
Hierarchically porous UiO-66 with tunable mesopores and oxygen vacancies for enhanced arsenic removal†
Rongming
Xu
 abc,
Qinghua
Ji
a,
Pin
Zhao
a,
Meipeng
Jian
d,
Chao
Xiang
a,
Chengzhi
Hu
*c,
Gong
Zhang
a,
Chaochun
Tang
b,
Ruiping
Liu
ce,
Xiwang
Zhang
*d and
Jiuhui
Qu
ace
abc,
Qinghua
Ji
a,
Pin
Zhao
a,
Meipeng
Jian
d,
Chao
Xiang
a,
Chengzhi
Hu
*c,
Gong
Zhang
a,
Chaochun
Tang
b,
Ruiping
Liu
ce,
Xiwang
Zhang
*d and
Jiuhui
Qu
ace
aCenter for Water and Ecology, State Key Joint Laboratory of Environment Simulation and Pollution Control, School of Environment, Tsinghua University, Beijing 100085, China
bSchool of Civil Engineering and Architecture, East China JiaoTong University, Nanchang 330013, Jiangxi, China
cState Key Laboratory of Environmental Aquatic Chemistry, Research Center for Eco-Environmental Sciences, Chinese Academy of Sciences, Beijing 100085, China. E-mail: czhu@rcees.ac.cn; Tel: +86 10 6284 9160
dDepartment of Chemical Engineering, Monash University, Clayton, VIC 3800, Australia. E-mail: xiwang.zhang@monash.edu; Tel: +61 3 9905 1867
eUniversity of Chinese Academy of Sciences, Beijing 100049, China
First published on 13th March 2020
Abstract
Metal–organic frameworks (MOFs) have attracted increasing interest for adsorption applications due to their ultrahigh porosity and specific surface area. However, their adsorption performance in liquids, particularly for water treatment, is significantly limited by the poor accessibility of micropores inside the MOF matrix to most molecules and deficient active sites. Herein, we use a facile strategy, ligand selective thermolysis, to create oxygen vacancies and construct mesopores in water stable UiO-66. The obtained hierarchically porous UiO-66 (HP-UiO-66) has tunable oxygen vacancies and mesopores, and outperforms the state-of-the-art MOF based adsorbents reported so far in removing arsenic, achieving an ultrahigh adsorption capacity of 248.75 mg g−1 under neutral conditions. Extended X-ray absorption fine structure (EXAFS) and X-ray absorption near-edge structure (XANES) analysis and density functional theory (DFT) calculations reveal that arsenic is mainly captured by forming bidentate Zr–O–As bonds between arsenate and HP-UiO-66 via occupying coordinatively unsaturated zirconium atoms. This study offers a new strategy for designing ultrahigh performance MOF based adsorbents.
1 Introduction
Arsenic contamination is one of the key global water issues, and threatens the health of more than 100 million people.1 Nowadays, adsorption is considered as a cost-effective technology for arsenic removal from contaminated water.2 Due to their low specific surface area and deficient active sites, arsenic adsorption capacities of conventional adsorbents are quite low.3 Metal–organic frameworks (MOFs) have been of great interest to researchers for various applications including adsorption because of their ultrahigh porosity (>50%) and specific surface area (up to 7140 m2 g−1).4 Among them, UiO-66 is most commonly used in aqueous environments due to its excellent water and chemical stability.5,6 Although a recent study showed that UiO-66 could capture As(V) up to 85 mg g−1, which was higher than most reported adsorbents,7 this adsorption capacity was much lower than expected considering its ultrahigh porosity and surface area. One of the reasons is that the pore diameter of UiO-66 is around 6 Å,8 which is smaller than the size of hydrated arsenate (≈7.7 Å).9 Hydrated arsenate cannot reach the inner channel of UiO-66, thus limiting its adsorption rate and capacity. In addition, the active sites for arsenate adsorption on metal oxides and MOFs are mainly coordinatively unsaturated metal atoms and surface hydroxyl.10,11 Zirconium atoms in normal UiO-66 are coordinately saturated with ligands, while the coordinatively unsaturated zirconium atoms only form at the structural defects.6 Thus, it is highly needed to enlarge the pore diameter and increase the coordinatively unsaturated zirconium atoms for enhancing the As(V) adsorption performance of UiO-66. Although a few recent investigations on MOF defects have suggested their preliminary influences on adsorption,10,12 to the best of our knowledge, it has never been experimentally explored to clarify how different levels of defects in MOFs systematically affect their adsorption performance.Recently, Zhou and his co-workers reported a simple strategy, namely linker labilization, to construct hierarchically porous metal–organic frameworks.13,14 Based on this, we synthesized a hierarchically porous UiO-66 with tunable defects via a facile selective ligand thermolysis. As shown in Scheme 1, UiO-66 with multivariate linkers was firstly synthesized using an ordinary ligand 1,4-benzenedicarboxylate (BDC) and a thermolabile ligand 2-amino-1,4-benzenedicarboxylate (NH2-BDC). Then, NH2-BDC was selectively removed by controlling the decomposition temperature. Abundant coordinatively unsaturated zirconium atoms were formed due to the absence of oxygen atoms in the decarboxylation process, namely, a large amount of oxygen vacancies generated after thermolabile ligand decomposition. Meanwhile, the crystallinity and chemical stability remained after thermolabile linker thermolysis. Due to the synergetic effect of hierarchical pores and oxygen vacancies, HP-UiO-66 achieved a high adsorption capacity (up to 248.75 mg g−1) with a fast adsorption rate under neutral conditions, which was higher than those of all previously reported MOF based absorbents.
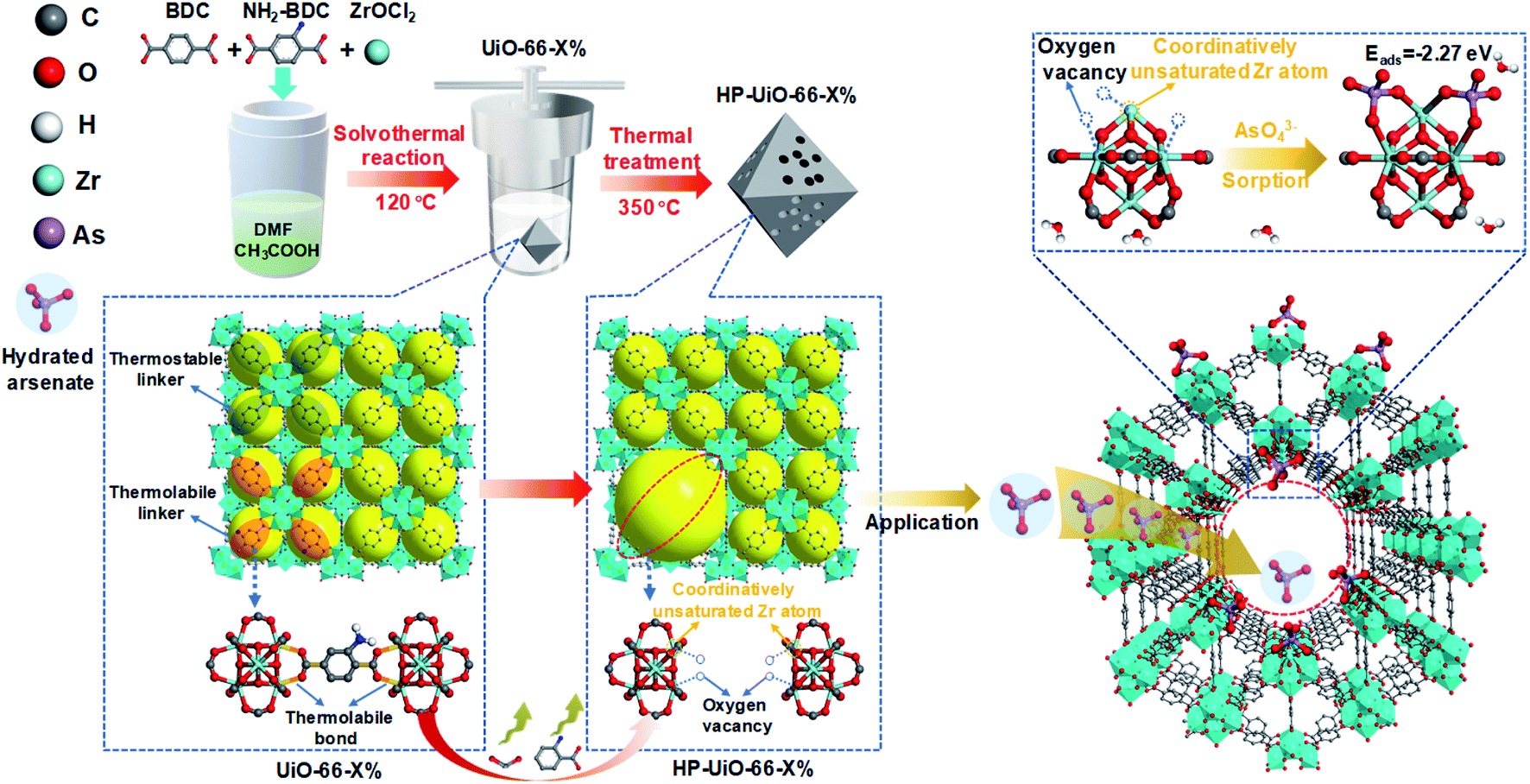 | ||
| Scheme 1 Schematic of the HP-UiO-66 synthesis procedure and arsenate adsorption. | ||
2 Experimental
2.1 Materials
ZrOCl2·8H2O (>99%), 1,4-benzenedicarboxylic acid (H2BDC) (>99%), 2-aminoterephthalic acid (H2BDC-NH2) (>99%), CH3COOH (HOAc) (>99%), and N,N-dimethylformamide (DMF) (>99%) were purchased from Sigma-Aldrich Company, USA. Ethanol (>99%), Na3AsO4·12H2O (>99%), KH2PO4 (>99%), NaNO3 (>99%), NaCl (>99%), Na2SO4 (>99%), NaHCO3 (>99%), Na2CO3 (>99%) and NaF (>99%) were purchased from Aladdin Chemical Company, Shanghai, China. Ultrapure water was obtained from a Millipak® Express 40 system (Merk-Millipore, Darmstadt, Germany) and used as a solvent for aqueous solution preparation.2.2 UiO-66-X% preparation
