Layer: Application to Magnesium Batteries, ACS Appl. Mater. Interfaces, 2021, 13, 8263–8273 CrossRef PubMed.
- A. Kopač Lautar, et al. Electrolyte Reactivity in the Double Layer in Mg Batteries: An Interface Potential-Dependent DFT Study, J. Am. Chem. Soc., 2020, 142, 5146–5153 CrossRef PubMed.
- M. Rashad, M. Asif, Y. Wang, Z. He and I. Ahmed, Recent advances in electrolytes and cathode materials for magnesium and hybrid-ion batteries, Energy Storage Mater., 2020, 25, 342–375 CrossRef.
- P. Jankowski, et al. Development of Magnesium Borate Electrolytes: Explaining the Success of Mg[B(hfip)4]2 Salt, Energy Storage Mater., 2022, 45, 1133–1143 CrossRef.
- S. B. Son, et al. An artificial interphase enables reversible magnesium chemistry in carbonate electrolytes, Nat. Chem., 2018, 10, 532–539 CrossRef CAS PubMed.
- Y. Sun, Q. Zou, W. Wang and Y. C. Lu, Non-passivating Anion Adsorption Enables Reversible Magnesium Redox in Simple Non-nucleophilic Electrolytes, ACS Energy Lett., 2021, 6, 3607–3613 CrossRef CAS.
- X. Liang, et al. A facile surface chemistry route to a stabilized lithium metal anode, Nat. Energy, 2017, 2, 17119 CrossRef CAS.
- S. Choudhury, et al. Electroless Formation of Hybrid Lithium Anodes for Fast Interfacial Ion Transport, Angew. Chem., Int. Ed., 2017, 56, 13070–13077 CrossRef CAS PubMed.
- Q. Pang, X. Liang, I. R. Kochetkov, P. Hartmann and L. F. Nazar, Stabilizing Lithium Plating by a Biphasic Surface Layer Formed In Situ, Angew. Chem., Int. Ed., 2018, 57, 9795–9798 CrossRef CAS PubMed.
- Z. Tu, et al. Fast ion transport at solid-solid interfaces in hybrid in hybrid battery anodes, Nat. Energy, 2018, 3, 310–316 CrossRef CAS.
- J. Touja, N. Louvain, L. Stievano, L. Monconduit and R. Berthelot, An Overview on Protecting Metal Anodes with Alloy-Type Coating, Batteries Supercaps, 2021, 4, 1252–1266 CrossRef CAS.
- R. Davidson, et al. Formation of Magnesium Dendrites during Electrodeposition, ACS Energy Lett., 2019, 4, 375–376 CrossRef CAS.
- R. Davidson, et al. Mapping mechanisms and growth regimes of magnesium electrodeposition at high current densities, Mater. Horiz., 2020, 7, 843–854 RSC.
- Y. Zhao, et al. Effect of Mg Cation Diffusion Coefficient on Mg Dendrite Formation, ACS Appl. Mater. Interfaces, 2022, 14(5), 6499–6506 CrossRef CAS PubMed.
- R. Lv, X. Guan, J. Zhang, Y. Xia and J. Luo, J. Enabling Mg Metal Anodes Rechargeable in Conventional Electrolytes by Fast Ionic Transport Interphase, Natl. Sci. Rev., 2020, 7(2), 333–341 CrossRef CAS PubMed.
- Y. Zhao, et al. A Bismuth-Based Protective Layer for Magnesium Metal Anode in Noncorrosive Electrolytes, ACS Energy Lett., 2021, 6, 2594–2601 CrossRef CAS.
- J. Zhang, et al. Rechargeable Mg metal batteries enabled by a protection layer formed in vivo, Energy Storage Mater., 2020, 26, 408–413 CrossRef.
- Y. Li, et al. , Formation of an Artificial Mg2+ -Permeable Interphase on Mg Anodes Compatible with Ether and Carbonate Electrolytes, ACS Appl. Mater. Interfaces, 2021, 13(21), 24565–24574 CrossRef CAS PubMed.
- Y. Li, et al. Reversible Mg metal anode in conventional electrolyte enabled by durable heterogeneous SEI with low surface diffusion barrier, Energy Storage Mater., 2022, 46, 1–9 CrossRef.
