
A comprehensive review on oxidative desulfurization catalysts targeting clean energy and environment
Antony
Rajendran
 ,
Tian-you
Cui
,
Hong-xia
Fan
,
Zhi-fen
Yang
,
Jie
Feng
and
Wen-ying
Li
*
,
Tian-you
Cui
,
Hong-xia
Fan
,
Zhi-fen
Yang
,
Jie
Feng
and
Wen-ying
Li
*
Training Base of State Key Laboratory of Coal Science and Technology Jointly Constructed by Shanxi Province, Ministry of Science and Technology, Taiyuan University of Technology, Taiyuan 030024, PR China. E-mail: ying@tyut.edu.cn; Fax: +86 351 6018453; Tel: +86 351 6018453
First published on 2nd January 2020
Abstract
Harvesting clean energy from fuel feedstocks is of paramount significance in the field of environmental science. In this dynamic area, desulfurization provides a valuable contribution by eliminating sulfur compounds from fuel feedstocks to ensure the utilization of fuels without the emission of toxic sulfur oxides (SOx gases). Nonetheless, the inadequacy of the current industrial technique (hydrodesulfurization, HDS) in the removal of refractory sulfur (RS) compounds and the stringent rules imposed on the fuel sulfur level have kindled research on other desulfurization methods like oxidative desulfurization (ODS). With the capacity of eliminating RS compounds under mild conditions, ODS is endorsed as a suitable replacement or complementary to HDS. ODS, in general, consists of two steps: (i) oxidation and (ii) extraction. The oxidation of sulfur compounds is carried out using a suitable catalyst (hereafter termed as an ODS catalyst) in the presence of an oxidant. Choosing a suitable ODS catalyst for industrial applications is still a quest among the various types of catalysts reported so far. With this outline, herein, all the types of ODS catalysts along with their synthetic methods, reactivity and mechanistic insights are reviewed. The activity of ODS catalysts could be influenced by factors like the type of RS compound, solvent, fuel, etc. and those factors are reviewed. The effects of ionic liquids, light, and ultrasound on the performance of ODS catalysts are also briefly summarized. The opportunities and challenges for ODS catalysts are comprehensively explicated in the end. Through this review, systematic information about the types of ODS catalysts including the basic definition, preparative methods, reactivity and mechanism can be comprehended. Furthermore, this review reveals the merits and demerits related to highlighting catalytic ODS as a replacement or complementary to HDS.
 Antony Rajendran | Antony Rajendran has been doing post-doctoral research in Prof. Wen-ying Li's research lab, Taiyuan University of Technology (TUT), China since October 2018. He obtained his PhD (Anna University) under Prof. Theodore David, India in March 2015. He worked as a post-doctoral fellow with Prof. Murugavel in the Department of Chemistry, IIT Bombay (December 2014–September 2018). Recently, he was selected for the International Young Scientist Research Fund by the National Natural Science Foundation of China. Antony's current research interests mainly focus on designing selective catalysts for ODS and HDS of different liquid fuels. |
 Hong-xia Fan | Hong-xia Fan received her bachelor's degree (2011) and PhD from TUT. During the course of her PhD studies, she worked in Prof. Qingfeng Ge's group, Southern Illinois University, Carbondale, USA as a jointly training doctor (October 2015–February 2017). Subsequently, she joined the research group of Prof. Wen-ying Li and has been working as a post-doctoral researcher. Her research interests are the design and preparation of catalysts for CO2 utilization, oxidative dehydrogenation of hydrocarbons, and refining of coal liquefaction oil. |
 Jie Feng | Jie Feng obtained his bachelor's degree (1989) and PhD (1998) in Chemical Engineering and Chemical Technology from TUT, China. Then, he spent a year as a post-doctoral researcher at Monash University, Australia. He was a senior research fellow (0.5 year) at Imperial College, London, UK. Subsequently, he worked at the University of Florida, USA for one year as a Senior Visiting Scholar. Then, he joined the faculty of Chemical Engineering, TUT where he has been working as a Professor. He works on the design of coal-to-liquid fuel processes and scaling-up the reactor for coal/biomass gasification, and optimizing traditional coal conversion processes. |
 Wen-ying Li | Wen-ying Li received her PhD from Dalian University of Technology in 1995. She was a post-doctoral fellow (1 year) at Imperial College, London, UK, a Senior Visiting Scholar (1 year) at the Max-Planck Institute for Coal Research, Germany, and a Senior Visiting Researcher (3 months) at Princeton University and at Southern Illinois University, Carbondale (3 months), USA. Since then, she has been working as a Professor of Chemical Engineering in TUT for about 20 years. She currently focuses on discovering and designing new processes and catalysts for clean conversion technology involving lower rank and poor-quality coals, clean and efficient catalytic pyrolysis, and the gasification of lignite and biomass. |
1. Introduction
Owing to the increasing demand for energy and dwindling fossil fuel reserves, the utility of low-quality fossil feedstocks (e.g. sour and heavy oil) has become inevitable. The combustion of liquid fuels derived from the low-quality fuel feedstocks leads to the augmented emission of unsolicited SOx and NOx gases.1 SOx emission is treated as a severe problem in the world as it adversely affects the environment through acid rain and human beings through various health issues including lung cancer. Furthermore, the sulfur compounds present in fuel feedstocks poison the costly noble metal catalysts in oil refining industries and thus, the profit is decreased.2 For these reasons, environmental protection agencies across the globe have imposed stringent conditions on the sulfur level of liquid fuels.2c Especially, for transportation purposes, countries covering ≈50% of the earth surface have forced themselves to utilize on-road diesel containing very limited sulfur (10–15 ppm) since 2018.3 Interestingly, these stringent conditions on the sulfur level have been tightening over the years, which can be realized from the trend of stringent rule implementation on the sulfur level of motor gasoline and diesel fuel across different regions (Fig. 1).3 No wonder the utilization of liquid fuels with zero sulfur may become mandatory in the coming years. Consequently, the efficient elimination of sulfur from fuel feedstocks has become a hot topic in research.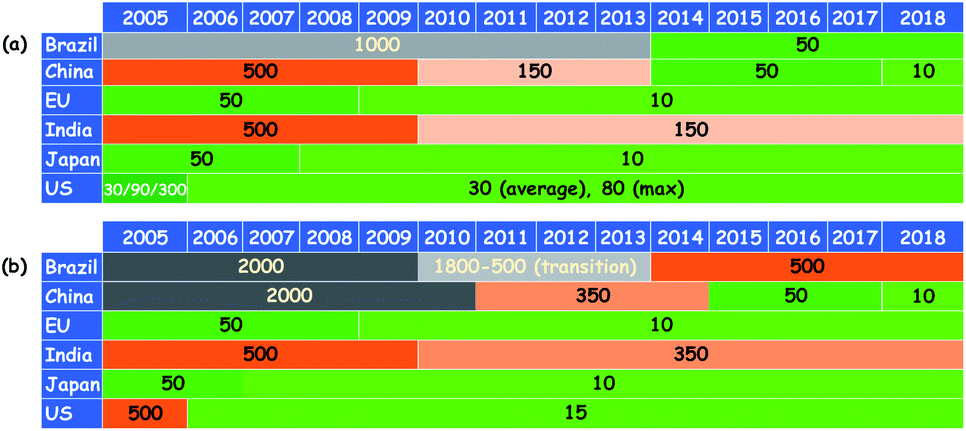 | ||
| Fig. 1 The trend of stringent rules imposed on the sulfur level of motor liquid fuels over the years in different regions ((a) gasoline and (b) diesel fuel). Adapted from ref. 3 with permission from The International Council on Clean Transportation. | ||
While the inorganic sulfur constituents such as elemental sulfur, H2S and pyrite (FeS2) are easily eliminated from the fuel feedstocks, the removal of organosulfur compounds is somewhat tricky. Irrespective of the sources (coal/crude oil), the fuel feedstocks invariably contain organosulfur compounds of different varieties (sulfides, disulfides, mercaptans and thiophenes) (Fig. 2), despite the difference in their quantities. In the industries, organosulfur compounds are traditionally removed from the fuel feedstocks by hydrodesulfurization (HDS) using a sulfided bimetallic catalyst (CoMo/Al2O3 or NiMo/Al2O3).4 Chemically, during HDS, sulfur compounds are converted into aliphatic hydrocarbons with the elimination of H2S gas. HDS is usually accomplished by following two reaction routes (direct desulfurization and hydrogenation), which have been extensively studied in previous reports.4,5 Though well optimized and industrialized, HDS has several pitfalls as listed below: (i) the necessity for high temperature (593–673 K) and pressure (20–60 bar), (ii) the need for expensive H2 gas in large quantities, (iii) the selectivity problems as the result of π-bond hydrogenation of olefins (leading to greater hydrogen requirement), and (iv) inefficiency in removing refractory sulfur (RS) compounds, which occupy the major portion of the total organosulfur content in the fuel feedstocks.6
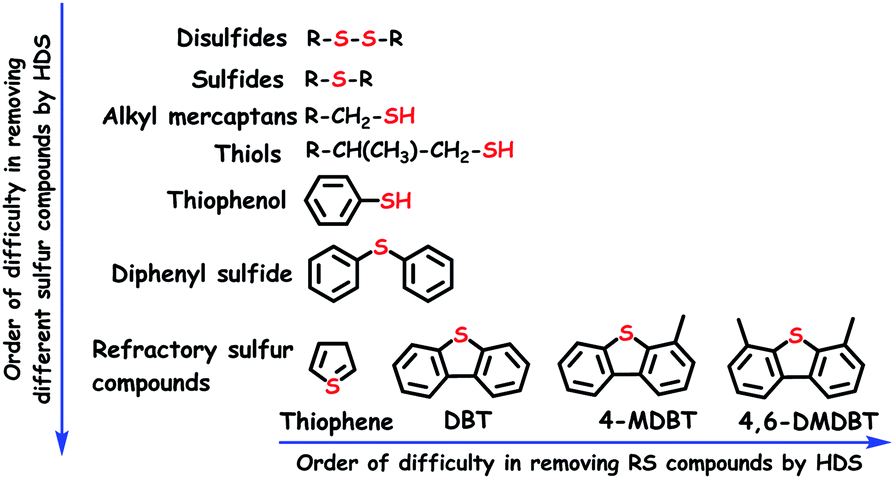 | ||
| Fig. 2 The list of commonly available sulfur compounds in the fuel feedstocks and the order of difficulty in removing different organosulfur compounds during HDS (DBT: dibenzothiophene, 4-MDBT: 4-methyl DBT and 4,6-DMDBT: 4,6-dimethyl DBT). | ||
The efficiency order of HDS towards the removal of different organosulfur compounds including RS compounds from the fuel feedstocks is given in Fig. 2. It shows that removing RS compounds is difficult by HDS as compared to other organosulfur compounds. The n-electrons located on the sulfur atom of RS compounds undergo delocalization with aromatic rings (Fig. 3), creating more resonance structures and extra stability. Though the resonance stabilization energy of RS compounds (120–130 kJ mol−1) is relatively low as compared to that of benzene (160–170 kJ mol−1), it is still sufficient to make HDS inefficient and energetically demanding to break the C–S bond of RS compounds.7 Apart from this, the resonance of n-electrons tunes the energy of the C–S bond to be practically identical to that of the C–C bond, which reduces the selectivity of HDS and promotes the hydrogenation of C–C π-bonds.6
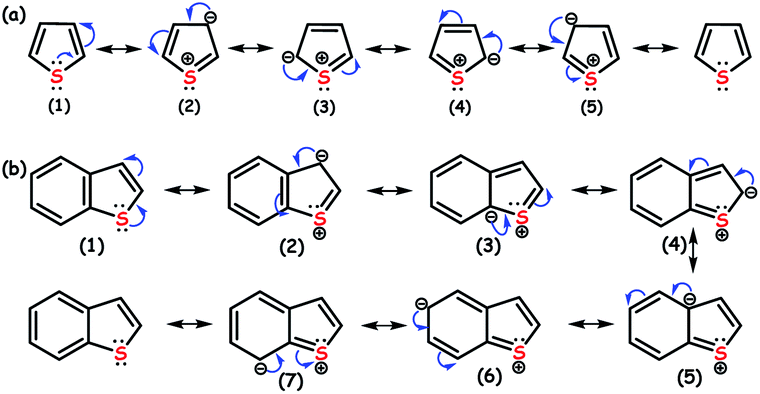 | ||
| Fig. 3 Formation of resonance structures promoting the stability of (a) thiophene (Th) and (b) benzothiophene (BT). | ||
In addition to the resonance structure, the greater electron density and steric factor around the sulfur atom also promote the stability of RS compounds and as a result, the current HDS has become inefficient to treat RS compounds. As a result, a portion of RS compounds remain in the liquid fuels which in turn leads to the emission of harmful SOx gases upon fuel combustion. For the complete removal of RS compounds, the traditional HDS requires more catalyst (3 times extra), an increase in initial temperature by 311 K and an increased amount of H2 gas (50–100% increase).8 However, these suggestions seem to be practically impossible in light of the modifications that would need to be made in the reactor design and economy. Thus, research into finding new HDS catalysts which could possibly achieve 100% removal of RS compounds has been promoted over the years; however, no alternative HDS catalysts have been industrialized till now. This has prompted researchers to find a different way to accomplish 100% removal of RS compounds from liquid fuels under mild conditions with the aim of lowering the final fuel price and economizing the desulfurization energy consumption.9
The literature has evidenced a lot of alternative methods to treat RS compounds such as extractive desulfurization, adsorptive desulfurization, oxidative desulfurization (ODS), bio-desulfurization, and alkylation-based, chlorinolysis-based and supercritical water-based desulfurization. All these types are well documented and explained in the literature elsewhere.7
Among these methods, ODS has been frequently suggested as the best technique due to the mild operation conditions and the ease of separation of oxidized sulfur compounds (sulfones). Since the first report of ODS published in 1967,10 a lot of attempts have been made at ODS. Significantly, the number of reports on ODS has steadily increased over the years with the motive of satisfying the needs of environmental protection agencies regarding the sulfur level in the liquid fuels (Fig. 4).
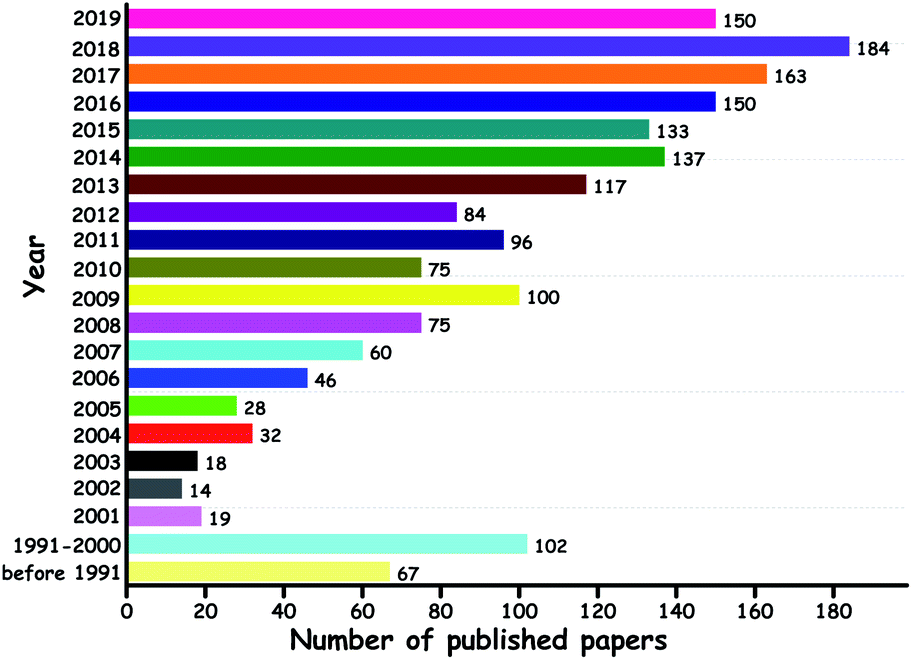 | ||
| Fig. 4 Papers published on ODS over the years (source: SCOPUS with the keyword “oxidative desulfurization”, dated 13th August 2019). | ||
Till now, different types of catalysts with different properties have been reported for the ODS technique. Knowing for a fact that a review that exclusively covers all the types of ODS catalysts is yet to be reported, in this review, we present and discuss all types of ODS catalysts under different classifications by analysing their activities and properties. The mechanistic action of the catalysts in ODS has been emphasized. Furthermore, the activity of ODS catalysts under ultrasonication and photochemical conditions is elaborated in this review. At the end of the review, the conversion and utility of oxidized sulfur compounds are discussed because they are also significant for recommending the ODS process to the industries.
2. Description of ODS
ODS is a process that removes sulfur compounds from fuel feedstocks through the oxidation reaction in the presence of a suitable oxidizing agent with/without a catalyst. In ODS, sulfur compounds of the fuel feedstocks undergo sequential oxidation to yield the end-product sulfones (initially to sulfoxide and finally to sulfone). During ODS, the oxidants chemically transfer their reactive oxygen species to the sulfur compounds for the formation of sulfones. Briefly, ODS consists of two stages: (i) the oxidation of sulfur compounds to sulfones and (ii) the removal of sulfones.4,6,9 The sharp changes in polarity and reduction in C–S bond energies (by 5.2 kcal mol−1 and by 11.8 kcal mol−1 for aliphatic and aromatic sulfides/RS compounds, respectively) of sulfur compounds after ODS make sulfones easily separable from the hydrocarbons (oil) and easily processable for further use.6 Sulfones can be separated from the fuel feedstocks through either solvent extraction, adsorption or thermal distillation. In solvent extraction, polar solvents are used to extract sulfones from non-polar oils that are rich in non-polar hydrocarbon compounds. Suitable sorbents that selectively adsorb sulfones are more desirable for the removal of sulfones through the adsorption method. Thermal distillation is a process of separating sulfones from fuel feedstocks on the basis of their boiling temperatures with the fact that sulfones have a higher boiling point than their parent sulfur compounds. Thus, by collecting only the lower boiling fractions, sulfones can be made to settle as residues.11 In the literature, solvent extraction is often utilized for the removal of sulfones because it is a relatively handy process. A schematic of the ODS process combined with solvent extraction is depicted in Fig. 5. According to Guth and Diaz, the solvents used for the extraction of sulfones should have the following characteristics: (a) immiscibility in oil, (b) ability to dissolve oxidized sulfur compounds, (c) relatively low boiling point for the easy separation of sulfones and oil, (d) inability to readily form an emulsion with the oil, (e) substantial density difference as compared to the oil for the ease of separation, (f) low price as compared to oil, and (g) ability to maintain the fuel properties of the oil.12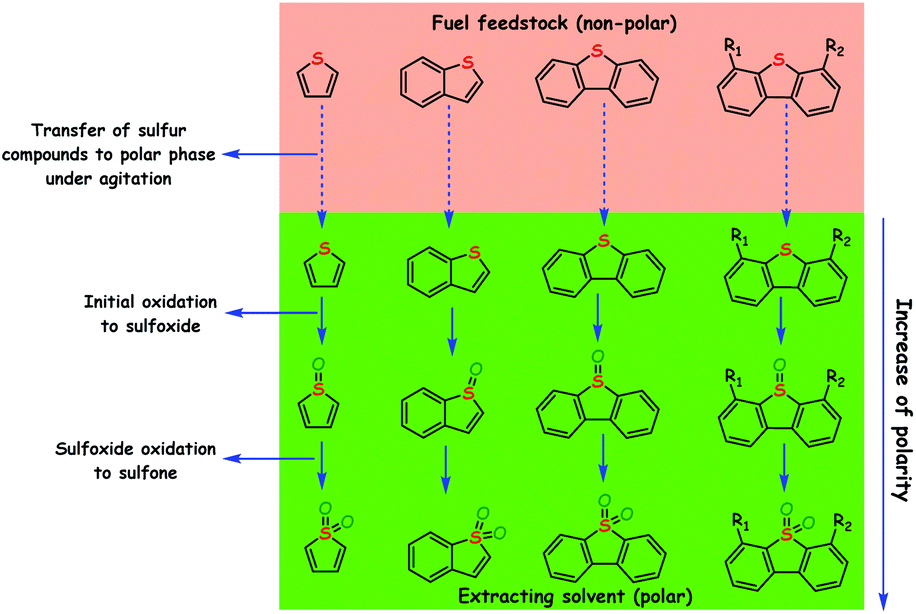 | ||
| Fig. 5 ODS of representative RS compounds (Th, BT, DBT and alkyl substituted DBTs) combined with solvent extraction working on the basis of polarity difference (note: the catalyst and oxidant are assumed to be in the polar phase). | ||
It is significant to note that the oxidation of olefins and other hydrocarbons should be carefully circumvented during ODS because it may negatively/positively influence the cetane/octane rating of diesel/gasoline.4,14 The added advantage of ODS is that the strong affinity of sulfur for oxygen and lower oxidation potential of sulfur compounds as compared to those of hydrocarbons are expected to promote the selective oxidation of ODS without damaging C–C and C–S bonds. Otsuki et al. have proposed a relationship between the electron density of sulfur in different sulfur compounds and the oxidation rate constant in the ODS process performed using HCOOH and H2O2. It is apparent that the compounds with greater electron density on sulfur are readily eliminated by ODS as evidenced by their oxidation rate constants (Fig. 6). Otsuki et al. have stated that the oxidation of sulfur compounds possessing the sulfur electron density in the range 5.696–5.716 is quite difficult under mild conditions (≤323 K).13 On the other hand, interestingly, Fig. 6 supports the view that the elimination of DBT based RS compounds, which is highly problematic in HDS, is viable and easy in ODS even under mild conditions.
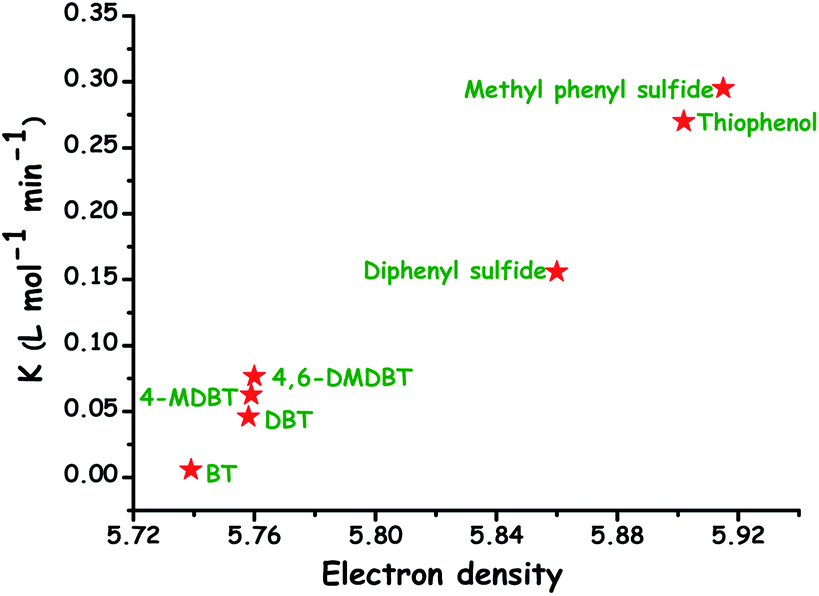 | ||
| Fig. 6 Relation between the electron density on the sulfur atom of different sulfur compounds and their oxidation rate constants in the H2O2/formic acid ODS system. Adapted from ref. 13 with permission from The American Chemical Society. | ||
Over the years, a variety of oxidants such as H2O2, molecular O2, organic peracids, tert-butylhydroperoxide (TBHP), cyclohexanone hydroperoxide, cumene hydroperoxide, ozone, nitrogen oxides and air have been reported in the literature.4,8,9 Organic peracids can be prepared in situ during ODS from the chemical reaction of an organic acid and H2O2.4,9 On the basis of oxidants used for ODS, the ODS system can be broadly classified into three types: two-phase liquid system using water-soluble oxidants, single-phase liquid system with oil-soluble oxidants and gas–liquid system with gas-phase oxidants.15 Despite the list of oxidants, H2O2 is repeatedly emphasized as a better oxidant as it has more active oxygen (47% by mass unit) and leaves only water.8,9 The mechanistic action, advantages and disadvantages of various oxidants in the ODS process have already been extensively discussed in previous review articles.4,8 The order of reactivity seems to vary with the oxidants, catalysts, and operating conditions; however, no major category of organosulfur compounds is unreactive during the ODS process.4 Though yet to be commercialized, due to the efficiency in oxidizing RS compounds to sulfones, which are easily separable from the fuel feedstocks, ODS is presumed to be a good alternative or complementary to HDS in the removal of RS compounds.
3. Catalytic ODS
ODS can also be performed by oxidants without the necessity of any catalyst but it often yields poor results and requires harsh conditions. Therefore, the need for a catalyst for the efficient and speedy ODS process was emphasized right in the first report of ODS published by J. F. Ford et al.10 In the same patent, it is also mentioned that the catalyst could promote selectively the oxidation of sulfur compounds. However, the progress of research on ODS catalysts reveals that catalytic designing is very important to accomplish the selective oxidation of sulphur compounds.16 In general, catalysts tend to promote the generation of reactive oxygen species (metal peroxo species) using the most wanted oxidants such as H2O2 and O2, and consequently, boost the speed of the ODS process. If a polyoxometalate (POM) with inherent metal peroxo groups is used, such metal peroxo groups could promote ODS using their catalytic oxygen without the need for an oxidant; however, the oxidant is needed to regenerate the metal peroxo species in the POM. According to the traditional catalytic approach, ODS catalysts provide a different energy profile (or reaction pathway) which minimizes the energy barrier associated with ODS and thereby increase the rate of ODS. The practical advantages of catalytic ODS are related to the high economic benefits that are obtained due to the mild operating conditions (low temperature and atmospheric pressure) and the better efficiency enabled using a catalyst which follows green chemistry principle nine.1a A literature survey has evidenced a wide range of catalysts finding applications in ODS, from simple acids to solid composites. Such ODS catalysts are classified into different types, reviewed and discussed in the following sections.4. Types of ODS catalysts
4.1. Metal oxides
With their facile synthesis, stability, easy availability and insolubility in common organic solvents, metal oxides are very attractive catalysts for a variety of organic reactions. The synthesis of metal oxides is straightforward and it generally involves precipitation followed by calcination. Note that pH maintenance is significant while preparing the different metal oxides. Few metal precursors with volatile ligands/anions/cations (e.g. ammonium molybdate and ammonium metatungstate) can be directly calcined to their corresponding metal oxides. Despite the fact that no support is needed for metal oxides to be used as heterogeneous catalysts,18 the support is highly solicited to achieve higher catalytic activity through the higher dispersion and accessibility of the metal oxide on the chosen support. The role of the support in metal oxide catalyzed ODS can be exemplified by the ODS activities of TiO2 (anatase), TiO2 (rutile) and TiO2@SiO2 in the presence of H2O2 under the same reaction conditions. TiO2@SiO2 yields better catalytic activity in comparison with the non-supported TiO2 in any form due to the high dispersion and accessibility of TiO2 particles over SiO2.19 Hitherto, for ODS, a large number of transition metal oxide catalysts have been utilized in the literature either with or without a support. The catalytic activity of any metal oxide in ODS is dependent on the nature of the metal. Amidst the different metal oxides reported so far, Mo,20 W,21 Ti,18b,22 and V23 based metal oxides are repeatedly used for catalytic ODS. Other metal oxides are also occasionally used as the catalysts in ODS.24 It is obvious that transition metal oxides are very attractive catalysts in ODS; however, SnO2 can also be an equally good catalyst in ODS due to its Lewis acidity.25 The variation in the ODS activity of the oxides of different metals can be observed from the work of D. Wang et al. In the ODS of a model oil (DBT in kerosene), the oxides of different metals show different activities under the same reaction conditions and MoO3/Al2O3 exhibits the highest ODS activity (Fig. 7).17 W. A. W. A. Bakar et al. have also emphasized the significance of choosing the oxide of the correct metal for efficient ODS by comparing the catalytic activity of different metal oxides in ODS of various sulfur compounds (Zn < Co < Fe < Mn < Mo).26 In addition to the metal type, there are other significant factors governing the overall catalytic activity of metal oxides in ODS. Those significant factors are discussed below.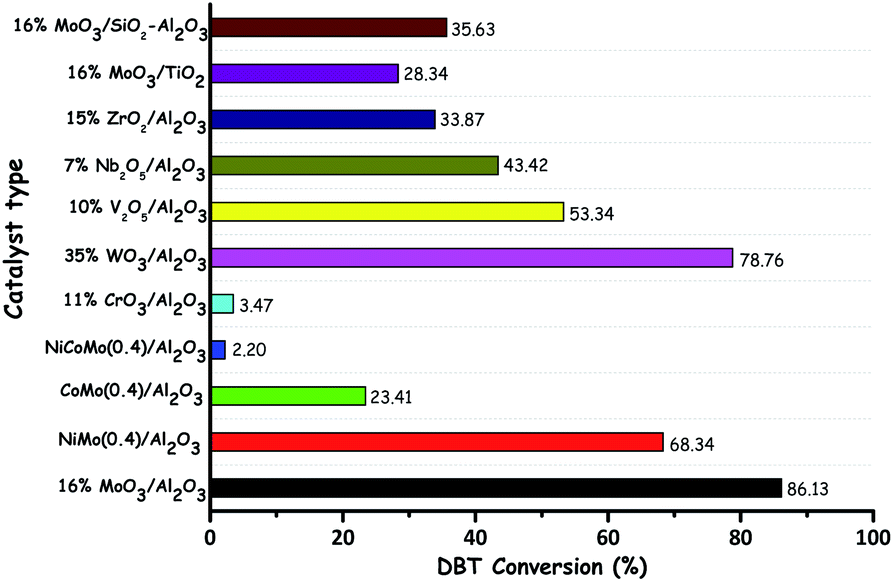 | ||
| Fig. 7 Comparison of the activity of different metal oxide catalysts in ODS of kerosene (DBT in kerosene). Reaction conditions: WHSV (60 h−1); TBHP (O/S = 1.5); catalyst (1 g); T (383 K); t (3 h). Adapted from ref. 17 with permission from Elsevier. | ||
For instance, α-MoO3 is supported on integrated three-dimensional networks of mesoporous SiO2 nanowires with tuneable porosity (19.0–32.5 nm) by impregnation followed by calcination, showing better ODS activity in comparison with other related catalysts (MoO3/SBA-15, MoO3–Al2O3 and MoO3/fumed SiO2). The superior activity is the outcome of the high specific surface area (951 m2 g−1) and highly accessible porosity of SiO2 nanowires that accomplish excellent mass transport during ODS. Advantageously, the oxidized products are effectively adsorbed on the surface of the catalyst, circumventing the requirement for an extractant and promoting green chemistry. The adsorbed oxidized compounds may be removed by solvent washing or thermal treatment from the catalyst surface before the catalyst is utilized in the next catalytic run. The loading of α-MoO3 in the bimodal porous structure of silica nanowires and the solvent-free ODS over α-MoO3 supported silica nanowires are shown in Fig. 8.33
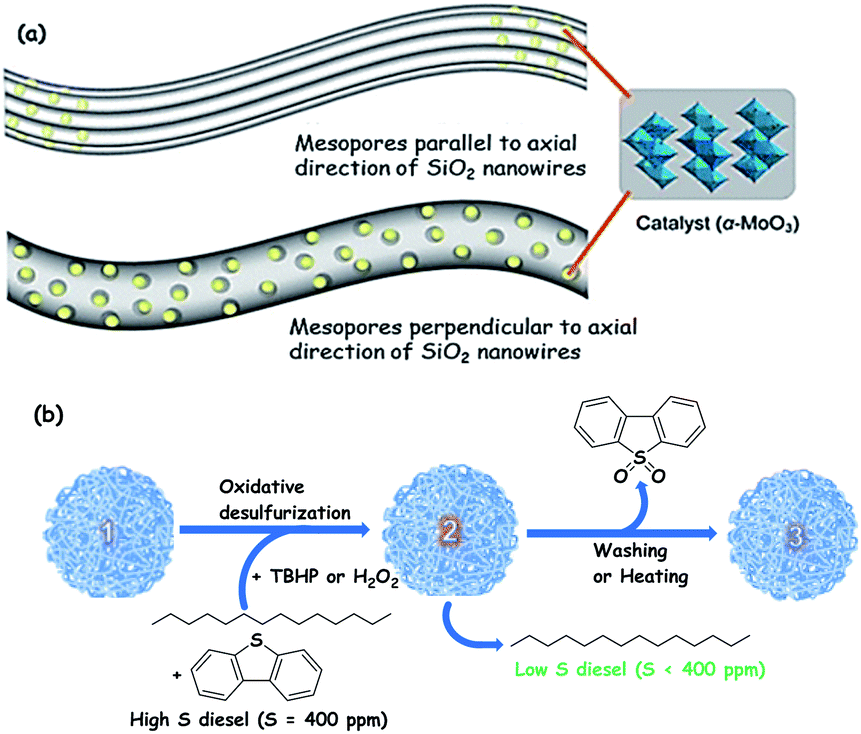 | ||
| Fig. 8 (a) Illustrations of catalyst nanoparticles loaded via two different routes in the mesoporous SiO2 nanowires (mesopores parallel to the axial direction of SiO2 nanowires and mesopores perpendicular to the axial direction of SiO2 nanowires). (b) Dual functions (catalyst + adsorbent) of Mo/mSiO2 (m denotes mesoporous) in the present investigation of the oxidative desulfurization process (1): fresh Mo/mSiO2 nanowires, (2): dibenzothiophene sulfone adsorbed Mo/mSiO2 nanowires, and (3): washed Mo/mSiO2 nanowires (i.e., equivalent to 1); intricate thin threads represent networked Mo/mSiO2 nanowires with mesopores perpendicular to the axial direction (which is detailed in (a)). Adapted from ref. 33 with permission from The American Chemical Society. | ||
A metal oxide supported over a mesoporous support possessing high surface area can be prepared either by impregnating the already prepared support with the precursors of the metal oxide or in situ preparation (both the metal oxide and support are formed together in one pot). The support with excellent textural properties is prepared using a suitable surfactant. Interestingly, it is possible to introduce different textural properties into a support simply by varying the surfactant during the preparation. In comparison, the in situ preparation is most wanted as it leads to higher dispersion without any agglomeration and is carried out by mixing a surfactant, and the precursors of the support and metal oxide in a solvent mixture (usually water–ethanol) under suitable pH conditions.35 Surprisingly, using nanocellulose as a template, D. Shen et al. prepared mesoporous WO3−x–SiO2 to perform catalytic ODS in which 100% sulfur removal is realized within just 15 minutes. As shown in Fig. 9, it leads to much better ODS activity as compared to that of bulk WO3. The faster ODS rate may be attributed to the efficient and quick mass transport accomplished by the textural properties of the WO3−x–SiO2 catalyst.34
