
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceReduced graphene oxide–silsesquioxane hybrid as a novel supercapacitor electrode†
Włodzimierz
Czepa
*ab,
Samanta
Witomska
ab,
Artur
Ciesielski
 *bc and
Paolo
Samorì
*c
*bc and
Paolo
Samorì
*c
aFaculty of Chemistry, Adam Mickiewicz University, ul. Uniwersytetu Poznańskiego 8, 61614 Poznań, Poland
bCenter for Advanced Technologies, Adam Mickiewicz University, Uniwersytetu Poznańskiego 10, 61614 Poznań, Poland
cUniversité de Strasbourg, CNRS, ISIS, 8 alleé Gaspard Monge, 67000 Strasbourg, France. E-mail: ciesielski@unistra.fr; samori@unistra.fr
First published on 12th August 2020
Abstract
Supercapacitor energy storage devices recently garnered considerable attention due to their cost-effectiveness, eco-friendly nature, high power density, moderate energy density, and long-term cycling stability. Such figures of merit render supercapacitors unique energy sources to power portable electronic devices. Among various energy storage materials, graphene-related materials have established themselves as ideal electrodes for the development of elite supercapacitors because of their excellent electrical conductivity, high surface area, outstanding mechanical properties combined with the possibility to tailor various physical and chemical properties via chemical functionalization. Increasing the surface area is a powerful strategy to improve the performance of supercapacitors. Here, modified polyhedral oligosilsesquioxane (POSS) is used to improve the electrochemical performance of reduced graphene oxide (rGO) through the enhancement of porosity and the extension of interlayer space between the sheets allowing efficient electrolyte transport. rGO–POSS hybrids exhibited a high specific capacitance of 174 F g−1, power density reaching 2.25 W cm−3, and high energy density of 41.4 mW h cm−3 endowed by the introduction of POSS spacers. Moreover, these electrode materials display excellent durability reaching >98% retention after 5000 cycles.
1. Introduction
The ever-increasing energy demand has triggered an intensive search for sustainable and renewable power sources which has led to the emergence of multiple technological solutions including clean energy conversion and storage devices. In fact, nowadays the most employed energy sources such as fossil fuels, coal, oil, and natural gasses are neither ecofriendly nor everlasting. Fossil fuel burning releases harmful gasses, resulting in continuous deterioration of the global environment. During the past decades, renewable energy sources have become extremely important because of their sustainability and efficiency combined with their pollution-free processability. Among them, supercapacitors (SCs) have received considerable attention as modern portable energy storage devices featuring high power density, ultrafast charge–discharge, long cycling life, high operating durability, and low maintenance.1,2 Since the first demonstration of SC in 1957 by Becker using porous carbon electrodes, much progress has been made in this research area. Albeit a vast number of electrode materials have been reported so far, carbon nanomaterials have attracted the greatest attention due to their unique physicochemical and electrochemical properties. In particular, the use of graphitic carbon nanomaterials yielded an increase in the overall performance of SCs. Moreover, carbonaceous electrodes represent an ecologically friendly alternative for traditional energy storage devices such as batteries.3,4 Considering the mechanism of charge storage, supercapacitors can be classified as electrical double layer capacitors (EDLCs) based on electrostatic adsorption of electrolyte ions onto the surface of electrodes and pseudocapacitors (PCs), additionally supported by faradaic redox reactions. Notably, EDLCs are beneficial especially in terms of higher power density and longer cyclic stability but they exhibit lower power density than PCs.5,6Among all carbon-related materials that can be used as electrodes for EDLCs, graphene possesses the largest surface area (up to 2700 m2 g−1). However, graphene-based supercapacitors exhibit significant quasi-rectangular curves at lower scan rates (1–10 mV s−1) due to their densely packed layered structure, kinetically limiting electrolyte ion penetration at higher scan rates, thereby resulting in overall electrochemical performance decrease.7
Three-dimensional (3D) graphene-based architectures have gathered remarkable attention for EDLC applications owing to their highly accessible surface area, low density, structural interconnectivity (micro-, meso- and macro-interconnected pores), excellent electrical conductivity, and good mechanical features. Overall, the supercapacitance performance of 3D graphene-based SCs has been correlated to the enhanced accessibility of the electrode surface to electrolyte ions, which also provides conductive channels for electron transfer. Such an increase in the dimensionality can be beneficial in terms of the physicochemical properties of hybrid materials.8–10 Therefore, the use of porous scaffolds can be an asset, especially in terms of specific surface area, the presence of the pores with controllable size, and effective charge transport.
