
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceChirality memory of α-methylene-π-allyl iridium species†
Yifan
Cui
ab,
Yizhan
Zhai‡
ab,
Junzhe
Xiao‡
 ab,
Can
Li‡
ab,
Wei-Feng
Zheng‡
c,
Chaofan
Huang‡
c,
Guolin
Wu‡
c,
Anni
Qin‡
c,
Jie
Lin‡
c,
Qi
Liu‡
ab,
Huanan
Wang‡
c,
Penglin
Wu‡
c,
Haibo
Xu‡
ab,
Yangguangyan
Zheng‡
c and
Shengming
Ma
*ac
ab,
Can
Li‡
ab,
Wei-Feng
Zheng‡
c,
Chaofan
Huang‡
c,
Guolin
Wu‡
c,
Anni
Qin‡
c,
Jie
Lin‡
c,
Qi
Liu‡
ab,
Huanan
Wang‡
c,
Penglin
Wu‡
c,
Haibo
Xu‡
ab,
Yangguangyan
Zheng‡
c and
Shengming
Ma
*ac
aState Key Laboratory of Organometallic Chemistry, Shanghai Institute of Organic Chemistry, Chinese Academy of Sciences, 345 Lingling Lu, Shanghai 200032, P. R. China. E-mail: masm@sioc.ac.cn; masm.fudan.edu.cn
bUniversity of Chinese Academy of Sciences, Beijing 100049, P. R. China
cResearch Center for Molecular Recognition and Synthesis, Department of Chemistry, Fudan University, 220 Handan Road, Shanghai 200433, P. R. China
First published on 21st July 2021
Abstract
Chirality is one of the most important types of steric information in nature. In addition to central chirality, axial chirality has been catching more and more attention from scientists. However, although much attention has recently been paid to the creation of axial chirality and the chirality transfer of allenes, no study has been disclosed as to the memory of such an axial chirality. The reason is very obvious: the chiral information is stored over three carbon atoms. Here, the first example of the memory of chirality (MOC) of allenes has been recorded, which was realized via an optically active alkylidene-π-allyl iridium intermediate, leading to a highly stereoselective electrophilic allenylation with amines. Specifically, we have established the transition metal-mediated highly stereoselective 2,3-allenylation of amines by using optically active 2,3-allenyl carbonates under the catalysis of a nonchiral iridium(III) complex. This method is compatible with sterically bulky and small substituents on both amines and 2,3-allenyl carbonates and furnishes the desired optically active products with a high efficiency of chirality transfer. Further mechanistic experiments reveal that the isomerization of the optically active alkylidene-π-allyl iridium intermediate is very slow.
Introduction
Memory of chirality (MOC) is a topic of ever-lasting interest in the storage/transmission of steric information, chiral recognition, and asymmetric syntheses.1 In addition to enolate chemistry,2 some examples of transition-metal catalyzed stereospecific functionalizations of optically active allylic derivatives have been recorded.3 However, reports on the memory of chirality of α-alkylidene-π-allyl metallic species are very limited: the rapid racemization of such chiral intermediate via σ–π–σ-rearrangements has been reported by Tsukamoto (Scheme 1a).4 Herein, we wish to report the observation of an excellent memory of chirality (MOC) in the highly stereoselective reaction of optically active allenylic methyl carbonates with amines affording 2,3-allenylic amines in high ee via α-alkylidene-π-allyl iridium species (Scheme 1b).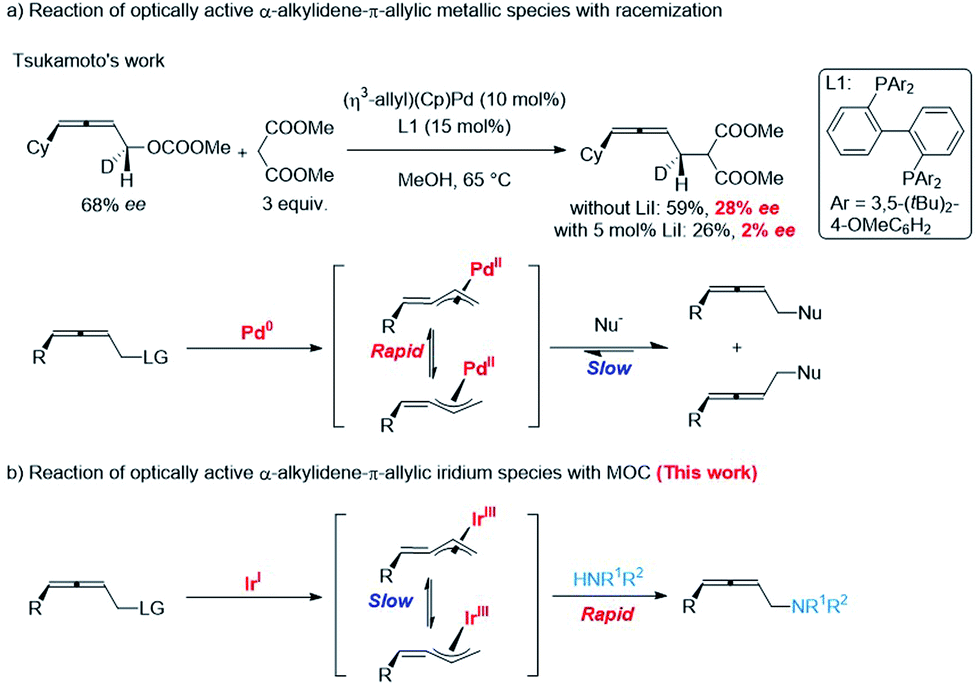 | ||
| Scheme 1 Reaction of optically active α-alkylidene-π-allylic metallic species. | ||
Results
Optimization of reaction conditions