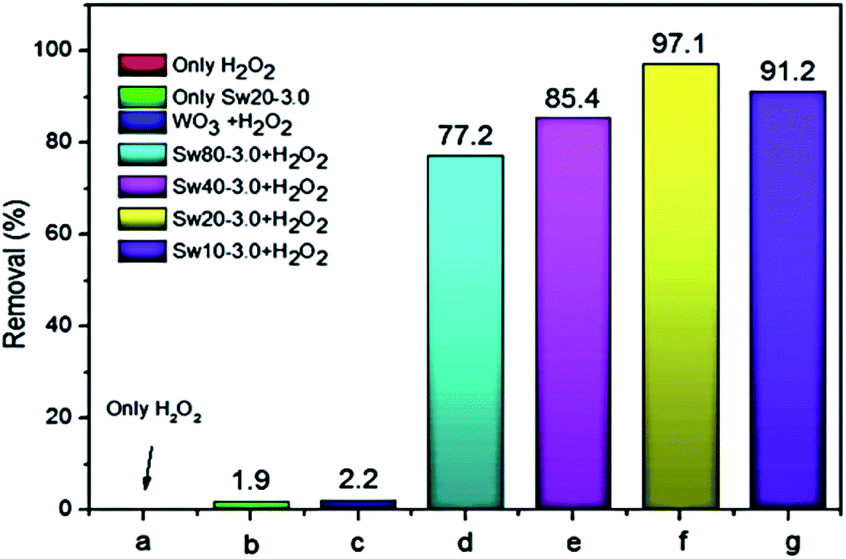 | ||
Fig. 9 ODS performance of DBT under different conditions with various catalyst Swx–y ratios (x = Si![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :W molar ratio and Y = nanocellulose:TEOS mass ratio) (where TEOS = tetraethyl orthosilicate). (a) Only H2O2, (b) only Sw20–3.0, (c) bulk WO3 and H2O2, (d) Sw80–3.0 and H2O2, (e) Sw40–3.0 and H2O2, (f) Sw20–3.0 and H2O2, (g) Sw10–3.0 and H2O2. Reaction conditions: T (323 K), H2O2 (O/S = 4); t (15 min). Adapted from ref. 34 with permission from Elsevier. :W molar ratio and Y = nanocellulose:TEOS mass ratio) (where TEOS = tetraethyl orthosilicate). (a) Only H2O2, (b) only Sw20–3.0, (c) bulk WO3 and H2O2, (d) Sw80–3.0 and H2O2, (e) Sw40–3.0 and H2O2, (f) Sw20–3.0 and H2O2, (g) Sw10–3.0 and H2O2. Reaction conditions: T (323 K), H2O2 (O/S = 4); t (15 min). Adapted from ref. 34 with permission from Elsevier. | ||
Advantageously, in a few cases, the metal precursor required for the preparation of the metal oxide also performs the role of surfactant, evading the need for a surfactant.21h,36 For example, S. Xun et al. prepared mesoporous WO3–SiO2 without using an external template. Briefly, a surfactant like a POM ([C16mim]4SiW12O40) and a silica precursor (TEOS) are mixed together at a certain pH. The obtained gel leads to mesoporous WO3–SiO2 after calcination, executing 100% DBT removal even after 6 consecutive ODS runs.21h,36b In this type of synthesis, POM precursors with higher alkyl chain lengths provide a greater number of mesoporous structures in better orderliness.36b Dispersion of MoOx inside the dendritic mesoporous SiO2 by the modified Stober synthesis leads to the ODS catalyst displaying very high surface area and porosity, which achieves 100% DBT removal within 40 minutes under the chosen reaction conditions. Also, with the use of this dendritic catalyst, no solvent is required and the oxidized sulfur compounds are efficiently adsorbed on the catalyst and removed by solvent washing before the catalyst is utilized in the next run.37
P. Wu et al. directly prepared WOx nanoparticles confined in graphene like boron nitride (g-BN) at higher temperature (1173 K). The synergistic effect is evolved between g-BN and WOx and significantly, WOx is formed in the size range of 4–5 nm inside g-BN. The synergistic effect and nanosize of WOx boost the ODS performance. To obtain the nanosized WOx, the amount of W loading in g-BN is very important because higher loading results in the aggregation of WOx particles, which forms the bulk WOx and limits the activity of the catalyst in ODS (Fig. 10). The confinement of WOx in g-BN is crucial to attain maximum ODS activity since the physical mixture of bulk WOx and g-BN shows relatively lower ODS performance.39 In the same way, nanosized MoOx is confined in g-BN and shows similar results during ODS to WOx/g-BN.40 L. P. Rivoira et al. emphasized the significance of the preparative method to derive nanosized TiO2 supported on mesoporous carbon (CMK-3). One pot synthesis provides a high dispersion of anatase TiO2 on CMK-3 in comparison with preparative methods involving impregnation or nano-casting and consequently, displays better activity in ODS.22c Adapting a reverse microemulsion method, MoO3 sub-nanoclusters are supported on ultrasmall mesoporous SiO2 nanoparticles, showing 100% ODS efficiency within 15 minutes with a turn over frequency of 53.3 h−1.41
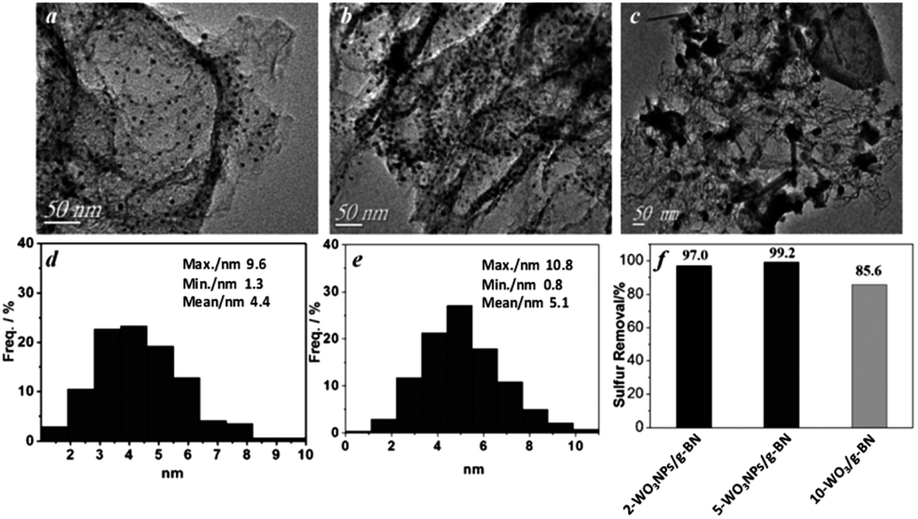 | ||
| Fig. 10 TEM images of (a) 2-WOx nanoparticles/g-BN, (b) 5-WOx nanoparticles and (c) 10-WOx/g-BN. (d and e) Particle size distributions of (a) and (b), respectively. (f) Catalytic oxidation ability of the three materials under the experimental conditions: m(cat) = 50 mg, V(H2O2) = 32 μL, V(oil) = 5 mL, T = 303 K, t = 80 min. Adapted from ref. 39 with permission from John Wiley & Sons. | ||
In addition to the size, the morphology could also influence the activity of metal oxides in ODS. For example, TiO2 nanotubes demonstrate 10% excess ODS efficiency as compared to TiO2 nanoparticles under the same reaction conditions.42 WOx nanobelts, with a width of 10–30 nm, length of 500 nm to a few micrometres and thickness of sub-1 nm, are prepared from solvothermal synthesis using a POM precursor under acidic conditions. With this very interesting morphology, the ultrathin WOx nanobelts display very good activity in ODS of a model oil (DBT in octane) in the presence of H2O2 at 323 K. In comparison with bulk WOx, the activity of WOx nanobelts is much higher in ODS.43 Hexagonal Ga2O3 nanoplates have shown heterogeneous ODS performance without the need for any additional support.44 M. A. Astle et al. encapsulated nanosized MoO2 inside hollow graphitic carbon nanofibers (MoO2@GNFs) following the principle of a “carbon nanosponge” that profitably combines the catalysis of organosulfur oxidation and sequestration of the products from the reaction mixtures without the need for any solvent. Fig. 11a shows the preparation of this new ODS catalyst (MoO2@GNF) that leads to 100% removal of DMDBT, DBT and BT in the order of 4,6-DMDBT > DBT > BT. The sulfur molecules are initially accessed by the internal channels of graphitic carbon nanofibers (Fig. 11b and c) and therein oxidized by MoO2. After the oxidation, the obtained sulfur products are very safely removed from the oil mixture due to the adsorption capacity of a catalyst nanosponge (MoO2@GNF). Reusability studies signify the importance of thermal treatment of the spent catalyst in the removal of the oxidized sulfur compounds. Without the thermal treatment, the oxidized sulfur compounds are continually deposited inside the channels of graphitic carbon nanofibers, blocking the active sites, and consequently, reduced catalytic activity is noted (Fig. 11d).45
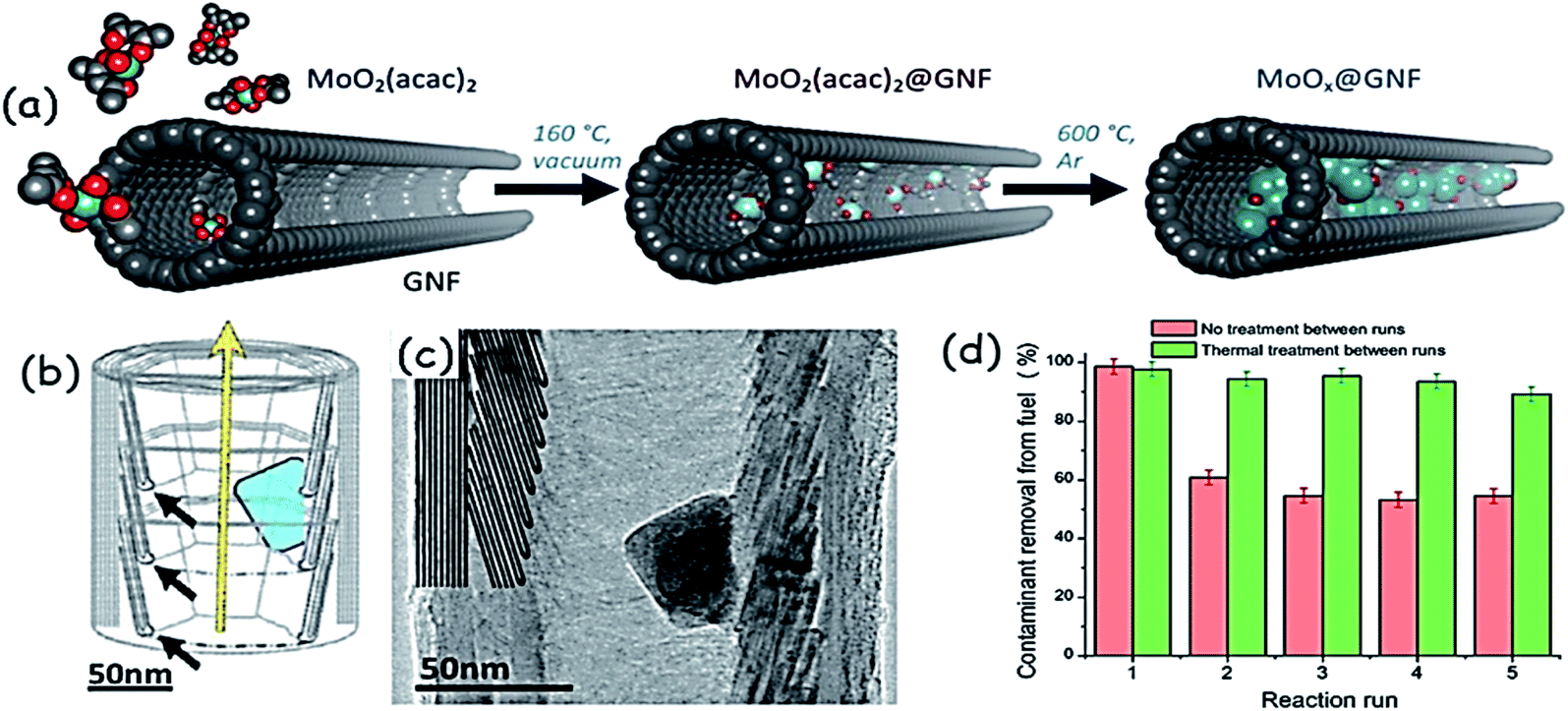 | ||
| Fig. 11 Schematic representation showing (a) the gas-phase encapsulation of MoO2(acac)2 and thermal growth of molybdenum oxide nanoparticles encapsulated within the hollow graphitized carbon nanofibers, and (b) the structure of graphitic carbon nanofibers (interior step-edges are denoted by black arrows; the yellow arrow signifies the direction of the nanofiber growth axis; the blue shape represents a MoO2 nanoparticle). (c) Bright field TEM image of MoO2@GNF. The internal step edges have been highlighted for clarity, with the MoO2 nanoparticle clearly shown residing at the interior step-edges. (d) Recycling experiments using MoO2@GNF with no catalyst treatment between runs for the ODS of DBT indicating a ≈40% lower sulfur removal capacity after the first use, with no significant changes noted over the next four uses. Adapted from ref. 45 with permission from John Wiley & Sons. | ||
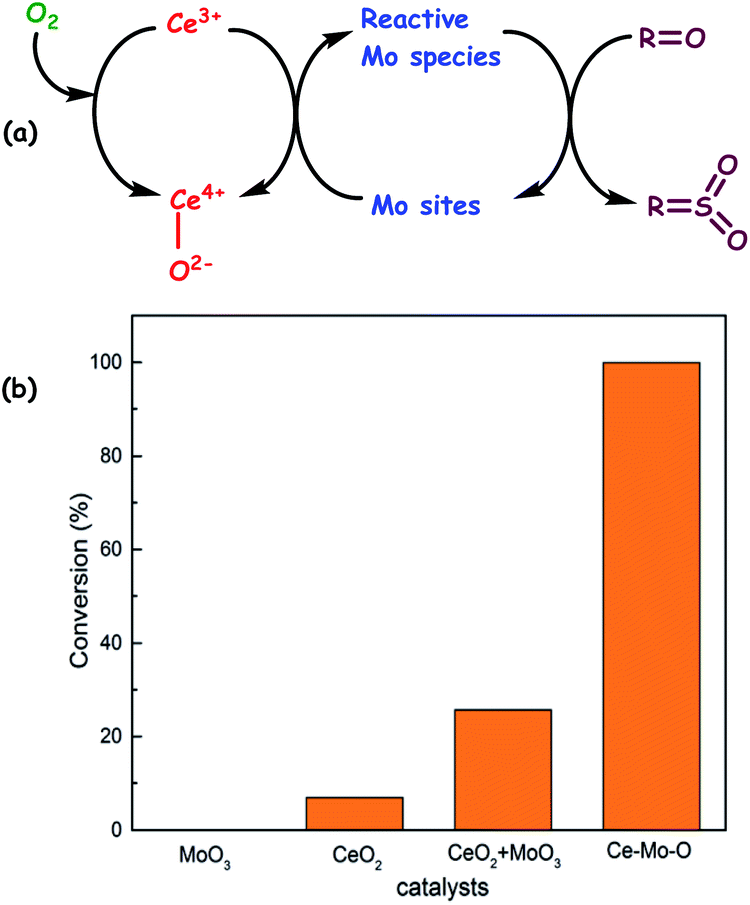 | ||
| Fig. 12 (a) The proposed mechanism for aerobic oxidation of sulfur compounds over the Ce–Mo–O catalyst. (b) Conversion of DBT over different catalysts. Reaction conditions: catalyst (0.1 g), DBT (500 ppmwS), decalin (20 mL), O2 (bubbled in at ambient pressure), T (373 K), t (6 h). Adapted from ref. 50 with permission from The Royal Society of Chemistry. | ||
Significantly, in the catalyst (MoOx–VOx/Al2O3), the interaction between MoOx and VOx promotes the dispersion of isolated vanadium sites on the support which is supported by the strong interaction of MoOx and weak interaction of VOx with the support.54 The addition of cerium/sodium into MoO3@SiO2 has established the electronic interaction between cerium/sodium and molybdenum, resulting in the formation of a small amount of highly active molybdenum(V) sites and better dispersion of MoO3. Thus, the obtained bimetallic catalyst shows improved activity in ODS than the corresponding monometallic oxide.55 Upon adding VOx into WOx–Al2O3, no formation of V–O–W bonds is realized; however, the catalytic activity of the resultant mixed metal oxide in ODS is remarkably enhanced owing to the ability of VOx to increase the catalytically more active WO6 species (increase of the WO6/WO4 ratio). The tungsten atom surrounded by more oxygen in WO6 becomes highly favourable for the nucleophilic attack of peroxide, in comparison with WO4 and thus, better activity is realized.56 While introducing iron into Ti-MCM-41 via impregnation followed by calcination, the derived bimetallic (Fe–Ti) oxide shows positive improvements in the surface oxygen reducibility and acidity which in turn leads to better ODS performance.57
Furthermore, the calcination temperature of metal oxide also plays an important role in defining the activity in ODS. The crystallinity, number of exposed catalytically active sites and mesoporosity of the metal oxide can be tuned simply by varying the calcination temperature, and thus, a change in ODS activity can be expected.25,58 Tuning the activity of a particular metal oxide in ODS is also possible with the choice of metal precursor and preparative method during the preparation of metal oxide. For instance, J. F. Palomeque-Santiago et al. prepared WOx–ZrO2 catalysts by two different methods. In the first method, tungstic acid is involved in an anion exchange with ZrOH and a subsequent calcination which results in more active tetrahedral tungsten sites with more Lewis and Brønsted acid sites. The second method involves the impregnation of ZrO2 with ammonium metatungstate and calcination, leading to less active octahedral W sites for ODS.21j
The real success of ODS in the presence of catalysts lies in the selectivity towards the oxidation of sulfur compounds without oxidizing the olefins of fuel oil. With the use of novel H-titanate nanotubes, selective ODS is carried out in the presence of different olefins. The obtained data are given in Table 1, highlighting that straight-chain alkenes, aromatics and cyclic aliphatics do not have any influence on the rate of ODS. The selectivity of H-titanate is due to the occurrence of the Ti-η1-hydroperoxide mechanism during ODS instead of the Ti-η2-hydroperoxide mechanism which is common in titanosilicate-1 (TS-1) catalysts.61 The Ti-η2-hydroperoxide mechanism favours olefin epoxidation due to the unsaturated coordination of Ti(IV) centres in TS-1 catalysts. As the peroxo ligand is pinned down in the η2-coordination, the proximal oxygen is exposed for the nucleophilic attack by alkenes (marked with * in Scheme 1), yielding an epoxide. However, in H-titanate nanotubes, TiO6 octahedra are connected with each other, forming layers with hydrogen ions existing within the layers. As a result, the already achieved hexacoordinate saturation state is attained and hence, Ti-η1-hydroperoxide becomes more feasible with H-titanate nanotubes in the presence of H2O2.61 Furthermore, certain metal oxides are well-known photocatalysts for ODS under the action of light.62
| Content | X/% | t/min | |||
|---|---|---|---|---|---|
| 0 | 10 | 20 | 30 | ||
|
a Reaction conditions: m (catalyst) = 0.1 g, T = 313 K, n (H2O2)/n (DBT) = 4, v (oil)/v (methanol) = 1:1, t = 30 min.
|
|||||
| None | — | 43.41 | 82.36 | 97.97 | 99.80 |
| 1-Octylene | 5 wt% | 42.53 | 82.97 | 97.98 | 99.31 |
| 10 wt% | 41.33 | 82.47 | 98.83 | 99.28 | |
| 15 wt% | 42.19 | 82.92 | 98.31 | 99.75 | |
| 20 wt% | 43.63 | 82.89 | 99.31 | 99.55 | |
| Xylene | 5 wt% | 42.31 | 79.64 | 97.55 | 99.53 |
| 10 wt% | 41.53 | 81.92 | 97.08 | 99.21 | |
| 15 wt% | 42.25 | 80.79 | 97.43 | 98.86 | |
| 20 wt% | 42.53 | 83.24 | 97.27 | 99.80 | |
| Cyclohexane | 5 wt% | 45.24 | 83.76 | 96.69 | 99.41 |
| 10 wt% | 43.52 | 81.76 | 97.06 | 98.93 | |
| 15 wt% | 44.00 | 82.77 | 98.69 | 99.77 | |
| 20 wt% | 43.12 | 83.17 | 97.12 | 99.29 | |
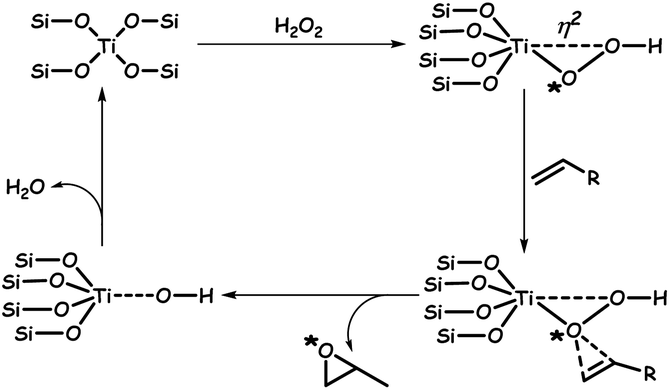 | ||
| Scheme 1 Mechanism for the epoxidation of olefins by TS-1 catalysts. Adapted from ref. 61 with permission from The Royal Society of Chemistry. | ||
4.2. Titanosilicate catalysts
The discovery of the first titanosilicate (TS-1) by Taramasso et al. has made a great impact in oxidation reactions due to its catalytic activity, especially in the presence of H2O2.63 Titanosilicates (TSs) are zeolitic metal oxides possessing no aluminium sites in their structure. Structurally, in TSs, silicon(IV) ions are isomorphously substituted by titanium(IV) ions. The hydrophobicity of the silicate framework in TSs sets a platform for the facile diffusion of non-polar substrates to the active sites and thus promotes the oxidation of organic substrates ranging from alkenes to sulfides.64 TSs are broadly classified into four types (conventional, hierarchical, lamellar and mesoporous TSs) in view of their textural properties, size and shape of the pores, and the location and content of Ti. More details about TSs and their role in oxidation reactions can be seen in a review article published by J. Přech.63bOwing to the above-stated benefits of TSs in oxidation reactions, ODS has embraced the catalytic utilities of TSs. In this section, the TS catalysts used so far in ODS are reviewed. Among the reported TSs, TS-1 (with MFI (Mobil Five) topology) is a familiar catalyst in ODS due to the ease of its well-established synthesis. The initially reported TS-1 is a microporous solid and thus, it could not exhibit better performance in ODS of bulky sulfur compounds like DBT. This leads to the appearance of new TS-1 ODS catalysts with mesoporosity or bimodal porosity (co-existence of microporosity and mesoporosity). Mesoporous TS-1 catalysts are generally prepared by soft templated or hard templated synthesis. For the soft templated synthesis, the structure directing agents namely surfactants are taken along with the titanium(IV) and silicon(IV) precursors in a definite ratio during the preparation.65 Of note, the type of surfactant and ratio between the reactants are the key factors during the preparation of porous TS-1 catalysts since they could influence the textural properties and porosity.
As an example of soft-templated synthesis of hierarchical TSs with a bimodal structure, Du et al. utilized a green and cheap surfactant (Triton X-100) under hydrothermal conditions (Fig. 13a). As expected, the adjustment of surfactant content in the preparative mixture has a significant impact that leads to the formation of three hierarchical TSs (HTS-1A, HTS-1B and HTS-1C) with different textural properties. Briefly, the optimized molar composition to obtain the three different hierarchical TSs (HTSs) is SiO2: 0.033TiO2:0.2 tetrapropylammonium hydroxide: 1.5CH3CH2OH:9H2O: nTriton X-100 (n = 0.102, 0.204 and 0.408). Significantly, the prepared HTSs showed bimodal porosity (micro and mesopores). While the obtained HTSs, due to their bimodal porosity, exhibit better performance in the removal of Th and DBT in ODS, the conventional TS-1 shows relatively poor performance in the removal of DBT under identical reaction conditions due to its microporosity (Fig. 13b and c).65d The added advantage of TS-1 catalysts with a bimodal porous structure is that the co-existence of micropores and meso/macropores could accomplish the best ODS performance invariably towards the small and bulky sulfur compounds that are present in fuel feedstocks.66 When a hard template (CMK-3) is used, the prepared TS-1 also displays a bimodal porous structure in which mesoporosity is introduced through the packing of individual TS-1 nanocrystals. As a result, the obtained TS-1 demonstrates 100% removal both in the case of Th and DBT within 1 h, highlighting the importance of the bimodal pore structure. The conventional TS-1 performs well in the ODS of Th (100%) but very poorly in the ODS of bulky DBT (0%) as it lacks a bimodal porous structure.67 Sugar molecules like caramel and sucrose initially act as the soft template and later change into the hard carbon template during the course of preparation of TSs, yielding hierarchical TS-1 with bimodal porosity.68 Essentially, in this method, two templates are used viz. tetrapropylammonium bromide and carbon material derived from the sugar carbonization for generating microporosity and mesoporosity, respectively.
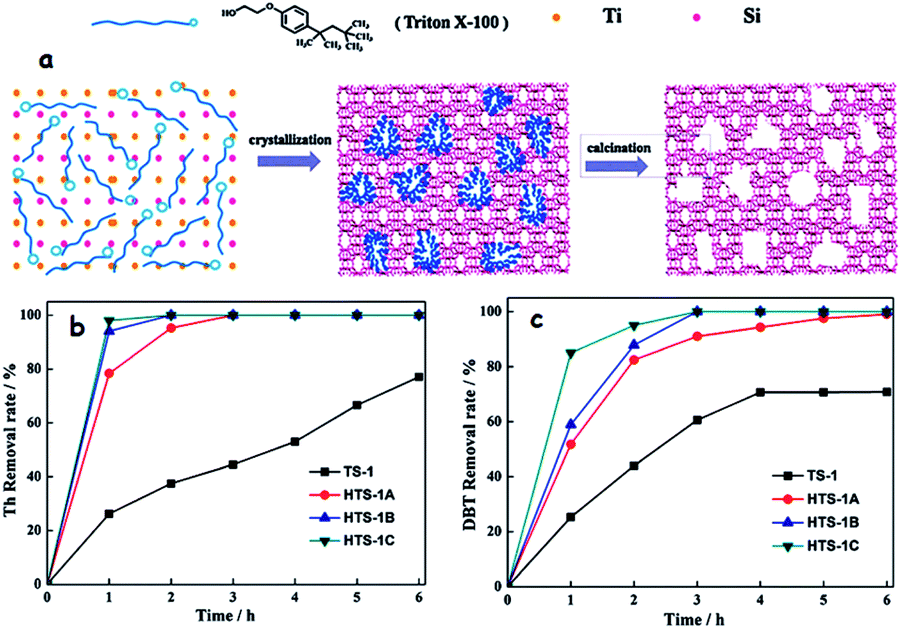 | ||
| Fig. 13 (a) Proposed formation process of hierarchically porous TS-1 using Triton X-100 surfactant as the mesoporous template. Oxidation of (b) Th and (c) DBT over hierarchically porous TS-1 and conventional TS-1 zeolites. ODS conditions: 10 mL model fuel (500 ppm sulfur compound in n-octane; 10 mL H2O; 30% H2O2 (O/S = 2); 50 mg catalyst and 333 K). Adapted from ref. 65d with permission from The Royal Society of Chemistry. | ||
In addition to the surfactants, controlling the crystallization conditions has a significant influence on fabricating TS-1 catalysts with desirable porosity which in turn promotes the ODS activity.65a,69 R. Bai et al. synthesized HTS-1 by a seed-assisted (TS-1(C3)) hydrothermal method with different crystallization times (Fig. 14A). While the zeolite HTS-1 obtained after intermediate crystallization (1 h) exhibits abundant intracrystalline and intercrystalline mesopores due to the aggregated nano-sized crystals (5–10 nm), the HTS-1 produced after complete crystallization (24 h) only displays abundant uniform intracrystalline mesopores (Fig. 14B). As a result, the HTS-1 (1 h/3 h) obtained by intermediate crystallization promotes 100% removal of DBT and 4,6-DMDBT during ODS but the HTS-1 (24 h) produced by complete crystallization leads to the removal of only 55.5% DBT and 50.4% 4,6-DMDBT under identical conditions (Fig. 14C).70 In other reports, multi-step crystallizations lead to the formation of TS-1 catalysts with a bimodal porous structure for ODS.69
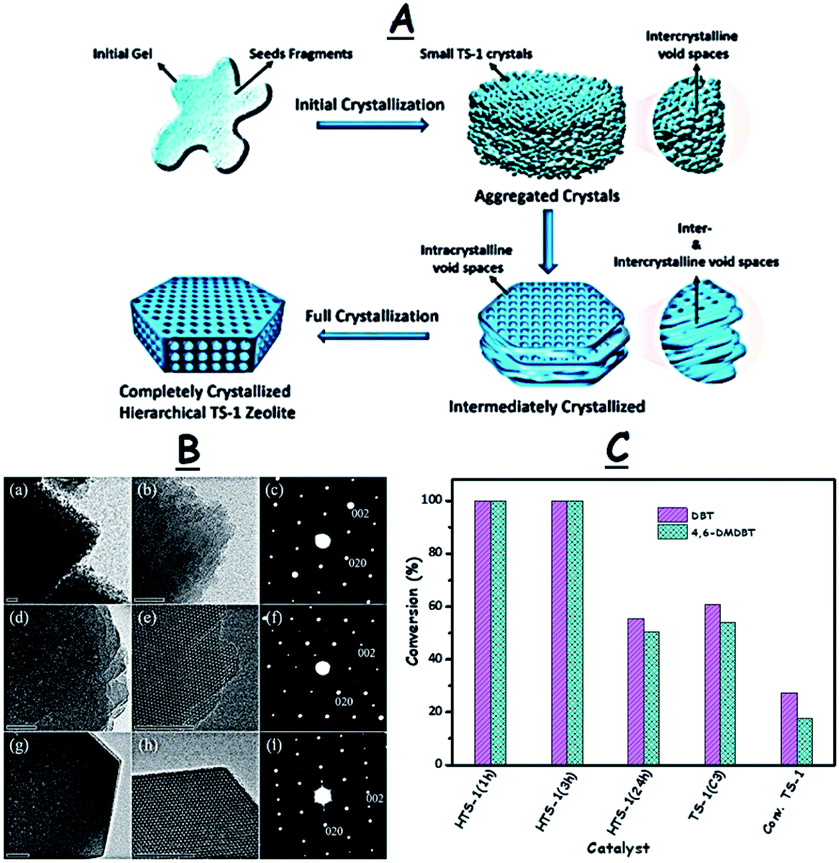 | ||
| Fig. 14 (A) Synthesis of the intermediately and completely crystallized zeolite catalysts. (B) TEM images and selected area electron diffraction patterns of nano-sized hierarchical HTS-1 catalysts prepared with different crystallization times, 1 h (a–c), 3 h (d–f), and 24 h (g–i) (scale bar, 20 nm). The SAED (c, f and i) patterns display the discrete spots indexed to the (002) and (020) planes corresponding to the MFI phase, indicating the single-crystalline nature of HTS catalysts. (C) ODS performance of different HTS catalysts. Reaction conditions: 10 mL model oil (500 ppm DBT in n-octane); TBHP (O/S = 0.5); T (333 K); t (30 min). Adapted from ref. 70 with permission from The Royal Society of Chemistry. | ||
HTS-1 having a bimodal structure was also prepared by a top-down approach involving the chemical etching of conventional TS-1. Two different etching methods were used. In the first method, the etching is carried out using a mixed aqueous solution of HF and NH4F. The second etching method involves a consecutive treatment of conventional TS-1 with NaOH and HF–NH4F aqueous solution. Among the two preparative etching methods, the second method yields HTS-1 with more open pores which thus shows better activity, especially in the later part of the catalytic run. HTS-1 prepared by the second method exhibits 100% DBT removal but the conventional TS-1 accomplishes only 40% DBT removal under identical reaction conditions.71
Apart from TS-1, a few other TSs possessing different topologies are reported as the catalysts for ODS such as Ti-MCM-41,72 Ti-MWW,73 Ti-SBA-16,74 Ti-SBA-15,75 and Ti-SBA-2. These TSs are synthesized via surfactant-mediated synthesis except Ti-MWW which involves a different strategy. Briefly, for being utilized as an ODS catalyst, TS-MWW is prepared in the presence of boric acid as reported by P. Wu et al.73a,76 Germanosilicates are familiar due to their inherent bimodal porosity that emerged due to the additional large pores formed during the construction of the zeolite framework. In fact, germanium atoms facilitate the construction of small building units similar to a double 4-ring (D4R) which is the reason for the emergence of those extra-large pores. Therefore, due to the intrinsic bimodal porosity, titanium-containing germanosilicates (Ti-UTL) might be the finest catalysts for the removal of all sulfur compounds, irrespective of the size, during ODS. However, the instability of Ge–O bonds in Ti-UTL may discount this choice. To overcome this barrier, a facile synthetic method is adopted in which stable Ti-UTL is realized under mild conditions with the assistance of ·OH radicals. Structurally, in this new method, increased isomorphous silicon substitution by germanium atoms takes place. Interestingly, the thus-prepared Ti-UTL removes a very bulky molecule (4,6 DMDBT) in ODS which is 70% higher than that removed by the conventional TS-1 under identical conditions.77
Irrespective of the type of TS, different catalytically active metal ions such as palladium,78 silver,79 gold73c,80 and copper81 are impregnated in the framework of TS to achieve better ODS performance. Very recently, G. Lv et al. synthesized tungsten-incorporated TS-1 not via the impregnation but by a one-pot method. This tungsten containing TS-1 shows more Lewis acid sites as well as high surface area and thus exhibits better ODS performance as compared to the conventional TS-1.82 By dispersing gold(0) nanoparticles over a TS (Ti-HMS), H.-Y. Song et al. accomplished the in situ generation of H2O2 from H2 and O2 which is later used in catalytic ODS.80 The in situ synthesis of H2O2 for ODS from H2 and O2 may be intriguing for future industrial applications as it evades the unsolicited storage of H2O2. In addition to introducing catalytically active metal ions, supporting TS over certain supports can also enhance the activity. Supporting TS-1 over porous glass beads enhances the surface area of the resultant supported TS-1 and hence, the removal of bulky sulfur compounds is enhanced during ODS. Such an improved activity is attributed to the larger surface area caused by TS-1 as a result of immobilization.83
The presence of free silanols in TS catalysts may diminish the lifetime of TS during the course of repeated use in ODS since the free silanols tend to adsorb sulfones (the oxidized sulfur compounds) which in turn deactivates the TS catalysts. However, this problem can be circumvented by silylating the surface silanols with suitable silylating agents. In the work of A. Chica et al., the silylated Ti-MCM-41 suppresses the adsorption of sulfones by 10-fold in comparison with the non-silylated Ti-MCM-41, leading to enhanced catalyst life.84 Like TS, vanadosilicate is also known to promote ODS.85
4.3. Polyoxometalates
Despite the fact that a polyoxometalate (POM) and heteropolyacid (HPA) are a little different (HPA is the acid form of its corresponding POM, and vice versa; a POM is the conjugate anion of HPA),86 in this review, they have been discussed together as POMs due to their similar structures and properties. POMs are molecular compounds comprising structurally diverse anionic metal–oxygen clusters that are mainly based on early transition metals. POMs are broadly classified into isopolyanions ([MmOy]q−), which have only one metallic element (M) and heteropolyanions ([XrMmOy]q−), which additionally have a heteroelement (X).87 Owing to the fast and reversible multi-electron properties, POMs are utilized as well-known catalysts for the oxidation of diverse organic substrates including alkanes, aromatics, alcohols, olefins and sulfides.88 For ODS, Dawson,89 Anderson,90 and Keggin91 type POMs are frequently used as catalysts. Keggin type POMs are very familiar among the POM ODS catalysts due to their high thermal stability and ease of synthesis.86 Lindqvist POMs,92 decavanadates,93 and decatungstates94 are also occasionally used as ODS catalysts. It is presumed that a discussion about the different types of POMs would be redundant here because they have already been extensively discussed.95 However, the basic structures of POMs (Keggin, Dawson, Anderson, Lindqvist, decatungstate and decavanadate) that are used as ODS catalysts are given in Fig. 15.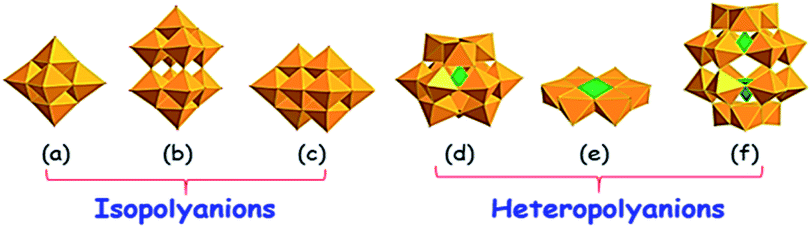 | ||
| Fig. 15 Representation of the typical POM structure of (a) Lindqvist ([M6O19]n−), (b) decatungstate ([W10O32]4), (c) decavanadate ([V10O28]6−), (d) Keggin ([XM12O40]n−), (e) Anderson ([Hx(M′O6)M6O18]n−) and (f) Dawson ([X2M18O62]n−). Adapted from ref. 95 with permission from Elsevier. | ||
In general, synthesis of POMs involves a very simple aqueous phase strategy. Basically, POMs are prepared from acid solution containing relevant metal-oxide anions (mostly molybdate and tungstate). In contrast, vanadate-based POMs are prepared at relatively higher pH.96 Though the synthesis of POMs needs a small number of steps or even just one step (“one-pot synthesis”), there are some synthetic variables holding the greatest importance such as concentration/type of metal-oxide anion, pH, ionic strength, heteroatom type/concentration, the presence of additional ligands, reducing agents, temperature of reaction and processing methods.95 Apart from the common synthesis, to look for POMs with very different properties, new synthetic methods have been attempted, especially with the use of protonated organic amine cations97 or mixed-solvents98 or hydrothermal process99 or ionic liquids (ILs) as solvent/cation directing species.100 POMs are largely used as ODS catalysts like metal oxides owing to their ease of synthesis, common availability and easily adjustable structure–activity relationship.
