
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
An efficient carbon-based ORR catalyst from low-temperature etching of ZIF-67 with ultra-small cobalt nanoparticles and high yield†
Jian
Guo
 a,
Srinivas
Gadipelli
*ab,
Yuchen
Yang
a,
Zhuangnan
Li
a,
Yue
Lu
a,
Dan J. L.
Brett
b and
Zhengxiao
Guo‡
*a
a,
Srinivas
Gadipelli
*ab,
Yuchen
Yang
a,
Zhuangnan
Li
a,
Yue
Lu
a,
Dan J. L.
Brett
b and
Zhengxiao
Guo‡
*a
aDepartment of Chemistry, University College London, 20 Gordon Street, London WC1H 0AJ, UK. E-mail: z.x.guo@ucl.ac.uk; gsrinivasphys@gmail.com
bElectrochemical Innovation Lab, Department of Chemical Engineering, University College London, London WC1E 7JE, UK
First published on 30th January 2019
Abstract
A facile and controllable low-temperature (450 °C) route is reported to produce ultra-small Co3O4/Co nanoparticles in nitrogen-doped hyperporous graphenic networks (Co3O4/Co@N-G-450). Firstly, a monolayer of ZIF-67 nanocrystals is directly grown in thermal-shock exfoliated graphene networks (EGO) of suitable porosity and pore-widths. Later, the ZIF-67 is etched by targeting the small concentrations of residual oxygen functionalities on EGO (≈13 atom%) under a nitrogen atmosphere at 450 °C. Therefore, the partial gasification of ZIF-67 followed by oxidation of the resultant open cobalt metal centres produces a highly active nanophase of Co3O4/Co@N-G in a mass yield of >65 wt%. The as-synthesised Co3O4/Co@N-G-450 catalyst, without any further acid washing or oxidation process, exhibits an outstanding ORR performance with a high onset (0.962 V vs. RHE) and half-wave (0.808 V vs. RHE) potential as well as limiting current density (5.2 mA cm−2) in 0.1 M KOH solution. These merits are comparable to those of commercial Pt/C and many ZIF-derived catalysts, synthesised under extended and complex chemical treatment. Moreover, the catalyst also exhibits fast reaction kinetics with a dominant 4-electron reaction pathway and high durability.
Fuel cells and metal–air batteries are promising power sources for zero/low-emission vehicles, drones and wearable microelectronic devices owing to their high energy conversion efficiency, theoretical energy density and reliability.1 Currently, one of the main challenges hindering their widespread application resides in developing high performance and cost-effective oxygen reduction reaction (ORR) electrocatalysts. Numerous studies demonstrate that heteroatom-doped porous carbons and their metal/oxide nanoparticle grafted analogues have the potential to replace the Pt-based noble-metal catalyst, Pt/C.2–6 For example, numerous carbon catalysts have been designed by utilizing biomass, carbon nanotubes (CNTs), graphene-oxide, metal–organic frameworks (MOFs) and polymers as precursors. These carbon catalysts show very different structure–activity properties due to the related structural characteristics. Here, for instance, the nature of the carbon skeleton/substrate (i.e., amorphous, graphitic and porosity) plays a critical role in achieving the desired functionalised surfaces to promote their overall catalytic activity. Moreover, the concentration of active sites/centres, such as dopants and metal/-oxides in the carbons varies significantly with respect to the starting precursor and processing method. Thus, these systems pose a great challenge in understanding for further developments, particularly for a generalized system-based process, viable for mass production and implementation of Pt-free fuel cells, metal–air batteries and electrolysers. The development of advanced synthesis methods with well-controllable structural features is therefore highly desirable.
Well-defined porous precursors, such as MOFs are highly attractive and can be designed to have the appropriate amounts of heteroatoms and metal centres.3,6–9 MOFs with their ordered porous structures are gaining tremendous attention as multifunctional materials for many green energy applications, including molecular adsorption and catalysis.10–14 Furthermore, MOFs can be tailored to develop an efficient electrocatalyst.3,4,6,15,16 A microporous carbon framework consisting of nitrogen dopant coordinated metal centers, i.e., the M–N–C (where M is a 3d transition metal/-oxide or combination of metals/-oxides, Mn, Fe, Co, and Ni) system, is proposed to be an ideal ORR candidate.4,17–19 Carbonization of MOFs can generate simultaneous incorporation of intrinsic ligand heteroatoms and metal centres within the carbon matrix. The calcination temperature and atmosphere also play a key role in designing catalyst derivatives.15,20,21 Accordingly, ZIF-67 (zeolitic imidazolate framework-67) with its intrinsic microporosity and imidazolate (C3H3N2) ligand heteroatoms (C and N) and Co metal centres is an ideal precursor to develop the M–N–C catalyst system through a suitable thermolysis route. During this process the framework undergoes complete structural transformation through decomposition followed by rearrangement of the ligands carbon and nitrogen with embedded cobalt-complexes. However, in the literature these