
Deep eutectic solvent as solvent and catalyst: one-pot synthesis of 1,3-dinitropropanes via tandem Henry reaction/Michael addition†
Greta
Colombo Dugoni
a,
Alessandro
Sacchetti
 *a and
Andrea
Mele
ab
*a and
Andrea
Mele
ab
aDepartment of Chemistry, Materials and Chemical Engineering “G. Natta”, Politecnico di Milano, via Mancinelli 7, 20131 Milano, Italy. E-mail: alessandro.sacchetti@polimi.it
bCNR-ICRM Istituto di Chimica del Riconoscimento Molecolare, “U.O.S. Milano Politecnico”, Via L. Mancinelli, 7, 20131 Milano, Italy
First published on 20th August 2020
Abstract
The Henry reaction was performed using microwave heating within the deep eutectic solvent (DES) choline chloride/urea (ChCl/urea) which acted as both the catalyst and solvent for the reaction. The optimisation of the conditions (temperature, heating mode, time, DES) allowed 1,3-dinitropropane derivatives to be obtained via tandem Henry reaction/Michael addition, in one step from a range of different aromatic aldehydes in high yields and under mild reaction conditions.
Introduction
In organic synthesis, reactions leading to the formation of carbon–carbon bonds are fundamental for the design of complex molecular systems. A large class of C–C bond formation reactions are based on the coupling between nucleophiles – typically carbanions or their equivalents – and electrophiles. Among the carbanions, nitronate ions, derived from the deprotonation of nitroalkanes, have the advantage of being easier and less demanding to prepare with respect to other classes of carbanions. Consequently, nitroalkanes1 appear to be the ideal synthetic building blocks due to their efficient and versatile reactivity profile. The Henry reaction,2 also known as the nitroaldol reaction, is a powerful C–C bond formation reaction leading to nitro-derivatives. The reaction consists of the coupling of a nucleophilic nitroalkane with an electrophilic aldehyde or ketone to produce β-nitro alcohol (Scheme 1). β-Nitro alcohols are considered valuable synthetic intermediates in the synthesis of polyaminoalcohols, polyhydroxylated amines and natural products.3 The Henry reaction usually occurs under basic catalysis. Common basic catalysts used are carbonates, alkali metal hydroxides, alkoxides, or organic nitrogen bases. The asymmetric Henry reaction has also been reported to occur using metal catalysis,4 organocatalysis5 and biocatalysis.6 The emerging trend of the chemical industry adopting more sustainable processes has stimulated many studies on the Henry reaction and the synthesis of nitro derivatives in an eco-friendly way. Proposed sustainable approaches include the use of synthetic routes featuring heterogeneous catalysis,7 green and non-toxic solvents8 and microwave irradiation.9 In general, the development of new efficient technologies with lower energy demand and more environmentally friendly processes is considered an important goal.10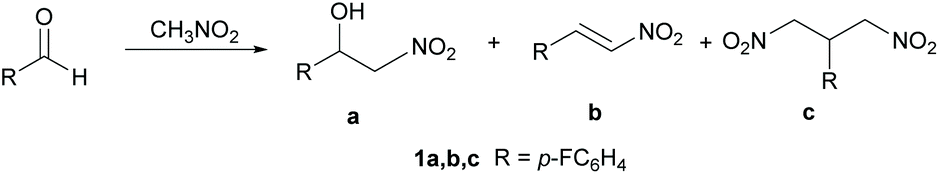 | ||
| Scheme 1 Model Henry reaction and possible products a–c. | ||
Two important issues should be considered when dealing with the Henry reaction: (i) Selectivity. The Henry reaction may in some cases lead to three different products: the β-nitro alcohol 1a, the dehydration product 1b and the 1,3-dinitropropane 1c (see Scheme 1). These latter compounds are important building blocks for many applications and their synthesis through other approaches is challenging. (ii) Catalysis. As mentioned above, the Henry reaction often requires metal catalysis, thus introducing in the synthetic cycle elements of potential environmental impact.
In this paper, we report on a synthetic approach designed to address the issues mentioned above. The method proposed here is based on a one-step synthesis mediated by microwave irradiation and carried out in some selected deep eutectic solvents (DES) as reaction media. Interestingly, the DES used went beyond the role of solvent, showing a catalytic effect too. Unexpectedly, the reaction conditions described here led to the serendipitous finding of a highly selective, mild and efficient synthetic route to challenging 1,3-dinitroderivatives (vide infra).
In recent years, DES were proposed as a new class of environmentally green solvent and extensively tested as reaction media.11 The concept of DES was first proposed by Abbott in 200312 as a mixture of Lewis and Brønsted acids and bases which led to significant melting point depression at the eutectic point.12
Compared to conventional organic solvents, DES show low melting points, low volatility, and thus are often non-flammable. In addition, DES can be designed to be biodegradable, non-toxic and inexpensive. The preparation of DES is often simple and seldom requires purification steps. These properties make DES good candidates as reaction media for synthesis,13 electrochemistry,14 nanomaterials fabrication,15 biochemistry,16 separation,17 biomass processing,18 and chemical analysis.19 The most popular DES are obtained by mixing ammonium or phosphonium salts as hydrogen bond acceptors (HBA), with a variety of hydrogen-bond donors (HBD). The most popular DES is a mixture of choline chloride (ChCl), a biodegradable and non-toxic ammonium salt, and urea in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 molar ratio. This DES is sometimes referred to as reline. The easy sample preparation has allowed a large library of DES to be obtained with the possibility of modulating the physical and chemical properties by appropriate selection of the two components, HBD and HBA.