Like the pure form of POMs, POM derivatives are also utilized as ODS catalysts. POM derivatives are usually prepared by altering the cation or anion of POMs. The structural and compositional modifications accomplished on POMs have led to interesting results in ODS. The simple cations (proton/sodium/potassium/ammonium) of POMs could be substituted by different organic cations in a facile method involving an ion-exchange reaction. For replacing the simple cations of POMs, organic salts such as surfactants,90b,90d,106 and ILs107 are favoured and generally taken in the form of quaternary ammonium salts during the ion-exchange process. Organic salts having cations of lengthy alkyl chains are preferred for the substitution as they can enhance the lipophilicity of the resultant POM derivatives. These POM derivatives are termed as amphiphilic catalysts due to their hydrophilic anion and lipophilic long chain alkyl cation. Due to the amphiphilicity of POM derivatives, the rate of ODS reactions has been remarkably improved. The working mechanism of amphiphilic POMs is that the lipophilic cation pulls the sulfide compounds from the oil phase towards the hydrophilic active sites. This, in turn, accelerates the mass transfer between the oil and aqueous phases and consequently, the ODS rate is enhanced. The hydrophilic walls of the glass ODS reactor incline to attract the hydrophilic active centres of the POM, placing the active sites far from the oil phase. This problem can be eliminated using POM catalysts possessing cations of lengthy alkyl chains. Therefore, it can be concluded that the lengthy alkyl chain of the cation in amphipathic POMs increases the ODS efficiency.108 However, in a very few cases, this trend is not followed due to various reasons.109 For instance, in the report of J. Xu et al., a decrease in ODS activity is noted when dimethyldodecylammonium is the cation of the POM. The reason is that, though lengthy, the double alkyl chain of dimethyldodecylammonium slows down the ODS rate, due to the steric hindrance induced by the double alkyl chains that limits the interaction between the active centres and sulfides.109d In other occasions, the effect of alkyl chain length on ODS activity is affected by the size of sulfide molecules, that is, the lengthy alkyl chain could not show a positive effect on ODS of smaller sulfide molecules.109b,109c In an amphiphilic catalyst, the functional groups (particularly sulfonic acid) available in the substituted cation of POMs are also found to influence the ODS activity.107a
For ODS, from the perspective of industrial applications, molecular O2 is a more preferred oxidant than H2O2. The disadvantage is that, unlike H2O2, harsh conditions or sacrificial agents are required when molecular O2 is used. With the use of an amphiphilic Anderson type POM ([(C18H37)2N(CH3)2]5IMo6O24) as a catalyst, the activation of O2 has been achieved under mild conditions (353 K and 1 atm) due to the electrons flowing in from the electron donating quaternary ammonium ions.90b This leads to the opportunity for more POMs to be utilized as ODS catalysts in the presence of O2 under mild conditions.90d,110 The ODS performance of Na3Co(OH)6Mo6O18 is dramatically enhanced from 5% to 100% during ODS of a model oil (500 ppm DBT in decalin) in the presence of O2 (1 atm) at 353 K by introducing amphiphilicity (((C18H37)2N(CH3)2)3[Co(OH)6Mo6O18]). Herein, the quaternary ion donates its electrons to promote the formation of active mixed valence molybdenum(V/VI) species.90d Recently, Zeng et al. reported an effective ODS system that works well with O2 even at 303 K using a choline substituted HPW catalyst ((HO(CH2)2N(CH3)3)3[PW12O40]) due to the improved electron–hole pair interaction.110a
Amphipathic catalysts are also known to form catalytic emulsion (water/oil) droplets during ODS reactions.89a–c,91e,94b,109d,111 The formation of emulsion droplets has led to the high dispersion of the POM catalyst throughout the oil phase, increasing ODS activity. These metastable emulsion droplets may be stabilized by the amphiphilic POM catalyst. Emulsions in ODS are made up of three regions: the interior of a droplet (aqueous solvent and anionic part of the amphipathic POM), the continuous phase (oil and cationic part of the amphipathic POM), and the interfacial membrane. The interfacial membrane is a very significant narrow region where the substrate, oxidant, and catalysts interact with each other to facilitate ODS.109d The emulsion system formed in ODS is pictorially presented in Fig. 16.
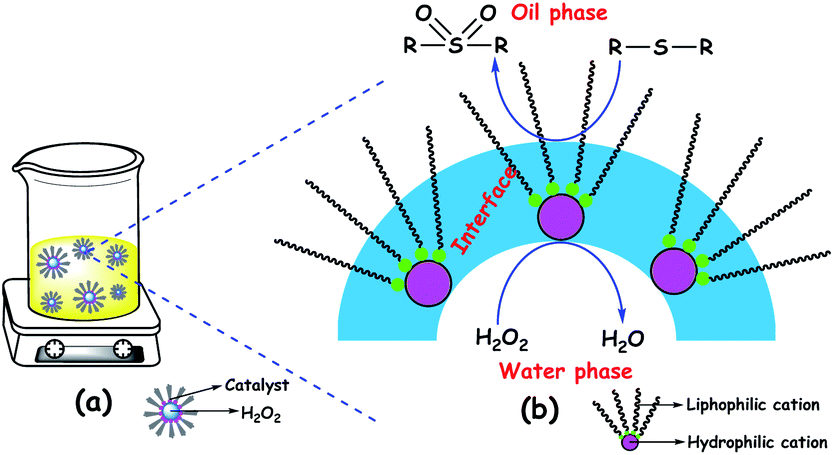 | ||
| Fig. 16 (a) Dispersed ODS emulsion catalyst: in the oil phase and (b) illustration of three regions of the emulsion catalyst. | ||
Regarding the substitution of the heteroelement in the anionic part of POMs, a variety of heteroelements have been utilized ranging from transition metals to lanthanides.109d,112 The change in ODS activity due to the heteroelement substitution in the anionic part of POMs is obvious. For example, in (TBA)3+x[PW12−xVxO40] (where x = 0–3, TBA = tetra-n-butylammonium), W. Trakarnpruk and K. Rujiraworawut replaced tungsten by vanadium which leads to the order of DBT oxidation as V3 (95%) > V2 (82%) > V1 (72%) > V0 (52%) due to the changes in reduction potential arising from vanadium substitution. The substitution of tungsten(VI) with vanadium(V) would also result in the generation of more reactive lattice oxygen associated with W–O–V species. It is also believed that the substitution of tungsten by vanadium can turn the acid-dominant properties of HPW into redox-dominant properties which is highly desirable for a POM to be a catalyst in the oxidation reactions.112a On the other hand, while substituting tungsten with different heteroelements (titanium, manganese, iron, cobalt, nickel and copper) in (C18H37N(CH3)3)[H2[PW12O40], the ODS activity is decreased due to the intact Keggin structure of PW11M which is not able to form the active polyperoxometalate in the presence of H2O2.91e In addition, to modify the ODS activity of a POM by adjusting its cation or anion, calcining at different temperatures could also influence the ODS activity. From the results of M.-W. Wan et al., it is evident that the calcination of HPW at higher temperature (673 K) leads to better activity in ODS of a model oil (DBT in toluene) using H2O2 and TOAB, as compared to calcination at relatively lower temperatures (473 and 573 K). This is because the water content and the crystallite size of HPW are decreased upon high temperature calcination.113 Very recently, a new POM, [3-(pyridine-1-ium-1-yl)propane-1-sulfonate]3(NH4)3Mo7O24·4H2O, was able to accomplish 99% ODS efficiency within 1 hour at 298 K in an ODS system consisting of a model oil (DBT in n-octane) and H2O2. This catalyst selectively promotes the oxidation of DBT in the presence of toluene and naphthalene, and accomplishes 99% DBT oxidation even at 273 K.16a Hence, we presume that this catalytic ODS system can be highly intriguing in the coming years due to its better activity and selectivity.
The mechanism for POM catalyzed ODS is generally proposed as shown in Scheme 2a. According to the mechanism, the oxidant initially attacks the metal oxo species to yield the active metal peroxo species which subsequently transfers oxygen to sulfur for the formation of sulfoxide. This sulfoxide is again converted to the final product (sulfone) by abstracting the reactive oxygen from the metal peroxo species. In the end, water is obtained as the by-product and the POM is regenerated for the successive ODS cycles. As given in Scheme 2b, oxidation of sulfides is also possible by the action of intrinsic metal peroxo species of POMs in which the reduced POM is regenerated by the oxidant.90b,114 In contrast to the usual ODS pathway, a different reaction pathway is followed when H8PV5Mo7O40 catalyzes the oxidation of DBT using O2 as the oxidant. Instead of being oxidized to sulfone, DBT is oxidized to water soluble sulfuric acid, sulfoacetic acid, and sulfobenzoic acid. Here, the advantage is that the environmentally friendly water is utilized as the extractant. However, the formed acidic intermediates (except oxalic acid) and products suppress the ODS activity of H8PV5Mo7O40 by rearranging the higher vanadium-substituted species into lower vanadium-substituted species and VO2+. To avoid this problem, at present, the nanofiltration technique is under investigation for the concurrent removal of acidic products. Oxalic acid, with its reducing effect, enhances the ODS activity of H8PV5Mo7O40 by enabling the formation of very reactive vanadium(V)-peroxo species.115
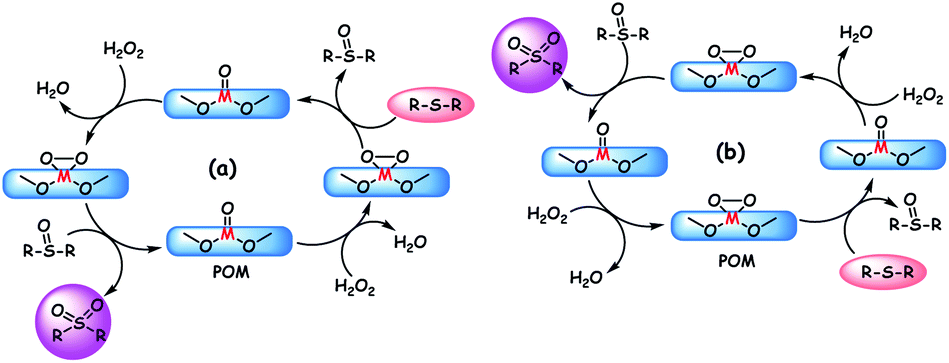 | ||
| Scheme 2 Two different mechanisms mainly proposed for the ODS process that is catalyzed by POMs in the presence of peroxide oxidants/O2. (a) Oxidation of sulfides by oxidant promoted metal-peroxo species; (b) oxidation of sulfides by native metal-peroxo species of POMs. | ||
A salient feature of POM catalyzed homogeneous ODS systems is that they can be reused, that is, these ODS systems combine the advantages of homogeneous and heterogeneous catalysis. Homogeneous ODS systems are generally reused simply by decanting the oil phase, and directly adding the model oil and oxidant in the next run.116 However, during the progress, the deposition of oxidized sulfur compounds as a white precipitate hinders the efficiency of the homogeneous ODS system. This problem can be eliminated using heterogeneous ODS catalysts. The amphipathic catalysts having the ability to form emulsions could be easily separated by centrifugation and utilized in the next run without any special treatment.89d,117
4.3.2.1. Het-POM catalysts prepared by precipitation and hybridization. The synthesis of het-POMs via precipitation with simple metal cations or hybridization with bulky organic/inorganic molecules is quite straightforward, and it usually involves a simple ion-exchange reaction. Metal ions as well as ammonium ions (NH4+) are reported for the preparation of het-POMs through precipitation. During the precipitation, the degree of ion-exchange between the POM and the used metal cation/NH4+ is significant as it remarkably affects the ODS activity.123 For example, when a het-POM ((NH4)x[H4xPMo11VO40] (x = 1, 2, 3, 4)) is used as the ODS catalyst, the order of ODS activity is (NH4+)3HPMo11VO40 > (NH4+)4PMo11VO40 > (NH4+)2H2PMo11VO40 > (NH4+)H3PMo11VO40 in ODS of DBT in the presence of polyethylene glycol under an O2 flow, signifying the degree of substitution.123b
In the literature, the hybrid POMs that are used as het-POM catalysts in ODS are prepared using bulky organic cations and metal complexes. The length of the alkyl chain is very crucial since a lengthy alkyl cation leads to the formation of a heterogeneous ODS catalyst while a shorter one can't.109c,125 For instance, S. O. Ribeiro et al. prepared two hybrid ODS catalysts, (TBA)5([PW11Zn(H2O)O39]) and (ODA)5([PW11Zn(H2O)O39]) (where ODA = trimethyloctadecylammonium), among which the latter shows the behaviour of heterogeneous catalysts during ODS in the presence of H2O2.109c Calix[4]arene ligands are effectively used for the in situ preparation of POM hybrids which find applications as het-POM ODS catalysts.124,126 M.-Y. Yu et al. prepared metal complex–POM hybrids (Fig. 17) where silicotungstic and molybdotungstic acids function as bridges in the isolated copper(I) and silver(I) thiacalix(4)arene dimers and are subsequently used as the heterogeneous ODS catalyst in the presence of TBHP.124
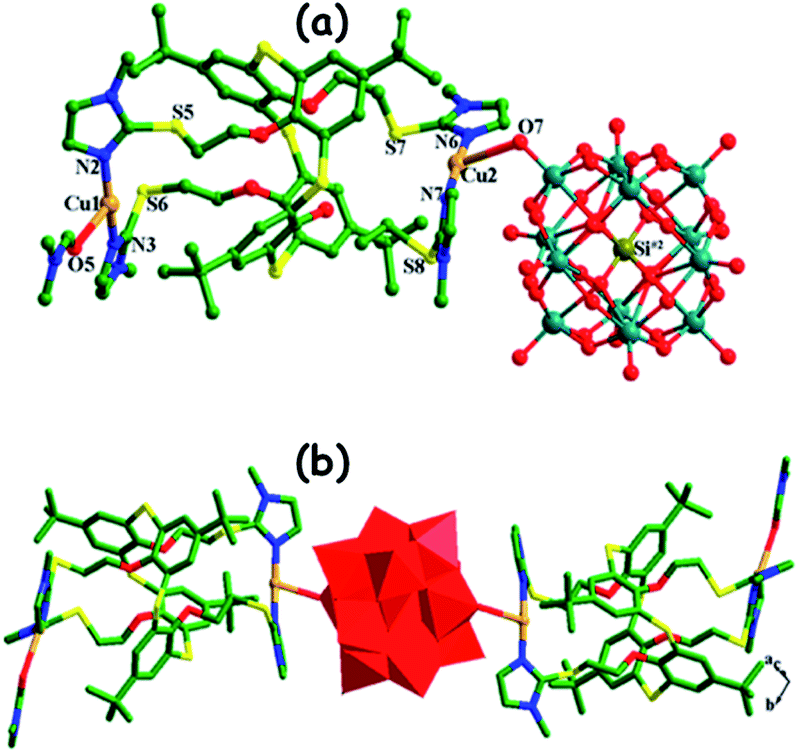 | ||
| Fig. 17 (a) Coordination spheres of Cu(I) cations and (b) POM-bridged Cu(I)-thiacalix[4]arene dimers. Adapted from ref. 124 with permission from The American Chemical Society. | ||
In another report, a POM hybrid is prepared via an ion-exchange method where the POM (Na9EuW10O36·32H2O) is electrostatically bound to IL grafted calix[4]arene. The advantage is that this POM–IL calix[4]arene hybrid reveals superhydrophobicity by which the mass transfer of sulfur compounds is prevalently enhanced via hydrophobic–hydrophobic interaction, resulting in better ODS performance in the presence of H2O2.126a During the synthesis of the organic–inorganic hybrid between the POM and 2,3-pyrazinedicarboxylic acid, POM clusters emerge as aggregates bridged by 2,3-pyrazinedicarboxylic acid and are utilized as heterogeneous ODS catalysts in ODS.127 While all the above-mentioned hybrid POMs or precipitated POMs preferentially follow the ODS mechanism as represented in Scheme 2a, silver decorated POM single-walled nanotubes follow a different mechanism. During ODS of DBT or diphenylsulfide in n-octane in the presence of H2O2, silver nanoparticles attract sulfur compounds and act as the promotor to accomplish 100% ODS of DBT or diphenylsulfide following the mechanism given in Scheme 3. Silver nanoparticles decorated inside the POM single-walled nanotubes have shown better results as compared to those decorated outside the POM single-walled nanotubes.128
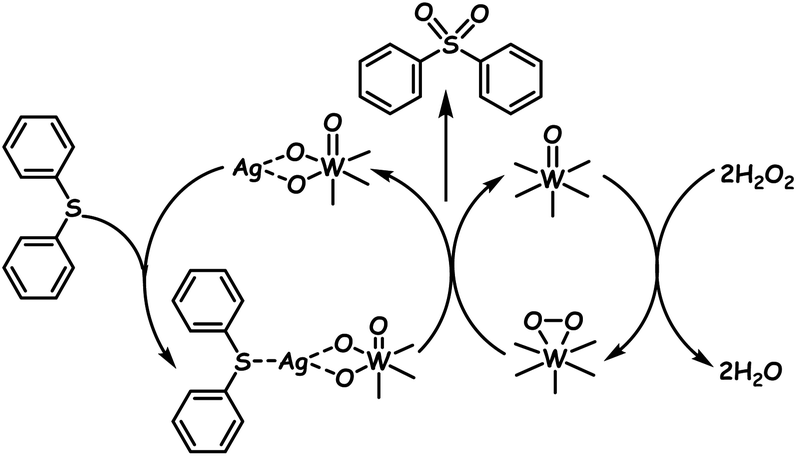 | ||
| Scheme 3 Proposed mechanism for ODS catalyzed by silver nanoparticle decorated POM single-walled nanotubes. Adapted from ref. 128 with permission from The Royal Society of Chemistry. | ||
4.3.2.2. Het-POM catalysts prepared by dispersion on supports. Dispersion of POMs over the chosen support can be carried out through one-step or multi-step synthesis. Al2O3,129 SiO2 in different forms,130 carbon in its various analogues,121,131 graphene oxide in different forms,132 TiO2 in different varieties,91b,133 BN,93,120,134 carbon nitride,135 clay minerals,136 lead oxide,137 zeolites,138 CeO2,139 polyvinyl alcohol140 and MgCu2O4-polyvinyl alcohol composites141 have been used till now as supports for the preparation of het-POM ODS catalysts via dispersion.
Among the above-reported supports, BN has been very special as it can pull the lone pair of electrons from a sulfur atom or the occupied π-electrons of planar structure DBT molecules to the virtual orbitals of boron, enhancing the absorbability of the het-POM catalyst towards sulfur compounds. As with sulfur compounds, BN shows the same behaviour towards the adsorption of peroxide oxidants on the catalyst's surface. As a result, collectively, an increased ODS rate is realized using the BN support.134d H. Li et al. proved the superiority of BN over other common supports. BN supported silicotungstic acid demonstrates very good ODS performance compared to other het-silicotungstic acids prepared using various supports including amorphous Al2O3, carbon nanotubes (CNTs), SBA-15 and SiO2 as shown in Fig. 18. The reason is that, unexpectedly, BN promotes the formation of low-valent tungsten(V) species which are very active in ODS.134a
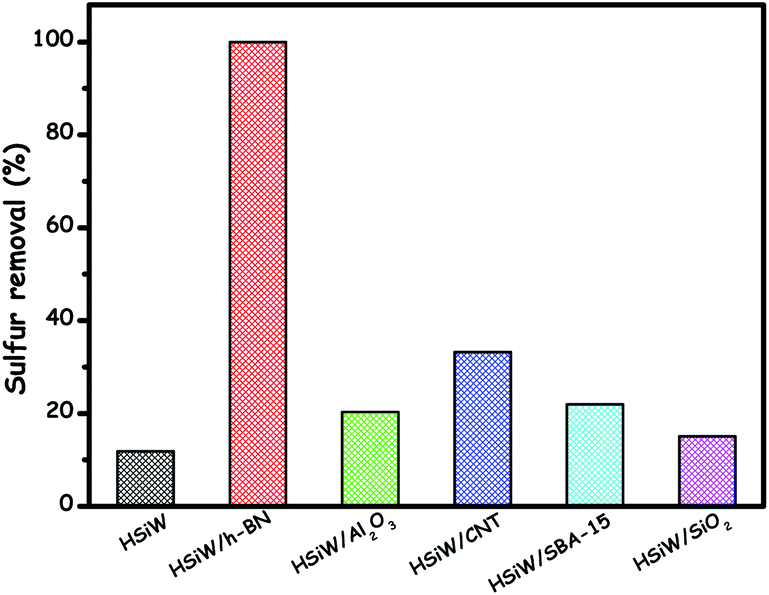 | ||
| Fig. 18 Comparison of ODS activity of different het-silicotungstic acid catalysts prepared using different supports in the ODS of DBT. Reaction conditions: T (313 K); H2O2 (O/S = 4); model oil (5 mL); catalyst (0.05 g); t (1 h). Adapted from ref. 134a with permission from Elsevier. | ||
S.-Y. Dou and R. Wang formulated a different ODS system where an aldehyde (n-octanal) is used along with a graphene oxide supported POM in the presence of air and model oil. Notably, in this ODS system, ODS is accomplished by the action of peroxy-acid formed from the catalytic oxidation of n-octanal. Surprisingly, instead of catalysing ODS by following the mechanism given in Scheme 2a, POM promotes the oxidation of n-octanal to octanoic acid which is then converted to peroxy-acid.132b,142
4.3.2.3. Het-POMs prepared by encapsulation. With the benefits of a porous and cage structure, metal organic frameworks (MOFs) are very attractive for encapsulating catalytically active entities like POMs, leading to effective heterogeneous catalysis. A list of MOFs finds applications as the support for the preparation of het-POMs (POM@MOF) in ODS by encapsulating POMs inside their pores and cages. A wide range of MOFs varying in their surface area, porosity and dimensionality are used as the host materials for POMs such as MIL-101(Cr),143 MIL-101(Al),144 NH2-MIL-101(Cr),145 NH2-MIL-53(Al),146 MIL-101(Fe)–NH2–Cl,147 HKUST-1,148 MIL-100(Fe),149 UiO-66,150 ZIF-8,150 MOF-199,151 and others.152 Profitably, POM@MOF catalysts can possibly achieve increased ODS activity owing to the cooperative catalysis of metal clusters in the host framework (MOF) and the guest POM molecules.153 Care must be taken to avoid the overloading of the POM inside the cages/pores of MOFs during the encapsulation since this overloading blocks the cages and windows of MOFs, inhibiting the accessibility of sulfur compounds to POM active sites during ODS.153 Furthermore, the accessibility of organosulfur compounds to the active POM sites can be facilitated and controlled by the choice of MOFs with a suitable window size and confined effect.146,150 This is exemplified when HPW is separately encapsulated in UiO-66, ZIF-8 and MIL-100(Fe). HPW@MIL-100(Fe) exhibits better ODS activity due to the large window size of MIL-100(Fe).150
X.-L. Hao et al. isolated a POM@MOF (phosphomolybdic acid encapsulated in a cationic triazole-based MOF) whose ODS activity is influenced by the pore (channel) size of the MOF (Fig. 19). This POM@MOF has two interlinked channels in which the straight channel (A) is occupied by the POM and the undulated channel B is occupied by lattice water. Though, during ODS, the sulfur compounds adsorbed in channel B are accessed by POMs located in channel A, the small size of channel B is not able to locate the big size DBT and 4,6-DMDBT. As a result, the occurrence of ODS for DBT and 4,6-DMDBT is preferred in the surface and not in the channels of POM@MOF, leading to less removal of DBT and 4,6-DMDBT relative to that of small sulfur molecules (thioanisole and methyl benzothiophene) at 323 K in the presence of TBHP.152a This further implies the importance of the pore size of MOFs in determining the ODS activity of POM@MOF catalysts. During the physical encapsulation of POMs using MOFs, the electrostatic or the covalent coordinate interaction can also be realized by POMs depending on the types of MOFs.147,154 This interaction may strengthen the recyclability of POM@MOFs in the ODS. One-pot preparation of POM@MOF is more solicited than the multi-step preparation (impregnation) to reach the maximum ODS activity.155 The encapsulated POM@MOF catalysts exhibit predominant ODS behaviour if they are exfoliated into nanosheets of atomic size thickness. L. Xu et al., when using exfoliated POM@MOF ([Co2(H2O)4(BTX)3][PMo12O40], BTX = 1,4-bis(1,2,4-triazol-1-ylmethyl)benzene) instead of bulk POM@MOF, realized a 14 fold increase of activity in ODS of DBT with H2O2 at 353 K. The reason is that the exfoliated nanosheets provide more accessible active sites which essentially control the mass-transfer issues, increasing ODS activity.154 MOFs constructed with hydrophobic ligands are fascinating because they promote the easy access of RS compounds to catalytic POM sites in the non-polar media (fuel oils) and thus increase the ODS performance of the resultant POM@MOF.156
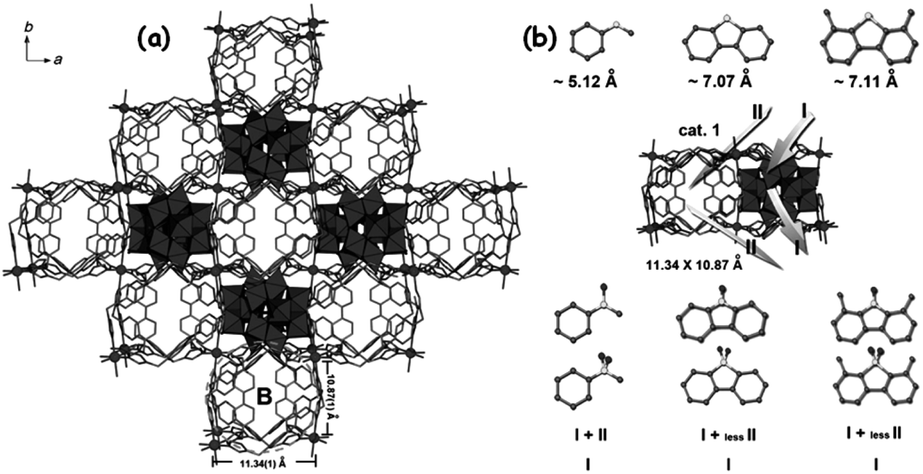 | ||
| Fig. 19 (a) Ball-and-stick and polyhedral view of the POM@MOF structure of [Co(BBPTZ)3][HPMo12O40]·24H2O [BBPTZ = 4,4′-bis(1,2,4-triazol-1-ylmethyl)biphenyl] viewed along the c axis. (b) Different catalytic reaction routes among the sulfide substrates and different catalysts, based on the catalytic results. Route I represent catalysis taking place on the surface of catalysts; route II represents catalysis taking place within the channels of catalysts. Adapted from ref. 152a with permission from John Wiley & Sons. | ||
Apart from MOFs, other supports including SiO2 pillared clays,91i,157 water-soluble cage compounds,158 porous organic polymers,159 β-cyclodextrin,160 core–shell models,161 ZSM-5,151b,162 and porous SiO2151a,c,d,163 can also be utilized for the encapsulation of POMs for the preparation of het-POM ODS catalysts. During the physical encapsulation of POMs in the preparation of het-POM ODS catalysts, the electrostatic interaction can also be realized by POMs depending on the nature and functional groups of supports.147,159 This interaction may strengthen the recyclability of POM@MOFs in ODS. An interesting surfactant type POM ([C16H33(CH3)2NOH]3(PO4[WO(O2)2]4)) encapsulated in porous SiO2 has turned out to be water soluble under the action of H2O2 during ODS and self-assembles back on the pore-walls of SiO2 after the consumption of H2O2, providing an opportunity for homogeneous catalysis under heterogeneous conditions.93
4.3.2.4. Covalently tethered het-POM catalysts. Covalent bonding is entertained between the support and POM in this type of het-POM ODS catalyst. R. Xia et al. prepared a het-POM by grafting HPW on a hydrophobic copolymer (derived by the radical copolymerization of 1,3,5-tris(4-vinylphenyl)-benzene and 4-vinyl pyridine) through the coordinate covalent bonds formed between HPW and pyridine nitrogen of the copolymer. The catalyst showed an amphipathic character with the contact angles of n-octane and water as 31.2 and 34.6°, respectively (Fig. 20a). As a result, this het-POM played multiple roles such as those of a catalyst, sorbent for sulfur compounds and stabilizer for the Pickering emulsion system as shown in Fig. 20d. Due to these multiple roles of het-POM, 100% sulfur removal is quickly reached (within 15 min) under emulsification conditions as compared to that under bulk conditions (30 min).164 Amine functionalized SiO2
165 and phosphazene functionalized SiO2166 are also reported in the literature to form het-POM ODS catalysts where the coordinate covalent bond is operative between the POM and functional groups. M. Carraro et al. prepared POM clusters possessing polymerizable ethylene bonds by which the POM clusters (Keggin type polyoxotungstates) are copolymerized with methyl methacrylate, resulting in het-POM copolymers in which POM is covalently grafted on the resultant copolymer.167
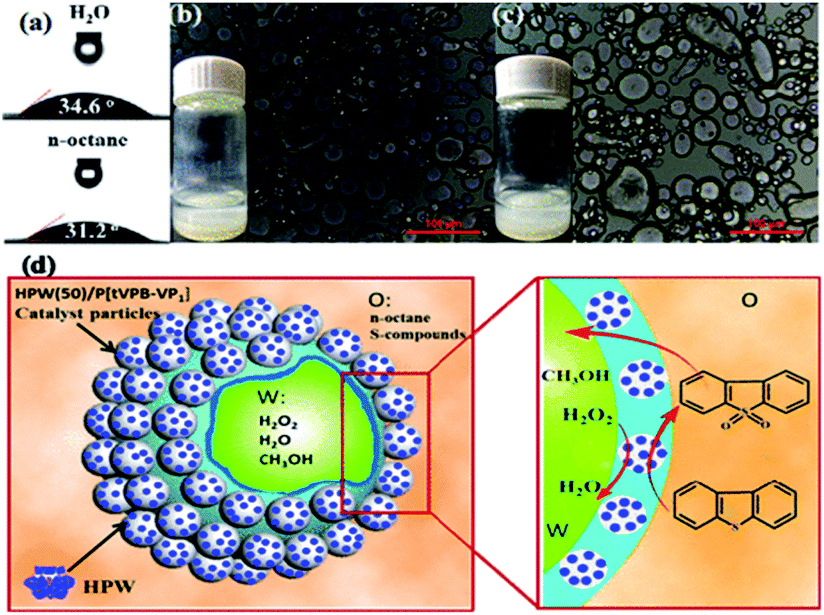 | ||
| Fig. 20 (a) Contact angle measurements of H2O and n-octane droplets on HPW(50)/P[tVPB-VP1] surfaces; (b and c) polarizing microscope images of H2O/n-octane catalytic emulsion stabilized by HPW(50)/P[tVPB-VP1] (the inset shows a picture of the emulsions 5 min after the emulsification at 333 K (b) and after 1 month at room temperature (c)); (d) schematic illustration of the Pickering emulsion catalytic system for simultaneous oxidation and extraction desulphurization. Adapted from ref. 164 with permission from The American Chemical Society. | ||
4.3.2.5. Non-covalently tethered het-POM catalysts. In this type of catalyst, non-covalent interactions such as ionic interactions operate between POMs and supports. With their readily exchangeable intrinsic anions, layered double hydroxides (LDHs) are special supports for the preparation of het-POM ODS catalysts through ion-exchange, inhibiting the leaching of active sites during the recycling studies.168 Due to the synergistic effect resulting from the confined space electron transfer and reduced electron–hole recombination state, POM intercalated LDHs can promote heterogeneous photocatalytic ODS.169 Using LDHs, an exfoliation–assembly method has been highly regarded for the preparation of het-POM catalysts for ODS.168a,169 Interestingly, Y. Xu et al. prepared an IL based het-POM using LDH by an exfoliation–grafting–assembly method (Fig. 21), showing 93% sulfur removal under the optimized reaction conditions even after 20 ODS cycles. Unlike other extractive catalytic ODS systems, this system does not require addition, separation and purification of ILs after every catalytic cycle.168a
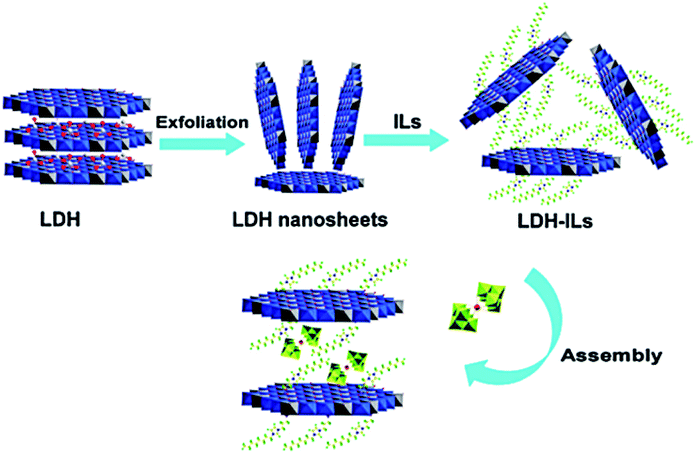 | ||
| Fig. 21 The synthetic procedure for the preparation of the designed het-POM catalyst (Mg3Al-IL-EuW10) by an exfoliation–grafting–assembly method using LDH. Adapted from ref. 168a with permission from The Royal Society of Chemistry. | ||
A variety of organic polymers have also been found to be suitable to prepare het-POMs through electrostatic interactions.170 Furthermore, SiO2 functionalized with different functional groups including amines, quaternary ammonium ions and ILs is also known to immobilize POMs through electrostatic interactions for the preparation of het-POM ODS catalysts.171 Note that the functional groups grafted on SiO2 to induce the interaction with POMs can significantly influence the ODS activity of functionalized SiO2 supported het-POM ODS catalysts.91g O. Ribeiro et al. formulated a solvent-free ODS system with het-POM prepared using amine functionalized SBA-15 in which the use of organic solvent is avoided and instead, water is utilized as the extractant, making the ODS system more environmentally sustainable.171m Similar to the emulsion depicted in Fig. 20, an emulsion ODS system can be designed using a het-POM catalyst where the amphipathic nature emerges from the hydrophobic aromatic/alkyl groups and hydrophilic ionic groups that are attached to any solid support. In these catalysts, the POM is involved in the typical ionic interaction with the ionic groups attached to the solid supports.172 H. Yang et al. prepared a core–shell structure by covering cellulose nanocrystals with a poly(ionic liquid) which is further used to immobilize [Co(OH)6Mo6O18]3−via an ion-exchange method. The obtained composite catalyst promotes 100% removal of various sulfur compounds (BT, DBT, and 4,6-DMDBT) in different reaction times under an air flow with the advantage of possessing a natural catalyst carrier (cellulose).173
4.4. Metal complexes
Metal complexes are generally prepared from the reaction of metal salts and organic ligands possessing donor atoms, either at room temperature or under refluxing conditions in a suitable common solvent. Owing to the switchable multiple oxidation states of the central metal atom/ion, metal complexes have attracted attention as oxidation catalysts. Consequently, a variety of metal complexes have been utilized as ODS catalysts in the literature, including both homogeneous and heterogeneous catalysts. Simple metal salts like cobalt acetate and Fenton reagents have also been tested in ODS as catalysts; however, their efficiency is poor as compared to that of their corresponding complexes under the same reaction conditions.174 The reason is that the ligands surrounding the metal ions in the complexes stabilize the metal ions in multiple oxidation states and tune the electronic properties of the central metal atom/ion. In fact, with the choice of ligand, the ODS activity of metal complexes can be tuned to a notable extent. Metal phthalocyanines, structural analogues of metal porphyrins, seem to be frequently reported metal complexes as ODS catalysts174a,175 for different reasons including their structure, thermal stability and ease of heterogenization.176 The substituents attached to the periphery of phthalocyanine (Pc) ligands exert a significant influence on the ODS activity of their complexes. The nature and number of electron-withdrawing substituents are significant in deciding the ODS activity of metal phthalocyanines. For instance, in ODS of DBT using air, cobalt(II) phthalocyanines demonstrate an increasing order of ODS activity with the number of chloride substituents: CoPc(Cl)4 (18%) < CoPc(Cl)8 (∼40%) < CoPc(Cl)12 (∼72%) < CoPc(Cl)16 (∼90%).175e On the other hand, R. Zhao et al. tuned the ODS activity of an iron(III) porphyrin complex with the rational design of axial ligands of the complex. Iron(III) porphyrin with a relatively weak axial ligand exhibits the maximum ODS of DBT under the optimized reaction conditions in the presence of H2O2 and the obtained reactivity order is FeIII(TPP)Cl (93.2%) < FeIII(TPP)BF4 (98.8%) < FeIII(TPP)PF6 (99.5%) (TPP: tetraphenylporphyrin).177 After phthalocyanine and porphyrin complexes, complexes of oxo ligands (especially vanadium, molybdenum and tungsten containing other ancillary ligands) seem to be the best choice as catalysts for ODS.178A variety of supports, following one of the three mentioned methods (Fig. 22), have been employed during the preparation of heterogeneous metal complexes for catalytic ODS.175a-c,178a,178b,179 D. Piccinino et al. prepared five different heterogeneous catalysts consisting of CNTs and a crosslinked polymer by different preparative methods as shown in Scheme 4 and subsequently utilized them as ODS catalysts. The different preparative pathways lead to different ODS activities. Pathway C is more successful in accomplishing maximum ODS activity because of the effective loading of methyltrioxorhenium(VII) noted in this method which may be due to the already established interaction (π–π) between the crosslinked polymer and CNTs. Especially, in pathway C, the highest active site loading (1.434 mmol g−1 of support, equivalent to 71.6% immobilization yield) is accomplished when poly(4-vinylpyridine)-N-oxide is used. This may be due to the oxophilicity of rhenium towards poly(4-vinylpyridine)-N-oxide, thus yielding better ODS activity.180
 | ||
| Fig. 22 Heterogenization methods of metal complexes for catalytic ODS: (a) electrostatic interaction, (b) covalent/covalent-coordination grafting and (c) encapsulation. Adapted from ref. 176 with permission from The American Chemical Society. | ||
 | ||
| Scheme 4 Preparation of heterogeneous rhenium(VII) complex ODS catalysts via different routes. Adapted from ref. 180 with permission from Elsevier. | ||
Like in POMs, solicited phase transfer ability can be induced in metal complexes using the cations of lengthy alkyl groups and ionic liquids.181 J.-K. Li et al. explained the dependence of ODS activity on the crystallite size of the metal complex ([Ni2(C2O4) (dpa)4][(C4H6O4)(VO2)]2·2[CH3OH]). After grinding for 10 min, the ODS activity improved from 75% to 100% in ODS of DBT with O2 due to the emergence of small crystallites that promote the efficient interaction of active sites with the substrate and oxidant.182 Steric hindrance around the central metal atom of the complex is not desirable as it limits the interaction of active sites with bulky RS compounds. D. Juliao et al. tuned the activity of an organometallic complex by simply varying the ancillary ligands of different sizes. When the complex of a small ancillary ligand is used, an improved ODS activity is noted as it reduces the steric factor around the central metal ion.183 While being used in ODS processes, the metal complexes usually follow one of the three general oxidation mechanisms where either oxo or peroxo or superoxo metal complexes are noted as the intermediates.175c,175f-i,178b,178d A brief summary of the metal complexes employed in catalytic ODS is given in Table 2.
