
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The use of bromopyridazinedione derivatives in chemical biology
Calise
Bahou
* and
Vijay
Chudasama
 *
*
UCL Department of Chemistry, 20 Gordon Street, London WC1H 0AJ, UK. E-mail: calise.bahou.12@ucl.ac.uk; v.chudasama@ucl.ac.uk
First published on 30th March 2022
Abstract
Tools that facilitate the chemical modification of peptides and proteins are gaining an increasing amount of interest across many avenues of chemical biology as they enable a plethora of therapeutic, imaging and diagnostic applications. Cysteine residues and disulfide bonds have been highlighted as appealing targets for modification due to the highly homogenous nature of the products that can be formed through their site-selective modification. Amongst the reagents available for the site-selective modification of cysteine(s)/disulfide(s), pyridazinediones (PDs) have played a particularly important and enabling role. In this review, we outline the unique chemical features that make PDs especially well-suited to cysteine/disulfide modification on a wide variety of proteins and peptides, as well as provide context as to the problems solved (and applications enabled) by this technology.
 Vijay Chudasama | Prof. Vijay Chudasama obtained his MSci degree and PhD from University College London (UCL) in 2008 and 2011, respectively. Following post-doctoral studies under the supervision of Prof. Stephen Caddick, Vijay obtained a Ramsay Memorial Fellowship. During this time, he was made Technical Director of a biotechnology spin-out (ThioLogics). In April 2015, he was appointed as a Lecturer at UCL Department of Chemistry, before being promoted to Reader (2017) and then Professor (2019) at the same institution. Vijay's research has been highlighted by Forbes, Scientific American, CNN News, Nature Chemistry and the Royal Society of Chemistry. |
1. Introduction
Over the last two decades, the functionalisation of peptides and proteins through bioconjugation reactions has become of great importance and interest to researchers across the breadth of chemical biology.1,2 Advancements in peptide and protein modification strategies have enabled new development in the fields of biopharmaceuticals (e.g. antibody–drug conjugates (ADCs)),3 diagnostic tools,4,5 tuning pharmacokinetic profiles of drugs (e.g. extending blood half-lives),6 and probing biological systems.7 One of the most successfully employed bioconjugation strategies to date comes in the form of cysteine modification, which enables the formation of well-defined, homogenous bioconjugates with pharmacokinetically superior profiles over heterogenous analogues.8–10 The low natural abundance of cysteine, in addition to it bearing a highly nucleophilic thiol side chain, makes this amino acid ideal to add functionality to a peptide/protein in a site-selective and controlled fashion.11,12 It is important to note that as cysteines are normally present in their oxidised disulfide form, or buried deep within the protein structure, bioconjugation strategies (including those alluded to within this review) will likely either (i) incorporate a mutant cysteine into the protein at a solvent accessible site (via site-directed mutagenesis) or (ii) reduce solvent accessible disulfide bonds and the modify the newly liberated cysteine residues (Fig. 1).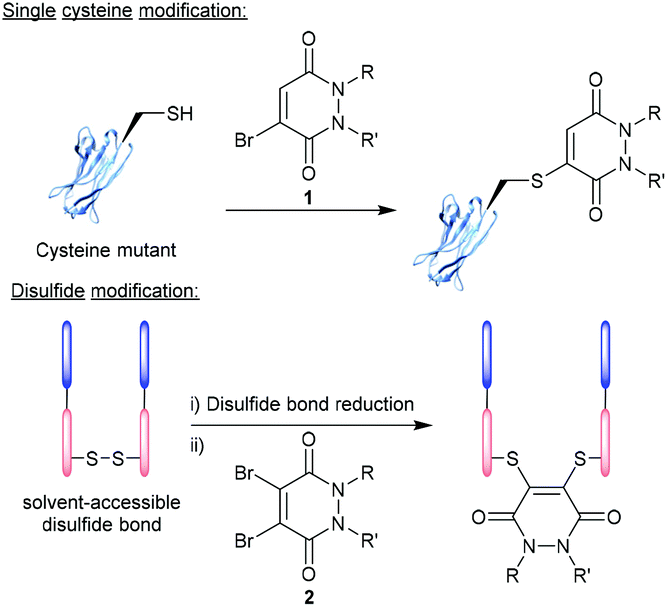 | ||
| Fig. 1 Modification of cysteine residues and disulfide bonds using BrPDs 1 and DiBrPDs 2, respectively. | ||
A plethora of exciting reagents and technologies exist that can facilitate the selective modification of cysteine residues and disulfide bonds, each displaying a unique set of properties and associated advantages and disadvantages.13–21 Amongst some of the most popular highly reactive cysteine-targeting technologies (e.g. maleimide and maleimide derivatives) issues can arise with regard to bioconjugate stability (i.e. to ensure a high degree of bioconjugate stability, maleimides need to be hydrolysed post-cysteine modification), cysteine selectivity and/or cross reactivity with reducing agents (required to liberate cysteine residues from disulfide bonds). The scope of this review will cover the development and application of pyridazinediones (PDs) – a particularly promising class of reagents that can facilitate cysteine and disulfide modification, whilst avoiding the associated disadvantages of many highly reactive (and in some cases less selective) cysteine-targeting methodologies. Bromopyridazinediones (BrPDs) 1 or dibromopyridazinediones (DiBrPDs) 2 are electrophilic in nature and act as a Michael acceptor in the reaction with cysteine residues (Fig. 1). The reaction between PDs and the thiol group present on cysteine residues produces a highly stable unsaturated thio-substituted structure (mechanistically thought to proceed via an addition–elimination pathway).22 The PDs six-membered heterocyclic structure is thought to be less electrophilic when compared to conventional Michael acceptors (e.g. maleimides), which results in a higher selectivity for cysteine residues (i.e. over lysine residues) and a greater associated stability of the bioconjugates formed. PDs, and the cysteine-bioconjugates they form, have shown to be stable to hydrolysis, acidic/basic conditions, and in blood serum.23,24 These highly desirable properties have led to the continued use of PDs throughout many avenues of chemical biology in the last decade. This review will explore the properties that make the PD scaffold unique and give an updated account of the novel modification strategies derived from PD-based technology – as well as the applications of the formed PD-conjugates.
2. Features of the pyridazinedione scaffold
