
Taking advantage of lithium monohalocarbenoid intrinsic α-elimination in 2-MeTHF: controlled epoxide ring-opening en route to halohydrins†
Laura
Ielo
 ad,
Margherita
Miele
a,
Veronica
Pillari
a,
Raffaele
Senatore
a,
Salvatore
Mirabile
b,
Rosaria
Gitto
b,
Wolfgang
Holzer
a,
Andrés R.
Alcántara
*c and
Vittorio
Pace
*ad
ad,
Margherita
Miele
a,
Veronica
Pillari
a,
Raffaele
Senatore
a,
Salvatore
Mirabile
b,
Rosaria
Gitto
b,
Wolfgang
Holzer
a,
Andrés R.
Alcántara
*c and
Vittorio
Pace
*ad
aUniversity of Vienna – Department of Pharmaceutical Chemistry, Althanstrasse, 14, 1090, Vienna, Austria. E-mail: vittorio.pace@univie.ac.at; vittorio.pace@unito.it; Web: http://drugsynthesis.univie.ac.at/
bUniversity of Messina – Department of Chemical, Biological, Pharmaceutical and Environmental Sciences, Viale Palatucci, 13, 98168 Messina, Italy
cComplutense University of Madrid – Department of Chemistry in Pharmaceutical Sciences, Plaza de Ramón y Cajal, s/n, Madrid, Spain. E-mail: andalcan@ucm.es; Web: https://www.ucm.es/qffa/transbiomat
dUniversity of Turin – Department of Chemistry, Via P. Giuria 7, 10125, Turin, Italy
First published on 8th January 2021
Abstract
The intrinsic degradative α-elimination of Li carbenoids somehow complicates their use in synthesis as C1-synthons. Nevertheless, we herein report how boosting such an α-elimination is a straightforward strategy for accomplishing controlled ring-opening of epoxides to furnish the corresponding β-halohydrins. Crucial for the development of the method is the use of the eco-friendly solvent 2-MeTHF, which forces the degradation of the incipient monohalolithium, due to the very limited stabilizing effect of this solvent on the chemical integrity of the carbenoid. With this approach, high yields of the targeted compounds are consistently obtained under very high regiocontrol and, despite the basic nature of the reagents, no racemization of enantiopure materials is observed.
Lithium monohalocarbenoids (LiCH2X, X = halogen) constitute highly versatile C1-synthons for accomplishing homologation processes in both nucleophilic and electrophilic regimes, the former being the usual mode of action for lithium derivatives.1 Accordingly, upon releasing the CH2X unit onto a proper electrophilic manifold, a plethora of useful synthetic transformations could be designed.2 The coexistence of C–Li and C–halogen bonds on the same carbon atom promotes an extremely high tendency to induce degradative Kirmse's α-elimination phenomenon, triggered by the strong Li–X internal coordination, with the final result of making the carbenoid unproductive for homologation purposes (Scheme 1a).1c,3 This constitutive aspect of carbenoid chemistry is profoundly reflected in the strict operational details requested for their correct use in batch mode:1e,4 (1) to overcome the intrinsic instability, the carbenoid generation event from a given precursor (dihalomethane, stannane, or sulfoxide) must be conducted under Barbier conditions to ensure that no degradation takes place; (2) running the process in coordinative solvents (THF and diethyl ether) in the presence of lithium halide salts contributes to attenuating (if not eliminating) Kirmse's α-elimination to a free carbene and a LiX species (Scheme 1b).5 Notably, these paradigms represent the necessary guidelines for the convenient application of carbenoid-mediated strategies in synthetic methodologies.6
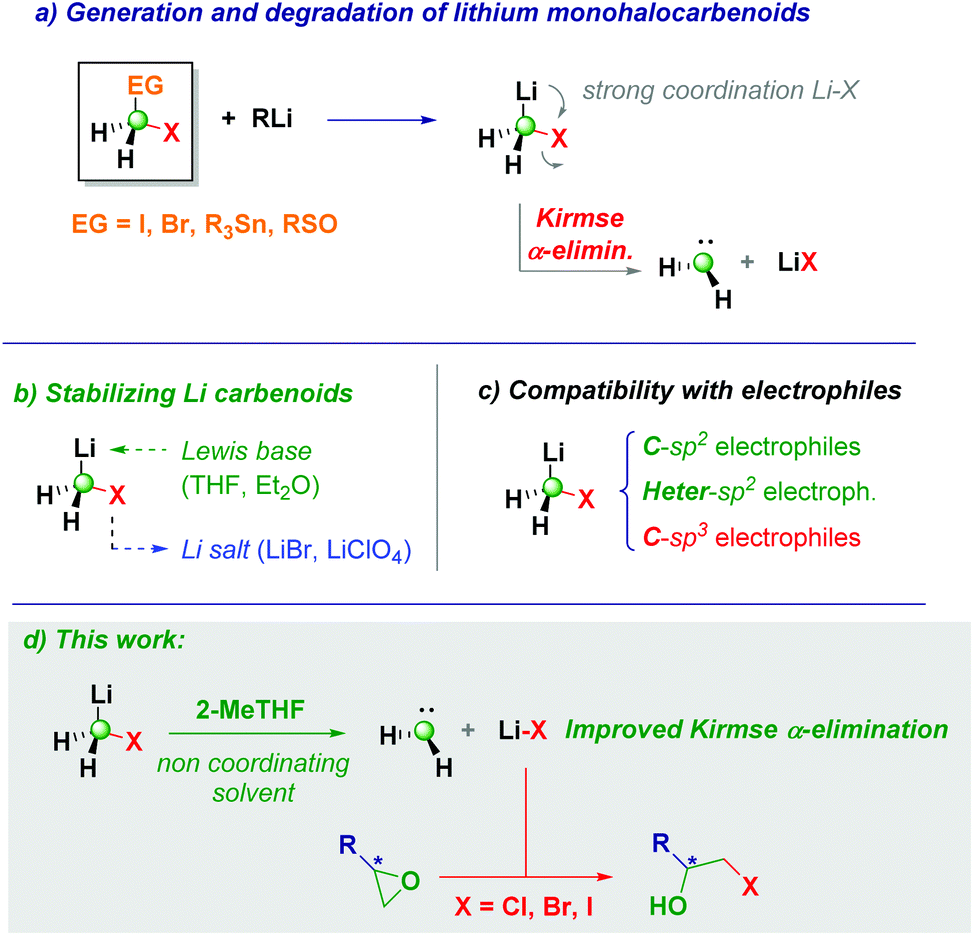 | ||
| Scheme 1 General context of the presented work. | ||
Due to our continued interest in this chemistry over the years, we wondered if Kirmse's elimination could have a synthetic significance behind the well-known degradative effect on the chemical integrity of these C1-synthons. The current state-of-the-art of carbenoid-guided processes, clearly evidences that sp2-hybridized carbon electrophiles are privileged platforms for accomplishing homologation-type transformations, as well as various heteroatoms (e.g. B,7 Sn,8inter alia). Unfortunately, the corresponding sp3-hybridized carbon electrophiles remain elusive materials for accomplishing homologations with LiCH2X, as documented in the seminal work by Huisgen9 and Hahn (Scheme 1c).10 In this context, our group very recently documented the formal homologation of C–X functionalities through a sequential installation of the carbenoid into a carbonyl group followed by Si–H-mediated deoxygenation.11