UiO-66-X% (X%, the ratio of NH2-BDC ligand mass to total ligand mass (mg mg−1), X = 0, 5, 15, 30, and 40) metal organic frameworks were prepared by a modified method reported in the literature.13 ZrOCl2·8H2O (542.3 mg), H2BDC-NH2 (0, 10, 30, 60, 80 mg), H2BDC (200, 190, 170, 140, 120 mg), CH3COOH (10 mL) and DMF (10 mL) were charged in a 50 mL Teflon lined steel reactor. The reactor was shaken for 2 min after sealing it. Then it was heated in an oven at 120 °C for 24 h. After cooling down to room temperature, the resulting powder was separated by centrifugation at 8000 rpm for 10 min and washed three times with DMF and ethanol, respectively. The resulting white sticky substance was transferred to a porcelain crucible and placed in a 150 °C vacuum oven for 12 h to remove the excess solvent. After cooling naturally, the final samples were placed in a 10 mL centrifuge tube for use.2.3 HP-UiO-66-X% preparation
Hierarchically porous metal organic frameworks HP-UiO-66-X% (X = 0, 5, 15, 30, and 40) were prepared on the basis of UiO-66-X%. The obtained pale-yellow powder samples UiO-66-X% were placed in a thermostatic furnace at 350 °C for 2 h under air. After cooling naturally, the samples were activated again at 150 °C for 6 h in a vacuum oven, and the final samples were placed in a 10 mL centrifuge tube for use. The synthesis process is shown in Fig. S1.†2.4 Characterization
The adsorbed solid sample was centrifuged at 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm for 10 min. Then, placed in a vacuum oven at 60 °C for 6 h. After cooling naturally, the adsorbed samples were placed in a 5 mL centrifuge tube for characterization. The PXRD pattern was obtained with an X-ray diffractometer (XRD, Bruker D8 focus, USA) equipped with a Cu sealed tube (λ = 1.54178 Å) at 40 kV and 30 mA. N2 adsorption measurements were conducted using an accelerated surface and porosimetry system (Micromeritics, ASAP 2020 Plus HD88, USA), and the relative pressure (P/P0) range was 0–0.992. The pore size distribution plots were obtained using the N2-DFT Model. Fine structures of UiO-66-X% and HP-UiO-66-X% were obtained by transmission electron microscopy (TEM, JEOL JEM-2100F, Japan) with an energy dispersive X-ray spectrometer (EDX). The thermogravimetric curve was obtained using a TGA Q5000 integration thermal analyzer from room temperature to 800 °C at a heating rate of 10 °C min−1 under an air flow of 20 mL min−1. Electron paramagnetic resonance (EPR) was applied to detect unpaired electrons in paramagnetic species, and is a powerful method to detect the oxygen vacancies. To obtain information about the oxygen vacancies, the EPR spectra were obtained using a BRUKER E500 EPR device. FT-IR spectra were obtained using a Thermo Nicolet 8700 infrared spectrometer. Dissolution/1H NMR spectra were obtained on a JEOL JNM-ECA600 (600 MHz) spectrometer using 1 M NaOH in D2O as a digestion medium. X-ray photoelectron spectroscopy (XPS, Thermo ESCALAB250Xi, USA) was used to investigate the chemical constitution, and an Al Kα (150 W) X-ray source at a chamber was used to excite photoelectrons. The obtained XPS spectra were processed using XPS peak (version 4.1) for fitting the peaks. The extended X-ray absorption fine structures (EXAFS) of the As K-edge spectra were collected in fluorescence mode using the XAFS station of the Beamline 1W1B at the Beijing Synchrotron Radiation Facility (BSRF), China. The obtained XAFS data were processed using Athena (version 0.9.25) for background, pre-edge line and post-edge line calibrations. Then Fourier transformed fitting was carried out using Artemis (version 0.9.25). The k3 weighting, the k-range of 3–11 Å−1 and the R range of 1–3.5 Å were used for the fitting. The model of AsO43− was used to calculate the simulated scattering paths. Four parameters, the coordination number (CN), the bond length (R), the Debye–Waller factor (σ2) and E0 shift (ΔE0), all fitted were unfixed, unconstrained, or uncorrelated. Feff 9.7.1 is used to calculate the As–K edge X-ray absorption near-edge structure (XANES). The unit cell of the T-6 model is used as the model. The full multiple scattering radius (FMS) is 6 angstroms. The edge is treated with the RPA core hole, and Hedin–Lundqvist self-energy is used to calculate the energy dependent exchange correlation potential.
000 rpm for 10 min. Then, placed in a vacuum oven at 60 °C for 6 h. After cooling naturally, the adsorbed samples were placed in a 5 mL centrifuge tube for characterization. The PXRD pattern was obtained with an X-ray diffractometer (XRD, Bruker D8 focus, USA) equipped with a Cu sealed tube (λ = 1.54178 Å) at 40 kV and 30 mA. N2 adsorption measurements were conducted using an accelerated surface and porosimetry system (Micromeritics, ASAP 2020 Plus HD88, USA), and the relative pressure (P/P0) range was 0–0.992. The pore size distribution plots were obtained using the N2-DFT Model. Fine structures of UiO-66-X% and HP-UiO-66-X% were obtained by transmission electron microscopy (TEM, JEOL JEM-2100F, Japan) with an energy dispersive X-ray spectrometer (EDX). The thermogravimetric curve was obtained using a TGA Q5000 integration thermal analyzer from room temperature to 800 °C at a heating rate of 10 °C min−1 under an air flow of 20 mL min−1. Electron paramagnetic resonance (EPR) was applied to detect unpaired electrons in paramagnetic species, and is a powerful method to detect the oxygen vacancies. To obtain information about the oxygen vacancies, the EPR spectra were obtained using a BRUKER E500 EPR device. FT-IR spectra were obtained using a Thermo Nicolet 8700 infrared spectrometer. Dissolution/1H NMR spectra were obtained on a JEOL JNM-ECA600 (600 MHz) spectrometer using 1 M NaOH in D2O as a digestion medium. X-ray photoelectron spectroscopy (XPS, Thermo ESCALAB250Xi, USA) was used to investigate the chemical constitution, and an Al Kα (150 W) X-ray source at a chamber was used to excite photoelectrons. The obtained XPS spectra were processed using XPS peak (version 4.1) for fitting the peaks. The extended X-ray absorption fine structures (EXAFS) of the As K-edge spectra were collected in fluorescence mode using the XAFS station of the Beamline 1W1B at the Beijing Synchrotron Radiation Facility (BSRF), China. The obtained XAFS data were processed using Athena (version 0.9.25) for background, pre-edge line and post-edge line calibrations. Then Fourier transformed fitting was carried out using Artemis (version 0.9.25). The k3 weighting, the k-range of 3–11 Å−1 and the R range of 1–3.5 Å were used for the fitting. The model of AsO43− was used to calculate the simulated scattering paths. Four parameters, the coordination number (CN), the bond length (R), the Debye–Waller factor (σ2) and E0 shift (ΔE0), all fitted were unfixed, unconstrained, or uncorrelated. Feff 9.7.1 is used to calculate the As–K edge X-ray absorption near-edge structure (XANES). The unit cell of the T-6 model is used as the model. The full multiple scattering radius (FMS) is 6 angstroms. The edge is treated with the RPA core hole, and Hedin–Lundqvist self-energy is used to calculate the energy dependent exchange correlation potential.