In 2011, BrPDs and DiBrPDs emerged onto the scene of cysteine bioconjugation, providing a unique level of bioconjugate stability when compared with conventional Michael acceptors (e.g. maleimides).25 In this work, simplistic diethyl BrPD 3 and DiBrPD 4 scaffolds were shown to quantitatively modify a model single cysteine containing protein Grb2-SH2 (SH2 domain of the Grb2 adaptor protein) and a disulfide containing peptide (somatostatin), respectively (Fig. 2). In this report, and in subsequent work,23 a large amount of evidence has been provided to establish that cysteine-PD based constructs are stable in terms of hydrolytic stability (a flaw associated with the commonly employed maleimide motif),26,27 towards blood thiols (at concentrations found in the blood) and in human serum.23 Additionally, two highly useful characteristics of the PD scaffold are eluded to in this work:25 (1) PD–cysteine conjugates can be cleaved from the cysteine residue making the process reversible, if desired; (2) PDs can have up to 4 points of chemical attachment, making this scaffold ideally suited to provide a platform for multi-functionality.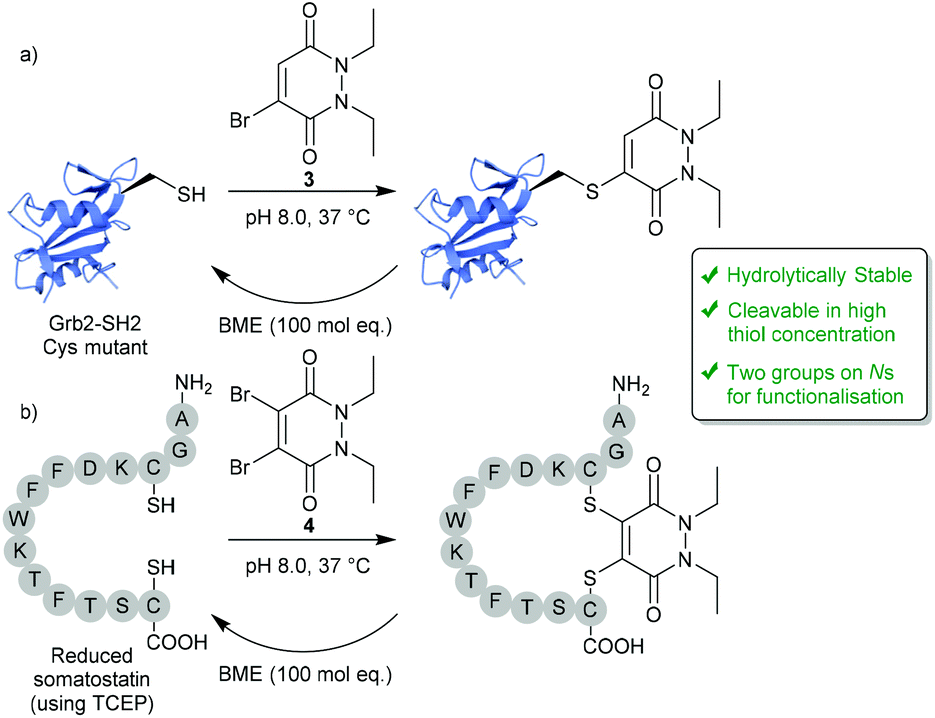 | ||
| Fig. 2 Reversible modification of mutant cysteine Grb2-SH2 and reduced somatostatin using BrPDs and DiBrPDs.25 | ||
2.1. Controlling reversibility
Whilst PDs and the cysteine-modified constructs they form are stable in blood, it has been demonstrated that the PD scaffold can also be removed from the cysteine residue, but only when exposed to very high concentrations of a competing thiol.25 Initial cleavage studies conducted on Grb2-SH2-PD conjugates and somatostatin-PD conjugates demonstrated the PD modification to be fully reversible upon addition of a high concentration of β-mercaptoethanol (BME) (Fig. 2). PD-based Grb2-SH2 conjugates were also shown to be susceptible to cleavage with GSH (1 mM, pH 7.2).25 This unique thiol-reactivity profile is exceptionally well-suited for use in vivo as the PD–cysteine construct is stable when exposed to low concentrations of thiol found in the blood but may be cleaved when exposed to the high thiol concentrations found in unique physiological environments (e.g. intracellularly or in some cases in a tumour environment).28 As most biopharmaceuticals used in vivo require payload release in these target environments (in this case the payload would be attached to the PD), the PD being cleaved under these conditions is considered to be a great potential advantage.One particularly exciting use case for the reversible nature of the PD scaffold was published in the context of small-molecule drug-conjugates (SMDCs).29 In this work presented by Fernandez et al., a platform was developed using the PD core to enable the irreversible attachment of tumour targeting ligands (via the N-positions) and reversible attachment of fluorophores/drugs (via the C-positions) (Fig. 3a). In this work, a small library of alkyl and aryl thio-substituted PDs were synthesised from BrPD and DiBrPDs, which were then subject to incubation with varying concentrations of glutathione (GSH). These studies were designed to mimic the thiol concentrations found in blood (pH 7.4, 5 μM GSH) and in cells (pH 6.5, 5 mM GSH). It was reported that whilst all synthesised constructs were stable in blood concentrations of GSH, the aryl thio-substituted PDs cleaved quickly and efficiently in the cell-like thiol environment. Following these results, a novel self-immolative PD linker 5 was designed and synthesised. It featured a turn-on fluorescence probe in thiol rich environments, through the controlled release of umbelliferone following thiol-PD cleavage (Fig. 3a). Furthermore, the N-positions of the PD core were also synthesised to harbour alkyne and azide groups, which could enable the facile and modular attachment of various small-molecule tumour targeting ligands (e.g. folic acid).30
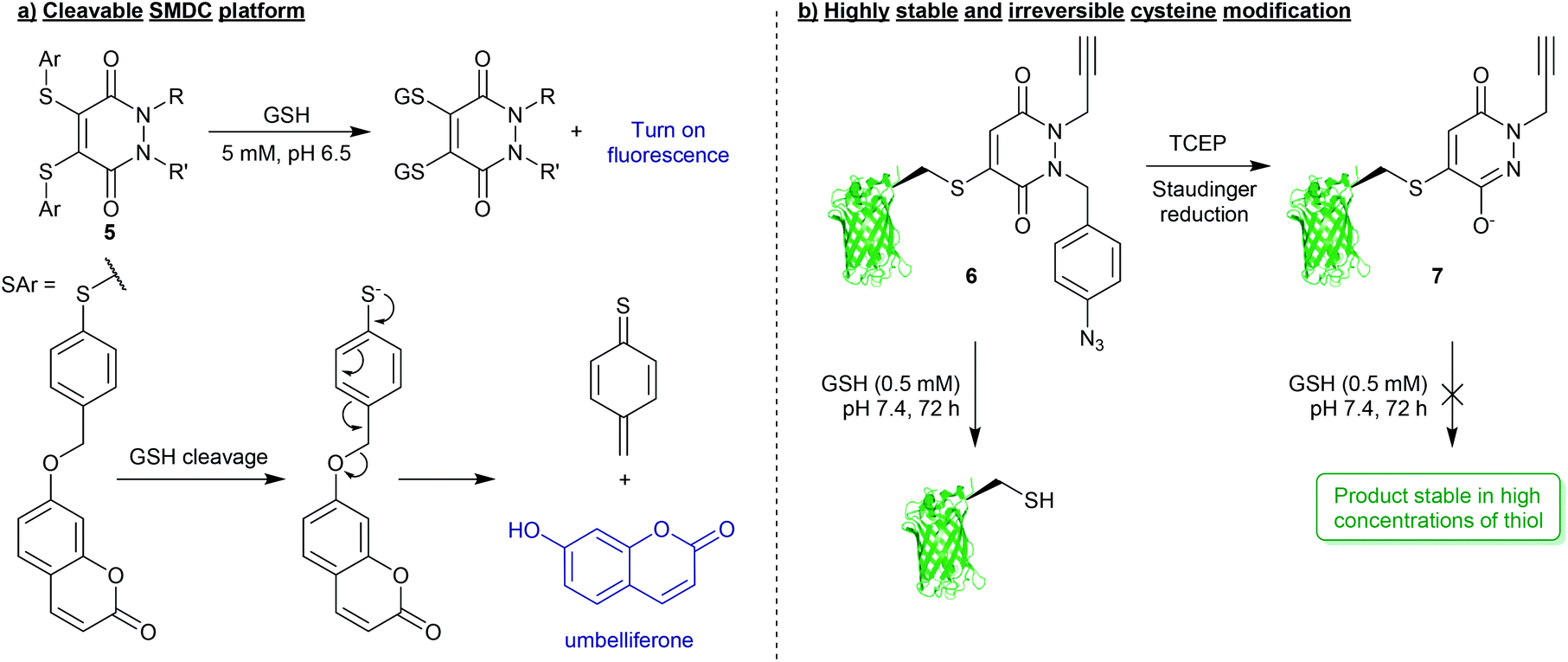 | ||
| Fig. 3 (a) Platform for the formation of multifunctional small-molecule drug-conjugates (SMDCs) 5 with turn on fluorescence mechanism achieved through PD-cleavage.29 (b) Bioconjugation strategy to secure highly robust and thiol-stable bioconjugates (7).33 | ||
For applications where PD-thiol cleavage is undesirable in highly thiol rich environments, and a scaffold is required to be stable in the presence of a large excess of thiol (e.g. applied materials, or biopharmaceuticals with extended blood half-lives),31,32 PD-based chemistries have been developed that can effectively completely “switch-off” reversibility.33 Work presented by Maurani et al. demonstrated the synthesis of a novel PD derivative that would yield bioconjugate 6 containing a para-azidobenzyl group attached at the N-position of the PD (Fig. 3b). This PD was used to quantitatively modify the mutant cysteine containing green fluorescent protein (GFPS147C). However, it was reported that by exposing the para-azidobenzyl group to a Staudinger reduction, facilitated by the mild reducing agent tris(2-carboxyethyl)phosphine (TCEP), the resultant para-aminobenzyl group participated in self-immolation to produce the highly conjugated species 7 (Fig. 3b). Due to increased pseudo-aromatic character of this species 7, it was proposed that this novel thiol-substituted species would be stable to subsequent thiol exchange. To appraise this, GFPS147C-conjugates 6 and 7 were subjected to GSH incubations (pH 7.4, 0.5 mM GSH, 37 °C, 72 h). Conjugates harbouring the para-azidobenzyl group showed efficient cleavage under these conditions, whereas GFPS147C conjugates that were treated with TCEP prior to the incubation (i.e. reducing the para-azidobenzyl group) were shown to undergo no reaction under analogous conditions (Fig. 3b). This work therefore demonstrates the unique level of control that can be gained over stability of PD-based conjugates towards thiols, which has exciting applications in the field of drug/linker release.
