
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSelf-assembly of copper nanoclusters: isomeric ligand effect on morphological evolution†
Sarita
Kolay
a,
Subarna
Maity
 a,
Dipankar
Bain
a,
Sikta
Chakraborty
a and
Amitava
Patra
*ab
a,
Dipankar
Bain
a,
Sikta
Chakraborty
a and
Amitava
Patra
*ab
aSchool of Materials Sciences, Indian Association for the Cultivation of Science, Jadavpur, Kolkata-700032, India. E-mail: msap@iacs.res.in; Fax: +91-33-2473-2805; Tel: +91-33-2473-4971
bInstitute of Nano Science and Technology, Knowledge City, Sector 81, Mohali 140306, India
First published on 9th August 2021
Abstract
Tailoring the hierarchical self-assembly of metal nanoclusters (NCs) is an emergent area of research owing to their precise structure and flexible surface environment. Herein, the morphological evolution from rods to platelets to ribbon-like structures through self-assembly of Cu7 NCs is dictated by the positional isomerism of the surface capping ligand, dimethylbenzenethiol (DMBT). Besides cuprophilic interaction, the interplay between π–π stacking and agostic interaction (Cu⋯H–C) directs the inter-NC organization into different ordered architectures. The excited-state relaxation dynamics of the red phosphorescent assembled structures has been correlated with their compactness and the degree of bonding interactions present.
Self-assembly is a recognized process associated with the spontaneous organization of components into ordered structures or aggregates.1–5 Notable progress in synthetic chemistry has allowed rapid growth in the self-assembly of organic molecules, metal nanoparticles, and ultrasmall nanomaterials (<2 nm).6–10 Metal NCs are promising candidates as the building blocks of assembled architectures to control the morphology by manipulating the driving forces which eventually control their properties.11–13 Although isolated metal NCs often exhibit lower quantum yield (QY), the self-assembly of NCs leads to bright and tunable luminescence properties.14 Several strategies have been used to design various assembled structures by tailoring the capping ligands and regulating temperature, pH, light, solvent, etc.15–19 Zhang et al. have designed self-assembled ribbons with tunable emission by changing the electron-donating groups of the capping ligand.20 It is reported that the solubility of ligands in different solvents controls the morphology of self-assembled structures. The nature of solvent influences the changes of the inter-cluster distance of assembly, which dictates the assembly structure.18,21 Molecular self-assembly involves non-covalent or weak covalent interactions such as van der Waals, electrostatic and hydrophobic interactions, and hydrogen and coordination bonds.22–24 The C–H⋯π interaction,5 aurophilic,25 argentophilic,26 and cuprophilic14 interactions are responsible for assembling metal NCs. Thus, the mechanism of formation of the self-assembled structures of metal NCs is not trivial, and a deep understanding is essential to control their properties. The formation of assembled structures is directed by the electronic charge and the functional groups of the ligands.20 The bulkiness and position of the substituents on the ligands may be decisive parameters for the self-assembly of metal NCs.
Photoluminescence (PL) of atomically precise metal NCs is the prime property required for optoelectronic and biomedical applications, and is governed by the metal-core and ligand shell.27–31 The assembly-induced emission enables them to be applied as phosphors to fabricate LEDs generated from strong metallophilic interaction depending on their packing. The relaxation dynamics of the excited state of the NCs is controlled by the ligand–ligand, ligand–metal, and metal–metal interactions.32–34 The restriction of the rotation and vibration of the ligands also influences the rate of non-radiative relaxation of the excited state.35–38
Herein, we design inter-NC assembly into ordered structures by the precise control of the surface protecting ligand. Although the compositions of the Cu NCs are identical, only the position of a –CH3 group in the DMBT ligand regulates the morphology of the assembled structures. The interplay of cuprophilic, π–π stacking and agostic interactions leads to the organization of Cu7 NCs into rods, platelets, and ribbon-like assemblies. A higher degree of bonding interaction provides a longer excited-state lifetime because of the rigidification of the surface ligands.
The one-pot synthesis procedure involves reductive decomposition of the Cu-thiolate complex to form self-assembled Cu NCs (for details, see the ESI†). In a MeOH/DCM mixed solvent, CuNO3·3H2O is dissolved and reacted with three isomers of DMBT in each synthesis process. The color of the reaction medium changes from sky blue to yellow ochre due to the complexation of Cu2+ with 2,4-DMBT. However, the complex formation with 2,5-DMBT and 2,6-DMBT leads to light yellow and curdy white suspensions. A secondary phosphine ligand (PPh4Br) is added, followed by the reduction with NaBH4. For the 2,4-isomer, we obtained a bright yellow-colored solution under ice-cold conditions after 5 hours. The same method provided a light yellow suspension for 2,5-DMBT capped self-assembled NCs, while the 2,6-DMBT capped NC assembly gave a muddy white suspension (Fig. S1†). Fig. 1A–C show the structures of isomeric 2,4-, 2,5- and 2,6-DMBT ligands. The extent of steric hindrance due to the two –CH3 groups adjacent to –SH in the 2,6-isomer is more significant than that of 2,4- and 2,5-isomers. The isomeric ligands may affect the morphology and optical properties of self-assembled structures. The formation of rod-like structures in the presence of 2,4-DMBT is seen clearly in Fig. 1D with an average length 66 ± 2.1 nm, whereas the width is 15 ± 1.1 nm (Fig. 1E).