2.5 Batch adsorption experiments
100 mg L−1 As(V) stock solution was prepared by dissolving 565.8 mg AsNa3O4·12H2O into a 1 L volumetric flask containing ultrapure water, and all the experimental solutions were diluted using a stock solution to obtain the corresponding concentrations. Batch adsorption experiments were performed in glass beakers under magnetic stirring, and solution pH was adjusted to desired values thereafter. All adsorption experiments in this study were performed at room temperature (25 °C). Samples were filtered through a 0.22 μm Millipore filter and the concentrations of As(V) were analyzed by inductively-coupled plasma optical-emission spectrometry (ICP-OES, Agilent 5110VDV, Japan) and LC/inductively-coupled plasma mass spectrometry (LC-ICP-MS, Agilent 7800, USA). The adsorbed solid sample was centrifuged at 10000 rpm for 10 min, lyophilized, and stored in a 5 mL air-tight centrifuge tube in the dark.
The adsorption kinetic experiments were performed by adding 50 mg L−1 adsorbent to a 20 mg L−1 solution of As(V) in a glass beaker at a pH of 6. The sampling time points were 2, 5, 10, 15, 30, 60, 120, 180, 240, 300, and 420 min, respectively. The results of adsorption kinetics were modelled using the pseudo-first-order model (eqn (1)), pseudo-second-order model (eqn (2)) and Weber and Morris intraparticle diffusion model (eqn (3)), and the equations can be expressed as below:
| ln(Qe − Qt) = lnQe − K1t | (1) |
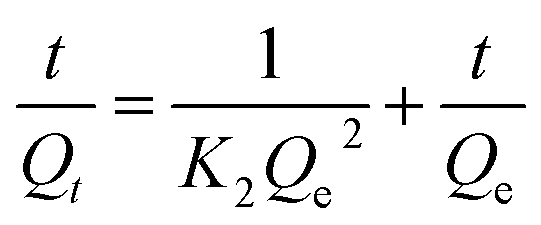 | (2) |
| Qt = Kit0.5 + C | (3) |
The adsorption isotherm experiments were carried out at a series of concentrations (5, 10, 25, 50, 80, and 100 mg L−1) of As(V) solutions with 200 mg L−1 adsorbent in a glass beaker under magnetic stirring for 8 h at a pH of 6. The results of the adsorption isotherm were modelled using the Langmuir model (eqn (4)) and the Freundlich model (eqn (5)):
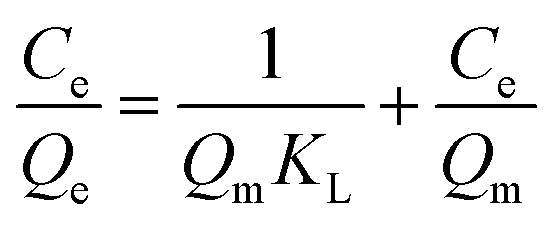 | (4) |
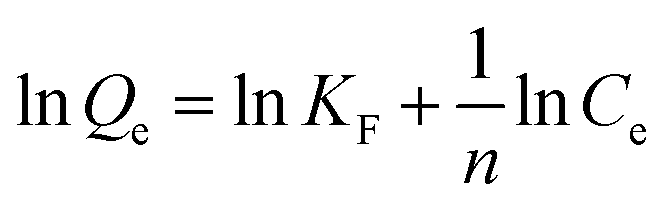 | (5) |
Low concentration As(V) adsorption experiments were performed by adding 50 mg L−1 adsorbent (HP-UiO-66-40%) to 100 μg L−1 solution of As(V) in a glass beaker in the pH range of 2–12 for 6 hours. The obtained liquid samples were filtered through a 0.22 μm Millipore filter and the concentrations of As(V) were analyzed by LC-ICP-MS.
The regeneration experiments were performed by adding 50 mg L−1 adsorbent (HP-UiO-66-40%) to 10 mg L−1 solution of As(V) in a glass beaker at pH of 6. The contact time of all experiments was 6 hours. The adsorbed solid sample was centrifuged at 10000 rpm for 10 min. Then the obtained solid sample was added into 0.1 M NaOH solution and stirred for 12 h, after that it was washed three times with deionized water. Finally, the sample was placed in a vacuum oven at 60 °C for 6 h. After cooling naturally, it was used for the next adsorption experiment.
The competitive uptake of AsO43− by the HP-UiO-66-40% adsorbent was investigated in an environment where different types of co-existing anions (NO3−, Cl−, SO42−, HCO3−, CO32−, PO43− and F−). The competitive adsorption experiments were performed by adding 50 mg L−1 adsorbent (HP-UiO-66-40%) to 10 mg L−1 solution of As(V) in a glass beaker at a pH of 6 for 6 hours. The initial concentrations of all co-existing anions were consistent with the As(V) initial concentration of 10 mg L−1.