Herein, we report the controlled ring-opening of epoxides with the lithium halide liberated during Kirmse's α-elimination for obtaining a high-yield of β-halohydrins (Scheme 1d). Pivotal for the success of the method is the generation and rapid degradation of the carbenoid in 2-MeTHF12 (a green solvent increasingly becoming more useful in classical organic chemistry and biotransformations, as well as for polymerization or extractive purposes),13 therefore leading to the degradation of the monohalolithium. We anticipate the genuine regioselectivity of the transformation harnessed on the formation of LiX which plays the dual role of Lewis acid (facilitating the C–O cleavage) and source of the attacking halide.14
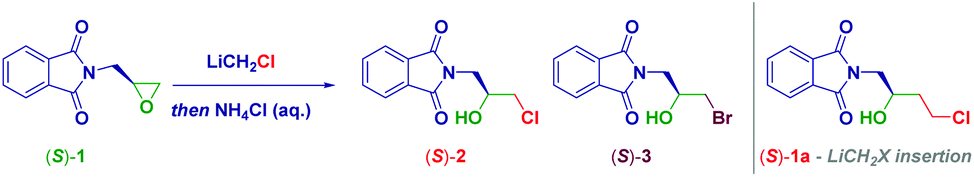
We selected the enantiomerically pure epoxide-containing phthalimide (S)-1 as the starting material to gather, additionally, information on the stereochemical outcome of the process. LiCH2Cl was generated under our previously established reaction conditions6d from ICH2Cl (3.0 equiv.) and MeLi–LiBr (2.8 equiv.) in THF at −78 °C (Table 1). Surprisingly, no attack of the nucleophile leading to the homologated adduct (S)-1a was observed, but rather a mixture of two enantiopure halohydrins [(S)-(2) and (S)-(3)] was recovered in an 84![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :16 ratio (entry 1). The subsequent HPLC analysis of both reaction products indicated the full preservation of the stereochemical information embodied, thereby confirming our previous findings on the retention of configuration in reactions involving basic α-substituted methyllithiums.2a,12k,15 This initial experiment was indicative of the ring-opening of the epoxide with a LiX salt.16 On the other hand, the simultaneous presence of the chloro (S)-(2) and bromo-(S)-(3) derivatives points towards a ring-opening mediated by two different species, namely LiCl and LiBr. Presumably, LiCl was formed during Kirmse's α-elimination of the carbenoid, thus yielding the chlorohydrin, whereas LiBr, complexing the metalating agent MeLi (i.e. MeLi-LiBr complex in Et2O), directly furnished bromohydrin (S)-3. To ascertain this hypothesis, LiX-free MeLi (MeLi in Et2O) was employed for generating the carbenoid under identical conditions: exclusively the chlorohydrin (S)-2 was formed, thus confirming that LiCl (carbenoid degradation product) was responsible for the attack on the oxirane (entry 2). Some important aspects of the process were deduced by running the reaction in different solvents: thus, decreasing the medium-coordination effect on the carbenoid allowed an increase of the yield of the chlorohydrin, as a consequence of a positive modulating effect on Kirmse's elimination. Accordingly, the formation of (S)-2 increased when using Et2O, CPME,17 and TBME (entries 3–5) and it was maximized in 2-MeTHF, giving an excellent yield of 93% (entry 6). Some additional points merit mention: (a) decreasing the loading of carbenoid to 2.0 equiv. has practically no effect (91% yield), thus suggesting that it is optimal in terms of efficiency-costs (entries 7 and 8); (b) the presence of the additive TMEDA did not improve the process (entry 9); (c) the reaction conducted in toluene considerably lowered the yield (entry 10); (d) by running the process at higher temperatures (−40 °C and −10 °C), the effectiveness decreased, probably because of the non-optimal generation of the carbenoid (prior to its decomposition, entries 11 and 12); (e) the process is independent of the pro-carbenoid source, as evidenced using a stannane or a sulfoxide for accomplishing the corresponding metal exchange (entries 13 and 14), though the procedure on ICH2Cl afforded the best results; (f) the other Kirmse's elimination product, CH2 (free carbene), had no effect on the whole transformation (vide infra); and (g) the adoption of non-Barbier-type conditions for forming the carbenoid did not provide any material because of the inadequate formation of the carbenoid (entry 15). Collectively, these results confirmed the previous evidence that LiCH2Cl is not a competent nucleophile for C-sp3 hybridized electrophiles.