Graphene-based materials, which are popular because of their exceptional electrical and mechanical properties, can be produced either via bottom-up or top-down strategies.11–14 Among the various GRMs, graphene oxide (GO) offers multiple advantageous features including good dispersibility in aqueous media and easy chemical functionalization.15,16 Notably, GO exhibits modest electrical properties compared to pristine graphene due to the existence of multiple structural defects in the sp2 lattice combined with the presence of various oxygen-containing groups.17,18 However, GO can be reduced leading to reduced graphene oxide (rGO). The latter, which can have a sheet resistance of a few Ω □−1, can be efficiently used as an electrode in different device types.19 In a simplistic model, the conductivity of graphene-based materials is proportional to the carbon to oxygen ratio. However, other parameters such as geometrical aspects, including sheet orientation and percolation effects, also play a non-negligible role.20–22 A plethora of reduction pathways to restore to a great extent the π-conjugated electronic structure were proposed and widely applied, including chemical, thermal, electrochemical, and microwave-assisted processes.20,23–26 Reduction is a viable approach to generate large quantities of high-quality rGO with moderate oxygen content, while the use of structural scaffolding might support electrochemical processes that require effective ion penetration.27 Chemically reduced GO was previously investigated as a neat component for energy storage applications reaching a moderate capacitance ranging from 67 to 100 F g−1, mainly due to the limited accessibility for electrolyte molecules. The more effective rGO-based electrodes usually exhibit lower cyclability and durability while tested in aqueous media or even require the use of organic electrolytes.28,29
Notably, chemically or thermally reduced graphene oxide sheets, which combine high electrical conductivity and electrochemical stability, are unfortunately prone to undergo a substantial restacking when producing macroscopic assemblies from a liquid suspension. This restacking drastically reduces the material's surface area leading to dramatic decreases in the energy density of the graphene film-based supercapacitors. However, the inhibition of the restacking of graphene sheets can be achieved by the use of molecular spacers to guarantee high specific surface porosity which is required for effective electrolyte penetration.30 A vast number of approaches using the covalent and non-covalent functionalizations of graphene-based materials have been explored leading to superior electrochemical performance.31–33 It was demonstrated that the appropriate selection of functional molecules allows fine-tuning the interlaminar distance between rGO sheets and might result in higher capacitance and improved durability of electrodes.34
Polyhedral oligomeric silsesquioxanes (POSSs) are inorganic molecules comprising a Si–O cage-like core surrounded by organic sites exposing the functional groups.35 By exploiting their well-defined structure, high stability and good solubility, POSSs have been used as attractive functional building blocks to develop nanohybrid materials, in particular when combined with porous carbon structures.36,37 The introduction of silsesquioxanes makes it possible to endow the materials with enhanced durability, rigidity, and higher specific surface area. Moreover, the presence of POSSs can enhance the electrochemical performance of graphene-based materials due to the pseudocapacitive co-contribution and expansion of the interlayer space of tightly packed nanosheets.