2.2. Developing multifunctional platforms
The 4-points of chemical attachment on the PD scaffold have been utilised to provide a platform to enable the facile attachment of multiple functionalities onto a protein via a single point of modification. Through applying bioorthogonal “click” chemistries, DiBrPDs were used as a platform for the modular attachment of commercially available, or easily obtainable modalities onto a protein.34 Through functionalising the nitrogens of the PD heterocycle with strained alkynes and terminal alkynes, azide-containing drugs (doxorubicin), fluorophores (sulfo-Cy5) and polyethylene glycol (PEG) chains were added in a quantitative fashion to the disulfide bond of trastuzumab fragments (Fig. 4a). The chemistry applied in this early work utilised the orthogonal strain-promoted azide–alkyne cycloaddition (SPAAC) and copper-catalysed azide–alkyne cycloaddition (CuAAC) reactions of the PD 8. There are now several more recent accounts in literature of other clickable handles being installed at this position on the PD for faster “click” kinetics (e.g. trans-cyclooctene (TCO), tetrazines, azides, etc.), which further emphasises the modularity of this approach.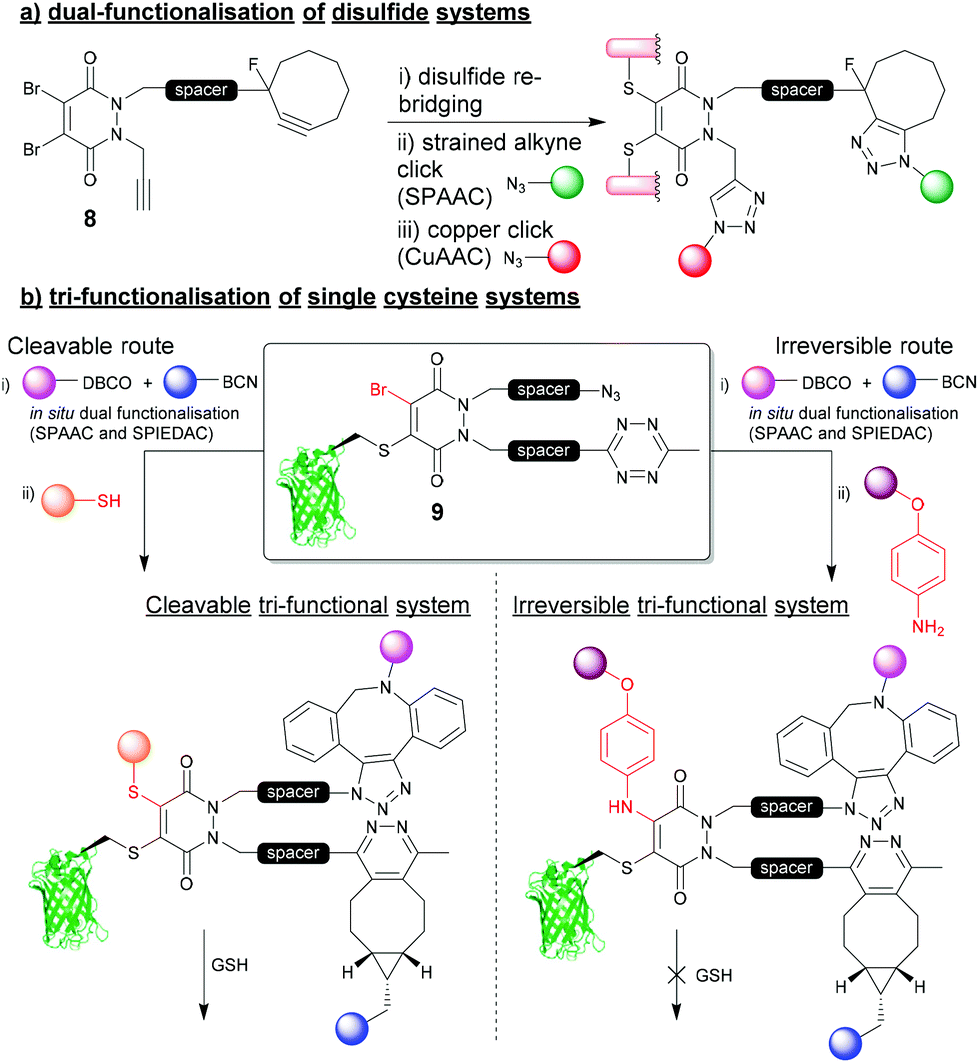 | ||
| Fig. 4 (a) Dual functionalisation approach to re-bridging disulfide bonds on proteins.34 (b) tri-functionalisation of single cysteine systems and controlling cleavability of multifunctional bioconjugates.35 | ||
Most recently, a novel bioconjugation approach was proposed which utilised a DiBrPD scaffold to enable the attachment of three functionalities to a single cysteine residue with additional control over the reversibility of the modification.35 In this work, a DiBrPD containing both azide and tetrazine functionality was used to quantitatively modify the cysteine mutant GFPS147C (to produce bioconjugate 9), leaving three chemical points available for subsequent functional additions (i.e. (1) azide reaction with DBCO, (2) tetrazine reaction with BCN, (3) PD core reaction with a thiol or amine to substitute the remaining Br on the PD core, Fig. 4b). Through utilising favourable “click” kinetics, a one-pot reaction was carried to functionalise the azide and tetrazine groups in one-pot, using a DBCO-PEG and a BCN-fluorophore reagent, respectively. Following this dual modification, the authors proceeded to modify the Br position of the PD with either a thiol (i.e. a peptide), or a very high concentration of amine (i.e. para-azido aniline) as a third point for functionalisation, which offered control over stability of the final construct towards an excess of thiol. Due to the increased level of electron donation into the heterocyclic structure the PD, aniline-modified PD–protein constructs were shown to be robustly stable, even in the presence of a large excess of thiol (5 mM GSH); perhaps serving as an alternative strategy to that described in Fig. 3b. This work is a key demonstration of the two features that make the PD moiety an attractive tool for cysteine modification, and how they have been further understood and developed over the last decade (i.e. controlling cleavability and providing a modular “click” platform for the addition of functionality to a protein).
Utilising the elaborate chemistries outlined above to form stable and multifunctional bioconjugates, the PD moiety has been used to enable a plethora of applications across chemical biology. A number of modification strategies and applications that employ BrPDs and DiBrPDs for modification of single cysteine residues and disulfide bonds will be presented herein.
3. Single-cysteine modification strategies using BrPDs
3.1. Protecting groups for peptide synthesis
In light of the substantial data presented over the last decade demonstrating the robustness and controlled cleavable nature of PD-thiol conjugates, a logical use case was presented for BrPDs in the form of protecting groups for peptide synthesis. In this work presented by Spears et al., a protected Fmoc-Cys(PD)-OH species 10 was synthesised and incorporated into short peptide sequences (e.g. oxytocin, Fig. 5a).24 The Cys-PD motif was found to be stable in a wide variety of conditions required for solid phase peptide synthesis (SPPS) including: (i) high concentration of base (i.e. piperidine) – required for Fmoc deprotection, (ii) high concentrations of acid (i.e. TFA) – required for peptide-resin cleavage, and (iii) conditions required for microwave-assisted peptide synthesis.24 Once PD-deprotection was required (i.e. following SPPS with cysteine(s) being protected by PDs), a cleavage cocktail comprising 10% DTT w/v in DMF![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :5 mM phosphate buffer (8:2) was used to efficiently cleave the PD from the peptide. Furthermore, as the PD displayed a unique UV absorbance value once cleaved from the resin, instantaneous quantification of PD deprotection was shown to be possible through facile UV analysis of cleavage washes. When compared with other state-of-the-art thiol-cleavable protecting groups, the PD motif presents an attractive option for SPPS and could likely be utilised as an orthogonal strategy in conjunction with other protecting groups (e.g. Trt) to yield complex peptides with multi-disulfide systems.24,36 Furthermore, whilst this proof-of-concept work features a simplistic diethyl PD model, the authors also allude to the advantages that could be obtained through utilising functional BrPDs in this space (i.e. PDs harbouring affinity/solubility tags).
:5 mM phosphate buffer (8:2) was used to efficiently cleave the PD from the peptide. Furthermore, as the PD displayed a unique UV absorbance value once cleaved from the resin, instantaneous quantification of PD deprotection was shown to be possible through facile UV analysis of cleavage washes. When compared with other state-of-the-art thiol-cleavable protecting groups, the PD motif presents an attractive option for SPPS and could likely be utilised as an orthogonal strategy in conjunction with other protecting groups (e.g. Trt) to yield complex peptides with multi-disulfide systems.24,36 Furthermore, whilst this proof-of-concept work features a simplistic diethyl PD model, the authors also allude to the advantages that could be obtained through utilising functional BrPDs in this space (i.e. PDs harbouring affinity/solubility tags).