:16 ratio (entry 1). The subsequent HPLC analysis of both reaction products indicated the full preservation of the stereochemical information embodied, thereby confirming our previous findings on the retention of configuration in reactions involving basic α-substituted methyllithiums.2a,12k,15 This initial experiment was indicative of the ring-opening of the epoxide with a LiX salt.16 On the other hand, the simultaneous presence of the chloro (S)-(2) and bromo-(S)-(3) derivatives points towards a ring-opening mediated by two different species, namely LiCl and LiBr. Presumably, LiCl was formed during Kirmse's α-elimination of the carbenoid, thus yielding the chlorohydrin, whereas LiBr, complexing the metalating agent MeLi (i.e. MeLi-LiBr complex in Et2O), directly furnished bromohydrin (S)-3. To ascertain this hypothesis, LiX-free MeLi (MeLi in Et2O) was employed for generating the carbenoid under identical conditions: exclusively the chlorohydrin (S)-2 was formed, thus confirming that LiCl (carbenoid degradation product) was responsible for the attack on the oxirane (entry 2). Some important aspects of the process were deduced by running the reaction in different solvents: thus, decreasing the medium-coordination effect on the carbenoid allowed an increase of the yield of the chlorohydrin, as a consequence of a positive modulating effect on Kirmse's elimination. Accordingly, the formation of (S)-2 increased when using Et2O, CPME,17 and TBME (entries 3–5) and it was maximized in 2-MeTHF, giving an excellent yield of 93% (entry 6). Some additional points merit mention: (a) decreasing the loading of carbenoid to 2.0 equiv. has practically no effect (91% yield), thus suggesting that it is optimal in terms of efficiency-costs (entries 7 and 8); (b) the presence of the additive TMEDA did not improve the process (entry 9); (c) the reaction conducted in toluene considerably lowered the yield (entry 10); (d) by running the process at higher temperatures (−40 °C and −10 °C), the effectiveness decreased, probably because of the non-optimal generation of the carbenoid (prior to its decomposition, entries 11 and 12); (e) the process is independent of the pro-carbenoid source, as evidenced using a stannane or a sulfoxide for accomplishing the corresponding metal exchange (entries 13 and 14), though the procedure on ICH2Cl afforded the best results; (f) the other Kirmse's elimination product, CH2 (free carbene), had no effect on the whole transformation (vide infra); and (g) the adoption of non-Barbier-type conditions for forming the carbenoid did not provide any material because of the inadequate formation of the carbenoid (entry 15). Collectively, these results confirmed the previous evidence that LiCH2Cl is not a competent nucleophile for C-sp3 hybridized electrophiles.
|
|
|||||
|---|---|---|---|---|---|
| Entry | Solvent | LiCH2Cl (equiv) | Ratio 2/3a |
Yield of (S)-2b (%) | er of (S)-2 |
| Unless otherwise stated, the carbenoid was generated under Barbier-type conditions starting from ICH2Cl and MeLi (Et2O solution 1.6 M) in THF at −78 °C.a The ratio has been calculated by 1H-NMR analysis using 1,3,5-trimethylbenzene as an internal standard.b Isolated yield.c MeLi–LiBr complex (Et2O solution 1.5 M) was used; compound (S)-3 was obtained in 12% yield and >99:1 er.d TMEDA (2.0 equiv.) was added.e Reaction was run at −40 °C.f Reaction was run at −10 °C.g The carbenoid was prepared from (n-Bu)3SnCH2Cl.h The carbenoid was prepared from PhS(O)CH2Cl.i The carbenoid was generated under non-Barbier conditions. |
|||||
| 1c | THF | 2.8 | 84:16 |
66 | >99:1 |
| 2 | THF | 2.8 | >99:1 |
74 | >99:1 |
| 3 | Et2O | 2.8 | >99:1 |
78 | >99:1 |
| 4 | CPME | 2.8 | >99:1 |
81 | 98:2 |
| 5 | TBME | 2.8 | >99:1 |
85 | 97:3 |
| 6 | 2-MeTHF | 2.8 | >99:1 |
93 | >99:1 |
| 7 | 2-MeTHF | 1.8 | >99:1 |
91 | >99:1 |
| 8 | 2-MeTHF | 1.5 | >99:1 |
77 | >99:1 |
| 9d | 2-MeTHF | 2.0 | >99:1 |
87 | >99:1 |
| 10 | Toluene | 2.0 | >99:1 |
61 | >99:1 |
| 11e | 2-MeTHF | 2.0 | >99:1 |
67 | >99:1 |
| 12f | 2-MeTHF | 2.0 | >99:1 |
48 | >99:1 |
| 13g | 2-MeTHF | 2.0 | >99:1 |
85 | >99:1 |
| 14h | 2-MeTHF | 2.0 | >99:1 |
79 | >99:1 |
| 15i | 2-MeTHF | 2.0 | — | — | — |
Having established the reaction conditions, we then studied the scope of the reaction (Scheme 2). The use of the other enantiomer of 1, i.e. [(R)-1], afforded the corresponding epimeric halohydrin (R)-2 with analogous efficiency and enantiomeric purity. This aspect was further evidenced in the case of different halocarbenoids, such as LiCH2Br [(S)-3 and (R)-3] and LiCH2I [(S)-4 and (R)-4], thus indicating the flexibility of the methodology. The procedure was then extended to rac-1 with the three different carbenoids confirming the average yields obtained with the chiral platform. Unfortunately, the methodology could not be applied for the LiF ring-opening en route to a fluorohydrin. Considering the key role of the solvent in generating LiCH2F,6b,c,18 the use of the optimal mixture THF:Et2O (1:1 v/v) was tested as an alternative to the herein employed 2-MeTHF, without observing the formation of the desired compound. Therefore, regardless of the proper generation of the unstable carbenoid, the degradation salt LiF may not be efficient in promoting the ring opening.19 Epoxides derived from aldehydes could also be subjected to the reaction conditions leading to decorated aromatic β-chlorohydrins (5–6). Analogously, disubstituted epoxides (derived from ketones) easily underwent the ring-opening despite the substitution pattern on the aromatic ring. Notably, the carbenoid formation was not affected by the presence of functionalities sensitive to the organolithium environment. Thus, bromo-(7), iodo-(8), fluoro-(9, 11, 12), chloro-(10) and trifluoromethyl-(13) β-chlorohydrins were smoothly prepared in 2-MeTHF. The additional presence of an acidic aromatic alcohol did not constitute a limitation for the method when deprotonation (with the same MeLi) was carried out prior to carbenoid formation (14). Epoxides formally derived from benzo- or acetophenones were amenable substrates for the reaction furnishing derivatives 15–16 and 17–18, respectively, in high yields. The use of an α-trifluoromethyl-epoxide enabled the straightforward formation of analogue 19, while, much to our delight, even a labile ester group was tolerated during the carbenoid generation–degradation sequence, giving compound 20. The installation of unsaturated fragments (alkyne and alkene) on the epoxide core was particularly attractive not only for the synthetic importance of the so-obtained halohydrins (21–22) but, more importantly, for constituting unambiguous proof of the lack of reactivity of the free carbene generated during the carbenoid degradation.18b,20 No cyclopropanation-like process was detected, as judged by NMR analysis of the crude product, thus confirming the initial hypothesis of the higher reactivity of the lithium halide salt.