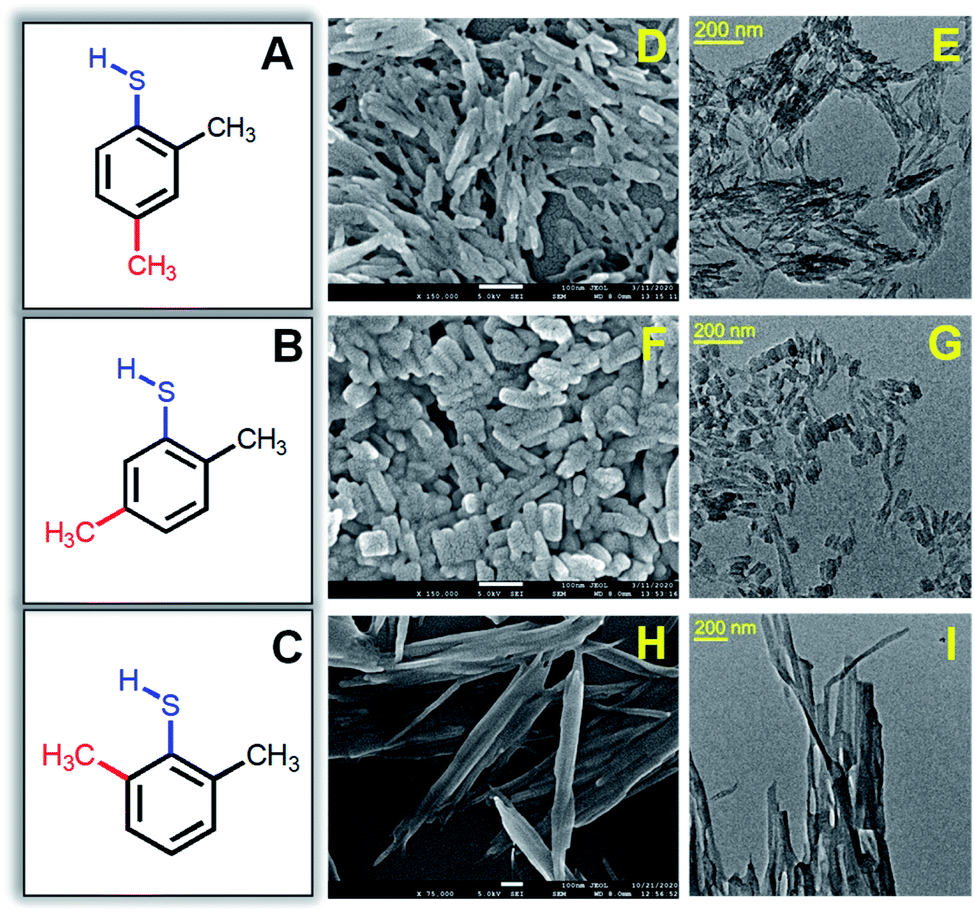 | ||
| Fig. 1 Structures of (A) 2,4- (B) 2,5- and (C) 2,6-DMBT ligands. FE-SEM and TEM images of rods (D and E), platelets (F and G) and ribbon-like (H and I) self-assemblies, respectively. The scale bar of the SEM images is 100 nm. | ||
The morphology of the assembled structures changes with changing the methyl position from fourth to fifth in 2,5-DMBT (Fig. 1F). The length is found to be 46 ± 2.5 nm which is lower than that of the assembled rods, while the width increases from 15 nm to 25 ± 1.8 nm (Fig. 1G). Thus, the change in the aspect ratio of the self-assemblies arises from the positional isomerism of the DMBT ligand. A ribbon-like morphology is found in the presence of the 2,6-DMBT ligand where the length is more than 1 μm and the width is 31 ± 2.5 nm (Fig. 1H and I). The hierarchical self-assembled structures are composed of ∼1 nm Cu NCs which is evident from the high-resolution TEM images in Fig. S2.† An even arrangement of the ribbon-like assemblies with lengths of several micrometers is observed (Fig. S2C†). Energy-dispersive X-ray (EDX) element mapping shows an even distribution of Cu and S across the self-assembled structures (Fig. S3†). The thickness of the assembled structures is measured using atomic force microscopy (AFM), and the heights are 34 nm, 16.5 nm, and 25 nm for rods, platelets, and ribbon-like assemblies, respectively (Fig. S4†). Compositional and structural analyses were done to get insights into the mechanism.