| Catalyst | RS compounds | Oxidant | O/S | T (K) | Time (min) | Conv. (%) | Ref. | ||
|---|---|---|---|---|---|---|---|---|---|
| Type | Amount (g L−1) | Type | Conc. (ppm) | ||||||
| a MTh: methyl thiophene; Pc: phthalocyanine; py: pyridine; BZA: benzylidene aniline; PAMAM: polyamidoamine; MSA: 5-methyl salicylaldehyde; PVPNO: polyvinylpyridine-N-oxide-2% cross linked divinylbenzene; MTO: methyltrioxorhenium; fsal: 3-formylsalicylic acid; dmen: N,N-dimethyl ethylenediamine; C4mim: 1-butyl-3-methylimidazolium; phen: 1,10-phenanthroline; CF: carbon fibres; D113: a type of resin; MSNP: magnetic silica nanoparticles; RGO: reduced graphene oxide; gly: glycine; dpa: di(2-pyridylamine); TPP: tetraphenylporphyrin; (PorCl4): tetra(ortho-chlorophenyl)porphyrin; PorMe4: tetra(ortho-methylphenyl)porphyrin; TPFPP: meso-tetrakis(pentafluorophenyl)porphyrin. b ODS in the presence of extractant IL. c ODS under visible light irradiation. d ODS under UV radiation. e The complexes utilized in the ODS of real fuels and multicomponent model fuel are not mentioned in the table. | |||||||||
| [CoPc(SO2NH2)4 | 2.1 | 4-MDBT, 4,6-DMDBT | 4950 | O2 | 313, 313 | 120, 120 | 100, 100 | 174a | |
| ZSM-5-Ce(Pc) (NO2)6(NH2)2 | 1 | Th | 1900 | O2 | 333 | 180 | 99.5 | 175a | |
| Ti-MCM-41-NH2PcFeb,c | 1 | DBT | 1000 | O2 | RT | 120 | 95.6 | 175b | |
| SG2.0-[VVO2-PAMAM-MSA] | 0.005 | DBT | 500 | TBHP | 3 | 363 | 60 | 86.1 | 178a |
| [CNT/PVPNO]/MTO | 0.276, 0.318, 0.222, 0.222, 0.166 | DBT, 4,6DMDBT, 2-MBT, 3-MBT, 3-MTh | 9200, 10600, 7410, 7410, 5535 |
H2O2 | 4 | 333 | 180 | >99, 80, 78, 51, 28 | 180 |
| PS-[VVO2(fsal-dmen)] | 7.5 | Th, BT, DBT, 2-MTh | 500 | H2O2 | 3 | 333 | 120 | 98.1, 98.3, 98.4, 98.8 | 178b |
| [C4mim]3Fe(CN)6b | 6, 6, 12 | BT, DBT, 4,6-DMDBT | 250, 500 | H2O2 | 4, 4, 12 | 313 | 300 | 91.1, 97.9, 90.2 | 181a |
| [WO(O2)2Phen]H2O, [MoO(O2)2Phen] | 0.411, 0.319 | DBT | 1000 | H2O2 | 10 | 343 | 180 | 98.6, 93 | 178c |
| FePc(NO2)3-CF | 10 | DBT | 500 | O2 | 403 | 180 | ∼92 | 175c | |
| VO(acac)2b | 0.216 | BT, DBT, 4,6-DMDBT | 250, 500, 250 | H2O2 | 5 | 303 | 120 | 56.8, 99.6, 18.4 | 184 |
| FePc(NO2)4, FePc(NO2)3NH2-D113 | 10, 10 | DBT | 500 | O2 | 373 | 120 | 98.7, ∼82 | 175d | |
| CoPc(Cl)16b | 3 | Th, MTh, BT, DBT, 4,6-DMDBT | 1000 | O2 | RT | 120 | 100, 81.9, 89.1, 90.4, 80 | 175e | |
| ZnPc/RGO | 0.2 | DBT | 800 | O2 | 333 | 30 | 97.51 | 175f | |
| FeC4Pc-MSNPd | 20 | BT | 600 | H2O2 | 20 | 333 | 30 | 94.5 | 175g |
| MoO(O2)2glyb | 8 | BT, DBT, 4,6-DMDBT | 1000 | H2O2 | 4 | 343 | 180 | 93.2, 99.2, 99.6 | 178d |
| [Ni2(C2O4)(dpa)4][(C4H6O4) (VO2)]2·2[CH3OH] | 0.8 | BT, DBT, 4,6-DMDBT | 500 | O2 | 373 | 420, 240, 300 | 100, 100, 100 | 182 | |
| FeIIITPP(PF6)b | 22 | DBT, 4-MDBT, 4,6-DMDBT | 500 | H2O2 | 3 | 333 | 240 | 99.5 | 177 |
| (PorCl4)FeCl | 1 | DBT, 4,6-DMDBT | 500 | O2 | 393 | 150, 180 | 100, 100 | 175h | |
| (PorMe4)FeCl | 28, 20, 18 | BT, DBT, 4,6-DMDBT | 500 | H2O2 | 4 | 333 | 360, 300, 240 | 94.7, 100, 100 | 175i |
| μ-O(FeTPFPP)2 | 1.5 | BT, DBT, 2,6-DMDBT | 500 | H2O2 | 9, 12, 14 | RT | 180 | 100, 100, ∼100 | 178e |
4.5. Metal organic frameworks
MOFs are crystalline porous hybrid materials originating from the pervasive coordination bonds formed between the metal nodes and organic linkers (ligands). Owing to the excellent tunability of pore size, shape and functionality, MOFs have received attention in a variety of applications ranging from gas storage to catalysis.185 The solvothermal method is a common synthetic method to prepare MOFs by taking a fixed ratio of metal precursors and organic linkers in a suitable solvent. Other synthetic methods are also occasionally reported. Catalytic applications of MOFs are mainly governed by the nature of metal nodes of MOFs. Titanium-based MOFs are among the fascinating ODS catalysts due to their proclivity to activate peroxides for oxidation, especially MIL-125(Ti) and its amine functionalized analogue (NH2-MIL-125(Ti)).186 However, the relatively small pores of MIL-125(Ti) suppress the oxidation activity when large molecules like 4,6-DMDBT are utilized as the substrate. N. D. McNamara and J. C. Hicks carried out a vapor-assisted crystallization (VAC) method to produce a fascinating mesoporous MIL-125(Ti) (VAC-meso-MIL-125) without the necessity for any chelating agent. Interestingly, VAC-meso-MIL-125 (kapp = 22.9 × 10−3 min−1) demonstrates higher performance during ODS of DBT as compared to VAC-micro-MIL-125 (kapp = 11.6 × 10−3 min−1) and conventional micro-MIL-125 (kapp = 11.8 × 10−3 min−1) prepared by a solvothermal method in the presence of TBHP at 353 K.186a Y. Zhang et al. stated that NH2-MIL-125(Ti) was the inferior ODS catalyst in comparison with MIL-125(Ti) because NH2 groups suppress the sulfur compounds from being accessed by the active titanium(IV) sites.186b Apart from titanium-based MOFs, other metal containing MOFs such as MIL-47(V),186c MFM-300(V),187 TMU-10(Co),188 TMU-12(Co),188 NH2-TMU-53(Co),189 MIL-101(Cr),190 and MIL-101(Fe)190 are also reported to be ODS catalysts under various reaction reactions.In MOF-based ODS catalysts, the more open sites (accessible catalytic sites for the oxidant and sulfur compounds) on metal centres are very important to achieve improved catalytic activity. S. Smolders et al. derived a titanium(IV)-4,4′-biphenyl dicarboxylate MOF (COK-47s) featuring defect-rich Ti–O sheets showing very good activity with a rate constant of 41.1 × 10−3 min−1. In Fig. 23, the reactivity of COK-47S is compared with those of COK-47L, MOF-808, MIL-25, Degussa P25 TiO2 and MIL-47. The higher activity of COK-47S compared to those of all other MOFs is governed by its more open sites (defects). The order of apparent rate constant in DBT oxidation with TBHP at 333 K is 41.1 × 10−3 min−1 (COK-47S) > 11.9 × 10−3 min−1 (MIL-47) > 4.3 × 10−3 min−1 (MOF-808) > 3.6 × 10−3 min−1 (COK-47L) > 2.7 × 10−3 min−1 (UiO-66) > 0.1 × 10−3 min−1 (P25 TiO2).186d The significance of the number of open sites in MOF catalysts for reaching the maximum ODS efficiency is further emphasized by H.-Q. Zheng et al. who compared the ODS activity of four different Zr-based MOFs (UiO-66, UiO-67, NU-1000, and MOF-808) in ODS of DBT with H2O2 at 323 K and without any surprise, MOF-808 shows better activity than other Zr-based MOFs because of its more defect sites.191
 | ||
| Fig. 23 Catalytic activity of different MOFs in ODS of DBT. Reaction conditions: 5 mL model oil (0.5 mmol DBT in toluene); catalyst (0.075 mmol); TBHP (1.25 mmol); T (333 K). Adapted from ref. 186d with permission from John Wiley & Sons. | ||
UiO-66 is seen as a stable material due to its excellent mechanical strength emerging from the strong ligand–metal coordination bonds of Zr6 secondary building units. Thus, it is assumed that employing UiO-66 is a promising strategy in ODS to ensure a highly reusable catalyst. However, the lack of defects leads to poor reactivity of UiO-66 in ODS. To make UiO-66 a very useful catalyst in ODS, defect engineering is accepted to be a prolific strategy that generates more Lewis acid sites (zirconium(IV) sites with unsaturated coordination) that are highly accessible to sulfur compounds and oxidants during ODS.192 Different techniques are found to be beneficial for improving the defects in UiO-66.
W. Xiao et al. modulated the synthesis time to create defects in UiO-66. The rapid synthesis of UiO-66 promises more defects and consequently improves the ODS activity under the optimized reaction conditions as illustrated in Fig. 24a.192 Adapting the non-modulated synthesis which provides less crystalline UiO-66 with more open zirconium(IV) sites is also a very good idea. The non-modulated synthesis reported by C. M. Granadeiro et al. yielded UiO-66 of poor crystallinity and more open sites (linker deficiencies in the framework) displaying higher ODS activity than that of UiO-66 prepared by the modulated synthesis in the presence of a crystallization agent (HCl) and/or modulator (trifluoroacetic acid) (Fig. 24b). UiO-66 prepared via the non-modulated synthesis accomplishes the removal of 81% of total sulfur compounds from real diesel containing sulfur compounds equal to 2300 ppm.193
 | ||
| Fig. 24 (a) Catalytic activities of UiO-66 with different synthesis times in ODS. Reaction conditions: 0.5 mL model oil (1000 ppm DBT in n-octane); 30% H2O2 (0.5 mL); catalyst (50 mg); T (333 K); acetonitrile (20 mL). Adapted from ref. 192 with permission from The Royal Society of Chemistry. (b) Catalytic activities of the different UiO-66(Zr) samples showing the initial extraction stage (before the dashed line) and the catalytic step (after the dashed line) in ODS. Reaction conditions: 0.75 mL multicomponent model fuel (mixture of DBT, 4-MDBT and 4,6-DMDBT in n-octane with a concentration of 500 ppm of sulfur from each compound); 30% H2O2 (75 μL); catalyst (15 mg); T (323 K); acetonitrile (0.75 mL). Adapted from ref. 193 with permission from The Royal Society of Chemistry. (c) Catalytic activity of various UiO-66 catalysts in ODS. Reaction conditions: 10 g model oil (1000 ppm DBT in n-octane); 30% H2O2 (195 μL); catalyst (50 mg); T (333 K); acetonitrile (10 g). Adapted from ref. 194 with permission from John Wiley & Sons. (d) Catalytic activities of various Zr-based catalysts in ODS. Reaction conditions: 10 mL model oil (1000 ppm DBT in n-octane); 30% H2O2 (O/S = 6); catalyst (0.184 mmol of zirconium); T (333 K); acetonitrile (10 mL). Adapted from ref. 195 with permission from The Royal Society of Chemistry. | ||
Y. Sun et al. improved the ODS activity of UiO-66 by the partial substitution of titanium in place of zirconium via an ion-exchange method. Not only was there an increase in the accessible active sites, but also in the pore volume as a result of titanium substitution. These positive changes improved the ODS activity of UiO-66. Methodically, UiO-66-D and UiO-66-H are prepared in the absence and presence of a crystallization agent (HCl), respectively, and are later subjected to partial titanium substitution. After titanium substitution, as given in Fig. 24c, the ODS activities of high crystallinity and low crystallinity MOFs have been remarkably improved. Especially in the case of UiO-66-H, the yield of DBTO2 is increased 11-fold, from 5.6% to 66.3%, after Ti-substitution.194 Partially titanium substituted UiO-66 showed better ODS activity in a continuous flow reactor compared to that in a batch reactor.196 Furthermore, the catalytic efficacy of UiO-66 can be tuned by decoration with functional groups. G. Ye et al. adapted a green and scalable synthesis of UiO-66, where neither a modulator nor a solvent is utilized. Interestingly, the textural properties of UiO-66 prepared in the green method are very much comparable to those of UiO-66 prepared by the conventional method. All the prepared UiO-66 samples are separately functionalized with –NH2 and –NO2 groups, and subsequently utilized in ODS of DBT under the same reaction conditions. UiO-66 prepared via the green method yields better ODS activity than that prepared by the conventional method. The –NO2 functionalization improves the ODS activity (Fig. 24d) by designing electron deficient zirconium(IV) centres. In contrast, the functionalization of UiO-66 with electron donating –NH2 groups increases the electron density on zirconium(IV) sites and thus, the ODS activity is decreased as depicted in Fig. 24d.195
According to R. Limvorapitux et al., more open sites could be introduced into UiO-66 by removing benzoic acid modulators that cap the Zr6-oxo-hydroxo cluster nodes. Briefly, HCl treatment is carried out on pristine UiO-66 in two different solvents (n-butanol and dimethylformamide) at 373 K for 24 h. Interestingly, while HCl treatment in n-butanol leads to more open sites, a decrease in open sites is noted after the HCl treatment in dimethylformamide due to the fact that high temperature acid hydrolysis of dimethylformamide results in the formation of capping ligands (HCOOH). The more open sites emerging after the HCl treatment in n-butanol are efficiently converted into the active Zr-μ1-OOH species in the presence of H2O2, increasing the ODS activity of UiO-66.197 Like UiO-66, crystal defect engineering is also beneficial in other zirconium-based MOFs (MOF-808 and UMCM-309) which are subjected to post-treatment involving the replacement of formate (HCOO−) ligands with methanol. This ligand exchange creates additional zirconium(IV) open sites and thereby promotes the ODS activity. Meticulously, three modified MOFs (MOF-808-M, UMCM-309-M1, and UMCM-309-M2) are synthesized; M1 and M2 denote the different amounts of methanol. CD3CN chemisorption monitored by Fourier-transform infrared spectroscopy (FTIR) proves the formation of additional zirconium(IV) open sites. Based on the intensity of the band at 2299 cm−1 (attributed to chemisorbed CD3CN), the number of accessible Lewis acid sites is calculated and it is found that MOF-808-M, UMCM-309-M1 and UMCM-M2 have a higher number of accessible Lewis acid sites than their pristine samples (Fig. 25). Note that a MOF's topology is also a key factor to generate more accessible Zr4+ sites with the fact that though MOF-808 has fewer missing formate ions as compared to UMCM-309, it shows larger zirconium(IV) open sites.185b In contrast to the above-discussed zirconium-based MOFS, a different zirconium-based MOF, namely NU-1000, could exhibit higher ODS activity without the necessity for any post-treatment because the linker (tetraethyl 4,4′,4′′,4′′′-(pyrene-1,3,6,8-tetrayl)tetrabenzoic acid) that is used for the preparation of NU-1000 leads to the maximum pore volume (30.0 Å), providing higher accessibility of the sulfur compounds and oxidant to the active sites during ODS.2c This again supports the fact that the topology of the MOF is significant in determining the total ODS activity.
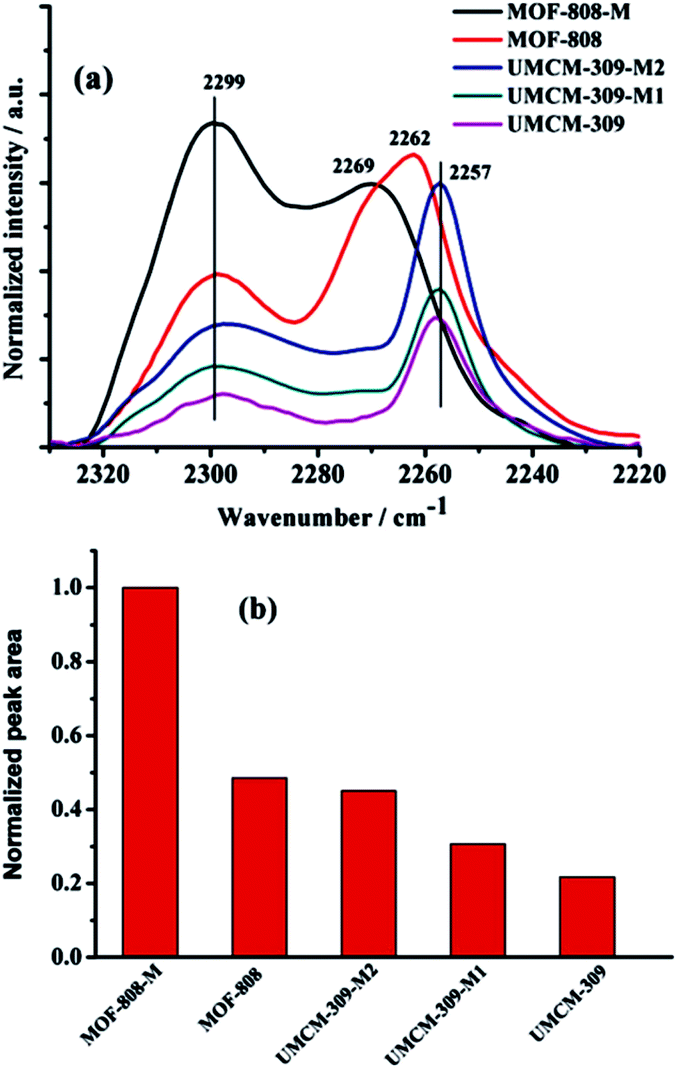 | ||
| Fig. 25 (a) FTIR spectra of CD3CN chemisorption on MOFs, normalized to the total amount of zirconium. CD3CN was adsorbed at room temperature and desorbed under vacuum (1 mbar) for 40 min. (b) The amount of CD3CN which remains adsorbed (as the normalized intensity of the peak at 2299 cm−1) after outgassing under vacuum. Adapted from ref. 185b with permission from John Wiley & Sons. | ||
Carbonization of MOFs by pyrolysis under an inert atmosphere provides interesting catalytically active MOF-based carbonaceous materials with high surface area and porosity. Mostly, zinc-based MOFs are used as the base MOF in which a secondary catalytically active metal ion such as titanium, cobalt or manganese is incorporated and subsequently pyrolyzed into the carbon materials. The pyrolysis of MOFs at 1273 K tends to yield better results which may be due to the evaporation of zinc at 1273 K. It is noted that the zinc content in the carbon material derived from the pyrolysis of MOFs at 1273 K is estimated to be either zero or very negligible.2b,198 At first, J. C. Hicks and co-workers used MOF-derived carbon materials in the ODS process as a catalyst. Briefly, they carried out the post-modification of zinc-based IRMOF-3 by titanium-incorporation using Ti(OiPr)4, forming a coordination bond with the amine groups of IRMOF-3 at the loss of one molecule of HOiPr. The pyrolysis of titanium-modified IRMOF-3 at 1273 K under inert conditions provides well-dispersed titanium nanoparticles on porous carbon, showing improved catalytic activity as compared to that of titanium-modified IRMOF-3 in oxidation of DBT with TBHP at 373 K. The proposed pyrolysis mechanism is shown in Fig. 26.198a
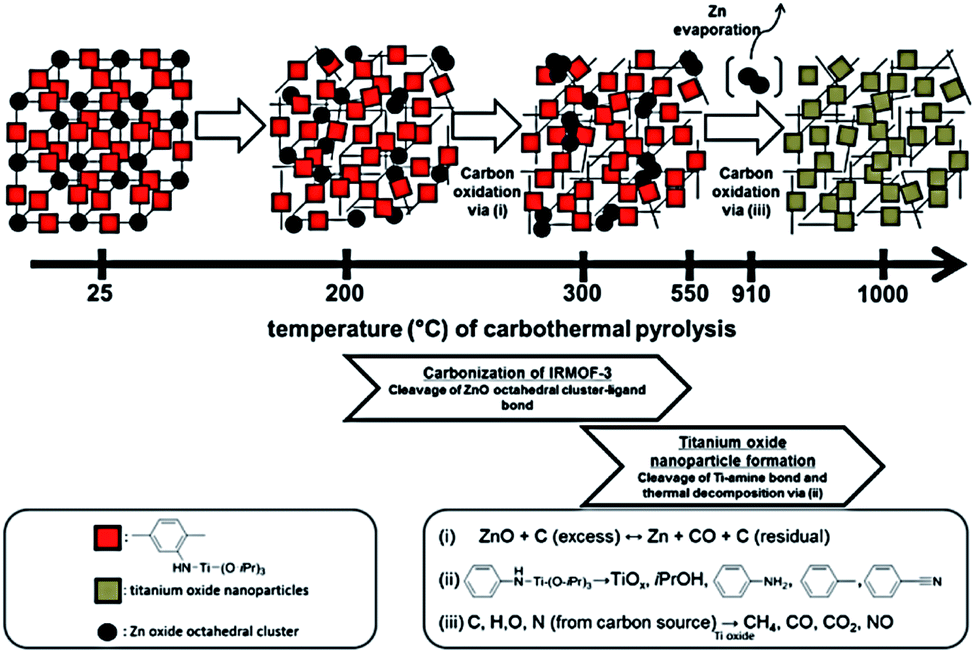 | ||
| Fig. 26 Pyrolysis mechanism of titanium-modified IRMOF-3 for the formation of titanium@carbon. Adapted from ref. 198a with permission from The American Chemical Society. | ||
With the novel post-modification strategy based on the principles of hydrophilicity, M. Sarker et al. incorporated titanium(IV) sites in two different zinc-based MOFs (MAF-6 and MOF-74) using the double-solvent method (Fig. 27a). Selective loading of titanium in a desired location related to MOFs defines the size of TiO2 particles formed after the pyrolysis. Titanium(IV) sites loaded inside MOFs generate small sized TiO2 particles after the pyrolysis, exhibiting better ODS performance as shown in Fig. 27b.199 In addition to the pyrolysis of zinc-based MOFs incorporated with other metal ions, pyrolyzing a MOF composite (MOF(Zn)@MOF) consisting of a zinc-based MOF is also a promising strategy to prepare active MOF-based carbon materials. B. N. Bhadra et al. prepared TiO2 containing carbon materials from pristine NH2-MIL-125 and its composite ZIF8@NH2-MIL-125. The pyrolysis of the composite MOF gives mesoporous carbon and highly dispersed small size TiO2 particles in comparison with the pyrolysis of NH2-MIL-125. This leads to improved catalytic performance during the ODS of DBT with H2O2 under the optimized reaction conditions and signifies the advantage of carbonization of MOF@MOF materials.200 Of note, when MOFs with no zinc content are pyrolyzed, a significant loss of surface area and porosity is noted in the resultant carbon materials. To overcome this, J. C. Hicks and co-workers pyrolyzed VAC-meso-MIL-125 which provides well dispersed titanium particles over the mesoporous carbon with improved surface area, showing very good ODS activity. This improved ODS activity, in comparison with that of the titanium–carbon material derived from the pyrolysis of micro-MIL-125, is attributed to the hierarchical mesoporosity of VAC-meso-MIL-125.201 When MIL-47(V) is pyrolyzed at various temperatures, vanadium oxides and vanadium carbides are the main products along with carbon. Vanadium@activated carbon obtained from the pyrolysis of vanadium impregnated activated carbon loses a total ∼80 mol% of its initial vanadium loading during the course of three consecutive reactions. Therefore, the activities of vanadium@activated carbon are presumed to reach zero within a short number of recycle runs. In contrast, C1000 (MIL-47 pyrolyzed at 1273 K) shows a consistent increase in activity and C1100 (MIL-47 pyrolyzed at 1373 K) maintains a consistent activity throughout the five consecutive runs. This is because the low valent inactive vanadium sites entrapped as carbides are oxidized into active high valent vanadium sites (vanadium(IV)/vanadium(V)) by TBHP during the progress of ODS.202
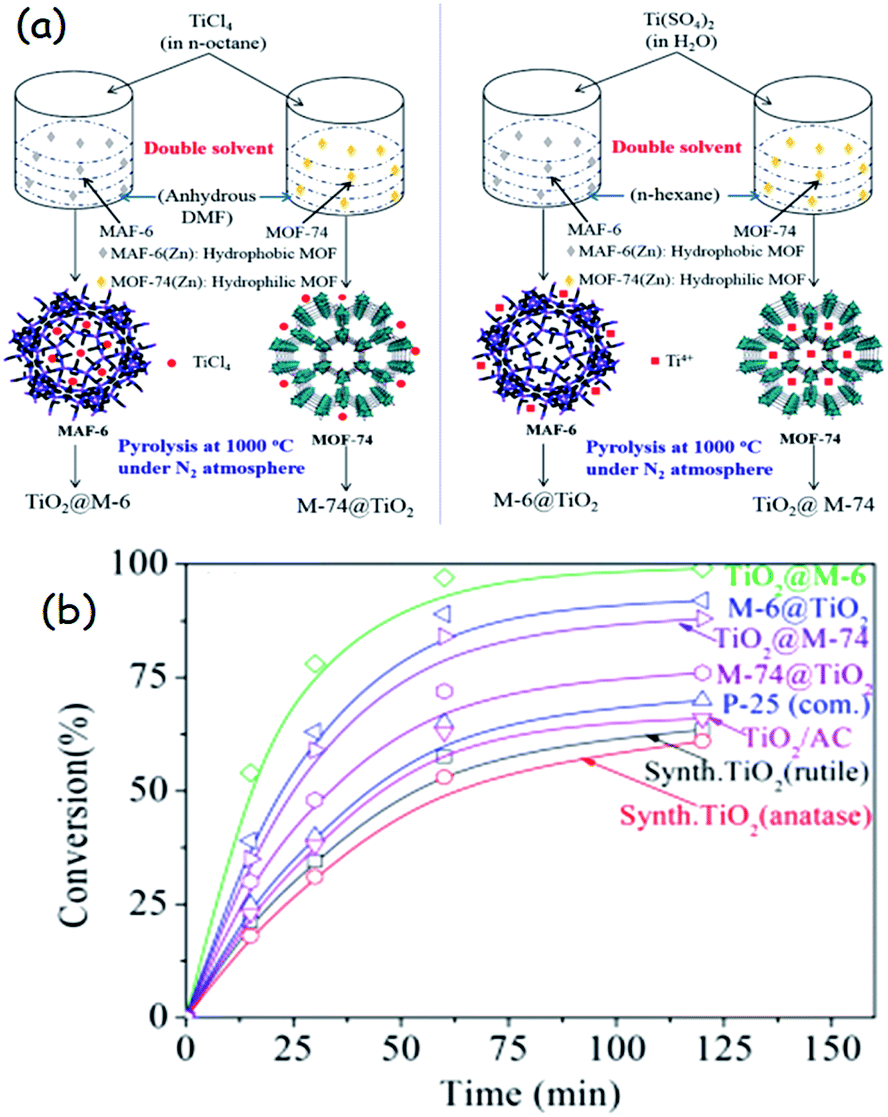 | ||
| Fig. 27 (a) Loading position of the titanium precursor on MOFs via the double-solvent method (the loading position can be selected by controlling the hydrophobic and hydrophilic properties of the MOF and solvent). (b) Activity of different catalysts during ODS of DBT in the presence of H2O2 at 353 K. Adapted from ref. 199 with permission from The American Chemical Society. | ||
In addition to showing intrinsic catalytic activity in ODS, MOFs are used as supports to encapsulate other catalytically active sites in their pores.91d,150,152a,203 Particularly, POMs are known for being encapsulated inside the pores of MOFs and used as POM@MOF catalysts in ODS. POM@MOFs have already been discussed in the previous section of the current review.