ZIF-derived carbon structures are far from the ideal system, commonly due to inadequate processing methods. The carbonization of ZIFs is frequently carried out at elevated temperature, beyond 600 °C and up to 1100 °C.3,4,16,20–28 This carbonization temperature is a critical parameter to control the respective elemental concentration in the targeted M–N–C system. The high-temperature treatment of the ZIF precursors usually induces a low catalyst yield with a mass loss of >70 wt%, mostly due to the dissociation of the highly desirable active –N–C, or hydrocarbon content.24,29 This also leads to a high concentration of inherent metal in the final product, which can amount to over 45 wt% in the case of ZIF-67.21,24 Moreover, the thermally induced reduction of metal cations followed by severe aggregation, and associated metal catalysed graphitization of ligand carbon, significantly reduces the porosity and mass activity of the final structures. Thus, often a post-synthesis acid washing procedure is required to remove the large metal clusters and excessive free metal in the carbon. Nevertheless, simply washing out the cobalt metal (about 30 wt% cobalt mass loss) is a great waste of the resources.20,24,30–34 Furthermore, a second oxidation step is necessary in order to achieve a metal-oxide or oxide-shell at the metal core in the system.35–37
In another approach, to mitigate this problem or for efficient use of inherent nitrogen and metal centres, the hybrid structures have been developed by using extra carbon substrates, such as graphene-oxide (GO) or reduced GO (rGO).29–31,38–43 For instance, Zhong et al. reported a nitrogen-doped carbon system by carbonization of ZIF-8/GO at 950 °C.29 It is worth noting that the precursors exhibit a significant mass loss with a catalyst yield of less than 30 wt%.29 Since the MOFs/ZIFs can be calcined at as low as 350 °C in ambient air, the high concentration of thermally decomposed oxygen functional groups (CO/CO2/H2O) from GO can cause significant gasification of the ZIF ligand, in the form of CO/CO2,44–46 at the expense of the mass yield of the catalyst. Furthermore, owing to the low specific surface area of GO (less than 100 m2 g−1),44 with its packed layer-structure, it is difficult to achieve a homogeneous hybrid system from the surface grown MOF/ZIF crystals.30,38,40,47 However, the derivatives of the ZIF@rGO framework with increased surface area can boost the overall activity by facilitating the accessibility and distribution of reactants and products.48
In principle, thermal-shock exfoliated GO (EGO) would be an ideal substrate, as it not only exhibits a high specific surface area (≈700 m2 g−1) but also has a flexible pore structure with an extensive network of hierarchical meso/macropores (up to 6 cm3 g−1; with sizes between 5 and 200 nm), for pore-confined growth of ZIF nanocrystals.44,45 Moreover, the lower oxygen content (≈13 atom%), with highly concentrated defects as anchoring sites can promote homogeneous crystal nucleation within its hyperporous nanospaces. The thermolysis of ZIFs/MOFs at mild temperatures before their full decomposition or carbonization at >600 °C has yielded a defect-rich active structure by preserving their intrinsic pore structure with highly exposed (open) metal centres from the partly decomposed framework links at a minimal loss of the sample.10,11 Therefore, such ZIF@EGO is expected to produce an active ORR catalyst in high mass yield at mild thermolysis temperatures, ≤500 °C. It is well known that the ORR performance of carbon-based catalysts is mainly determined by the exposed active sites, mass transport and electron transfer.6
Hence, to realise this, for the first time, we demonstrate the in situ growth of ZIF-67 nanocrystals at a high concentration within the nanospaces of hyperporous graphenic networks (ZIF-67@EGO) (Fig. 1). Firstly, the precursor GO is produced by a modified Hummers method followed by thermal-shock exfoliation at 400 °C for ≈5 minutes (see the Experimental section). Subsequently, the ZIF-67@EGO is synthesised by homogenising EGO with the cobalt and imidazole precursor containing methanol solutions under continuous shaking overnight at room temperature. The scanning and transmission electron microscopy (SEM and TEM) as well as powder X-ray diffraction (PXRD) and Fourier-transform infrared spectroscopy (FTIR) reveal that the polyhedral ZIF-67 crystals with an average size around 30–40 nm are uniformly decorated in the networked graphene sheets (Fig. 1a–f and S1–S3†). This is further revealed using atomic force microscopy (Fig. S3†). The structural characteristics and mass tracking before and after synthesis confirm a ZIF loading of 60 wt% at EGO (Fig. 1g, h and S4†). It is worth noting that there is no free ZIF-67 (free of EGO substrate) detected under such controlled synthesis conditions. The surface oxygen functional groups and defect sites in the EGO can act as anchoring sites for ZIF crystals to nucleate and grow on its surface.29,30,48 However, here a controlled synthesis, i.e., controlling the concentration of precursors is important. Any excess ZIF growth leads to free and agglomerated particles due to the second or multilayer formation to reduce the EGO surface interaction. The sample with an increased concentration of ZIF at EGO (for a loading of 80 wt%) shows surface free ZIF agglomerated crystals (Fig. S5†). The hybrid exhibits a combined highly hierarchical accessible porosity of abundant micropores from ZIF-67 and meso- and macropores from EGO (Fig. 1h, S6 and S7†).