- Z. Meng, et al. Surface Engineering of a Mg Electrode via a New Additive to Reduce Overpotential, ACS Appl. Mater. Interfaces, 2021, 13, 37044–37051 CrossRef CAS PubMed.
- G. He, Q. Li, Y. Shen and Y. Ding, Flexible Amalgam Film Enables Stable Lithium Metal Anodes with High Capacities, Angew. Chem., Int. Ed., 2019, 58, 18466–18470 CrossRef CAS PubMed.
- Q. Zhang, et al. A thermodynamically stable quasi-liquid interface for dendrite-free sodium metal anodes, J. Mater. Chem. A, 2020, 8, 6822–6827 RSC.
- Q. Yang, Y. Ding and G. He, An amalgam route to stabilize potassium metal anodes over a wide temperature range, Chem. Commun., 2020, 56, 3512–3515 RSC.
- R. R. Moskalyk, Gallium: The backbone of the electronics industry, Miner. Eng., 2003, 16, 921–929 CrossRef CAS.
- S. Mir, A. Vaishampayan and N. Dhawan, A Review on Recycling of End-of-Life Light-Emitting Diodes for Metal Recovery, JOM, 2022, 74, 599–611 CrossRef CAS.
- Z. Zhao, Y. Yang, Y. Xiao and Y. Fan, Recovery of gallium from Bayer liquor: A review, Hydrometallurgy, 2012, 125–126, 115–124 CrossRef CAS.
- J. Niu, Z. Zhang and D. AurbachAlloy Anode Materials for Rechargeable Mg Ion Batteries, 2020, vol. 1–33, p. 2000697 Search PubMed.
- L. Wang, et al. High-Rate and Long Cycle-Life Alloy-Type Magnesium-Ion Battery Anode Enabled Through (De)magnesiation-Induced Near-Room-Temperature Solid–Liquid Phase Transformation, Adv. Energy Mater., 2019, 9, 1902086 CrossRef CAS.
- Y. Yu, et al. Instability at the Electrode/Electrolyte Interface Induced by Hard Cation Chelation and Nucleophilic Attack, Chem. Mater., 2017, 29, 8504–8512 CrossRef CAS.
- O. Tutusaus, R. Mohtadi, N. Singh, T. S. Arthur and F. Mizuno, Study of Electrochemical Phenomena Observed at the Mg Metal/Electrolyte Interface, ACS Energy Lett., 2017, 2, 224–229 CrossRef CAS.
- J. G. Connell, et al. Tuning the Reversibility of Mg Anodes via Controlled Surface Passivation by H2O/Cl– in Organic Electrolytes, Chem. Mater., 2016, 28, 8268–8277 CrossRef CAS.
- Q. Yan, et al. A Perspective on interfacial engineering of lithium metal anodes and beyond, Appl. Phys. Lett., 2020, 117, 080504 CrossRef CAS.
- G. Whang, et al. Avoiding dendrite formation by confining lithium deposition underneath Li–Sn coatings, J. Mater. Res., 2021, 36, 797–811 CrossRef CAS.
- M. D. Levi, et al. Kinetic and Thermodynamic Studies of Mg2 + and Li + Ion Insertion into the Mo6 S 8 Chevrel Phase, J. Electrochem. Soc., 2004, 151, A1044–A1051 CrossRef CAS.
- E. Levi, et al. Phase Diagram of Mg Insertion into Chevrel Phases, MgxMo6T8 (T = S, Se). 1. Crystal Structure of the Sulfides, Chem. Mater., 2006, 18, 5492–5503 CrossRef CAS.
- Z. Zhao-Karger and M. Fichtner, Magnesium-Sulfur battery: its beginning and recent progress, MRS Commun., 2017, 1–15 Search PubMed.
- L. Kong, et al. A Review of Advanced Energy Materials for Magnesium–Sulfur Batteries, Energy Environ. Mater., 2018, 1, 100–112 CrossRef CAS.
- M. Mao, T. Gao, S. Hou and C. Wang, A critical review of cathodes for rechargeable Mg batteries, Chem. Soc. Rev., 2018, 47, 8804–8841 RSC.