:2 molar ratio. This DES is sometimes referred to as reline. The easy sample preparation has allowed a large library of DES to be obtained with the possibility of modulating the physical and chemical properties by appropriate selection of the two components, HBD and HBA.
Stimulated by the positive features of the DES summarized above, and during the study of the Henry reaction in DES, we envisaged the possible use of DES as solvent and catalyst by choosing the DES appropriate formulation. In a first report, the Henry reaction has been investigated by Shankarling with the use of catalytic amounts of ChCl/urea in methanol.20 When 20% of DES ChCl/urea was employed, the expected nitroaldol adduct was obtained in good yields from different aromatic aldehydes (Fig. 1). Later, Zheng21 reported the enzyme-catalyzed Henry reaction using DES as co-catalysts. The lipase from Aspergillus Niger (lipase AS) showed excellent catalytic activity towards aromatic aldehydes in water containing the presence of 30% of ChCl/glycerol in the molar ratio 1:2 (Fig. 1). In both works, the reaction was run in molecular solvent and the DES was introduced only in catalytic amounts. Starting from these findings, we studied the Henry reaction in pure DES without other solvents, thus combining the catalytic and solvent role.
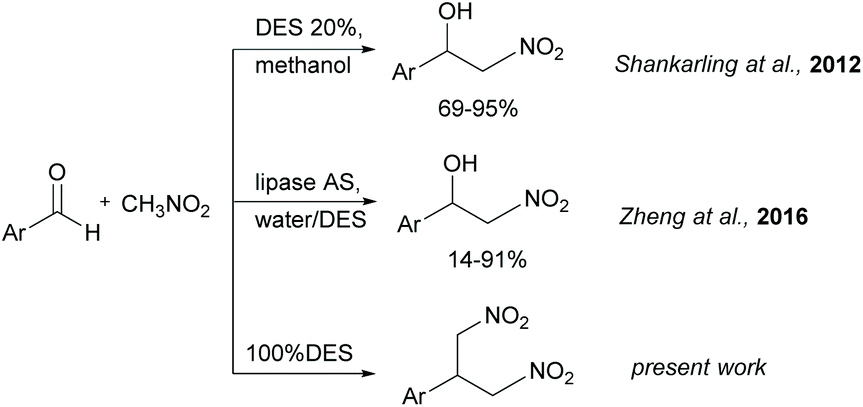 | ||
| Fig. 1 Application of DES in the Henry reaction. | ||
The reaction between 4-fluorobenzaldehyde and nitromethane in pure DES ChCl/urea (Fig. 2) is used here as a model reaction. DES ChCl/urea is expected to act as a catalyst in this reaction due to the presence of urea in its formulation. In fact, it is known from the literature that urea derivatives can efficiently promote the Henry reaction through the activation of nitromethane.22 Further examples of reaction conditions described in this work include: (i) the use of different temperatures, (ii) the presence of triethylamine (TEA), (iii) the use of substituted aldehydes, (iv) tests on selected ketones, (v) tests on ChCl based DES with different HBD and (vi) tests on DES based on choline acetate (ChOAc). Finally, the possibility of DES recycling is reported and briefly discussed.
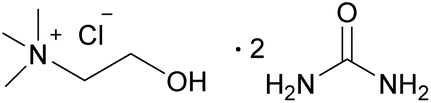 | ||
| Fig. 2 Structure of DES ChCl/urea, (choline chloride/urea 1:2). | ||
Results and discussion
In this study, the DES ChCl/urea was prepared according to the procedures reported in the literature. Briefly, the preparation consisted of mixing ChCl or ChOAc (HBA) with different HBDs, urea or glycolic acid (GlyA). The mixture was stirred at 80 °C for 30 min until a homogeneous and transparent solution was formed. The prepared DES was cooled and used for the Henry reaction without any purification.Scheme 1 sketches the three different products that in principle can be obtained from the reaction of a generic aromatic aldehyde with nitromethane: (i) the expected nitroaldol adduct 1a, (ii) the β-nitrostyrene derivative 1b, resulting from dehydration of 1a, and (iii) the bis-adduct 1c, obtained by the addition of a second molecule of nitromethane to intermediate 1b. In a first attempt, p-fluorobenzaldehyde was reacted with 5 equivalents of nitromethane in 10 equivalents of DES ChCl/urea for 24 h. The effect of temperature was investigated by running the reaction at 20 °C (rt) and after heating to 50 °C and 80 °C. Results are reported in Table 1.