In the beginning, due to the importance of amines,5 we were studying the synthesis of racemic 2,3-allenyl amines. After trial and error, we observed some very attractive results with iridium-catalysis for the allenylation of benzylamine 2a with racemic methyl dodeca-2,3-dienyl carbonate 1a. With [Ir(COD)Cl]2 as the catalyst, a variety of phosphine ligands was screened. PPh3 (Table 1, entry 1) afforded a 13% yield of 3aa, 1% of bis-allenylation product 4aa, and 85% recovery of 1a. Further screening of other phosphine ligands led to poor results (Table 1, entries 2–10). To our delight, the utilization of a known cationic Ir(III) pre-catalyst A developed by Hartwig and co-workers6 yielded 26% of 3aa and 25% of 4aa with no recovery of 1a (Table 1, entry 11).|
|
||||
|---|---|---|---|---|
| Entry | Ligand (mmol%) | Yield of 3aab (%) | Yield of 4ab (%) | Recovery of 1ab (%) |
| a The reaction was conducted using 1a (0.2 mmol) and BnNH2 (0.3 mmol) in 0.4 mL of THF. b Determined by 1H NMR analysis of the crude reaction mixture using 1,3,5-trimethylbenzene as an internal standard. | ||||
| 1 | PPh3 (7) | 13 | 1 | 85 |
| 2 | dppm (3.5) | 2 | — | 95 |
| 3 | DPEphos (3.5) | 2 | — | 99 |
| 4 | P(OPh)3 (7) | 1 | — | 96 |
| 5 | dppe (3.5) | — | — | 97 |
| 6 | dppp (3.5) | — | — | 99 |
| 7 | dppf (3.5) | — | — | 100 |
| 8 | dppbe (3.5) | — | — | 99 |
| 9 | Xantphos (3.5) | — | — | 100 |
| 10 | BINAP (3.5) | — | — | 99 |
| 11 | Ir pre-catalyst A (3.5) | 26 | 25 | — |
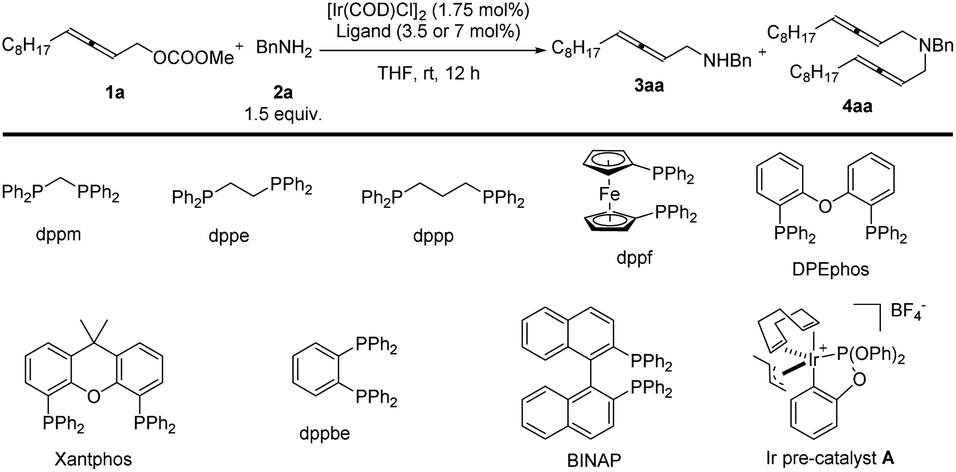
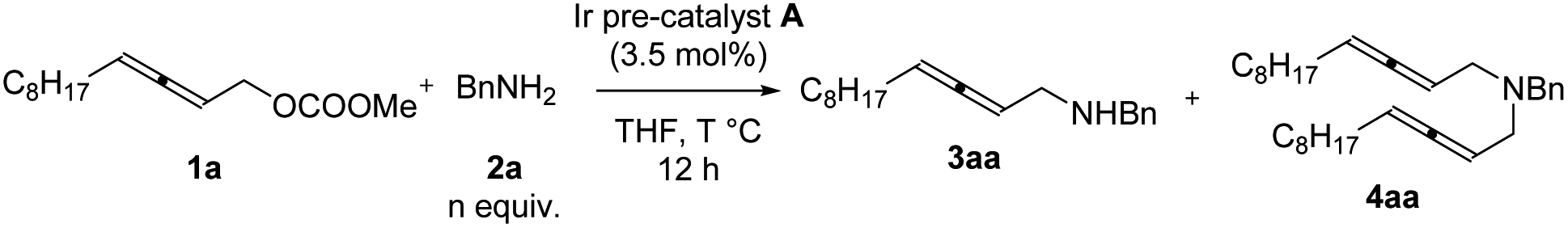
Further optimization was focused on the selectivity of 3aa/4aa. We firstly increased the amount of Ir pre-catalyst A to 7 mol%, but no improvement was observed (Table 2, entry 2). As expected, the loading of benzylamine greatly affected the selectivity of 3aa/4aa: the addition of 4 equiv. of benzylamine provided 41% of 3aa, 6% of 4aa, and 38% recovery of 1a (Table 2, entry 4). By running the reaction at 50 °C, the yield was improved to 70% and no recovery of 1a was detected (Table 2, entry 7). The reaction at a higher temperature resulted in an erosion of yield (Table 2, entry 8).
|
|
|||||
|---|---|---|---|---|---|
| Entry | n | T/°C | Yield of 3aab (%) | Yield of 4aab (%) | Recovery of 1ab (%) |
| a The reaction was conducted using 1a (0.2 mmol) in 0.4 mL of THF. b Determined by 1H NMR analysis of the crude reaction mixture using 1,3,5-trimethylbenzene as an internal standard. c 7 mol% Ir pre-catalyst A was used. d The reaction was conducted using 1a (0.2 mmol) in 1.0 mL of THF. | |||||
| 1 | 1.5 | r.t. | 26 | 25 | — |
| 2c | 1.5 | r.t. | 24 | 27 | — |
| 3 | 2 | r.t. | 35 | 28 | — |
| 4 | 4 | r.t. | 41 | 6 | 38 |
| 5d | 4 | r.t. | 45 | 7 | 45 |
| 6d | 4 | 40 | 64 | 8 | 11 |
| 7d | 4 | 50 | 70 | 8 | — |
| 8d | 4 | 60 | 65 | 9 | — |
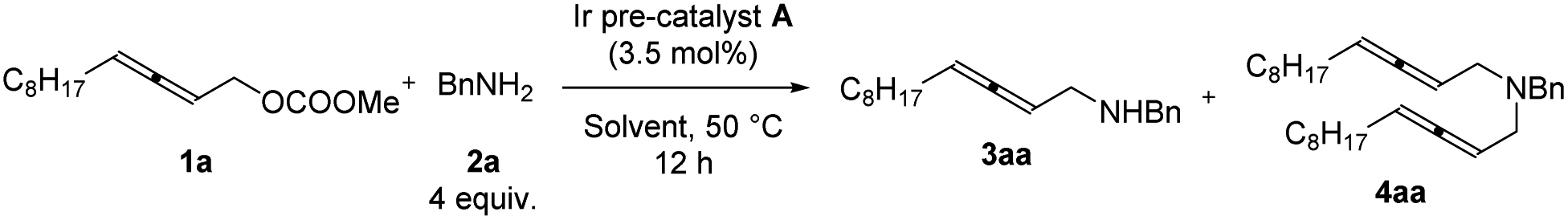
The reactions in other ethers (Table 3, entries 1–3) and chlorinated solvents (Table 3, entries 4 and 5) led to a lower selectivity of 3aa/4aa. Increasing the polarity of aprotic solvents displayed decreasing yields (Table 3, entries 6–8). Toluene provided a very similar yield and selectivity (Table 3, entry 9). Scaling the reaction up to 1.0 mmol furnished similar results with 3aa isolated in a 71% yield together with 9% of the bis-allenylation product (Table 3, entries 10 and 11). Thus, 1a (1 equiv.), 2a (4 equiv.), and Ir pre-catalyst A (3.5 mol%) in THF at 50 °C was defined as the optimized reaction conditions for further study.