4.6. Metal-free catalysts
Metal-free catalysts are highly applauded for any reactions as they circumvent the use of costly and often toxic metals. The simple Brønsted acids, chiefly HCOOH and CH3COOH, have been known for years to promote ODS in the presence of H2O2 through the formation of active peroxyacids (Scheme 5a). Therefore, these acids may be regarded as metal-free and relatively inexpensive catalysts, despite the risk factors associated with their acidity.13,105,204 However, due to the lower activity, risk factor and non-reusability of simple Brønsted acids, new metal-free catalysts that avoid these demerits are highly necessitated. In a few reports, these simple acids have been used along with activated carbon, especially after acid treatment, which could enhance the ODS performance due to the sulfur adsorption phenomenon.205 M. T. Timko et al. exclusively investigated the role of different activated carbons in promoting the performance of a COOH–H2O2 ODS system. It is found that, in addition to the surface area and pore volume, the surface acidity, surface oxygen and more defects in the basal plane of activated carbon can also influence the ODS performance. The adsorption of sulfur compounds is very much favourable to the basal planes of carbon due to the maximizing effect of π–π interaction in the basal planes.206 Furthermore, the Brønsted acid catalyzed ODS system can be made more intriguing by using the acid groups (–COOH) grafted on supports, promoting the ODS via the formation of peroxy-acids and sulfur adsorption.207 Brønsted acids of IL types have also been studied as metal-free catalyst with the advantage that they can aid the extraction of sulfur compounds from the oil, enhancing the rate of ODS in the presence of H2O2.208 Despite the Brønsted acids' poor separation from the ODS system, they can be made handy by being heterogenized over the support which adsorbs the sulfur compounds, and utilized as the ODS catalyst in the presence of H2O2.209 Very differently to all the above reports, A. D. Bokare and W. Choi utilized sodium bicarbonate which forms the active peroxycarbonate in the presence of H2O2 as an ODS catalyst at 298 K. Peroxycarbonate species are also formed by purging CO2 in an ODS system containing H2O2.210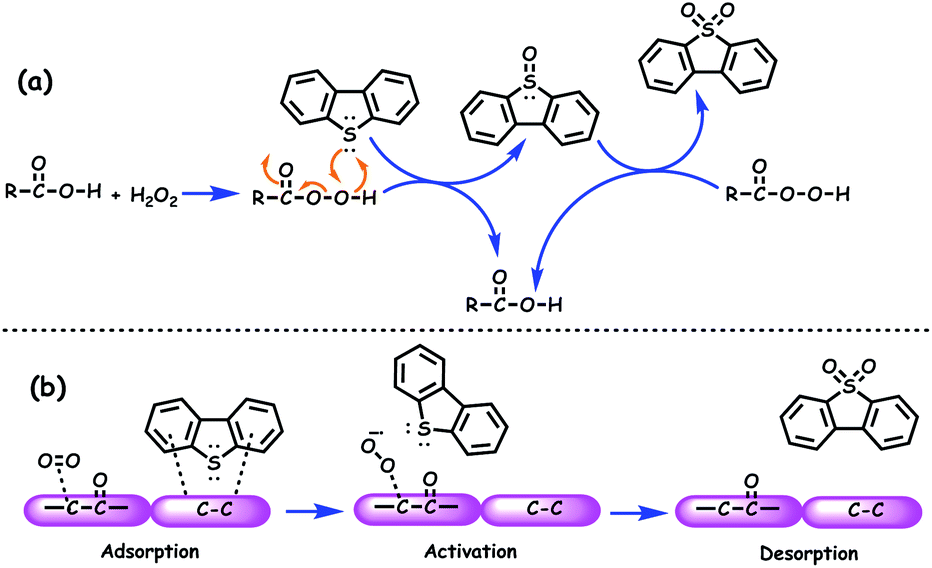 | ||
| Scheme 5 The ODS mechanism catalyzed by (a) simple carboxylic acids and (b) carbonyl groups containing carbon materials. | ||
In the process of searching for reusable, effective and risk-free metal-free ODS catalysts, carbon-based materials such as graphene, CNTs, carbon nitride (C3N4) and graphene oxide have emerged. Note that these carbon materials are often found to work in the presence of molecular O2 that undergoes the generation of the active superoxide radical anion.211 The carbonyl groups that are present in carbon-based catalysts play a vital role, that is, the carbon atom located adjacent to carbonyl groups activates O2 and generates the active superoxide radical anion (O2˙−) (Scheme 5b).211a,211b On the other hand, while using CNTs as the catalyst, the observed mechanism is that the hydroxyl groups of CNTs donate their electrons to molecular O2 which subsequently generates the reactive oxygen species that is responsible for ODS.212 Graphene like carbon nitride (g-C3N4) shows ODS activity in the presence of molecular O2; however, better performance is noted only in the presence of light;211c otherwise it exhibits lower ODS activity.211d However, doping g-C3N4 with Lewis acid sites like boron enhances the ODS activity by facilitating the interaction between superoxide radicals and sulfur compounds.211d
Like g-C3N4, g-h-BN nanosheets also find catalytic applications in ODS in the presence of O2 at 423 K. When solvents with low boiling points are used in the preparation, g-h-BN nanosheets with a larger surface area and more crystal defects are obtained, demonstrating improved ODS activity.213 The major drawback of an ODS system working with metal-free catalysts (graphene oxide, CNTs, g-C3N4 and g-h-BNs) in the presence of O2 is that it requires high temperature (>373 K) for the O2 activation, which may reduce the yield of low boiling fuels (e.g. diesel) due to their volatilization at higher temperatures.214 L. Lu et al. recently reported that doping g-h-BN nanosheets with carbon atoms leads to the ODS system working efficiently in the presence of O2 at relatively low temperature due to the bonding between doped carbon atoms and g-h-BN's nitrogen. Particularly, this C–N bonding promotes the formation and delocalization of π-electrons, facilitating the electron transfer from g-h-BN to O2. Eventually, the O2 activation and ODS performance are promoted even at relatively low temperature.214
4.7. Other catalysts
Other than those in the above-listed categories, there are a few other catalysts known to perform ODS and in particular, metal nanoparticles. Incorporating copper(0) nanoparticles into g-C3N4 boosts the electron mobility of g-C3N4 by which the aerobic ODS performance of g-C3N4 is increased to 100% from 60 and 65% in the removal of DBT and 4,6-DMDBT, respectively.215 Red mud can be used as a catalyst but its higher hydrophilicity and non-magnetic behaviour are disadvantages. Through the reduction of iron(III) to iron(II) and carbon coating, Fe3O4 can be made magnetic and amphipathic, respectively. Over this advantageous red mud, gold(0) nanoparticles are dispersed, which leads to the synergy between gold(0) and iron(II). The cores of iron(0) and Fe3O4 in the surface of reduced red mud produce ˙OH radicals according to the Fenton reaction mechanisms. With the use of the thus-produced ˙OH radicals, gold(0) nanoparticles, due to their affinity for sulfur, facilitate the ODS reaction by adsorbing more sulfur compounds. The synergy between gold(0) and iron(II), and the consequent improved ODS are pictorially presented in Fig. 28.216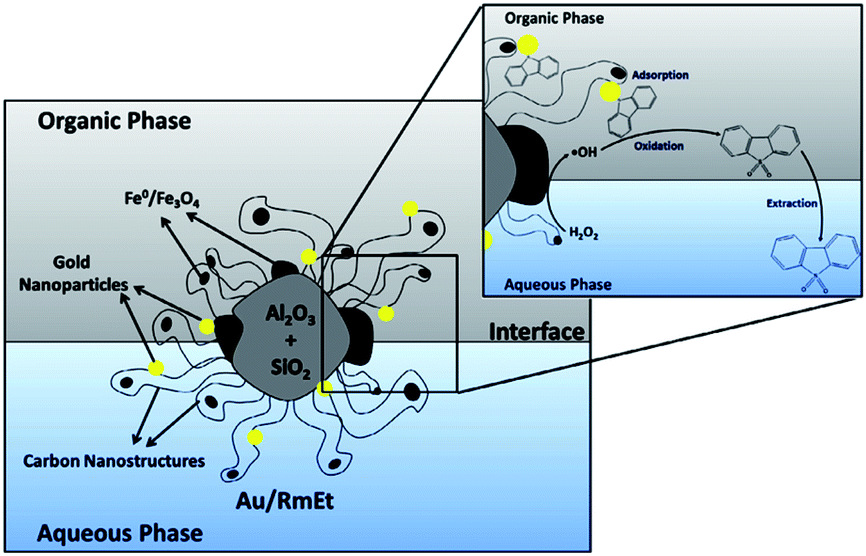 | ||
| Fig. 28 Mechanism of synergy between Au and Fe during the ODS. Adapted from ref. 216 with permission from Elsevier. | ||
Removing the barriers in the synthesis of bulk crystalline tungsten nitride (W2N) through combined pyrolysis of HPW and polyaniline at ≥1073 K, N. A. Khan et al. prepared crystalline W2N@porous carbon which displays excellent ODS activity with a faster DBT conversion rate (1.1 × 10−1 min−1) in the presence of H2O2 at 333 K.217 L. Wu et al. carried out ODS just using two-layer SiO2 gels as the catalyst without the addition of any common catalyst using cumene hydroperoxide as the oxidant (Fig. 29a). In their study, the following commercial SiO2 gels are employed: different thin layer chromatography SiO2 gels (T1: Type-G-1, T2: Type-G-2 and T3: Type-G-3), column chromatography silica gels of different mesh sizes (C1: 100–200, C2: 200–300 and C3: 300–400) and amorphous fumed silicas of different surface areas (F1: 175–225, F2: 300–350, and F3: 350–420 m2 g−1). The reason for the amazing catalytic activity of SiO2 gels is attributed to the presence of inherent Lewis acid sites (titanium(IV)) even in a small amount (≤0.018 wt%). As column chromatography SiO2 gel possesses relatively more Lewis acid sites, it demonstrates relatively better performance in ODS. On the other hand, amorphous fumed SiO2 exhibits very poor ODS activity due to its lack of Lewis acid sites. The incorporation of 0.02 wt% titanium(IV) dramatically boosts the activity of amorphous fumed SiO2 (F3) from 7.38% to 98.8%. The variation of ODS activity with the total Lewis acid sites and titanium content is shown in Fig. 29b. Interestingly, the textural properties of these commercial SiO2 gels do not show a significant influence as compared to the Lewis acid sites. We believe that this commercial cheap SiO2 gel catalyst may be highly intriguing for ODS applications in the coming years if it works together with environmentally friendly oxidants (H2O2/O2).218
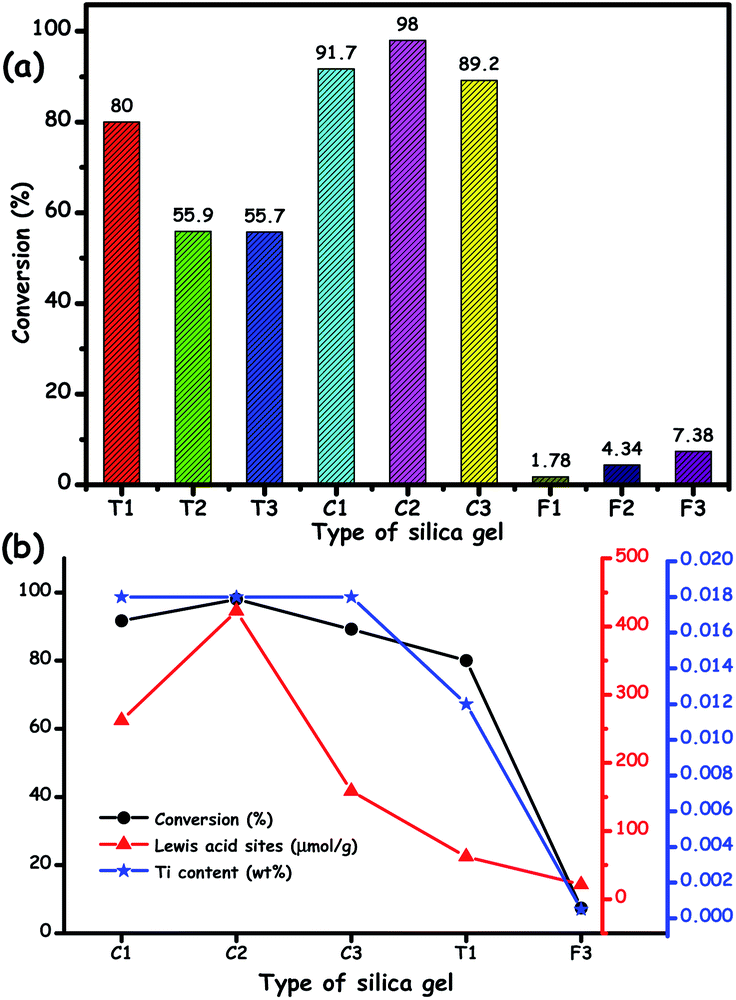 | ||
| Fig. 29 (a) DBT conversion with different SiO2 gels. Reaction conditions: catalyst (1.5 mg mL−1), cumene hydroperoxide (O/S = 3); t (3 h); T (373 K). (b) Catalytic activity of different SiO2 gels and its dependency on the total Lewis acidity, and titanium content. Adapted from ref. 218 with permission from The American Chemical Society. | ||
5. Influence of reaction conditions on ODS activity
There are a few significant reaction conditions that influence the activity of ODS catalysts either directly or indirectly and such factors are described in this section.5.1. Temperature and pressure
It is generally possible to control the activity of any catalyst with the reaction temperature and thus, ODS catalysts behave differently at various temperatures. In line with the common fact, ODS catalysts require a particular temperature to ensure the maximum collision from the reactants/solvents for accomplishing better results. Mostly, temperatures of 333–353 K seem to be the optimum reaction temperature range to achieve better ODS performance in the presence of catalysts, since the boiling point of the common solvents either used to prepare the model oil (n-hexane or n-heptane or n-octane) or to construct the biphasic system (acetonitrile) falls in the range of 333–353 K. Notably, temperatures above 353 K are not desirable when H2O2 is utilized as the oxidant due to the fact that the higher temperatures promote faster degradation of H2O2, which leads to the improper use of H2O2. On the other hand, if air or molecular O2 is the chosen oxidant, the ODS catalyst requires a higher temperature (≥373 K) since higher thermal energy is required for the activation of O2. With liquid oxidants like H2O2, ODS catalysts usually show the maximum performance at atmospheric pressure. However, during the use of gaseous oxidants like O2, high pressure is sometimes required depending on the nature of the catalysts.115c,122,175c,175d For example, Fig. 30 illustrates the activity variation of FePc(NO2)4 under different initial pressure conditions during ODS. Of note, the hindering of sulfone precipitation due to the high pressure emphasizes the significance related to controlling the pressure in ODS.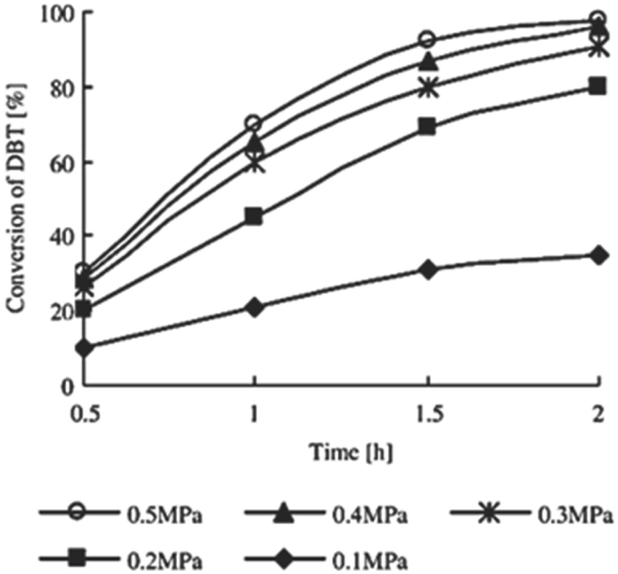 | ||
| Fig. 30 Effect of initial pressure on the conversion of DBT at 373 K with 1 wt% FePc(NO2)4 over the whole solution. Adapted from ref. 175d with permission from Elsevier. | ||
5.2. Solvent and oxidant
Solvents play a crucial role in chemical reactions by creating a phase for multiple reactants to have the maximum interaction with each other. The ODS system is mostly biphasic in nature (polar and non-polar) and thus, the role of solvents is understood differently in comparison with that in common monophasic reactions. As the non-polar medium (oil phase) is the real liquid fuel, the influence of solvents on the activity of ODS catalysts is assumed to be induced by the polar solvent. However, for the ODS of model oils, though no obvious change is noted in the relative ODS performance of the catalyst, the change in reaction rate is apparent while using non-polar solvents with different carbon numbers. Among the three different non-polar solvents (n-hexane, n-heptane and n-octane), n-hexane supports a faster DBT conversion during ODS with HPW. This highlights the advantage of non-polar solvents with a lower carbon number.108 Typically, catalysts and oxidants are expected to be present in the polar solvent during ODS. During the course of ODS, the polar solvents could extract a certain amount of sulfur compounds from the non-polar oil phase and for this, they are termed as the extractants. Interestingly, the polar solvents accomplish the effortless recovery of the formed sulfones from the ODS system. The bi-phasic system and the extraction of sulfones in ODS is depicted in Fig. 5. Using an aprotic polar solvent is beneficial since aprotic solvents maintain the stability of HO− and HO2− radicals that produced from the dissociation of H2O and H2O2. This increases the probability of interaction between HO2− and HO− groups to yield more superoxide radical O2−˙ (reactive oxygen species).21i In this context, being a polar and aprotic solvent, acetonitrile is very familiar in ODS and exhibits a good cooperativity with almost all the types of ODS catalysts. In addition to the polarity and aprotic nature of the solvents, the synergy of the solvents with oxidants and catalysts could also influence the ODS activity.21i This synergy is exemplified (Fig. 31) by comparing the DBT removal accomplished by simple solvent extraction and extraction–oxidation using 16% MoO3@γ-Al2O3. The solvent with the best extraction ability fails to accomplish the best results during the extraction–oxidation process, emphasizing the significance of synergy between the solvent and catalyst.20c It is assumed that the solvents should not occupy the vacant coordination site of the catalyst which is essentially allotted for the oxidant and sulfur compound.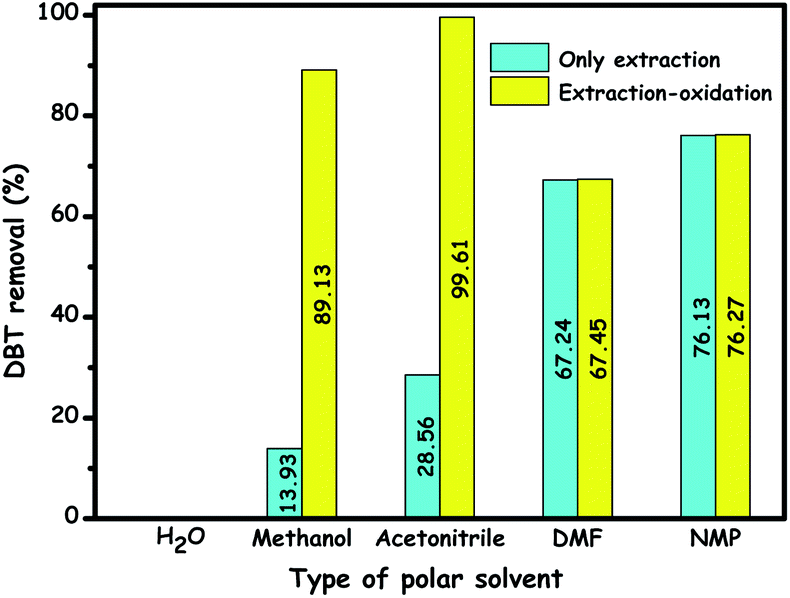 | ||
| Fig. 31 Effects of different polar solvents (extractants) on the removal of DBT. Reaction conditions: T (333 K); 30% H2O2 (O/S = 2.3); CDBT (320 ppmw); catalyst (0.2 g); t (1 h); v(oil)/v(extractant) (3:1). Note: For the simple extraction, neither the catalyst nor the oxidant is used. Adapted from ref. 20c with permission from The American Chemical Society. | ||
In the literature, despite their high cost, ILs are highly recommended owing to their thermal stability, poorly combustible nature and solvation properties towards a wide range of polar and non-polar compounds. A variety of ILs are known as solvents for catalytic ODS applications. It is intriguing that specially designed ILs can also act as catalysts. Such IL-based catalysts are randomly mentioned in this review under POM ODS catalysts.107b,133c P. Yuan et al. theoretically gave significant insights into the interaction of ILs with Th/sulfone. These proposed interactions strongly support the use of ILs as solvents for the catalytic ODS. It is proposed that both the cationic and anionic ILs and ion-pair of ILs show noticeable interaction with Th as well as sulfone. Sulfone involves a much stronger interaction than Th due to the electronegative oxygen atoms. ILs can possibly decrease the energy barrier associated with the oxidation of Th and thus are highly useful compared to the usual solvents like acetonitrile.219 The multiple roles of ILs in desulfurization can be better understood from earlier reviews.220
Oxidants leaving no unwanted side-products are more preferable for the catalytic ODS. For instance, TBHP or cumene hydroperoxides produce unsolicited wastes after ODS and thus are not advisable despite their facile mass transport across the polar and non-polar media during the biphasic ODS. On the other hand, molecular O2 and H2O2 are very attractive in view of green chemistry as they yield only water as the side-product. Advantageously, these oxidants are very cheap and hence are highly recommended for ODS applications. The merits and demerits of various oxidants in ODS have already been debated in many reports, which can be referred to for further discussion.9,221 In terms of determining ODS activity, the interaction between the oxidant and ODS catalysts is more significant and it should lead to faster generation and utilization of reactive oxygen species. However, this significant interaction is generally dependent on the nature and electronic properties of the ODS catalysts. Of note, oil-soluble oxidants (e.g. TBHP) are intriguing for carrying out ODS without the use of any extractant but are not advised for extraction–ODS.186b
5.3. Ultrasonication and photochemical conditions
Irrespective of the type of catalyst, the role of ultrasonication in influencing ODS activity is based on a few important acoustic processes associated with fluidic actions such as cavitation and micro-streaming. Micro-streaming induces emulsification, which in turn produces a higher interfacial area between the fuel and oxidant. As a result of cavitation, the generation of highly active radicals is realized.222 However, cavitation sometimes produces a negative effect on the net ODS activity due to the fact that the active radicals generated by cavitation scavenge the HO2˙ radicals. Especially, the generated chemical species like hydrogen and carbon monoxide consume the oxidant species.223 The power of the ultrasound is generally directly proportional to the performance of catalytic ODS. Despite yielding fruitful results, the high energy utilization and high capital cost due to the sono-reactor, amplifier and function generator make the practical application of ultrasound-assisted catalytic ODS questionable.222aThe performance of light-assisted catalytic ODS (catalytic photochemical ODS) greatly depends on the nature of catalysts, generally defined as “the photocatalysts”. A variety of metal oxides have shown the maximum ODS activity in the presence of light due to their unique electronic structures that facilitate the generation of photoinduced electrons and holes.224 Especially, TiO2 is a very familiar photocatalyst due to its abundance, low cost and chemical stability. However, the wide band gap of bare TiO2 is only for harvesting UV light which is a very small portion of sunlight.224c For catalytic photochemical ODS applications under visible light, TiO2-based composites are prepared using certain additives like noble metals and employed as catalysts for harvesting visible light.225 Furthermore, utilizing such composites is beneficial for preventing the recombination of electron–hole pairs and increasing the light absorption. In this context, a lot of metal oxide based composite materials have been reported as catalysts for catalytic photochemical ODS such as Nb2O5/Bi2WO6,226 Ag/TiO2@porous glass,225 TiO2/multi-walled CNTs,227 ZnPc/SnO2,228 Pt–RuO2/TiO2,229 Ni–CuO/BiVO4,62h Bi2S3/Bi2WO6,230 Au/TiO2,231 carbon/TiO2@MCM-41,232 Cu–Fe/TiO2,233 mixed metal oxide(CoAl)/BiVO4,62b CuW/TiO2–graphene oxide,234 FePc-NH2/Ti-MCM-41,175b BiVO4/C3N4@SiO2,235 and CoPc/La0.8Ce0.2NiO3.236 Dispersing the catalysts over the supports for the catalytic photochemical ODS avoids the agglomeration of the catalysts, yielding better performance.175b,229,232,235
The common mechanism for the catalytic photochemical ODS is given in Fig. 32 based on photochemical ODS catalyzed by Ag/TiO2. The photoinduced free electrons react with O2 or H2O2 to produce O2−˙ or ·OH and −OH, respectively. On the other hand, the holes (h+) react with −OH and H2O to produce ·OH. Subsequently, these photogenerated radicals accomplish the oxidation of sulfur compounds to sulfones in ODS.225 The oxygen vacancies in the photocatalysts facilitate the electron jump from the valence band (VB) to the conduction band (CB) by acting as the drawing board. Significantly, this promotes the separation of the photogenerated electron–hole pairs, favouring an improved ODS performance. The heterojunction created in the photocatalysts ensures the sufficient release of photoinduced electrons by evading the recombination of electron–hole pairs.225
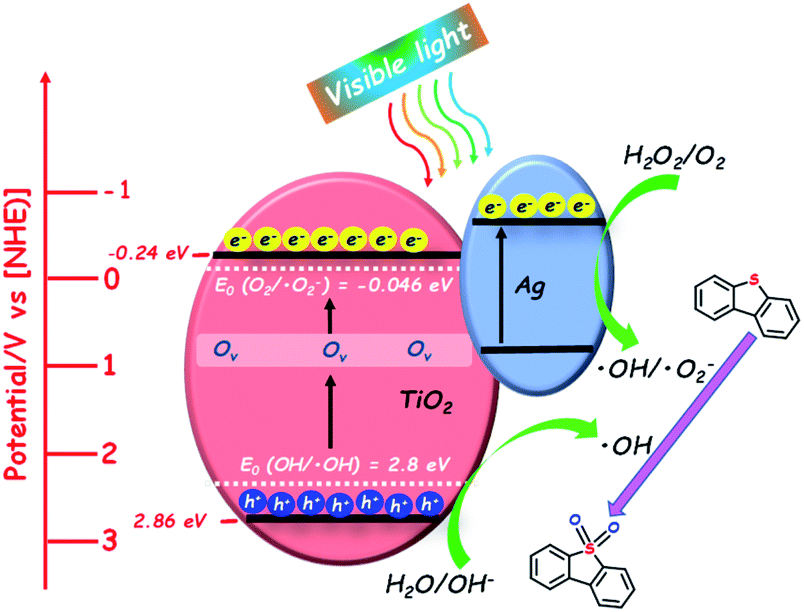 | ||
| Fig. 32 Main concept of the catalytic photochemical ODS system based on the catalyst Ag/TiO2 which illustrates the possible separation and transfer of charge carriers in Ag/TiO2@porous glass under visible light. Adapted from ref. 225 with permission from The American Chemical Society. | ||
Recently, graphene oxide and its related materials have emerged as catalysts for ODS under photochemical conditions owing to their layer structure, electron transfer capacity and ability to form π-complexes with sulfur compounds.211c,237 In addition, POMs238 and Schiff base complexes179a can also serve as catalysts for the photochemical ODS. Executing the catalytic photochemical ODS circumvents the need for thermal energy for the activation, which means that such activation energy can be acquired from natural sunlight. A. S. Morshedy et al. compared the removal of sulfur from real diesel under visible light obtained from a linear halogen lamp and natural sunlight using a CdO catalyst. Interestingly, the reaction performed under natural sunlight reduces the sulfur content of real diesel from 11500 to 217 ppm, while under the visible light from the halogen lamp, the sulfur content is only reduced to 270 ppm. These results suggest the economic and environment benefits related to the catalytic photochemical ODS that could be performed at room temperature using natural solar energy in the presence of environmentally friendly oxidants (H2O2/O2). The same authors also found that the catalytic photochemical ODS (catalyst + oxidant + light) shows better results than photocatalytic ODS (catalyst + light + no oxidant) and photochemical ODS (oxidant + light + no catalyst). This shows the significance of integrating the photocatalytic and photochemical ODS processes.224d
6. Influence of feedstock characteristics on ODS activity
Only a very few reports are available on the catalytic ODS of real oil feedstocks. With such reports, it seems difficult to derive the direct relation between the types of real oil feedstocks and the catalytic ODS performance. Of course, expecting the same activity for an ODS catalyst in two different real oil feedstocks is practically not possible. ODS of light oil types, e.g. diesel, is relatively easy in comparison with that of heavy oil which has a higher density and catalyst fouling nature. The density of real oil may affect the mass transport and consequently the ODS performance.7 By elucidating the behaviour of alkenes/aromatics/nitrogen compounds during the ODS, we may indirectly relate how the real oils, according to their composition, influence the catalytic ODS processes.6.1. Fuel constituents (alkenes/aromatics/nitrogen compounds)
Alkenes, alkanes and aromatics are the main constituents and building networks of real oil feedstocks. Like sulfur compounds, a considerable amount of nitrogen compounds is also invariably present in almost all fuel feedstocks. Thus, it is noteworthy to analyse the interference of these compounds during the catalytic ODS. With the vulnerability to oxidation reactions, alkenes/alkanes/aromatics/nitrogen compounds are generally expected to influence ODS. In 2001, for the first time, Otsuki et al. studied the effects of alkane (n-pentadecane), alkene (diisobutylene), aromatic (xylene) and nitrogen compounds (indole) on ODS of a model oil (DBT in decalin) catalysed by 3%-Pt/Al2O3 in the presence of t-BuOCl oxidant. Despite the absence of any significant effects being noted during the addition of n-pentadecane and xylene, the addition of indole and diisobutylene (alkene) to the model oil notably retards the ODS activity. This is attributed to the better reactivity of indole and diisobutylene (alkene) than DBT under the catalytic oxidation conditions.239 Subsequently, more reports have agreed with and supported findings indicating the interference of alkenes with the catalytic ODS activity due to the olefin oxidation.110a,240 Y.-K. Lee and co-workers suggested that the polyaromatic compounds of fuel feedstocks, due to their ability to dissolve sulfones, can assist the ODS catalysts to maintain constant activity during the course of ODS. Significantly, these polyaromatic compounds are assumed to circumvent the deposition of oxidized sulfur compounds over the catalyst surface.16c,241 Unfortunately, these polyaromatic compounds could not show the same positive effect during the catalytic ODS under photochemical conditions due to the interference of polyaromatic compounds with the energy transition process associated with sulfur oxidation. This indicates that the catalytic photochemical ODS is very effective only for fuel feedstocks with low polyolefin and polyaromatic contents (e.g. straight-run gasoline).110aConcerning nitrogen compounds, L. C. Caero et al. compared the influence of different nitrogen compounds (indole, carbazole and quinoline) on the activity of V2O5/Al2O3 and V2O5/TiO2 during ODS of the model oil in the presence of TBHP oxidant. Carbazole and quinoline do not diminish the catalytic ODS activity through their competitive oxidation but through the adsorption on the chosen catalyst surface which poisons the active sites. As a consequence, the decomposition of the oxidant is suppressed and thus, the ODS activity is decreased.242 In contrast to this, when H2O2 is used as the oxidant, the addition of carbazole/quinoline has shown positive effects on ODS activity of the catalyst. The reason is that the usual decomposition of H2O2 is usually uncontrolled under thermal and catalytic conditions. The presence of carbazole and quinoline type nitrogen compounds assists the proper utilization of H2O2 and consequently increases the catalytic ODS activity in the presence of quickly decomposable H2O2.130c However, unlike carbazole and quinoline, the presence of indole severely diminishes the catalytic ODS activity of the catalysts due to its high electron density as compared to that of all the RS compounds. This negative effect of indole during the catalytic ODS is also acknowledged in various occasions where different catalysts are used.85,218,241
Very recently, L. Wu et al. performed a systematic analysis regarding the effects of different fuel components (m-xylene, 1-hexene, 1-isopropylnaphthalene, methyl tert-butyl ether (MTBE), and indole) on the ODS activity of titanium-containing commercial chromatography SiO2 gel with 200–300 mesh size. This investigation, apart from giving valuable insights into the effects of fuel components on catalytic ODS activity, also suggests routes to accomplish better ODS results in the presence of these interferents. As shown in Fig. 33, the catalytic ODS activity, even in the presence of the tested fuel components, is increased by utilizing the oxidants excessively. The effect of MTBE can be made negligible at a high O/S ratio because common fuels contain only less than 7 vol% MTBE. On the other hand, the presence of 150 ppm indole severely diminishes the ODS activity. Even increasing the O/S ratio from 2:1 to 5:1 is not found to be helpful as it could only improve the ODS activity from 8.37 to 9.05%.218
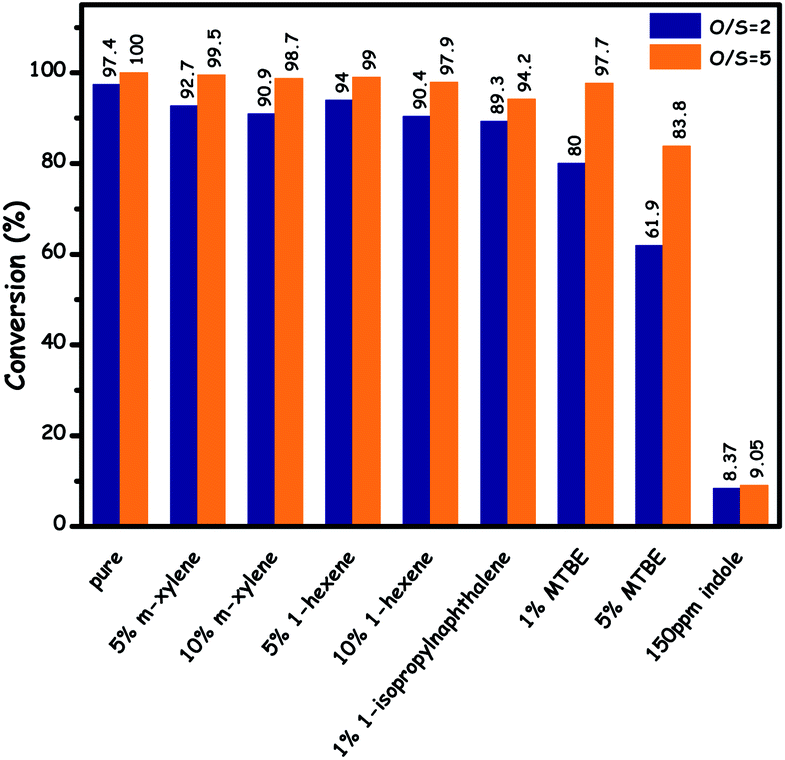 | ||
| Fig. 33 DBT conversion using commercial column chromatography SiO2 gel as the catalyst at 373 K under two different O/S ratio conditions in the presence of different fuel components. Reaction conditions: catalyst (2.5 mg mL−1); cumene hydroperoxide (O/S = 2 and 5); T (373 K); and t (2.5 h). Adapted from ref. 218 with permission from The American Chemical Society. | ||
Based on the above facts related to the effects of fuel components on the catalytic ODS activity, the following key points are summarized. The effect of alkanes and aromatics on the catalytic ODS activity is usually ignorable or nullified with the addition of excessive oxidants. Though the competitive oxidation of olefins looks quite problematic, it could be solved by utilizing the oxidants in greater quantities. Furthermore, the issues caused by olefins can easily be rectified with the application of selective catalysts. The real challenge is to overcome the effects of indole on ODS activity. With high electron density and facile oxidation to indigo dyestuff, indole causes serious trouble during the catalytic ODS. Thus, as of now, the selective extraction of all nitrogen compounds from fuel feedstocks before performing the ODS is highly recommended for better results and practical applications.
6.2. Types of sulfur compounds
Sulfur compounds of real fuel feedstocks are generally categorized into a few significant types as given in Fig. 2. It is very rare for any catalyst to show the same rate of ODS towards two different RS compounds. The removal of RS compounds is possible in ODS even under mild conditions due to their electron density. It is beneficial that the common fuel feedstocks mainly contain RS compounds. The common reactivity order of catalytic ODS towards different RS compounds without the catalysts is 4,6-DMDBT > MDBT > DBT > BT > Th according to the order of electron density.13 But the same order cannot be realized in the presence of catalysts because the different catalysts, depending upon their properties, follow different reactivity orders. With catalysts, in most of the cases, the highest ODS performance is noted towards DBT than 4,6-DMDBT due to the fact that the steric hindrance of bulky 4,6-DMDBT prevents the catalytically active sites from freely accessing the sulfur atom.17,116,133b,135a,170c,181a,243 Depending on the type of catalyst, the ODS activity in the removal of 4,6-DMDBT is sometimes even less than that of BT.146,181a,243b Considering DBTs with more alkyl substituents, the rate of catalytic ODS depends on both the number and position of alkyl substituents present in the alkyl DBTs.244Significantly, when porous catalysts like MOFs or POM@MOFs are used, the order of ODS reactivity towards the different RS compounds will be different. In these cases, the window size of the catalysts plays a decisive role in determining the catalytic ODS activity.153 For instance, the ODS activity of different HPW@MOF catalysts of different window sizes towards BT, DBT and 4,6-DMDBT is compared in Fig. 34. It shows that HPW@UiO-66 displays lower activity in the removal of 4,6-DMDBT, despite showing better activity towards BT, because its window size is smaller than the size of 4,6-DMDBT. In contrast, HPW@MIL-100(Fe) shows better activity in the case of 4,6-DMDBT owing to its big window size. On the other hand, HPW@ZIF-8 could not show better ODS activity in any of the tested RS compounds due to its very small, inaccessible window. For catalysts with big window sizes, the active sites located on the outer surfaces and inside the pores are accessible to 4,6-DMDBT. But only the active sites located on the outer surface are accessible to the bulky RS compounds if catalysts with small window sizes are employed.150 The above discussion emphasizes that the relation between the ODS catalysts and the reactivity order of different RS compounds is primarily dependent on the types and characteristics of the chosen ODS catalysts.
