
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Diindolocarbazole – achieving multiresonant thermally activated delayed fluorescence without the need for acceptor units†‡
David
Hall
*ab,
Kleitos
Stavrou
 c,
Eimantas
Duda
d,
Andrew
Danos
c,
Sergey
Bagnich
d,
Stuart
Warriner
e,
Alexandra M. Z.
Slawin
a,
David
Beljonne
b,
Anna
Köhler
*d,
Andrew
Monkman
*c,
Yoann
Olivier
*f and
Eli
Zysman-Colman
*a
c,
Eimantas
Duda
d,
Andrew
Danos
c,
Sergey
Bagnich
d,
Stuart
Warriner
e,
Alexandra M. Z.
Slawin
a,
David
Beljonne
b,
Anna
Köhler
*d,
Andrew
Monkman
*c,
Yoann
Olivier
*f and
Eli
Zysman-Colman
*a
aOrganic Semiconductor Centre, EaStCHEM School of Chemistry, University of St Andrews, St Andrews, KY16 9ST, UK. E-mail: eli.zysman-colman@st-andrews.ac.uk; Web: http://www.zysman-colman.com
bLaboratory for Chemistry of Novel Materials, University of Mons, 7000, Mons, Belgium
cDepartment of Physics, Durham University, Durham, DH1 3LE, UK. E-mail: a.p.monkman@durham.ac.uk
dSoft Matter Optoelectronics, BIMF & BPI, University of Bayreuth, Universitätsstraße 30, Bayreuth 95447, Germany. E-mail: anna.koehler@uni-bayreuth.de
eSchool of Chemistry, University of Leeds, Woodhouse Lane, Leeds, UK
fLaboratory for Computational Modeling of Functional Materials, Namur Institute of Structured Matter, University of Namur, Rue de Bruxelles, 61, Namur 5000, Belgium. E-mail: yoann.olivier@unamur.be
First published on 24th January 2022
Abstract
In this work we present a new multi-resonance thermally activated delayed fluorescence (MR-TADF) emitter paradigm, demonstrating that the structure need not require the presence of acceptor atoms. Based on an in silico design, the compound DiICzMes4 possesses a red-shifted emission, enhanced photoluminescence quantum yield, and smaller singlet-triplet energy gap, ΔEST, than the parent indolocarbazole that induces MR-TADF properties. Coupled cluster calculations accurately predict the magnitude of the ΔEST when the optimized singlet and triplet geometries are used. Slow yet optically detectable reverse intersystem crossing contributes to low efficiency in organic light-emitting diodes using DiICzMes4 as the emitter. However, when used as a terminal emitter in combination with a TADF assistant dopant within a hyperfluorescence device architecture, maximum external quantum efficiencies of up to 16.5% were achieved at CIE (0.15, 0.11). This represents one of the bluest hyperfluorescent devices reported to date. Simultaneously, recognising that MR-TADF emitters do not require acceptor atoms reveals an unexplored frontier in materials design, where yet greater performance may yet be discovered.
New conceptsThermally activated delayed fluorescence (TADF) compounds have generated tremendous interest over the past decade for use as emitters in organic light-emitting diodes (OLEDs). The conventional donor–acceptor (D–A) design presented in the literature shows broad and unstructured emission, resulting in poor colour purity in the devices. A sub-class of TADF materials, so-called multi-resonant TADF (MR-TADF) overcomes this issue, emitting with very narrow spectra. Compared to D–A TADF materials there are relatively few MR-TADF materials. This is in part due to the poor in silico modelling of these compounds. We recently reported how wavefunction-based methods were necessary to accurately model them. We have since exploited this computational methodology towards the design of a number of novel MR-TADF compounds. In previous MR-TADF designs, acceptor atoms or functionalities were essential components in the polycyclic aromatic hydrocarbon compounds to achieve TADF. In the present contribution we demonstrate conclusively that this is not the case and MR-TADF with no acceptor groups is possible. We report a tetramesitylated diindolocarbazole emitter that shows TADF. We further report one of the deepest blue hyperfluorescent OLEDs using this compound as a terminal emitter. |
Introduction
The organic light-emitting diode (OLED) field has taken another step forward with the introduction of multiresonant thermally activated delayed fluorescent (MR-TADF) materials.1 As with conventional donor–acceptor (D–A) TADF emitters, MR-TADF compounds possess suitably small singlet–triplet energy gaps (ΔEST) to permit triplet excitons to be up-converted to singlets by reverse intersystem crossing (RISC), unlocking considerably improved device efficiency in OLEDs2–4 alongside applications in several other optoelectronic contexts.5–8 RISC is achieved in D–A TADF materials through the reduction of the exchange integral by electronically decoupling the donor and acceptor moieties as a result of a highly twisted conformation,2,4 with the HOMO situated on the donor and LUMO on the acceptor, combined with vibronic coupling between local and charge transfer triplet states to facilitate spin orbit coupling.9 Due to the conformational flexibility inherent in these classes of emitter, the charge-transfer emission bands are particularly broad, resulting in poor colour purity of the resulting OLEDs and extreme challenges in achieving deep-blue colour coordinates.10For MR-TADF emitters the HOMO–LUMO separation and thus small ΔEST are achieved via a complementary pattern of the electron density distribution on adjacent atoms within the molecule1 between HOMO and LUMO states, made possible by the incorporation of suitably positioned electron-donating and electron-withdrawing atoms (or functional groups). The reorganization of the electron density upon excitation is relatively localized, so that the lowest singlet and triplet excited states possess short-range charge transfer (SRCT) character.11 The small exchange integral in MR-TADF compounds is best illustrated with difference density plots (Fig. 1). Seemingly paradoxical for charge-transfer states, there is also a suitably large overlap of the excited and ground state wavefunctions, leading to larger oscillator strengths for the S1–S0 transition and thus fast radiative decay rates, kr. We note that the wavefunction will be of mixed locally excited (LE) and CT character, and in this case the LE contribution appears to dominate, thus coupling to the ground state is high but electron exchange energy remains sizeable. Combined with conformationally rigid structures these SRCT states confer a very narrow emission spectrum with full width at half maxima (FWHM) below 30 nm,1 leading to much greater colour purity, which is required for high-definition displays and advantageous for achieving deep-blue emission.12
 | ||
| Fig. 1 Evolution of MR-TADF emitters including simplified difference density plots for each core. | ||
MR-TADF materials DABNA-1 and DABNA-2 were first reported in 2016 by Hatakeyama and co-workers.13 These compounds contain a central accepting boron atom and para-disposed donating nitrogen atoms that achieve the desired alternating pattern of the electron density distribution. OLEDs employing DABNA-1 and DABNA-2 showed maximum external quantum efficiencies, EQEmax, of 13.5% and 20.2% with Commission Internationale de l’Éclairage, CIE, coordinates of (0.13, 0.09) and (0.12, 0.13), respectively. Low RISC rates (compared to contemporaneous D–A–D emitters)14–17 in this early work resulted in severe efficiency roll-off though, with efficiencies at 1000 cd m−2 not reported. A large number of other MR-TADF materials using this same design template have since been reported.18–20
Dramatic improvements in both EQEmax and efficiency roll-off were recently reported for the extended system v-DABNA (Fig. S1, ESI‡).21 This compound contains two DABNA-1 units that are fused together, extending the π conjugation (Fig. 1), which results in a smaller ΔEST from 0.18 eV in DABNA-1 (in 1 wt% mCBP)13 to 0.02 eV (in 1 wt% DOBNA-OAr).21 OLEDs made with this emitter show an outstanding EQEmax of 34.4% at CIE (0.12, 0.11), assisted in part by spontaneous emitter alignment improving device optical outcoupling, as confirmed by angularly-resolved emission measurements in a later work.22 Further, due in part to its small ΔEST and class-leading RISC rates, the device shows superior efficiency roll-off, with an EQE at 1000 cd m−2 of 26.1% (Table S1, ESI‡).