Here, we report the modification of GO with OctaAmmonium-POSS followed by the chemical reduction of the hybrid to generate rGO–POSS electrodes for supercapacitors. The latter device exhibited an enhanced specific capacitance of 173 F g−1, thus comparable to the state-of-the-art chemical rGO materials. The introduction of POSS resulted in a porous structure with a specific surface area of 180 m2 g−1, with a moderate pore size of 4 nm allowing effective electrolyte penetration and thereby ensuring effective electrochemical performance and exceptional cyclability. The prepared electrodes exhibited high durability and excellent capacitance retention (>98%) after 5000 cycles. Moreover, the use of molecular spacers provided a high volumetric energy density of 41.4 mW h cm−3. These results provide clear evidence regarding the beneficial role of POSS in improving the electrochemical performance via simple chemical functionalization of rGO.
2. Results and discussion
2.1 Synthesis and characterizations
The synthesis of rGO–POSS was performed by using the condensation reaction between primary amine moieties, which are generated upon the hydrolysis of ammonium chloride on the termini of OctaAmmonium POSS and epoxides present on the GO surface, mainly via the nucleophilic ring opening reaction yielding covalent cross-links of the two components (Fig. 1). Subsequently, the isolated product was chemically reduced with hydrazine to get rid of the excess of oxygen species and non-covalently attached POSS molecules.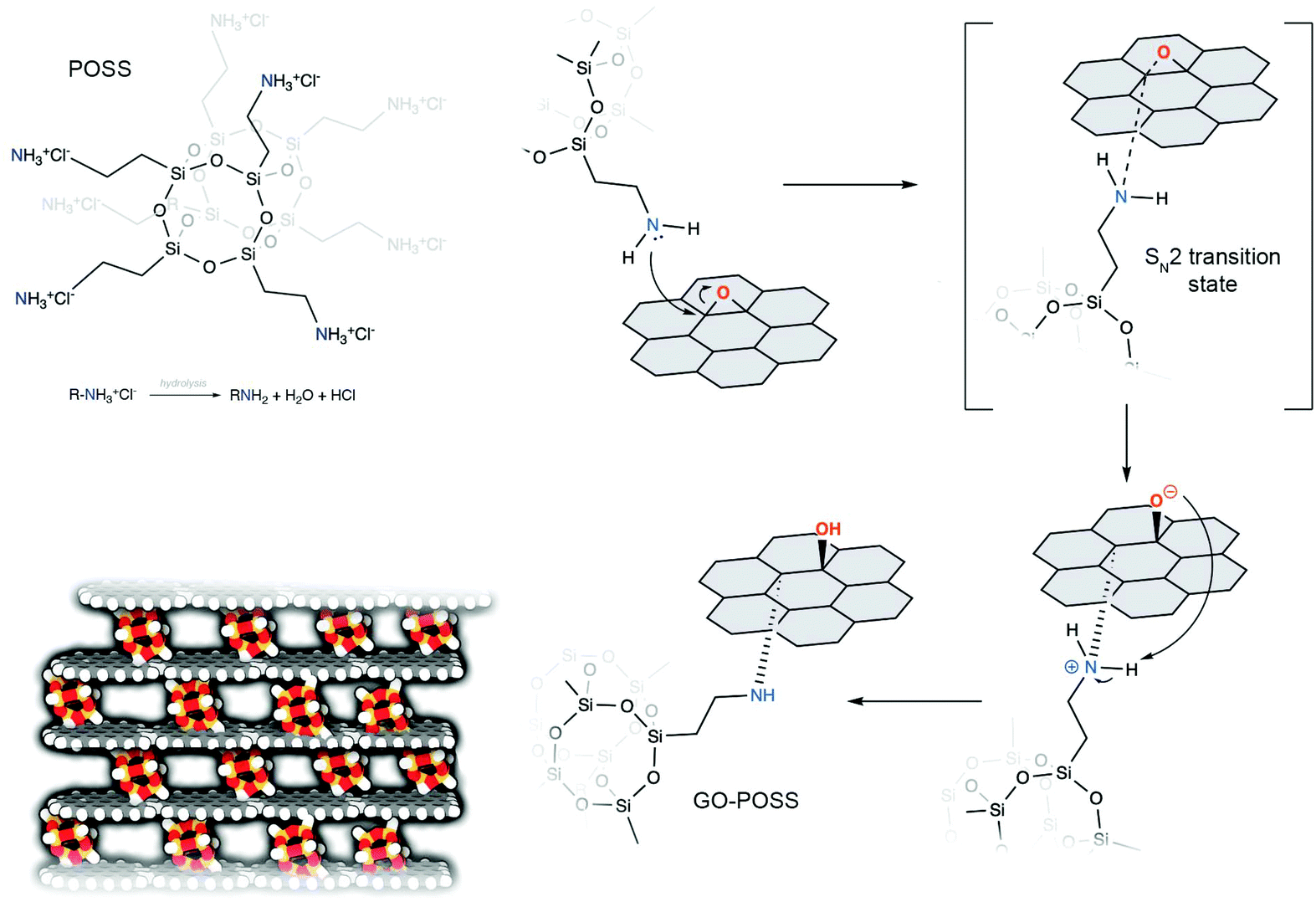 | ||
| Fig. 1 Scheme representing the mechanism of functionalization of GO with OctaAmmonium POSS. | ||
Scanning electron microscopy (SEM) and energy-dispersive X-ray spectroscopy (EDS) analysis made it possible to gain insight into the morphology and elemental distribution in the final material. Fig. 2a reveals that rGO–POSS possesses a folded structure typical of graphene oxide-based hybrids.38–40 Elemental mapping confirms the homogeneous distribution of the elements in the final material i.e. silicon atoms dispersed in the matrix of rGO, thereby excluding the formation of aggregates (Fig. S1†). High-resolution transmission electron microscopy (HR-TEM) was performed to reveal the porosity provided by the evenly spread POSS cages in the matrix of reduced graphene oxide (Fig. 2b). The estimated average POSS loading amounts to 1 POSS for each 10 nm2. The Fourier transform infrared spectroscopy (FT-IR) spectra of GO, POSS, GO–POSS, and rGO–POSS portrayed in Fig. S2† provide evidences for the covalent functionalization of rGO with POSS. The spectra of pristine GO displays typical peaks associated with C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching at 1724 cm−1, CC stretching at 1624 cm−1, C–O stretching of epoxy groups at 1216 and 1054 cm−1, and a broad peak between 3000 and 3500 cm−1 corresponding to hydroxyl species stretching. The functionalization with POSS molecules determined the appearance of a strong absorption peak at 1573 cm−1 corresponding to N–H stretching vibration, and a broad peak at 3276 cm−1 corresponding to N–H vibration of secondary amines suggesting that most of the –NH2 groups took part in covalent bonding to the surface of graphene oxide.41 Moreover, due to hydrazine reduction, we can observe a significant decrease in hydroxyl species around 3500 cm−1. Notably, the decreased intensity of the peaks corresponding to the C–O bonds results from the functionalization and chemical reduction of the final material and was previously reported while cross-linking GO with amine-based functional molecules.42,43 In addition, the asymmetric peaks at 2937 and 2870 cm−1 are assigned to the C–H stretching vibrations of the alkyl group –CH2. The formation of the rGO–POSS composite was further evidenced by the appearance of bands characteristic of siloxane groups i.e. Si–O–Si (1130 cm−1) and Si–C (908 cm−1) coming from the core of the POSS cage.