Thermogravimetric analysis (TGA) reveals a mass loss of 65% in each case due to the decomposition of organic ligands as gaseous products, where the two –CH3 groups are the most labile to dissociate first (Fig. S5†). The dissociation of the two –CH3 groups happens at <260 °C, which leads to a 14% mass loss. In the next stage, the phenyl rings are likely to decompose within 260–320 °C resulting in 35.5% loss of mass. In the last step, 15% mass loss is observed as the S atoms of the ligand dissociate, which are covalently coordinated with Cu. The firmly bound S atoms dissociate at >320 °C. It is noted that the matrix-assisted laser desorption ionization (MALDI) mass spectrometric study confirmed the formation of Cu7L6 for all three cases (Fig. 2A). The mass loss corresponds to a Cu/DMBT molar ratio of 1.162/1. In MALDI mass spectra, a small peak at m/z = 1267 is identified as a molecular ion peak along with a few peaks at m/z 1090, 1076, and 1012 due to fragmentation. Based on MALDI-MS and TGA analyses, the structure of the NCs is assigned as Cu7(DMBT)6. The peak at m/z = 1090 is ascribed to Cu6(DMBT)5 + Na+, which arises from the dissociation of Cu-DMBT from Cu7(DMBT)6. The peak at m/z = 1076 Da is assigned to [Cu7(DMBT)4 + Br− + 2H+], which arises from the dissociation of two DMBT ligands. Again, the most intense peak at m/z = 1012 Da is ascribed to [Cu6(DMBT)4 + Br− + 2H+], which arises due to the dissociation of Cu(DMBT)2 from Cu7(DMBT)6. As the composition of the building blocks of all three assembled structures is the same (Cu7L6), the compositional effect on the morphology is eliminated. Therefore, the formation of self-assemblies is a surface phenomenon that arises due to inter-NC bonding interactions. The appearance of a covalent Cu–S bond is confirmed by FTIR spectroscopy. The S–H vibration in free DMBT arises at 2525 cm−1, which then disappears after self-assembly of Cu NCs. The absence of S–H stretching frequency confirms the Cu–S covalent bond formation between Cu and DMBT (Fig. S6†).
 | ||
| Fig. 2 (A) Positive mode MALDI-TOF mass spectra, (B) XPS spectra for S 2p and (C) powder X-ray diffraction patterns of self-assembled (a) rods, (b) platelets, and (c) ribbons. | ||
To assemble into the ordered structures, each individual Cu NC must experience inter-NC non-covalent interactions. The DMBT ligands do not have any electron-donating or withdrawing group with active conjugation. So, the key driving force may be metallophilic interaction as well as π–π stacking which is governed by the nature of the ligand. XPS analysis is used to understand the Cu(I)–Cu(I) cuprophilic interaction. The peaks at 932.7 and 952.6 eV arise due to Cu 2p3/2 and 2p1/2, respectively, in self-assembled rods and platelets (Fig. S7†). The Cu peaks for self-assembled ribbons show a negligible shift to a lower energy value (932.56 and 952.4 eV). Cu(II) is not present in the core as there is no satellite peak at 942 eV.39
There is no significant change in Cu 2p binding energy in rods, platelets, and ribbon self-assemblies, indicating the presence of the same cuprophilic interaction for all cases. The peaks for S 2p3/2, and 2p1/2 appear at 162.4 and 163.6 eV, respectively, for both rods and platelets (Fig. 2B). A dramatic decrease in S 2p binding energy is evident in ribbons as the peaks show up at 162.2 and 163.3 eV. The lower binding energy of S 2p indicates more inter NC surface interaction for ribbons than for the rods and platelets. The long-range non-covalent inter-NC interactions must govern the ligand-dependent self-assembly formation. Here, the close alignment of the Cu NCs may be due to the cuprophilic interaction. The drastic difference in morphology of the self-assemblies is not attributed to the metallophilic interaction as there is no significant change in the binding energies of Cu 2p for rods and platelets.
Powder X-ray diffraction (XRD) measurements were carried out to understand the packing of NCs into the assembled structures. The Cu NCs exhibit a highly ordered arrangement within the self-assembled structures, giving intense peaks in powder XRD (Fig. 2C). The XRD pattern of rod-like assemblies shows a single peak at 6.53°, whereas three distinct diffraction peaks at 6.63°, 7.83°, and 10.5° are obtained for the platelets. On the other hand, ribbon-like assemblies exhibit one distinct peak at 7.66°. For rods and ribbon-like assembled structures, the sole peak indicates only one dominant distance between the individual NCs. The appearance of three peaks in platelets indicates three different distances among the adjacent NCs.24 Of the two peak positions for platelets, one has a close resemblance to that of rods and another to that of ribbon-like assemblies. It reveals that the platelet-like assemblies have grown along longitudinal as well as the transverse direction.