|
|
||||
|---|---|---|---|---|
| Entry | Solvent | Yield of 3aab (%) | Yield of 4aab (%) | Recovery of 1ab (%) |
| a The reaction was conducted using 1a (0.2 mmol) and BnNH2 (0.8 mmol) in 1.0 mL of THF. b Determined by 1H NMR analysis of the crude reaction mixture using 1,3,5-trimethylbenzene as an internal standard. c The reaction was conducted using allene 1a (1.0 mmol), BnNH2 (4.0 mmol) in 5 mL of THF. d Isolated yield. | ||||
| 1 | Ethyl ether | 54 | 15 | 3 |
| 2 | DME | 64 | 10 | — |
| 3 | MTBE | 57 | 13 | — |
| 4 | DCM | 50 | 17 | — |
| 5 | DCE | 51 | 18 | — |
| 6 | MeCN | 50 | 18 | — |
| 7 | DMF | 35 | 21 | — |
| 8 | DMSO | 19 | — | 53 |
| 9 | Toluene | 70 | 9 | — |
| 10 | THF | 70 | 8 | — |
| 11c | THF | 71d | 9 | — |
Allenylation of amines
With the optimal conditions in hand, diverse 2,3-allenyl carbonates and amines were investigated to demonstrate the scope of this reaction (Scheme 2). Terminal 2,3-allenyl carbonates showed slightly lower reactivities (products 3bb–3be) (part I). 4-Mono-substituted 2,3-allenyl carbonates were then examined (part II): R1 may be a 1°-alkyl group, such as n-C8H17 (1a), n-C5H11 (1d), n-C6H13 (1t), or a 2°-alkyl group, i-Pr (1e) and Cy (1f), smoothly affording products 3aa, 3af, 3ag, 3ah, 12, 3tx, 3dj, 3dk, 3el, 3fm, and 3fn with the corresponding amines. Benzoxy (1c) and phenyl group (1u) were tolerated (products 3ci, 3uy, and 4uz).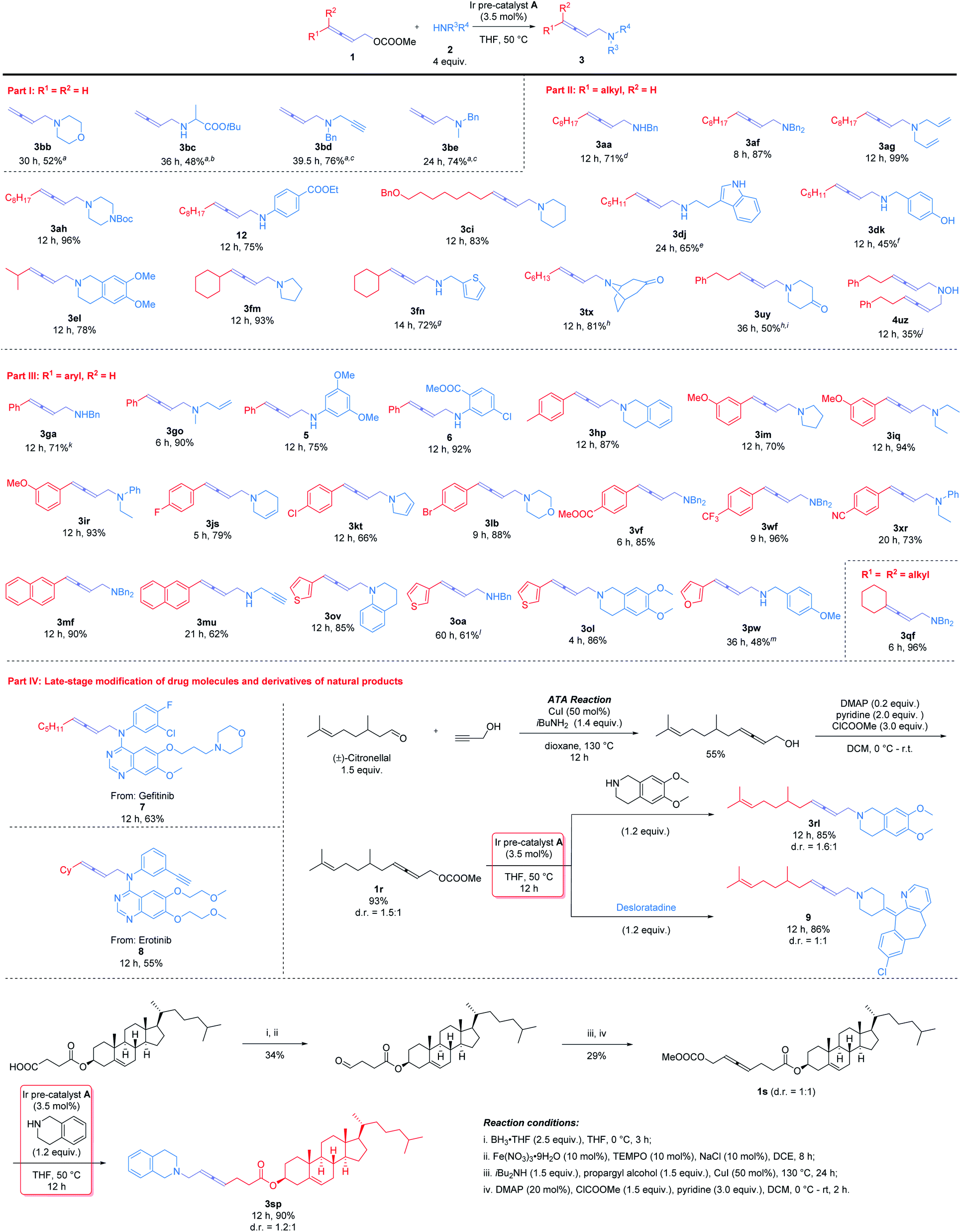 | ||
| Scheme 2 Substrate scope of Ir-catalyzed 2,3-allenylation of amines. Standard conditions: Ir pre-catalyst A (3.5 mmol%), allene (1.0 mmol) and amine (4.0 mmol) in 5 mL of THF. a5.3 mol% Ir pre-catalyst A was used. b32% of 1b was recovered. c10% of 1b was recovered. d9% NMR yield of 4aa. e5.3 mol% Ir pre-catalyst A was used and the reaction was conducted at 60 °C. f17% NMR yield of 4dk. g7% NMR yield of 4fn. hThe reaction was conducted using allene (1.0 mmol), amine·HCl (1.2 mmol), and NaHCO3 (1.2 mmol) in 5 mL of THF. i7 mol% Ir pre-catalyst A was used. jThe reaction was conducted using allene (1.0 mmol), NH2OH·HCl (4.0 mmol), and NaHCO3 (4.0 mmol) in 5 mL of THF and 6% NMR yield of 3uz was detected. k6% NMR yield of 4ga was detected. l15% of 1o was recovered and 3% NMR yield of 4oa was detected. m7 mol% Ir pre-catalyst A was used and the reaction was conducted at 60 °C. | ||