O stretching at 1724 cm−1, CC stretching at 1624 cm−1, C–O stretching of epoxy groups at 1216 and 1054 cm−1, and a broad peak between 3000 and 3500 cm−1 corresponding to hydroxyl species stretching. The functionalization with POSS molecules determined the appearance of a strong absorption peak at 1573 cm−1 corresponding to N–H stretching vibration, and a broad peak at 3276 cm−1 corresponding to N–H vibration of secondary amines suggesting that most of the –NH2 groups took part in covalent bonding to the surface of graphene oxide.41 Moreover, due to hydrazine reduction, we can observe a significant decrease in hydroxyl species around 3500 cm−1. Notably, the decreased intensity of the peaks corresponding to the C–O bonds results from the functionalization and chemical reduction of the final material and was previously reported while cross-linking GO with amine-based functional molecules.42,43 In addition, the asymmetric peaks at 2937 and 2870 cm−1 are assigned to the C–H stretching vibrations of the alkyl group –CH2. The formation of the rGO–POSS composite was further evidenced by the appearance of bands characteristic of siloxane groups i.e. Si–O–Si (1130 cm−1) and Si–C (908 cm−1) coming from the core of the POSS cage.
 | ||
| Fig. 2 (a) SEM picture of rGO–POSS and (b) HR-TEM investigation of rGO–POSS hybrid material. | ||
X-ray photoelectron spectroscopy (XPS) was exploited to obtain information on the chemical composition of materials by identifying the relevant chemical elements and bonds present in the neat and functionalized GO (see Fig. 3). The C 1s spectra of GO show peaks corresponding to the C–C (284.6 eV), C–O (286.4 eV), CO (287.5 eV), and COOH (288.3 eV) bonds. Comparably, the C 1s spectra of the functionalized GO–POSS appear differently and feature peaks at 284.6 eV, 285.5 eV, 286.4 eV, and 287.5 eV, which can be assigned to C–C, C–N, C–O, and CO bonds, respectively. However, the C 1s spectra of rGO–POSS exhibit a significant decrease of C–O bonds indicating the successful reduction of unreacted oxygen group on the surface of GO-POSS (Fig. 3c). As expected, the high-resolution N 1s spectra of the GO sample does not show the presence of a nitrogen signal. Conversely, signals observed in the GO-POSS sample can be deconvoluted into two peaks, i.e. at 398.9 eV and 401.2 eV, which can be attributed to the secondary (N-sp3 bonds) and protonated primary amines, respectively.27 Notably, the existence of the external –NH3+ groups suggests that only the majority of amines undergo covalent grafting onto GO sheets with the coexistence of electrostatic interactions mostly at the edges of the GO surface and form the ammonium-carboxylate species. Interestingly, the non-covalent grafted molecules were easily removed from the GO-POSS during the reduction step, which was previously reported in the case of functionalization and reduction of graphene-based materials.44 Moreover, the N 1s spectra of rGO–POSS reveal a decrease in the signal corresponding to the protonated primary amines (401.2 eV). In addition, a new peak at 400.2 eV on the N 1s spectra of GO–POSS after hydrazine treatment indicates minor nitrogen inclusion mostly on the edges of the GO sheets forming N-sp2 species.45 Furthermore, determination of the C/N ratio, which amounts to 10.2 and 25.4 for GO–POSS and rGO–POSS, respectively, confirms the removal of non-covalently attached POSS molecules.44
 | ||
| Fig. 3 Comparison of XPS high-resolution C 1s and N 1s spectra of (a and d) GO, (b and e) GO–POSS, and (c and f) rGO–POSS. | ||
Thermogravimetric analysis was performed to investigate the thermal stability and composition of GO, POSS, and rGO–POSS. The GO curve displays about 45% weight loss in the range of 150–300 °C due to oxygen functional group degradation. POSS exhibits good thermal stability up to 340° when the organic aminopropyl-species start to decompose with increasing temperature leading to the formation of amorphous SiO2.46,47 The rGO–POSS curve shows similar mass drops corresponding to organic functional group degradation with the 20% mass residue coming from the POSS Si–O core.
Powder X-ray diffraction investigations were performed to study POSS intercalation into the 3D structure and the structural orientation changes due to functionalization. Wide-angle X-ray scattering (WAXS) results of the pristine GO display only a typical sharp peak at ≈10.01° (see Fig. S3†), corresponding to an interlayer spacing of 0.87 nm due to the (002) reflection of the stacked GO sheets, in accordance with the previously reported value.48,49 Interestingly, after functionalization with POSS, a significant shift of the peak towards lower angle values (7.65°) was observed, the latter corresponding to an increased interlayer spacing of 1.15 nm. The latter can be assigned to the intercalation of POSS particles with sizes of ca. 0.5 nm (core–shell), which vary depending on the substituents.50 Moreover, due to functionalization, a new broad peak at 21.03° coming from POSS is observed. After reduction with hydrazine, a new peak at 27.96° appears suggesting the presence of new crystalline domains, which corresponds to the partially restored graphitic domains and π–π interactions with a significant shift compared to rGO that resulted from the presence of POSS species.44 Moreover, chemical reduction provides the removal of non-covalently attached POSS cages in accordance with XPS results. The irregular intercalation of POSS between graphene sheets results in broadening of the peak towards the 2ϴ range of 6–11° and broad signals around 20° and 30° corresponding to the high-intensity peaks of crystalline POSS itself (Fig. S4†).