| Entry | Temperature | Producta yield%b | ||
|---|---|---|---|---|
| 1a | 1b | 1c | ||
| Reaction conditions: aldehyde 0.4 mmol, nitromethane 2.0 mmol, DES 4.0 mmol, 24 h.a Ar = p-FC6H4.b Yield was evaluated by 1H-NMR on the crude (see Experimental section for details).c Formation of unidentified by-products.d Reaction with 2 equiv. of nitromethane. | ||||
| 1 | 20 °C | 0 | 0 | 17 |
| 2 | 50 °C | 0 | 0 | 90 |
| 3 | 80 °C | ndc | ndc | Ndc |
| 4d | 50 °C | 31 | 0 | 28 |
Table 1 shows some unprecedented results: the Henry reaction can be driven to the dinitro-derivatives with excellent selectivity and conversion with the only product observed being 1c. Neither the expected nitroaldol 1a nor the elimination product 1b were detected in the reaction mixture. Compound 1c is the result of a tandem Henry reaction/Michael addition between p-fluorobenzaldehyde and nitromethane. Product 1a is first formed according to the classical Henry reaction. 1a then spontaneously undergoes the elimination of water leading to the intermediate β-nitrostyrene 1b which, in turn, undergoes a Michael addition of a second molecule of nitromethane to the double bond to afford the 1,3-dinitropropane derivative 1c. This mechanism is independent of the reaction temperature: at 20 °C only 1c is formed in 17% yield. At 50 °C the 1,3-dinitropropane product is obtained in excellent yield (90%) after 24 h. Increasing the temperature to 80 °C for 24 h produced an inseparable mixture of unidentified by-products. When 2 equiv. of nitromethane were used, the selectivity decreased yielding an almost equimolar mixture of 1a and 1c. Notably, 1b could not be detected in the mixture. This observation suggests that once 1b is formed, it rapidly reacts with nitromethane to afford 1c.
This highly selective route towards the synthesis of the 1,3-dinitropropane derivatives is indeed interesting. 1,3-Dinitropropane derivatives are building blocks with a versatile reactivity profile making them ideal synthetic precursors for diverse functional and structural groups. The reduction of the nitro moiety produces 1,3 diamines, important structural motives existing in many natural products and active pharmaceutical ingredients.23 1,3-Dinitropropane derivatives are precursors of different targets such as heterocycles,24 benzene derivatives,25 carbohydrates,26 and cyclohexane derivatives.27 The use of 1,3-dinitropropane derivatives as key building blocks for biologically active substances, including a novel oxazolidinone antibacterial candidate, has also been reported.28 Several examples of the synthesis of 1,3-dinitroalkanes can be found in the literature. The conventional synthesis of 1,3-dinitroalkanes was performed under basic condition via Michael addition of nitroalkanes to nitroolefins under basic catalysis, usually affording the products in low or moderate yields29 due to the formation of polymeric byproducts. Some attempts at developing one-pot synthetic procedures starting from the aldehyde have also been reported. Good results were obtained in the presence of different heterogeneous catalysts like basic alumina,30 KF-NaHCO3,31 silica-alumina supported amines32 or, alternatively, with Ni-phosphine species33 or by electrochemical synthesis.34
Stimulated by these findings, we tried to improve the procedure to shorten the reaction time and to achieve higher yields. To this end, we investigated the use of microwave irradiation to heat the reaction mixture.
Microwave (MW) assisted chemistry offers several advantages with respect to conventional heating methods, such as reduced processing time and lower energy costs. For MW-assisted heating, the use of strong MW absorbing solvents is mandatory. Additionally, physical and chemical properties like high thermal stability and low vapour pressure are desirable, due to the risks associated with the use of pressurised reaction vessels.35 DESs are in general good solvents for microwave heating.36 DES ChCl/urea, is particularly suitable due its ionic nature and very low vapour pressure.
The influence of microwave irradiation on the reaction between nitromethane and p-fluorobenzaldehyde was thus investigated, starting with 5 equivalents of nitromethane in 10 equivalents of DES ChCl/urea, at different temperatures and reaction times. The aim was to determine the conditions that could achieve the highest yield of the product 1c in the shortest time. Indeed, the reaction time could be reduced from 24 h (thermal heating) to only 2 h (MW heating). The results are reported in Table 2.
At 50 °C after 1 h a poor 9% yield was obtained which could be improved to 96% at 80 °C after only 2 h. It is worth mentioning that, although the formation of a complex mixture of products was observed to take place under conventional heating at 80 °C for 24 h (Table 1, entry 3), the use of MW led to shortened reaction time, thereby preserving the integrity of the system.
In another experiment, we investigated the use of triethylamine (TEA) as an additional basic catalyst to facilitate the formation of the nitroaldol adduct, the first step towards the formation of the 1,3-dinitropropane derivatives. 5 equivalents of TEA were added to the reaction mixture at 50 °C (entry 3) and 80 °C (entry 4). Under these conditions, 1a was isolated in 38% and 29% yield, respectively, together with 1c as the major product (58% and 72% yield). These results suggest that the amine interacts with the DES in some way to decrease its catalytic activity. The result is an evident decrease of the reaction selectivity. We then tried to completely reverse the selectivity towards 1a by using a stoichiometric amount of nitromethane (entry 5), but a mixture of 1a and 1c was obtained. Interestingly, 1b was not detected in any circumstances, thus suggesting that, once the β-nitrostyrene is formed, the second addition of nitromethane is very fast.