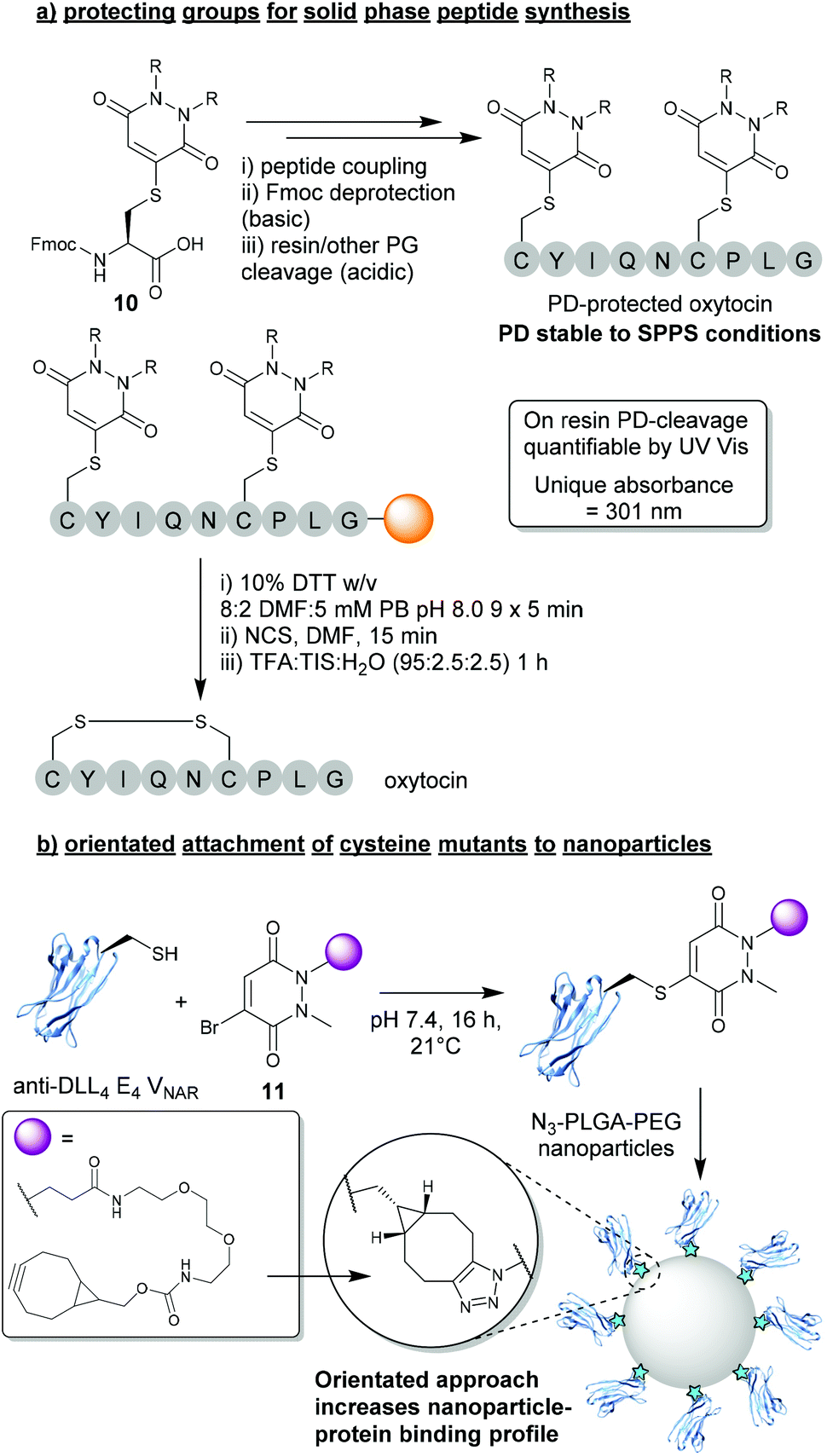 | ||
| Fig. 5 (a) BrPDs as protecting groups for solid phase peptide synthesis (SPPS).24 (b) BrPDs facilitate addition of cysteine mutants to the surface of nanoparticles.37 | ||
3.2. Orientated attachment of cysteine mutants to nanoparticles
Another exciting application that can be achieved through utilising BrPDs was presented by Nogueira et al., in the formation of protein–nanoparticle conjugates.37 In this work, BrPDs were used to facilitate the addition of an anti-DLL4 E4 Variable New Antigen Receptor (VNAR) to an azide-harbouring polylactic acid-co-glycolic acid–polyethylene glycol (PLGA–PEG) nanoparticle scaffold. To achieve this, a cysteine mutant anti-DLL4 E4 VNAR was expressed containing an additional Ala-Cys-Ala sequence inserted at the C-terminus, which was then site-selectively modified with BrPD 11 (i.e. a BrPD harbouring a strained alkyne handle). The strained alkyne-functionalised protein was then loaded onto the surface of the PLGA-PEG-azide nanoparticle through efficient SPAAC “click” chemistry (Fig. 5b).37 This approach of site-selectively modifying proteins through cysteine modification, prior to loading onto the nanoparticle surface, yielded highly effective VNAR–nanoparticle conjugates that were far superior when compared with a non-site-selective lysine modification strategy. Site-selectively modified VNAR–nanoparticle conjugates (i.e. an orientated protein approach) were shown to outperform lysine-modified VNAR–nanoparticle conjugates (i.e. a randomly orientated protein approach), with equal protein loading, in binding assays towards DLL4, by several orders of magnitude. This work not only showcases the versatile nature of using PDs to “click” proteins to nanoparticles, but also a significant advantage of a site-selective protein modification strategy.4. Disulfide (antibody) modification strategies using DiBrPDs
Despite BrPDs and DiBrPDs showing similar chemistries and features, to date, the DiBrPD scaffold has been the most extensively utilised PD technology due to their ability to efficiently re-bridge disulfide bonds, especially those present on antibodies and antibody fragments.38,39 The disulfide re-bridging approach offers a site-selective method to attach functionality to a protein, whilst also retaining the inherent structural function that stems from covalently cross-linking proteins. This approach is most widely applied in the context of antibody modification, as many subtypes of antibody (e.g. IgG1, IgG2, IgG4 etc.) have solvent accessible disulfide bonds that can be modified to produce homogenous antibody conjugates with well-defined loadings of functionalities.9,40–43 However, this approach to antibody modification has limitations, especially in the context of forming antibody–drug conjugates (ADCs). Firstly, in the most commonly used antibody subtype IgG1, there are just four disulfide bonds that are solvent accessible and available to modify (i.e. 2 in the hinge region and 2 in the Fab region), but to date there is no reliable method to functionally distinguish between these two types of disulfides. This presents an inherent challenge as there are just four available points for modification. When all four disulfide bonds are modified (which is required to form homogenous products as it is not possible to reliably reduce a sub-set of these 4 inter-strand disulfide bonds), the resultant antibody-conjugates are typically limited to a drug-to-antibody ratio (DAR) of 4 (Fig. 6). The two hinge region disulfide bonds being in close proximity presents a second challenge, as these are close enough such that many re-bridging reagents (e.g. divinyltriazines, dibromomaleimides and arylene-dipropiolonitriles) can staple together intrachain disulfides in the non-native confirmation and produce appreciable quantities of a species referred to as “half-antibody” (Fig. 6).44–46 This disulfide scrambling process not only produces isomers (and therefore lowers homogeneity), but the presence of the “half-antibody” species formed is thought to affect secondary antibody interactions (e.g. antibody-dependant cell-mediated cytotoxicity (ADCC)).47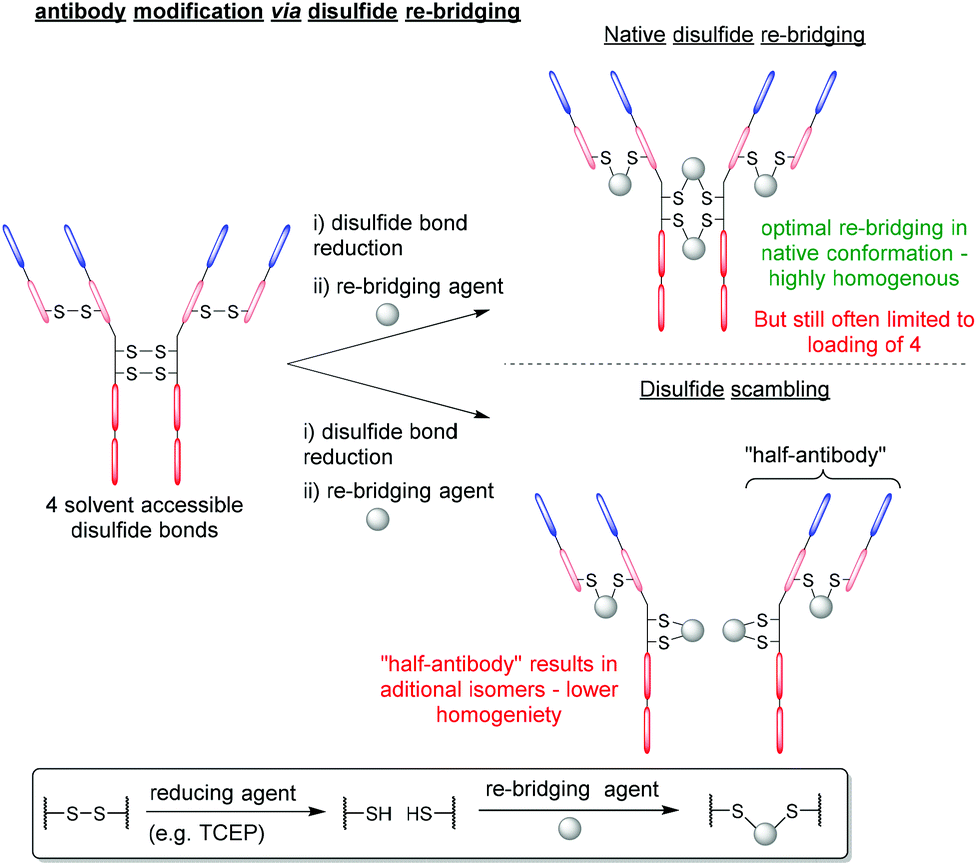 | ||
| Fig. 6 General antibody re-bridging strategy and associated limitations. | ||
Antibody modification strategies that employ the DiBrPD scaffold have revolutionised the disulfide re-bridging approach, as they offer two distinct advantages over conventional methods: (i) the antibody conjugates formed are highly homogenous in nature, can be tuned to avoid the formation of the aforementioned “half-antibody” species (see Fig. 6) and do not any require additional stabilisation (e.g. hydrolysis) steps to be stable in blood, and (ii) through employing unique PD-based chemistries, antibody conjugates can be formed with well-defined loadings, combined with click chemistry to enable multi-modification in a controlled and an overall provide exquisite level of control over DAR.