Further insights into the morphologies of rGO–POSS were obtained by quantifying the specific surface area (SSA), pore size, and pore distribution with the Brunauer–Emmett–Teller (BET) model, which relies on N2 adsorption desorption. Our hybrid material exhibited a large SSA reaching 180 m2 g−1, which is roughly twice the accessible surface area of pristine GO (Fig. S5†). Notably, the material was finally reduced, a reaction which typically is accompanied by a decrease in the surface area. However, in our case, the presence of mesopores during the functionalization made it possible to maintain the porous structure. The type IV hysteresis loop (Fig. S5b†) confirms the presence of capillary condensation occurring in mesopores with new types of pores of size 4 nm and an average pore volume of 0.54 cm3 g−1, which clearly differ morphologically from the materials used for the synthesis (Table S1†), enabling effective electrolyte transport during the electrochemical measurements.51–53
2.2 Electrochemical study
Cyclic voltammetry (CV) was employed to probe the electrochemical behaviour of the rGO–POSS electrode in aqueous H2SO4 (1 M) electrolyte at a potential window from 0 to 1 V. The working electrode was investigated at scan rates ranging from 1 mV s−1 to 1000 mV s−1 as shown on Fig. 4. At lower scan rate values (1–100 mV s−1), the CV curves display a quasi-rectangular shape confirming effective electrolyte ion transport resulting in good electrochemical performance. This phenomenon can be attributed to the presence of the stable porous structure provided by POSS scaffolding which allows achieving effective capacitance at higher scan rates, which usually drastically drops over 20 mV s−1 for graphene-based materials.54,55 A specific capacitance of 179 F g−1 was obtained at a scan rate of 1 mV s−1 and represents a relatively high result considering that chemically reduced graphene oxide-based materials, including the reference hydrazine-reduced graphene oxide, reach 94 F g−1 (Fig. S10†). In addition, voltamperograms recorded at low scan rates revealed peaks around 0.4 V suggesting the slight co-participation of pseudocapacitance in the electrochemical behaviour of electrode which might be due to the presence of heteroatoms (such as nitrogen, oxygen, and silicon).56,57 With the scan rate increase, an obvious drop in specific capacitance was observed, e.g. 86 F g−1 at 50 mV s−1, sustaining good electrochemical performance (Fig. 5a). Further experiments show a significant drop in specific capacitance comparable to the pristine rGO electrode. Notably, the prepared electrode also exhibited good areal capacitance of 350 mF cm−2 and volumetric capacitance reaching 115 F cm−3. | ||
| Fig. 4 Cyclic voltammetry curves of rGO–POSS electrode at different scan rates (1–1000 mV s−1). | ||
 | ||
| Fig. 5 (a) The dependence of specific capacitance and power density at various scan rates for rGO–POSS. (b) Cyclic stability of electrodes at 100 mV s−1 for rGO–POSS and h-rGO. (c) Comparison of Nyquist plots for rGO–POSS and hydrazine-reduced graphene oxide. | ||
The prepared electrode displayed a power density of 2.25 W cm−3 at an operating voltage of 1 V s−1 (Fig. 5a). Notably, the prepared electrode revealed excellent cyclability: its electrochemical capacitance remains at level over 98% of initial capacitance after 5000 cycles while tested at a scan rate of 100 mV s−1 in aqueous media which is superior when compared to the reference rGO material, which drops to 95% under the same conditions (Fig. 5b, Fig. S9†). Galvanostatic charge–discharge curves at the current density of 1 A g−1 reveal a significant difference in the cycle time for rGO–POSS (290 s) when compared to reference rGO (190 s). Electrochemical impedance spectroscopy (EIS) was employed to gain kinetic information of the electrode materials wherein the data was obtained in the form of Nyquist plots where the real part of impedance was plotted against the imaginary part of the impedance. The Warburg resistance (a slope of 45° region of the plots) of the rGO–POSS electrode was estimated as 1.75 Ohm, indicating fast charge transfer during the energy storage process and good conductivity and better ion transfer between the electrolyte–electrode than that of hydrazine-reduced graphene oxide (Fig. 5c). The volumetric energy density (E) constitutes an important parameter, especially for real applications in electric vehicles and mobile devices. Our electrodes exhibited energy density as high as 41.4 mW h cm−3.