4-Aryl-2,3-allenyl carbonates were next exposed to the optimized reaction conditions (part III): 4-phenyl-2,3-allenyl carbonate 1g furnished a similar result as compared with 1a (product 3ga). Substrates with a wide range of functional groups, such as p-Me (1h), m-OMe (1i), p-F (1j), p-Cl (1k), p-Br (1l), p-COOMe (1v), p-CF3 (1w), and p-CN (1x), all exhibited a decent reactivity under the standard conditions (products 3hp, 3im–3ir, 3js, 3kt, 3lb, 3vf, 3wf, and 3xr). 2-Naphthyl (1m) and 3-thienyl (1o) were also accommodated to afford the target products 3mf, 3mu, 3ov, and 3oa in 61–90% yields; 3-furyl substituted allenyl carbonate 1p turned out to be less reactive and reacted with p-methoxybenzylamine 2w to yield product 3pw in 48% isolated yield at 60 °C with 7 mol% Ir catalyst. 4,4-Disubstituted allenyl carbonate 1q also worked to afford product 3qf.
A wide range of amines were also tested. Acyclic amines bearing different alkyl (2e, 2o, 2q, and 2r), phenyl (12, 5, 6, and 2r), benzyl (2a, 2f, 2k, and 2w), and synthetically useful functional groups, such as the allylic (2g and 2o), propargyl (2d and 2u) and ester (2c) groups, all furnished the corresponding products 3be, 3go, 3iq, 12, 5, 6, 3ir, 3xr, 3aa, 3ga, 3vf, 3wf, 3mf, 3oa, 3pw, 3ag, 3go, 3bd, 3mu, and 3bc in moderate to quantitative yields (48–99%). Tryptamine 2j, which contains 3 potential reaction sites, was detected to be allenylated exclusively on the primary amino group as judged by 1H NMR analysis of the crude product(s). Under standard reaction conditions, it is not necessary to protect the hydroxyl group (2k) to afford product 3dk.
Cyclic amines act as widespread skeletons in drug molecules and natural products. Cyclic amines, such as morpholine 2b, pyrrolidine 2m, pyrroline 2t, piperidine 2i, piperazine 2h, 1,2,3,6-tetrahydropyridine 2s, tetrahydroisoquinoline (2l and 2p), and tetrahydroquinoline 2v, may all be modified with the allene unit furnishing the desired products 3bb, 3lb, 3fm, 3im, 3kt, 3ci, 3ah, 3js, 3el, 3hp, 3ol, and 3ov smoothly in 52–96% yields. Quaternary amine hydrochlorides exist widely in medical and pharmaceutical science due to their superior water solubility, absorption, and ease of formulation. Both nortropinone (2x) and 4-piperidinone (2y) furnished the desired products 3tx and 3uy in the presence of NaHCO3. Hydroxylamine hydrochloride 2z could also be allenylated, affording bis-allenylation product 4uz.
In order to demonstrate the scope of the current Ir-catalyzed 2,3-allenylation reaction, we applied this strategy for the late-stage modification of drug molecules and derivatives of natural products (part IV). Two commercial drugs for cancer treatment, Gefitinib and Erlotinib,7 could be directly modified with an allene unit (products 7 and 8). Furthermore, the ATA reaction8 of (±)-citronellal and cholesterol with propargyl alcohol conveniently gave the desired allenols, which were treated with chloroformate to afford the carbonates 1r and 1s. Subsequent Ir-catalyzed reactions with tetrahydroisoquinolines and desloratadine9 afforded 2,3-allenyl amines for 3rl, 9, and 3sp with 85–90% yields.