We then explored the general applicability of our approach by treating different aromatic aldehydes under the optimized reaction conditions (Table 3).
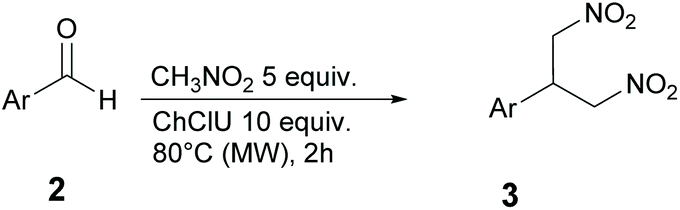
The 1,3-dinitropropane derivative 3 was obtained as the main product in all of the cases. This finding confirms the intrinsic selectivity of the Henry reaction in the DES ChCl/urea with MW irradiation. Excellent yields were observed with benzaldehyde and halogen-substituted aromatic aldehydes (entries 1, 2 and 4). As expected, the presence of substituents with +I or +M electronic effects on the aromatic ring made the substrates less reactive, leading to lower yields (entries 5 and 7), with the exception of the phenol derivative 3c (entry 3). Ortho substituted aldehydes suffered from steric hindrance, affording the product 3h in moderate yield (entry 8). Unfortunately, the reaction carried out on aliphatic aldehydes isovaleraldehyde and butyraldehyde gave only an inseparable mixture of byproducts.
When nitropropane was used instead of nitromethane, compound 4 was obtained in 88% yield as a syn/anti 1:1 mixture of diastereoisomers (Scheme 2).
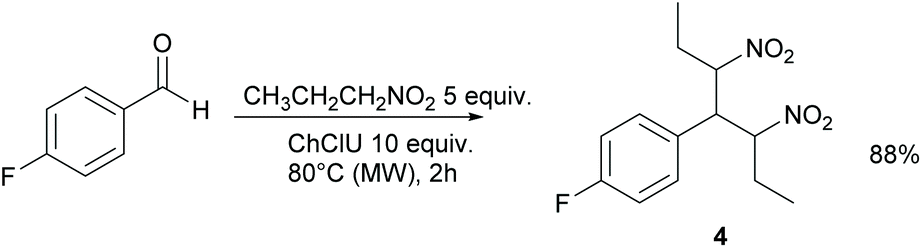 | ||
| Scheme 2 Reaction between p-fluorobenzaldehyde and nitropropane. | ||
The reactivity towards ketones was also investigated. Usually, ketones are much less reactive than aldehydes. Nevertheless, good yields can be obtained from the Henry reaction with the activated trifluoromethylketones as substrates,37 particularly with the use of MW irradiation.38 We then reacted nitromethane with both acetophenone and 2,2,2-trifluoroacetophenone under optimized conditions. No reaction occurred with acetophenone, whereas the reaction of the activated 2,2,2-trifluoroacetophenone produced the nitroaldol 5a in excellent yield (92%) (Scheme 3). In any case no trace of the dinitro adduct was detected. Excellent yields (90% yield, Scheme 3) were also achieved in the reaction of 1,1,1-trifluorobutan-2-one with nitromethane to afford the nitroaldol 5b. Isatin, a non-enolizable activated ketone, was subjected to the same reaction conditions but in this case no significant reaction occurred with only traces of the nitroaldol derivatives detected in the crude by 1H NMR.
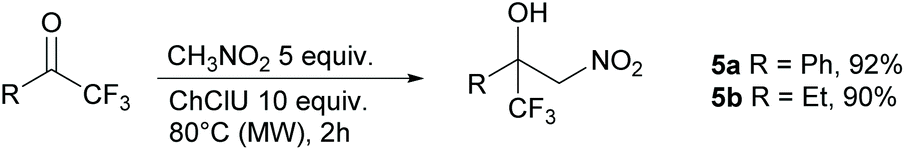 | ||
| Scheme 3 Reaction between nitromethane and trifluoromethylketones. | ||
A preliminary exploration of the role of the HBA (chloride or acetate ions present in ChCl and ChOAc, respectively) and HBD (urea or glycolic acid) of the DES on the outcome of the reaction was also carried out. The possible combinations of the two components to form a DES are indicated in Table 4. The use of the DES in Table 4 is expected to provide a first indication of the influence of HBA and HBD on the reaction selectivity and yields.
| Entry | HBA | HBD | Molar ratio | Abbreviation |
|---|---|---|---|---|
| 1 | Choline chloride | Urea | 1:2 |
ChCl/urea |
| 2 | Choline acetate | Urea | 1:2 |
ChOAc/urea |
| 3 | Choline chloride | Glycolic acid | 1:1 |
ChCl/GlyA |
| 4 | Choline acetate | Glycolic acid | 1:1 |
ChOAc/GlyA |
The results of these investigations are summarized in Table 5. The replacement of chloride ions with acetate (ChOAc/urea, entry 2) led to an unwanted mixture of by-products. A tentative explanation could be that the presence of acetate ions could change the reaction mechanism and inhibit the selective formation of 1,3 dinitro compound. The tests on choline chloride–glycolic acid (ChCl/GlyA, entry 3) and choline acetate–glycolic acid (ChOAc/GlyA, entry 4) confirmed the importance of the presence of urea for the catalytic activity (no Henry-adduct observed). Interestingly, the use of ChOAc/GlyA produced the partial oxidation of the aldehyde to the corresponding carboxylic acid. Studies on this result are ongoing in our laboratories.