- J. Bitenc, et al. Anthraquinone-Based Polymer as Cathode in Rechargeable Magnesium Batteries, ChemSusChem, 2015, 8, 4128–4132 CrossRef CAS PubMed.
- J. Bitenc, et al. Poly(hydroquinoyl-benzoquinonyl sulfide) as an active material in Mg and Li organic batteries, Electrochem. commun., 2016, 69, 1–5 CrossRef CAS.
- X. Fan, et al. A Universal Organic Cathode for Ultrafast Lithium and Multivalent Metal Batteries, Angew. Chem., Int. Ed., 2018, 57, 7146–7150 CrossRef CAS PubMed.
- J. Bitenc, et al. Electrochemical Performance and Mechanism of Calcium Metal-Organic Battery, Batteries Supercaps, 2021, 4, 214–220 CrossRef CAS.
- H. Dong, et al. High-power Mg batteries enabled by heterogeneous enolization redox chemistry and weakly coordinating electrolytes, Nat. Energy, 2020, 5, 1043–1050 CrossRef CAS.
- B. Pan, et al. Polyanthraquinone-Based Organic Cathode for High-Performance Rechargeable Magnesium-Ion Batteries, Adv. Energy Mater., 2016, 6, 2–4 Search PubMed.
- T. Pavčnik, J. Bitenc, K. Pirnat and R. Dominko, Electrochemical Performance of Mg Metal-Quinone Battery in Chloride-Free Electrolyte, Batteries Supercaps, 2021, 4, 815–822 CrossRef.
- Y. Han, et al. High-performance Mg–organic batteries based on hybrid MgCl2–LiCl/THF electrolytes, Energy Storage Mater., 2022, 46, 300–312 CrossRef.
- T. Gao, et al. Reversible S0/MgSx Redox Chemistry in a MgTFSI2/MgCl2/DME Electrolyte for Rechargeable Mg/S Batteries, Angew. Chem., 2017, 129, 13711–13715 CrossRef.
- T. Gao, et al. Thermodynamics and Kinetics of Sulfur Cathode during Discharge in MgTFSI2 -DME Electrolyte, Adv. Mater., 2018, 30 CAS.
- Z. Meng, et al. Alloys to Replace Mg Anodes in Efficient and Practical Mg-Ion/Sulfur Batteries, ACS Energy Lett., 2019, 4, 2040–2044 CrossRef CAS.
- M. Song, et al. A self-healing room-temperature liquid eutectic GaSn anode with improved wettability for advanced Mg ion batteries, Chem. Eng. J., 2022, 435, 134903 CrossRef CAS.
- S.-H. Choi, et al. Role of Cu in Mo6S8 and Cu Mixture Cathodes for Magnesium Ion Batteries, ACS Appl. Mater. Interfaces, 2015, 7, 7016–7024 CrossRef CAS PubMed.
- Z. Song, H. Zhan and Y. Zhou, Anthraquinone based polymer as high performance cathode material for rechargeable lithium batteries, Chem. Commun., 2009, 448–450, 10.1039/B814515F.
- G. Kresse and J. Hafner, Ab initio molecular dynamics for liquid metals, Phys. Rev. B, 1993, 47, 558–561 CrossRef CAS PubMed.
- P. E. Blöchl, Projector augmented-wave method, Phys. Rev. B, 1994, 50, 17953–17979 CrossRef PubMed.
- J. P. Perdew, K. Burke and M. Ernzerhof, Generalized Gradient Approximation Made Simple, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- K. Mathew, R. Sundararaman, K. Letchworth-Weaver, T. A. Arias and R. G. Hennig, Implicit solvation model for density-functional study of nanocrystal surfaces and reaction pathways, J. Chem. Phys., 2014, 140, 84106 CrossRef PubMed.
- A. Kopač Lautar, A. Hagopian and J.-S. Filhol, Modeling interfacial electrochemistry: concepts and tools, Phys. Chem. Chem. Phys., 2020, 22, 10569–10580 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See https://doi.org/10.1039/d2ta02083a |
| This journal is © The Royal Society of Chemistry 2022 |