Another family of MR-TADF compounds, introduced by Zhang et al. in 2019,23 relies on boron and nitrogen atoms to direct the electron density pattern, but these compounds incorporate fused donor units, such as in CzBN. The fused system results in increased electronic delocalisation, thus stabilizing S1, which produces a red-shift in the emission (Fig. 1). The first series of emitters incorporated peripheral electron-withdrawing groups onto the CzBN core, which led to a further red-shift of the emission. OLEDs with 2F-BN, 3F-BN and 4F-BN showed EQEmax of 22.0%, 22.7% and 20.9%, respectively, at CIE of (0.16, 0.60), (0.20, 0.58) and (0.12, 0.48), representing the first examples of green-emitting MR-TADF OLEDs. Following this early work a range of materials based on this molecular design have been reported;23–28 the OLED with BBCz-G showing the highest efficiencies, with an EQEmax of 31.8% at CIE of (0.26, 0.68).29 This design was also used to produce the first examples of red-emitting MR-TADF compounds, BBCz-R29 and R-(T)BN30 (Fig. S1, ESI‡). The red-shifted emission in both examples results from the positioning of the donating nitrogen atoms, and thus the withdrawing boron atoms, para to each other, thereby inducing partial bonding and antibonding character and resulting in a smaller HOMO–LUMO gap, ΔE, and a much lower energy emissive S1 state.30 Devices with these three emitters showed EQEmax surpassing 20% at CIE coordinates of (0.67, 0.33)29 and (0.72, 0.18).30 Another route to colour tuning has been the addition of electron donating and withdrawing substituents on the CzBN core, with blue- or red-shifts of the emission possible depending on the position and number of additional donors and acceptors.27,29,31,32
An alternative core unit based on DABNA-1 was presented firstly by Oda et al.,33 where the positions of boron and nitrogen atoms are switched (ADBNA-Me-Mes and ADBNA-Me-Tip, Fig. 1 and Fig. S1, ESI‡). Having two boron and one nitrogen atoms results in a smaller ΔE compared to that of DABNA-1; however, there is minimal impact on ΔEST (Table S1, ESI‡). The resulting OLEDs showed CIE of (0.10, 0.27) and (0.11, 0.29) for ADBNA-Me-Mes and ADBNA-Me-Tip, respectively. The EQEmax for these devices are 16.2% and 21.4%, respectively, while efficiency roll-off is modest, with EQE100 of 11.2% and 15.4%.
Another family of MR-TADF compounds contains carbonyl groups in lieu of boron atoms as the electron acceptor. The first example, QAO, reported in 2019, translated into devices with an EQEmax 19.4% at CIE (0.13, 0.18).34 We showed that decoration of this core with mesityl groups, Mes3DiKTa, can mitigate aggregation induced quenching (AIQ),20 which is a common problem with these planar molecules. With this emitter, the OLED showed the highest EQEmax for this family of compounds of 21.1% at CIE of (0.12, 0.32). A phenyl-substituted structure, 3MTPTOAT, based on a related core, TOAT, which itself has previously been reported as a room temperature phosphorescent emitter,35 was used as the emitter in an OLED that showed a very high EQEmax of 31.2%.36 A range of emitters has now been reported incorporating carbonyl groups within the molecular design;33,34,37 however, most of these emitters show relatively large ΔEST and the devices often show EQEmax values inferior to 20%. A full summary of the discussed literature emitters including structures, photophysical data and OLED device performances can be found in Fig. S1 and Table S1 (ESI‡).
Despite the excellent characteristics of MR-TADF emitters, the majority of MR-TADF emitters have a low kRISC, with most around 104 s−1 (Table S2, ESI‡). The slow kRISC has proved detrimental to device performance with most OLEDs using the MR-TADF compound as an emitter suffering from large efficiency roll-off.13 In the literature only two examples exist where kRISC surpasses 106 s−1 (Fig. S2, ESI‡), m-CzBNCz27 and BSBS-N1,38 where kRISC reaches 1.08 and 1.90 × 106 s−1, respectively. Even direct comparison between reported RISC rates from different research teams is challenging though, due to the plurality of reported methods for determining its value39,40 and subtle yet important practical concerns.41 The MR-TADF emitters with the fastest kRISC are nevertheless two orders of magnitude slower than the best performing D–A TADF emitter (Fig. S2, ESI‡). This was predicted by Northey and Penfold42 and experimentally shown by Stavrou et al.,43 that the RISC mechanism in MR-TADF systems occurs through crossing between T1 and an upper triplet state via reverse internal conversion. This involves closely-lying triplet states and requires new design rules for new chemical structures with optimal efficiency. A large factor in this apparent gap in RISC rates is that the chemical space explored for MR-TADF emitters remains small compared to the thousands of donor–acceptor TADF compounds reported. Furthermore, we recently demonstrated11 that time-dependent DFT calculations, which are commonly used to predict the nature and the energies of the excited singlet and triplet states of D–A TADF compounds,44 do not accurately predict these parameters for MR-TADF compounds, thus hindering computationally guided molecular design. We have shown repeatedly that coupled cluster calculations,33,45–47 which include double excitation contributions, perform significantly more accurately, albeit at a higher computational cost.
Here, we apply the same coupled cluster methodology to guide the design of a new class of MR-TADF materials, which surprisingly do not require an electron-accepting functionality within the compound. Despite the lack of acceptor atom, a complimentary pattern of increasing and decreasing electron density is achieved for S1 (but not necessarily for T1) compared to S0 in this class of emitters. DiICzMes4 was also compared to two smaller reference emitters, ICz and ICzMes3, with mesityl groups in DiICzMes4 intended to suppress AIQ.33 Compared to ICz and ICzMes3, the expansion of the π-system in DiICzMes4 ensures a further decrease of the HOMO–LUMO overlap and results in a much smaller ΔEST, reduced from 0.47 eV in ICz to 0.26 eV in DiICzMes4 (in toluene). Further, there is a desirable increase in ΦPL across the series from 37%, 56% and 67%, accompanied with a red-shift in the emission maximum, λPL, from 374 nm, 387 nm and 441 nm in 3 wt% PMMA films, for ICz, ICzMes3 and DiICzMes4, respectively, all in agreement with recent SCS-CC2 calculations for B/N-doped nanographenes.11
Crucially, although the core DiICz structure decorated with tBu groups has recently been reported,48,49 its identity as a TADF emitter – confirmed here by time-resolved photophysical measurements – was overlooked until recently.50 An analogous non-fused tricarbazole-amine system (TCA_C4) had previously been shown to have a small singlet triplet gap, 0.21 eV, and gives moderate TADF via a reverse internal conversion (rIC), upper triplet state crossing mechanism.51 It is only recently that the MR-TADF mechanism has been elucidated to take place through a similar rIC mechanism in v-DABNA,43 and presumably also other MR-TADF emitters. Nonetheless, in both previous reports of the DilCz structure the compound was presented as a purely fluorescent system (named pICz48 and 549), with relatively large ΔEST of 0.29 eV. Recently, a similar derivative, tPBisICz, was introduced as a MR-TADF emitter, and the authors contended that RISC proceeds between T2 and S1.50 The device showed an EQEmax of 23.1% at CIE (0.15, 0.05); however, efficiency roll-off was severe (Table S3, ESI‡) and this is likely due to the inefficient kRISC of 1.4 × 103 s−1. Although the RISC rate for DiICzMes4 is slow (similar to that of TCA_C451), this work supports the existence of an entirely new subcategory of ‘acceptor-free’ MR-TADFs, which may yield improved performance in device applications in future.