Urea-based catalysts are known to work in these reactions by hydrogen bonding catalysis.39 On the bases of the results previously discussed, a possible mechanism for the formation of the 1,3-dinitropropane derivative can be postulated. The proposed mechanism is provided in Scheme 4. In the first step, urea activates nitromethane by forming a hydrogen-bonded complex, thus promoting the formation of the nitronate ion which reacts witht he aldehyde to form the intermediate β-nitro alcohol. This step is also facilitated by the basic environment provided by the DES ChCl/urea.39 In the same way, the H-bond interactions between urea and the aldehyde increase the electrophilicity of the carbonyl carbon atom.
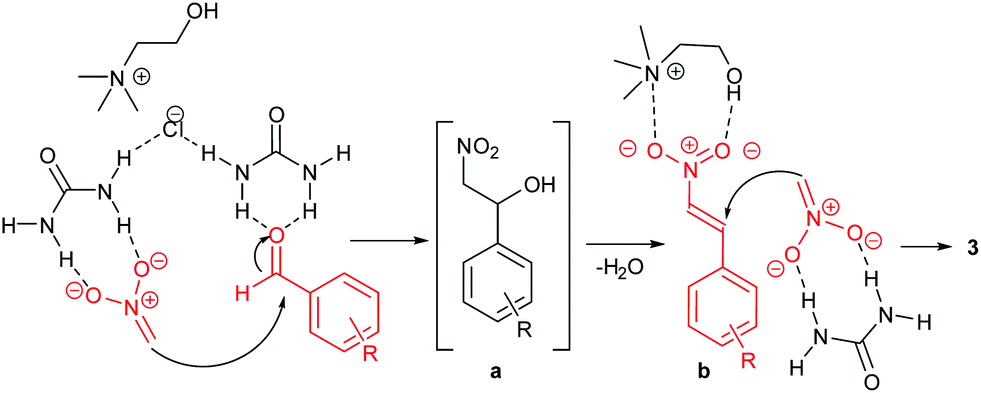 | ||
| Scheme 4 Proposed mechanism for the Henry reaction in the DES ChCl/urea.18 The β-nitro alcohol a intermediate is dehydrated to the β-nitrostyrene b which undergoes a Michael addition of a nitronate ion. | ||
After the β-nitrostyrene b is formed by elimination of water, a possible cooperative action of both the urea and the choline components of the DES is proposed to explain the high reactivity towards the formation of the 1,3-dinitropropane product. Choline can activate β-nitrostyrene by coordination of both the ammonium and the hydroxy group. Similar interactions have been observed in the solid state.40
Finally, the ability to recycle the DES was studied. The recycling of a catalyst and solvent is a key variable for both the economic and environmental sustainability of the process. The tests on DES recycling were carried out on the model reaction between p-fluorobenzaldehyde and nitromethane in ChCl/urea. The reaction was performed under the optimized conditions (1:5:10 equiv. at 80 °C under MW irradiation for 2 h). The product was recovered by extraction with dichloromethane. The DES was separated from the organic phase, recovered, and re-used as a solvent for a subsequent reaction by the addition of new reagents. Its purity was checked by 1H NMR spectroscopy. As shown in Fig. 3, there are no differences between the fresh and the recycled DESs. Throughout the four cycles, ChCl/urea maintained its chemical structure and its ability to catalyse the reaction. Moreover, there was no evidence of any reagents or products in the recycled solvent.
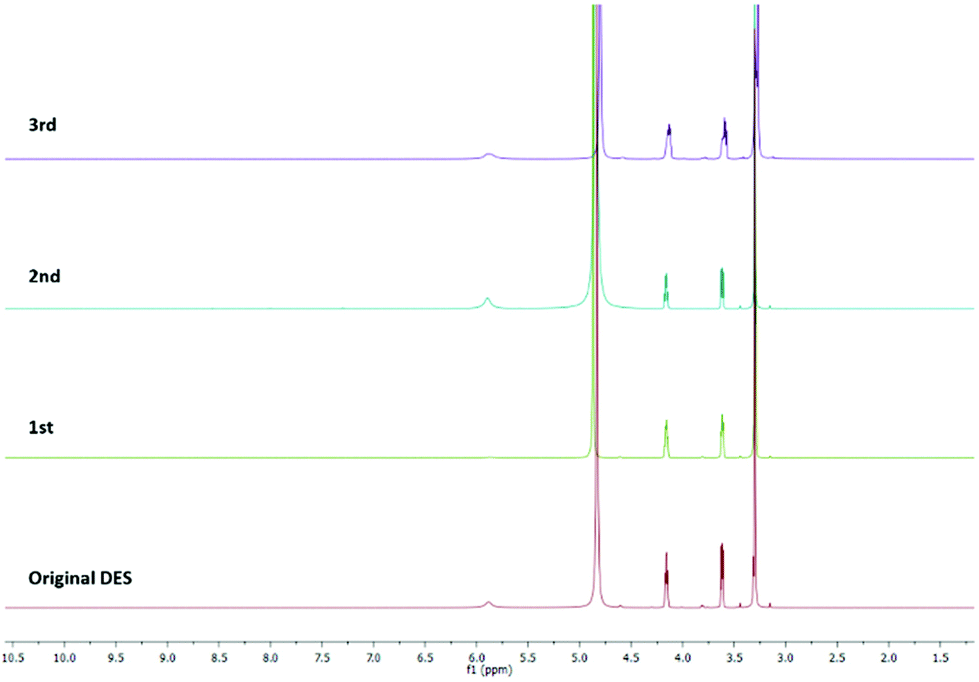 | ||
| Fig. 3 1H NMR spectra of fresh and the recycled DES after different reaction cycles. | ||
In Fig. 4 the performance and the recovery of the DES are reported for four cycles. As can be seen, the ability to catalyse the reaction is maintained (95–84%). The mass of recovered DES remains relatively high with about 90% being retained in the fourth cycle.