4.1. Forming homogenous disulfide-modified antibody-conjugates
The first significant advantage that PDs offer over traditional re-bridging reagents (e.g. vinyl sulfones), is that PDs generally tend to re-bridge hinge region disulfides in the native confirmation, reducing the amount of “half-antibody” present.34 The high levels of homogeneity obtained stem from applying an “in situ” protocol, which features the one-pot addition of TCEP and DiBrPDs to reduce and re-bridge antibody disulfides, respectively (Fig. 7a).48 Such a protocol is thought to only be possible when levels of cross-reactivity between TCEP and the re-bridging agent are low or non-existent. The robust and stable nature of DiBrPDs makes them ideally poised for incubation with phosphine-based reducing agents, and suitable for employment in “in situ” protocols, whereas traditional (more reactive) reagents are not (e.g. maleimide derivatives). The most recently reported optimised PD–antibody conjugation protocol produced antibody conjugates with a homogeneity of (>90%), which was obtained by using widely accessible reducing agents (i.e. TCEP) and DiBrPD derivatives at lower temperatures (i.e. 4 °C).48 It is also worth noting that in this work, the authors reported an optimised and more adoptable synthesis for a diverse array of functional PD linkers.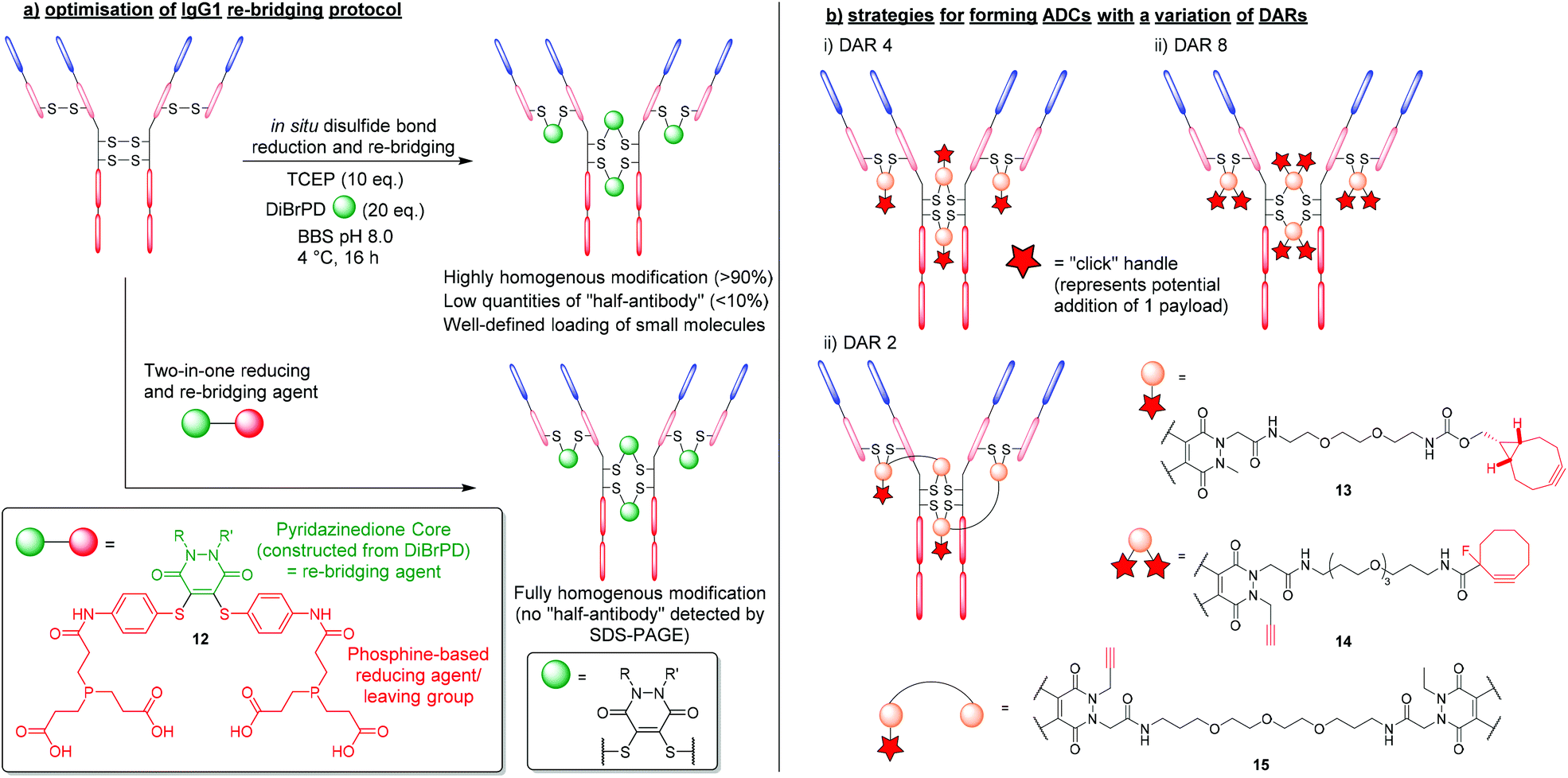 | ||
| Fig. 7 (a) Efficient and homogenous antibody modification using DiBrPDs and phosphine-containing PD 12.48,49 (b) controlling payload loading in the formation of antibody-conjugates using DiBrPDs.23,34,50 | ||
This in situ approach has also been exploited further by incorporating a reducing agent directly into the PD structure to form a two-in-one reducing and re-bridging reagent; this is believed to be the first example of the construction of such a reagent. The stable profile of the PD core enabled the design of this first-in-class reagent that contained both a Michael acceptor and phosphine-based reagent capable of reducing disulfide bonds.49 The phosphine-based PD 12 was reported to produce re-bridged bioconjugates with exceptional efficiency (i.e. quantitative conversion), circumventing the requirement to subject peptides and proteins to reducing conditions (Fig. 7a). Additionally, when the two-in-one reagent 12 was reacted with an IgG1 antibody scaffold (i.e. trastuzumab), a fully homogenous antibody conjugate was formed yielding a bioconjugate with only natively re-bridge disulfide bonds (i.e. no half-antibody was observed by SDS-PAGE). The authors deduced that the tendency of the IgG1 disulfide bonds to bridge in the native confirmation correlates well with the residency times of the thiols once liberated from the disulfide bond (i.e. thiols that spend longer in their reduced state have an increased chance of re-bridging in the non-native confirmation and form bioconjugates with more “half-antibody”). This data also concurs with the optimised conditions reported for producing homogenous antibody-conjugates via the aformentioned in situ reduction/re-bridging protocol.
4.2. Controlling drug loading of disulfide modified antibody-conjugates
As previously outlined, the up to 4-points of possible chemical attachment of the PD scaffold enables their extensive use in forming multifunctional bioconjugates. For this reason, DiBrPDs are ideally poised to solve a major limitation associated with the disulfide re-bridging approach, in that they are ideal for making conjugates bearing more than one functional group per disulfide bond; this provides great flexibility. Over the years, variations of the DiBrPD scaffold have been designed such that IgG1 antibody scaffolds (i.e. containing 4 disulfide bonds) can be fully modified to form homogenous products with a well-defined loading of 2, 4 and 8 (or 4 + 4) functional modalities, including via click chemistry.23,34,50The most straightforward antibody conjugate to form with a well-defined functional modality loading is when synthesising a species with 4 functionalities per antibody. As there are 4 solvent accessible disulfide bonds available on conventional IgG1 antibodies, these can be reduced and subsequently modified with a DiBrPD species, as alluded to in the aforementioned work.25 By using a PD with a single functional moiety attached via the nitrogen of the PD's heterocyclic structure, a payload can be attached either pre- or post- disulfide modification through employing commercially available reagents to carry out efficient click chemistries (Fig. 7bi). One reported example in the literature was presented by Robinson et al. where the authors modify the disulfide bonds of trastuzumab with a DiBrPD-drug molecule to form an anti-HER2 monomethyl auristatin E (MMAE) ADC.23 Extensive characterisation using mass spectrometry and hydrophobic interact chromatography (HIC), revealed antibody conjugates to display an average DAR of 4, with minimal amounts of other DAR species being observed. The PD-based ADC formed in this work was shown to be highly active against HER2-positive human breast cancer cells (i.e. in the sub nM range), and also showed excellent efficiency in a mouse xenograph model.
Through utilising the multifunctional nature of the PD scaffold, the facile formation of ADCs with higher DARs can be achieved through reported “dual-click” strategies. By adding two “clickable” moieties onto the PD scaffold, literature reports have showed the synthesis of antibody-conjugates with a loading of 8, in a “plug-and-play” fashion (Fig. 7bii).34 Furthermore, through utilising orthogonal “click” chemistries (i.e. SPAAC followed by CuAAC reactions), two groups of 4 moieties were added in succession, offering a unique level of modularity and control over antibody modification.
Inherently, the most challenging antibody-conjugates to synthesise via disulfide re-bridging are those harbouring a functional modality loading of less than 4 (e.g. DAR 2 species). To produce bioconjugates with exceptional levels of homogeneity, all solvent accessible disulfide bonds must be modified (i.e. when targeting less than 4 disulfide bonds, there is no control over where the payload will re-bridge, and numerous isomers will be produced). Work presented by Lee et al. provided a route to a lower loading of functional modalities per antibody through the use of a bis-DiBrPD that could re-bridge a pair of disulfide bonds to produce the antibody conjugate 15 (Fig. 7biii).50 By utilising the 4 points of chemical attachment unique to the PD scaffold, two DiBrPD cores were connected by a small linker – only one of which was functionalised with a “clickable” handle (i.e. an alkyne for CuAAC reactions). Once antibody-conjugate 15 was formed, through re-bridging all 4 solvent accessible disulfide bonds available on the IgG1 antibody trastuzumab (each small molecule reacting with a pair of reduced disulfide bonds), the 2 newly installed alkyne handles were reacted with Alexa Fluor® 488 azide. The resultant labelled conjugate was confirmed to have a fluorophore loading of 2.0, with all disulfide bonds fully modified (analysed by mass spectrometry and UV-vis) and showed a high level of homogeneity.