3. Experimental
3.1 Materials
A 4 mg mL−1 aqueous dispersion of graphene oxide (GO) was acquired from Graphenea. Hydrazine monohydrate and absolute ethanol (Sigma Aldrich) were used as received without purification. OctaAmmonium POSS (AM0285) was purchased from Hybrid Plastics Inc. Carbon Black Super P (H30253) was acquired from Alfa Aesar. All the solutions were prepared using Milli-Q water.3.2 Preparation of rGO–POSS hybrid
A batch of rGO–POSS was prepared as follows: a mixture of ethanol (100 ml) and GO (100 ml, 4 mg mL−1) was sonicated for 15 min and added to a solution of POSS (1 g in 200 mL of DI water). The mixture was vigorously stirred overnight under reflux. After 24 hours, the precipitate was filtrated, rinsed several times with water and ethanol and then the obtained GO–POSS was dried under vacuum. 570 mg of GO-POSS was dispersed in water and 570 μL of hydrazine was added and stirred for another 6 hours. The precipitate was then filtered and rinsed with a large amount of water and ethanol. The resulting black powder was then freeze-dried for 24 h under vacuum.3.3 Characterization techniques
FT-IR spectra were studied within the mid-IR range (400–4000 cm−1) using a PerkinElmer spectrometer (Spectrum Two) equipped with an ATR Diamond. X-ray photoelectron spectroscopy (XPS) analyses were carried out using a Thermo Scientific K-Alpha X-ray photoelectron spectrometer with a basic chamber pressure of ∼10−9 mbar and an Al anode as the X-ray source (X-ray radiation of 1486 eV). X-ray powder diffraction (XRD) experiments were conducted on powder specimens using a Bruker ASX D8 Advanced diffractometer equipped with Cu anode and Kα radiation (λ = 1.5418 Å). Diffraction patterns were collected at room temperature in the scattered angular range between 6° and 40° with an angular resolution of 0.02° per step and a typical counting time 4 of 10 s per step. The specific surface area was measured using a Micromeritics ASAP 2050 surface area and porosity analyzer. Prior to the BET measurements, the samples were outgassed for 12 hours at 100 °C. Adsorption isotherms were calculated for nitrogen adsorption at 77 K and pressures up to 1 bar. Scanning electron microscopy (SEM) characterization was carried out by means of an FEI Quanta 250 FEG instrument using energy-dispersive X-ray (EDX) analyses. High-resolution transmission electron microscopy (HR-TEM) was performed with a Hitachi HT7700 transmission electron microscope.3.4 Electrochemical measurements
The working electrode (disc with a diameter of 0.7 cm and a thickness of 30 μm) was prepared by vacuum filtration of rGO–POSS dispersed in ethanol and carbon black (as a conducting additive, 10 wt% ratio of the total mass of the film). After drying, a free-standing pastille was deposited on Au-covered polyethylene terephthalate (PET) substrate by pressing under a pressure of 10 MPa for 24 hours (Fig. S5†). A platinum wire was employed as the counter electrode and an Ag/AgCl electrode in KCl was used as the reference electrode. 1 M H2SO4 aqueous solution was used as the electrolyte for CV measurements. The CV curves were performed under a potential window from 0 to 1 V at different scan rates ranging from 1 to 1000 mV s−1. The capacitance was calculated from the CV curves using the eqn (1):58 | (1) |
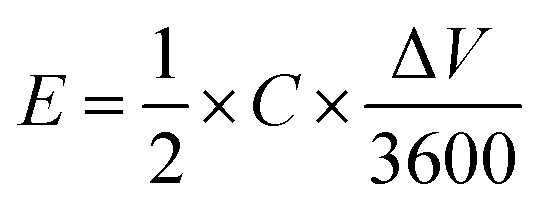 | (2) |
 | (3) |
4. Conclusions
In conclusion, we have demonstrated a facile approach for covalent functionalization of graphene oxide with silsesquioxane (POSS) followed by chemical reduction and exploited the hybrid structure as an electrode for supercapacitors. Due to the introduction of POSS cages and optimization of oxygen content, the final material exhibited a three-dimensional porous structure that was fully characterized in terms of morphology, chemical composition, and electrochemical behaviour. The prepared electrode exhibited high specific, areal, and volumetric capacitances (171 F g−1, 350 mF cm−2, and 115 F cm−3, respectively). The introduction of POSS molecules provided optimal pores allowing efficient electrolyte penetration while the electrochemical processes resulted in excellent performance over a wide range of scan rates. Moreover, the electrode revealed excellent durability and cyclability maintaining >98% capacitance retention after 5000 cycles showing superior electrochemical performance when compared to rGO. This work enriches the current family of modern molecularly engineered graphene-based energy storage materials. Such an approach enables the tuning of a variety of physicochemical properties of the hybrid system towards the generation of high-performing multifunctional electrode materials exhibiting enhanced stability.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the National Science Center (Grant No. 2019/35/B/ST5/01568 and Grant No. 2019/33/N/ST5/00052), co-financed by the European Union through the European Social Fund under the Operational Program Knowledge Education Development (Grant No. POWR.03.02.00-00-I026/16) and the EC–Graphene Flagship Core 3 project (GA-881603), the Agence Nationale de la Recherche through the Labex project CSC (ANR-10-LABX-0026 CSC) within the Investissement d'Avenir program (ANR-10-120 IDEX-0002-02) and the International Center for Frontier Research in Chemistry (icFRC). S.W. acknowledges the support from the Foundation for Polish Science (FNP) in the form of a scholarship.Notes and references
- F. X. Wang, X. W. Wu, X. H. Yuan, Z. C. Liu, Y. Zhang, L. J. Fu, Y. S. Zhu, Q. M. Zhou, Y. P. Wu and W. Huang, Chem. Soc. Rev., 2017, 46, 6816–6854 RSC.
- N. Choudhary, C. Li, J. Moore, N. Nagaiah, L. Zhai, Y. Jung and J. Thomas, Adv. Mater., 2017, 29, 30 Search PubMed.
- Z. Liu, H. Zhang, M. Eredia, H. Qiu, W. Baaziz, O. Ersen, A. Ciesielski, M. Bonn, H. I. Wang and P. Samorì, ACS Nano, 2019, 13, 9431–9441 CrossRef CAS.
- Z. P. Song and H. S. Zhou, Energy Environ. Sci., 2013, 6, 2280–2301 RSC.
- Z. N. Yu, L. Tetard, L. Zhai and J. Thomas, Energy Environ. Sci., 2015, 8, 702–730 RSC.
- C. Zhong, Y. D. Deng, W. B. Hu, J. L. Qiao, L. Zhang and J. J. Zhang, Chem. Soc. Rev., 2015, 44, 7484–7539 RSC.