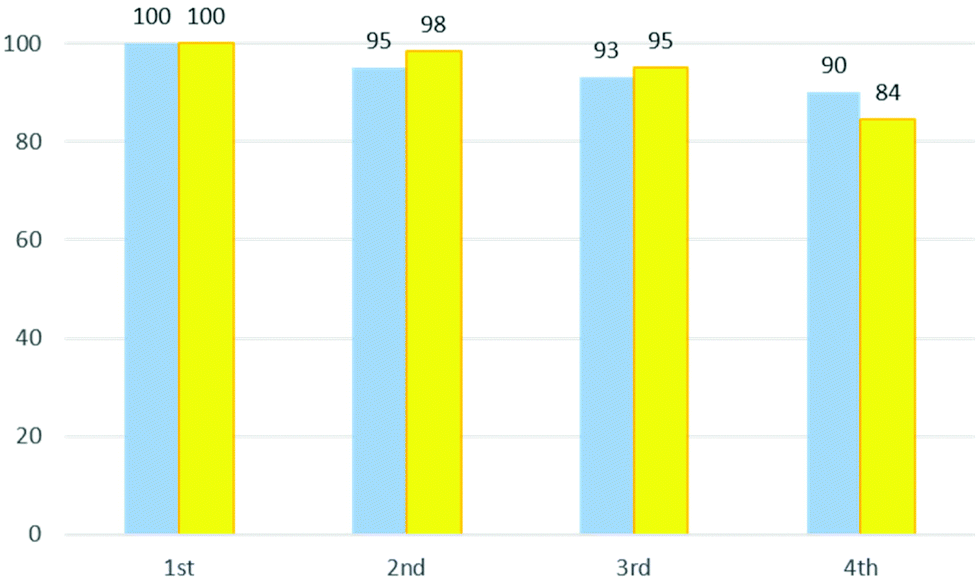 | ||
| Fig. 4 Recovery yield (%) of ChCl/urea for four cycles of the reaction (blue bars) and catalyst performance of DES (%) (yellow bars). | ||
Conclusions
In conclusion, an efficient and straightforward synthesis of symmetric 1,3-dinitropropanes in DES ChCl/urea using MW irradiation was presented in this work. The reaction products were obtained in high yields for several aldehydes, with both nitromethane and nitropropane. The proposed approach appears to be less efficient with ketones, except for activated trifluoromethyl ketones which were converted into the nitroaldol adduct in very high yields. The reaction outcomes suggest a reaction mechanism that involves both choline and urea to explain the high reactivity of the system. The serendipitous finding of excellent selectivity and yields of the tandem Henry reaction/Michael addition under DES-MW conditions represents a mild, efficient and facile synthesis to challenging 1,3-dinitropropane derivatives.Experimental
General information
All reagents and solvents were purchased from commercial sources and used without further purification. The reactions were carried out under atmospheric air.The 1H and 13C NMR spectra were recorded on a Bruker 400 spectrometer (1H NMR, 400 MHz; 13C NMR, 100 MHz). The spectra were recorded at room temperature, unless otherwise indicated, in CDCl3, with tetramethylsilane (TMS, δ = 0.0 ppm) used as the internal standard.
DESs preparation
DESs were prepared according to one of the most used procedures reported in the literature. Briefly, the preparation involved the combination of choline chloride (HBA) with urea or glycolic acid (HBDs), according to the molar ratio reported in Table 4, at 80 °C. This mixture was stirred for 30 min until a homogeneous and transparent solution was formed. The prepared DESs were cooled and used for our solubility tests without any purification. The water content of freshly prepared DES was determined using a Karl Fischer (KF) coulometric titrator from Mettler Toledo and was found to be 0.96% for ChCl/urea, 1.40% for ChCl/GlyA, 0.54% for ChOAc/urea and 0.74% for ChOAc/GlyA.General procedure for Henry reactions
In a typical procedure, the desired DES (10 equiv.) was transferred to a 5 mL vessel. Nitromethane or nitropropane (5 equiv.) and the aldehyde or ketone (1 equiv.) were then added in one portion. The reaction vessel was irradiated with MW and maintained at the indicated temperature for the desired time. At the end of the reaction, the mixture was extracted with dichloromethane (3 × 5 mL). The collected organic phases were washed with water (2 × 15 mL) to eliminate traces of DES. The combined organic phases were dried over Na2SO4 and the solvent was removed under reduced pressure. The resulting crude mixture was dissolved in CDCl3 for 1H-NMR analysis. Conversions were calculated from 1H-NMR on the crude and then converted in yields (as calculated from the total amount of material recovered after reaction work-up).When necessary, the crude was purified by column chromatography (hexane/ethyl acetate 8:2).