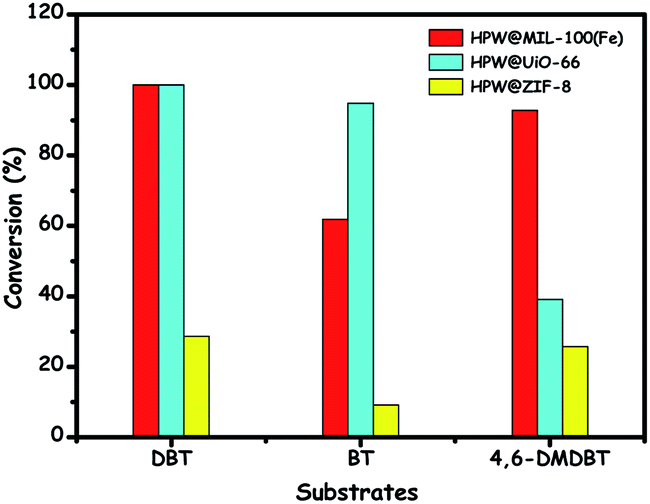 | ||
| Fig. 34 ODS of different RS compounds using catalysts with different window sizes. Reaction conditions: model oil (2 mL), catalyst (0.6 mol%), CH3CN (2 mL), H2O2 (O/S = 4), T (343 K), t (24 h). Adapted from ref. 150 with permission from John Wiley & Sons. | ||
7. Conversion and utility of sulfones
ODS causes the loss of carbon content in the resultant fuel which in turn disturbs the chemical potential of the fuel. This problem would be severe in the ODS of sulfur-rich fuel feedstocks.245 Therefore, to rectify this issue, the conversion of sulfones into hydrocarbons has drawn interest along with ODS. In the literature, sulfones are generally subjected to thermal decomposition for the production of hydrocarbons with the elimination of SO2 as a by-product. R. Weh and A. de Klerk analyzed the thermal decomposition products of different sulfones such as acyclic aliphatic, cyclic aliphatic, acyclic aromatic, and aromatic ones. From this study, two significant observations have been recognized. One is that the thermal decomposition of acyclic aliphatic sulfones and sulfones attached to two aromatic groups begins at higher temperature (>350 °C). The other one is that the thermal decomposition of five-membered cyclic sulfones in a terminal ring, no matter whether it is aliphatic or aromatic, starts at lower temperature (<300 °C). This is due to the fact that the addition reaction that occurs during the pyrolysis of five-membered cyclic sulfones in a terminal ring promotes the more facile elimination of SO2.245Highly basic alkali and alkaline earth metal-based compounds such as their hydroxides,247 layered double hydroxides,248 and oxides246,249 have been utilized to facilitate the conversion of sulfones into hydrocarbons by thermal decomposition with the elimination of SO2. Note that under the action of these highly basic compounds, the decomposition of the parent sulfur compounds is very difficult as compared to that with their oxidized counterparts (sulfones) under the same reaction conditions (Fig. 35), as published by R. Sundararaman and C. Song.246 The alkali and alkaline metal oxides used in the thermal decomposition of sulfones can serve as sorbents to capture the eliminated SO2. Interestingly, after the sorption of SO2, the formed MxSO3 (x = 1 (alkaline earth metals) or 2 (alkali metals)) can be easily regenerated in the presence of an inert sweep gas.246
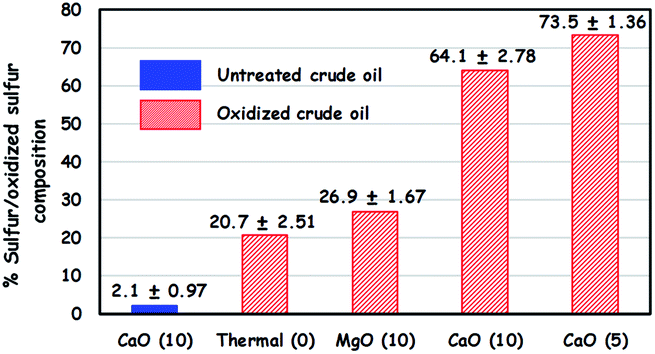 | ||
| Fig. 35 Effect of basic oxides on decomposition of sulfur and oxidized sulfur compounds in untreated and oxidized crude oils (numbers in parentheses indicate the feed/alkaline earth metal oxide ratio on a weight basis). The error bound is the standard deviation over three runs (reaction conditions: temperature, 633 K; time, 480 s). Thermal (0) denotes the absence of sorbent during the decomposition. Adapted from ref. 246 with permission from The American Chemical Society. | ||
Even though the thermal decomposition in the presence of alkali/alkaline earth metal-based compounds is familiar, the requirement for very high temperature has shed light on the discovery of new methods and catalysts which can convert sulfones to hydrocarbons relatively better under mild conditions. In this context, the photochemical conversion of sulfones to hydrocarbons was accomplished at room temperature by Y. Shiraishi et al.250 Recently, a nanosized Fe2O3 was used as the catalyst for enabling the sulfone conversion to hydrocarbons at 90 °C under atmospheric pressure conditions.251 However, irrespective of the catalysts and methods, the complete conversion of sulfones with 100% selectivity towards hydrocarbons is yet to be established. It is suggested that the sulfones need to be extracted from the feedstocks before their treatment. If not, the core components of the fuel feedstocks like aromatics may yield some unsolicited side products during the conversion of sulfones to hydrocarbons. To the best of our knowledge, in this field, apart from converting sulfones to hydrocarbons, other possible approaches are still under consideration.
8. Reactors for catalytic ODS
ODS reactions, in the literature, are frequently carried out in batch reactors which are not generally viable for industries because fixed-bed flow reactors are considered to be suitable and profitable for large scale and long run industrial applications. Till now, only a very few reports are available on catalytic ODS processes performed in bed reactors.24a,84,252 Depending upon the nature of the oxidant (H2O2 or molecular O2), the design of these bed reactors may vary slightly. Furthermore, the reactor design will be more challenging if photocatalysts are used since an additional energy source (light) needs to be implemented in the reactor. The same challenge is expected while designing the reactor for the ultrasound assisted ODS in the presence of any solid catalysts. Despite the availability of already reported reactors for the catalytic ODS, we assume that, for realizing industrial scale ODS applications, the reactor design is still under progress and requires a lot of developments along with data derived from process modelling applications like Aspen Plus.252a An ODS catalyst possibly displays different catalytic behaviours when being separately utilized under batch reactor conditions and continuous flow bed reactor conditions. Therefore, designing pilot scale flow reactors is desirable and it may quicken the process of recommending any ODS catalyst for industrial applications and two such reactors are presented in Fig. 36 and 37.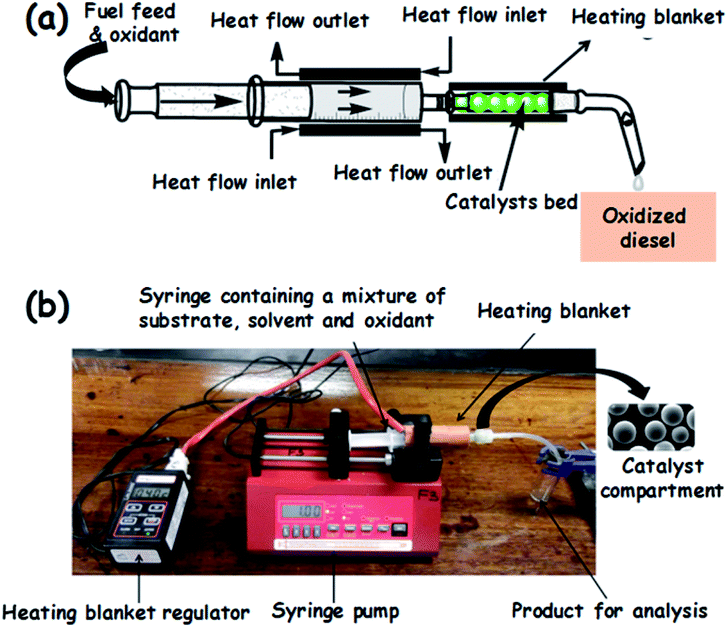 | ||
| Fig. 36 (a) Schematic representation of the setup of a continuous flow system for the catalytic ODS and (b) the real experimental setup. Adapted from ref. 252b with permission from The American Chemical Society. | ||
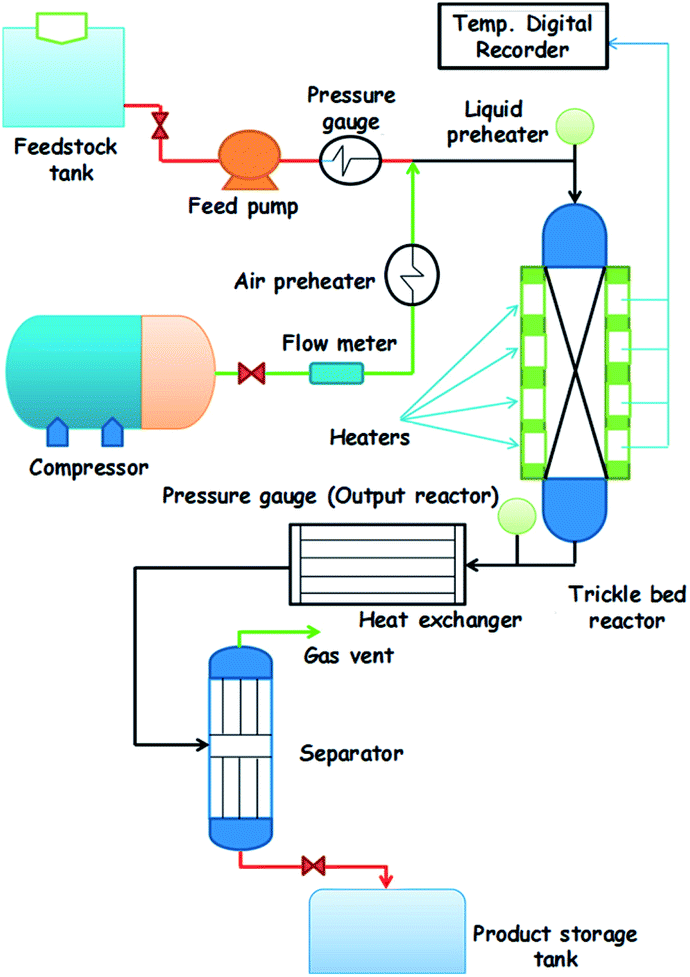 | ||
| Fig. 37 Schematic representation of the trickle bed reactor proposed by A. T. Nawaf et al. Adapted from ref. 252c with permission from The American Chemical Society. | ||
9. ODS of compounds (H2S, CS2 and COS) other than RS compounds
Although this present review is entirely focused on the catalysts used in the ODS of RS compounds present in liquid fuels, for a comparison and better understanding, catalysts reported for the ODS of hydrogen sulfide (H2S), carbon disulphide (CS2) and carbonyl sulfide (COS) are briefly discussed in this section. Unlike RS compounds, H2S, CS2 and COS are either gaseous (H2S and COS) or highly volatile liquid (CS2) compounds and vastly present in natural gas, petroleum gas and industrial tail gas. HDS of RS compounds also leaves H2S as the major side-product. Due to the physical forms of H2S, CS2 and COS, catalysts with more adsorption capacity are preferable. Thus, the ODS catalysts with greater surface area, porosity and interesting morphology are very intriguing. ODS of H2S is relatively easy as compared to that of CS2 and COS. Therefore, in the literature, CS2 and COS are initially hydrolyzed into H2S and CO2. Subsequently, the formed H2S undergoes the ODS process that yields elemental sulfur and water.253 The hydrolysis of CS2 and COS into H2S, and the ODS of H2S are presented in Scheme 6.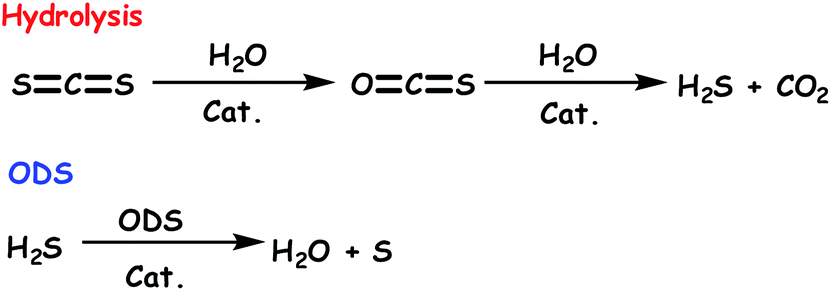 | ||
| Scheme 6 Hydrolysis of CS2 and COS into H2S, and ODS of H2S into sulfur. | ||
Though the Claus process is currently operative, it cannot accomplish 100% oxidation of H2S to sulfur. As a result, new catalysts have been proposed for 100% ODS of H2S to sulfur. Interestingly, catalysts performing partial oxidation of H2S are highly preferential because such catalysts could yield sulfur with 100% selectivity during ODS. Otherwise, the formed sulfur may be oxidized further to SOx gases which are the intermediates in the production of H2SO4. The literature evidences a range of catalysts for the ODS of H2S including oxides,254 mixed oxides,255 MOFs,256 carbon-based materials,257 ILs,258 and organic polymers with basic sites.253a However, nitrogen containing/doped carbon materials have recently gained much interest due to their electron richness and basic surface functionalities.259 It should be emphasized that the application of nitrogen containing/doped carbon materials in ODS of H2S is highly focused in materials chemistry as the synthetic methods and morphologies of such materials greatly influence the ODS of H2S. For example, G. Lei et al. devised a new synthetic strategy (Fig. 38a) to prepare g-C3N4 possessing nanosheet morphology (Fig. 38b–d) by which carbon species are introduced into the g-C3N4 framework and the carbon conjugation is induced. As a result, better H2S conversion with 100% sulfur selectivity is attained in comparison with bulk g-C3N4 and nitrogen doped graphitic carbon.259b Apart from the carbon-based materials, the morphology also plays a pivotal role in determining the catalytic performance of metal oxides during the ODS of H2S. X. Zheng et al. prepared CeO2 in different morphologies which create the variation in the oxygen vacancies of CeO2. Due to the more oxygen vacancies, the rod shaped CeO2 demonstrates superior activity in the ODS of H2S as compared to CeO2 with other morphologies.260 For more details about catalysts for the ODS of H2S, a review article published by X. Zhang et al. may be useful.257b
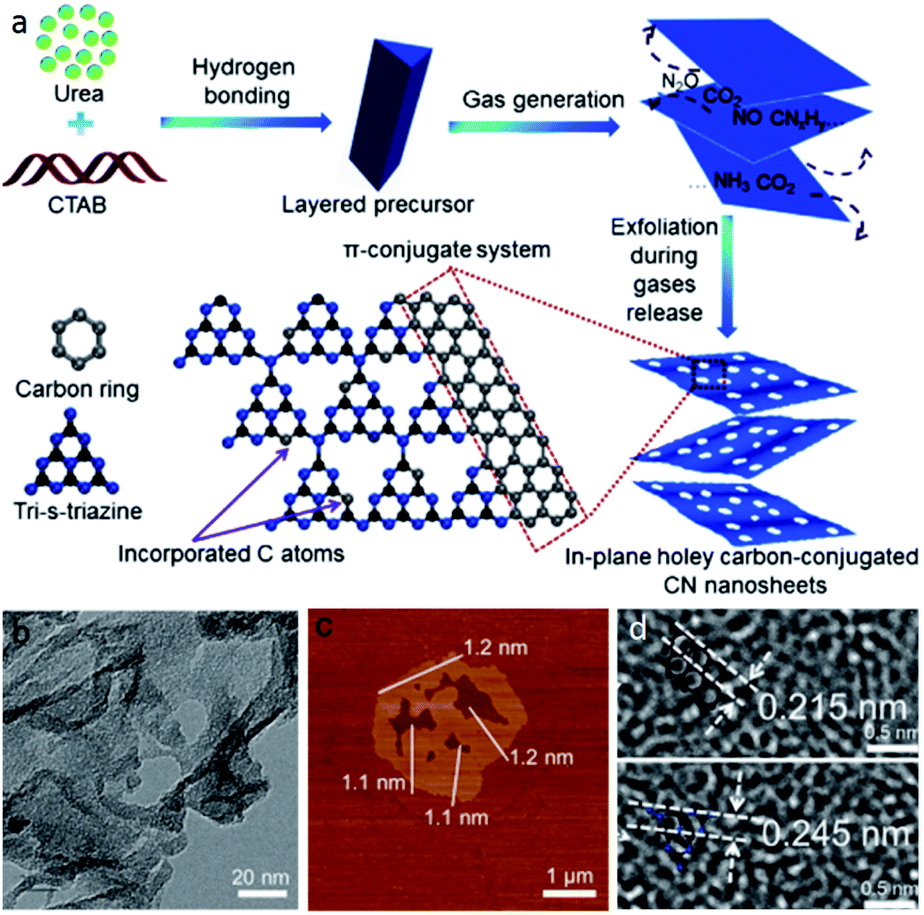 | ||
| Fig. 38 (a) Schematic illustration for the generation of in-plane holey carbon-conjugated g-C3N4 nanosheets. (b) TEM image, (c) AFM image and (d) HR-TEM image of an in-plane holey carbon-conjugated g-C3N4 nanosheet. Adapted from ref. 259b with permission from Elsevier. | ||
10. Summary and outlook
In this review, the ODS catalysts reported so far have been categorized and reviewed as metal oxides, TSs, POMs, metal complexes, MOFs, and metal-free catalysts along with the brief general description about ODS. The synthetic types, properties, reactivity and the factors influencing the reactivity of the ODS catalysts have been comprehensively discussed. Advantageously, most of these reviewed catalysts have been very suitable for working with environmentally friendly oxidants such as H2O2 and molecular O2. In general, heterogeneous ODS catalysts have been greatly focused on as compared to homogeneous catalysts due to their reusability. Though metal oxides such as MoO3 and WO3 were initially utilized, POMs have received much attention and have evolved as better catalysts because IL type POMs having an amphiphilic character can be easily prepared for accomplishing better mass transport. MOFs have recently emerged as ODS catalysts and also as supports for other ODS catalysts. But the costly organic ligand precursors required to prepare MOFs may suppress their repeated use. Significantly, for catalytic ODS applications, the porosity of MOFs is found to be equally important as the nature of metal sites since MOFs with small size pores have not been able to demonstrate better ODS activity in the case of bulky substrates like 4,6-DMDBT. Thus, catalysts with bimodal porosity have been greatly intriguing due to their ability to access both small and bulky sulfur compounds. Especially, bimodal porosity has been very familiar in TS-1 catalysts since the traditional TS-1 catalyst is microporous, due to which it is not at all suitable for ODS of bulky RS compounds. To be effective reusable heterogeneous ODS catalysts, metal complexes need to be anchored on supports like SiO2; however, their leaching from the support is found to be inevitable. Thus, metal complexes may not be suitable for industrial applications. Reusable solid metal-free ODS catalysts like graphene oxide, g-C3N4 and g-B3N4 have been recently investigated in the presence of molecular O2. The disadvantage is that these metal-free catalysts require high temperature (>100 °C) for the O2 activation. At these high temperatures, the low boiling fuels may boil off, resulting in a lower fuel yield. Harvesting sunlight using photocatalysts has been found to be useful to accomplish the catalytic photochemical ODS. Doped TiO2 is the most commonly reported photocatalyst for ODS applications. In future outlook, either metal oxides or amphiphilic POMs may gain a place in industrial catalytic ODS applications. Very recently, a small amount of Ti (≤0.018 wt%) present in commercial column chromatography SiO2 gel successfully catalyzed ODS but with cumene hydroperoxide. If this silica gel works efficiently in the presence of H2O2 or O2, it may be highly intriguing for future applications due to its low price, reusability and easy availability.218Of course, due to its ability to remove RS compounds under mild conditions, catalytic ODS has a lot of opportunities. With the choice of a suitable catalyst–oxidant combination, the catalytic ODS can accomplish selective removal of sulfur compounds in the midst of alkene compounds. Moreover, the catalytic photochemical ODS offers the use of sunlight to activate molecular O2 for the efficient removal of sulfur compounds. This looks very attractive because molecular O2 is activated by natural solar energy without the need for any thermal energy. Designing a suitable, cheap and efficient reactor may make the catalytic photochemical ODS suitable for industrial applications in the near future. ODS can be treated either as a complementary or replacement to HDS. Relatively, designing ODS as a complementary to HDS may lead to sulfur-free fuels (zero-sulfur). The temperature, pressure and H2 requirement of HDS can be adjusted to remove all the aliphatic sulfur compounds, thiophene and BT except DBTs which require very harsh reaction conditions. Profitably, softening of the harsh HDS conditions may circumvent the unsolicited side reactions that negatively affect significant fuel properties like the octane/cetane number. During HDS, nitrogen compounds like indole can also possibly be eliminated through a process called hydrodenitrogenation, and olefins and aromatic rings can be partially hydrogenated. These processes may be advantageous for performing the catalytic ODS with high selectivity with the consumption of a very small quantity of oxidant. On the other hand, while designing catalytic ODS as a replacement to HDS, the accomplishment of zero-sulfur fuel may not be realized. As a concluding remark, searching for an ODS catalyst that removes all the types of sulfur compounds effortlessly and selectively under mild conditions would be the immediate goal for researchers working in this area.
Though it ensures the effortless removal of RS compounds, catalytic ODS has a list of drawbacks as listed below.
(i) The poor mass transport of the sulfur compounds in the biphasic catalytic ODS suppresses the efficiency. Amphipathic catalysts are known to solve this problem; however, this would be fruitful only for light oils. For heavy oils, due to the high density, ultrasonication assisted catalytic ODS is recommended to accomplish better mass transport. However, the need for an ultrasound generator causes the reactor design to be more complicated.
(ii) The polar solvent used to extract the oxidized sulfur compounds can also extract a few other components like olefins. This decreases the carbon contents in the fuels and consequently, the chemical potential energy of the fuel is reduced.
(iii) The influence of other components like alkenes and indole will create more problems. Thus, a very selective ODS catalyst is highly required. Even then, due to the greater electron density of indole, it is very difficult to avoid its competitive influence on the oxidation of sulfur compounds during ODS.
(iv) The storage of H2O2 in large quantities is difficult and not safe. To avoid this, in situ generation of H2O2 may be helpful. Utilizing molecular O2 is also a good choice but requires relatively higher temperature. Sunlight can be used to activate O2 without the need for higher temperature under catalytic photochemical conditions. However, the presence of polyaromatic compounds in the oils affects the energy transition of sulfur compounds only to reduce the catalytic ODS efficiency. Therefore, a very efficient catalyst that activates molecular O2 at lower temperature is needed for industrial applications.
(v) The reactor design is still under consideration for finding an efficient and economically viable reactor.
(vi) Till now, the efficiency of ODS using different catalysts has mostly been investigated in model oils consisting of DBTs. But data on catalytic ODS with real oils are scarce and hence, in the coming years, research on the catalytic ODS of real oils is greatly required to yield more insights which will be very useful for the practical applications.
Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
The financial support from the National Natural Science Foundation of China (grant numbers: 21950410511; U1810128; U1610221) is greatly acknowledged. Dr Antony Rajendran thanks the Taiyuan University of Technology, PR China for providing a post-doctoral fellowship.References
- (a) S. Houda, C. Lancelot, P. Blanchard, L. Poinel and C. Lamonier, Catalysts, 2018, 8, 344 CrossRef; (b) A. Marafi, H. Albazzaz and M. S. Rana, Catal. Today, 2019, 329, 125 CrossRef CAS; (c) A. S. Ramadhas, Alternative Fuels for Transportation, CRC Press, Boca Raton, USA, 2011 Search PubMed.
- (a) V. Chandra Srivastava, RSC Adv., 2012, 2, 759 RSC; (b) B. N. Bhadra, N. A. Khan and S. H. Jhung, J. Mater. Chem. A, 2019, 7, 17823 RSC; (c) L. Hao, M. J. Hurlock, X. Li, G. Ding, K. W. Kriegsman, X. Guo and Q. Zhang, Catal. Today, 2019 DOI:10.1016/j.cattod.2019.04.012..
- The International Council on Clean Transportation, https://theicct.org/sites/default/files/publications/ICCTupdate_CH_fuelsulfur_mar2013_rev.pdf, (accessed March 2013).
- Z. Ismagilov, S. Yashnik, M. Kerzhentsev, V. Parmon, A. Bourane, F. M. Al-Shahrani, A. A. Hajji and O. R. Koseoglu, Catal. Rev.: Sci. Eng., 2011, 53, 199 CrossRef CAS.
- (a) S. K. Bej, S. K. Maity and U. T. Turaga, Energy Fuels, 2004, 18, 1227 CrossRef CAS; (b) O. Y. Gutiérrez, S. Singh, E. Schachtl, J. Kim, E. Kondratieva, J. Hein and J. A. Lercher, ACS Catal., 2014, 4, 1487 CrossRef; (c) J. Liu, W.-y. Li, J. Feng, X. Gao and Z.-y. Luo, Chem. Eng. Sci., 2019, 207, 1085 CrossRef CAS.
- A. Attar and W. H. Corcoran, Ind. Eng. Chem. Prod. Res. Dev., 1978, 17, 102 CrossRef CAS.
- R. Javadli and A. Klerk, Appl. Petrochem. Res., 2012, 1, 3 CrossRef CAS.
- F. S. Mjalli, O. U. Ahmed, T. Al-Wahaibi, Y. Al-Wahaibi and I. M. AlNashef, Rev. Chem. Eng., 2014, 30, 337 CAS.
- J. M. Campos-Martin, M. C. Capel-Sanchez, P. Perez-Presas and J. L. G. Fierro, J. Chem. Technol. Biotechnol., 2010, 85, 879 CrossRef CAS.
- J. F. Ford, T. A. Rayne and D. G. Adiington, US Pat., US3341448A, 1967.
- F. M. Collins, A. R. Lucy and C. Sharp, J. Mol. Catal. A: Chem., 1997, 117, 397 CrossRef CAS.
- E. Guth and A. Diaz, KVB Engineering, Inc., Justin, Calif., 1974, p. 1 Search PubMed.
- S. Otsuki, T. Nonaka, N. Takashima, Q. Weihua, A. Ishihara, T. Imai and T. Kabe, Energy Fuels, 2000, 14, 1232 CrossRef CAS.
- (a) J. Xiao, L. Wu, Y. Wu, B. Liu, L. Dai, Z. Li, Q. Xia and H. Xi, Appl. Energy, 2014, 113, 78 CrossRef CAS; (b) M. Ja'fari, S. L. Ebrahimi and M. R. Khosravi-Nikou, Ultrason. Sonochem., 2018, 40, 955 CrossRef PubMed.
- M. A. Betiha, A. M. Rabie, H. S. Ahmed, A. A. Abdelrahman and M. F. El-Shahat, Egypt. J. Pet., 2018, 27, 715 CrossRef.
- (a) L. Hao, L. Sun, T. Su, D. Hao, W. Liao, C. Deng, W. Ren, Y. Zhang and H. Lü, Chem. Eng. J., 2019, 358, 419 CrossRef CAS; (b) H. Li, W. Zhu, S. Zhu, J. Xia, Y. Chang, W. Jiang, M. Zhang, Y. Zhou and H. Li, AlChE J., 2016, 62, 2087 CrossRef CAS; (c) G.-N. Yun and Y.-K. Lee, Fuel Process. Technol., 2013, 114, 1 CrossRef CAS; (d) K.-E. Jeong, T.-W. Kim, J.-W. Kim, H.-J. Chae, C.-U. Kim, Y.-K. Park and S.-Y. Jeong, Korean J. Chem. Eng., 2013, 30, 509 CrossRef CAS.
- D. Wang, E. W. Qian, H. Amano, K. Okata, A. Ishihara and T. Kabe, Appl. Catal., A, 2003, 253, 91 CrossRef CAS.
- (a) L. P. Rivoira, M. L. Martínez, H. Falcón, A. R. Beltramone, J. M. Campos-Martin, J. L. Fierro and P. Tartaj, ACS Omega, 2017, 2, 2351 CrossRef CAS PubMed; (b) M. Zuo, X. Huang, J. Li, Q. Chang, Y. Duan, L. Yan, Z. Xiao, S. Mei, S. Lu and Y. Yao, Catal. Sci. Technol., 2019, 9, 2923 RSC.
- C. Piscopo, J. Tochtermann, M. Schwarzer, D. Boskovic, R. Maggi, G. Maestri and S. Loebbecke, React. Chem. Eng., 2018, 3, 13 RSC.
- (a) N. Ghorbani and G. Moradi, Chin. J. Chem. Eng., 2019, 27, 2759 CrossRef; (b) Z. Xinrui, G. Hongtao, W. Jing, S. ZHANG, Y. Jinzong and S. ZHANG, Chin. J. Chem. Eng., 2009, 17, 189 CrossRef; (c) Y. Tian, Y. Yao, Y. Zhi, L. Yan and S. Lu, Energy Fuels, 2015, 29, 618 CrossRef CAS; (d) L. Qiu, Y. Cheng, C. Yang, G. Zeng, Z. Long, S. Wei, K. Zhao and L. Luo, RSC Adv., 2016, 6, 17036 RSC; (e) L. Kang, H. Liu, H. He and C. Yang, Fuel, 2018, 234, 1229 CrossRef CAS; (f) L. Seifikar Gomi and M. Afsharpour, Appl. Organomet. Chem., 2019, 33, e4830 CrossRef; (g) Q. Yang, J. Wang, W.-H. Wang and M. Bao, RSC Adv., 2019, 9, 21473 RSC; (h) B. Mokhtari, A. Akbari and M. Omidkhah, Energy Fuels, 2019, 33, 7276 CrossRef CAS; (i) G. Yang, X. Zhang, H. Yang, Y. Long and J. Ma, J. Colloid Interface Sci., 2018, 532, 92 CrossRef CAS PubMed; (j) Z. Dini, M. Afsharpour and K. Tabar-Heydar, Diamond Relat. Mater., 2019, 91, 237 CrossRef CAS; (k) J. Chang, A. Wang, J. Liu, X. Li and Y. Hu, Catal. Today, 2010, 149, 122 CrossRef CAS; (l) L. Kamel and M. Anbia, Silicon, 2019, 1 Search PubMed.
- (a) C. Wang, A. Li, J. Xu, J. Wen, H. Zhang and L. Zhang, J. Chem. Technol. Biotechnol., 2019, 94, 3403 CrossRef CAS; (b) R. Ma, J. Guo, D. Wang, M. He, S. Xun, J. Gu, W. Zhu and H. Li, Colloids Surf., A, 2019, 572, 250 CrossRef CAS; (c) R. Goyal, D. Dumbre, L. S. Konathala, M. Pandey and A. Bordoloi, Catal. Sci. Technol., 2015, 5, 3632 RSC; (d) E. Torres-García, A. Galano and G. Rodriguez-Gattorno, J. Catal., 2011, 282, 201 CrossRef; (e) Z. Feng, Y. Zhu, Q. Zhou, Y. Wu and T. Wu, Mater. Sci. Eng., B, 2019, 240, 85 CrossRef CAS; (f) E. Torres-Garcia, G. Canizal, S. Velumani, L. Ramirez-Verduzco, F. Murrieta-Guevara and J. Ascencio, Appl. Phys. A, 2004, 79, 2037 CrossRef CAS; (g) Z. Li, C. Li, S. B. Park, G. H. Hong, J. S. Park, B. J. Song, C. W. Lee and J. M. Kim, Res. Chem. Intermed., 2018, 44, 3687 CrossRef CAS; (h) S. Xun, W. Zhu, F. Zhu, Y. Chang, D. Zheng, Y. Qin, M. Zhang, W. Jiang and H. Li, Chem. Eng. J., 2015, 280, 256 CrossRef CAS; (i) L. Ramirez-Verduzco, E. Torres-Garcia, R. Gomez-Quintana, V. Gonzalez-Pena and F. Murrieta-Guevara, Catal. Today, 2004, 98, 289 CrossRef CAS; (j) J. F. Palomeque-Santiago, R. López-Medina, R. Oviedo-Roa, J. Navarrete-Bolaños, R. Mora-Vallejo, J. A. Montoya-de la Fuente and J. M. Martínez-Magadán, Appl. Catal., B, 2018, 236, 326 CrossRef CAS.
- (a) L. Cedeño-Caero, M. Ramos-Luna, M. Méndez-Cruz and J. Ramírez-Solís, Catal. Today, 2011, 172, 189 CrossRef; (b) D. Zheng, W. Zhu, S. Xun, M. Zhou, M. Zhang, W. Jiang, Y. Qin and H. Li, Fuel, 2015, 159, 446 CrossRef CAS; (c) L. P. Rivoira, B. C. Ledesma, J. M. Juárez and A. R. Beltramone, Fuel, 2018, 226, 498 CrossRef CAS; (d) C. Shen, Y. Wang, J. Xu and G. Luo, Green Chem., 2016, 18, 771 RSC.
- (a) H. Gómez-Bernal, L. Cedeño-Caero and A. Gutiérrez-Alejandre, Catal. Today, 2009, 142, 227 CrossRef; (b) M. I. de Mello, E. V. Sobrinho, V. Teixeira da Silva and S. B. Pergher, Ind. Eng. Chem. Res., 2018, 57, 15663 CrossRef CAS; (c) L. Rivoira, J. Juárez, H. Falcón, M. G. Costa, O. Anunziata and A. Beltramone, Catal. Today, 2017, 282, 123 CrossRef CAS; (d) L. Fabián-Mijangos and L. Cedeno-Caero, Ind. Eng. Chem. Res., 2010, 50, 2659 CrossRef.
- (a) A. T. Nawaf, A. T. Jarullah, S. A. Gheni and I. M. Mujtaba, Ind. Eng. Chem. Res., 2015, 54, 12503 CrossRef CAS; (b) S. Jatav and V. C. Srivastava, Pet. Sci. Technol., 2019, 37, 633 CrossRef; (c) A. T. Nawaf, S. A. Gheni, A. T. Jarullah and I. M. Mujtaba, Fuel Process. Technol., 2015, 138, 337 CrossRef CAS; (d) L. P. Rivoira, V. A. Valles, M. L. Martínez, Y. Sa-ngasaeng, S. Jongpatiwut and A. R. Beltramone, Catal. Today, 2019 DOI:10.1016/j.cattod.2019.08.005.
- A. Liu, M. Zhu and B. Dai, Appl. Catal., A, 2019, 583, 117134 CrossRef CAS.
- W. A. W. A. Bakar, R. Ali, A. A. A. Kadir and W. N. A. W. Mokhtar, Fuel Process. Technol., 2012, 101, 78 CrossRef CAS.
- L. Cedeño-Caero, H. Gomez-Bernal, A. Fraustro-Cuevas, H. D. Guerra-Gomez and R. Cuevas-Garcia, Catal. Today, 2008, 133, 244 CrossRef.
- L. Rivoira, J. Juárez, M. L. Martínez and A. Beltramone, Catal. Today, 2018 DOI:10.1016/j.cattod.2018.05.030.
- (a) X. Han, A. Wang, X. Wang, X. Li, Y. Wang and Y. Hu, Catal. Commun., 2013, 42, 6 CrossRef CAS; (b) L. Rivoira, M. L. Martínez, O. Anunziata and A. Beltramone, Microporous Mesoporous Mater., 2017, 254, 96 CrossRef CAS.
- K. Chen, X.-M. Zhang, X.-F. Yang, M.-G. Jiao, Z. Zhou, M.-H. Zhang, D.-H. Wang and X.-H. Bu, Appl. Catal., B, 2018, 238, 263 CrossRef CAS.
- W. Jiang, J. Xiao, L. Dong, C. Wang, H. Li, Y. Luo, W. Zhu and H. Li, ACS Sustainable Chem. Eng., 2019, 7, 15755 CrossRef CAS.
- C. Wang, Z. Chen, X. Yao, W. Jiang, M. Zhang, H. Li, H. Liu, W. Zhu and H. Li, RSC Adv., 2017, 7, 39383 RSC.
- J. Dou and H. C. Zeng, ACS Catal., 2014, 4, 566 CrossRef CAS.
- D. Shen, Y. Dai, J. Han, L. Gan, J. Liu and M. Long, Chem. Eng. J., 2018, 332, 563 CrossRef CAS.
- (a) G. Estephane, C. Lancelot, P. Blanchard, V. Dufaud, S. Chambrey, N. Nuns, J. Toufaily, T. Hamiye and C. Lamonier, Appl. Catal., A, 2019, 571, 42 CrossRef CAS; (b) Y. Li, M. Zhang, W. Zhu, M. Li, J. Xiong, Q. Zhang, Y. Wei and H. Li, RSC Adv., 2016, 6, 68922 RSC; (c) M. Zhang, W. Zhu, H. Li, S. Xun, W. Ding, J. Liu, Z. Zhao and Q. Wang, Chem. Eng. J., 2014, 243, 386 CrossRef CAS; (d) M. Zhang, W. Zhu, S. Xun, J. Xiong, W. Ding, M. Li, Q. Wang and H. Li, Chem. Eng. Technol., 2015, 38, 117 CrossRef CAS.
- (a) Y. Qin, S. Xun, L. Zhan, Q. Lu, M. He, W. Jiang, H. Li, M. Zhang, W. Zhu and H. Li, New J. Chem., 2017, 41, 569 RSC; (b) S. Xun, C. Hou, H. Li, M. He, R. Ma, M. Zhang, W. Zhu and H. Li, J. Mater. Sci., 2018, 53, 15927 CrossRef CAS; (c) Q. Gu, W. Zhu, S. Xun, Y. Chang, J. Xiong, M. Zhang, W. Jiang, F. Zhu and H. Li, Fuel, 2014, 117, 667 CrossRef CAS; (d) M. Zhang, W. Zhu, H. Li, S. Xun, M. Li, Y. Li, Y. Wei and H. Li, Chin. J. Catal., 2016, 37, 971 CrossRef CAS; (e) L. Yang, J. Xiong, H. Li, M. Zhang, W. Jiang, H. Liu, W. Zhu and H. Li, RSC Adv., 2015, 5, 104322 RSC.
- M. Zhang, J. Liu, J. Yang, X. Chen, M. Wang, H. Li, W. Zhu and H. Li, Inorg. Chem. Front., 2019, 6, 451 RSC.
- B. Saha, S. Kumar and S. Sengupta, Chem. Eng. Sci., 2019, 199, 332 CrossRef CAS.
- P. Wu, W. Zhu, A. Wei, B. Dai, Y. Chao, C. Li, H. Li and S. Dai, Chem.–Eur. J., 2015, 21, 15421 CrossRef CAS PubMed.