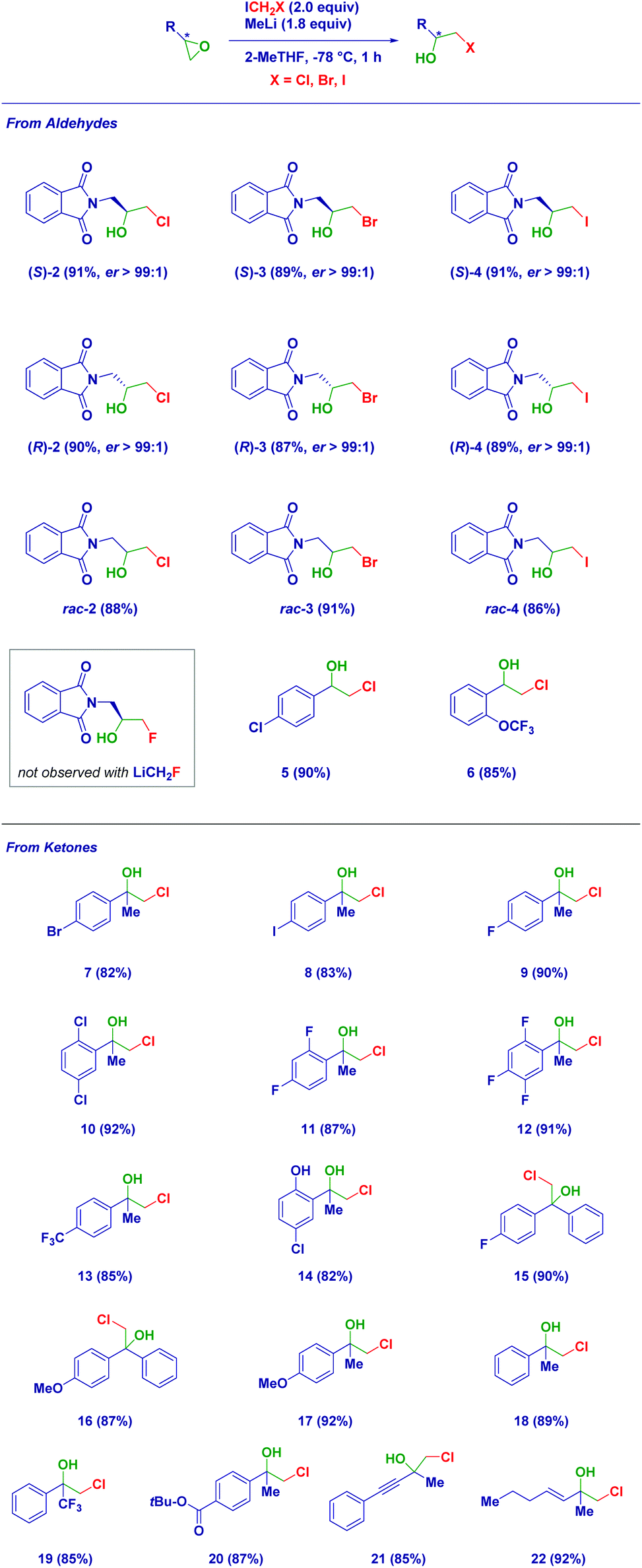 | ||
| Scheme 2 Scope of the reaction. | ||
With the aim to gather useful insights into the mechanistic aspects of the transformation, a 1:1 molar mixture of epoxide rac-1 and an aromatic aldehyde was reacted with LiCH2Cl (1.8 equiv.) in 2-MeTHF (Scheme 3a). Two halohydrins were obtained upon the attack of the carbenoid on the aldehyde (major compound, 5) and the attack of the degradation product (LiCl) on the epoxide (minor compound, rac-2). This fact can be rationalized taking into account the higher electrophilicity of the carbonyl group compared to that of the epoxide: although not optimal for stabilizing LiCH2Cl, the carbonyl nucleophilic addition still takes place, as the predominant process, in 2-MeTHF. Concomitantly, the epoxide ring-opening product was observed, though in a lesser yield (21% yield of rac-2), promoting itself the degradation of the carbenoid. Notably, when only 1 equiv. of LiCH2Cl was used, no significant differences in the ratio of rac-2 and 5 were observed, probably indicating that, due to its short life, the carbenoid reacted (again) with the more activated substrate (aldehyde) before the decomposition took place, so that the degradation product (LiCl) attacked the epoxide. However, when the reaction was carried out in the carbenoid stabilizing THF, the attack of 1 equiv. of LiCH2Cl on the aldehyde was a practically uniquely occurring phenomenon, since no significant Kirmse's elimination took place. Finally, as an additional proof of the formation of LiX, we carried out an experiment using the labelled carbenoid LiCD2I (formed upon treatment of CD2I2 with MeLi),6d observing the iodohydrin rac-4, as the sole product (Scheme 3b). This could be explained considering that Kirmse's elimination of LiCD2I provided CD2 (unreactive) and LiI, responsible for the oxirane opening.