5. DiBrPDs in the generation of biopharmaceuticals and diagnostic tools
The DiBrPD scaffold and the bioconjugation strategies that utilise this motif have been extensively developed over the years to provide a valuable tool to the chemical biologist's “toolbox”, especially within the field of antibody modification.51 The reported applications that have stemmed from the use of this technology are discussed herein.5.1. Clinically relevant antibody-conjugates
Due to the high levels of homogeneity that can be obtained, and the modular way in which products can be formed, DiBrPDs have become an attractive tool in the synthesis of antibody–drug conjugates. To date, several antibody platforms have been shown to be compatible with the DiBrPD re-bridging approach including IgG1, IgG2a (mouse), IgG2b (rat), and IgG4.52–55 In addition, a wide variety of functionalities have been added through efficient and quantitative “click” reactions (e.g. Doxorubicin, MMAE, porphyrins, etc.),23,34,56–58 demonstrating the chemical compatibility between the PD core and a plethora of functional motifs.One particularly promising class of ADC synthesised using the DiBrPD motif was presented by Javaid et al. and was targeted towards leucine-rich alpha-2-glycoprotein 1 (LRG1).55 The anti-LRG1-MMAE ADC, synthesised using a “pre-click” strategy, was formed with relatively high levels of homogeneity, and a well-defined PD-loading of 4. For this approach, a strained alkyne-functionalised DiBrPD was reacted with a commercially available vc-PABC-MMAE azide (i.e. through SPAAC chemistry), prior to antibody functionalisation. The resultant species was subsequently used to re-bridging the IgG4 antibody Magacizumab to form the desired anti-LRG1-MMAE ADC (Fig. 8a). The synthesised ADC was shown to retain binding, and to be effective in targeting the non-internalising LRG1 target in vitro. Treatment with the novel ADC also resulted in an increase in survival in vivo (human LRG1 knock-in mice) when compared to antibody/chemotherapy treatments alone, whilst also avoiding undesired side-effects. The authors also highlight the stability of the antibody-conjugate in conditions designed to mimic the in vivo environment (i.e. in human serum) which, when combined with the versatile/homogenous synthesis of this ADC, highlights significant advantages of using DiBrPDs for antibody modification.
 | ||
| Fig. 8 (a) Formation of anti-LRG1-MMAE ADC using DiBrPDs to re-bridge disulfide bonds.55 (b) Using DiBrPDs and click chemistry to chemically construct bi-specific antibodies.59 (c) Employing DiBrPDs to re-bridge antibody fragments for the modular formation of oriented antibody–nanoparticle conjugates.66 | ||
5.2. Chemically constructed bi-specific antibodies
Using the PD scaffold to facilitate the modular attachment of various modalities onto proteins is indeed a well-received concept within the field of bioconjugation, especially when applied to ADC synthesis. However, recent work has attempted to take this methodology one step further and achieve the highly ambitious task of chemically constructing bispecific antibodies (bsAbs) and bispecific antibody–drug conjugates (bsADCs).59 Chemical methods that can facilitate the construction of bsAbs and bsADCs are highly sought after as they offer significant advantages in terms of reproducibility and modularity (i.e. a large library of antibody fragments can be brought together to form bsAbs in a systematic way, which is not reliant on expression systems).60 In this work, a small library of model antibodies (trastuzumab, cetuximab and rituximab) were enzymatically digested into smaller fragments that contained single accessible disulfide residues (i.e. fragment antigen-binding region (Fab) fragments).59 These Fab fragments (FabTra, FabCet and FabRit) were then reduced with TCEP, to liberate two cysteine residues, and then subsequently modified with DiBrPDs to form antibody conjugates 16 and 17, containing a strained alkyne and tetrazine functionality, respectively (Fig. 8b). Through strain-promoted inverse electron-demand Diels–Alder cycloaddition (SPIEDAC) “click” chemistries, the two PD-modified antibody fragments were covalently linked with a previously unseen level of efficiency (a yield ranging from 55–78% was observed which far exceeds previous chemical-construction attempts). In this case, it was observed that bringing two proteins together significantly affected the “click” kinetics involved, therefore the highly efficient SPIEDAC tetrazine-strained alkyne reaction was employed to compensate in this regard. This work is also therefore a key example of how orthogonal “click” chemistries are fine-tuned for the desired purpose, and a model derived from the modular PD scaffold proved essential in enabling efficient technologies.5.3. Orientated nanoparticle–antibody decoration
Another key application that has benefited from employing PD-based bioconjugation strategies, is the formation of nanoparticle–antibody conjugates.5,61,62 DiBrPDs in particular have been used extensively to site-selectively re-bridge antibody-fragments, which can then be attached to the surface of nanoparticles through efficient copper free “click” chemistries. Previous reports have shown that antibody–nanoparticle conjugates synthesised with antibody fragments (e.g. Fab fragments), show an increase in selectivity, antigen binding, and generally have more favourable pharmacokinetics when compared with the full antibody equivalent.63–65 A novel approach that utilised site-selective protein modification to add antibody-fragments onto nanoparticles in an orientated fashion (through disulfide re-bridging) was pioneered by Richards et al.66 In this work, the IgG1 antibodies trastuzumab and cetuximab were digested into smaller Fab fragments. DiBrPDs were then used to effectively modify both fragments in a site-specific fashion (as Fab fragments contain a single solvent accessible disulfide bond), to produce a Fab species harbouring strained alkyne functionality (Fig. 8c). Fab fragments were then added to azide-functionalised PLGA–PEG nanoparticles, and the resultant antibody–nanoparticle conjugates were assessed in terms of conjugation efficiency and antigen-binding. When compared with nanoparticles decorated with non-site-selectively modified protein (i.e. modified through lysine-NHS ester reactions), the site-selective approach yielded a significantly higher conjugation efficiency and a vast increase in antigen-binding capability. This work provides substantial evidence to suggest that a higher antibody-fragment loading can be achieved when attaching proteins in an orientated fashion, which can be achieved through modifying and attaching antibody-fragments via a single point. This approach of digesting antibodies and re-bridging the resultant Fab fragments with DiBrPDs therefore provides a highly modular and attractive route to nanoparticle decoration when compared with conventional strategies. In subsequent work, bromopyridazinedione derivatives used in this context have enabled the development of highly efficacious therapeutics (i.e. through decorating nanoparticles with VNAR proteins – see section 3.2),37 and diagnostic tools (i.e. through decorating gold nanoparticles with IgG antibodies).5 This work, and the technology that it enables, demonstrates significant advantages of the oriented approach to nanoparticle–protein synthesis, with bromopyridazinedione derivatives playing a key facilitating role.6. Synthesis of bromopyridazinedione derivatives
The synthesis of pyridazinediones was first reported by Mizzoni et al.67 in 1951, but their importance as scaffolds in chemical biology (as discussed in this review) has only been recently established. Their preparation is straightforward and relatively simple to conduct using commercially available or easily obtainable starting blocks such as monobromo or dibromo maleic anhydrides 18 or N-methoxycarbonylmaleimides 19, which react with hydrazines 20 under acidic or basic conditions (respectively, see Fig. 9a).48,68 These reactions yield thiol-reactive PDs comprising up to two distinct groups in a facile manner, this is especially useful as dually functional hydrazines are readily obtainable (an example is provided in Fig. 9b).34 Owing to the facile and modular manner in which bromopyridazinediones can be synthesised, various substituted PDs can be obtained in a straightforward manner. This attribute has, in part, contributed to the various applications of bromopyridazinediones, i.e. as the synthesis of bromopyridazinediones can be readily adapted for the synthesis of complex multi-clickable PDs, bisPDs with various linkers, etc. they can be tuned for use in specific applications.35,50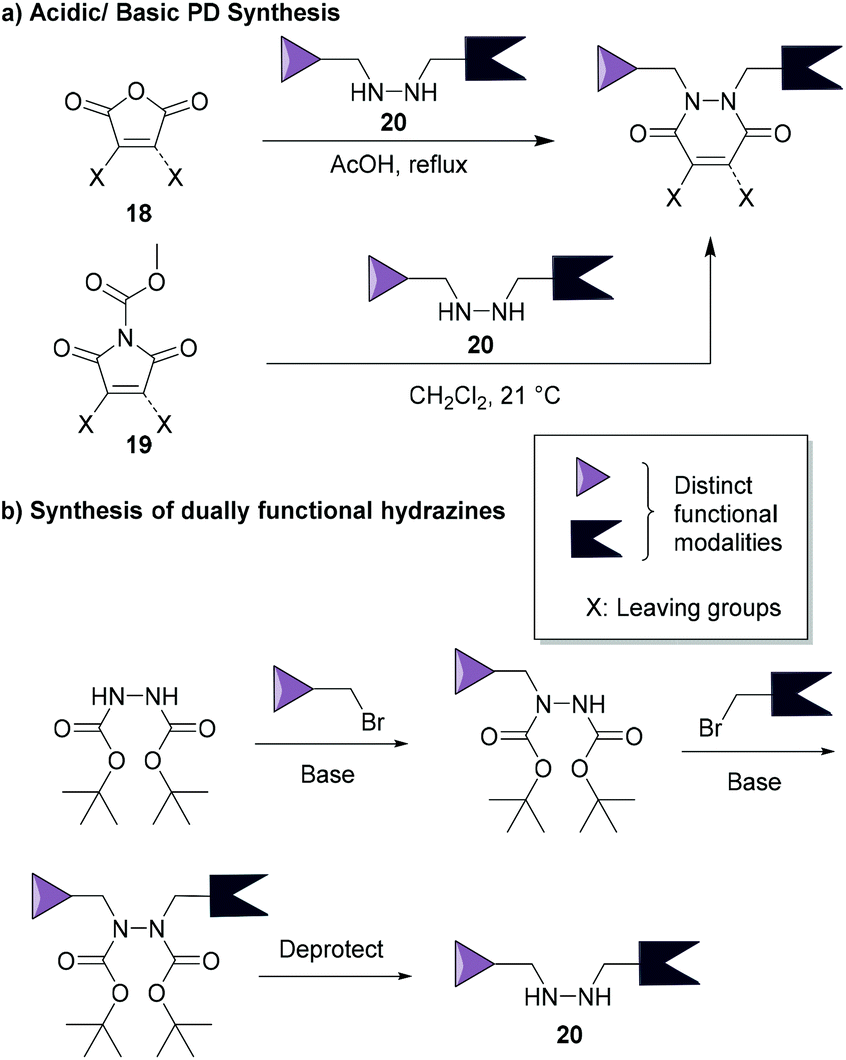 | ||
| Fig. 9 (a) Most common synthetic routes for PD synthesis.46,68 (b) Example of a common route to make dually functionalised hydrazines.34 | ||
7. Conclusions
The site-selective chemical modification of peptides and proteins is an important tool for probing living systems, developing biopharmaceuticals, and improving diagnostic techniques. Key methodologies have been reported in the literature that enable functionalisation of cysteine residues and disulfide bonds, and one promising class of small molecules that can achieve this are pyridazinediones (PDs). Since 2011, bromopyridazinediones (BrPDs) and dibromopyridazinediones (DiBrPDs) have enabled the quantitative modification of various cysteine residues and disulfide bonds, respectively, on various peptides and proteins. The PD scaffold has 4 points of chemical attachment, which has been shown to facilitate the addition of multiple functionalities to a peptide or protein in a facile and modular manner. This multifunctional, site-selective bioconjugation approach has been shown to display an exquisite level of control over cleavability in concentrated thiol environments. By default, PD-based bioconjugates are stable in blood, but can be cleaved in very high concentrations of thiol (i.e. bioconjugates are designed to cleave intracellularly), or can be tuned to infer complete thiol stability even in extreme thiol rich environments (i.e. using a self-immolative cleavable linker or addition of p-anisdine). In addition, the robust nature of the PD scaffold and thiol-PD conjugates thereof (i.e. stable in a wide range of pH, towards hydrolysis etc.) has allowed PDs to be utilised for applications not explored for traditional cysteine-modification reagents (e.g. as protecting groups for peptide synthesis).DiBrPDs have innovated in the field of disulfide re-bridging methodologies, especially with respect to antibody modification, through delivering solutions to the key limitations in the field. Bioconjugates formed through disulfide modification using DiBrPDs have shown to be of exceptional quality in terms of both homogeneity and well-defined functional modality loadings. Furthermore, bioconjugation strategies have been presented that allow exceptional control over drug-to-antibody ratios (DARs), offering solutions to form ADCs with a potential of DAR of 2, 4 and 8 (or 4 + 4). PD-based modification of disulfide bonds has also enabled several applications including: (i) the synthesis of clinically relevant ADCs with well-defined DARs; (ii) the chemical construction of bi-specific antibodies and bispecific ADCs; and (iii) the site-specific modification and oriented attachment of antibody/protein conjugates to nanoparticle scaffolds for application in therapeutics and diagnostics.