Results and discussion
Modelling
Initial ground state optimisation followed by vertical excitation were performed at the SCS-CC2/cc-pVDZ level of theory. Indolocarbazole (ICz) has been frequently used by the TADF community, able to act as both a donor or acceptor52 depending on to the nature of the substituents. ΔEST was predicted to be 0.33 eV, which is high for TADF materials but rationalized by the different nature of S1 and T1 excited states. Indeed, S1 displays a typical difference density pattern characteristic of a SRCT excited state while T1 exhibits a locally excited (LE)-like pattern, with the latter more stabilized, hence the large ΔEST (Fig. 2). It has been inferred previously that extending the MR-TADF electronic delocalisation could be a viable strategy to decrease ΔEST at the same time as increasing the oscillator strength.11 Based on this hypothesis, four derivatives of ICz were modelled, with differing patterns of the relative position of the nitrogen atoms: DiICz-m-1, DiICz-m-2, DiICz-p-1 and DiICz-p-2. Compared to the parent ICz, each of these four emitters has a stabilized S1 state, decreasing from 3.78 eV for ICz to 3.58 eV, 3.57 eV, 3.36 eV and 3.32 eV for DiICz-m-1, DiICz-m-2, DiICz-p-1 and DiICz-p-2, respectively, the result of delocalization of the S1 wavefunction (see Fig. 2). As previously reported for other MR-TADF emitters,30 when the donating nitrogen atoms are located para to each other the red-shift is the largest. The para-derivatives here also have the smallest predicted ΔEST of 0.17 eV and 0.15 eV for DiICz-p-1 and DiICz-p-2, respectively, while ΔEST is 0.30 eV and 0.32 eV for DiICz-m-1 and DiICz-m-2.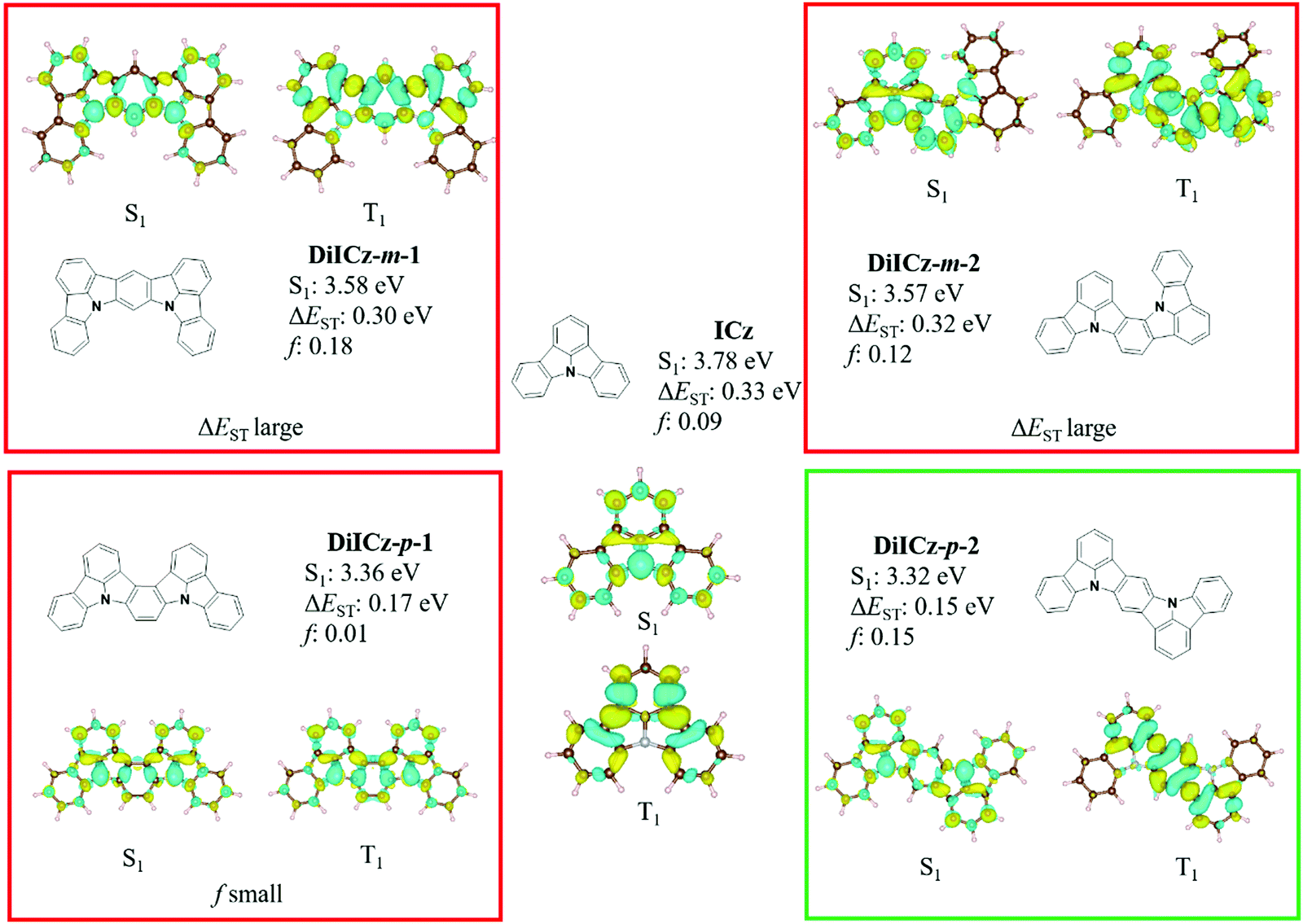 | ||
| Fig. 2 S1 and T1 difference density patterns, ΔEST, S1 energy and oscillator strength, f, for the proposed DiICz units. | ||
Of DiICz-p-1 and DiICz-p-2, DiICz-p-2 has a considerably larger oscillator strength of 0.15 compared to 0.01 in DiICz-p-1 and thus this motif was assessed as the most promising. Furthermore, we have previously demonstrated that addition of mesityl groups can mitigate AIQ,33 which plagues MR-TADF materials43 (and many other similar systems53,54) owing to their planar and electron-rich geometries. With this in mind, we designed the mesityl derivative of ICz, ICzMes3. In this compound the mesityl groups have the added benefit of reducing ΔEST (calculated for vertical transitions from the ground state geometry) from 0.33 eV to 0.21 eV. The decrease in ΔEST is essentially the result of preferential stabilization of S1 while the energy of T1 energy is only minimally affected (Fig. S31, ESI‡). The small stabilization of T1 in ICzMes3 can be explained by the absence of significant orbital contributions from the carbon atoms connecting the mesityl groups in the T1 difference density pattern (Fig. S31, ESI‡). We also investigated the role that decoration with mesityl groups would play on the core structure of DiICz-p-2, which together form the target material DiICzMes4. In DiICzMes4 (Fig. S31, ESI‡); the mesityl substitution helps to reduce the predicted ΔEST from 0.15 eV to 0.13 eV for similar reasons as described for ICzMes3. Due to the close energy of the LE T1 and the SCRT T2 states of the DiICz-p-2, substitution by the four mesityl groups allows inversion between the two. T1 becomes SCRT in DiICzMes4 possessing similar, yet slightly different character than S1. The literature emitter tBisICz was also modelled using the same approach and the ΔEST of 0.14 eV was found to be larger. Further, the energy gap between S1 and T2 is also larger at 0.07 eV compared to 0.05 eV in DiICzMes4. Owing to these moderate differences in the energy landscape of the excited states, DiICzMes4 is expected to show improved RISC rates.
In contrast to previously investigated MR-TADF emitters, we see large changes when comparing ΔEST computed from vertical excitation from the ground state geometry and experiments due to the different nature of T1 and S1 states (see difference density plots in Fig. S29 and S30, ESI‡). In such a case, relaxation of the excited states could be key to reach quantitative agreement with the experiments. We thus optimized both the S1 and T1 states within the TDA using PBE0 functional and 6-31G(d,p) basis set, and computed the T1 and S1 excited state energies at the SCS-CC2/cc-PVDZ level of theory for ICz, ICzMes3 and DiICzMes4 as well as for three literature MR-TADF compounds, DABNA-1, BCzBN and DiKTa. Quantitative agreement with the experiments is reached with ΔEST increasing for ICz, ICzMes3 and DiICzMes4, to 0.59 eV, 0.45 eV and 0.29 eV (Fig. 3), respectively, caused by a larger relaxation energy of the T1 state in line with a greater LE character for this state (Tables S9–S11, ESI‡). Interestingly, such an increase in ΔEST does not manifest for DABNA-1, BCzBN and DiKTa, wherein ΔEST is only shifted by a maximum of 0.04 eV, owing to the similar SRCT nature of T1 and S1 (see Tables S9–S11 and Fig. S30 and S32, ESI‡). The similar orbital character of S1 and T1 in many previous emitters, and the ones presented here, implies that RISC between these two states is not symmetry allowed according to El Sayed's rules.9 Thus, RISC must occur via a spin-vibronic mechanism involving intermediate triplet states lying between S1 and T1. Irrespective of the starting geometry, a close lying triplet state of different orbital type is present, whose involvement has been shown to contribute to the MR-TADF RISC mechanism.43 Both smaller ΔEST and ΔET2T1 were observed, decreasing across the series from ICz, ICzMes3 and DiICzMes4. We observed again a decreased ΔEST upon incorporation of mesityl groups from 0.59 eV for ICz to 0.45 eV ICzMes3. Unlike previously reported MR-TADF emitters that contain acceptor atoms/groups, for this class it is essential to optimise the excited states in order to achieve quantitative agreement with experimental ΔEST.