2.6 Density functional theory (DFT) calculation details
First-principles calculations have been performed using spin-polarized DFT as implemented in the Vienna Ab initio Simulation Package (VASP). The ionic potentials are described by the projector augmented wave (PAW) pseudopotential 3 with valence configurations of 1s1, 2s22p2, 2s22p4, 4s24p3 and 5s24s2 for H, C, O, As and Zr atoms, respectively. The cutoff energy for the plane-wave basis set was chosen to be 450 eV. Brillouin-zone integration was performed on Monkhorst–Pack grids 4 with a 2 × 2 × 1 mesh where a Gaussian-smearing approach with σ = 0.02 eV is used during the ionic optimization, whereas total energies and densities of states (DOS) were calculated using the tetrahedron method with Blöchl corrections. The surface has been modeled using a symmetric slab with a (1 × 1) unit cell, which is based on cubic-phase UiO-66 (a = 20.889 Å). The slabs are separated by a vacuum space of approximately 15 Å. In order to maintain the bulk behavior below the surface, the bottom layers were kept fixed at the bulk crystal positions during the structural optimization while the top layers of the slab were allowed to relax. The ion positions were optimized using a conjugate-gradient algorithm until the Hellmann–Feynman forces became less than 0.05 eV Å−1 and energy convergence was reached within 10−6 eV.3 Results and discussion
3.1 Characterization of HP-UiO-66-X%
The hierarchically porous UiO-66 samples named HP-UiO-66-X% (X% represents the percentage of NH2-BDC over the sum of NH2-BDC and BDC) were firstly characterized by transmission electron microscopy (TEM). As shown in these TEM images (Fig. 1a–e), a large number of mesoporous defects occurred inside HP-UiO-66-X%, and such defects increased with the increase of the ratio of NH2-BDC. Furthermore, in spite of the formation of a large number of defects inside HP-UiO-66-X%, all samples show similar PXRD patterns, which match well with the simulated one (CCDC, 837796)15 (Fig. S2†). It proves that the crystallinity remained after thermolabile linker thermolysis. Notably, the influence of partial structural collapse on the chemical stability of HP-UiO-66-X% has been demonstrated to be negligible (Fig. S3†). Moreover, electron paramagnetic resonance (EPR) has now been widely employed to detect the coordinatively unsaturated defects.16 For example. Li et al.17 used the EPR technique to investigate the properties and quantity of oxygen vacancies in NH2-UiO-66. S. Smolders et al.18 use EPR to determine the number of coordinatively unsaturated titanium atoms in Ti-MOF. Therefore, the EPR technique has been used as a reliable method for characterizing the defects of MOFs.19 As shown in Fig. 1f, a symmetrical signal at g = 2.003 appeared in the EPR spectra of the HP-UiO-66-X% samples, and can be assigned to oxygen vacancies,20 and the intensity of oxygen vacancies increases with the ratio of NH2-BDC. Obviously, the absence of oxygen atoms in UiO-66 must be accompanied by the formation of coordinatively unsaturated zirconium atoms (Scheme 1). Namely, the concentration of oxygen vacancies can represent the number of coordinatively unsaturated zirconium atoms in this work. Thus, the EPR characterization results prove that HP-UiO-66-X% has plentiful coordinatively unsaturated zirconium atoms, and the number of coordinatively unsaturated zirconium atoms is tunable.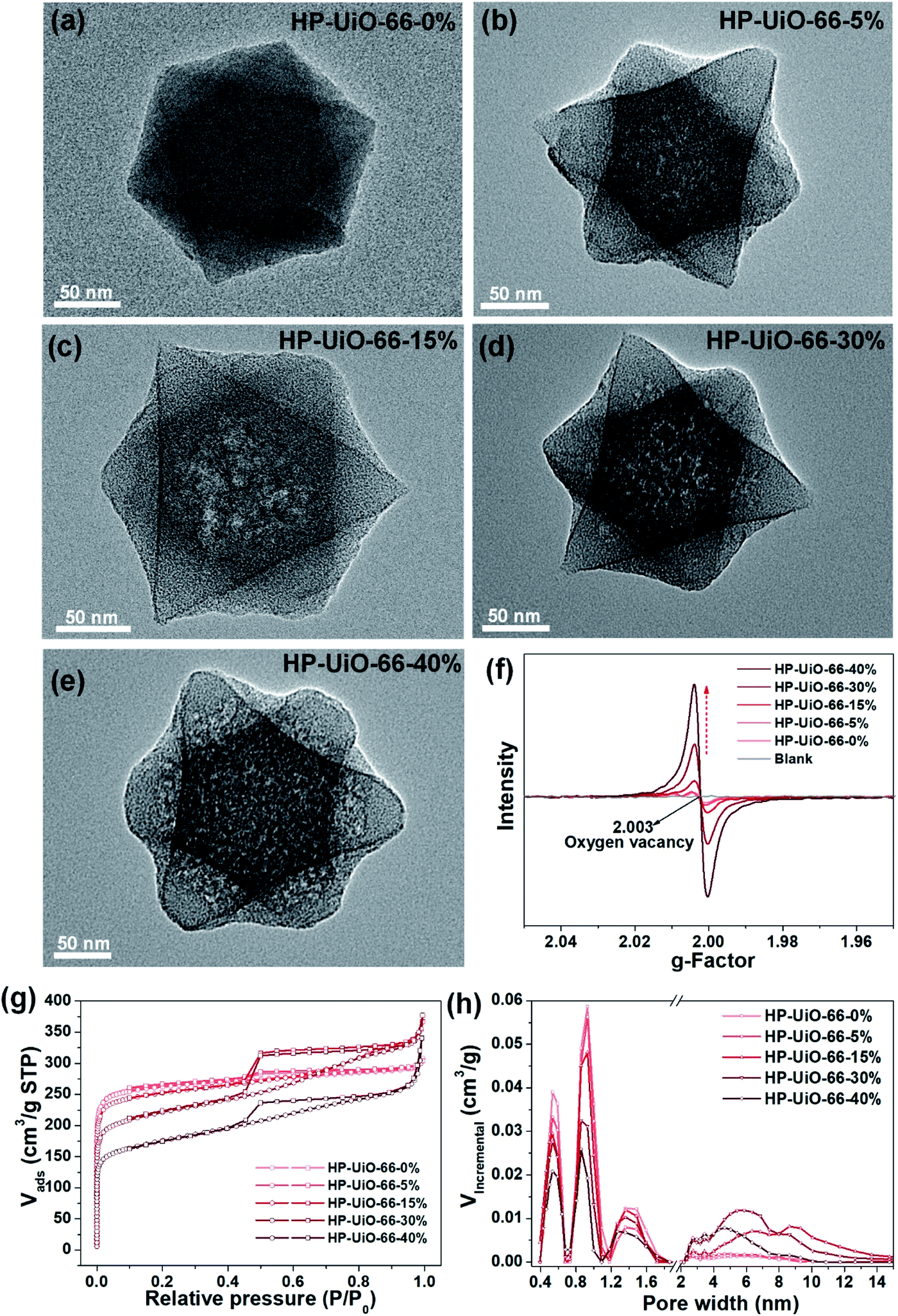 | ||
| Fig. 1 (a–e) TEM images, (f) EPR spectra, (g) N2 adsorption/desorption isotherms, (h) pore size distributions based on density-functional theory (DFT) analysis of HP-UiO-66-X% (X = 0, 5, 15, 30, 40). | ||
On the other hand, as illustrated by the N2 adsorption–desorption isotherm curves in Fig. 1g, with the introduction of NH2-BDC, the micropore volume decreased and the isotherm curves changed from type I to type IV. The significant hysteresis loop is associated with capillary condensation taking place in mesopores. Furthermore, the pore size distributions (Fig. 1h) indicated that the mesopores increased with the enhanced ratio of NH2-BDC. The proportion of mesopores increases from 23.7% to 61% as the ratio of NH2-BDC increases from 0% to 40% (Table S1†). These results show that the defects in MOFs caused the partial structural collapse and enabled the transformation from micropores into mesopores. More importantly, the number of mesopores is controllable. Based on the above analysis, the chemically stable HP-UiO-66-X% MOFs with large diameter pores (2–10 nm) and plenty of coordinatively unsaturated zirconium atoms have been successfully synthesized.