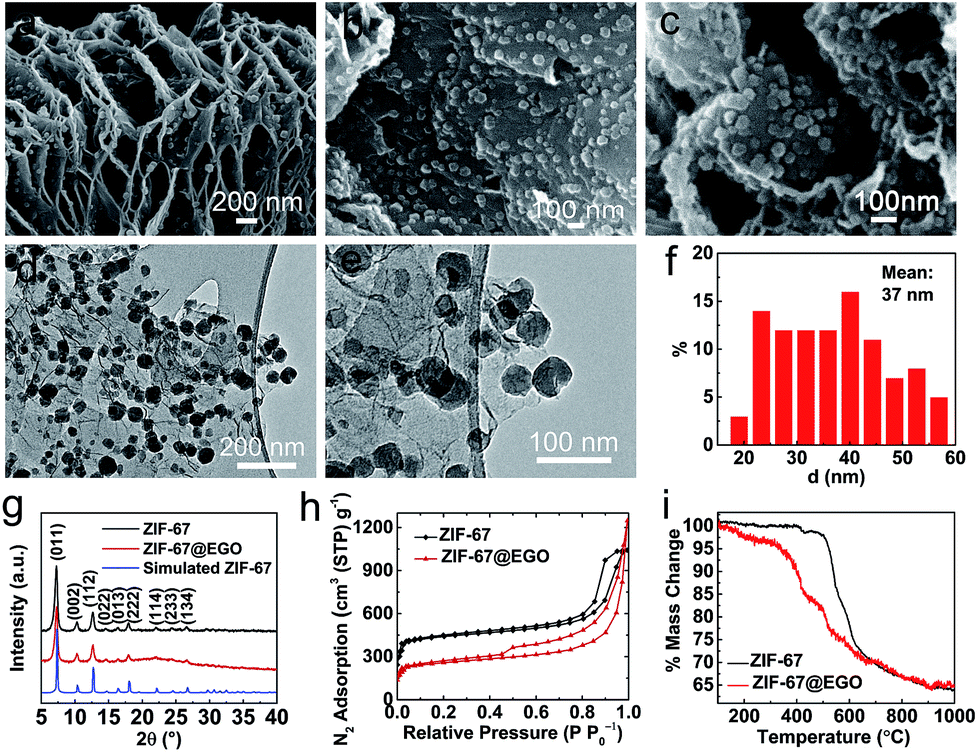 | ||
| Fig. 1 Structural characteristics of the as-synthesised hybrid, ZIF-67@EGO: (a–c) SEM and (d and e) TEM images; (f) particle size distribution of ZIF-67 on the surface of EGO; (g) PXRD patterns; (h) porosity, N2 adsorption–desorption isotherms and (i) TG curves of ZIF-67 and ZIF-67@EGO. | ||
The combined thermogravimetry and mass-spectrometry (TG-MS) of ZIF-67 and ZIF-67@EGO reveal further information necessary for developing efficient hybrid catalyst systems (Fig. 1i and S8†). Compared with pristine ZIF-67, ZIF-67@EGO exhibits different mass-loss behaviour – partly due to the dissociation of residual oxygen groups on the EGO (at >200 °C) followed by further oxidation associated gasification of ZIF-67 (at ≥350 °C).39,46 This partial decomposition of the ligand leads to breakdown of the ZIF framework structure to create active unsaturated cobalt metal centres (also called ‘open metal’ centres). Thus, this active nanophase readily reacts with the outgoing residual oxygen fragments on the EGO to form surface oxidised cobalt centres. This phase transition process strongly depends on the liberated oxygen functional groups from the EGO and thermolysis temperature. It should be noted that the actual framework and ligand decomposition of the ZIF-67 (without the EGO) occurs at >500 °C, and thus most of the literature uses relatively high carbonization temperatures.11,21,24 Further to this, the literature reveals that such elevated thermolysis temperatures lead to severe metal aggregation, with a significant loss of active nitrogen content and mass yield of the catalyst.24,38,41
In contrast, as discussed below, this newly developed hybrid, the ZIF-67@EGO sample with the above characteristic transformative insights into the optimised catalyst derivative, Co3O4/Co@N-G-450, is designed by carrying out thermolysis in the mild temperature region of 400–500 °C. The synthesis at different temperatures is carried out in a tube furnace under a nitrogen atmosphere (see the Experimental section). The mass tracking of the precursor and product before and after thermolysis at 450 °C, respectively accounts for a high mass yield of ≈65 wt%. The as-produced samples are investigated directly for all of the characterization techniques, including electrochemical tests, without any further chemical treatment, such as acid treatment or secondary oxidation and/or heat treatment.20,30,36 Detailed insight into the structural transformation with respect to the thermolysis temperature is obtained by electron microscopy (Fig. 2a–f and S9†). For instance, at 400 °C, the ZIF-67@EGO sample exhibits a polyhedral ZIF-67 structure with a slight reduction of the average size induced by the partial gasification (as detailed in Fig. 1). A considerable morphological change and full fragmentation of ZIF-67 can be seen for the sample produced at 450 °C. The mean size of the nanoparticles is now only 5 nm and they are finely distributed in the graphene networks. High-resolution TEM (HR-TEM) and elemental mapping clearly demonstrate the uniform distribution of C, N, O and Co elements (Fig. 2g–m). It is worth noting that the two main phases of the dispersed nanoparticles are metallic Co and Co3O4. Further growth of these nanoparticles with a mean size of 22 nm is observed when the synthesis temperature is increased to 500 °C. All these observations suggest that full fragmentation of ZIF-67 occurs to produce ultra-small Co3O4/Co metal nanoparticles at around 450 °C. After this point, the increase of the thermolysis temperature can cause more dissociation of hydrocarbon and Co–N linker coordination. This induces a further oxidation of cobalt, and rapid metal migration and crystallinity, thus eventually leading to a bigger size of nanoparticles.21,24,41
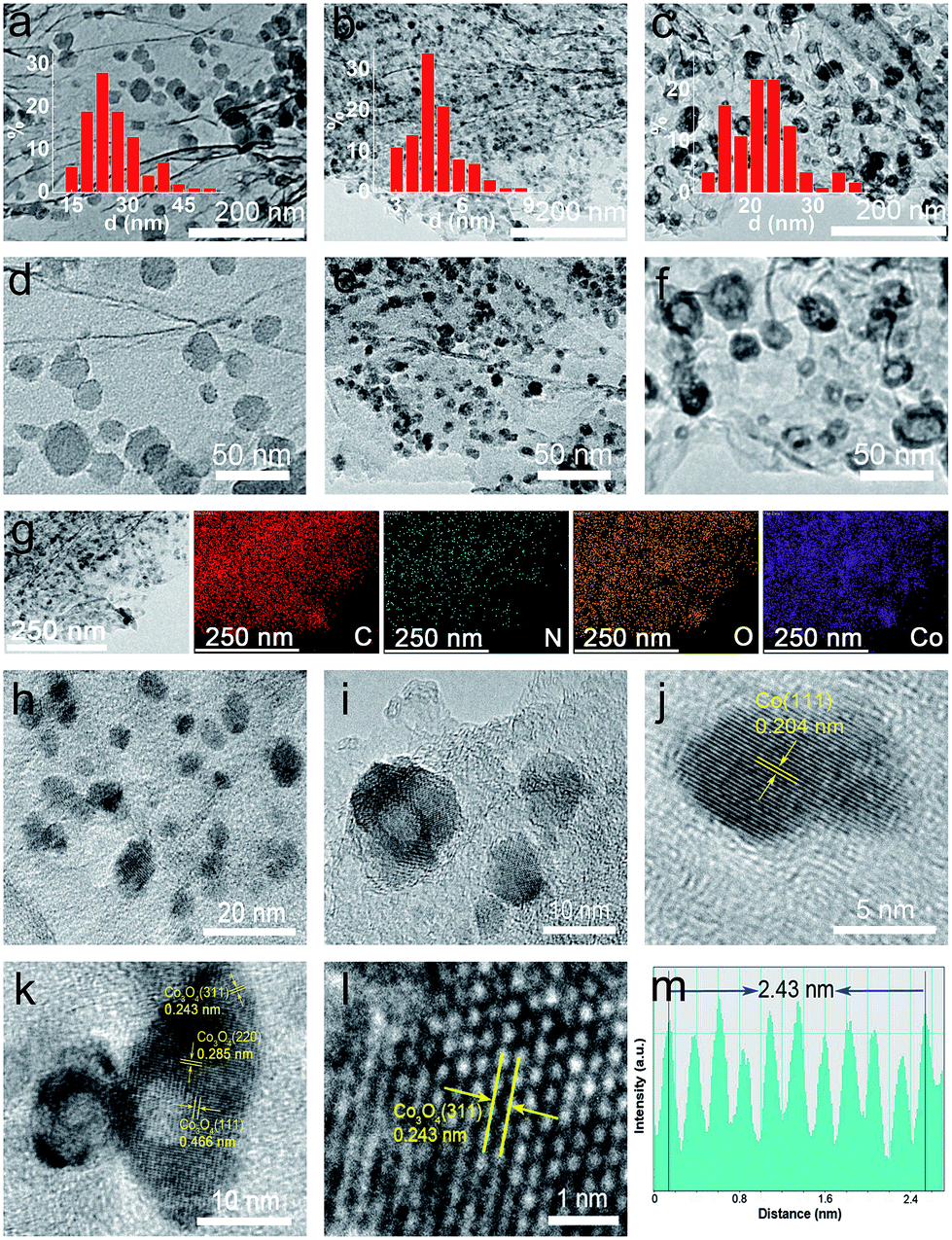 | ||
| Fig. 2 TEM images of ZIF-67@EGO derived products at 400 °C (a and d), 450 °C (b and e), and 500 °C (c and f). The inset shows a corresponding particle size distribution of ZIF-67 and newly developed Co3O4/Co nanoparticles in the graphene networks (corresponding mean sizes are 26 nm (a), 5 nm (b) and 22 nm (c)). TEM characterization of Co3O4/Co@N-G-450: (g) TEM and corresponding elemental mapping images, (h and i) HR-TEM images, and (j) HR-TEM images of a single Co nanoparticle and (k) Co3O4 nanoparticle, (l) HR-TEM images of the Co3O4 phase and (m) the corresponding lattice spacing line profile. | ||