 | ||
| Fig. 3 Structures, excited state energies and difference density plots of each S1 and T1 for ICz (left panel), ICzMes3 (central panel) and DiICzMes4 (right panel) from excited state optimized geometry. | ||
Synthesis
The materials were synthesised through a multistep reaction sequence outlined in Fig. 4. Carbazole was coupled to 2-bromofluorobenzene under SNAr conditions at elevated temperatures in an excellent yield of 96%. Intramolecular oxidative ring closing using Pd(OAc)2 afforded ICz in a good yield of 85%. Subsequent electrophilic bromination using NBS afforded intermediate ICzBr3 in 79% yield, which was then decorated with mesityl groups using a Suzuki–Miyaura coupling reaction, producing ICzMes3 in a good yield of 69%. A similar Suzuki–Miyaura coupling was employed to obtain CzMes2 from dibromocarbazole in 62% following a literature procedure.55 Intermediate 2 was obtained in 75% via an SNAr reaction that proceeded at lower temperature (50 °C). Double oxidative cyclization using Pd(OAc)2 generated DiICzMes4 in 59% yields. Crystals of ICzMes3 and DiICzMes4 were grown from slow evaporation of methanol into a saturated solution of toluene over several days. Packing in ICzMes3 is primarily governed by π–π stacking interactions between mesityl groups on adjacent molecules (Fig. S25, ESI‡). For DiICzMes4 π–π stacking occurs between the mesityl group of one molecule and the DiICz core of an adjacent molecule. The ICz unit in both compounds was not perfectly flat (Fig. 4). Thermogravimetric analysis (TGA) of ICzMes3 and DiICzMes4 (Fig. S24, ESI‡) reveals good thermal stability for both compounds with Td, the temperature representing 5% weight loss, of, respectively, 374 °C and 450 °C. | ||
| Fig. 4 Synthesis of (a) the emitters and crystal structures of (b) ICzMes3 and (c) DiICzMes4. | ||
Optoelectronic characterization
The electrochemical properties were investigated using cyclic voltammetry (CV) and differential pulse voltammetry (DPV) in DCM for oxidation and DMF for reduction (Fig. 5a), with the electrochemical potentials reported versus SCE (Table 1). ICz showed irreversible oxidation and reduction waves with the former appearing to undergo polymerisation, which has been previously reported for ICz56 and seen in other carbazole-containing emitters.57 Addition of the mesityl groups in ICzMes3 renders the oxidation pseudoreversible in a similar manner to what was previously observed for Mes3DiKTa,33 with Eox at 1.45 V versus 1.43 V for ICzMes3. Indeed, McNab et al. had demonstrated that the electrochemical instability of ICz is associated with dimer formation centred at the para positions.56 There is likewise little change in the irreversible reduction waves with reduction potentials of these two compounds, Ered, at −2.21 V and −2.16 V for ICz and ICzMes3, respectively. By contrast, both oxidation and reduction waves for DiICzMes4 are largely reversible. The oxidation wave is cathodically shifted to 1.11 V while the reduction wave is anodically shifted to −1.92 V, both a reflection of the larger conjugation length of this molecule compared to ICz and ICzMes3. This produced a significant reduction in the redox gap, ΔEredox, in agreement with calculations, where the calculated ΔE decreases from 4.65 eV and 4.50 eV for ICz and ICzMes3 to 3.86 eV in DiICzMes4. The trends in HOMO and LUMO values are corroborated at the DFT level (Tables S5 and S12, ESI‡). | ||
| Fig. 5 Solution-state optoelectronic data. (a) CV (solid line) and DPV (dashed) where the anodic scan is in DCM and the cathodic scan is in DMF, with 0.1 M [nBu4N]PF6 as the supporting electrolyte and Fc/Fc+ as the internal reference (0.46 V for DCM and 0.45 for DMF vs. SCE);58 (b) steady-state PL in toluene, at concentrations of 0.6–2.0 ×10−5 M, where λexc = 320 nm for ICz and ICzMes3 and λexc = 380 nm for DiICzMes4, and pictures of each in solution (ICz, ICzMes3 and DiICzMes4 left to right). | ||
| Compound | λ abs (ε)a/nm (×104 M−1 cm−1) | λ PL , (FWHM)/nm | Φ PL N2 (air)/% | S1d/eV | T1d/eV | ΔESTe/eV | τ p /ns | HOMOg/eV | LUMOg/eV |
|---|---|---|---|---|---|---|---|---|---|
| a Toluene at 300 K. b Values in parentheses are the FWHM. c In degassed toluene measured with an integrating sphere under N2, values in parentheses are in air-saturated solution, λexc = 330 nm. d From the onset of the steady-state emission and phosphorescence in toluene glass at 77 K, λexc = 330 nm. e Energy difference between the onset of the steady-state and phosphorescence at 77 K. f λ exc = 355 nm. g The HOMO and LUMO energies were determined according to EHOMO/LUMO = −(Eoxonset/Eredonset + 4.8) eV.59 h λ exc = 320 nm. i λ exc = 380 nm. j λ exc = 350 nm. k λ exc = 350 nm. | |||||||||
| ICz | 285 (31), 392 (10), 309 (6), 320 (7), 350 (6), 364 (9) | 374 (21)h | 58 (30) | 3.35 | 2.88 | 0.47 | 15 | −5.79 | −2.14 |
| ICzMes3 | 291 (40), 300 (14), 318 (6), 330 (8), 363 (6), 379 (8) | 387 (21)h | 66 (43) | 3.24 | 2.85 | 0.39 | 22 | −5.77 | −2.19 |
| DiICzMes4 | 302 (58), 307 (62), 316 (59), 345 (19), 365 (39), 410 (8), 431 (11) | 441 (17)i | 70 (47)j | 2.83k | 2.57k | 0.26 | 41 | −5.45 | −2.43 |
We next investigated the photophysical properties of the three emitters in solution. The UV-Vis absorption data in toluene (PhMe), 2-methyltetrahydrofuran (2-MeTHF), ethyl acetate (EtOAc), dichloromethane (DCM) and dimethylformamide (DMF) can be found in Fig. S33 and Tables S13–S15 (ESI‡). The polarity of the solvent had minimal impact on the absorption spectra, with nearly identical absorption maxima, λabs, and molar absorptivity values, ε, regardless of solvent. Using the representative data in toluene (Table 1), there is a high intensity, low energy band at 364 nm, 379 nm and 431 nm for ICz, ICzMes3, and DiICzMes4, respectively, assigned by calculations to a SRCT band; there is a second distinguishable band at smaller ε at 350 nm, 363 nm and 410 nm, respectively, that is likely due to a transition to a different vibronic level of the S1 state based on the ca. 0.15 eV energy gap between these two bands. Both ICz and ICzMes3 possess higher energy bands at 320 nm and 330 nm of similar ε, which we assign to transitions to the S2 state. The similar ε values are captured at the SCS-CC2 level where both S1 and S2 have similar oscillator strengths, f, of 0.10 and 0.09 for ICz and 0.14 and 0.13 for ICzMes3. A far greater oscillator strength of 0.66 is predicted for the transition to S2 for DiICzMes4 compared to that to S1 (f = 0.21). Indeed, the band at 365 nm possesses a significantly larger ε of 39 × 104 M−1 cm−1 compared to that at 431 nm (ε = 11 × 104 M−1 cm−1), suggesting a greater degree of LE character for the transition associated with this band.
Minimal changes in emission energy and band shape were observed upon modulation of the solvent polarity (Fig. S33 and Tables S13–S15, ESI‡). Such behaviour is characteristic of MR-TADF emitters, which undergo emission from a SRCT excited state.11 Owing to their rigid nature, the emission is narrow and the Stokes shifts are small (10, 8, and 10 nm, respectively, for ICz, ICzMes3 and DiICzMes4) reflecting the very small reorganisation energy between the ground and excited state. The corresponding FWHM for the PL spectra in toluene are 21 nm, 21 nm and 17 nm for ICz, ICzMes3 and DiICzMes4, respectively. There are low energy shoulders apparent in the steady-state PL of all three emitters. This shoulder is assigned to a vibronic shoulder (vide infra) (Fig. S34, ESI‡).