- X. Yao, C. Wang, H. Liu, H. Li, P. Wu, L. Fan, H. Li and W. Zhu, Ind. Eng. Chem. Res., 2018, 58, 863 CrossRef.
- J. Wang, W. Wu, H. Ye, Y. Zhao, W.-H. Wang and M. Bao, RSC Adv., 2017, 7, 44827 RSC.
- M. Salmasi, S. Fatemi and Y. Mortazavi, J. Ind. Eng. Chem., 2016, 39, 66 CrossRef CAS.
- J. He, H. Liu, B. Xu and X. Wang, Small, 2015, 11, 1144 CrossRef CAS.
- Z. Yang, L. X. Song, Y. Q. Wang, M. M. Ruan, Y. Teng, J. Xia, J. Yang, S. S. Chen and F. Wang, J. Mater. Chem. A, 2018, 6, 2914 RSC.
- M. A. Astle, G. A. Rance, H. J. Loughlin, T. D. Peters and A. N. Khlobystov, Adv. Funct. Mater., 2019, 29, 1808092 CrossRef.
- X. Li, L. Liu, A. Wang, M. Wang, Y. Wang and Y. Chen, Catal. Lett., 2014, 144, 531 CrossRef CAS.
- J. He, P. Wu, Y. Wu, H. Li, W. Jiang, S. Xun, M. Zhang, W. Zhu and H. Li, ACS Sustainable Chem. Eng., 2017, 5, 8930 CrossRef CAS.
- H. Sun, P. Wu, J. He, M. Liu, L. Zhu, F. Zhu, G. Chen, M. He and W. Zhu, Pet. Sci. Technol., 2018, 15, 849 CrossRef CAS.
- J. González, J. Wang, L. Chen, R. Limas, R. Manzo, J. V. Rodríguez and O. G. Vargas, Mater. Lett., 2018, 220, 70 CrossRef.
- Y. Shi, G. Liu, B. Zhang and X. Zhang, Green Chem., 2016, 18, 5273 RSC.
- Q. Zhang, J. Zhang, H. Yang, Y. Dong, Y. Liu, L. Yang, D. Wei, W. Wang, L. Bai and H. Chen, Catal. Sci. Technol., 2019, 9, 2915 RSC.
- A. Teimouri, M. Mahmoudsalehi and H. Salavati, Int. J. Hydrogen Energy, 2018, 43, 14816 CrossRef CAS.
- W. N. A. W. Mokhtar, W. A. W. A. Bakar, R. Ali and A. A. A. Kadir, Arabian J. Chem., 2018, 11, 1201 CrossRef CAS.
- M. A. Alvarez-Amparán and L. Cedeño-Caero, Catal. Today, 2017, 282, 133 CrossRef.
- (a) J. Zhang, B. Xiumei, L. Xiang, W. Anjie and M. Xuehu, Chin. J. Catal., 2009, 30, 1017 CrossRef CAS; (b) Y. Tu, T. Li, G. Yu, L. Wei, L. Ta, Z. Zhou and Z. Ren, Energy Fuels, 2019, 33, 8503 CrossRef.
- M. L. Luna, M. A. Alvarez-Amparán and L. Cedeño-Caero, J. Taiwan Inst. Chem. Eng., 2019, 95, 175 CrossRef CAS.
- U. Arellano, J. Wang, L. Chen, G. Cao, M. Asomoza and S. Cipagauta, J. Mol. Catal. A: Chem., 2016, 421, 66 CrossRef CAS.
- (a) Z. Hasan, J. Jeon and S. H. Jhung, J. Hazard. Mater., 2012, 205, 216 CrossRef PubMed; (b) J. Luo, W. Liu, J. Xiong, L. Yang, H. Li, S. Yin, W. Zhu and H. Li, J. Chin. Adv. Mater. Soc., 2018, 6, 44 CrossRef CAS.
- R. Sundararaman and C. Song, Ind. Eng. Chem. Res., 2014, 53, 1890 CrossRef CAS.
- I. R. Guimarães, A. S. Giroto, W. F. de Souza and M. C. Guerreiro, Appl. Catal., A, 2013, 450, 106 CrossRef.
- S.-x. Lu, H. Zhong, D.-m. Mo, Z. Hu, H.-l. Zhou and Y. Yao, Green Chem., 2017, 19, 1371 RSC.
- (a) X. Li, S. Ma, H. Qian, Y. Zhang, S. Zuo and C. Yao, Powder Technol., 2019, 351, 38 CrossRef CAS; (b) L. Yun, Z. Yang, Z.-B. Yu, T. Cai, Y. Li, C. Guo, C. Qi and T. Ren, RSC Adv., 2017, 7, 25455 RSC; (c) C. Hitam, A. Jalil, S. Triwahyono, A. Ahmad, N. Jaafar, N. Salamun, N. Fatah, L. Teh, N. Khusnun and Z. Ghazali, RSC Adv., 2016, 6, 76259 RSC; (d) T. Geng, J. He, L. Hu and J. Li, Inorg. Chem. Commun., 2019, 101, 103 CrossRef CAS; (e) L. Li, J. Zhang, C. Shen, Y. Wang and G. Luo, Fuel, 2016, 167, 9 CrossRef CAS; (f) F. Lin, Z. Jiang, N. Tang, C. Zhang, T. Liu and B. Dong, Appl. Catal., B, 2016, 188, 253 CrossRef CAS; (g) F.-t. Li, Y. Liu, Z.-m. Sun, Y. Zhao, R.-h. Liu, L.-j. Chen and D.-s. Zhao, Catal. Sci. Technol., 2012, 2, 1455 RSC; (h) F. Lin, Z. Shao, P. Li, Z. Chen, X. Liu, M. Li, B. Zhang, J. Huang, G. Zhu and B. Dong, RSC Adv., 2017, 7, 15053 RSC.
- (a) M. Taramasso, G. Perego and B. Notari, US Pat., US4410501A, 1983; (b) J. Přech, Catal. Rev.: Sci. Eng., 2018, 60, 71 CrossRef.
- R. Murugavel and H. W. Roesky, Angew. Chem., Int. Ed., 1997, 36, 477 CrossRef CAS.
- (a) Q. Du, Y. Guo, P. Wu and H. Liu, Microporous Mesoporous Mater., 2018, 264, 272 CrossRef CAS; (b) C. Shi, W. Wang, N. Liu, X. Xu, D. Wang, M. Zhang, P. Sun and T. Chen, Chem. Commun., 2015, 51, 11500 RSC; (c) Y. Zhu, Z. Hua, X. Zhou, Y. Song, Y. Gong, J. Zhou, J. Zhao and J. Shi, RSC Adv., 2013, 3, 4193 RSC; (d) S. Du, F. Li, Q. Sun, N. Wang, M. Jia and J. Yu, Chem. Commun., 2016, 52, 3368 RSC; (e) G. Lv, S. Deng, Y. Zhai, Y. Zhu, H. Li, F. Wang and X. Zhang, Appl. Catal., A, 2018, 567, 28 CrossRef CAS; (f) C. Jin, G. Li, X. Wang, L. Zhao, L. Liu, H. Liu, Y. Liu, W. Zhang, X. Han and X. Bao, Chem. Mater., 2007, 19, 1664 CrossRef CAS; (g) S.-T. Yang, K.-E. Jeong, S.-Y. Jeong and W.-S. Ahn, Mater. Res. Bull., 2012, 47, 4398 CrossRef CAS; (h) S. Du, Q. Sun, N. Wang, X. Chen, M. Jia and J. Yu, J. Mater. Chem. A, 2017, 5, 7992 RSC.
- C. Jin, G. Li, X. Wang, L. Zhao, Y. Wang and D. Sun, Top. Catal., 2008, 49, 118 CrossRef CAS.
- Y. Fang and H. Hu, Catal. Commun., 2007, 8, 817 CrossRef CAS.
- (a) W. Wang, G. Li, W. Li and L. Liu, Chem. Commun., 2011, 47, 3529 RSC; (b) X. Wang, G. Li, W. Wang, C. Jin and Y. Chen, Microporous Mesoporous Mater., 2011, 142, 494 CrossRef CAS; (c) W. Wang, G. Li, L. Liu and Y. Chen, Microporous Mesoporous Mater., 2013, 179, 165 CrossRef CAS.
- (a) Q. Du, Y. Guo, H. Duan, H. Li, Y. Chen and H. Liu, Fuel, 2017, 188, 232 CrossRef CAS; (b) C. Jin, G. Li, X. Wang, Y. Wang, L. Zhao and D. Sun, Microporous Mesoporous Mater., 2008, 111, 236 CrossRef CAS.
- R. Bai, Q. Sun, Y. Song, N. Wang, T. Zhang, F. Wang, Y. Zou, Z. Feng, S. Miao and J. Yu, J. Mater. Chem. A, 2018, 6, 8757 RSC.
- S. Du, X. Chen, Q. Sun, N. Wang, M. Jia, V. Valtchev and J. Yu, Chem. Commun., 2016, 52, 3580 RSC.
- J. Zhang, D. Zhao, Z. Ma and Y. Wang, Catal. Lett., 2010, 138, 111 CrossRef CAS.
- (a) G. Gao, S. Cheng, Y. An, X. Si, X. Fu, Y. Liu, H. Zhang, P. Wu and M. Y. He, ChemCatChem, 2010, 2, 459 CrossRef CAS; (b) L. Wang, Y. Wang, Y. Liu, L. Chen, S. Cheng, G. Gao, M. He and P. Wu, Microporous Mesoporous Mater., 2008, 113, 435 CrossRef CAS; (c) X. Si, S. Cheng, Y. Lu, G. Gao and M.-Y. He, Catal. Lett., 2008, 122, 321 CrossRef CAS.
- (a) A. T. Shah, B. Li and Z. E. A. Abdalla, J. Colloid Interface Sci., 2009, 336, 707 CrossRef CAS PubMed; (b) L. P. Rivoira, V. A. Valles, B. C. Ledesma, M. V. Ponte, M. L. Martinez, O. A. Anunziata and A. R. Beltramone, Catal. Today, 2016, 271, 102 CrossRef CAS.
- R. D. Andrei, N. Cambruzzi, M. Bonne, B. Lebeau and V. Hulea, J. Porous Mater., 2019, 26, 533 CrossRef CAS.
- P. Wu, T. Tatsumi, T. Komatsu and T. Yashima, Chem. Lett., 2000, 29, 774 CrossRef.
- D. Shi, L. Xu, P. Chen, T. Ma, C. Lin, X. Wang, D. Xu and J. Sun, Chem. Commun., 2019, 55, 1390 RSC.
- C. Shi, B. Zhu, M. Lin and J. Long, Catal. Today, 2010, 149, 132 CrossRef CAS.
- L. Kong, G. Li, X. Wang and B. Wu, Energy Fuels, 2006, 20, 896 CrossRef CAS.
- H.-Y. Song, G. Li, X.-S. Wang and Y.-J. Xu, Catal. Today, 2010, 149, 127 CrossRef CAS.
- N. Jose, S. Sengupta and J. Basu, Fuel, 2011, 90, 626 CrossRef CAS.
- G. Lv, S. Deng, Z. Yi, X. Zhang, F. Wang, H. Li and Y. Zhu, Chem. Commun., 2019, 55, 4885 RSC.
- C. Shen, Y. Wang, J. Xu and G. Luo, Chem. Eng. J., 2015, 259, 552 CrossRef CAS.
- A. Chica, A. Corma and M. E. Dómine, J. Catal., 2006, 242, 299 CrossRef CAS.
- Y. Shiraishi, T. Naito and T. Hirai, Ind. Eng. Chem. Res., 2003, 42, 6034 CrossRef CAS.
- A. Enferadi-Kerenkan, T.-O. Do and S. Kaliaguine, Catal. Sci. Technol., 2018, 8, 2257 RSC.
- N. I. Gumerova and A. Rompel, Nat. Rev. Chem., 2018, 2, 0112 CrossRef CAS.
- J. J. Ye and C. D. Wu, Dalton Trans., 2016, 45, 10101 RSC.
- (a) F. Banisharif, M. R. Dehghani, M. C. Capel-Sanchez and J. M. Campos-Martin, J. Ind. Eng. Chem., 2017, 47, 348 CrossRef CAS; (b) Y. Zhang, L. Wang, Y. Zhang, Z. Jiang and C. Li, Chin. J. Catal., 2011, 32, 235 CrossRef CAS; (c) F. Banisharif, M. R. Dehghani, M. Capel-Sánchez and J. M. Campos-Martin, Ind. Eng. Chem. Res., 2017, 56, 3839 CrossRef CAS; (d) F. Banisharif, M. R. Dehghani and J. M. Campos-Martin, Energy Fuels, 2017, 31, 5419 CrossRef CAS.
- (a) L. Sun, T. Su, P. Li, J. Xu, N. Chen, W. Liao, C. Deng, W. Ren and H. Lü, Catal. Lett., 2019, 149, 1888 CrossRef CAS; (b) H. Lü, Y. Zhang, Z. Jiang and C. Li, Green Chem., 2010, 12, 1954 RSC; (c) H. Lü, W. Ren, H. Wang, Y. Wang, W. Chen and Z. Suo, Appl. Catal., A, 2013, 453, 376 CrossRef; (d) H. Lü, W. Ren, W. Liao, W. Chen, Y. Li and Z. Suo, Appl. Catal., B, 2013, 138–139, 79 CrossRef; (e) H. Lü, P. Li, Y. Liu, L. Hao, W. Ren, W. Zhu, C. Deng and F. Yang, Chem. Eng. J., 2017, 313, 1004 CrossRef.
- (a) H. Wang, R. Wang, Y. Zhang, S. Dou, S. Olga and K. Vladimir, Catal. Lett., 2018, 148, 2501 CrossRef CAS; (b) Z. Yu, D. Wang, S. Xun, M. He, R. Ma, W. Jiang, H. Li, W. Zhu and H. Li, Pet. Sci., 2018, 15, 870 CrossRef CAS; (c) A. H. Jin, B. S. Li and Z. J. Dai, Pet. Sci. Technol., 2010, 28, 700 CrossRef CAS; (d) S. Ribeiro, A. D. S. Barbosa, A. C. Gomes, M. Pillinger, I. S. Gonçalves, L. Cunha-Silva and S. S. Balula, Fuel Process. Technol., 2013, 116, 350 CrossRef CAS; (e) Y. Zhang, H. Lü, L. Wang, Y. Zhang, P. Liu, H. Han, Z. Jiang and C. Li, J. Mol. Catal. A: Chem., 2010, 332, 59 CrossRef CAS; (f) S. O. Ribeiro, B. Duarte, B. de Castro, C. M. Granadeiro and S. S. Balula, Materials, 2018, 11, 1196 CrossRef PubMed; (g) S. O. Ribeiro, C. M. Granadeiro, P. L. Almeida, J. Pires, M. C. Capel-Sanchez, J. M. Campos-Martin, S. Gago, B. de Castro and S. S. Balula, Catal. Today, 2019, 333, 226 CrossRef CAS; (h) J. Ding, Y. Zhang and R. Wang, New J. Chem., 2019, 43, 7363 RSC; (i) N. Wu, B. Li, Z. Liu and C. Han, Catal. Commun., 2014, 46, 156 CrossRef CAS; (j) E. Rafiee and N. Nobakht, J. Mol. Catal. A: Chem., 2015, 398, 17 CrossRef CAS.
- J.-K. Li, J. Dong, C.-P. Wei, S. Yang, Y.-N. Chi, Y.-Q. Xu and C.-W. Hu, Inorg. Chem., 2017, 56, 5748 CrossRef CAS PubMed.
- C. Wang, Z. Chen, X. Yao, Y. Chao, S. Xun, J. Xiong, L. Fan, W. Zhu and H. Li, Fuel, 2018, 230, 104 CrossRef CAS.
- (a) H. Li, X. Jiang, W. Zhu, J. Lu, H. Shu and Y. Yan, Ind. Eng. Chem. Res., 2009, 48, 9034 CrossRef CAS; (b) X. Jiang, H. Li, W. Zhu, L. He, H. Shu and J. Lu, Fuel, 2009, 88, 431 CrossRef CAS.
- M. Hutin, M. H. Rosnes, D. L. Long and L. Cronin, in Comprehensive Inorganic Chemistry II, 2013, p. 241 Search PubMed.
- D. L. Long, E. Burkholder and L. Cronin, Chem. Soc. Rev., 2007, 36, 105 RSC.
- C. Ritchie, E. Burkholder, P. Kogerler and L. Cronin, Dalton Trans., 2006, 1712 RSC.
- C. Fleming, D. L. Long, N. McMillan, J. Johnston, N. Bovet, V. Dhanak, N. Gadegaard, P. Kogerler, L. Cronin and M. Kadodwala, Nat. Nanotechnol., 2008, 3, 289 CrossRef PubMed.
- C.-Y. Sun, S.-X. Liu, D.-D. Liang, K.-Z. Shao, Y.-H. Ren and Z.-M. Su, J. Am. Chem. Soc., 2003, 131, 1883 CrossRef PubMed.
- M. Ammam and J. Fransaer, J. Solid State Chem., 2011, 184, 818 CrossRef CAS.
- (a) G. Liu, Y. Cao, R. Jiang, L. Wang, X. Zhang and Z. Mi, Energy Fuels, 2009, 23, 5978 CrossRef CAS; (b) K. Yazu, T. Furuya, K. Miki and K. Ukegawa, Chem. Lett., 2003, 32, 920 CrossRef CAS; (c) Y. Kazumasa, Y. Yorihiro, F. Takeshi, M. Keiji and U. Koji, Energy Fuels, 2001, 15, 1535 CrossRef; (d) L.-L. Chuang, J.-F. Huang, W.-H. Lo and G.-T. Wei, J. Chin. Chem. Soc., 2012, 59, 324 CrossRef CAS.
- (a) M. L. Samaniego, M. D. G. de Luna, D. C. Ong, M.-W. Wan and M.-C. Lu, Energy Fuels, 2019, 33, 1098 CrossRef CAS; (b) M. D. G. de Luna, M.-W. Wan, L. R. Golosinda, C. M. Futalan and M.-C. Lu, Energy Fuels, 2017, 31, 9923 CrossRef CAS.
- D. K. Bal and J. B. Bhasarkar, Asia-Pac. J. Chem. Eng., 2018, 14, e2271 CrossRef.
- J. Li, L. N. Yang and J. Shen, Pet. Sci. Technol., 2011, 29, 247 CrossRef CAS.
- T. Mure, C. Fairbridge and Z. Ring, Appl. Catal., A, 2001, 219, 267 CrossRef.
- (a) B. Zhang, W. Jiang, X. Fan, M. Zhang, Y. Wei, J. He, C. Li, H. Li and W. Zhu, Pet. Sci., 2018, 15, 890 CrossRef CAS; (b) M. Tao, H. Zheng, J. Shi, S. Wang, X. Wang and G. Huang, Catal. Surv. Asia, 2015, 19, 257 CrossRef CAS; (c) J. Ge, Y. Zhou, Y. Yang and M. Xue, Ind. Eng. Chem. Res., 2011, 50, 13686 CrossRef CAS; (d) F. Zou, X. Y. Wu, W. S. Zhu, H. M. Li, D. Xu and H. Xu, Pet. Sci. Technol., 2011, 29, 1113 CrossRef CAS; (e) T. Wang, Y. Lu, H. Wu and E. Wang, Inorg. Chim. Acta, 2016, 446, 13 CrossRef CAS; (f) F. Yu and R. Wang, Chem. Lett., 2014, 43, 834 CrossRef CAS; (g) D. Hao, W. You, P. Liu, C. Li, Q. Wu and H. Lü, Phosphorus, Sulfur Silicon Relat. Elem., 2016, 191, 30 CrossRef CAS; (h) D. Huang, Z. Zhai, Y. C. Lu, L. M. Yang and G. S. Luo, Ind. Eng. Chem. Res., 2007, 46, 1447 CrossRef CAS; (i) X. Zhang, Y. Shi and G. Liu, Catal. Sci. Technol., 2016, 6, 1016 RSC; (j) D. Julião, A. C. Gomes, L. Cunha-Silva, R. Valença, J. C. Ribeiro, M. Pillinger, B. de Castro, I. S. Gonçalves and S. S. Balula, Appl. Catal., A, 2019, 589, 117154 CrossRef.
- (a) W. Jiang, L. Dong, W. Liu, T. Guo, H. Li, M. Zhang, W. Zhu and H. Li, RSC Adv., 2017, 7, 55318 RSC; (b) A. Li, H. Song, H. Meng, Y. Lu and C. Li, Chem. Eng. J., 2020, 380 Search PubMed; (c) L. Li, Y. Lu, H. Meng and C. Li, Fuel, 2019, 253, 802 CrossRef CAS; (d) F. Mirante, N. Gomes, M. C. Corvo, S. Gago and S. S. Balula, Polyhedron, 2019, 170, 762 CrossRef CAS; (e) W. Huang, W. Zhu, H. Li, H. Shi, G. Zhu, H. Liu and G. Chen, Ind. Eng. Chem. Res., 2010, 49, 8998 CrossRef CAS; (f) W. Zhu, G. Zhu, H. Li, Y. Chao, M. Zhang, D. Du, Q. Wang and Z. Zhao, Fuel Process. Technol., 2013, 106, 70 CrossRef CAS; (g) W. Zhu, Y. Ding, H. Li, J. Qin, Y. Chao, J. Xiong, Y. Xu and H. Liu, RSC Adv., 2013, 3, 3893 RSC.
- D. Huang, Z. Zhai, Y. C. Lu, L. M. Yang and G. S. Luo, Ind. Eng. Chem. Res., 2007, 46, 1447 CrossRef CAS.
- (a) J. Chen, C. Chen, R. Zhang, L. Guo, L. Hua, A. Chen, Y. Xiu, X. Liu and Z. Hou, RSC Adv., 2015, 5, 25904 RSC; (b) J. Qiu, G. Wang, D. Zeng, Y. Tang, M. Wang and Y. Li, Fuel Process. Technol., 2009, 90, 1538 CrossRef CAS; (c) S. O. Ribeiro, D. Julião, L. Cunha-Silva, V. F. Domingues, R. Valença, J. C. Ribeiro, B. de Castro and S. S. Balula, Fuel, 2016, 166, 268 CrossRef CAS; (d) J. Xu, S. Zhao, Y. Ji and Y. F. Song, Chem.–Eur. J., 2013, 19, 709 CrossRef CAS PubMed.
- (a) X. Zeng, X. Xiao, J. Chen and H. Wang, Appl. Catal., B, 2019, 248, 573 CrossRef CAS; (b) C. Jiang, J. Wang, S. Wang, H. y. Guan, X. Wang and M. Huo, Appl. Catal., B, 2011, 106, 343 CrossRef CAS.
- (a) Y. Ding, W. Zhu, H. Li, W. Jiang, M. Zhang, Y. Duan and Y. Chang, Green Chem., 2011, 13, 1210 RSC; (b) J. Gao, S. Wang, Z. Jiang, H. Lu, Y. Yang, F. Jing and C. Li, J. Mol. Catal. A: Chem., 2006, 258, 261 CrossRef CAS; (c) Y. Xu, W.-W. Ma, A. Dolo and H. Zhang, RSC Adv., 2016, 6, 66841 RSC; (d) H. Lu, J. Gao, Z. Jiang, F. Jing, Y. Yang, G. Wang and C. Li, J. Catal., 2006, 239, 369 CrossRef; (e) H. Lu, J. Gao, Z. Jiang, Y. Yang, B. Song and C. Li, Chem. Commun., 2007, 150 RSC.
- (a) W. Trakarnpruk and K. Rujiraworawut, Fuel Process. Technol., 2009, 90, 411 CrossRef CAS; (b) C. Komintarachat and W. Trakarnpruk, Ind. Eng. Chem. Res., 2006, 45, 1853 CrossRef CAS; (c) X. Dong, C. Yu, D. Wang, Y. Zhang, P. Wu, H. Hu and G. Xue, Mater. Res. Bull., 2017, 85, 152 CrossRef CAS.
- M.-W. Wan, M. D. G. de Luna, L. R. Golosinda, C. M. Futalan, P. Phatai and M.-C. Lu, Clean Technol. Environ. Policy, 2019, 21, 1459 CrossRef CAS.
- (a) A. M. Al-Ajlouni, T. M. Daiafla and M. El-Khateeb, J. Mol. Catal. A: Chem., 2007, 275, 139 CrossRef CAS; (b) M. Te, C. Fairbridge and Z. Ring, Appl. Catal., A, 2001, 219, 267 CrossRef CAS.
- (a) B. Bertleff, J. Claussnitzer, W. Korth, P. Wasserscheid, A. Jess and J. Albert, ACS Sustainable Chem. Eng., 2017, 5, 4110 CrossRef CAS; (b) B. Bertleff, J. Claussnitzer, W. Korth, P. Wasserscheid, A. Jess and J. Albert, Energy Fuels, 2018, 32, 8683 CrossRef CAS; (c) B. Bertleff, R. Goebel, J. Claussnitzer, W. Korth, M. Skiborowski, P. Wasserscheid, A. Jess and J. Albert, ChemCatChem, 2018, 10, 4602 CrossRef CAS.
- L. Sun, T. Su, J. Xu, D. Hao, W. Liao, Y. Zhao, W. Ren, C. Deng and H. Lü, Green Chem., 2019, 21, 2629 RSC.
- C. Li, Z. Jiang, J. Gao, Y. Yang, S. Wang, F. Tian, F. Sun, X. Sun, P. Ying and C. Han, Chem.–Eur. J., 2004, 10, 2277 CrossRef CAS PubMed.
- E. Rafiee and S. Eavani, RSC Adv., 2016, 6, 46433 RSC.
- L. Liu, Y. Zhang and W. Tan, Front. Chem. Sci. Eng., 2013, 7, 422 CrossRef CAS.
- W. Zhu, B. Dai, P. Wu, Y. Chao, J. Xiong, S. Xun, H. Li and H. Li, ACS Sustainable Chem. Eng., 2014, 3, 186 CrossRef.
- R. Wang, F. Yu, G. Zhang and H. Zhao, Catal. Today, 2010, 150, 37 CrossRef CAS.
- S. Lu, H. Zhang, D. Wu, X. Han, Y. Yao and Q. Zhang, RSC Adv., 2016, 6, 79520 RSC.
- (a) Y. Zhang, Y. Gu, X. Dong, P. Wu, Y. Li, H. Hu and G. Xue, Catal. Lett., 2017, 147, 1811 CrossRef CAS; (b) X. Liao, D. Wu, B. Geng, S. Lu and Y. Yao, RSC Adv., 2017, 7, 48454 RSC.
- M. Y. Yu, T. T. Guo, X. C. Shi, J. Yang, X. Xu, J. F. Ma and Z. T. Yu, Inorg. Chem., 2019, 58, 11010 CrossRef CAS PubMed.
- (a) F. Mirante, L. Dias, M. Silva, S. O. Ribeiro, M. C. Corvo, B. de Castro, C. M. Granadeiro and S. S. Balula, Catal. Commun., 2018, 104, 1 CrossRef CAS; (b) Y. Li, H. Zhang, Y. Jiang, M. Shi, M. Bawa, X. Wang and S. Zhu, Fuel, 2019, 241, 861 CrossRef CAS.
- (a) X.-M. Zhou, W. Chen and Y.-F. Song, Eur. J. Inorg. Chem., 2014, 2014, 812 CrossRef CAS; (b) M. Y. Yu, J. Yang, X. Xu, J. F. Ma and Z. Wang, Appl. Organomet. Chem., 2019, 33, e5169 CrossRef.
- Y. Niu, Q. Xu, Y. Wang, Z. Li, J. Lu, P. Ma, C. Zhang, J. Niu and J. Wang, Dalton Trans., 2018, 47, 9677 RSC.
- H. Zhang, X. Xu, H. Lin, M. A. Ud Din, H. Wang and X. Wang, Nanoscale, 2017, 9, 13334 RSC.
- (a) J. L. García-Gutiérrez, G. A. Fuentes, M. E. Hernández-Terán, F. Murrieta, J. Navarrete and F. Jiménez-Cruz, Appl. Catal., A, 2006, 305, 15 CrossRef; (b) L. Hui-Peng, S. Jian and Z. Hua, Pet. Chem., 2007, 47, 452 CrossRef.
- (a) Z. E. A. Abdalla and B. Li, Chem. Eng. J., 2012, 200–202, 113 CrossRef CAS; (b) L. Tang, G. Luo, M. Zhu, L. Kang and B. Dai, J. Ind. Eng. Chem., 2013, 19, 620 CrossRef CAS; (c) J. Zhang, A. Wang, X. Li and X. Ma, J. Catal., 2011, 279, 269 CrossRef CAS; (d) D. Nedumaran and A. Pandurangan, Mater. Res. Bull., 2015, 61, 1 CrossRef CAS; (e) D. Yue, J. Lei, Z. Lina, G. Zhenran, X. Du and J. Li, Catal. Lett., 2018, 148, 1100 CrossRef CAS; (f) Y. Tian, G. Wang, J. Long, J. Cui, W. Jin and D. Zeng, Chin. J. Catal., 2016, 37, 2098 CrossRef CAS; (g) X. Chen, M. Zhang, Y. Wei, H. Li, J. Liu, Q. Zhang, W. Zhu and H. Li, Inorg. Chem. Front., 2018, 5, 2478 RSC; (h) X. Zhang, Y. Zhu, P. Huang and M. Zhu, RSC Adv., 2016, 6, 69357 RSC; (i) J. Qiu, G. Wang, Y. Zhang, D. Zeng and Y. Chen, Fuel, 2015, 147, 195 CrossRef CAS; (j) A. Najafi Chermahini, M. Rafiee and S. Shaybanizadeh, J. Porous Mater., 2019, 26, 1691 CrossRef CAS; (k) M. Chamack, A. R. Mahjoub and H. Aghayan, Chem. Eng. J., 2014, 255, 686 CrossRef CAS; (l) G. Wang, Y. Han, F. Wang, Y. Chu and X. Chen, React. Kinet., Mech. Catal., 2015, 115, 679 CrossRef CAS; (m) Y. Yao, H. Zhang, Y. Lu, Y. Zhi and S. Lu, React. Kinet., Mech. Catal., 2016, 118, 621 CrossRef CAS.
- (a) W. Jiang, D. Zheng, S. Xun, Y. Qin, Q. Lu, W. Zhu and H. Li, Fuel, 2017, 190, 1 CrossRef CAS; (b) R. Ghubayra, C. Nuttall, S. Hodgkiss, M. Craven, E. F. Kozhevnikova and I. V. Kozhevnikov, Appl. Catal., B, 2019, 253, 309 CrossRef CAS; (c) Y. Gao, R. Gao, G. Zhang, Y. Zheng and J. Zhao, Fuel, 2018, 224, 261 CrossRef CAS; (d) E. Rafiee, M. Joshaghani and P. Ghaderi-Shekhi Abadi, J. Saudi Chem. Soc., 2017, 21, 599 CrossRef CAS.
- (a) X. Liao, Y. Huang, Y. Zhou, H. Liu, Y. Cai, S. Lu and Y. Yao, Appl. Surf. Sci., 2019, 484, 917 CrossRef CAS; (b) S.-y. Dou and R. Wang, New J. Chem., 2019, 43, 3226 RSC.
- (a) M. A. Rezvani and M. Shaterian, Inorg. Nano-Met. Chem., 2019, 49, 23 CrossRef CAS; (b) D. Yue, J. Lei, Y. Peng, J. Li and X. Du, Fuel, 2018, 226, 148 CrossRef CAS; (c) S. Xun, D. Zheng, S. Yin, Y. Qin, M. Zhang, W. Jiang, W. Zhu and H. Li, RSC Adv., 2016, 6, 42402 RSC; (d) H. Gao, X. Wu, D. Sun, G. Niu, J. Guan, X. Meng, C. Liu, W. Xia and X. Song, Dalton Trans., 2019, 48, 5749 RSC; (e) W. Ma, Y. Xu, K. Ma and H. Zhang, Appl. Catal., A, 2016, 526, 147 CrossRef CAS; (f) W. Ma, Y. Xu, K. Ma, Y. Luo, Y. Liu and H. Zhang, Mol. Catal., 2017, 433, 28 CrossRef CAS; (g) X.-M. Yan, P. Mei, J. Lei, Y. Mi, L. Xiong and L. Guo, J. Mol. Catal. A: Chem., 2009, 304, 52 CrossRef CAS; (h) J. Fu, Y. Guo, W. Ma, C. Fu, L. Li, H. Wang and H. Zhang, J. Mater. Sci., 2018, 53, 15418 CrossRef CAS.
- (a) H. Ji, J. Sun, P. Wu, Y. Wu, J. He, Y. Chao, W. Zhu and H. Li, Fuel, 2018, 213, 12 CrossRef CAS; (b) H. Ji, H. Ju, R. Lan, P. Wu, J. Sun, Y. Chao, S. Xun, W. Zhu and H. Li, RSC Adv., 2017, 7, 54266 RSC; (c) H. Ji, J. Sun, P. Wu, B. Dai, Y. Chao, M. Zhang, W. Jiang, W. Zhu and H. Li, J. Mol. Catal. A: Chem., 2016, 423, 207 CrossRef CAS; (d) B. Dai, P. Wu, W. Zhu, Y. Chao, J. Sun, J. Xiong, W. Jiang and H. Li, RSC Adv., 2016, 6, 140 RSC.
- (a) Y. Zhu, M. Zhu, L. Kang, F. Yu and B. Dai, Ind. Eng. Chem. Res., 2015, 54, 2040 CrossRef CAS; (b) E. Rafiee and M. Khodayari, Chin. J. Catal., 2017, 38, 458 CrossRef CAS.
- P. Huang, G. Luo, L. Kang, M. Zhu and B. Dai, RSC Adv., 2017, 7, 4681 RSC.
- M. A. Rezvani, S. Khandan and N. Sabahi, Energy Fuels, 2017, 31, 5472 CrossRef CAS.
- T. F. Cai, H. P. Li and H. Zhao, Pet. Sci. Technol., 2014, 32, 1713 CrossRef CAS.
- M. Zhang, W. Zhu, S. Xun, H. Li, Q. Gu, Z. Zhao and Q. Wang, Chem. Eng. J., 2013, 220, 328 CrossRef CAS.
- M. A. Rezvani, M. Oveisi and M. A. Nia Asli, J. Mol. Catal. A: Chem., 2015, 410, 121 CrossRef CAS.
- M. A. Rezvani, M. Shaterian, F. Akbarzadeh and S. Khandan, Chem. Eng. J., 2018, 333, 537 CrossRef CAS.
- S. Y. Dou and R. Wang, Appl. Organomet. Chem., 2019, 33, e4924 CrossRef.
- S. Ribeiro, C. M. Granadeiro, P. Silva, F. A. A. Paz, F. F. de Biani, L. Cunha-Silva and S. S. Balula, Catal. Sci. Technol., 2013, 3, 2404 RSC.
- L. Zhang, S. Wu, Y. Liu, F. Wang, X. Han and H. Shang, Appl. Organomet. Chem., 2016, 30, 684 CrossRef CAS.
- X. S. Wang, Y. B. Huang, Z. J. Lin and R. Cao, Dalton Trans., 2014, 43, 11950 RSC.
- C. M. Granadeiro, L. S. Nogueira, D. Julião, F. Mirante, D. Ananias, S. S. Balula and L. Cunha-Silva, Catal. Sci. Technol., 2016, 6, 1515 RSC.
- Y. Zhang, J. Wan, Y. Wang and Y. Ma, RSC Adv., 2015, 5, 97589 RSC.
- E. Rafiee and N. Nobakht, Korean J. Chem. Eng., 2015, 33, 132 CrossRef.
- D. Zhang, H. Song and D. Yuan, Chem. Eng. Commun., 2019, 206, 1706 CrossRef CAS.
- X.-S. Wang, L. Li, J. Liang, Y.-B. Huang and R. Cao, ChemCatChem, 2017, 9, 971 CrossRef CAS.
- (a) S.-W. Li, Z. Yang, R.-M. Gao, G. Zhang and J.-s. Zhao, Appl. Catal., B, 2018, 221, 574 CrossRef CAS; (b) S.-W. Li, R.-M. Gao, W. Zhang, Y. Zhang and J.-s. Zhao, Fuel, 2018, 221, 1 CrossRef CAS; (c) S.-W. Li, J.-R. Li, Q.-P. Jin, Z. Yang, R.-L. Zhang, R.-M. Gao and J.-s. Zhao, J. Hazard. Mater., 2017, 337, 208 CrossRef CAS PubMed; (d) S.-W. Li, R.-M. Gao, R.-L. Zhang and J.-s. Zhao, Fuel, 2016, 184, 18 CrossRef CAS.