Apart from dominating cuprophilic interaction, aromatic thiol ligands experience significant π–π stacking interaction, which depends on the bulkiness of each isomer. We have observed broad XRD peaks at 2θ ∼ 25–27° arising from the π–π stacking of the self-assembled nanostructures (Fig. S8†).40,41 The cuprophilic interaction is further reinforced by π–π stacking between ligands. Previously, isomeric benzenethiols were used to synthesize various magic-sized AuNCs by controlling the interfacial Au–S bonds.42 Depending on the one –CH3 group position, the steric hindrance on the ligand is localized in one position for 2,4- and 2,6-DMBT ligands. The localized steric hindrance exerted by the methyl groups dictates the growth of self-assemblies predominantly in a single direction, leading to the formation of rods and ribbon-like assemblies.
On the other hand, two –CH3 and –SH binding sites are distributed over the aromatic ring in the 2,5-DMBT ligand. The steric hindrance-controlled π–π stacking partially explains the formation of different self-assembled structures. The difference in morphology between self-assembled rods and ribbons must be attributed to another non-covalent interaction. However, the nature of π–π stacking interaction in 2,4- and 2,6-DMBT is similar. Apart from cuprophilic and π–π stacking interaction, the Cu⋯H–C, namely agostic interaction, is the crucial factor in dictating the morphology. The pattern of ribbon-like assemblies indicates their growth along the longitudinal direction, which is controlled by the maximum extent of agostic interaction (Cu⋯H–C) on the surface. There are two kinds of M⋯H–C interactions, i.e., anagostic and agostic, depending on M⋯H distances and M–H–C angles, differentiated from 1H NMR spectroscopy.43 The Au⋯H–C anagostic interaction in o-substituted benzenethiol capped Au38 NCs imparts an effect on their stability.44 The chemical shift of agostic hydrogen atoms generally appears as an upfield shift compared to the uncoordinated hydrogen, whereas anagostic hydrogen shows a downfield shift. 1H NMR spectroscopic study is performed to understand the agostic or anagostic interaction for assembly formation. The 1H NMR spectra of DMBT exhibit broad peaks in the range of 7.19 to 6.89 ppm associated with aromatic protons.
In the 2,4-DMBT ligand, two methyl (–CH3) groups appear at 2.32 and 2.29 ppm. Upon self-assembled rod formation, the methyl protons appear at 2.36 and 2.28 ppm (Fig. S9†). The two –CH3 groups of 2,5-DMBT show up as two singlets at 2.47 and 2.44 ppm, whereas the –CH3 protons in platelets experience an upfield shift (2.37–2.25 ppm) compared to the uncoordinated ones, indicating the presence of agostic interaction (Fig. S10†). For 2,6-DMBT, the chemical environment of the two –CH3 groups is the same, leading to the appearance of a single peak at 2.38 ppm. The 1H NMR spectrum shows two different singlet peaks at 2.17 and 2.36 ppm in ribbon-like assembly formation (Fig. S11†). In self-assembled structures, the oxidation state of Cu is between 0 and 1. It creates a δ+ charge, whereas the H atom of the –CH3 group experiences a δ− charge, leading to an increase in the electron density. That causes an upfield shift of the hydrogen after coordinating with the metal. The large upfield shift of protons of one –CH3 indicates the presence of maximum agostic interaction in 2,6-DMBT capped ribbon-like NC assemblies. This result reflects in the TGA analysis (Fig. S5†). Although the total mass loss is the same for all the assemblies, the dissociation behavior is different. For the rod and platelet-like assemblies, a step-wise dissociation is observed below 260 °C, indicating different dissociation energies for the two –CH3 groups. On the other hand, the complete dissociation of the methyl groups requires a higher temperature than 260 °C, which indicates a greater extent of agostic interaction in the ribbon-like assemblies. Additionally, no steplike dissociation curve is observed, which means that the two –CH3 groups have similar dissociation energies in 2,6-DMBT capped self-assembled Cu NCs.
Fig. 3 shows a schematic illustration of self-assembly evolution from the individual Cu7(DMBT)6 into a ribbon-like assembly. In the case of the rod-like assemblies, relatively less agostic interaction occurs because the 2,4-isomer has only one ortho-substituted methyl. It favors longitudinal directional growth with a maximum length of 66 nm. One ortho-substituted thiol group is present in the 2,5-isomer also, which will not allow the self-assemblies to grow longer in the longitudinal direction. The para position of thiol is free and may form strong π–π interaction, causing the assemblies to grow in the transverse direction. A length of >1 μm for ribbons indicates maximum longitudinal growth, whereas the rod-like and plate-like assemblies show a much smaller average length of 66 nm and 46 nm, respectively. This clearly shows that the extent of agostic interaction is larger for the 2,6-isomer capped self-assembly structures as both the ortho positions are substituted, supported by the maximum upfield shift in NMR spectrum. The agostic-interaction-assisted self-assembly formation and the morphological evolution significantly affect the PL behaviour of the NCs.