The energies of the singlet and triplet states, and hence, ΔEST, were determined based on the high-energy onset of the prompt fluorescence and phosphorescence spectra obtained at 77 K in toluene glass (Fig. S34, ESI‡). In all cases, the phosphorescence is very well vibrationally structured and characteristic of a carbazole moiety in strong contrast with respect to the fluorescence, supporting the different nature of the S1 and T1 excited states. There is a progressive decrease in ΔEST of 0.47 eV, 0.39 eV and 0.26 eV for ICz, ICzMes3 to DiICzMes4, respectively, a trend that is well reproduced by the SCS-CC2 calculations when considering the optimized excited state structures (Table S11, ESI‡). We simulated the vibronically resolved fluorescence and phosphorescence spectra for DiCz-p-2 (we omitted the mesityl groups from DiICzMes4 to avoid spurious negative vibration modes) and obtain excellent agreement with the corresponding experimental spectra of DiICzMes4 (see Fig. S35, ESI‡). The lower energy shoulder of the fluorescence spectrum observed experimentally is attributed to a vibronic transition based on the cross-comparison with the simulated one. This shoulder disappears with increasing concentration when aggregate emission begins to contribute significantly to the emission spectrum (Fig. S37d, ESI‡). Furthermore, the simulated vibronically-resolved phosphorescence spectrum is also in excellent agreement with the experiment. Interestingly, there is an enhanced vibronic intensity associated with high-frequency (1200–1600 cm−1) vibrations in the phosphorescence spectrum in comparison to fluorescence spectrum. This reflects the more pronounced geometric relaxation taking place in T1 compared so S1, which translates into a larger adiabatic ΔEST in comparison to the vertical ΔEST (Table S11, ESI‡) and provides clear spectroscopic evidence for the different character of the S1 and T1 excited states. This behaviour is again in strong contrast with most of the MR-TADF emitters previously reported in the literature.
The solution photoluminescence quantum yields increase from 58%, 66% and 70% for ICz, ICzMes3 to DiICzMes4, respectively, again reflecting expected trends in the calculations.11 Time-resolved PL decays revealed prompt CT lifetimes of 15.0 ns, 21.6 ns and 40.5 ns for ICz, ICzMes3 and DiICzMes4, respectively. A small contribution of delayed emission was observed for ICz, which was ascribed to originate from TTA (Fig. S34b, ESI‡), while no delayed emission was observed for either ICzMes3 or DiICzMes4 (Fig. S34d and f, ESI‡).
We next investigated the solid-state PL behaviour in a wide bandgap host, PMMA at 3 wt% doping of emitter. This and subsequent wide bandgap (high triplet energy) OLED hosts were selected to strongly exclude the possibility of guest-to-host triplet quenching in both optical and device investigations. The λPL are 377 nm, 391 nm and 442 nm for ICz, ICzMes3 and DiICzMes4, respectively, values that are modestly red-shifted compared to those in toluene. The ΔEST values are similar to those measured in toluene at 0.50 eV, 0.41 eV and 0.29 eV for ICz, ICzMes3 and DiICzMes4, respectively, and align with the calculated ΔEST using optimized excited state structures. The ΦPL are similar to those in toluene at 37%, 58% and 67%, for ICz, ICzMes3 and DiICzMes4, respectively. Again, a red-shifted emission, a decreased ΔEST and an improved ΦPL are observed across the series from ICz to ICzMes3 and to DiICzMes4 (Fig. S34, ESI‡). Owing to their large ΔEST and excessive S1 energies, the photophysical properties of ICz and ICzMes3 were not investigated in other hosts.
We next investigated the photophysical properties of DiICzMes4 in mCP as this OLED-compatible host matrix has a suitably large T1 energy of 2.9 eV.60 The optimum doping concentration as a function of ΦPL was determined (Fig. 6a and Table S16, ESI‡). No AIQ was observed up to 3 wt%, with ΦPL maintained at 82%; beyond this concentration the ΦPL decreases, with neat films showing a ΦPL of 30% (Fig. 6a). The FWHM of a drop-cast 3 wt% doped film in mCP is larger at 40 nm; a low-energy shoulder increases in intensity with increasing doping, which we assigned to an emission from an aggregate (Fig. S37d, ESI‡). However, when films were spin-coated, the aggregate formation could be suppressed, with 3 wt% spin-coated films having a FWHM of 21 nm at λPL 451 nm. At this concentration the ΔEST is 0.26 eV, leading to a long τd of 433 μs but with a delayed emission suppressed at low temperatures (Fig. 6c). A similar behaviour exists when DiICzMes4 is doped in DPEPO at 5 wt% where the delayed emission is no longer observed below 80 K (Fig. S38b, ESI‡). In the time-resolved measurements the spectra of the delayed emission match that of the prompt fluorescence, and thus can be assigned to emission from the S1 state rather than any room temperature phosphorescence, which has been observed in other rigid systems61 (Fig. S37b, ESI‡). TTA was ruled out as the emission mechanism owing to the linear power dependence of the emission intensity (Fig. 6d).62
 | ||
| Fig. 6 Solid-state photophysics of DiICzMes4. (a) ΦPL as a function of doping concentration, calculated using an integrating sphere. An exponential decay has been fitted to guide the reader, λexc = 350 nm; (b) steady-state (SS) emission spectra at RT (λexc = 330 nm) and 77 K and phosphorescence spectrum at 77 K in 3 wt% mCP films, (λexc = 350 nm); (c) temperature-dependent time-resolved PL decays in 3 wt% mCP films, λexc = 355 nm; (d) intensity dependence as a function of laser power in 3 wt% mCP, λexc = 355 nm. | ||
The contribution of the delayed emission to the overall emission is often small in MR-TADF emitters, reflecting the efficient kSr (and small ΦISC) and the slow kRISC. For instance, for DABNA-1 and DiKTa the Φd is around 4% for 1 wt% DABNA-113 in mCBP and 1% for DiKTa in toluene.33 For DiICzMes4, the Φd is 1.2%, and thus kRISC is slow in this emitter, at 1.8 × 102 s−1 following the methodology of Masui et al.63 This is substantially slower than most MR-TADF emitters, but similar to tPBisICz and tBisICz, which were reported as 1.4 and 0.14 × 103 s−1, respectively, in 1 wt% mCP:TSPO1 films.50 In this work, neither ICz nor ICzMes3 show TADF due to their too large ΔEST of 0.47 eV and 0.39 eV, respectively, measured in toluene glass; however, DiICzMes4 shows weak (though unambiguous) TADF as its ΔEST of 0.26 eV is much smaller (Table 2).
| Compounda | λ PL (FWHM)b/nm | Φ PL /% | S1d/eV | T1d/eV | ΔESTe/eV | τ p /ns | τ d /μs |
|---|---|---|---|---|---|---|---|
| a In 3 wt% doped PMMA films. b λ exc = 330 nm, where value in parentheses is FWHM. c Determined using an integrating sphere, λexc = 330 nm. d S1 and T1 determined from the onset of the steady-state and phosphorescence spectra, respectively, at 77 K, λexc = 330 nm. e Calculated from the energy difference between the energies of the S1 and T1 states at 77 K. f λ exc = 355 nm. g λ exc = 350 nm. h λ exc = 350 nm. i In 3 wt% doped mCP films. | |||||||
| ICz | 377 (29) | 37 | 3.38 | 2.88 | 0.50 | N/A | N/A |
| ICzMes3 | 391 (28) | 58 | 3.26 | 2.85 | 0.41 | N/A | N/A |
| DiICzMes4 | 442 (20)g | 67 | 2.86h | 2.57h | 0.29 | N/A | N/A |
| DiICzMes4 | 451 (22)g | 82f | 2.82h | 2.56h | 0.26 | 14 | 433 |
In anticipation of OLED applications, additional time-resolved emission decays were also collected for DiICzMes4 in a wide range of suitably high-triplet OLED hosts (Fig. S38, ESI‡). For these experiments 10% loading in drop-cast films was used, improving the overall signal but also enhancing the emission detectable from red-shifted dimer or excimer species, as evident in the individual normalised spectra (contour plots, Fig. S38c, ESI‡). In line with the stationary emission spectra of MR-TADF materials in varying solvent polarity (Fig. S33, ESI‡), we observe only minor differenced in the time-resolved spectra and decays regardless of host.