Memory of chirality
Nowadays, optically active 2,3-allenols are readily available from the EATA reaction.10 Thus, after unveiling the scope of this protocol, we were anxious to investigate the memory of chirality by applying optically active 2,3-allenyl carbonates. Our first attempt was the reaction of (S)-1m with dibenzylamine 2f at room temperature with 3.5 mol% Ir pre-catalyst A (eqn (1)). The target product could be obtained with 95% yield and an excellent efficiency of chirality transfer (99% es), i.e., the axial chirality of the allene unit was passed to the axial chirality in the allenylic amines. To our knowledge, this is the first example of such transition metal-involved chirality memory.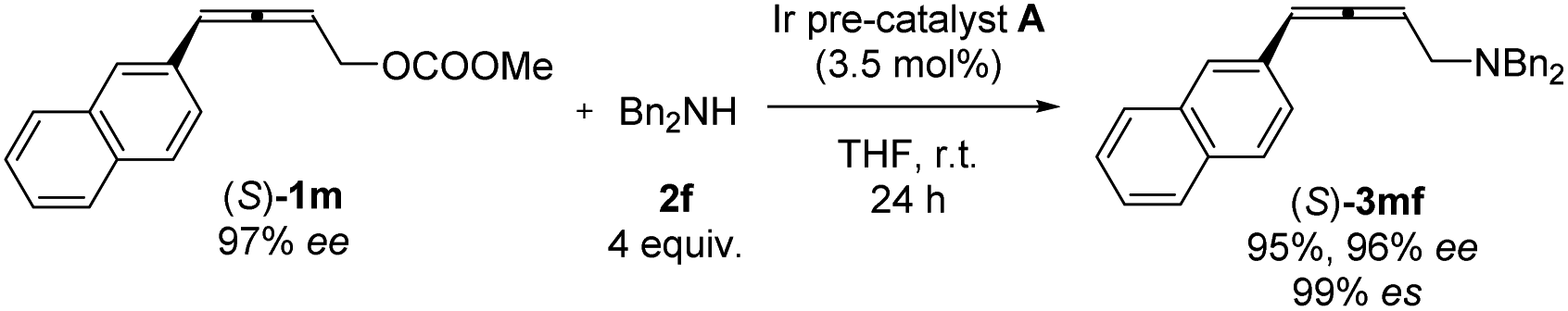 | (1) |
Encouraged by this result, we further examined the scope of memory of chirality (Scheme 3). For 4-alkyl-substituted allenyl carbonate (S)-1a, reactions afforded (S)-3aa in 71% yield and 98% es on a gram-scale with benzylamine 2a and (S)-12 in 85% yield and 95% es with 4-ethoxycarbonyl aniline, respectively. The more sterically hindered (S)-1f yielded (S)-3fn in 95% es. Various 4-aryl-2,3-allenyl carbonates were next investigated: different substituents on benzene ring, including F ((S)-1j), Cl ((S)-1k), and Br ((S)-1l), were intact under the reaction conditions (products (S)-3js, (S)-3kt, and (S)-3lb). Different types of acyclic (2a, 2n, 2o, and 2f) and cyclic amines (2s, 2t, and 2b) showed little influence on the yields and stereoselectivity. Paroxetine hydrochloride, a drug used against depression and social phobia,11 could also be modified with optically active 2,3-allenyl carbonate ((S)-1d) efficiently to afford the desired product (Sa,S,R)-10 in high es. Late-stage modification of optically active derivatives of natural products, (S)-citronellal ((Sa)-1r) and cholesterol ((Sa)-1s), were sequentially performed, furnishing the corresponding products (Sa)-3rl and (Sa)-3sp smoothly in high yields and excellent d.r. (Scheme 3).
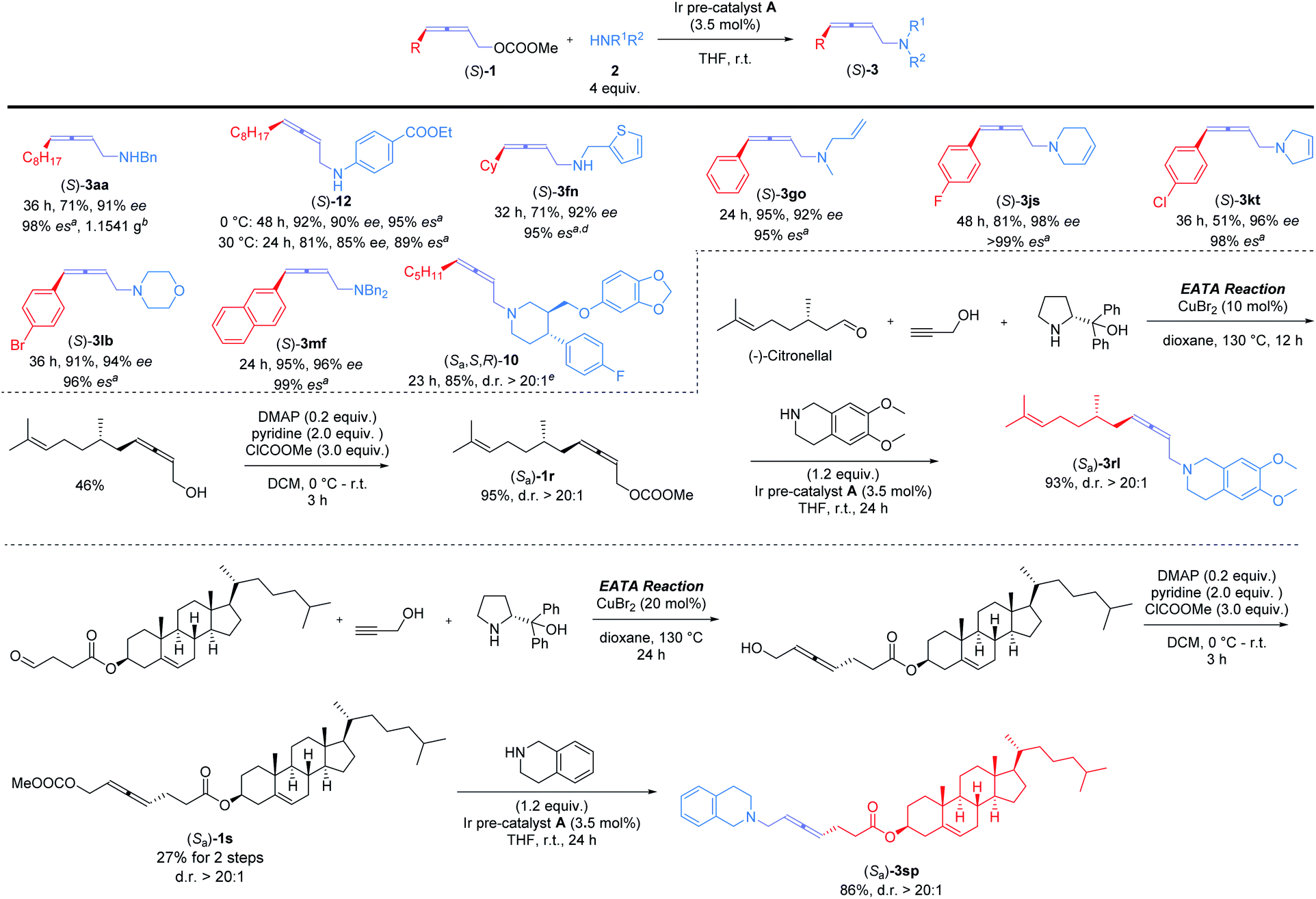 | ||
| Scheme 3 Ir-catalyzed 2,3-allenylation of optically active 2,3-allenyl carbonates with amines. Reaction procedure: to a flame-dried Schlenk tube (25 mL) were added Ir pre-catalyst A (0.035 mmol)/THF (3 mL), amine (4.0 mmol)/THF (1 mL), and allene (1.0 mmol)/THF (1 mL) sequentially under an Ar atmosphere. The resulting mixture was stirred at r.t. aEnantioselectivity of the transformation based on the ee of the staring material. bThe reaction was conducted using allene (6.0 mmol) and amine (24.0 mmol) in 30 mL of THF at 40 °C with a 10% NMR yield of (S,S)-4aa. cThe reaction was conducted at 0 °C. d8% NMR yield of (S,S)-4fn was detected.e The reaction was conducted using (S)-1d (1.0 mmol), paroxetine·HCl·½ H2O (1.2 mmol), and NaHCO3 (1.2 mmol) in 10 mL of THF. | ||
Mechanistic studies