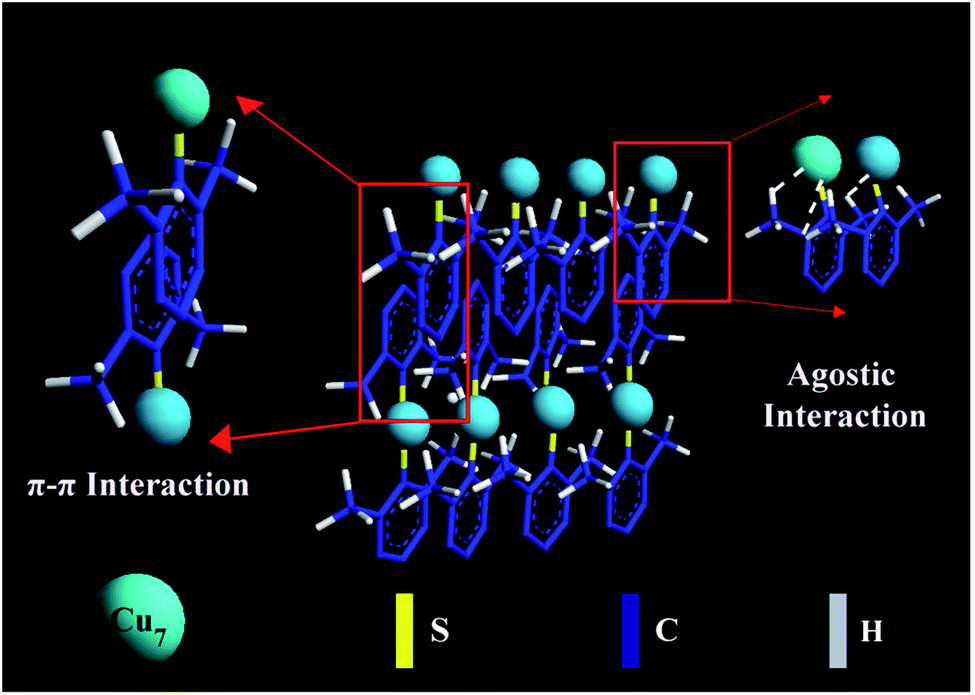 | ||
| Fig. 3 Schematic illustration of self-assembly evolution from the individual Cu7(DMBT)6 into a ribbon-like assembly. A few ligands in the Cu NCs are omitted for simplicity. | ||
The assembled or aggregated NCs exhibit intense PL in comparison to isolated NCs due to the hindrance of rotational and vibrational motions in compact structures.37,45,46 The assembled NCs showed absorption humps around 360 nm while the ribbon-like NC assemblies exhibit an additional hump at 320 nm, which is due to the electronic transition from DMBT to the Cu core (Fig. 4A). The peak around 330 nm signifies the presence of π–π stacking between the adjacent benzene rings.47 The PL excitation maxima are also in good agreement with the absorption spectra (Fig. S12†). All the assembled structures exhibit red emission in solid as well as solution states (inset of Fig. 4B). The emission maximum for the 2,4-DMBT capped NC assemblies is centered at 650 nm while it is 655 nm for platelets. The rod-like assemblies are found to have a QY of 1.1%, while it is 2.5% for the platelet-like assemblies. The ribbon-like assemblies of Cu NCs capped with 2,6-DMBT show a blue-shifted emission maximum at 640 nm and the highest QY of 7.8% (Fig. 4B). The microsecond scale lifetime and large Stokes shift (>200 nm) suggest that the emission of self-assembled NCs arises from a triplet state involving ligand to metal charge transfer (LMCT) or ligand to metal–metal charge transfer (LMMCT) from the sulfur atoms of the DMBT ligand to the Cu core.48 Platelet-like assemblies exhibit red-shifted emission from the low-lying triplet state as the π–π stacking interaction is the maximum. On the other hand, agostic interaction dominates over π–π stacking in ribbon-like assemblies. Thus the PL arises from a relatively high-lying triplet state which causes a blue-shifted emission maximum.