- Z. B. Lei, L. Lu and X. S. Zhao, Energy Environ. Sci., 2012, 5, 6391–6399 RSC.
- H. Ren, H. Tian, C. L. Gardner, T.-L. Ren and J. Chae, Nanoscale, 2016, 8, 3539–3547 RSC.
- J. Y. Chen, P. Xie and Z. P. Zhang, Chem. Eng. J., 2019, 361, 615–624 CrossRef CAS.
- F. Wang, Z. Hu, L. Mao and J. Mao, J. Power Sources, 2020, 450, 227692 CrossRef CAS.
- C. Moreno, M. Vilas-Varela, B. Kretz, A. Garcia-Lekue, M. V. Costache, M. Paradinas, M. Panighel, G. Ceballos, S. O. Valenzuela, D. Pena and A. Mugarza, Science, 2018, 360, 199–203 CrossRef CAS.
- H. J. Jiang, Small, 2011, 7, 2413–2427 CAS.
- A. Ciesielski and P. Samori, Chem. Soc. Rev., 2014, 43, 381–398 RSC.
- V. Eswaraiah, S. S. J. Aravind and S. Ramaprabhu, J. Mater. Chem., 2011, 21, 6800–6803 RSC.
- D. R. Dreyer, S. Park, C. W. Bielawski and R. S. Ruoff, Chem. Soc. Rev., 2010, 39, 228–240 RSC.
- Y. Zhu, S. Murali, W. Cai, X. Li, J. W. Suk, J. R. Potts and R. S. Ruoff, Adv. Mater., 2010, 22, 3906–3924 CrossRef CAS.
- S. H. Dave, C. Gong, A. W. Robertson, J. H. Warner and J. C. Grossman, ACS Nano, 2016, 10, 7515–7522 CrossRef CAS.
- C. Punckt, F. Muckel, S. Wolff, I. A. Aksay, C. A. Chavarin, G. Bacher and W. Mertin, Appl. Phys. Lett., 2013, 102, 023114 CrossRef.
- S. J. Rowley-Neale, E. P. Randviir, A. S. Abo Dena and C. E. Banks, Appl. Mater. Today, 2018, 10, 218–226 CrossRef.
- C. K. Chua and M. Pumera, Chem. Soc. Rev., 2014, 43, 291–312 RSC.
- S. Eigler, S. Grimm, M. Enzelberger-Heim, P. Müller and A. Hirsch, Chem. Commun., 2013, 49, 7391–7393 RSC.
- M. J. Fernández-Merino, L. Guardia, J. I. Paredes, S. Villar-Rodil, P. Solís-Fernández, A. Martínez-Alonso and J. M. D. Tascón, J. Phys. Chem. C, 2010, 114, 6426–6432 CrossRef.
- S. Pei and H.-M. Cheng, Carbon, 2012, 50, 3210–3228 CrossRef CAS.
- X. Gao, J. Jang and S. Nagase, J. Phys. Chem. C, 2010, 114, 832–842 CrossRef CAS.
- D. Voiry, J. Yang, J. Kupferberg, R. Fullon, C. Lee, H. Y. Jeong, H. S. Shin and M. Chhowalla, Science, 2016, 353, 1413–1416 CrossRef CAS.
- L. G. Guex, B. Sacchi, K. F. Peuvot, R. L. Andersson, A. M. Pourrahimi, V. Ström, S. Farris and R. T. Olsson, Nanoscale, 2017, 9, 9562–9571 RSC.
- S. Park, J. An, J. R. Potts, A. Velamakanni, S. Murali and R. S. Ruoff, Carbon, 2011, 49, 3019–3023 CrossRef CAS.
- P. K. Jha, S. K. Singh, V. Kumar, S. Rana, S. Kurungot and N. Ballav, Chem, 2017, 3, 846–860 CAS.
- C. Zhao, K. Shu, C. Wang, S. Gambhir and G. G. Wallace, Electrochim. Acta, 2015, 172, 12–19 CrossRef CAS.
- H. J. Kim, S.-Y. Lee, L. H. Sinh, C. S. Yeo, Y. R. Son, K. R. Cho, Y. Song, S. Ju, M. K. Shin, S.-J. Park and S. Park, J. Power Sources, 2017, 346, 113–119 CrossRef CAS.
- H. H. Shi, S. Jang, A. Reza-Ugalde and H. E. Naguib, ACS Appl. Energy Mater., 2020, 3, 987–997 CrossRef CAS.
- Z. Lei, N. Christov and X. S. Zhao, Energy Environ. Sci., 2011, 4, 1866–1873 RSC.
- V. H. Luan, H. N. Tien, L. T. Hoa, N. T. M. Hien, E.-S. Oh, J. Chung, E. J. Kim, W. M. Choi, B.-S. Kong and S. H. Hur, J. Mater. Chem. A, 2013, 1, 208–211 RSC.
- H. Kang, C. Zhang, Y. Xu, W. Zhang, J. Jiao, Z. Li, L. Zhu and X. Liu, RSC Adv., 2020, 10, 1507–1513 RSC.
- K. Tanaka and Y. Chujo, J. Mater. Chem., 2012, 22, 1733–1746 RSC.