Further to the above microscopic analysis, the structural transitions are evaluated by diffraction and spectroscopy (Fig. 3). The sample produced at 400 °C, as the on-set decomposition point, preserves the majority of the ZIF-67 crystallinity and framework structure. This is evidenced by characteristic diffraction peaks and metal–ligand (Co–N) and/or ligand (C–N) bond IR active vibrational modes at ≈424 cm−1, ≈1566 cm−1 and ≈755 cm−1, respectively (Fig. 3a and b). There is no identifiable evidence for cobalt-oxide (CoO or Co3O4) at this stage (PXRD and Raman). In good agreement with the electron microscopy, the sample thermolysed at 450 °C shows complete framework decomposition of ZIF-67, as observed by the disappearance of corresponding diffraction and IR peaks. Under such conditions, the product is mostly amorphous in nature, but a new set of weak and broad diffraction peaks emerge at ≈36.8° and ≈44.2° of 2θ, which can be attributed to the (311) plane of the cubic spinel structure of Co3O4 and the (111)/(200) plane of the fcc Co/CoO in their nanophase (Fig. 3a). The development of cobalt and cobalt-oxide phases is clearly seen in the sample derived at 500 °C. The diffraction peaks are well resolved for three components of Co, CoO and Co3O4, due to the increased oxidation, crystallinity and particle size, as observed by TEM. The new twin IR absorption peaks at ≈660 cm−1 and ≈563 cm−1 (originating from the Co–O vibration in Co3O4) and the new Raman peaks located at ≈193 cm−1, ≈474 cm−1, ≈519 cm−1, ≈617 cm−1 and ≈683 cm−1 (assigned to the F32g, Eg, F22g, F12g, and A1g vibration modes of Co3O4) are in good agreement with the PXRD data to suggest that Co3O4 is the dominant phase (Fig. 3c).39
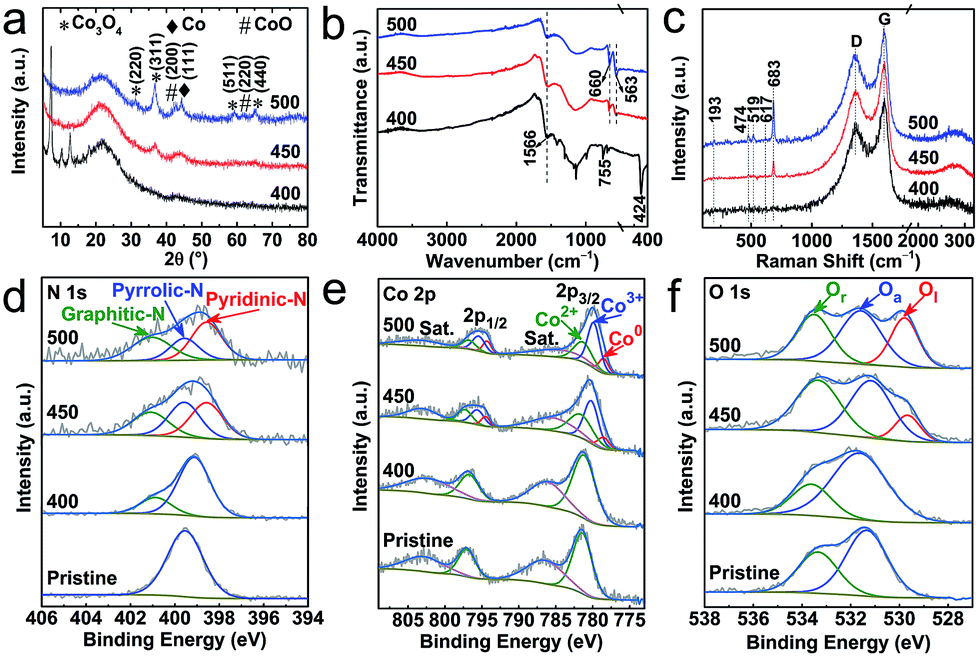 | ||
| Fig. 3 Structural characterization of the ZIF-67@EGO derived products at 400 °C, 450 °C and 500 °C: (a) PXRD patterns, (b) FTIR spectra, and (c) Raman spectra. (d–f) XPS spectra of N 1s, Co 2p and O 1s for pristine ZIF-67@EGO are shown for comparison. The full survey spectra and related elemental analysis are provided in Fig. S10 and S11, and Table S1.† | ||
More insights on the chemical nature of the derivatives, transformation of the coordination and associated atomic level information between Co/(-oxide)–N–C, are revealed by X-ray photoemission spectroscopy (XPS, Fig. 3d–f, S10–S12 and Table S1†). The oxidation associated on-set and full framework decomposition can be understood by relative changes of the C 1s and N 1s spectra. Specifically, the dissociation of C–N bonds in the ligand imidazolate ring can be seen by the narrowing of the shoulder at a high binding energy of the C 1s peak. Furthermore, the appearance of a new peak at high binding energy, at the expense of the symmetric N 1s peak indicates the loss of Co–N framework coordination (Fig. 3d, S12 and S13†). A clear conversion of ligand pyrrolic type nitrogen to a graphitic-N and pyridinic-N is seen as dissociated active nitrogen radicals can readily react with the exposed defects (on oxygen dissociated points) in the EGO. Note that such extensive conversion of ligand nitrogen to a graphitic-N and pyridinic-N is not evidenced in the ZIF-67 alone derived product under similar synthesis conditions (Fig. S14†). Such framework decomposition leads to open metal centres in ZIFs and MOFs.49,50 The active cobalt centres then readily react with thermally liberated oxygen from the EGO substrate. Here, it is worth noting that EGO exhibits limited residual oxygen content (as it is already exfoliated by thermal-shock at 400 °C),44,45 and the temperature plays a critical role in dissociating these remaining oxygen functional groups from EGO. Due to this, the hybrid, ZIF-67@EGO treated at 400 °C shows little evidence for the oxidation of cobalt metal centres. As there is minimal framework decomposition, most of the Co–N4 coordination remains intact (see PXRD and FTIR at Fig. 3a and b). With increasing temperature, the Co 2p peaks gradually shift towards lower binding energy due to the loss of Co–N4 followed by oxidation (Fig. 3e). Specifically, the Co 2p3/2 spectra can be deconvoluted into three characteristic peaks of Co2+ (≈781.6 eV), Co3+ (≈780.2 eV) and metallic Co (Co0, ≈778.5 eV).32 Furthermore, the relative atomic ratio of Co2+/Co3+ is ≈0.9 and ≈0.6 in the samples derived at 450 °C and 500 °C respectively, indicating an increased level of oxidization with temperature. In ZIF-67 the cobalt–metal centers (Co2+) are fully coordinated with four nitrogen units from imidazolate linkers. The carbonization of the ZIF under an inert atmosphere leads to carbon confined cobalt particles without any oxidation. However, in the case of ZIF@EGO, controlled etching of ZIF at 400–450 °C creates partially exposed cobalt sites. Under such conditions with the given low-concentration of liberated oxygen from EGO, these cobalt centers cannot be fully converted to cobalt oxide. Thus, this shows the existence of Co and its partially oxidized components in the form of CoO and Co3O4. This oxidation level is enhanced with increasing the temperature to 500 °C, due to the more dissociated oxygen functional groups from the EGO.