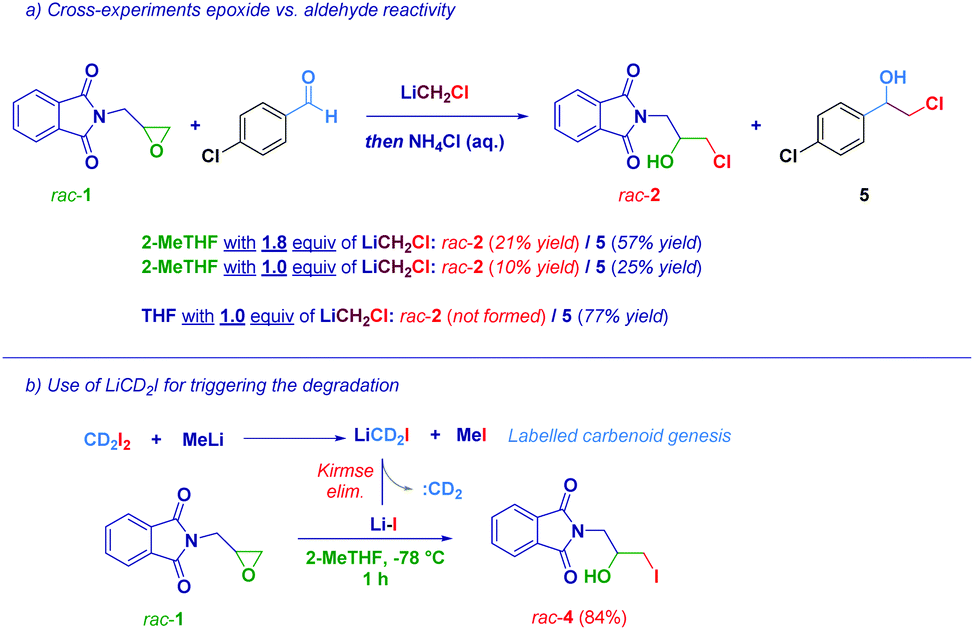 | ||
| Scheme 3 Additional mechanistic proof. | ||
In summary, we have developed a straightforward preparation of different β-halohydrins (chloro, bromo, and iodo) through boosted Kirmse's elimination of the corresponding lithium monohalocarbenoids starting from epoxide. The degradative process – usually conceived as problematic in canonical homologation chemistry – is herein implemented in the eco-friendly and non-coordinating solvent 2-MeTHF. Accordingly, the controlled formation of LiX salts is triggered, leading ultimately to the ring-opening of the epoxides. The uniformly high-yield, the full preservation of the embodied stereochemical information and the high chemocontrol – deduced by selectively preparing variously decorated motifs – further document the potential of this operationally simple and intuitive methodology.
Experimental part
General procedure 1
To a cooled (−78 °C) solution of the suitable epoxide (1.0 equiv.) in dry 2-MeTHF was added iodochloromethane (2.0 equiv.). After 2 min, an ethereal solution of MeLi (1.8 equiv., 1.6 M) was added dropwise, using a syringe pump (flow: 0.200 mL min−1). The resulting solution was stirred for one hour at −78 °C. A saturated solution of NH4Cl was added (2 mL mmol−1 substrate), and then extracted with Et2O (2 × 5 mL) and washed with water (5 mL) and brine (10 mL). The organic phase was dried over anhydrous Na2SO4, filtered and, after removal of the solvent under reduced pressure, the so-obtained crude mixture was subjected to chromatography on silica gel to afford pure compounds.:50 v/v, n-hexane/diethyl ether). 1H NMR (400 MHz, CDCl3) δ: 7.81 (m, 2H, Phthal H-4,7), 7.70 (m, 2H, Phthal H-5,6), 4.16 (brs, 1H, C![[H with combining low line]](https://www.rsc.org/images/entities/char_0048_0332.gif) OH), 3.91 (dd, J = 14.3, 7.4 Hz, 1H, NCH2), 3.83 (dd, J = 14.3, 4.4 Hz, 1H, NCH2), 3.64 (dd, J = 11.5, 4.7 Hz, 1H, CH2Cl), 3.59 (dd, J = 11.5, 5.5 Hz, 1H, CH2Cl), 3.18 (brs, 1H, OH). 13C NMR (100 MHz, CDCl3) δ: 168.6 (Phthal C-1,3), 134.2 (Phthal C-5,6), 131.7 (Phthal C-3a,7a), 123.4 (Phthal C-4,7), 69.5 (CHOH), 47.2 (CH2Cl) 41.5 (NCH2). HRMS (ESI), m/z: calcd for C11H11ClNO3+: 240.0422 [M + H]+; found: 240.0426.
:10 v/v, n-hexane/diethyl ether). 1H NMR (400 MHz, CDCl3) δ: 7.70 (m, 2H, Ph H-3,5), 7.21 (m, 2H, Ph H-2,6), 3.79 (A-part of an AB-system, 2JAB = 11.2 Hz, 1H, CH2Cl), 3.73 (B-part of an AB-system, 2JAB = 11.2 Hz, 1H, CH2Cl), 2.60 (brs, 1H, OH), 1.60 (s, 3H, CH3). 13C NMR (100 MHz, CDCl3) δ: 144.0 (Ph C-1), 137.5 (Ph C-3,5), 127.1 (Ph C-2,6), 93.2 (Ph C-4), 73.7 (COH), 55.0 (CH2Cl), 27.3 (CH3). HRMS (ESI), m/z: calcd for C9H11ClIO+: 296.9538 [M + H]+; found: 296.9542.
:10 v/v, n-hexane/diethyl ether). 1H NMR (400 MHz, CDCl3) δ: 7.52 (ddd, J = 11.5, 9.0, 7.3 Hz, 1H, Ph H-6), 6.91 (m, 1H, Ph H-3), 4.01 (d, J = 11.2 Hz, 1H, CH2Cl), 3.86 (dd, J = 11.2, 1.1 Hz, 1H, CH2Cl), 2.79 (brs, 1H, OH), 1.64 (d, J = 1.2 Hz, 3H, CH3). 13C NMR (100 MHz, CDCl3) δ: 153.8 (ddd, J = 244.1, 9.3, 2.9 Hz, Ph C), 149.3 (ddd, J = 251.5, 14.6, 12.8 Hz, Ph C), 146.8 (ddd, J = 244.6, 12.0, 3.4 Hz, Ph C), 127.9 (dt, Jd = 15.0 Hz, Jt = 4.4 Hz, Ph C-1), 116.4 (ddd, J = 21.4, 5.9, 1.3 Hz, Ph C-6), 106.4 (dd, J = 29.9, 21.0 Hz, Ph C-3), 72.6 (d, J = 4.7 Hz, COH), 53.4 (d, J = 6.5 Hz, CH2Cl), 25.8 (d, J = 3.5 Hz, CH3). 19F NMR (376 MHz, CDCl3) δ: −141.9 (m, F), −134.7 (m, F), −115.7 (m, F). HRMS (ESI), m/z: calcd for C9H9ClF3O+: 225.0289 [M + H]+; found: 225.0286.