Devices
Regioisomeric derivatives of DiICz have been reported in the form of 4,49m-FLDID64 and tDIDCz,65 where each was described as a traditional fluorescent emitter. Large experimentally determined ΔEST values of 0.36 eV and 0.44 eV were reported for m-FLDID64 and tDIDCz,65 respectively, in frozen THF glass. The corresponding UV-emitting OLEDs showed EQEmax of 5.2% and 3.3% at CIE of (0.16, 0.03) and (0.16, 0.02), respectively. Having confirmed the previously overlooked though admittedly weak TADF activity of DiICzMes4, its use as an emitter in OLEDs was assessed. Devices using a stack of ITO (HIL/anode) |NPB (HTL, 40 nm)|TSBPA (EBL, 10 nm)|DiICzMes4:DPEPO 10% (EML, 30 nm)|DPEPO (HBL, 10 nm)|TBPi (ETL, 40 nm)|LiF (EIL, 1 nm)|Al (cathode, 100 nm) were fabricated (Fig. 7), with representative performance shown in Fig. 8. These results show that the low rate of RISC in DiICzMes4 is insufficient to enable efficient triplet harvesting even at the lowest current densities (and corresponding lowest brightness, ∼10 cd m−2) investigated here. The resulting low EQEmax values are consistent with the DiICzMes4 acting akin to a fluorescent dopant, only able to harvest singlet excitons for emission with an upper limit of EQEmax < 5%. This result is in-line with what was observed for previous acceptor-free rIC DF material TCA_C4.51 The OLED shows CIE coordinates of (0.15, 0.11). Despite the higher EQEmax observed for tPBisICz at very low brightnesses, the efficiency roll-off of that device was severe with the EQE at 100 cd m−2 only about 5%. Comparing our OLED results at equivalent brightnesses reveals similar overall performance metrics with the previously reported work (Table S3, ESI‡).50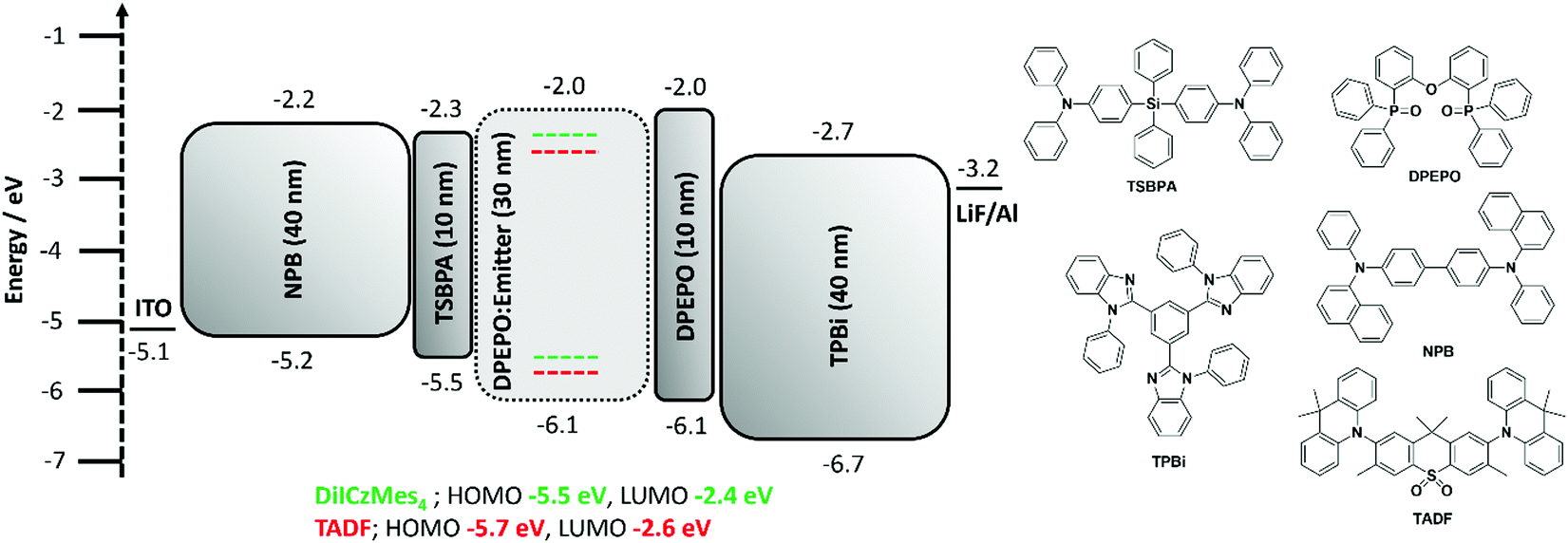 | ||
| Fig. 7 Device architecture and chemical structures of materials employed. | ||
 | ||
Fig. 8 OLED performance for different EMLs. (a) EL spectra of TADF D–A–D 35% (red), HF OLEDs 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :35 wt% (blue) in DPEPO host, absorption (black dotted) and PL (green) spectra of DiICzMes4 1 wt% in mCP for comparison. (b) CIE coordinates, where square (DiICzMes4 only) and circle (HF) overlap, (c) JVL curves, and (d) EQE vs. current density, where fitting has been applied (dotted line) to guide the reader. :35 wt% (blue) in DPEPO host, absorption (black dotted) and PL (green) spectra of DiICzMes4 1 wt% in mCP for comparison. (b) CIE coordinates, where square (DiICzMes4 only) and circle (HF) overlap, (c) JVL curves, and (d) EQE vs. current density, where fitting has been applied (dotted line) to guide the reader. | ||
Additional devices using a different stack consisting of ITO|NPB (HTL, 40 nm)|mCBP (EBL, 10 nm)|DiICzMes4:host X% (EML, 30 nm)|T2T (HBL, 10 nm)|T2T:LiQ 45% (EIL/ETL, 35 nm)|Al (cathode, 100 nm) were also fabricated. In these the concentration of the dopant was varied (5, 12, and 20 wt%) and different EML hosts additionally investigated: mCBP (hole transporting), DPEPO (electron transporting). Representative device performance and spectra are shown in Fig. S39 (ESI‡), with no significant improvement compared to the results in Fig. 8. With increasing concentration no difference was observed in current density–voltage–luminance (JVL) and EQE as a function of current density although a broadening in the electroluminescence (EL) spectrum was observed. The broadening is assigned to the dimer/excimer contribution as shown from the previous photophysical results (Fig. S37d and S38c, ESI‡).
In order to compensate for the low RISC rate of DiICzMes4 we also applied it as a terminal emitter in hyperfluorescent OLEDs (HF OLEDs) with a D–A–D TADF co-host. In order to ensure good spectral overlap necessary for energy transfer, we employed a dimethylacridine-tetramethylthioxanthene-S,S-dioxide (identified as TADF in Fig. 7) based TADF previously reported to give high EQEs and blue emission [CIE of (0.15, 0.19)] in the same OLED stack.14 This D–A–D TADF was co-evaporated at 35% in the EML, alongside 1% DiICzMes4 and bulk host DPEPO. The resulting OLEDs possessed good efficiency, with an EQEmax > 16% and CIE of (0.15, 0.11) enabled by triplet harvesting of the D–A–D co-host, while outputting narrow blue emission from the DiICzMes4. The HF OLED showed relatively lower efficiency roll-off, offering a practical strategy to circumvent large efficiency roll-off resulting from inefficient kRISC of the MR-TADF emitter (Fig. 8).
As our integrating sphere system is not sensitive to very low luminances, we do not observe the same high maximum EQEs (∼32%) previously reported using a similar hyperfluorescence approach with pICz.48 However, comparing our device data at equivalent brightnesses reveals improved performance (Fig. S40, ESI‡), which we infer is due to the improved efficiency roll-off of our D–A–D co-host. Indeed, this performance at higher brightnesses is amongst some of the best reported for HF OLEDs at this colour coordinate (Table S18, ESI‡). The previously reported DPAc-DtCzBN:PPF co-host has a similar intrinsic maximum efficiency as ours, and with slightly blue shifted EL spectrum should also enjoy marginally improved FRET overlap with the MR-TADF emitter in the device. Despite the adequate FRET overlap in both devices, a subtle shoulder can still be observed in our EL spectra, indicating residual emission from the D–A–D co-host. As hyperfluorescence applications of MR-TADF emitters become increasingly popular to circumvent their low RISC rates,22,66–68 engineering both their PL spectra (for ideal-blue emission), as well as their absorption spectra (for minimal Stokes shift, enabling broad compatibility with D–A–D TADF co-hosts)69 take on equally important roles for applications. The latter of these can significantly alleviate the requirement for D–A–D co-hosts with deep blue EL, which remain challenging to design despite nearly a decade of intense global research in this direction.
We finally note that in both our hyperfluorescence devices and those previously reported, inclusion of the MR-TADF leads to significantly worse efficiencies at reasonable brightnesses compared to the D–A–D co-host alone. While the DiICzMes4 would be expected to increase device performance due to spontaneously emitter dipole alignment and improved outcoupling,48 other detrimental processes must also be at play to result in an overall detriment to performance. These may include charge trapping or Dexter transfer to the slow-RISC MR-TADF dopant, although these processes have proven to be incredibly challenging to even quantify by traditional means.70 Therefore while the improvement in colour coordinate offered by the MR-TADF hyperfluorescence strategy is welcomed, it is clear that a deeper understanding of the relevant in operando mechanism and processes is required to unlock their full potential.