Conflicts of interest
V.C is a director of the spin-out ThioLogics, but there are no competing financial interests to declare.Acknowledgements
We gratefully acknowledge the Leverhulme Trust (RPG-2020-010) for funding C.B.References
- C. D. Spicer and B. G. Davis, Nat. Commun., 2014, 5, 4740 CrossRef CAS PubMed.
- O. Boutureira and G. J. L. Bernardes, Chem. Rev., 2015, 115, 2174–2195 CrossRef CAS PubMed.
- S. J. Walsh, J. D. Bargh, F. M. Dannheim, A. R. Hanby, H. Seki, A. J. Counsell, X. Ou, E. Fowler, N. Ashman, Y. Takada, A. Isidro-Llobet, J. S. Parker, J. S. Carroll and D. R. Spring, Chem. Soc. Rev., 2021, 50, 1305–1353 RSC.
- C. N. Loynachan, M. R. Thomas, E. R. Gray, D. A. Richards, J. Kim, B. S. Miller, J. C. Brookes, S. Agarwal, V. Chudasama, R. A. McKendry and M. M. Stevens, ACS Nano, 2018, 12, 279–288 CrossRef CAS PubMed.
- D. A. Richards, M. R. Thomas, P. A. Szijj, J. Foote, Y. Chen, J. C. F. Nogueira, V. Chudasama and M. M. Stevens, Nanoscale, 2021, 13, 11921–11931 RSC.
- A. Wall, K. Nicholls, M. B. Caspersen, S. Skrivergaard, K. A. Howard, K. Karu, V. Chudasama and J. R. Baker, Org. Biomol. Chem., 2019, 17, 7870–7873 RSC.
- E. Weerapana, C. Wang, G. M. Simon, F. Richter, S. Khare, M. B. D. Dillon, D. A. Bachovchin, K. Mowen, D. Baker and B. F. Cravatt, Nature, 2010, 468, 790–795 CrossRef CAS PubMed.
- J. M. Chalker, G. J. L. Bernardes, Y. A. Lin and B. G. Davis, Chem.– Asian J., 2009, 4, 630–640 CrossRef CAS PubMed.
- G. Badescu, P. Bryant, M. Bird, K. Henseleit, J. Swierkosz, V. Parekh, R. Tommasi, E. Pawlisz, K. Jurlewicz, M. Farys, N. Camper, X. Sheng, M. Fisher, R. Grygorash, A. Kyle, A. Abhilash, M. Frigerio, J. Edwards and A. Godwin, Bioconjugate Chem., 2014, 25, 1124–1136 CrossRef CAS PubMed.
- J. R. Junutula, H. Raab, S. Clark, S. Bhakta, D. D. Leipold, S. Weir, Y. Chen, M. Simpson, S. P. Tsai, M. S. Dennis, Y. Lu, Y. G. Meng, C. Ng, J. Yang, C. C. Lee, E. Duenas, J. Gorrell, V. Katta, A. Kim, K. McDorman, K. Flagella, R. Venook, S. Ross, S. D. Spencer, W. L. Wong, H. B. Lowman, R. Vandlen, M. X. Sliwkowski, R. H. Scheller, P. Polakis and W. Mallet, Nat. Biotechnol., 2008, 26, 925–932 CrossRef CAS PubMed.
- M. T. Petersen, P. H. Jonson and S. B. Petersen, Protein Eng., 1999, 12, 535–548 CrossRef CAS PubMed.
- S. G. Tajc, B. S. Tolbert, R. Basavappa and B. L. Miller, J. Am. Chem. Soc., 2004, 126, 10508–10509 CrossRef CAS PubMed.
- G. J. L. Bernardes, J. M. Chalker, J. C. Errey and B. G. Davis, J. Am. Chem. Soc., 2008, 130, 5052–5053 CrossRef CAS PubMed.
- S. J. Walsh, S. Omarjee, F. M. Dannheim, D.-L. Couturier, D. Bexheti, L. Mendil, G. Cronshaw, T. Fewster, C. Gregg, C. Brodie, J. L. Miller, R. Houghton, J. S. Carroll and D. R. Spring, Chem. Commun., 2022, 58, 1962–1965 RSC.
- H. Seki, S. J. Walsh, J. D. Bargh, J. S. Parker, J. Carroll and D. R. Spring, Chem. Sci., 2021, 12, 9060–9068 RSC.
- B. Bernardim, P. M. S. D. Cal, M. J. Matos, B. L. Oliveira, N. Martínez-Sáez, I. S. Albuquerque, E. Perkins, F. Corzana, A. C. B. Burtoloso, G. Jiménez-Osés and G. J. L. Bernardes, Nat. Commun., 2016, 7, 13128 CrossRef CAS PubMed.
- V. F. C. Ferreira, B. L. Oliveira, A. D'Onofrio, C. M. Farinha, L. Gano, A. Paulo, G. J. L. Bernardes and F. Mendes, Bioconjugate Chem., 2021, 32, 121–132 CrossRef CAS PubMed.
- R. Tessier, R. K. Nandi, B. G. Dwyer, D. Abegg, C. Sornay, J. Ceballos, S. Erb, S. Cianférani, A. Wagner, G. Chaubet, A. Adibekian and J. Waser, Angew. Chem., Int. Ed., 2020, 59, 10961–10970 CrossRef CAS PubMed.
- L. Xu, M. J. S. A. Silva, P. M. P. Gois, S. L. Kuan and T. Weil, Chem. Sci., 2021, 12, 13321–13330 RSC.
- M. E. B. Smith, F. F. Schumacher, C. P. Ryan, L. M. Tedaldi, D. Papaioannou, G. Waksman, S. Caddick and J. R. Baker, J. Am. Chem. Soc., 2010, 132, 1960–1965 CrossRef CAS PubMed.
- C. Bahou, R. J. Spears, A. E. Aliev, A. Maruani, M. Fernandez, F. Javaid, P. A. Szijj, J. R. Baker and V. Chudasama, Chem. Commun., 2019, 55, 14829–14832 RSC.
- M. T. W. Lee, A. Maruani and V. Chudasama, J. Chem. Res., 2016, 40, 1–9 CrossRef CAS.
- E. Robinson, J. P. M. Nunes, V. Vassileva, A. Maruani, J. C. F. Nogueira, M. E. B. Smith, R. B. Pedley, S. Caddick, J. R. Baker and V. Chudasama, RSC Adv., 2017, 7, 9073–9077 RSC.
- R. J. Spears, C. McMahon, M. Shamsabadi, C. Bahou, I. A. Thanasi, L. N. C. Rochet, N. Forte, F. Thoreau, J. R. Baker and V. Chudasama, Chem. Commun., 2022, 58, 645–648 RSC.
- V. Chudasama, M. E. B. Smith, F. F. Schumacher, D. Papaioannou, G. Waksman, J. R. Baker and S. Caddick, Chem. Commun., 2011, 47, 8781 RSC.