:10 v/v, n-hexane/diethyl ether). 1H NMR (400 MHz, CDCl3) δ: 7.43 (m, 2H, Ph2 H-2,6), 7.42 (m, 2H, Ph1 H-2,6), 7.36 (m, 2H, Ph2 H-3,5), 7.30 (m, 1H, Ph2 H-4), 7.03 (m, 2H, Ph1 H-3,5), 4.18 (A-part of an AB-system, 2JAB = 11.7 Hz, 1H, CH2Cl), 4.16 (B-part of an AB-system, 2JAB = 11.7 Hz, 1H, CH2Cl), 3.17 (brs, 1H, OH). 13C NMR (100 MHz, CDCl3) δ: 162.2 (d, J = 246.9 Hz, Ph1 C-4), 143.0 (Ph2 C-1), 139.1 (d, J = 3.2 Hz, Ph1 C-1), 128.4 (Ph2 C-3,5), 128.3 (d, J = 8.2 Hz, Ph1 C-2,6), 127.9 (Ph2 C-4), 126.3 (Ph2 C-2,6), 115.2 (d, J = 21.4 Hz, Ph1 C-3,5), 77.5 (COH), 53.1 (CH2Cl). 19F NMR (376 MHz, CDCl3) δ: −114.7 (m, F). HRMS (ESI), m/z: calcd for C14H13ClFO+: 251.0633 [M + H]+; found: 251.0659.
OH), 3.91 (dd, J = 14.3, 7.4 Hz, 1H, NCH2), 3.83 (dd, J = 14.3, 4.4 Hz, 1H, NCH2), 3.64 (dd, J = 11.5, 4.7 Hz, 1H, CH2Cl), 3.59 (dd, J = 11.5, 5.5 Hz, 1H, CH2Cl), 3.18 (brs, 1H, OH). 13C NMR (100 MHz, CDCl3) δ: 168.6 (Phthal C-1,3), 134.2 (Phthal C-5,6), 131.7 (Phthal C-3a,7a), 123.4 (Phthal C-4,7), 69.5 (CHOH), 47.2 (CH2Cl) 41.5 (NCH2). HRMS (ESI), m/z: calcd for C11H11ClNO3+: 240.0422 [M + H]+; found: 240.0426.
:10 v/v, n-hexane/diethyl ether). 1H NMR (400 MHz, CDCl3) δ: 7.70 (m, 2H, Ph H-3,5), 7.21 (m, 2H, Ph H-2,6), 3.79 (A-part of an AB-system, 2JAB = 11.2 Hz, 1H, CH2Cl), 3.73 (B-part of an AB-system, 2JAB = 11.2 Hz, 1H, CH2Cl), 2.60 (brs, 1H, OH), 1.60 (s, 3H, CH3). 13C NMR (100 MHz, CDCl3) δ: 144.0 (Ph C-1), 137.5 (Ph C-3,5), 127.1 (Ph C-2,6), 93.2 (Ph C-4), 73.7 (COH), 55.0 (CH2Cl), 27.3 (CH3). HRMS (ESI), m/z: calcd for C9H11ClIO+: 296.9538 [M + H]+; found: 296.9542.
:10 v/v, n-hexane/diethyl ether). 1H NMR (400 MHz, CDCl3) δ: 7.52 (ddd, J = 11.5, 9.0, 7.3 Hz, 1H, Ph H-6), 6.91 (m, 1H, Ph H-3), 4.01 (d, J = 11.2 Hz, 1H, CH2Cl), 3.86 (dd, J = 11.2, 1.1 Hz, 1H, CH2Cl), 2.79 (brs, 1H, OH), 1.64 (d, J = 1.2 Hz, 3H, CH3). 13C NMR (100 MHz, CDCl3) δ: 153.8 (ddd, J = 244.1, 9.3, 2.9 Hz, Ph C), 149.3 (ddd, J = 251.5, 14.6, 12.8 Hz, Ph C), 146.8 (ddd, J = 244.6, 12.0, 3.4 Hz, Ph C), 127.9 (dt, Jd = 15.0 Hz, Jt = 4.4 Hz, Ph C-1), 116.4 (ddd, J = 21.4, 5.9, 1.3 Hz, Ph C-6), 106.4 (dd, J = 29.9, 21.0 Hz, Ph C-3), 72.6 (d, J = 4.7 Hz, COH), 53.4 (d, J = 6.5 Hz, CH2Cl), 25.8 (d, J = 3.5 Hz, CH3). 19F NMR (376 MHz, CDCl3) δ: −141.9 (m, F), −134.7 (m, F), −115.7 (m, F). HRMS (ESI), m/z: calcd for C9H9ClF3O+: 225.0289 [M + H]+; found: 225.0286.
:10 v/v, n-hexane/diethyl ether). 1H NMR (400 MHz, CDCl3) δ: 7.43 (m, 2H, Ph2 H-2,6), 7.42 (m, 2H, Ph1 H-2,6), 7.36 (m, 2H, Ph2 H-3,5), 7.30 (m, 1H, Ph2 H-4), 7.03 (m, 2H, Ph1 H-3,5), 4.18 (A-part of an AB-system, 2JAB = 11.7 Hz, 1H, CH2Cl), 4.16 (B-part of an AB-system, 2JAB = 11.7 Hz, 1H, CH2Cl), 3.17 (brs, 1H, OH). 13C NMR (100 MHz, CDCl3) δ: 162.2 (d, J = 246.9 Hz, Ph1 C-4), 143.0 (Ph2 C-1), 139.1 (d, J = 3.2 Hz, Ph1 C-1), 128.4 (Ph2 C-3,5), 128.3 (d, J = 8.2 Hz, Ph1 C-2,6), 127.9 (Ph2 C-4), 126.3 (Ph2 C-2,6), 115.2 (d, J = 21.4 Hz, Ph1 C-3,5), 77.5 (COH), 53.1 (CH2Cl). 19F NMR (376 MHz, CDCl3) δ: −114.7 (m, F). HRMS (ESI), m/z: calcd for C14H13ClFO+: 251.0633 [M + H]+; found: 251.0659.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
L. I. and M. M. contributed equally to the work. We thank the University of Vienna, the University of Turin, the Complutense University of Madrid and Fondazione RiMed (Palermo, Italy) for generous support. M. M. acknowledges OEAD for a Blau grant. V. Pillari thanks the University of Vienna for a Uni:docs fellowship. S. M. thanks the University of Messina for a visiting pre-doctoral grant.Notes and references
- For reviews on carbenoid chemistry, see: (a) M. Braun, in The Chemistry of Organolithium Compounds, ed. Z. Rappoport and I. Marek, John Wiley and Sons, Chichester, 2004, vol. 1, pp. 829 Search PubMed; (b) V. H. Gessner, Chem. Commun., 2016, 52, 12011 RSC; (c) V. Capriati and S. Florio, Chem. – Eur. J., 2010, 16, 4152 CrossRef CAS; (d) V. Pace, Aust. J. Chem., 2014, 67, 311 CrossRef CAS; (e) V. Pace, W. Holzer and N. De Kimpe, Chem. Rec., 2016, 16, 2061 CrossRef CAS.