- (a) X. L. Hao, Y. Y. Ma, H. Y. Zang, Y. H. Wang, Y. G. Li and E. B. Wang, Chem.–Eur. J., 2015, 21, 3778 CrossRef CAS PubMed; (b) Y.-Y. Ma, H.-Q. Tan, Y.-H. Wang, X.-L. Hao, X.-J. Feng, H.-Y. Zang and Y.-G. Li, CrystEngComm, 2015, 17, 7938 RSC.
- Z. J. Lin, H. Q. Zheng, J. Chen, W. E. Zhuang, Y. X. Lin, J. W. Su, Y. B. Huang and R. Cao, Inorg. Chem., 2018, 57, 13009 CrossRef CAS PubMed.
- L. Xu, Y. Wang, T. Xu, S. Liu, J. Tong, R. Chu, X. Hou and B. Liu, ChemCatChem, 2018, 10, 5386 CrossRef CAS.
- D. Julião, A. C. Gomes, M. Pillinger, R. Valença, J. C. Ribeiro, B. de Castro, I. S. Gonçalves, L. Cunha Silva and S. S. Balula, Eur. J. Inorg. Chem., 2016, 2016, 5114 CrossRef.
- Y. Liu, S. Liu, S. Liu, D. Liang, S. Li, Q. Tang, X. Wang, J. Miao, Z. Shi and Z. Zheng, ChemCatChem, 2013, 5, 3086 CrossRef CAS.
- (a) B. Li, Z. Liu, C. Han, J. Liu, S. Zuo, Z. Zhou and X. Pang, J. Mol. Catal. A: Chem., 2011, 348, 106 CrossRef CAS; (b) B. Li, Z. Liu, C. Han, W. Ma and S. Zhao, J. Colloid Interface Sci., 2012, 377, 334 CrossRef CAS PubMed.
- L. X. Cai, S. C. Li, D. N. Yan, L. P. Zhou, F. Guo and Q. F. Sun, J. Am. Chem. Soc., 2018, 140, 4869 CrossRef CAS PubMed.
- J. Song, Y. Li, P. Cao, X. Jing, M. Faheem, Y. Matsuo, Y. Zhu, Y. Tian, X. Wang and G. Zhu, Adv. Mater., 2019, e1902444 CrossRef PubMed.
- M. A. Rezvani, S. Khandan, N. Sabahi and H. Saeidian, Chin. J. Chem. Eng., 2019, 27, 2418 CrossRef.
- X. Zhang, G. Luo, M. Zhu, L. Kang, F. Yu and B. Dai, RSC Adv., 2015, 5, 76182 RSC.
- Y. Zhang, W. Zhang, J. Zhang, Z. Dong, X. Zhang and S. Ding, RSC Adv., 2018, 8, 31979 RSC.
- M. Zhang, M. Li, Q. Chen, W. Zhu, H. Li, S. Yin, Y. Li and H. Li, RSC Adv., 2015, 5, 76048 RSC.
- R. Xia, W. Lv, K. Zhao, S. Ma, J. Hu, H. Wang and H. Liu, Langmuir, 2019, 35, 3963 CrossRef CAS PubMed.
- Z. Hu, S. Lu, X. Huang, J. Li, Y. Duan, L. Yan, Y. Yao and X. Liao, Appl. Organomet. Chem., 2018, 32, e4521 CrossRef.
- M. Craven, D. Xiao, C. Kunstmann-Olsen, E. F. Kozhevnikova, F. Blanc, A. Steiner and I. V. Kozhevnikov, Appl. Catal., B, 2018, 231, 82 CrossRef CAS.
- M. Carraro, G. Fiorani, L. Mognon, F. Caneva, M. Gardan, C. Maccato and M. Bonchio, Chem.–Eur. J., 2012, 18, 13195 CrossRef CAS PubMed.
- (a) Y. Xu, W. Xuan, M. Zhang, H. N. Miras and Y. F. Song, Dalton Trans., 2016, 45, 19511 RSC; (b) Y. Guo, J. Fu, L. Li, X. Li, H. Wang, W. Ma and H. Zhang, Inorg. Chem. Front., 2018, 5, 2205 RSC; (c) P. Huang, A. Liu, L. Kang, M. Zhu and B. Dai, New J. Chem., 2018, 42, 12830 RSC; (d) Z. Yao, H. N. Miras and Y.-F. Song, Inorg. Chem. Front., 2016, 3, 1007 RSC.
- Y. Cai, H. Song, Z. An, X. Xiang, X. Shu and J. He, Green Chem., 2018, 20, 5509 RSC.
- (a) B. Jiang, Z. Sun, L. Zhang, Y. Sun, H. Zhang and H. Yang, J. Appl. Polym. Sci., 2018, 135 CAS; (b) R. Yahya, M. Craven, E. F. Kozhevnikova, A. Steiner, P. Samunual, I. V. Kozhevnikov and D. E. Bergbreiter, Catal. Sci. Technol., 2015, 5, 818 RSC; (c) L. Zhang, S. Song, N. Yang, X. Tantai, X. Xiao, B. Jiang and Y. Sun, Ind. Eng. Chem. Res., 2019, 58, 3618 CrossRef CAS; (d) Y. Chen, H.-y. Song, Y.-z. Lu, H. Meng, C.-x. Li, Z.-g. Lei and B.-h. Chen, Ind. Eng. Chem. Res., 2016, 55, 10394 CrossRef CAS; (e) M. A. Rezvani and O. F. Miri, Chem. Eng. J., 2019, 369, 775 CrossRef CAS.
- (a) F. Mirante, S. O. Ribeiro, B. de Castro, C. M. Granadeiro and S. S. Balula, Top. Catal., 2017, 60, 1140 CrossRef CAS; (b) J. Xiong, W. Zhu, W. Ding, L. Yang, Y. Chao, H. Li, F. Zhu and H. Li, Ind. Eng. Chem. Res., 2014, 53, 19895 CrossRef CAS; (c) J. Zhuang, X. Jin, X. Shen, J. Tan, L. Nie, J. Xiong and B. Hu, Bull. Korean Chem. Soc., 2015, 36, 1784 CrossRef CAS; (d) X. N. Pham, D. L. Tran, T. D. Pham, Q. M. Nguyen, V. T. T. Thi and H. D. Van, Adv. Powder Technol., 2018, 29, 58 CrossRef CAS; (e) X. Li, J. Zhang, F. Zhou, Y. Wang, X. Yuan and H. Wang, Mol. Catal., 2018, 452, 93 CrossRef CAS; (f) F. Mirante, N. Gomes, L. C. Branco, L. Cunha-Silva, P. L. Almeida, M. Pillinger, S. Gago, C. M. Granadeiro and S. S. Balula, Microporous Mesoporous Mater., 2019, 275, 163 CrossRef CAS; (g) L. S. Nogueira, S. Ribeiro, C. M. Granadeiro, E. Pereira, G. Feio, L. Cunha-Silva and S. S. Balula, Dalton Trans., 2014, 43, 9518 RSC; (h) J. Xiong, W. Zhu, W. Ding, L. Yang, M. Zhang, W. Jiang, Z. Zhao and H. Li, RSC Adv., 2015, 5, 16847 RSC; (i) Y. Chen and Y.-F. Song, ChemPlusChem, 2014, 79, 304 CrossRef CAS; (j) F. Banisharif, M. R. Dehghani, M. C. Capel-Sanchez and J. M. Campos-Martin, Catal. Today, 2019, 333, 219 CrossRef CAS; (k) Y. Chen, S. Zhao and Y.-F. Song, Appl. Catal., A, 2013, 466, 307 CrossRef CAS; (l) D. Julião, F. Mirante, S. O. Ribeiro, A. C. Gomes, R. Valença, J. C. Ribeiro, M. Pillinger, B. de Castro, I. S. Gonçalves and S. S. Balula, Fuel, 2019, 241, 616 CrossRef; (m) S. O. Ribeiro, L. S. Nogueira, S. Gago, P. L. Almeida, M. C. Corvo, B. d. Castro, C. M. Granadeiro and S. S. Balula, Appl. Catal., A, 2017, 542, 359 CrossRef CAS; (n) M. Zhu, G. Luo, L. Kang and B. Dai, RSC Adv., 2014, 4, 16769 RSC.
- (a) L. Xia, H. Zhang, Z. Wei, Y. Jiang, L. Zhang, J. Zhao, J. Zhang, L. Dong, E. Li, L. Ruhlmann and Q. Zhang, Chem.–Eur. J., 2017, 23, 1920 CrossRef CAS PubMed; (b) J. Yang, D. Hu, W. Li and S. Yi, Chem. Eng. J., 2015, 267, 93 CrossRef CAS.
- H. Yang, Q. Zhang, J. Zhang, L. Yang, Z. Ma, L. Wang, H. Li, L. Bai, D. Wei and W. Wang, J. Colloid Interface Sci., 2019, 554, 572 CrossRef CAS PubMed.
- (a) T. Rao, P. Krishna, D. Paul, B. Nautiyal, J. Kumar, Y. Sharma, S. Nanoti, B. Sain and M. Garg, Pet. Sci. Technol., 2011, 29, 626 CrossRef CAS; (b) J. Zhang, W. Zhu, H. Li, W. Jiang, Y. Jiang, W. Huang and Y. Yan, Green Chem., 2009, 11, 1801 RSC; (c) W. Zhu, H. Li, X. Jiang, Y. Yan, J. Lu, L. He and J. Xia, Green Chem., 2008, 10, 641 RSC; (d) Y. Dai, Y. Qi, D. Zhao and H. Zhang, Fuel Process. Technol., 2008, 89, 927 CrossRef CAS.
- (a) Y. Zhang, D. Wang, R. Zhang, J. Zhao and Y. Zheng, Catal. Commun., 2012, 29, 21 CrossRef CAS; (b) R. Liu, J. Zhang, Z. Xu, D. Zhao and S. Sun, J. Mater. Sci., 2018, 53, 4927 CrossRef CAS; (c) S. Chen, W. Lu, Y. Yao, H. Chen and W. Chen, React. Kinet., Mech. Catal., 2014, 111, 535 CrossRef CAS; (d) X. Zhou, J. Li, X. Wang, K. Jin and W. Ma, Fuel Process. Technol., 2009, 90, 317 CrossRef CAS; (e) J. Zhang, J. Li, T. Ren, Y. Hu, J. Ge and D. Zhao, RSC Adv., 2014, 4, 3206 RSC; (f) G. Zhang, B. Liu, H. Zhou, Y. Yang, W. Chen and J. Zhao, Appl. Organomet. Chem., 2018, 32, e4477 CrossRef; (g) F. Wang, G. Wang, W. Sun, T. Wang and X. Chen, Microporous Mesoporous Mater., 2015, 217, 203 CrossRef CAS; (h) X. Zhou, S. Lv, H. Wang, X. Wang and J. Liu, Appl. Catal., A, 2011, 396, 101 CrossRef CAS; (i) R. Zhao, J. Wang, D. Zhang, Y. Sun, B. Han, N. Tang, N. Wang and K. Li, Appl. Catal., A, 2017, 532, 26 CrossRef CAS.
- A. B. Sorokin, Chem. Rev., 2013, 113, 8152 CrossRef CAS PubMed.
- R. Zhao, J. Wang, D. Zhang, Y. Sun, B. Han, N. Tang, J. Zhao and K. Li, ACS Sustainable Chem. Eng., 2017, 5, 2050 CrossRef CAS.
- (a) L. Junbo, L. Xiwen, C. Cancan, G. Jia and P. Zhiquan, Chin. J. Chem. Eng., 2013, 21, 860 CrossRef; (b) M. R. Maurya, A. Arya, A. Kumar, M. L. Kuznetsov, F. Avecilla and J. o. Costa Pessoa, Inorg. Chem., 2010, 49, 6586 CrossRef CAS PubMed; (c) W. Zhu, H. Li, X. Jiang, Y. Yan, J. Lu and J. Xia, Energy Fuels, 2007, 21, 2514 CrossRef CAS; (d) W. Zhu, H. Li, Q. Gu, P. Wu, G. Zhu, Y. Yan and G. Chen, J. Mol. Catal. A: Chem., 2011, 336, 16 CrossRef CAS; (e) A. Aguiar, S. Ribeiro, A. M. Silva, L. Cunha-Silva, B. de Castro, A. M. Silva and S. S. Balula, Appl. Catal., A, 2014, 478, 267 CrossRef CAS.
- (a) L. Wang, S. Li, H. Cai, Y. Xu, X. Wu and Y. Chen, Fuel, 2012, 94, 165 CrossRef CAS; (b) A. Di Giuseppe, M. Crucianelli, F. De Angelis, C. Crestini and R. Saladino, Appl. Catal., B, 2009, 89, 239 CrossRef CAS; (c) A. S. Ogunlaja, W. Chidawanyika, E. Antunes, M. A. Fernandes, T. Nyokong, N. Torto and Z. R. Tshentu, Dalton Trans., 2012, 41, 13908 RSC.
- D. Piccinino, I. Abdalghani, G. Botta, M. Crucianelli, M. Passacantando, M. L. Di Vacri and R. Saladino, Appl. Catal., B, 2017, 200, 392 CrossRef CAS.
- (a) W. Jiang, W. Zhu, Y. Chang, H. Li, Y. Chao, J. Xiong, H. Liu and S. Yin, Energy Fuels, 2014, 28, 2754 CrossRef CAS; (b) H. Gobara, M. Nessim, M. Zaky, F. Khalil and A. Anisimov, Catal. Lett., 2014, 144, 1043 CrossRef CAS.
- J.-K. Li, Y.-Q. Xu and C.-W. Hu, Inorg. Chem. Commun., 2015, 60, 12 CrossRef CAS.
- D. Julião, A. C. Gomes, M. Pillinger, R. Valença, J. C. Ribeiro, I. S. Gonçalves and S. S. Balula, Appl. Catal., B, 2018, 230, 177 CrossRef.
- W. Zhu, D. Xu, H. Li, Y. Ding, M. Zhang, H. Liu and Y. Chao, Pet. Sci. Technol., 2013, 31, 1447 CrossRef CAS.
- (a) H.-L. Jiang and Q. Xu, Chem. Commun., 2011, 47, 3351 RSC; (b) G. Fu, B. Bueken and D. De Vos, Small Methods, 2018, 2, 1800203 CrossRef; (c) J.-S. Qin, S. Yuan, Q. Wang, A. Alsalme and H.-C. Zhou, J. Mater. Chem. A, 2017, 5, 4280 RSC; (d) B. N. Bhadra and S. H. Jhung, Appl. Catal., B, 2019, 259, 118021 CrossRef CAS.
- (a) N. D. McNamara and J. C. Hicks, ACS Appl. Mater. Interfaces, 2015, 7, 5338 CrossRef CAS PubMed; (b) Y. Zhang, G. Li, L. Kong and H. Lu, Fuel, 2018, 219, 103 CrossRef CAS; (c) N. D. McNamara, G. T. Neumann, E. T. Masko, J. A. Urban and J. C. Hicks, J. Catal., 2013, 305, 217 CrossRef CAS; (d) S. Smolders, T. Willhammar, A. Krajnc, K. Sentosun, M. T. Wharmby, K. A. Lomachenko, S. Bals, G. Mali, M. B. Roeffaers and D. E. De Vos, Angew. Chem., Int. Ed., 2019, 131, 9260 CrossRef.
- X. Li, Y. Gu, H. Chu, G. Ye, W. Zhou, W. Xu and Y. Sun, Appl. Catal., A, 2019, 584, 117152 CrossRef CAS.
- M. Y. Masoomi, M. Bagheri and A. Morsali, Inorg. Chem., 2015, 54, 11269 CrossRef CAS PubMed.
- R. Abazari, S. Sanati, A. Morsali, A. Slawin and C. L. Carpenter-Warren, ACS Appl. Mater. Interfaces, 2019, 11, 14759 CrossRef CAS PubMed.
- A. Gómez-Paricio, A. Santiago-Portillo, S. Navalón, P. Concepción, M. Alvaro and H. Garcia, Green Chem., 2016, 18, 508 RSC.
- H.-Q. Zheng, Y.-N. Zeng, J. Chen, R.-G. Lin, W.-E. Zhuang, R. Cao and Z.-J. Lin, Inorg. Chem., 2019, 58, 6983 CrossRef CAS PubMed.
- W. Xiao, Q. Dong, Y. Wang, Y. Li, S. Deng and N. Zhang, CrystEngComm, 2018, 20, 5658 RSC.
- C. M. Granadeiro, S. O. Ribeiro, M. Karmaoui, R. Valença, J. C. Ribeiro, B. de Castro, L. Cunha-Silva and S. S. Balula, Chem. Commun., 2015, 51, 13818 RSC.
- G. Ye, H. Qi, X. Li, K. Leng, Y. Sun and W. Xu, ChemPhysChem, 2017, 18, 1903 CrossRef CAS PubMed.
- G. Ye, H. Qi, W. Zhou, W. Xu and Y. Sun, Inorg. Chem. Front., 2019, 6, 1267 RSC.
- C. Piscopo, L. Voellinger, M. Schwarzer, A. Polyzoidis, D. Bošković and S. Loebbecke, ChemistrySelect, 2019, 4, 2806 CrossRef CAS.
- R. Limvorapitux, H. Chen, M. L. Mendonca, M. Liu, R. Q. Snurr and S. T. Nguyen, Catal. Sci. Technol., 2019, 9, 327 RSC.
- (a) J. Kim, N. D. McNamara, T. H. Her and J. C. Hicks, ACS Appl. Mater. Interfaces, 2013, 5, 11479 CrossRef CAS PubMed; (b) B. N. Bhadra and S. H. Jhung, Nanoscale, 2018, 10, 15035 RSC.
- M. Sarker, B. N. Bhadra, S. Shin and S. H. Jhung, ACS Appl. Nano Mater., 2018, 2, 191 CrossRef.
- B. N. Bhadra, J. Y. Song, N. A. Khan and S. H. Jhung, ACS Appl. Mater. Interfaces, 2017, 9, 31192 CrossRef CAS PubMed.
- N. D. McNamara, J. Kim and J. C. Hicks, Energy Fuels, 2015, 30, 594 CrossRef.
- J. Kim, N. D. McNamara and J. C. Hicks, Appl. Catal., A, 2016, 517, 141 CrossRef CAS.
- (a) Y.-Y. Liu, K. Leus, Z. Sun, X. Li, H. Depauw, A. Wang, J. Zhang and P. Van Der Voort, Microporous Mesoporous Mater., 2019, 277, 245 CrossRef CAS; (b) D. Julião, A. C. Gomes, M. Pillinger, L. Cunha-Silva, B. de Castro, I. S. Gonçalves and S. S. Balula, Fuel Process. Technol., 2015, 131, 78 CrossRef.
- (a) P. De Filippis and M. Scarsella, Energy Fuels, 2003, 17, 1452 CrossRef CAS; (b) W.-H. Lo, H.-Y. Yang and G.-T. Wei, Green Chem., 2003, 5, 639 RSC; (c) E. Krivtsov and A. Golovko, Pet. Chem., 2014, 54, 51 CrossRef CAS; (d) C. Mao, R. Zhao, X. Li and X. Gao, RSC Adv., 2017, 7, 12805 RSC.
- (a) M. T. Timko, E. Schmois, P. Patwardhan, Y. Kida, C. A. Class, W. H. Green, R. K. Nelson and C. M. Reddy, Energy Fuels, 2014, 28, 2977 CrossRef CAS; (b) G. Yu, H. Chen, S. Lu and Z. Zhu, Front. Chem. Eng. China, 2007, 1, 162 CrossRef CAS; (c) G. Yu, S. Lu, H. Chen and Z. Zhu, Carbon, 2005, 43, 2285 CrossRef CAS.
- M. T. Timko, J. A. Wang, J. Burgess, P. Kracke, L. Gonzalez, C. Jaye and D. A. Fischer, Fuel, 2016, 163, 223 CrossRef CAS.
- G. Abdi, M. Ashokkumar and A. Alizadeh, Fuel, 2017, 210, 639 CrossRef CAS.
- (a) H. Gao, C. Guo, J. Xing, J. Zhao and H. Liu, Green Chem., 2010, 12, 1220 RSC; (b) J. Wu, Y. Gao, W. Zhang, Y. Tan, A. Tang, Y. Men and B. Tang, RSC Adv., 2014, 4, 58800 RSC.
- P. Wu, B. Dai, Y. Chao, Y. Wu, W. Jiang, C. Li, W. Zhu and H. Li, Mol. Catal., 2017, 436, 53 CrossRef CAS.
- A. D. Bokare and W. Choi, J. Hazard. Mater., 2016, 304, 313 CrossRef CAS PubMed.
- (a) Q. Gu, G. Wen, Y. Ding, K.-H. Wu, C. Chen and D. Su, Green Chem., 2017, 19, 1175 RSC; (b) J. He, P. Wu, L. Lu, H. Sun, Q. Jia, M. Hua, M. He, C. Xu, W. Zhu and H. Li, Energy Fuels, 2019, 33, 8302 CrossRef; (c) Y. Zhu, X. Li and M. Zhu, Catal. Commun., 2016, 85, 5 CrossRef CAS; (d) Q. Jia, J. He, P. Wu, J. Luo, Y. Wei, H. Li, S. Xun, W. Zhu and H. Li, Mol. Catal., 2019, 468, 100 CrossRef CAS.
- W. Zhang, H. Zhang, J. Xiao, Z. Zhao, M. Yu and Z. Li, Green Chem., 2014, 16, 211 RSC.
- P. Wu, W. Zhu, Y. Chao, J. Zhang, P. Zhang, H. Zhu, C. Li, Z. Chen, H. Li and S. Dai, Chem. Commun., 2016, 52, 144 RSC.
- L. Lu, J. He, P. Wu, Y. Wu, Y. Chao, H. Li, D. Tao, L. Fan, H. Li and W. Zhu, Green Chem., 2018, 20, 4453 RSC.
- P. Wu, W. Zhu, B. Dai, Y. Chao, C. Li, H. Li, M. Zhang, W. Jiang and H. Li, Chem. Eng. J., 2016, 301, 123 CrossRef CAS.
- A. A. Oliveira, D. A. Costa, I. F. Teixeira and F. C. Moura, Appl. Catal., B, 2015, 162, 475 CrossRef CAS.
- N. A. Khan, B. N. Bhadra, S. W. Park, Y. S. Han and S. H. Jhung, Small, 2019, 1901564 CrossRef PubMed.
- L. Wu, G. Miao, X. Dai, L. Dong, Z. Li and J. Xiao, Energy Fuels, 2019, 33, 7287 CrossRef CAS.
- P. Yuan, T. Zhang, A. Cai, C. Cui, H. Liu and X. Bao, RSC Adv., 2016, 6, 74929 RSC.
- (a) P. S. Kulkarni and C. A. Afonso, Green Chem., 2010, 12, 1139 RSC; (b) D. Zolotareva, A. Zazybin, K. Rafikova, V. M. Dembitsky, A. Dauletbakov and V. Yu, Vietnam J. Chem., 2019, 57, 133 CAS.
- (a) Z. Jiang, H. LÜ, Y. Zhang and C. Li, Chin. J. Catal., 2011, 32, 707 CrossRef CAS; (b) E. W. Qian, J. Jpn. Pet. Inst., 2008, 51, 14 CrossRef CAS.
- (a) A. E. S. Choi, S. Roces, N. Dugos and M.-W. Wan, Fuel, 2016, 180, 127 CrossRef CAS; (b) J. B. Bhasarkar, M. Singh and V. S. Moholkar, RSC Adv., 2015, 5, 102953 RSC; (c) M. K. Bolla, H. A. Choudhury and V. S. Moholkar, Ind. Eng. Chem. Res., 2012, 51, 9705 CrossRef CAS; (d) K. Keynejad, M. Nikazar and B. Dabir, Pet. Sci. Technol., 2018, 36, 718 CrossRef CAS.
- J. B. Bhasarkar, S. Chakma and V. S. Moholkar, Ind. Eng. Chem. Res., 2013, 52, 9038 CrossRef CAS.
- (a) X. Gao, J. Fei, Y. Shang and F. Fu, Chin. J. Chem. Eng., 2018, 26, 1508 CrossRef CAS; (b) H. Sahragard, M. R. Khosravi-Nikou and M. H. Maddahi, Pet. Sci. Technol., 2017, 35, 1879 CrossRef CAS; (c) X.-j. Wang, F.-t. Li, J.-x. Liu, C.-g. Kou, Y. Zhao, Y.-j. Hao and D. Zhao, Energy Fuels, 2012, 26, 6777 CrossRef CAS; (d) A. S. Morshedy, A. M. El Naggar, S. M. Tawfik, O. I. Sif El-Din, S. I. Hassan and A. I. Hashem, J. Phys. Chem. C, 2016, 120, 26350 CrossRef CAS.
- Y. Chen, C. Shen, J. Wang, G. Xiao and G. Luo, ACS Sustainable Chem. Eng., 2018, 6, 13276 CrossRef CAS.
- J. Wu, J. Li, J. Liu, J. Bai and L. Yang, RSC Adv., 2017, 7, 51046 RSC.
- T. H. T. Vu, T. T. T. Nguyen, P. H. T. Nguyen, M. H. Do, H. T. Au, T. B. Nguyen, D. L. Nguyen and J. S. Park, Mater. Res. Bull., 2012, 47, 308 CrossRef CAS.
- G. Zhang, J. Ren, B. Liu, M. Tian, H. Zhou and J. Zhao, Inorg. Chim. Acta, 2018, 471, 782 CrossRef CAS.
- F. Lin, Y. Zhang, L. Wang, Y. Zhang, D. Wang, M. Yang, J. Yang, B. Zhang, Z. Jiang and C. Li, Appl. Catal., B, 2012, 127, 363 CrossRef CAS.
- L. Guo, Q. Zhao, C. Wang, H. Shen, X. an Han, K. Zhang, D. Wang and F. Fu, Mater. Res. Bull., 2019, 110520 CrossRef CAS.
- S. Khayyat and L. S. Roselin, J. Saudi Chem. Soc., 2017, 21, 349 CrossRef CAS.
- M. Zarrabi, M. H. Entezari and E. K. Goharshadi, RSC Adv., 2015, 5, 34652 RSC.
- H. F. M. Zaid, F. K. Chong and M. I. A. Mutalib, Fuel, 2015, 156, 54 CrossRef.
- S. Li, N. Mominou, Z. Wang, L. Liu and L. Wang, Energy Fuels, 2016, 30, 962 CAS.
- G. Miao, D. Huang, X. Ren, X. Li, Z. Li and J. Xiao, Appl. Catal., B, 2016, 192, 72 CrossRef CAS.
- S.-W. Li, Y.-Y. Li, F. Yang, Z. Liu, R.-M. Gao and J.-s. Zhao, J. Colloid Interface Sci., 2015, 460, 8 CrossRef CAS PubMed.
- (a) X. Zeng, X. Xiao, Y. Li, J. Chen and H. Wang, Appl. Catal., B, 2017, 209, 98 CrossRef CAS; (b) X. Li, W. Zhu, X. Lu, S. Zuo, C. Yao and C. Ni, Chem. Eng. J., 2017, 326, 87 CrossRef CAS; (c) X. Zhang, H. Song, C. Sun, C. Chen, F. Han and X. Li, Mater. Chem. Phys., 2019, 226, 34 CrossRef CAS.
- (a) C. Yang, H. Ji, C. Chen, W. Ma and J. Zhao, Appl. Catal., B, 2018, 235, 207 CrossRef CAS; (b) L. Wang, Y. Chen, L. Du, S. Li, H. Cai and W. Liu, Fuel, 2013, 105, 353 CrossRef CAS.
- S. Otsuki, T. Nonaka, W. Qian, A. Ishihara and T. Kabe, J. Jpn. Pet. Inst., 2001, 44, 18 CrossRef CAS.
- (a) A. Akopyan, E. Ivanov, P. Polikarpova, A. Tarakanova, E. Rakhmanov, O. Polyakova, A. Anisimov, V. Vinokurov and E. Karakhanov, Pet. Chem., 2015, 55, 571 CrossRef CAS; (b) K. M. Dooley, D. Liu, A. M. Madrid and F. C. Knopf, Appl. Catal., A, 2013, 468, 143 CrossRef CAS.
- K.-S. Cho and Y.-K. Lee, Appl. Catal., B, 2014, 147, 35 CrossRef CAS.
- L. C. Caero, F. Jorge, A. Navarro and A. Gutiérrez-Alejandre, Catal. Today, 2006, 116, 562 CrossRef.
- (a) J. Zhang, A. Wang, Y. Wang, H. Wang and J. Gui, Chem. Eng. J., 2014, 245, 65 CrossRef CAS; (b) L. C. Caero, E. Hernández, F. Pedraza and F. Murrieta, Catal. Today, 2005, 107, 564 CrossRef.
- (a) M. A. Safa, R. Bouresli, R. Al-Majren, T. Al-Shamary and X. Ma, Fuel, 2019, 239, 24 CrossRef CAS; (b) J. T. Sampanthar, H. Xiao, J. Dou, T. Y. Nah, X. Rong and W. P. Kwan, Appl. Catal., B, 2006, 63, 85 CrossRef CAS.
- R. Weh and A. de Klerk, Energy Fuels, 2017, 31, 6607 CrossRef CAS.
- R. Sundararaman and C. Song, Energy Fuels, 2013, 27, 6372 CrossRef CAS.
- T. Wallace and B. Heimlich, Tetrahedron, 1968, 24, 1311 CrossRef CAS.
- M. J. Kim, H. Kim, K.-E. Jeong, S.-Y. Jeong, Y. K. Park and J.-K. Jeon, J. Ind. Eng. Chem., 2010, 16, 539 CrossRef CAS.
- (a) Y. K. Park, S. Y. Kim, H. J. Kim, K. Y. Jung, K.-E. Jeong, S.-Y. Jeong and J.-K. Jeon, Korean J. Chem. Eng., 2010, 27, 459 CrossRef CAS; (b) H. Kim, H. S. Kim, K.-E. Jeong, S.-Y. Jeong, Y.-K. Park and J.-K. Jeon, Res. Chem. Intermed., 2010, 36, 669 CrossRef CAS; (c) E. Karakhanov, A. Akopyan, O. Golubev, A. Anisimov, A. Glotov, A. Vutolkina and A. Maximov, ACS Omega, 2019, 4, 12736 CrossRef CAS PubMed.
- Y. Shiraishi, K. Tachibana, T. Hirai and I. Komasawa, Energy Fuels, 2003, 17, 95 CrossRef CAS.
- A. Lyadov, A. Kochyubeev, O. Parenago and S. Khadzhiev, Pet. Chem., 2018, 58, 548 CrossRef CAS.
- (a) S. Gao, X. Chen, X. Xi, M. Abro, W. Afzal, R. Abro and G. Yu, ACS Sustainable Chem. Eng., 2019, 7, 5660 CrossRef CAS; (b) A. S. Ogunlaja, R. S. Walmsley, C. du Sautoy, N. Torto and Z. R. Tshentu, Energy Fuels, 2013, 27, 7714 CrossRef CAS; (c) A. T. Nawaf, S. A. Gheni, A. T. Jarullah and I. M. Mujtaba, Energy Fuels, 2015, 29, 3366 CrossRef CAS; (d) A. Ishihara, D. Wang, F. Dumeignil, H. Amano, E. W. Qian and T. Kabe, Appl. Catal., A, 2005, 279, 279 CrossRef CAS.
- (a) J. Mi, F. Liu, W. Chen, X. Chen, L. Shen, Y. Cao, C. Au, K. Huang, A. Zheng and L. Jiang, ACS Appl. Mater. Interfaces, 2019, 11, 29950 CrossRef CAS PubMed; (b) S. Zhao, H. Yi, X. Tang, D. Kang, H. Wang, K. Li and K. Duan, Appl. Clay Sci., 2012, 56, 84 CrossRef CAS; (c) X. Sun, H. Ruan, X. Song, L. Sun, K. Li, P. Ning and C. Wang, RSC Adv., 2018, 8, 6996 RSC; (d) W. Li, W. Diyong, W. Shudong and Y. Quan, J. Environ. Sci., 2008, 20, 436 CrossRef.
- (a) Y. Cao, X. Zheng, Z. Du, L. Shen, Y. Zheng, C. Au and L. Jiang, Ind. Eng. Chem. Res., 2019, 58, 19353 CrossRef CAS; (b) L. Shen, X. Zheng, G. Lei, X. Li, Y. Cao and L. Jiang, Chem. Eng. J., 2018, 346, 238 CrossRef CAS.
- (a) X. Zhang, Z. Wang, N. Qiao, S. Qu and Z. Hao, ACS Catal., 2014, 4, 1500 CrossRef CAS; (b) W. Zhao, X. Zheng, S. Liang, X. Zheng, L. Shen, F. Liu, Y. Cao, Z. Wei and L. Jiang, Green Chem., 2018, 20, 4645 RSC.
- (a) X. Zheng, L. Zhang, Z. Fan, Y. Cao, L. Shen, C. Au and L. Jiang, Chem. Eng. J., 2019, 374, 793 CrossRef CAS; (b) X.-X. Zheng, L.-J. Shen, X.-P. Chen, X.-H. Zheng, C.-T. Au and L.-L. Jiang, Inorg. Chem., 2018, 57, 10081 CrossRef CAS PubMed.
- (a) X. Wu, V. Schwartz, S. H. Overbury and T. R. Armstrong, Energy Fuels, 2005, 19, 1774 CrossRef CAS; (b) X. Zhang, Y. Tang, S. Qu, J. Da and Z. Hao, ACS Catal., 2015, 5, 1053 CrossRef CAS.
- Z. Guo, T. Zhang, T. Liu, J. Du, B. Jia, S. Gao and J. Yu, Environ. Sci. Technol., 2015, 49, 5697 CrossRef CAS PubMed.
- (a) C. Lei, W. Zhou, L. Shen, X. Zheng, Q. Feng, Y. Liu, Y. Lei, S. Liang, D. Zhang and L. Jiang, ACS Sustainable Chem. Eng., 2019, 7, 16257 CrossRef CAS; (b) G. Lei, Z. Dai, Z. Fan, X. Zheng, Y. Cao, L. Shen, Y. Xiao, C. Au and L. Jiang, Carbon, 2019, 155, 204 CrossRef CAS; (c) G. Lei, Y. Cao, W. Zhao, Z. Dai, L. Shen, Y. Xiao and L. Jiang, ACS Sustainable Chem. Eng., 2019, 7, 4941 CrossRef CAS; (d) X. Kan, X. Chen, W. Chen, J. Mi, J.-Y. Zhang, F. Liu, A. Zheng, K. Huang, L. Shen and C. Au, ACS Sustainable Chem. Eng., 2019, 7, 7609 CrossRef CAS; (e) L. Shen, G. Lei, Y. Fang, Y. Cao, X. Wang and L. Jiang, Chem. Commun., 2018, 54, 2475 RSC; (f) D.-V. Cuong, L. Truong-Phuoc, T. Tran-Thanh, J.-M. Nhut, L. Nguyen-Dinh, I. Janowska, D. Begin and C. Pham-Huu, Appl. Catal., A, 2014, 482, 397 CrossRef CAS; (g) E. Ghasemy, H. B. M. Emrooz, A. Rashidi and T. Hamzehlouyan, Sci. Total Environ., 2019, 134819 CrossRef PubMed; (h) F. Sun, J. Liu, H. Chen, Z. Zhang, W. Qiao, D. Long and L. Ling, ACS Catal., 2013, 3, 862 CrossRef CAS; (i) Y. Liu, C. Duong-Viet, J. Luo, A. Hébraud, G. Schlatter, O. Ersen, J. M. Nhut and C. Pham-Huu, ChemCatChem, 2015, 7, 2957 CrossRef CAS.
- X. Zheng, Y. Li, L. Zhang, L. Shen, Y. Xiao, Y. Zhang, C. Au and L. Jiang, Appl. Catal., B, 2019, 252, 98 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2020 |