Analytical data for compounds 3a–e41 and 542 were in agreement with the literature.
:1 mixture of syn/anti stereoisomers. 1H NMR (CDCl3), A + B isomers: δ(ppm) 7.03 (m, 4H, Ar![[H with combining low line]](https://www.rsc.org/images/entities/char_0048_0332.gif) ), 5.00 (td, J = 10.2, 3.4 Hz, 1H, B), 4.63 (ddd, J = 10.9, 7.8, 2.6 Hz, 1H, A), 4.55 (dt, J = 9.4, 4.7 Hz, 1H, B), 3.95 (t, J = 7.8 Hz, 1H, A), 3.69 (dd, J = 10.5, 4.6 Hz,1H, B), 1.94–1.49 (m, 2H, A,B), 1.34–0.80 (t, J = 7.8 Hz, 3H, A,B). 13C NMR (CDCl3), A + B isomers: δ(ppm) 164.0–161.5 (A + B), 130.5 (A + B), 128.4 (A + B), 116.15 (A + B), 90.64 (A), 89.90–89.34 (B), 51.38 (A), 50.64 (B), 26.12–24.39 (B), 23.58 (A), 10.58 (A), 10.21–9.65 (B). Anal. calcd for C13H17FN2O4: C, 54.92; H, 6.03; F, 6.68; N, 9.85; O, 22.51. Found: C, 54.78; H, 6,12; N, 9,92.
), 5.00 (td, J = 10.2, 3.4 Hz, 1H, B), 4.63 (ddd, J = 10.9, 7.8, 2.6 Hz, 1H, A), 4.55 (dt, J = 9.4, 4.7 Hz, 1H, B), 3.95 (t, J = 7.8 Hz, 1H, A), 3.69 (dd, J = 10.5, 4.6 Hz,1H, B), 1.94–1.49 (m, 2H, A,B), 1.34–0.80 (t, J = 7.8 Hz, 3H, A,B). 13C NMR (CDCl3), A + B isomers: δ(ppm) 164.0–161.5 (A + B), 130.5 (A + B), 128.4 (A + B), 116.15 (A + B), 90.64 (A), 89.90–89.34 (B), 51.38 (A), 50.64 (B), 26.12–24.39 (B), 23.58 (A), 10.58 (A), 10.21–9.65 (B). Anal. calcd for C13H17FN2O4: C, 54.92; H, 6.03; F, 6.68; N, 9.85; O, 22.51. Found: C, 54.78; H, 6,12; N, 9,92.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
G. C. D. thanks Politecnico di Milano for her Inter-Departments PhD scholarship. The authors acknowledge Barbara Corsico Piccolino for help in experimental work and Dr Cameron Weber (School of Chemical Sciences, The University of Auckland, New Zealand) for discussion and revision of the manuscript.Notes and references
-
(a)
N. Ono, The Nitro Group in Organic Synthesis, John Wiley & Sons, Inc., 2001 CrossRef
; (b) G. Rosini and R. Ballini, Synthesis, 1988, 833–847 CrossRef CAS
- L. Henry, Bull. Acad. R. Med. Belg., 1896, 32, 33 Search PubMed
- F. A. Luzzio, Tetrahedron, 2001, 57, 915–945 CrossRef CAS
-
(a) S. Saranya, N. A. Harry, S. M. Ujwaldev and G. Anilkumar, Asian J. Org. Chem., 2017, 6, 1349–1360 CrossRef CAS
-
(a)
T. Marcelli, R. N. S. Van Der Haas, J. H. Van Maarseveen and H. Hiemstra, Angew. Chem., Int. Ed, 2006, 45, 929–931 Search PubMed
- S. E. Milner, T. S. Moody and A. R. Maguire, Eur. J. Org. Chem., 2012, 2012, 3059–3067 CrossRef CAS
- M. Mokhtar, B. F. A. Alhashedi, H. A. Kashmery, N. S. Ahmed, T. S. Saleh and K. Narasimharao, ACS Omega, 2020, 5, 6532–6544 CrossRef CAS PubMed
-
(a) T. Jiang, H. Gao, B. Han, G. Zhao, Y. Chang, W. Wu, L. Gao and G. Yang, Tetrahedron Lett., 2004, 45, 2699–2701 CrossRef CAS
- R. S. Vanna, R. Dahiya and S. Kumar, Tetrahedron Lett., 1997, 38, 5131–5134 CrossRef
-
(a) L. Vaccaro, Eur. J. Org. Chem., 2020, 28, 4273–4283 CrossRef
-
(a) Q. Zhang, K. De Oliveira Vigier, S. Royer and F. Jérôme, Chem. Soc. Rev., 2012, 41, 7108–7146 RSC
-
(a) A. P. Abbott, D. Boothby, G. Capper, D. L. Davies and R. K. Rasheed, J. Am. Chem. Soc., 2004, 126, 9142–9147 CrossRef CAS PubMed
- S. Khandelwal, Y. K. Tailor and M. Kumar, J. Mol. Liq., 2016, 215, 345–386 CrossRef CAS