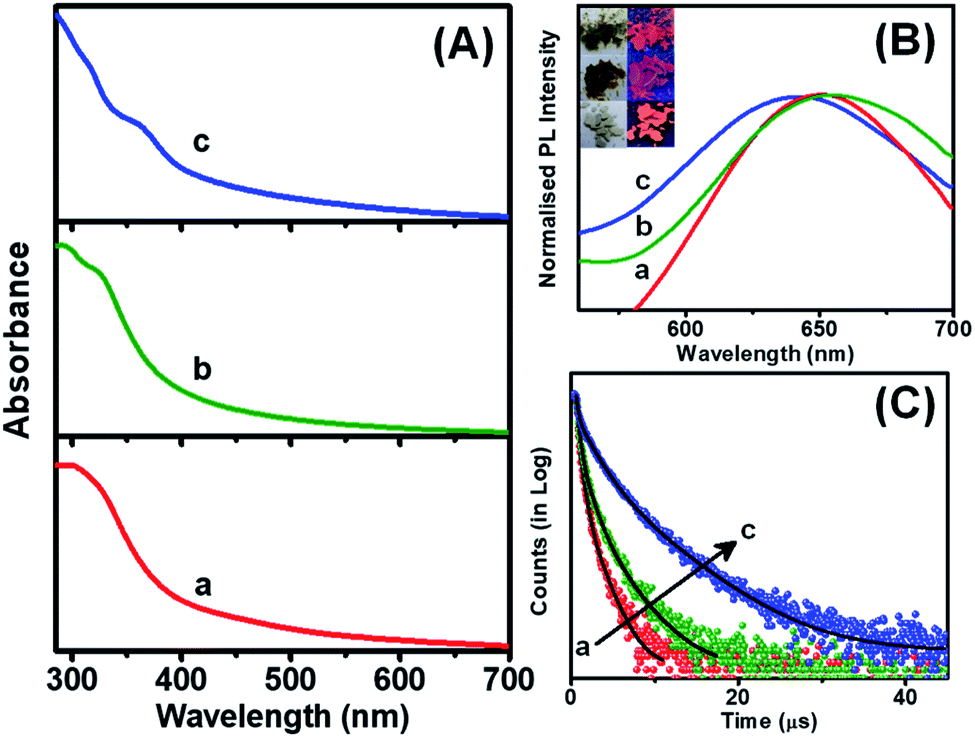 | ||
| Fig. 4 (A) UV-vis absorption, (B) PL emission spectra, and (C) PL decay curves of the self-assembled rods (a), platelets (b), and ribbons (c). The inset shows the digital photographs of self-assembled Cu NCs in daylight and under UV (365 nm) excitation. | ||
The PL decay time of each self-assembly is fitted with a tri-exponential fitting, and the average decay time varies from nanoseconds to a few microseconds (Fig. 4C). The detailed fitting parameters are summarized in Table S1 in the ESI.† The rod-like assemblies exhibit an average decay time of 0.35 μs, while the platelets show an increase in decay time to 0.65 μs. The rise in QY and average decay time for the 2,5-DMBT protected NC assemblies indicates increased radiative relaxation processes. Since the assembly of NCs forms platelets in two directions, the motion of surface ligands is suppressed in comparison with 2,4-DMBT capped rod-like assemblies. This indicates that the loose packing of NCs in rod-like assemblies makes it difficult to suppress the non-radiative relaxation process arising from the vibration and rotation of the ligands. The unidirectional assembly in rods causes a substantial motion of ligands leading to a greater extent of non-radiative processes during excited state relaxation.
On the other hand, the ribbon-like assemblies exhibit nearly one order of magnitude increase of the lifetime to 1.68 μs. Although the π–π stacking interaction is similar to that in the 2,4-DMBT capped rod-like NC assemblies, the manifestation of agostic interaction with o-CH3 groups creates the long-range assembly. Since the maximum QY is obtained for ribbon-like assemblies, it suggests that the higher order of assembly reduces the motion of ligands and rigidifies the structure, enhancing the decay time and blue shift in the emission wavelength. The significant Stokes shift and microsecond decay time suggest that phosphorescence largely depends on the inter-NC interactions. Therefore, the morphology and optical properties of self-assembled NCs can be regulated by rational tuning of the capping ligand. The design of NC-based luminescent self-assembled materials will be advantageous in the development of light-emitting phosphors and catalysis.
Conclusions
In summary, the DMBT-capped Cu7 NCs are organized into customizable superstructures from rods to platelets to ribbons. Although the cuprophilic interaction is indistinguishable, only the difference in the –CH3 position of the isomeric DMBT ligands significantly alters the extent of π–π stacking and agostic interaction. The more of o-CH3 present, the more vital the agostic interaction, which eventually culminates into the most extended ribbon-like assembly of 2,6-DMBT capped Cu7 NCs. The red emission is correlated with the compactness and effective bonding interactions in the assembled structures, leading to the most extended excited-state lifetime of ribbon-like assemblies. The insights into the bonding interactions in the self-assembled metal NCs will pave the way towards designing ordered architectures with potential applicability in optoelectronics, catalysis, and bio-imaging.Author contributions
SK contributed to the investigation, methodology, data curation, and writing of the original draft. SM helped in conceptualization and review, and editing the draft. DB contributed to editing and visualization. SC helped in data curation. AP contributed to supervision, validation, project administration, and final review.Conflicts of interest
There are no conflicts to declare.Acknowledgements
SERB-DST is gratefully acknowledged for financial support. SK acknowledges UGC, and SM thanks CSIR for awarding a fellowship. DB acknowledges INST, and SC thanks IACS for research support. We thank IACS for its support throughout our experiment.Notes and references
- G. M. Whitesides and B. Grzybowski, Science, 2002, 295, 2418 CrossRef CAS PubMed.