The absolute configuration of (S)-3mf was confirmed by preparing this same product by following a known procedure.12 To gain insight into the mechanism, a series of experiments was carried out.Firstly, we left a period of time for the racemization of the optically active alkylidene-π-allyl iridium intermediate as (S)-1m was treated with different amounts of Ir pre-catalyst A for as long as 12 h followed by the addition of the nucleophile dibenzylamine 2f. As showed in Scheme 4a, the level of racemization is linear with the loading of the Ir pre-catalyst A. With the increased loading of the Ir pre-catalyst A, the decarboxylation product, 2,3-allenyl methyl ether 11 was also generated with the same level of ee as compared to that of (S)-3mf. It is notable that the treatment of (S)-1m with 27% of pre-catalyst A for 12 h followed by the addition of dibenzylamine still formed (S)-3mf in 74% ee, which may be attributed to the much slower rate of racemization (Scheme 4b). Malcolmson et al.13 had reported that the rapid racemization of optically active 2,3-allenyl amine happened even in the presence of a chiral palladium catalyst (Scheme 4c and d). However, the treatment of the optically active product (S)-3mf under the standard reaction conditions did not lead to racemization (Scheme 4e). Moreover, no racemization of (S)-3mf or generation of (S)-3mr was observed in the scrambling experiment, indicating that the nucleophilic attack of amine is irreversible under the current Ir-catalyzed reaction conditions (Scheme 4f).
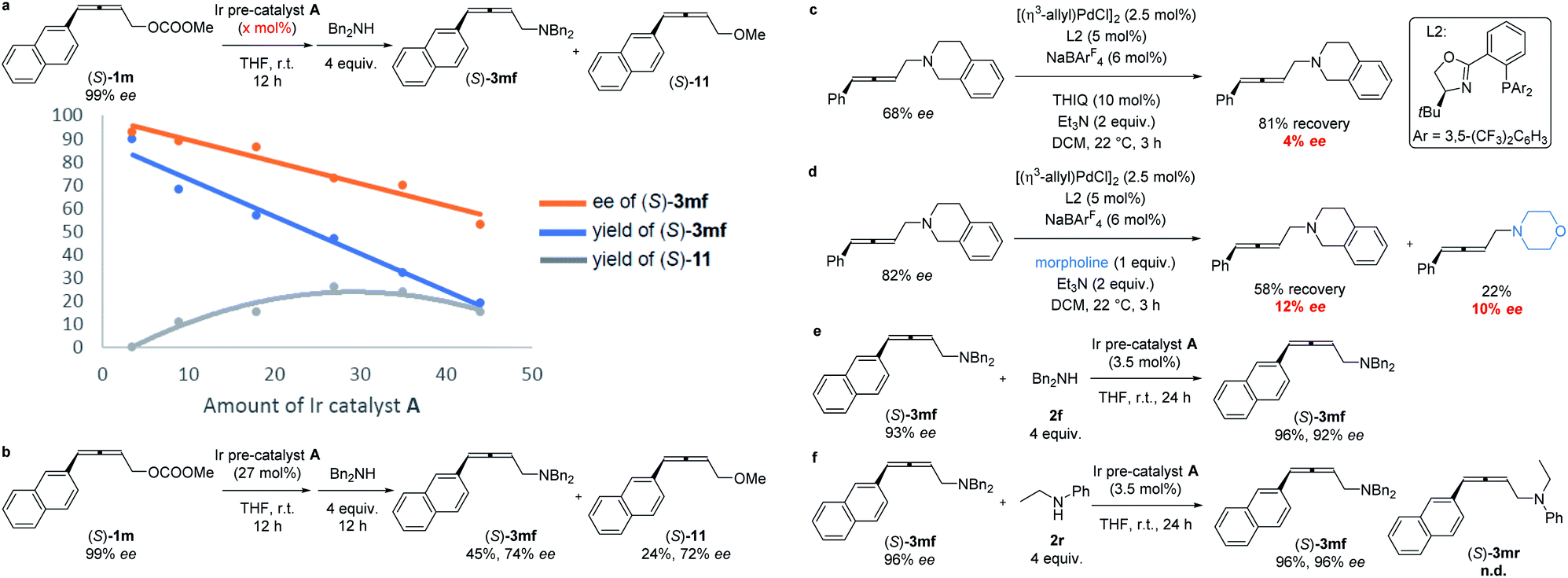 | ||
| Scheme 4 Mechanistic studies. The reaction procedure for the data in (a): to a flame-dried Schlenk tube were added Ir pre-catalyst A (x mol%)/THF (1.5 mL), and (S)-1m (0.5 mmol)/THF (0.5 mL) sequentially under an Ar atmosphere. The resulting mixture was stirred at room temperature for 12 h. Dibenzyl amine 2f (2.0 mmol)/THF (0.5 mL) were then added sequentially under an Ar atmosphere. The resulting mixture was stirred at room temperature. | ||
Based on the mechanistic experiments, a catalytic cycle is proposed as shown in Scheme 5. At first, the oxidative addition of Ir(I)-η2-(S)-1I leads to the formation of optically active alkylidene-π-allyl Ir(III) II, accompanied by the release of a carbonate anion. The chirality is memorized in this intermediate since the isomerization to form the allylic Ir intermediate V is very slow. The subsequent irreversible attack of amine results in the formation of Ir(I)-η2-2,3-allenyl aminylium ion III, which is much faster than the attack of methoxy in the carbonate anion after releasing carbon dioxide. The subsequent coordination of another (S)-1 with Ir(I) in III regenerates I and releases the 2,3-allenyl ammonium ion VI to complete the catalytic cycle. With the aid of a carbonate anion, the final product (S)-3 could be afforded by the deprotonation of VI, accompanied by the release of carbon dioxide and methanol.