- C. M. A. Brett, Curr. Opin. Electrochem., 2018, 10, 143–148 CrossRef CAS
- D. V. Wagle, H. Zhao and G. A. Baker, Acc. Chem. Res., 2014, 47, 2299–2308 CrossRef CAS PubMed
- Y. P. Mbous, M. Hayyan, A. Hayyan, W. F. Wong, M. A. Hashim and C. Y. Looi, Biotechnol. Adv., 2017, 35, 105–134 CrossRef CAS PubMed
-
(a) M. Vilková, J. Płotka-Wasylka and V. Andruch, J. Mol. Liq., 2020, 304, 112747 CrossRef
-
(a) H. Xu, J. Peng, Y. Kong, Y. Liu, Z. Su, B. Li, X. Song, S. Liu and W. Tian, Bioresour. Technol., 2020, 310, 123416 CrossRef CAS PubMed
-
(a) S. C. Cunha and J. O. Fernandes, TrAC, Trends Anal. Chem., 2018, 105, 225–239 CrossRef CAS
- B. S. Singh, H. R. Lobo and G. S. Shankarling, Catal. Commun., 2012, 24, 70–74 CrossRef CAS
- X. Tian, S. Zhang and L. Zheng, J. Microbiol. Biotechnol., 2016, 26, 80–88 CrossRef CAS PubMed
-
(a) R. Pedrosa, J. M. Andrés, D. P. Ávila, M. Ceballos and R. Pindado, Green Chem., 2015, 17, 2217–2225 RSC
- X. Ji and H. Huang, Org. Biomol. Chem., 2016, 14, 10557–10566 RSC
- F. C. Escribano, M. P. Derri Alcántara and A. Gómez-Sánchez, Tetrahedron Lett., 1988, 29, 6001–6004 CrossRef
-
(a) R. Ballini, L. Barboni, D. Fiorini, G. Giarlo and A. Palmieri, Chem. Commun., 2005, 2633–2634 RSC
- D. P. Pham-Huu, M. Petrušová, J. N. BeMiller and L. Petruš, Tetrahedron Lett., 1999, 40, 3053–3056 CrossRef CAS
-
(a) R. Ballini, L. Barboni, D. Fiorini, G. Giarlo and A. Palmieri, Green Chem., 2005, 7, 828–829 RSC
- T. Yang, J. X. Chen, Y. Fu, K. Chen, J. He, W. Ye, Z. Sang and Y. Luo, Org. Process Res. Dev., 2014, 18, 511–519 CrossRef CAS
-
(a) P. Bora, P. P. Bora, B. Wahlang and G. Bez, Can. J. Chem., 2017, 95, 1261–1266 CrossRef CAS
- R. Ballini, G. Bosica, D. Fiorini and A. Palmieri, Synthesis, 2004, 1938–1940 CrossRef CAS
- A. Fierro, M. C. Rezende, S. Sepúlveda-Boza, M. Reyes-Parada and B. K. Cassels, J. Chem. Res., 2001, 2001, 294–296 CrossRef
- K. Motokura, M. Tada and Y. Iwasawa, Angew. Chem., Int. Ed., 2008, 47, 9230–9235 CrossRef CAS PubMed
- M. Gao and Y.-P. Wei, J. Chem. Res., 2013, 37, 146–148 CrossRef CAS
- Z. I. Niazimbetova, D. H. Evans, L. M. Liable-Sands and A. L. Rheingold, J. Electrochem. Soc., 2000, 147, 256 CrossRef CAS
-
(a) D. Dallinger and C. O. Kappe, Chem. Rev., 2007, 107, 2563–2591 CrossRef CAS PubMed
-
(a) J. González-Rivera, E. Husanu, A. Mero, C. Ferrari, C. Duce, M. R. Tinè, F. D'Andrea, C. S. Pomelli and L. Guazzelli, J. Mol. Liq., 2020, 300, 112357 CrossRef
-
(a) F. Tur and J. M. Saá, Org. Lett., 2007, 9, 5079–5082 CrossRef CAS PubMed
- C. Gan, X. Chen, G. Lai and Z. Wang, Synlett, 2006, 387–390 CAS
- F. S. Mjalli and O. U. Ahmed, Asia-Pac. J. Chem. Eng., 2016, 11, 549–557 CrossRef CAS
- J. L. Wardell and E. R. T. Tiekink, Acta Crystallogr., Sect. E: Crystallogr. Commun., 2018, 74, 1735–1740 CrossRef CAS PubMed
- K. Tanemura and T. Suzuki, Tetrahedron Lett., 2018, 59, 392–396 CrossRef CAS
- J. Otevrel, D. Svestka and P. Bobal, Org. Biomol. Chem., 2019, 17, 5244–5248 RSC
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d0ob01516d |
| This journal is © The Royal Society of Chemistry 2020 |