- K. J. M. Bishop, C. E. Wilmer, S. Soh and B. A. Grzybowski, Small, 2009, 5, 1600–1630 CrossRef CAS PubMed.
- D. Luo, C. Yan and T. Wang, Small, 2015, 11, 5984–6008 CrossRef CAS PubMed.
- M. Grzelczak, J. Vermant, E. M. Furst and L. M. Liz-Marzán, ACS Nano, 2010, 4, 3591–3605 CrossRef CAS PubMed.
- C. Zeng, Y. Chen, K. Kirschbaum, K. J. Lambright and R. Jin, Science, 2016, 354, 1580 CrossRef CAS PubMed.
- Z. Xie, P. Sun, Z. Wang, H. Li, L. Yu, D. Sun, M. Chen, Y. Bi, X. Xin and J. Hao, Angew. Chem., Int. Ed., 2020, 59, 9922–9927 CrossRef CAS PubMed.
- L. Zang, Y. Che and J. S. Moore, Acc. Chem. Res., 2008, 41, 1596–1608 CrossRef CAS PubMed.
- H.-Y. Lee, S. H. R. Shin, A. M. Drews, A. M. Chirsan, S. A. Lewis and K. J. M. Bishop, ACS Nano, 2014, 8, 9979–9987 CrossRef CAS PubMed.
- X. Ouyang, M. Wang, L. Guo, C. Cui, T. Liu, Y. Ren, Y. Zhao, Z. Ge, X. Guo, G. Xie, J. Li, C. Fan and L. Wang, Angew. Chem., Int. Ed., 2020, 59, 11836–11844 CrossRef CAS PubMed.
- D. Zhou, M. Liu, M. Lin, X. Bu, X. Luo, H. Zhang and B. Yang, ACS Nano, 2014, 8, 10569–10581 CrossRef CAS PubMed.
- Z. Wu, Q. Yao, S. Zang and J. Xie, ACS Mater. Lett., 2019, 1, 237–248 CrossRef CAS.
- S. Hossain, Y. Imai, Y. Motohashi, Z. Chen, D. Suzuki, T. Suzuki, Y. Kataoka, M. Hirata, T. Ono, W. Kurashige, T. Kawawaki, T. Yamamoto and Y. Negishi, Mater. Horiz., 2020, 7, 796–803 RSC.
- X. Kang and M. Zhu, Coord. Chem. Rev., 2019, 394, 1–38 CrossRef CAS.
- Z. Wu, J. Liu, Y. Gao, H. Liu, T. Li, H. Zou, Z. Wang, K. Zhang, Y. Wang, H. Zhang and B. Yang, J. Am. Chem. Soc., 2015, 137, 12906–12913 CrossRef CAS PubMed.
- J. Liu, Y. Tian, L. Ai, Z. Wu, D. Yao, Y. Liu, B. Yang and H. Zhang, Langmuir, 2020, 36, 14614–14622 CrossRef CAS PubMed.
- X. Su and J. Liu, ACS Appl. Mater. Interfaces, 2017, 9, 3902–3910 CrossRef CAS PubMed.
- L. Ai, Y. Li, Z. Wu, J. Liu, Y. Gao, Y. Liu, Z. Lu, H. Zhang and B. Yang, J. Phys. Chem. C, 2016, 120, 24427–24436 CrossRef CAS.
- L. Ai, Z. Liu, D. Zhou, J. Liu, H. Zou, Z. Wu, Y. Liu, H. Zhang and B. Yang, Nanoscale, 2017, 9, 18845–18854 RSC.
- X. Jia, X. Yang, J. Li, D. Li and E. Wang, Chem. Commun., 2014, 50, 237–239 RSC.
- L. Ai, W. Jiang, Z. Liu, J. Liu, Y. Gao, H. Zou, Z. Wu, Z. Wang, Y. Liu, H. Zhang and B. Yang, Nanoscale, 2017, 9, 12618–12627 RSC.
- Q. Yao, Y. Yu, X. Yuan, Y. Yu, D. Zhao, J. Xie and J. Y. Lee, Angew. Chem., Int. Ed., 2015, 54, 184–189 CrossRef CAS PubMed.
- Z. Wu, Y. Li, J. Liu, Z. Lu, H. Zhang and B. Yang, Angew. Chem., Int. Ed., 2014, 53, 12196–12200 CrossRef CAS PubMed.