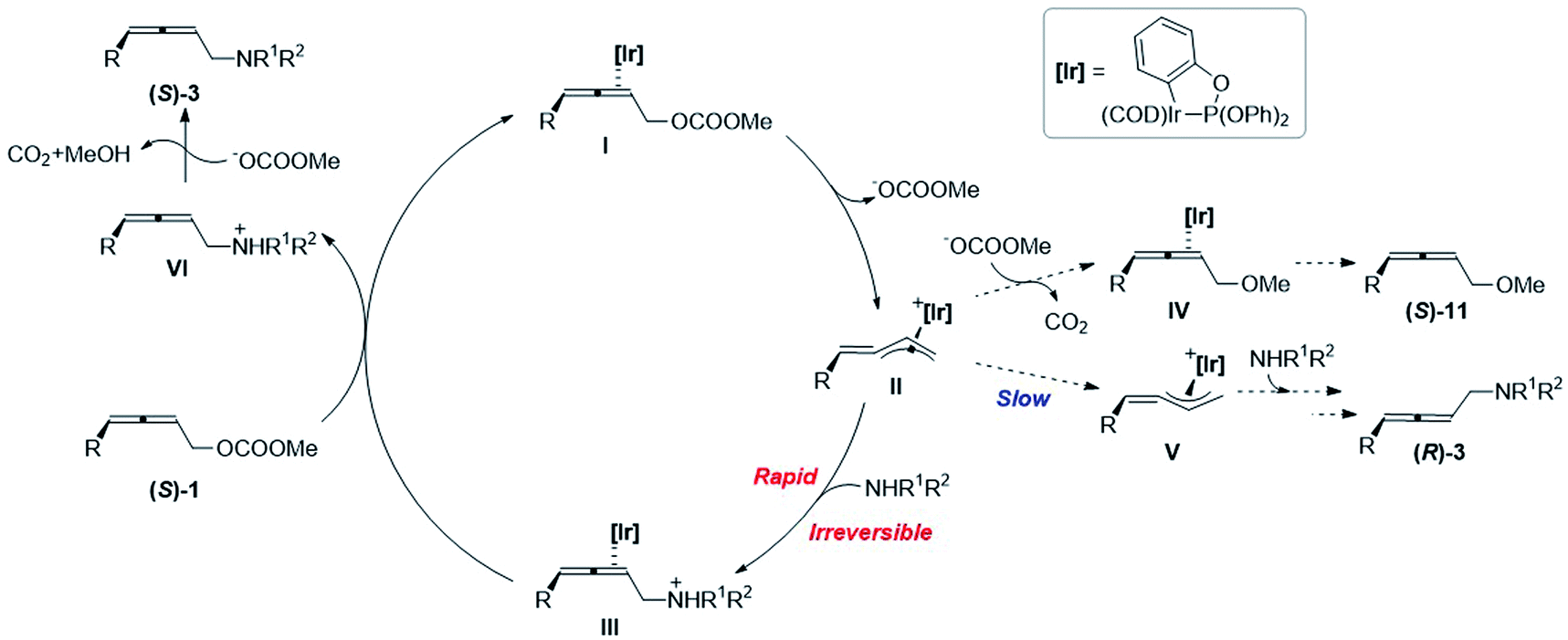 | ||
| Scheme 5 Proposed catalytic cycle. | ||
Conclusions
In conclusion, although racemizations of the optically active alkylidene-π-allylic transition metal complexes are very common, leading to the erasure of the chiral information in the starting materials and have been extensively applied to the asymmetric syntheses of optically active allenes,14 we have recorded here the first example of chirality memory involving alkylidene-π-allylic transition metal species and developed a robust highly stereoselective approach for asymmetric allene synthesis. Such a chirality memory involving iridium will be critical for the storage and transmission of the axial chirality in optically active allenols, which are readily available from propargylic alcohols and aldehydes. Further studies with other nucleophiles in this area are being actively pursued in our laboratory.Data availability
The ESI† include experimental details, specific rotation data, HPLC data, NMR data, MS data, IR data, elemental analysis data, and all the spectra.Author contributions
S. M. directed the research and developed the concept of the reaction with Y. C., who also performed most of the experiments and prepared the ESI.† Y. Z., J. X., C. L., W. Z., C. H., G. W., A. Q., J. L., Q. L., H. W., P. W., H. X. and Y. Z., who also performed part of the experiments, contributed equally to this work. Y. C. and S. M. checked the experimental data. Y. C. and S. M. wrote the manuscript with contributions from the other authors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support from National Natural Science Foundation of China (Grant No. 21690063 & 21988101) is greatly appreciated.References
- (a) T. Kawabata, K. Yahiro and K. Fuji, J. Am. Chem. Soc., 1991, 113, 9694 CrossRef CAS; (b) K. Fuji and T. Kawabata, Chem.–Eur. J., 1998, 4, 373 CrossRef CAS; (c) T. Kawabata, J. Chen, H. Suzuki and K. Fuji, Synthesis, 2005, 37, 1368 CrossRef; (d) H. Zhao, D. C. Hsu and P. R. Carlier, Synthesis, 2005, 37, 1 Search PubMed; (e) D. Campolo, S. Gastaldi, C. Roussel, M. P. Bertrand and M. Nechab, Chem. Soc. Rev., 2013, 42, 8434 RSC; (f) S. Oliver and P. A. Evans, Synthesis, 2013, 45, 3179 CrossRef CAS; (g) V. Alezra and T. Kawabata, Synthesis, 2016, 48, 2997 CrossRef CAS; (h) C. S. Gloor, F. Dénès and P. Renaud, Free Radical Res., 2016, 50, S102 CrossRef CAS PubMed; (i) K. Yamamoto, M. Kuriyama and O. Onomura, Acc. Chem. Res., 2020, 53, 105 CrossRef CAS PubMed.