Note that these new Co3O4/CoO nanophases are not in a fully crystalline state, and thus such an amorphous phase consists of more surface oxygen vacancies, particularly in the sample derived at 450 °C.32,51 This is also further well supported by the O 1s spectra. The precursor hybrid consists of two components, representing the surface residual oxygen functional groups in the form of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O (≈531.2 eV) and C–O (≈533.4 eV) in EGO (Fig. 3f).45 This has a considerable changeover exhibiting three peak behaviours for the samples derived at 450–500 °C.32,39 The lattice oxygen (≈529.5 eV) emerges at 450 °C and becomes more prominent at 500 °C, indicating the crystallization of surface cobalt-oxide. A similar situation is observed during the calcination of MOFs to produce metal-oxide nanoparticles.10,12,52,53 During the thermolysis the transformation of oxygen functional groups in the EGO to adsorbed oxygen on the surface oxidised cobalt-oxide can be seen with corresponding changes of the peak and position (531.7 ± 0.1 eV).52–54 Further to this, the decomposition of the ZIF-framework by oxygen etching shows a strong C–O peak owing primarily to the oxygen binding to the defective linker carbon.54,55 Here it is worth noting that simple surface-adsorbed oxygen without strong chemical interactions shows a symmetric peak in the pristine ZIF at a binding energy of ≈532 eV.11
O (≈531.2 eV) and C–O (≈533.4 eV) in EGO (Fig. 3f).45 This has a considerable changeover exhibiting three peak behaviours for the samples derived at 450–500 °C.32,39 The lattice oxygen (≈529.5 eV) emerges at 450 °C and becomes more prominent at 500 °C, indicating the crystallization of surface cobalt-oxide. A similar situation is observed during the calcination of MOFs to produce metal-oxide nanoparticles.10,12,52,53 During the thermolysis the transformation of oxygen functional groups in the EGO to adsorbed oxygen on the surface oxidised cobalt-oxide can be seen with corresponding changes of the peak and position (531.7 ± 0.1 eV).52–54 Further to this, the decomposition of the ZIF-framework by oxygen etching shows a strong C–O peak owing primarily to the oxygen binding to the defective linker carbon.54,55 Here it is worth noting that simple surface-adsorbed oxygen without strong chemical interactions shows a symmetric peak in the pristine ZIF at a binding energy of ≈532 eV.11
Overall, it is suggested that the slow release of the small concentrations of the residual oxygen functionalities on the EGO is responsible for the controlled etching of the ZIF structure. Thus, this, in turn, produces an ultra-small amorphous and N-doped cobalt-oxide nanostructure at a relatively mild-thermolysis temperature of ≈450 °C. This has not been achieved before in any type of ZIF/MOF/GO system. Control over dissociation of the framework, Co–N bonds and ligands, imidazolate ring in ZIF-67 is the key to produce a nanophase of N-doped Co3O4/Co. This overview of the structural manipulation mechanism under mild oxidation conditions (EGO exhibits a very low amount of oxygenated groups compared to that of pristine GO, 13 atom% to 35 atom%)44,45 is schematically represented in Scheme 1. Such structures in the hyperporous graphene networks should provide extensive surface accessibility and enhanced mass activity of N-doped Co3O4/Co (or CoO/Co) centres for oxygen reduction electrocatalysis.
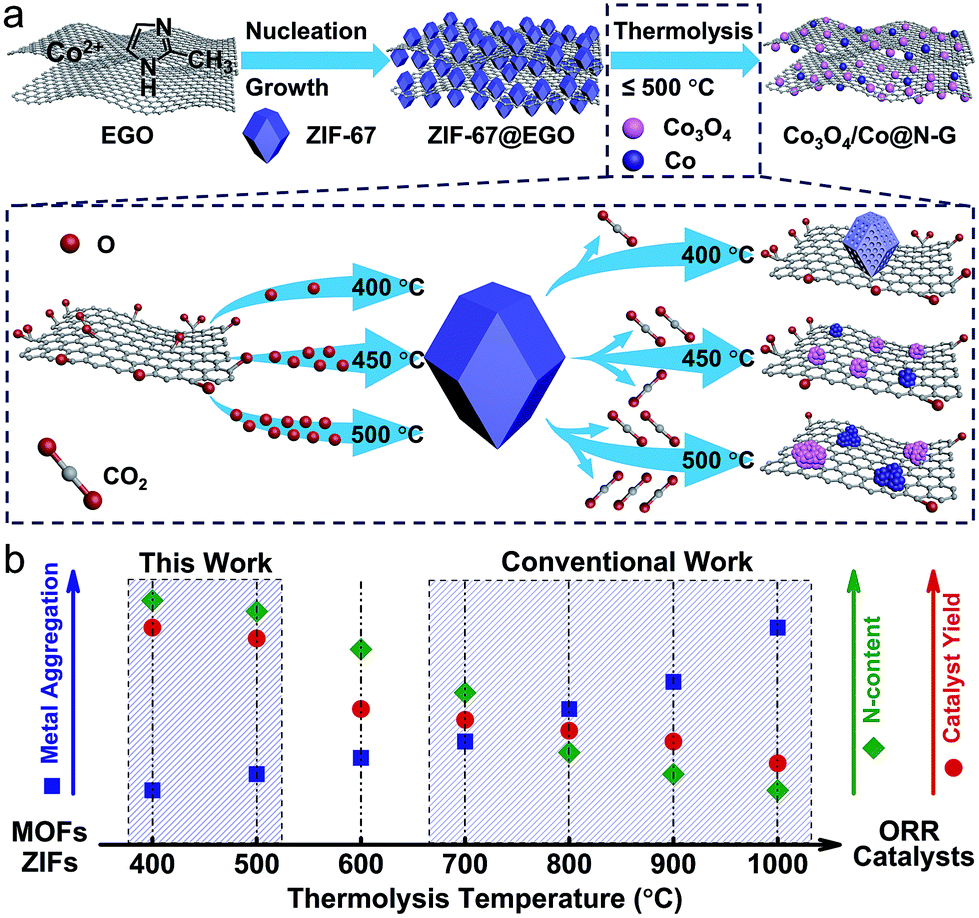 | ||
| Scheme 1 (a) Schematic illustration of the synthesis of the ZIF-67@EGO hybrid structure and further thermolysis to produce the catalyst, Co3O4/Co@N-G. The framework decomposition of the ZIF and associated thermal reconstruction mechanism under limited oxidation conditions and at mild temperatures is illustrated in the dashed box. (b) Qualitative illustration of the degree of metal aggregation, concentration of the nitrogen (N–) content and mass yield of the catalyst against the conventionally used thermolysis temperatures, including redesigned temperature in this work is highlighted. | ||
The ORR electrocatalytic performance of the as-synthesised samples is first investigated by cyclic voltammetry (CV) in O2-saturated 0.1 M KOH under a three-electrode configuration. The development of the cathodic peak is indicative of the emerging ORR activity.20,48 Compared with the constituent derivatives (ZDC-450 and EGO-450 from pure ZIF-67 and pure EGO precursors), the Co3O4/Co@N-G samples exhibit a prominent ORR activity with a desirable positive reduction peak potential (0.823 V vs. RHE) and higher cathodic current density (Fig. 4a). The linear sweep voltammetry (LSV) curves by a rotating disk electrode (RDE) technique show a remarkable activity variation among samples (Fig. 4b). The step-like polarization curves with more positive reduction potentials, a steep slope (also known as minimum half-wave potentials, E1/2) and a large current response (‘limiting current density’, J) toward negative potential are the main required characteristics for efficient ORR activity. Accordingly, the reference Pt/C with such characteristics was made as a benchmark material (Fig. S15†).56–58 In agreement with the CV data, the ZIF-67 alone derived Co@N-containing amorphous framework (ZDC-450) shows a negligible ORR activity. This can be ascribed to the more resistive amorphous structure due to the insufficient carbonization at 450 °C. This is one of the reasons why most of the literature has carried out the thermolysis at much higher temperatures, at least ≥700 °C.20,21,36 Also, in the case of calcination of ZIF-67 in air, the