3.2 Formation mechanism of coordinatively unsaturated zirconium atoms and mesopores
Several techniques have been adopted to investigate the formation mechanism of coordinatively unsaturated zirconium atoms and mesopores. First, UiO-66 with multivariate linkers named UiO-66-X% (X = 5, 15, 30, and 40) synthesized at different NH2-BDC concentrations without thermal treatment were characterized by the dissolution/1H NMR technique, an effective tool to identify the ratios between the NH2-BDC linker and BDC coordinated to Zr clusters (Fig. 2a). A significant signal enhancement assignable to NH2-BDC can be observed in the dissolution/1H NMR spectra of digested UiO-66-X% with gradually increased X values, suggesting the ratio of the NH2-BDC linker in UiO-66-X% is consistent with the concentrations of the NH2-BDC ligand we added. Secondly, according to the TGA curves of both the pure ligand MOF (UiO-66-0%) and the multivariate ligand MOF (UiO-66-5, 15, 30, and 40%) (Fig. 2b), the thermolabile ligand (NH2-BDC) can be easily removed, while the thermostable ligand (BDC) remains at a decomposition temperature of 350 °C. This result is also confirmed by the dissolution/1H NMR spectra (Fig. 2c), the signals of H1, H2, and H3 from the NH2-BDC linker disappeared but H4 from the BDC linker remained after thermal treatment at 350 °C. The PXRD patterns of HP-UiO-66-30% remain intact with UiO-66-30%, suggesting that UiO-66 has high tolerance for structural defects (Fig. S5†). Moreover, comparing the FT-IR spectra of HP-UiO-66-30% to those of UiO-66-30% (Fig. 2d), the peaks at 1430 and 1582 cm−1 corresponding to the COO− group13 have a slight blue shift and become weak, suggesting that there occurs decarboxylation in the thermal treatment process and most of the carboxyl groups remained. Meanwhile, the weakening of Zr–(OC) bond (555 cm−1)21 signal strength indicated that Zr–O bonds were broken after thermal treatment, which correspond to the reduction of the C–O–Zr band22 in the O 1s XPS spectra (Fig. S6†). Additionally, the peaks in the range 650 to 780 cm−1 correspond to the derivatives of the benzene ring23 showing visible changes after decomposition. These results propose that the thermal treatment caused decarboxylation and the terminal ligand was removed. More importantly, the split of Zr–O bonds leads to the formation of oxygen vacancies and coordinatively unsaturated zirconium atoms. Thus, the signal of oxygen vacancies has significantly enhanced (Fig. S7†). Furthermore, the signals of UiO-66-X% are much lower than those of HP-UiO-66-X%. It indicates that the oxygen vacancies were generated during thermal treatment, rather than coming from the inherent defects. Notably, this study provides a novel and simple method of oxygen vacancy formation and accurate regulation. Finally, the TEM images of UiO-66-30% and HP-UiO-66-30% show a significant difference, and a large number of mesopores formed inside UiO-66-30% after thermal treatment (Fig. S8†). The appearance of mesopores can be clearly seen from the pore size distribution curve (Fig. S8d†). Particularly, a pore diameter of 1.1–1.9 nm can be attributed to “Reo defects”, which are very common in Zr-MOFs,24 while the small amount of structural defects in the range of 2–4 nm corresponds to the “etched nanodomain”.25 In addition, by comparing the N2 adsorption–desorption isotherms of UiO-66-X% (Fig. 1g) and HP-UiO-66-X% (Fig. S9†), we can be sure that the mesoporous structure of H-UiO-66-X% is derived from NH2-BDC decomposition. Notably, as described by previous research, the ordered distribution of the thermolabile ligands cannot form mesopores.26 Hence, the unordered distribution of thermolabile ligands could be the reason for the formation of mesopores. In addition, the unevenness increases with the increase of thermolabile ligands, resulting in the increased mesopores (Table S1†). The possible formation mechanism of coordinatively unsaturated zirconium atoms and mesopores is shown in Scheme 1.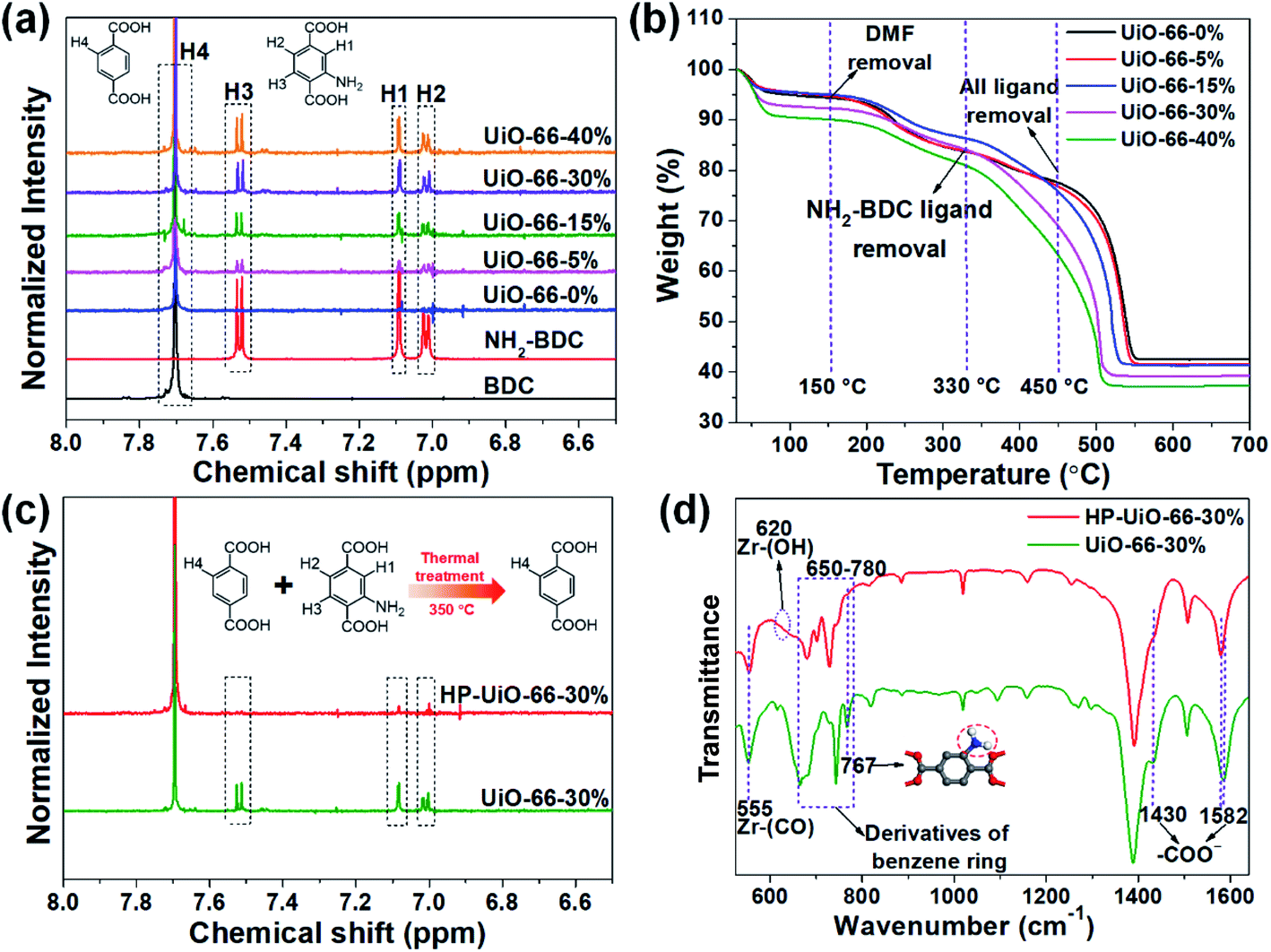 | ||
| Fig. 2 (a) Dissolution/1H NMR spectra and (b) thermogravimetric analyses (TGA) of UiO-66-X% (X = 0, 5, 15, 30, and 40). (c) Dissolution/1H NMR spectra and (d) FT-IR spectra of UiO-66-30% and HP-UiO-66-30%. | ||
3.3 As(V) adsorption performance of HP-UiO-66-X%