Conclusions
We have designed and investigated an MR-TADF compound that does not contain any explicit electron-acceptor units, opening a new design paradigm for MR-TADF emitters. SCS-CC2 calculations guided the design, confirming a strategy to coincidentally decrease ΔEST and improve oscillator strength with increasing electronic delocalization. Photophysical measurements revealed a reduced ΔEST and increased ΦPL were observed in both solution and doped films for DiICzMes4 compared to ICz and ICzMes3. Although ΔEST was rather large at 0.26 eV in mCP, TADF was nonetheless observed in this and other solid-state hosts. Activation of TADF occurs through the involvement of higher-lying triplet states of different orbital types to S1, resulting in non-negligible SOC.42,43 Owing to inefficient RISC, simple guest–host OLEDs showed low efficiency, although hyperfluorescent devices achieved good EQEmax of 16.5%, at deep-blue colour coordinates (0.15, 0.11) with improved relative efficiency roll-off. Discovery of new regions of chemical space suitable for the development of MR-TADF emitters thus opens new paths towards understanding their optical properties and improving their performance.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The St Andrews team would like to thank the Leverhulme Trust (RPG-2016-047) for financial support. Computational resources have been provided by the Consortium des Équipements de Calcul Intensif (CÉCI), funded by the Fonds de la Recherche Scientifiques de Belgique (F. R. S.-FNRS) under Grant No. 2.5020.11, as well as the Tier-1 supercomputer of the Fédération Wallonie-Bruxelles, infrastructure funded by the Walloon Region under the grant agreement n1117545. We acknowledge support from the European Union's Horizon 2020 research and innovation programme under the ITN TADFlife (GA 812872). Y.O. acknowledges funding by the Fonds de la Recherche Scientifique-FNRS under Grant no. F.4534.21 (MIS-IMAGINE). D. B. is a FNRS Research Director. EZ-C is a Royal Society Leverhulme Trust Senior Research fellow (SRF\R1\201089).References
- S. Madayanad Suresh, D. Hall, D. Beljonne, Y. Olivier and E. Zysman-Colman, Adv. Funct. Mater., 2020, 30, 1908677 CrossRef CAS.
- M. Y. Wong and E. Zysman-Colman, Adv. Mater., 2017, 29, 1605444 CrossRef PubMed.
- Y. Liu, C. Li, Z. Ren, S. Yan and M. R. Bryce, Nat. Rev. Mater., 2018, 3, 18020 CrossRef CAS.
- Z. Yang, Z. Mao, Z. Xie, Y. Zhang, S. Liu, J. Zhao, J. Xu, Z. Chi and M. P. Aldred, Chem. Soc. Rev., 2017, 46, 915 RSC.
- F. Ni, N. Li, L. Zhan and C. Yang, Adv. Opt. Mater., 2020, 8, 1902187 CrossRef CAS.
- W. Chen and F. Song, Chin. Chem. Lett., 2019, 30, 1717 CrossRef CAS.
- M. A. Bryden and E. Zysman-Colman, Chem. Soc. Rev., 2021, 50, 7587 RSC.
- C. Adachi and S. D. Sandanayaka Atula, CCS Chem., 2020, 2, 1203 CrossRef CAS.
- M. K. Etherington, J. Gibson, H. F. Higginbotham, T. J. Penfold and A. P. Monkman, Nat. Commun., 2016, 7, 13680 CrossRef CAS PubMed.
- I. A. Wright, A. Danos, S. Montanaro, A. S. Batsanov, A. P. Monkman and M. R. Bryce, Chemistry, 2021, 27, 6545 CrossRef CAS PubMed.
- A. Pershin, D. Hall, V. Lemaur, J.-C. Sancho-Garcia, L. Muccioli, E. Zysman-Colman, D. Beljonne and Y. Olivier, Nat. Commun., 2019, 10, 597 CrossRef CAS PubMed.
- X. Qiu, G. Tian, C. Lin, Y. Pan, X. Ye, B. Wang, D. Ma, D. Hu, Y. Luo and Y. Ma, Adv. Opt. Mater., 2020, 9, 2001845 CrossRef.
- T. Hatakeyama, K. Shiren, K. Nakajima, S. Nomura, S. Nakatsuka, K. Kinoshita, J. Ni, Y. Ono and T. Ikuta, Adv. Mater., 2016, 28, 2777 CrossRef CAS PubMed.
- P. Stachelek, J. S. Ward, P. L. Dos Santos, A. Danos, M. Colella, N. Haase, S. J. Raynes, A. S. Batsanov, M. R. Bryce and A. P. Monkman, ACS Appl. Mater. Interfaces, 2019, 11, 27125 CrossRef CAS PubMed.
- Y. Wada, H. Nakagawa, S. Matsumoto, Y. Wakisaka and H. Kaji, Nat. Photonics, 2020, 14, 643 CrossRef CAS.
- E. Zysman-Colman, Nat. Photonics, 2020, 14, 593 CrossRef CAS.
- L.-S. Cui, A. J. Gillett, S.-F. Zhang, H. Ye, Y. Liu, X.-K. Chen, Z.-S. Lin, E. W. Evans, W. K. Myers, T. K. Ronson, H. Nakanotani, S. Reineke, J.-L. Bredas, C. Adachi and R. H. Friend, Nat. Photonics, 2020, 14, 636 CrossRef CAS.
- N. Ikeda, S. Oda, R. Matsumoto, M. Yoshioka, D. Fukushima, K. Yoshiura, N. Yasuda and T. Hatakeyama, Adv. Mater., 2020, 32, e2004072 CrossRef PubMed.
- K. Matsui, S. Oda, K. Yoshiura, K. Nakajima, N. Yasuda and T. Hatakeyama, J. Am. Chem. Soc., 2018, 140, 1195 CrossRef CAS PubMed.
- S. Oda, B. Kawakami, R. Kawasumi, R. Okita and T. Hatakeyama, Org. Lett., 2019, 21, 9311 CrossRef CAS PubMed.
- Y. Kondo, K. Yoshiura, S. Kitera, H. Nishi, S. Oda, H. Gotoh, Y. Sasada, M. Yanai and T. Hatakeyama, Nat. Photonics, 2019, 13, 678 CrossRef CAS.
- C.-Y. Chan, M. Tanaka, Y.-T. Lee, Y.-W. Wong, H. Nakanotani, T. Hatakeyama and C. Adachi, Nat. Photonics, 2021, 15, 203 CrossRef CAS.
- Y. Zhang, D. Zhang, J. Wei, Z. Liu, Y. Lu and L. Duan, Angew. Chem., Int. Ed., 2019, 58, 16912 CrossRef CAS PubMed.
- Y. T. Lee, C. Y. Chan, M. Tanaka, M. Mamada, U. Balijapalli, Y. Tsuchiya, H. Nakanotani, T. Hatakeyama and C. Adachi, Adv. Electrode Mater., 2021, 7, 2001090 CrossRef CAS.
- Y. Xu, Z. Cheng, Z. Li, B. Liang, J. Wang, J. Wei, Z. Zhang and Y. Wang, Adv. Opt. Mater., 2020, 8, 1902142 CrossRef CAS.
- S. Oda, W. Kumano, T. Hama, R. Kawasumi, K. Yoshiura and T. Hatakeyama, Angew. Chem., Int. Ed., 2021, 60, 2882 CrossRef CAS PubMed.
- Y. Xu, C. Li, Z. Li, Q. Wang, X. Cai, J. Wei and Y. Wang, Angew. Chem., Int. Ed., 2020, 59, 17442 CrossRef CAS PubMed.
- Y. Zhang, D. Zhang, J. Wei, X. Hong, Y. Lu, D. Hu, G. Li, Z. Liu, Y. Chen and L. Duan, Angew. Chem., Int. Ed., 2020, 59, 17499 CrossRef CAS PubMed.
- M. Yang, I. S. Park and T. Yasuda, J. Am. Chem. Soc., 2020, 142, 19468 CrossRef CAS PubMed.
- Y. Zhang, D. Zhang, T. Huang, A. J. Gillett, Y. Liu, D. Hu, L. Cui, Z. Bin, G. Li, J. Wei and L. Duan, Angew. Chem., Int. Ed., 2021, 60, 2049 Search PubMed.
- X. Cai, Y. Xu, Q. Wang, C. Li and Y. Wang, ChemRxiv. Preprint, 2021 DOI:10.26434/chemrxiv.14371073.v1.
- C. Li, Y. Wang, Y. Xu, Z. Li, J. Wang, J. Xue, Q. Wang and X. Cai, 2021, ChemRxiv. Preprint DOI:10.26434/chemrxiv.14050712.v1.