derived Co3O4 alone does not exhibit desirable electron transfer and activity characteristics,46,59 and thus it is required to disperse it on conducting carbon nanostructures to enhance the accessibility and transportation of the reactants and products.42 In many cases, to achieve the best ORR activity, ZIF-67 or ZIF-67/GO or their extra heteroatom complexes are thermolysed at elevated temperatures followed by acid washing of excessive cobalt metal in the products and often subjected to a secondary oxidation step. The qualitative behaviour of the LSV curve of Co3O4/Co@N-G-450 shows the best ORR performance (Fig. 4c). The onset potential (Eonset, defined as the crossover potential at a current density of 0.1 mA cm−2) of ≈0.962 V is close to that of a commercial Pt/C standard of ≈0.952 V (Fig. S15†). A highly favourable E1/2 (≈0.808 V vs. RHE) and J are also comparable to those of the Pt/C standard (≈0.819 V vs. RHE). Not much activity performance enhancement can be seen in the sample, Co3O4/Co@N-G-500. This can be directly attributed to the increased particle size and loss of the active amorphous phase thus reducing its reactant adsorption binding and mass activity (Fig. 2).21,32,50,51,59 Clearly, the structures derived at >600 °C show cobalt metal particles of over 100 nm in size, and can lead to micron sized particles.38,41 The ultra-small nanoparticles and their amorphous type should exhibit more exposed edges, defects/vacancies to make the surface more heterogeneous for efficient oxygen adsorption and catalysis.21,32,49–51 Furthermore, the nitrogen functional groups can enhance the active heterogeneous surface and electron transfer.21 The Co3O4/Co@N-G-450 sample with a relatively high concentration of graphitic-N and pyridinic-N (Fig. 3d, S13 and S14†) should contribute to the enhancement of the overall electrocatalytic performance.43,60–62 The literature shows that nitrogen dopants in the carbon structures can enhance the heterogeneity of the charged carbon surface due to the lone pair electron transfer from nitrogen to carbon. Thus, the dopants can create positive cores to enhance the oxygen binding. Theoretical calculations further show that the associated reaction surface is exothermic.21 Metal or metal-oxide in the carbons can further enhance the active site density by enhancing the electrostatic attraction of molecular oxygen. Again these structures show further reduction in the activation barrier for the intermediate formation thus favoring the overall reaction pathway.21 Furthermore, the content of cobalt in Co3O4 can improve the activity. This can be ascribed to the more conductive nature of the metals than their oxidized counterparts. It was shown that the gradual increase of surface oxidation of cobalt in the ZIF-67 derived Co–N–C leads to reduced ORR performance.21,63 Nevertheless, the CoOx@Co core is favorably considered as an active ORR material due to the presence of the Co2+/Co3+ redox couple for electron transfer.42 Another important kinetic parameter (n, electron transfer number) for the ORR is analysed by the Koutecky–Levich (K–L) plots (Fig. 4d and S16†). Parallel linear fitting lines indicate first-order reaction kinetics and mostly a potential-independent electron transfer number, which is in the range of 3.91–3.96, close to the dominant four-electron pathway of Pt/C (i.e., O2 is reduced to OH−). This four-electron reaction pathway is considered to be favourable compared to the two-electron pathway for efficient ORR activity (ESI†).64 It is worth noting that the samples derived from ZIF-67 and EGO alone, and the hybrid thermolysed at 400 °C are less likely to show this four-electron activity (Fig. S17†). The steeper slopes in the kinetic region with a corresponding smaller Tafel slope of ≈62 mV dec−1 in Co3O4/Co@N-G-450 compared with ≈86 mV dec−1 of Pt/C (Fig. S18†) further confirm the excellent ORR activity. A dual-site mechanism was proposed for a composite ORR catalyst material, featuring Co nanoparticles coated with Co oxides and Co2+ species associated with N–C moieties, by which oxygen is reduced to peroxide at Co–N–C sites and further reduced to OH− at CoxOy/Co sites.42,65 The long term stability is another important characteristic of the catalyst. The chronoamperometry tests (current response with respect to time under a fixed potential) show comparatively enhanced stability with a current response activity retention of over 85% compared with 50% for Pt/C over 20 h operation (Fig. S19†). An activity retention of 75% can be seen after 50 h operation. This catalyst sample, before and after the ORR stability test, further probed by XPS indicates a slight change in its respective Co2+ and Co3+ components. As expected the sample after the ORR stability test shows enhanced Co3+ with a Co2+/Co3+ ratio of ≈0.6 compared to ≈0.9 in the sample before the ORR stability test (Fig. S20†). It was reported that a high surface Co2+ density can enhance the ORR activity of Co3O4 nanocrystals.64 Accordingly, this transition from Co2+ to Co3+ during the test can reduce the activity current response to show decreased stability.
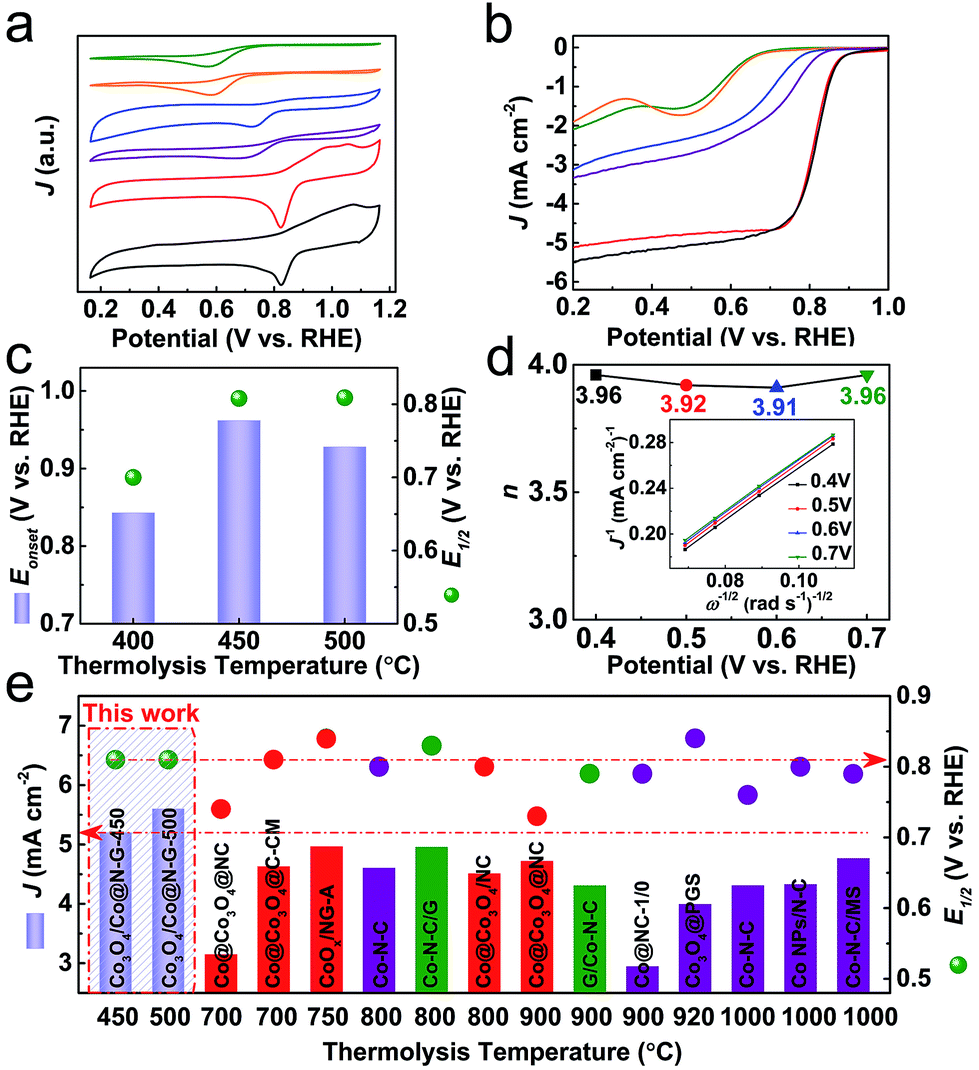 | ||