The adsorption performance of As(V) was investigated by batch adsorption experiments, in which the Weber–Morris model was applied to study the intraparticle diffusion process. As illustrated in Fig. 3a and Table S2† (the fitting results of the Weber–Morris model), the liquid film diffusion rate (Ki1) increases with the increase of the ratio of NH2-BDC. As the oxygen vacancy is an ionized donor atom with positive charge,27 more positive charges accumulated on the material surface (Fig. S10†), which leads to the stronger electrostatic attraction between As(V) and HP-UiO-66-X%. In addition, the internal diffusion rate (Ki2) enhanced and internal diffusion time also shortened significantly (Fig. 3a), resulting from the increase of mesopores. More importantly, because more coordinatively unsaturated zirconium atoms are exposed to the inner and outer surfaces of HP-UiO-66-X%, the saturated adsorption capacity for As(V) increased from 84.03 mg g−1 (HP-UiO-66-0%) to 248.75 mg g−1 (HP-UiO-66-40%) (Fig. 3b, Table S4†). It is much higher than those of the previously reported MOF based adsorbents under neutral conditions (Table S5†). In order to eliminate the influence of the inherent properties of UiO-66-X% on its adsorption performance, As(V) uptake capacities of UiO-66-X% and HP-UiO-66-X% were compared. As shown in Fig. 3c, UiO-66-X% had similar uptake capacities for different NH2-BDC ratios. In contrast, the uptake capacities of HP-UiO-66-X% increased significantly. This demonstrates the improvement in the uptake capacity of UiO-66-X% can be achieved by increasing the coordinatively unsaturated zirconium atoms and mesopores. More importantly, for the low concentration arsenic-containing water, HP-UiO-66-40% can easily reduce the remaining concentration below the WHO's drinking water limit (<10 μg L−1)28 in a wide pH range of 2–10 (Fig. 3d). The competitive adsorption experiments were conducted in the presence of different competitive anions (NO3−, Cl−, SO42−, HCO3−, CO32−, PO43− and F−) respectively. As shown in Fig. S11a,† only F− and PO43− could hinder As(V) adsorption seriously. The reasons for PO43− hindering the adsorption of arsenic is the similarity of PO43− and AsO43− in molecular chemistry (in the same column of the periodic table), PO43− has highly competitive adsorption with AsO43−, which caused the deficiency. In terms of F−, zirconium-based adsorbents generally exhibit a desirable removal of fluoride ions, owing to the high affinity of fluoride on zirconium.29 For instance, He et al.30 concluded that UiO-66 shows excellent fluorine adsorption capability, and the main active site is zirconium. Therefore, fluorine ions hinder the adsorption of arsenic in the mixture solution. We use 0.1 M NaOH as the desorption solution to investigate the reusability of HP-UiO-66-40%. As illustrated in Fig. S11b,† after five cycles, the removal efficiency of AsO43− was 65%, suggesting that the adsorbent exhibits good regeneration performance. In addition, according to the results of kinetic and isotherm analysis, the pseudo-second-order kinetic models and Langmuir isotherm models could better describe the kinetics and isotherms data (Fig. 3b, S12 and Tables S3 and S4†). It indicates that the monolayer chemisorption plays a dominating role in As(V) removal, and the speed-limiting step is adsorption itself instead of diffusion.31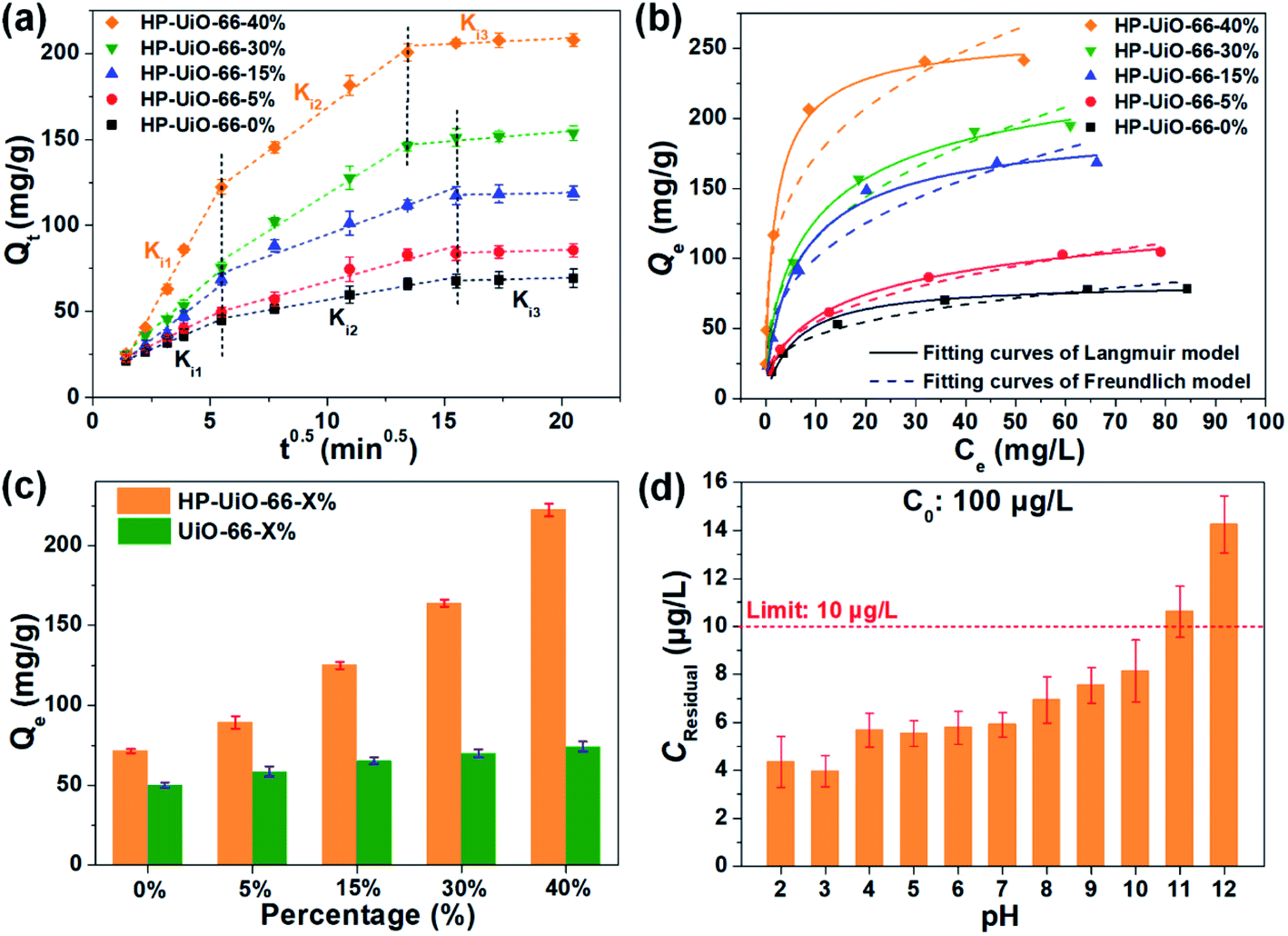 | ||
| Fig. 3 (a) The As(V) adsorption kinetic curves by Weber–Morris model fitting, (b) the As(V) adsorption isotherms of HP-UiO-66-X%. (c) The uptake capacity comparisons between UiO-66-X% and HP-UiO-66-X%. (d) The effect of pH on low concentration As(V) adsorption. | ||
3.4 As(V) adsorption mechanism of HP-UiO-66-X%
To explore the As(V) adsorption mechanism of HP-UiO-66-X%, the pristine and adsorbed samples of HP-UiO-66-0% and HP-UiO-66-40% were investigated. As illustrated in Fig. S14,† after As(V) adsorption, a broad peak appearing in the range of 810–890 cm−1 on the FT-IR spectra can be attributed to the As–O bond stretching.3,32 Meanwhile, the signal of the Zr–(OC) bond decreased significantly. This indicates that the arsenate splits the Zr–O bond, and then forms strong Zr–O–As bonds with UiO-66. This result was also confirmed by the high-resolution XPS spectra of O 1s, the signal at 531.7 eV corresponding to C–O–Zr bonds22 decreased significantly after adsorption (Fig. 4b and c). Additionally, as illustrated in Fig. 4a, the intensity of the oxygen vacancy signal in HP-UiO-66-40% weakened visibly after As(V) adsorption due to the exposed oxygen vacancies being occupied by As–O. Notably, as most of the oxygen vacancies are present inside and are inaccessible to arsenate molecules, the signal strength of HP-UiO-66-0% does not appear to change. This is consistent with the O 1s high-resolution XPS spectral analysis results, the strength of Zr–O–Zr bonds weakened visibly in HP-UiO-66-40% but remained unchanged in HP-UiO-66-0% (Fig. 4b and c). Furthermore, by comparing the thermogravimetric curves of HP-UiO-66-15% before and after adsorption (Fig. S13†), we found that the residual mass increased after adsorption, which can be attributed to the formation of Zr–O–As(V) covalent bonds. However, the pyrolysis temperature of the sample after arsenic adsorption shifted slightly to the left, indicating that the thermal stability of the adsorbent became worse. Based on the above analysis, the possible adsorption process is shown in Fig. 4h.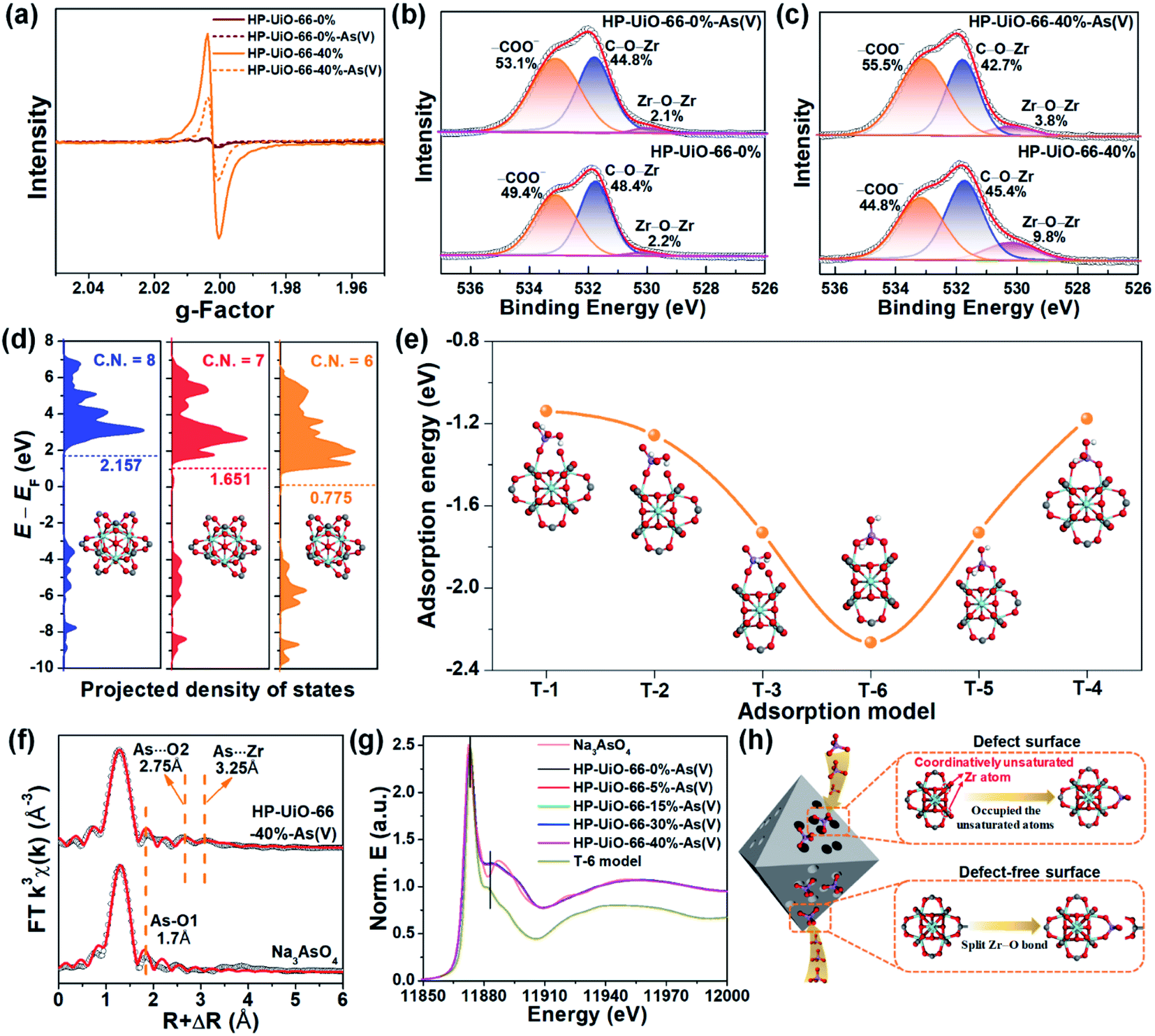 | ||