- Z. Wu, J. Liu, Y. Li, Z. Cheng, T. Li, H. Zhang, Z. Lu and B. Yang, ACS Nano, 2015, 9, 6315–6323 CrossRef CAS PubMed.
- Z. Wu, C. Dong, Y. Li, H. Hao, H. Zhang, Z. Lu and B. Yang, Angew. Chem., Int. Ed., 2013, 52, 9952–9955 CrossRef CAS PubMed.
- Z. Wu, Y. Du, J. Liu, Q. Yao, T. Chen, Y. Cao, H. Zhang and J. Xie, Angew. Chem., Int. Ed., 2019, 58, 8139–8144 CrossRef CAS PubMed.
- P. Sun, Z. Wang, Y. Bi, D. Sun, T. Zhao, F. Zhao, W. Wang and X. Xin, ACS Appl. Nano Mater., 2020, 3, 2038–2046 CrossRef CAS.
- S. Maity, D. Bain, S. Chakraborty, S. Kolay and A. Patra, ACS Sustain. Chem. Eng., 2020, 8, 18335–18344 CrossRef CAS.
- S. Maity, D. Bain and A. Patra, Nanoscale, 2019, 11, 22685–22723 RSC.
- D. Bain, S. Maity and A. Patra, Phys. Chem. Chem. Phys., 2019, 21, 5863–5881 RSC.
- A. Baghdasaryan and T. Bürgi, Nanoscale, 2021, 13, 6283–6340 RSC.
- X. Kang and M. Zhu, Chem. Soc. Rev., 2019, 48, 2422–2457 RSC.
- Z. Luo, X. Yuan, Y. Yu, Q. Zhang, D. T. Leong, J. Y. Lee and J. Xie, J. Am. Chem. Soc., 2012, 134, 16662–16670 CrossRef CAS PubMed.
- D. Bain, S. Maity and A. Patra, Chem. Commun., 2020, 56, 9292–9295 RSC.
- Z. Wu, H. Liu, T. Li, J. Liu, J. Yin, O. F. Mohammed, O. M. Bakr, Y. Liu, B. Yang and H. Zhang, J. Am. Chem. Soc., 2017, 139, 4318–4321 CrossRef CAS PubMed.
- J. Liang, Z. Chen, J. Yin, G.-A. Yu and S. H. Liu, Chem. Commun., 2013, 49, 3567–3569 RSC.
- X. Jia, J. Li and E. Wang, Small, 2013, 9, 3873–3879 CrossRef CAS PubMed.
- S. Maity, D. Bain and A. Patra, J. Phys. Chem. C, 2019, 123, 2506–2515 CrossRef CAS.
- R. Jin, C. Zeng, M. Zhou and Y. Chen, Chem. Rev., 2016, 116, 10346–10413 CrossRef CAS PubMed.
- X. Gao, Y. Lu, M. Liu, S. He and W. Chen, J. Mater. Chem. C, 2015, 3, 4050–4056 RSC.
- K. Dey, H. S. Kunjattu, A. M. Chahande and R. Banerjee, Angew. Chem., Int. Ed., 2020, 59, 1161–1165 CrossRef CAS PubMed.
- A. K. Mohammed, S. Usgaonkar, F. Kanheerampockil, S. Karak, A. Halder, M. Tharkar, M. Addicoat, T. G. Ajithkumar and R. Banerjee, J. Am. Chem. Soc., 2020, 142, 8252–8261 CrossRef CAS PubMed.
- Y. Chen, C. Zeng, D. R. Kauffman and R. Jin, Nano Lett., 2015, 15, 3603–3609 CrossRef CAS PubMed.
- M. Brookhart, M. L. H. Green and G. Parkin, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 6908 CrossRef CAS PubMed.
- Y. Li, R. Juarez-Mosqueda, Y. Song, Y. Zhang, J. Chai, G. Mpourmpakis and R. Jin, Nanoscale, 2020, 12, 9423–9429 RSC.
- J. Mei, N. L. C. Leung, R. T. K. Kwok, J. W. Y. Lam and B. Z. Tang, Chem. Rev., 2015, 115, 11718–11940 CrossRef CAS PubMed.
- M. Sugiuchi, J. Maeba, N. Okubo, M. Iwamura, K. Nozaki and K. Konishi, J. Am. Chem. Soc., 2017, 139, 17731–17734 CrossRef CAS PubMed.
- L. Li and Q. Wang, ACS Nano, 2013, 7, 3053–3060 CrossRef CAS PubMed.
- K. Pyo, V. D. Thanthirige, K. Kwak, P. Pandurangan, G. Ramakrishna and D. Lee, J. Am. Chem. Soc., 2015, 137, 8244–8250 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Details of synthesis procedures, TEM images, AFM images, TGA, FTIR, XPS, 1H NMR spectra and additional figures. See DOI: 10.1039/d1na00446h |
| This journal is © The Royal Society of Chemistry 2021 |