- P. A. Szijj, C. Bahou and V. Chudasama, Drug Discovery Today: Technol., 2018, 30, 27–34 CrossRef PubMed.
- R. E. Morgan, V. Chudasama, P. Moody, M. E. B. Smith and S. Caddick, Org. Biomol. Chem., 2015, 13, 4165–4168 RSC.
- S. Santra, C. Kaittanis, O. J. Santiesteban and J. M. Perez, J. Am. Chem. Soc., 2011, 133, 16680–16688 CrossRef CAS PubMed.
- M. Fernández, A. Shamsabadi and V. Chudasama, Chem. Commun., 2020, 56, 1125–1128 RSC.
- M. Fernández, F. Javaid and V. Chudasama, Chem. Sci., 2018, 9, 790–810 RSC.
- R. P. Lyon, J. R. Setter, T. D. Bovee, S. O. Doronina, J. H. Hunter, M. E. Anderson, C. L. Balasubramanian, S. M. Duniho, C. I. Leiske, F. Li and P. D. Senter, Nat. Biotechnol., 2014, 32, 1059–1062 CrossRef CAS PubMed.
- L. A. Sawicki and A. M. Kloxin, Biomater. Sci., 2014, 2, 1612–1626 RSC.
- A. Maruani, S. Alom, P. Canavelli, M. T. W. Lee, R. E. Morgan, V. Chudasama and S. Caddick, Chem. Commun., 2015, 51, 5279–5282 RSC.
- A. Maruani, M. E. B. Smith, E. Miranda, K. A. Chester, V. Chudasama and S. Caddick, Nat. Commun., 2015, 6, 6645 CrossRef CAS PubMed.
- C. Bahou, P. A. Szijj, R. J. Spears, A. Wall, F. Javaid, A. Sattikar, E. A. Love, J. R. Baker and V. Chudasama, Bioconjugate Chem., 2021, 32, 672–679 CrossRef CAS PubMed.
- M. Góngora-Benítez, J. Tulla-Puche, M. Paradís-Bas, O. Werbitzky, M. Giraud and F. Albericio, Biopolymers, 2011, 96, 69–80 CrossRef PubMed.
- J. C. F. Nogueira, M. K. Greene, D. A. Richards, A. O. Furby, J. Steven, A. Porter, C. Barelle, C. J. Scott and V. Chudasama, Chem. Commun., 2019, 55, 7671–7674 RSC.
- C. R. Behrens, E. H. Ha, L. L. Chinn, S. Bowers, G. Probst, M. Fitch-Bruhns, J. Monteon, A. Valdiosera, A. Bermudez, S. Liao-Chan, T. Wong, J. Melnick, J.-W. Theunissen, M. R. Flory, D. Houser, K. Venstrom, Z. Levashova, P. Sauer, T.-S. Migone, E. H. van der Horst, R. L. Halcomb and D. Y. Jackson, Mol. Pharm., 2015, 12, 3986–3998 CrossRef CAS PubMed.
- N. Forte, V. Chudasama and J. R. Baker, Drug Discovery Today: Technol., 2018, 30, 11–20 CrossRef PubMed.
- H. Liu and K. May, mAbs, 2012, 4, 17–23 CrossRef PubMed.
- J. P. M. Nunes, M. Morais, V. Vassileva, E. Robinson, V. S. Rajkumar, M. E. B. Smith, R. B. Pedley, S. Caddick, J. R. Baker and V. Chudasama, Chem. Commun., 2015, 51, 10624–10627 RSC.
- E. A. Hull, M. Livanos, E. Miranda, M. E. B. Smith, K. A. Chester and J. R. Baker, Bioconjugate Chem., 2014, 25, 1395–1401 CrossRef CAS PubMed.
- V. Chudasama, A. Maruani and S. Caddick, Nat. Chem., 2016, 8, 114–119 CrossRef CAS PubMed.
- S. J. Walsh, S. Omarjee, W. R. J. D. Galloway, T. T. L. Kwan, H. F. Sore, J. S. Parker, M. Hyvönen, J. S. Carroll and D. R. Spring, Chem. Sci., 2019, 10, 694–700 RSC.
- M. Morais, J. P. M. Nunes, K. Karu, N. Forte, I. Benni, M. E. B. B. Smith, S. Caddick, V. Chudasama and J. R. Baker, Org. Biomol. Chem., 2017, 15, 2947–2952 RSC.
- O. Koniev, I. Dovgan, B. Renoux, A. Ehkirch, J. Eberova, S. Cianférani, S. Kolodych, S. Papot and A. Wagner, Medchemcomm, 2018, 9, 827–830 RSC.
- C. Bahou, E. A. Love, S. Leonard, R. J. Spears, A. Maruani, K. Armour, J. R. Baker and V. Chudasama, Bioconjugate Chem., 2019, 30, 1048–1054 CrossRef CAS PubMed.
- C. Bahou, D. A. Richards, A. Maruani, E. A. Love, F. Javaid, S. Caddick, J. R. Baker and V. Chudasama, Org. Biomol. Chem., 2018, 16, 1359–1366 RSC.
- M. T. W. Lee, A. Maruani, J. R. Baker, S. Caddick and V. Chudasama, Chem. Sci., 2016, 7, 799–802 RSC.
- M. T. W. Lee, A. Maruani, D. A. Richards, J. R. Baker, S. Caddick and V. Chudasama, Chem. Sci., 2017, 8, 2056–2060 RSC.
- S. L. Kuan, T. Wang and T. Weil, Chem.– Eur. J., 2016, 22, 17112–17129 CrossRef CAS PubMed.
- M. Sadraeian, C. Bahou, E. F. da Cruz, L. M. R. Janini, R. S. Diaz, R. W. Boyle, V. Chudasama and F. E. G. Guimarães, Int. J. Mol. Sci., 2020, 21, 9151 CrossRef CAS PubMed.
- M. Sadraeian, E. F. da Cruz, R. W. Boyle, C. Bahou, V. Chudasama, L. M. R. Janini, R. S. Diaz and F. E. G. Guimarães, ACS Omega, 2021, 6, 16524–16534 CrossRef CAS PubMed.
- A. N. Marquard, J. C. T. Carlson and R. Weissleder, Bioconjugate Chem., 2020, 31, 1616–1623 CrossRef CAS PubMed.
- F. Javaid, C. Pilotti, C. Camilli, D. Kallenberg, C. Bahou, J. Blackburn, J. R. Baker, J. Greenwood, S. E. Moss and V. Chudasama, RSC Chem. Biol., 2021, 2, 1206–1220 RSC.
- F. Bryden, A. Maruani, J. M. M. Rodrigues, M. H. Y. Cheng, H. Savoie, A. Beeby, V. Chudasama and R. W. Boyle, Bioconjugate Chem., 2018, 29, 176–181 CrossRef CAS PubMed.
- A. Maruani, H. Savoie, F. Bryden, S. Caddick, R. Boyle and V. Chudasama, Chem. Commun., 2015, 51, 15304–15307 RSC.
- M. K. Greene, T. Chen, E. Robinson, N. L. Straubinger, C. Minx, D. K. W. Chan, J. Wang, J. F. Burrows, S. Van Schaeybroeck, J. R. Baker, S. Caddick, D. B. Longley, D. E. Mager, R. M. Straubinger, V. Chudasama and C. J. Scott, Br. J. Cancer, 2020, 123, 1502–1512 CrossRef CAS PubMed.
- A. Maruani, P. A. Szijj, C. Bahou, J. C. F. Nogueira, S. Caddick, J. R. Baker and V. Chudasama, Bioconjugate Chem., 2020, 31, 520–529 CrossRef CAS PubMed.
- P. Szijj and V. Chudasama, Nat. Rev. Chem., 2021, 5, 78–92 CrossRef CAS.
- J. C. F. Nogueira, K. Paliashvili, A. Bradford, F. Di Maggio, D. A. Richards, R. M. Day and V. Chudasama, Org. Biomol. Chem., 2020, 18, 2215–2218 RSC.
- M. K. Greene, J. C. F. Nogueira, S. R. Tracey, D. A. Richards, W. J. McDaid, J. F. Burrows, K. Campbell, D. B. Longley, V. Chudasama and C. J. Scott, Nanoscale, 2020, 12, 11647–11658 RSC.
- D. A. Richards, A. Maruani and V. Chudasama, Chem. Sci., 2017, 8, 63–77 RSC.
- W. W. K. Cheng and T. M. Allen, J. Controlled Release, 2008, 126, 50–58 CrossRef CAS PubMed.
- P. Sapra, E. H. Moase, J. Ma and T. M. Allen, Clin. Cancer Res., 2004, 10, 1100–1111 CrossRef CAS PubMed.
- M. K. Greene, D. A. Richards, J. C. F. Nogueira, K. Campbell, P. Smyth, M. Fernández, C. J. Scott and V. Chudasama, Chem. Sci., 2018, 9, 79–87 RSC.
- R. H. Mizzoni and P. E. Spoerri, J. Am. Chem. Soc., 1951, 73, 1873–1874 CrossRef CAS.
- L. Castañeda, Z. V. F. Wright, C. Marculescu, T. M. Tran, V. Chudasama, A. Maruani, E. A. Hull, J. P. M. Nunes, R. J. Fitzmaurice, M. E. B. Smith, L. H. Jones, S. Caddick and J. R. Baker, Tetrahedron Lett., 2013, 54, 3493–3495 CrossRef PubMed.
| This journal is © The Royal Society of Chemistry 2022 |