- S. K. Yadav, S. S. Mahapatra, H. J. Yoo and J. W. Cho, Nanoscale Res. Lett., 2011, 6, 122 CrossRef.
- Y. Xue, Y. Liu, F. Lu, J. Qu, H. Chen and L. Dai, J. Phys. Chem. Lett., 2012, 3, 1607–1612 CrossRef CAS.
- D. Pakulski, W. Czepa, S. Witomska, A. Aliprandi, P. Pawluć, V. Patroniak, A. Ciesielski and P. Samorì, J. Mater. Chem. A, 2018, 6, 9384–9390 RSC.
- P. M. Sudeep, T. N. Narayanan, A. Ganesan, M. M. Shaijumon, H. Yang, S. Ozden, P. K. Patra, M. Pasquali, R. Vajtai, S. Ganguli, A. K. Roy, M. R. Anantharaman and P. M. Ajayan, ACS Nano, 2013, 7, 7034–7040 CrossRef CAS.
- Y. Qin, J. Yuan, J. Li, D. Chen, Y. Kong, F. Chu, Y. Tao and M. Liu, Adv. Mater., 2015, 27, 5171–5175 CrossRef CAS.
- T. Kolev, M. Spiteller and B. Koleva, Amino Acids, 2010, 38, 45–50 CrossRef CAS.
- D. M. El-Gendy, N. A. A. Ghany, E. E. F. El Sherbini and N. K. Allam, Sci. Rep., 2017, 7, 43104 CrossRef CAS.
- H.-L. Ma, H.-B. Zhang, Q.-H. Hu, W.-J. Li, Z.-G. Jiang, Z.-Z. Yu and A. Dasari, ACS Appl. Mater. Interfaces, 2012, 4, 1948–1953 CrossRef CAS.
- C.-B. Huang, S. Witomska, A. Aliprandi, M.-A. Stoeckel, M. Bonini, A. Ciesielski and P. Samorì, Adv. Mater., 2019, 31, 1804600 CrossRef.
- S. Park, Y. Hu, J. O. Hwang, E.-S. Lee, L. B. Casabianca, W. Cai, J. R. Potts, H.-W. Ha, S. Chen, J. Oh, S. O. Kim, Y.-H. Kim, Y. Ishii and R. S. Ruoff, Nat. Commun., 2012, 3, 638 CrossRef.
- O. Bianchi, G. B. Repenning, L. B. Canto, R. S. Mauler and R. V. B. Oliveira, Polym. Test., 2013, 32, 794–801 CrossRef CAS.
- I. Penso, E. A. Cechinatto, G. Machado, C. Luvison, C. H. Wanke, O. Bianchi and M. R. F. Soares, J. Non-Cryst. Solids, 2015, 428, 82–89 CrossRef CAS.
- S. Chen, J. Zhu, X. Wu, Q. Han and X. Wang, ACS Nano, 2010, 4, 2822–2830 CrossRef CAS.
- S. Witomska, Z. Liu, W. Czepa, A. Aliprandi, D. Pakulski, P. Pawluć, A. Ciesielski and P. Samorì, J. Am. Chem. Soc., 2019, 141, 482–487 CrossRef CAS.
- A. Strąkowska, S. Członka and K. Strzelec, Polymers, 2019, 11, 1092 CrossRef.
- J. H. Bae, J.-H. Han and T. D. Chung, Phys. Chem. Chem. Phys., 2012, 14, 448–463 RSC.
- B. Moyo, D. Momodu, O. Fasakin, A. Bello, J. Dangbegnon and N. Manyala, J. Mater. Sci., 2018, 53, 5229–5241 CrossRef CAS.
- H. Lu, K. Kim, Y. Kwon, X. Sun, R. Ryoo and X. S. Zhao, J. Mater. Chem. A, 2018, 6, 10388–10394 RSC.
- A. Sumboja, C. Y. Foo, X. Wang and P. S. Lee, Adv. Mater., 2013, 25, 2809–2815 CrossRef CAS.
- Z. Lei, L. Lu and X. S. Zhao, Energy Environ. Sci., 2012, 5, 6391–6399 RSC.
- C. Romanitan, P. Varasteanu, I. Mihalache, D. Culita, S. Somacescu, R. Pascu, E. Tanasa, S. A. V. Eremia, A. Boldeiu, M. Simion, A. Radoi and M. Kusko, Sci. Rep., 2018, 8, 9654 CrossRef.
- Y. J. Oh, J. J. Yoo, Y. I. Kim, J. K. Yoon, H. N. Yoon, J.-H. Kim and S. B. Park, Electrochim. Acta, 2014, 116, 118–128 CrossRef CAS.
- M. D. Stoller, S. Park, Y. Zhu, J. An and R. S. Ruoff, Nano Lett., 2008, 8, 3498–3502 CrossRef CAS.
- Z. S. Wu, K. Parvez, X. Feng and K. Müllen, Nat. Commun., 2013, 4, 2487 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: SEM/EDX picture, FT-IR spectra, XRD patterns, BET isotherms, charge–discharge curves, stability investigations, and comparison of rGO-based electrode properties. See DOI: 10.1039/d0nr05226d |
| This journal is © The Royal Society of Chemistry 2020 |