- (a) A. Mambrini, D. Gori, R. Guillot, C. Kouklovsky and V. Alezra, Chem. Commun., 2018, 54, 12742 RSC; (b) A. Mambrini, D. Gori, C. Kouklovsky, H. Kim, J. I. Yoshida and V. Alezra, Eur. J. Org. Chem., 2018, 2018, 6754 CrossRef CAS; (c) V. Veeraswamy, G. Goswami, S. Mukherjee, K. Ghosh, M. L. Saha, A. Sengupta and M. K. Ghorai, J. Org. Chem., 2018, 83, 1106 CrossRef CAS PubMed; (d) J. H. Kim, I. Kim, Y. Song, M. J. Kim and S. Kim, Angew. Chem., Int. Ed., 2019, 58, 11018 CrossRef CAS; (e) S. Tan, F. Li, S. Park and S. Kim, J. Org. Chem., 2019, 84, 14436 CrossRef CAS; (f) J. H. Kim, Y. Chung, H. Jeon, S. Lee and S. Kim, Org. Lett., 2020, 22, 3989 CrossRef CAS PubMed.
- (a) J. L. Renaud, B. Demerseman, M. D. Mbaye and C. Bruneau, Curr. Org. Chem., 2006, 10, 115 CAS; (b) B. Breit and Y. Schmidt, Chem. Rev., 2008, 108, 2928 CrossRef CAS; (c) S. R. Harutyunyan, T. den Hartog, K. Geurts, A. J. Minnaard and B. L. Feringa, Chem. Rev., 2008, 108, 2824 CrossRef CAS PubMed; (d) S. M. Pound and M. P. Watson, Chem. Commun., 2018, 54, 12286 RSC; (e) B. W. H. Turnbull and P. Andrew Evans, J. Org. Chem., 2018, 83, 11463 CrossRef CAS PubMed; (f) M. B. Thoke and Q. Kang, Synthesis, 2019, 51, 2585 CrossRef CAS; (g) S. Akkarasamiyo, S. Ruchirawat, P. Ploypradith and J. S. M. Samec, Synthesis, 2020, 52, 645 CrossRef CAS; (h) H. S. Um, J. Min, T. An, J. Choi and C. Lee, Org. Chem. Front., 2018, 5, 2158 RSC; (i) Y. Sempere, J. L. Alfke, S. L. Rössler and E. M. Carreira, Angew. Chem., Int. Ed., 2019, 58, 9537 CrossRef CAS; (j) C. Casalta and S. Bouzbouz, Org. Lett., 2020, 22, 2359 CrossRef CAS PubMed; (k) C. Chen, H. Wang, Y. Sun, J. Cui, J. Xie, Y. Shi, S. Yu, X. Hong and Z. Lu, iScience, 2020, 23, 100985 CrossRef CAS PubMed; (l) M. J. Tom and P. A. Evans, J. Am. Chem. Soc., 2020, 142, 11957 CrossRef CAS PubMed.
- H. Tsukamoto, T. Konno, K. Ito and T. Doi, Org. Lett., 2019, 21, 6811 CrossRef CAS.
- (a) J. D. Scott and R. M. Williams, Chem. Rev., 2002, 102, 1669 CrossRef CAS PubMed; (b) A. L. Harvey, Drug Discovery Today, 2008, 13, 894 CrossRef CAS PubMed; (c) S. D. Roughley and A. M. Jordan, J. Med. Chem., 2011, 54, 3451 CrossRef CAS PubMed; (d) E. Vitaku, D. T. Smith and J. T. Njardarson, J. Med. Chem., 2014, 57, 10257 CrossRef CAS PubMed.
- S. T. Madrahimov, Q. Li, A. Sharma and J. F. Hartwig, J. Am. Chem. Soc., 2015, 137, 14968 CrossRef CAS.
- W. D. Travis, E. Brambilla, M. Noguchi, A. G. Nicholson, K. R. Geisinger, Y. Yatabe, D. G. Beer, C. A. Powell, G. J. Riely, P. E. Van Schil, K. Garg, J. H. M. Austin, H. Asamura, V. W. Rusch, F. R. Hirsch, G. Scagliotti, T. Mitsudomi, R. M. Huber, Y. Ishikawa, J. Jett, M. Sanchez-Cespedes, J.-P. Sculier, T. Takahashi, M. Tsuboi, J. Vansteenkiste, I. Wistuba, P.-C. Yang, D. Aberle, C. Brambilla, D. Flieder, W. Franklin, A. Gazdar, M. Gould, P. Hasleton, D. Henderson, B. Johnson, D. Johnson, K. Kerr, K. Kuriyama, J. S. Lee, V. A. Miller, I. Petersen, V. Roggli, R. Rosell, N. Saijo, E. Thunnissen, M. Tsao and D. Yankelewitz, J. Thorac. Oncol., 2011, 6, 244 CrossRef.
- J. Kuang, H. Luo and S. Ma, Adv. Synth. Catal., 2012, 354, 933 CrossRef CAS.
- D. V. Wallace, M. S. Dykewicz, D. I. Bernstein, J. Blessing-Moore, L. Cox, D. A. Khan, D. M. Lang, R. A. Nicklas, J. Oppenheimer, J. M. Portnoy, C. C. Randolph, D. Schuller, S. L. Spector and S. A. Tilles, J. Allergy Clin. Immunol., 2008, 122, S1 CrossRef PubMed.
- (a) X. Huang and S. Ma, Acc. Chem. Res., 2019, 52, 1301 CAS; (b) X. Huang, T. Cao, Y. Han, X. Jiang, W. Lin, J. Zhang and S. Ma, Chem. Commun., 2015, 51, 6956 RSC.
- K. M. Bertelsen, K. Venkatakrishnan, L. L. Von Moltke, R. S. Obach and D. J. Greenblatt, Drug Metab. Dispos., 2003, 31, 289 CrossRef CAS.
- J. Ye, W. Fan and S. Ma, Chem.–Eur. J., 2013, 19, 716 CrossRef CAS.
- N. J. Adamson, H. Jeddi and S. J. Malcolmson, J. Am. Chem. Soc., 2019, 141, 8574 CrossRef CAS PubMed.
- (a) Y. Imada, K. Ueno, K. Kutsuwa and S.-I. Murahashi, Chem. Lett., 2002, 31, 140 CrossRef; (b) B. M. Trost, D. R. Fandrick and D. C. Dinh, J. Am. Chem. Soc., 2005, 127, 14186 CrossRef CAS PubMed; (c) S. Song, J. Zhou, C. Fu and S. Ma, Nat. Commun., 2019, 10, 507 CrossRef; (d) F. Glatz, D. A. Petrone and E. M. Carreira, Angew. Chem., Int. Ed., 2020, 59, 16404 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1sc02636d |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2021 |