- D. Hall, S. M. Suresh, P. L. dos Santos, E. Duda, S. Bagnich, A. Pershin, P. Rajamalli, D. B. Cordes, A. M. Z. Slawin, D. Beljonne, A. Köhler, I. D. W. Samuel, Y. Olivier and E. Zysman-Colman, Adv. Opt. Mater., 2020, 8, 1901627 CrossRef CAS.
- Y. Yuan, X. Tang, X. Y. Du, Y. Hu, Y. J. Yu, Z. Q. Jiang, L. S. Liao and S. T. Lee, Adv. Opt. Mater., 2019, 7, 1801536 CrossRef.
- E. Hamzehpoor and D. F. Perepichka, Angew. Chem., Int. Ed., 2020, 59, 9977 CrossRef CAS PubMed.
- K. Wang, X.-C. Fan, Y. Tsuchiya, Y.-Z. Shi, M. Tanaka, Z. Lin, Y.-T. Lee, X. Zhang, W. Liu, G.-L. Dai, J. Chen, B. Wu, J. Zhong, J.-Y. Yuan, C.-J. Zheng, J. Yu, C. Lee, C. Adachi and X.-H. Zhang, ChemRxiv. Preprint, 2021 DOI:10.26434/chemrxiv.13699057.v1.
- H. Min, I. S. Park and T. Yasuda, Angew. Chem., Int. Ed., 2021, 60, 7643 CrossRef CAS PubMed.
- M. Nagata, H. Min, E. Watanabe, H. Fukumoto, Y. Mizuhata, N. Tokitoh, T. Agou and T. Yasuda, Angew. Chem., Int. Ed., 2021, 60, 20820 CrossRef PubMed.
- N. Haase, A. Danos, C. Pflumm, A. Morherr, P. Stachelek, A. Mekic, W. Brütting and A. P. Monkman, J. Phys. Chem. C, 2018, 122, 29173 CrossRef CAS.
- Y. Tsuchiya, S. Diesing, F. Bencheikh, Y. Wada, P. L. dos Santos, H. Kaji, E. Zysman-Colman, I. D. W. Samuel and C. Adachi, J. Phys. Chem. A, 2021, 125, 8074–8089 CrossRef CAS PubMed.
- T. Serevicius, R. Skaisgiris, G. Kreiza, J. Dodonova, K. Kazlauskas, E. Orentas, S. Tumkevicius and S. Jursenas, J. Phys. Chem. A, 2021, 125, 1637 CrossRef CAS PubMed.
- T. Northey and T. J. Penfold, Org. Electron., 2018, 59, 45 CrossRef CAS.
- K. Stavrou, A. Danos, T. Hama, T. Hatakeyama and A. Monkman, ACS Appl. Mater. Interfaces, 2021, 13, 8643 CrossRef CAS PubMed.
- Y. Olivier, B. Yurash, L. Muccioli, G. D’Avino, O. Mikhnenko, J. C. Sancho-García, C. Adachi, T. Q. Nguyen and D. Beljonne, Phys. Rev. Mater., 2017, 1, 075602 CrossRef.
- D. Sun, S. M. Suresh, D. Hall, M. Zhang, C. Si, D. B. Cordes, A. M. Z. Slawin, Y. Olivier, X. Zhang and E. Zysman-Colman, Mater. Chem. Front., 2020, 4, 2018 RSC.
- J. A. Knoller, G. Meng, X. Wang, D. Hall, A. Pershin, D. Beljonne, Y. Olivier, S. Laschat, E. Zysman-Colman and S. Wang, Angew. Chem., Int. Ed., 2020, 59, 3156 CrossRef PubMed.
- S. M. Suresh, E. Duda, D. Hall, Z. Yao, S. Bagnich, A. M. Z. Slawin, H. Bassler, D. Beljonne, M. Buck, Y. Olivier, A. Kohler and E. Zysman-Colman, J. Am. Chem. Soc., 2020, 142, 6588 CrossRef CAS PubMed.
- J. Wei, C. Zhang, D. Zhang, Y. Zhang, Z. Liu, Z. Li, G. Yu and L. Duan, Angew. Chem., Int. Ed., 2021, 60, 12269 CrossRef CAS PubMed.
- T. Taniguchi, Y. Itai, Y. Nishii, N. Tohnai and M. Miura, Chem. Lett., 2019, 48, 1160 CrossRef CAS.
- V. V. Patil, H. L. Lee, I. Kim, K. H. Lee, W. J. Chung, J. Kim, S. Park, H. Choi, W. J. Son, S. O. Jeon and J. Y. Lee, Adv. Sci., 2021, e2101137 CrossRef PubMed.
- P. Pander, R. Motyka, P. Zassowski, M. K. Etherington, D. Varsano, T. J. da Silva, M. J. Caldas, P. Data and A. P. Monkman, J. Phys. Chem. C, 2018, 122, 23934 CrossRef CAS.
- J. A. Seo, Y. Im, S. H. Han, C. W. Lee and J. Y. Lee, ACS Appl. Mater. Interfaces, 2017, 9, 37864 CrossRef CAS PubMed.
- D. G. Congrave, B. H. Drummond, V. Gray, A. D. Bond, A. Rao, R. H. Friend and H. Bronstein, Polym. Chem., 2021, 12, 1830 RSC.
- E. Aksoy, A. Danos, C. Li, A. P. Monkman and C. Varlikli, J. Phys. Chem. C, 2021, 125, 13041 CrossRef CAS.
- C. Maeda, T. Todaka and T. Ema, Org. Lett., 2015, 17, 3090 CrossRef CAS PubMed.
- S. I. Wharton, J. B. Henry, H. McNab and A. R. Mount, Chem. – Eur. J., 2009, 15, 5482 CrossRef CAS PubMed.
- K. Karon and M. Lapkowski, J. Solid State Electrochem., 2015, 19, 2601 CrossRef CAS.
- N. G. Connelly and W. E. Geiger, Chem. Rev., 1996, 96, 877 CrossRef CAS PubMed.
- C. M. Cardona, W. Li, A. E. Kaifer, D. Stockdale and G. C. Bazan, Adv. Mater., 2011, 23, 2367 CrossRef CAS PubMed.
- S. H. Kim, J. Jang and J. Y. Lee, Appl. Phys. Lett., 2007, 90, 223505 CrossRef.
- J. S. Ward, R. S. Nobuyasu, A. S. Batsanov, P. Data, A. P. Monkman, F. B. Dias and M. R. Bryce, Chem. Commun., 2016, 52, 2612 RSC.
- F. B. Dias, T. J. Penfold and A. P. Monkman, Methods Appl. Fluoresc., 2017, 5, 012001 CrossRef PubMed.
- K. Masui, H. Nakanotani and C. Adachi, Org. Electron., 2013, 14, 2721 CrossRef CAS.
- V. V. Patil, J. Lim and J. Y. Lee, ACS Appl. Mater. Interfaces, 2021, 13, 14440 CrossRef CAS PubMed.
- H. L. Lee, W. J. Chung and J. Y. Lee, Small, 2020, 16, e1907569 CrossRef PubMed.
- D. Zhang, X. Song, A. J. Gillett, B. H. Drummond, S. T. E. Jones, G. Li, H. He, M. Cai, D. Credgington and L. Duan, Adv. Mater., 2020, 32, e1908355 CrossRef PubMed.
- S. H. Han, J. H. Jeong, J. W. Yoo and J. Y. Lee, J. Mater. Chem. C, 2019, 7, 3082 RSC.
- S. O. Jeon, K. H. Lee, J. S. Kim, S.-G. Ihn, Y. S. Chung, J. W. Kim, H. Lee, S. Kim, H. Choi and J. Y. Lee, Nat. Photonics, 2021, 15, 208 CrossRef CAS.
- A. Monkman, ACS Appl. Mater. Interfaces, 2021 Search PubMed.
- N. Haase, A. Danos, C. Pflumm, P. Stachelek, W. Brütting and A. P. Monkman, Mater. Horiz., 2021, 8, 1805 RSC.
Footnotes |
| † The research data supporting this publication can be accessed at https://doi.org/10.17630/915048c3-9d32-43ee-a18d-b49f95042a55 |
| ‡ Electronic supplementary information (ESI) available: 1H and 13C NMR, and HRMS spectra of all new compounds and HPLC traces of all the emitters; computational data and coordinates; photophysical data; device data; crystallographic data for ICzMes3 and DiICzMes4. CCDC 2104486 and 2104487. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1mh01383a |
| This journal is © The Royal Society of Chemistry 2022 |