| Fig. 4 (a) EPR spectra of HP-UiO-66-0% and HP-UiO-66-40% before and after adsorption. O 1s high-resolution XPS spectra of (b) HP-UiO-66-0%, (c) HP-UiO-66-40% before and after adsorption. (d) The projected electronic density of states of the d-band for the surface Zr atoms in UiO-66 with different coordination numbers. The horizontal dashed lines indicate the calculated d-band center. (e) The adsorption energy of two different coordination structures in three different surfaces. (f) Observed (black circles) and fitted (red lines) FT magnitudes of the standard sample (Na3AsO4) and UiO-66-HP-40% loaded As(V). (g) The XANES spectra of HP-UiO-66-X%-H2AsO4− and the theoretical spectra of the T-6 model. (h) The possible As(V) adsorption mechanism of HP-UiO-66-X%. | ||
To investigate the effect of coordinatively unsaturated zirconium atoms on the adsorption reaction and interaction of arsenate with HP-UiO-66-X% surfaces, density functional theory (DFT) was applied for the geometry optimization and density of states (DOS) and adsorption energy calculations. The optimized surfaces of UiO-66 with different zirconium coordination numbers are shown in Fig. S15.† As shown in Fig. 4d, the calculated results demonstrate that the transferred electrons fill the d-band of the surface Zr atoms after decarboxylation, shifting the center of the band towards negative energy. The downshift of the d-band center reveals that the d-orbital energy of the Zr atom decreases. Moreover, the characteristic peak in the Zr 3d XPS spectra (Fig. S16†) shifts to the low binding energy direction as the NH2-BDC ratio increases, which is consistent with the calculated results. The reduction of d-orbital energy leads to the valence band (VB) of the HP-UiO-66-X% surface being more likely to react with the lowest unoccupied molecular orbital (LUMO) of the H2AsO4− molecules.33 The possible complexation models of the H2AsO4− (see Fig. S17,† As(V) exists mainly in the form of H2AsO4− at a pH of 6) molecules and the surface of UiO-66 with different Zr coordination numbers were established. The optimized monodentate and bidentate coordination models are shown in Fig. S18,† and the adsorption energies corresponding to each model are present in Fig. 4e. As illustrated in Fig. 4e, the increase of oxygen vacancies causes a significant decrease of the adsorption energy, and the bidentate coordination has a lower adsorption energy than that of monodentate coordination under the same conditions. The above analysis indicates that the decrease of the Zr coordination number reduces the d-orbital energy of Zr atoms, which is beneficial to the adsorption of H2AsO4− on the surface of UiO-66. Meanwhile, the bidentate coordination plays a dominating role in the complexation of H2AsO4− on the surface of HP-UiO-66-X%.
To confirm the accuracy of the coordination model obtained by DFT calculations, the extended X-ray absorption fine structure (EXAFS) and X-ray absorption near-edge structure (XANES) were used to characterize the local coordination environment of H2AsO4− on the surface of HP-UiO-66-X%. The experimental and fitted k3-weighted EXAFS spectra and their corresponding magnitude part of Fourier transformed (FT) R-space are shown in Fig. 4f and S21,† and the fitting results are presented in Table S7.† The first coordination shell of As is composed of the oxygen atoms inherent to arsenate with a distance of 1.70 Å and a coordination number (CN) of 3.2–4.2. Notably, compared with the standard sample (Na3AsO4), there are two new peaks at 2.75 and 3.25 Å. Thus, the second FT peak can be attributed to As⋯O single scattering with a distance of 2.75 Å, and the third FT peak at 3.25 Å can be ascribed to a As⋯Zr bidentate binuclear coordination with a CN of 1.3–3.1. From the comparison of the DFT calculation results of the T-1–T-6 models (Table S6†), the results of the T-6 model were found to be in good agreement with the EXAFS results. Furthermore, we obtained the possible As local coordination environment of the T-6 model through the finite difference method for near-edge structure. As shown in Fig. 4g, the theoretical XANES spectra of the T-6 model match well with the observed XANES spectra of HP-UiO-66-X%-H2AsO4−. It means that the DFT calculation results are in good agreement with the actual As local coordination environment. Based on the above analysis results, it can be concluded that the T-6 coordination model is suitable to describe the local coordination environment of H2AsO4− on HP-UiO-66-X%, and H2AsO4− links Zr atoms to form a bidentate covalent bond coordination structure, in which the angles are similar to those of the carboxyl in UiO-66 (Fig. S22†).