- (a) L. Castoldi, S. Monticelli, R. Senatore, L. Ielo and V. Pace, Chem. Commun., 2018, 54, 6692 RSC; (b) L. Ielo, V. Pillari, M. Miele, D. Castiglione and V. Pace, Synlett, 2020 DOI:10.1055/s; (c) V. Pace, L. Castoldi, S. Monticelli, M. Rui and S. Collina, Synlett, 2017, 879 CrossRef CAS.
- For stability and degradative studies on carbenoids, see: (a) W. Kirmse, Angew. Chem., Int. Ed. Engl., 1965, 4, 1 CrossRef; (b) S. Molitor and V. H. Gessner, Angew. Chem., Int. Ed., 2016, 55, 7712 CrossRef CAS; (c) S. Molitor and V. H. Gessner, Chem. – Eur. J., 2013, 19, 11858 CrossRef CAS; (d) C. Kupper, S. Molitor and V. H. Gessner, Organometallics, 2014, 33, 347 CrossRef CAS; (e) S. Molitor and V. H. Gessner, Synlett, 2015, 861 CAS; (f) F. M. Bickelhaupt, H. L. Hermann and G. Boche, Angew. Chem., Int. Ed., 2006, 45, 823 CrossRef CAS.
- S. Monticelli, M. Rui, L. Castoldi, G. Missere and V. Pace, Monatsh. Chem., 2018, 149, 1285 CrossRef CAS.
- R. Tarhouni, B. Kirschleger, M. Rambaud and J. Villieras, Tetrahedron Lett., 1984, 25, 835 CrossRef CAS.
- For recent work with carbenoids from our group, see: (a) S. Touqeer, R. Senatore, M. Malik, E. Urban and V. Pace, Adv. Synth. Catal., 2020, 362, 5056 CrossRef CAS; (b) L. Ielo, L. Castoldi, S. Touqeer, J. Lombino, A. Roller, C. Prandi, W. Holzer and V. Pace, Angew. Chem., 2020, 59, 20852 CrossRef CAS; (c) L. Ielo, S. Touqeer, A. Roller, T. Langer, W. Holzer and V. Pace, Angew. Chem., Int. Ed., 2019, 58, 2479 CrossRef CAS; (d) V. Pace, L. Castoldi, E. Mazzeo, M. Rui, T. Langer and W. Holzer, Angew. Chem., Int. Ed., 2017, 56, 12677 CrossRef CAS.
- For illuminating examples, see: (a) D. S. Matteson, J. Org. Chem., 2013, 78, 10009 CrossRef CAS; (b) D. S. Matteson and D. Majumdar, J. Am. Chem. Soc., 1980, 102, 7588 CrossRef CAS; (c) S. Balieu, G. E. Hallett, M. Burns, T. Bootwicha, J. Studley and V. K. Aggarwal, J. Am. Chem. Soc., 2015, 137, 4398 CrossRef CAS; (d) G. Casoni, M. Kucukdisli, J. M. Fordham, M. Burns, E. L. Myers and V. K. Aggarwal, J. Am. Chem. Soc., 2017, 139, 11877 CrossRef CAS; (e) J. Wu, P. Lorenzo, S. Zhong, M. Ali, C. P. Butts, E. L. Myers and V. K. Aggarwal, Nature, 2017, 547, 436 CrossRef CAS; (f) A. Fawcett, A. Murtaza, C. H. U. Gregson and V. K. Aggarwal, J. Am. Chem. Soc., 2019, 141, 4573 CrossRef CAS.
- (a) S. Touqeer, L. Castoldi, T. Langer, W. Holzer and V. Pace, Chem. Commun., 2018, 54, 10112 RSC For analogous operations leading to stable a-selenomethyl halides, see: (b) R. Senatore, M. Malik, E. Urban, W. Holzer and V. Pace, Tetrahedron, 2021 DOI:10.1016/j.tet.2021.131921.
- R. Huisgen and U. Burger, Tetrahedron Lett., 1970, 11, 3053 CrossRef.
- R. C. Hahn and J. Tompkins, Tetrahedron Lett., 1990, 31, 937 CrossRef CAS.
- M. Miele, A. Citarella, T. Langer, E. Urban, M. Zehl, W. Holzer, L. Ielo and V. Pace, Org. Lett., 2020, 22, 7629 CrossRef CAS.
- For reviews and recent selected applications of 2-MeTHF in synthesis, see: (a) V. Pace, P. Hoyos, L. Castoldi, P. Domínguez de María and A. R. Alcántara, ChemSusChem, 2012, 5, 1369 CrossRef CAS; (b) V. Pace, Aust. J. Chem., 2012, 65, 301 CrossRef CAS; (c) D. F. Aycock, Org. Process Res. Dev., 2007, 11, 156 CrossRef CAS; (d) P. Lei, Y. Ling, J. An, S. P. Nolan and M. Szostak, Adv. Synth. Catal., 2019, 361, 5654 CrossRef CAS; (e) E. Bisz and M. Szostak, ChemSusChem, 2018, 11, 1290 CrossRef CAS; (f) F. F. Mulks, L. J. Bole, L. Davin, A. Hernán-Gómez, A. Kennedy, J. García-Álvarez and E. Hevia, Angew. Chem., 2020, 59, 19021 CrossRef CAS; (g) M. Fairley, L. J. Bole, F. F. Mulks, L. Main, A. R. Kennedy, C. T. O'Hara, J. García-Alvarez and E. Hevia, Chem. Sci., 2020, 11, 6500 RSC; (h) V. Pace, P. Hoyos, M. Fernández, J. V. Sinisterra and A. R. Alcántara, Green Chem., 2010, 12, 1380 RSC; (i) V. Pace, L. Castoldi, P. Hoyos, J. V. Sinisterra, M. Pregnolato and J. M. Sánchez-Montero, Tetrahedron, 2011, 67, 2670 CrossRef CAS; (j) V. Pace, P. Hoyos, A. R. Alcántara and W. Holzer, ChemSusChem, 2013, 6, 905 CrossRef CAS; (k) L. Castoldi, L. Ielo, P. Hoyos, M. J. Hernáiz, L. De Luca, A. R. Alcántara, W. Holzer and V. Pace, Tetrahedron, 2018, 74, 2211 CrossRef CAS.