| Fig. 4 ORR activity characteristics and comparative data of the samples in O2-saturated 0.1 M KOH: (a) CV curves (at 10 mV s−1) and (b) LSV curves (at 10 mV s−1 and 1600 rpm): ZDC-450 (olive), ZDC-450-A (orange), EGO-450 (blue), ZIF-67@EGO-400 (purple), Co3O4/Co@N-G-450 (red) and Co3O4/Co@N-G-500 (black). (c) Comparison of Eonset and E1/2 values against the thermolysis temperature. (d) Electron transfer number (n) of Co3O4/Co@N-G-450 as a function of potential (inset: K–L plots at different potentials calculated from Fig. S16†). (e) Comparative ORR activity data: J and E1/2 values against the thermolysis temperature for the catalysts in this work and other ZIF-derivatives, produced at elevated temperatures, taken from the literature (samples with second heat treatment and/or oxidation, and further acid washing are represented by red and olive colours, respectively).21,22,24,26,30,32,33,35–37,40,48 See Table S2 in the ESI† for further details. | ||
Finally, the performance of many recently reported ZIF-derived ORR electrocatalysts is summarized and compared with that of Co3O4/Co@N-G in terms of J and E1/2 against the thermolysis temperature for the catalysts (Fig. 4e, Table S2†). The low-temperature designed Co3O4/Co@N-G exhibits better performance metrics as indicated by J and E1/2. It is worth noting that those literature catalysts were derived at much higher synthesis temperatures, and also with extensive chemical treatment, such as acid treatment and/or second-step thermolysis, or other functional additives. The pure ZIF-derived metal nanoparticle encapsulated in carbon nanostructures usually involves carbonization in the presence of hazardous H2 gas and further oxidation to form metal@metal-oxide heterostructures.20,36,37 In other cases, the ZIF/GO hybrid structures often suffer from significant mass loss due to the abundant oxygen functional groups in GO and an additional long-term acid leaching procedure to remove highly aggregated metal nanoparticles, which would result in a very low mass yield of the catalyst.29,30,38,40 Therefore, the present low-temperature synthesis strategy is not only a facile approach to produce high-performance ORR catalysts with higher catalyst yield and lower thermal energy consumption, but also an effective way of controlling the metal nanoparticle growth and surface oxidation.
Conclusions
In summary, a facile and highly controllable strategy is presented that can produce ultra-small Co3O4/Co nanoparticles in nitrogen-doped hierarchical carbon networks for a high-performance oxygen reduction reaction. The residual oxygen groups in the thermal-shock exfoliated graphene-oxide lead to controlled gasification of ZIF-67 followed by oxidation of the open active cobalt metal centres at mild thermolysis temperatures of ≈450 °C. Such conditions facilitate a high mass yield of the catalyst over 65 wt% compared to the commonly reported carbon-based catalyst yield of ≈30 wt% or less, obtained through harsh synthesis and high-temperature thermolysis routes. The proposed low-temperature approach can also favour the amorphous phase of Co3O4/CoO/Co to exhibit an active surface for the catalysis reaction. Thus, the as-prepared Co3O4/Co@N-G delivers a high onset and half-wave potential, comparable with those of the commercial platinum/carbon standard. Furthermore, the activity of the Co3O4/Co@N-G is superior to those of several ZIF-67 based derivatives, which are produced by extended chemical manipulations, including addition of extra heteroatom precursors, thermolysis at elevated temperatures followed by acid etching of metal particles, and/or secondary heat treatment or chemical modification and doping. Therefore, these systems pose a great challenge in understanding for further developments. This currently proposed low-temperature thermolysis route is a new and cost-effective approach to design and produce diverse MOF (ZIF)-derived metal/metal-oxides in carbon-based hybrid structures for electrochemical catalysis reactions.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the EPSRC (Grant No. EP/L018330/1 and EP/K002252/1). Jian Guo would like to thank the University College London and China Scholarship Council for a joint UCL-CSC PhD scholarship.Notes and references
- M. Lefèvre, E. Proietti, F. Jaouen and J.-P. Dodelet, Science, 2009, 324, 71–74 CrossRef PubMed.
- C. Yuan, H. Bin Wu, Y. Xie and X. W. Lou, Angew. Chem., Int. Ed., 2014, 53, 1488–1504 CrossRef CAS PubMed.
- S. Fu, C. Zhu, J. Song, D. Du and Y. Lin, Adv. Energy Mater., 2017, 7, 1700363 CrossRef.
- L. Yang, X. Zeng, W. Wang and D. Cao, Adv. Funct. Mater., 2018, 28, 1704537 CrossRef.
- Y. Jiao, Y. Zheng, M. Jaroniec and S. Z. Qiao, Chem. Soc. Rev., 2015, 44, 2060–2086 RSC.
- J.-C. Li, P.-X. Hou and C. Liu, Small, 2017, 13, 1702002 CrossRef PubMed.
- S. Dang, Q. L. Zhu and Q. Xu, Nat. Rev. Mater., 2017, 3, 17075 CrossRef.
- X. Cao, C. Tan, M. Sindoro and H. Zhang, Chem. Soc. Rev., 2017, 46, 2660–2677 RSC.
- Z. Liang, C. Qu, W. Guo, R. Zou and Q. Xu, Adv. Mater., 2017, 30, 1702891 CrossRef PubMed.
- S. Gadipelli and Z. Guo, Chem. Mater., 2014, 26, 6333–6338 CrossRef CAS.
- S. Gadipelli, W. Travis, W. Zhou and Z. Guo, Energy Environ. Sci., 2014, 7, 2232–2238 RSC.
- G. Srinivas, V. Krungleviciute, Z. X. Guo and T. Yildirim, Energy Environ. Sci., 2014, 7, 335–342 RSC.
- X. Zhang, Y. Yang, L. Song, J. Chen, Y. Yang and Y. Wang, J. Hazard. Mater., 2019, 365, 597–605 CrossRef CAS PubMed.
- N. Liu, W. Huang, M. Tang, C. Yin, B. Gao, Z. Li, L. Tang, J. Lei, L. Cui and X. Zhang, Chem. Eng. J., 2019, 359, 254–264 CrossRef CAS.
- X. Zhang, H. Li, F. Hou, Y. Yang, H. Dong, N. Liu, Y. Wang and L. Cui, Appl. Surf. Sci., 2017, 411, 27–33 CrossRef CAS.
- G. Zhong, D. Liu and J. Zhang, J. Mater. Chem. A, 2017, 6, 1887–1899 RSC.
- Y. J. Sa, D.-J. Seo, J. Woo, J. T. Lim, J. Y. Cheon, S. Y. Yang, J. M. Lee, D. Kang, T. J. Shin, H. S. Shin, H. Y. Jeong, C. S. Kim, M. G. Kim, T.-Y. Kim and S. H. Joo, J. Am. Chem. Soc., 2016, 138, 15046–15056 CrossRef CAS PubMed.