4 Conclusions
In summary, we have successfully designed and synthesized an adsorbent with plentiful mesopores and active sites of uncoordinated Zr atoms, which overcomes the shortcomings such as the poor specific surface area accessibility and the lack of active sites of the common UiO-66 based adsorbent. The water stable HP-UiO-66-X% has an ultrahigh As(V) uptake capacity (248.75 mg g−1) and is applicable in a wide pH range (2–12). The adsorption mechanism analysis results reveal that H2AsO4− was immobilized by forming Zr–O–As bonds (T-6 model) through splitting the Zr–O bonds in the crystal structure and occupied the uncoordinated Zr atoms in the defect position. The local coordination environment of H2AsO4− on the surface of HP-UiO-66-X% was obtained by DFT calculations, EXAFS and XANES. The T-6 coordination model is suitable to describe the As local coordination environment on HP-UiO-66-X%. This study highlights that the enhancement of coordinatively unsaturated metal atoms and their channel diameter can significantly improve the adsorption performance of oxyanion pollutants (e.g. AsO43−, PO43−, and SeO42−, which have a similar molecular radius and chemical properties). This finding provides a new idea for the design of ultrahigh uptake capacity MOF based adsorbents.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financially supported by the Joint Research Fund for Overseas Chinese Scholars and Scholars in Hong Kong and Macao (51729801) awarded by the National Natural Science Foundation of China (No. 5181001028 and 51438011) and Youth Innovation Promotion Association (CAS).References
- A. van Geen, B. C. Bostick, T. K. Pham, M. L. Vi, M. Nguyen-Ngoc, D. M. Phu, H. V. Pham, K. Radloff, Z. Aziz, J. L. Mey, M. O. Stahl, C. F. Harvey, P. Oates, B. Weinman, C. Stengel, F. Frei, R. Kipfer and M. Berg, Nature, 2013, 501, 204–207 CrossRef CAS PubMed
; J. W. Stuckey, M. V. Schaefer, B. D. Kocar, S. G. Benner and S. Fendorf, Nat. Geosci., 2015, 9, 70–76 CrossRef
- Q. Zhang, S. Bolisetty, Y. Cao, S. Handschin, J. Adamcik, Q. Peng and R. Mezzenga, Angew. Chem., Int. Ed., 2019, 58, 6012–6016 CrossRef CAS PubMed
- C. Wang, J. Luan and C. Wu, Water Res., 2019, 158, 370–382 CrossRef CAS
- H. Furukawa, K. E. Cordova, M. O'Keeffe and O. M. Yaghi, Science, 2013, 341, 1230444 CrossRef PubMed
- X. Ma, L. Wang, Q. Zhang and H. L. Jiang, Angew. Chem., Int. Ed., 2019, 58, 12175–12179 CrossRef CAS PubMed
- C. A. Trickett, K. J. Gagnon, S. Lee, F. Gandara, H. B. Burgi and O. M. Yaghi, Angew. Chem., Int. Ed., 2015, 54, 11162–11167 CrossRef CAS PubMed
- A. Chakraborty, S. Bhattacharyya, A. Hazra, A. C. Ghosh and T. K. Maji, Chem. Sci., 2016, 52, 2831–2834 CAS
- X. Liu, N. K. Demir, Z. Wu and K. Li, J. Am. Chem. Soc., 2015, 137, 6999–7002 CrossRef CAS PubMed
- J. E. R. Nightingale, J. Phys. Chem., 1959, 63, 1381–1387 CrossRef
- P. Shao, L. Ding, J. Luo, Y. Luo, D. You, Q. Zhang and X. Luo, ACS Appl. Mater. Interfaces, 2019, 11, 29736–29745 CrossRef CAS
- A. R. Head, R. Tsyshevsky, L. Trotochaud, Y. Yu, L. Kyhl, O. Karslıoǧlu, M. M. Kuklja and H. Bluhm, J. Phys. Chem. C, 2016, 120, 29077–29088 CrossRef CAS
- K. Wang, C. Li, Y. Liang, T. Han, H. Huang, Q. Yang, D. Liu and C. Zhong, Chem. Eng. J., 2016, 289, 486–493 CrossRef CAS
- L. Feng, S. Yuan, L. L. Zhang, K. Tan, J. L. Li, A. Kirchon, L. M. Liu, P. Zhang, Y. Han, Y. J. Chabal and H. C. Zhou, J. Am. Chem. Soc., 2018, 140, 2363–2372 CrossRef CAS
- S. Yuan, L. Zou, J. S. Qin, J. Li, L. Huang, L. Feng, X. Wang, M. Bosch, A. Alsalme, T. Cagin and H. C. Zhou, Nat. Commun., 2017, 8, 15356 CrossRef CAS PubMed
- B. Ghalei, K. Wakimoto, C. Y. Wu, A. P. Isfahani, T. Yamamoto, K. Sakurai, M. Higuchi, B. K. Chang, S. Kitagawa and E. Sivaniah, Angew. Chem., Int. Ed., 2019, 58, 19034–19040 CrossRef CAS PubMed
- S. M. Wu, X. L. Liu, X. L. Lian, G. Tian, C. Janiak, Y. X. Zhang, Y. Lu, H. Z. Yu, J. Hu, H. Wei, H. Zhao, G. G. Chang, G. Van Tendeloo, L. Y. Wang, X. Y. Yang and B. L. Su, Adv. Mater., 2018, 30, e1802173 CrossRef CAS PubMed
- C. Li, J. Huang, H. Zhu, L. Liu, Y. Feng, G. Hu and X. Yu, Sens. Actuators, B, 2017, 253, 275–282 CrossRef CAS
- S. Smolders, T. Willhammar, A. Krajnc, K. Sentosun, M. T. Wharmby, K. A. Lomachenko, S. Bals, G. Mali, M. B. J. Roeffaers, D. E. De Vos and B. Bueken, Angew. Chem., Int. Ed., 2019, 58, 9160–9165 CrossRef CAS PubMed
- Z. Fang, B. Bueken, D. E. De Vos and R. A. Fischer, Angew. Chem., Int. Ed., 2015, 54, 7234–7254 CrossRef CAS PubMed
- J. Li, M. Zhang, Z. Guan, Q. Li, C. He and J. Yang, Appl. Catal., B, 2017, 206, 300–307 CrossRef CAS
- Y. Han, M. Liu, K. Li, Y. Zuo, Y. Wei, S. Xu, G. Zhang, C. Song, Z. Zhang and X. Guo, CrystEngComm, 2015, 17, 6434–6440 RSC
- Y. Wang, L. Li, P. Dai, L. Yan, L. Cao, X. Gu and X. Zhao, J. Mater. Chem. A, 2017, 5, 22372–22379 RSC
- J. Wei, W. Zhang, W. Pan, C. Li and W. Sun, Environ. Sci.: Nano, 2018, 5, 1441–1453 RSC
- M. J. Cliffe, W. Wan, X. Zou, P. A. Chater, A. K. Kleppe, M. G. Tucker, H. Wilhelm, N. P. Funnell, F. X. Coudert and A. L. Goodwin, Nat. Commun., 2014, 5, 4176 CrossRef CAS PubMed
- X. Kong, H. Deng, F. Yan, J. Kim, J. A. Swisher, B. Smit, O. M. Yaghi and J. A. Reimer, Science, 2013, 341, 882–885 CrossRef CAS
- S. Yuan, Y. P. Chen, J. S. Qin, W. Lu, L. Zou, Q. Zhang, X. Wang, X. Sun and H. C. Zhou, J. Am. Chem. Soc., 2016, 138, 8912–8919 CrossRef CAS
- J. H. Kim, Y. J. Jang, J. H. Kim, J. W. Jang, S. H. Choi and J. S. Lee, Nanoscale, 2015, 7, 19144–19151 RSC
- N. R. Glasser, P. H. Oyala, T. H. Osborne, J. M. Santini and D. K. Newman, Proc. Natl. Acad. Sci. U. S. A., 2018, 115, E8614–E8623 CrossRef CAS PubMed
- M. Wang, X. Yu, C. Yang, X. Yang, M. Lin, L. Guan and M. Ge, Chem. Eng. J., 2017, 322, 246–253 CrossRef CAS
- J. He, X. Cai, K. Chen, Y. Li, K. Zhang, Z. Jin, F. Meng, N. Liu, X. Wang, L. Kong, X. Huang and J. Liu, J. Colloid Interface Sci., 2016, 484, 162–172 CrossRef CAS PubMed
- P. Zhao, M. Jian, Q. Zhang, R. Xu, R. Liu, X. Zhang and H. Liu, J. Mater. Chem. A, 2019, 7, 16598–16621 RSC
- D. Wang, S. E. Gilliland, X. Yi, K. Logan, D. R. Heitger, H. R. Lucas and W. N. Wang, Environ. Sci. Technol., 2018, 52, 4275–4284 CrossRef CAS PubMed
- L. Yan, J. Song, T. Chan and C. Jing, Environ. Sci. Technol., 2017, 51, 6335–6341 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available: Details of materials and methods, DFT calculations, EXAFS and XANES. See DOI: 10.1039/c9ta13747e |
| This journal is © The Royal Society of Chemistry 2020 |