- (a) A. R. Alcántara and P. Dominguez de Maria, Curr. Green Chem., 2018, 5, 85 Search PubMed; (b) A. Pellis, F. P. Byrne, J. Sherwood, M. Vastano, J. W. Comerford and T. J. Farmer, Green Chem., 2019, 21, 1686 RSC; (c) G. Englezou, K. Kortsen, A. A. C. Pacheco, R. Cavanagh, J. C. Lentz, E. Krumins, C. Sanders-Velez, S. M. Howdle, A. J. Nedoma and V. Taresco, J. Polym. Sci., 2020, 58, 1571 CrossRef CAS.
- (a) M. Pineschi, Eur. J. Org. Chem., 2006, 4979 CrossRef CAS; (b) A. K. Yudin, Aziridines and Epoxides in Organic Synthesis, Wiley-VCH, Weinheim, 2006 CrossRef.
- For recent works from our group with carbenoid-like reagents, see: (a) M. Miele, R. D'Orsi, V. Sridharan, W. Holzer and V. Pace, Chem. Commun., 2019, 55, 12960 RSC; (b) V. Pace, L. Castoldi and W. Holzer, Chem. Commun., 2013, 49, 8383 RSC; (c) V. Pace, I. Murgia, S. Westermayer, T. Langer and W. Holzer, Chem. Commun., 2016, 52, 7584 RSC; (d) V. Pace, L. Castoldi, S. Monticelli, S. Safranek, A. Roller, T. Langer and W. Holzer, Chem. – Eur. J., 2015, 21, 18966 CrossRef CAS; (e) S. Monticelli, W. Holzer, T. Langer, A. Roller, B. Olofsson and V. Pace, ChemSusChem, 2019, 12, 1147 CrossRef CAS; (f) R. Senatore, L. Ielo, E. Urban, W. Holzer and V. Pace, Eur. J. Org. Chem., 2018, 2466 CrossRef CAS; (g) K. de la Vega-Hernández, R. Senatore, M. Miele, E. Urban, W. Holzer and V. Pace, Org. Biomol. Chem., 2019, 17, 1970 RSC; (h) V. Pace, S. Monticelli, K. de la Vega-Hernández and L. Castoldi, Org. Biomol. Chem., 2016, 14, 7848 RSC; (i) V. Pace, K. de la Vega-Hernández, E. Urban and T. Langer, Org. Lett., 2016, 18, 2750 CrossRef CAS; (j) R. Senatore, L. Castoldi, L. Ielo, W. Holzer and V. Pace, Org. Lett., 2018, 20, 2685 CrossRef CAS; (k) M. Miele, A. Citarella, N. Micale, W. Holzer and V. Pace, Org. Lett., 2019, 21, 8261 CrossRef CAS.
- LiBr has been used for the epoxide ring-opening; see: (a) S. R. Asit, K. Chakraborti and A. Kondaskar, Eur. J. Org. Chem., 2004, 3597 Search PubMed. Previous studies dealing with epoxide ring opening conducted with lithium halides documented non-optimal regiocontrol, see for example: (b) J. S. Bajwa and R. C. Anderson, Tetrahedron Lett., 1991, 32, 3021 CrossRef CAS (LiX-AcOH, up to 24 h reaction time); (c) M. A. Reddy, K. Surendra, N. Bhanumathi and K. R. Rao, Tetrahedron, 2002, 58, 6003 CrossRef CAS (LiX -β-cyclodextrin in water).
- U. Azzena, M. Carraro, L. Pisano, S. Monticelli, R. Bartolotta and V. Pace, ChemSusChem, 2019, 12, 40 CrossRef CAS.
- For the use of LiCH2F and related fluorinated reagents, see: (a) G. Parisi, M. Colella, S. Monticelli, G. Romanazzi, W. Holzer, T. Langer, L. Degennaro, V. Pace and R. Luisi, J. Am. Chem. Soc., 2017, 139, 13648 CrossRef CAS; (b) M. Colella, A. Tota, A. Großjohann, C. Carlucci, A. Aramini, N. S. Sheikh, L. Degennaro and R. Luisi, Chem. Commun., 2019, 55, 8430 RSC; (c) S. Monticelli, M. Colella, V. Pillari, A. Tota, T. Langer, W. Holzer, L. Degennaro, R. Luisi and V. Pace, Org. Lett., 2019, 21, 584 CrossRef CAS; (d) M. Colella, A. Tota, Y. Takahashi, R. Higuma, S. Ishikawa, L. Degennaro, R. Luisi and A. Nagaki, Angew. Chem., 2020, 59, 10924 CrossRef CAS.
- The ring opening of epoxides with fluorides is a particularly challenging reaction requiring drastic conditions. For an example, see: V. Pace and W. Holzer, Tetrahedron Lett., 2012, 53, 5106 CrossRef CAS.
- For cyclopropanation-type processes conducted with carbenoid reagents, see for example: (a) M. J. Duran-Pena, M. E. Flores-Giubi, J. M. Botubol-Ares, L. M. Harwood, I. G. Collado, A. J. Macias-Sanchez and R. Hernandez-Galan, Org. Biomol. Chem., 2016, 14, 2731 RSC; (b) Z. Ke, Y. Zhou, H. Gao, C. Zhao and D. L. Phillips, Chem. – Eur. J., 2007, 13, 6724 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ob02407d |
| This journal is © The Royal Society of Chemistry 2021 |