- Y. Zheng, Y. Jiao, Y. Zhu, Q. Cai, A. Vasileff, L. H. Li, Y. Han, Y. Chen and S.-Z. Qiao, J. Am. Chem. Soc., 2017, 139, 3336–3339 CrossRef CAS PubMed.
- H. Wu, H. Li, X. Zhao, Q. Liu, J. Wang, J. Xiao, S. Xie, R. Si, F. Yang, S. Miao, X. Guo, G. Wang and X. Bao, Energy Environ. Sci., 2016, 9, 3736–3745 RSC.
- B. Y. Xia, Y. Yan, N. Li, H. Bin Wu, X. Wen, D. Lou and X. Wang, Nat. Energy, 2016, 1, 15006 CrossRef CAS.
- S. Gadipelli, T. Zhao, S. A. Shevlin and Z. Guo, Energy Environ. Sci., 2016, 9, 1661–1667 RSC.
- G. Jia, W. Zhang, G. Fan, Z. Li, D. Fu, W. Hao, C. Yuan and Z. Zou, Angew. Chem., Int. Ed., 2017, 56, 13781–13785 CrossRef CAS PubMed.
- H. Zhang, X. Liu, Y. Wu, C. Guan, A. K. Cheetham and J. Wang, Chem. Commun., 2018, 54, 5268–5288 RSC.
- X. Wang, J. Zhou, H. Fu, W. Li, X. Fan, G. Xin, J. Zheng and X. Li, J. Mater. Chem. A, 2014, 2, 14064–14070 RSC.
- J. Tang, R. R. Salunkhe, J. Liu, N. L. Torad, M. Imura, S. Furukawa and Y. Yamauchi, J. Am. Chem. Soc., 2015, 137, 1572–1580 CrossRef CAS PubMed.
- P. Yin, T. Yao, Y. Wu, L. Zheng, Y. Lin, W. Liu, H. Ju, J. Zhu, X. Hong, Z. Deng, G. Zhou, S. Wei and Y. Li, Angew. Chem., Int. Ed., 2016, 55, 10800–10805 CrossRef CAS PubMed.
- S. Gadipelli, Z. Li, T. Zhao, Y. Yang, T. Yildirim and Z. Guo, J. Mater. Chem. A, 2017, 5, 24686–24694 RSC.
- H. Zhang, S. Hwang, M. Wang, Z. Feng, S. Karakalos, L. Luo, Z. Qiao, X. Xie, C. Wang, D. Su, Y. Shao and G. Wu, J. Am. Chem. Soc., 2017, 139, 14143–14149 CrossRef CAS.
- H. X. Zhong, J. Wang, Y. W. Zhang, W. L. Xu, W. Xing, D. Xu, Y. F. Zhang and X. B. Zhang, Angew. Chem., Int. Ed., 2014, 53, 14235–14239 CrossRef CAS PubMed.
- J. Wei, Y. Hu, Y. Liang, B. Kong, J. Zhang, J. Song, Q. Bao, G. P. Simon, S. P. Jiang and H. Wang, Adv. Funct. Mater., 2015, 25, 5768–5777 CrossRef CAS.
- L. Gao, S. Chen, R. Cai, Q. Zhao, X. Zhao and D. Yang, Global Challenges, 2018, 2, 1700086 CrossRef.
- Y. Jiang, Y. P. Deng, J. Fu, D. U. Lee, R. Liang, Z. P. Cano, Y. Liu, Z. Bai, S. Hwang, L. Yang, D. Su, W. Chu and Z. Chen, Adv. Energy Mater., 2018, 8, 1702900 CrossRef.
- Y. Li, B. Jia, Y. Fan, K. Zhu, G. Li and C. Y. Su, Adv. Energy Mater., 2018, 8, 1702048 CrossRef.
- W. Zhang, X. Jiang, X. Wang, Y. V. Kaneti, Y. Chen, J. Liu, J. Sen Jiang, Y. Yamauchi and M. Hu, Angew. Chem., Int. Ed., 2017, 56, 8435–8440 CrossRef CAS PubMed.
- W. Xia, R. Zou, A. Li, D. Xia and S. Guo, Energy Environ. Sci., 2015, 8, 568–576 RSC.
- A. Aijaz, J. Masa, C. Rösler, W. Xia, P. Weide, A. J. R. Botz, R. A. Fischer, W. Schuhmann and M. Muhler, Angew. Chem., Int. Ed., 2016, 55, 4087–4091 CrossRef CAS PubMed.
- Z. Guo, F. Wang, Y. Xia, J. Li, A. G. Tamirat, Y. Liu, L. Wang, Y. Wang and Y.-Y. Xia, J. Mater. Chem. A, 2017, 6, 1443–1453 RSC.
- J. Wei, Y. Hu, Z. Wu, Y. Liang, S. Leong, B. Kong, X. Zhang, D. Zhao, G. P. Simon and H. Wang, J. Mater. Chem. A, 2015, 3, 16867–16873 RSC.
- Z. Wu, L.-P. Sun, M. Yang, L.-H. Huo, H. Zhao and J.-C. Grenier, J. Mater. Chem. A, 2016, 4, 13534–13542 RSC.
- J. Wei, Y. Hu, Y. Liang, B. Kong, Z. Zheng, J. Zhang, S. P. Jiang, Y. Zhao and H. Wang, J. Mater. Chem. A, 2017, 5, 10182–10189 RSC.
- X. Huang, J. Wang, H. Bao, X. Zhang and Y. Huang, ACS Appl. Mater. Interfaces, 2018, 10, 7180–7190 CrossRef CAS PubMed.
- Y. Liang, Y. Li, H. Wang, J. Zhou, J. Wang, T. Regier and H. Dai, Nat. Mater., 2011, 10, 780–786 CrossRef CAS PubMed.
- P. Zhang, F. Sun, Z. Xiang, Z. Shen, J. Yun and D. Cao, Energy Environ. Sci., 2014, 7, 442–450 RSC.
- Z. Li, S. Gadipelli, Y. Yang and Z. Guo, Small, 2017, 13, 1702474 CrossRef PubMed.
- S. Gadipelli, Y. Lu, N. T. Skipper, T. Yildirim and Z. Guo, J. Mater. Chem. A, 2017, 5, 17833–17840 RSC.
- R. Zhang, T. Zhou, L. Wang and T. Zhang, ACS Appl. Mater. Interfaces, 2018, 10, 9765–9773 CrossRef CAS PubMed.
- Y. Hao, Y. Xu, J. Liu and X. Sun, J. Mater. Chem. A, 2017, 5, 5594–5600 RSC.
- W. Xia, C. Qu, Z. Liang, B. Zhao, S. Dai, B. Qiu, Y. Jiao, Q. Zhang, X. Huang, W. Guo, D. Dang, R. Zou, D. Xia, Q. Xu and M. Liu, Nano Lett., 2017, 17, 2788–2795 CrossRef CAS PubMed.
- L. Tao, C. Y. Lin, S. Dou, S. Feng, D. Chen, D. Liu, J. Huo, Z. Xia and S. Wang, Nano Energy, 2017, 41, 417–425 CrossRef CAS.
- S. Dou, C. L. Dong, Z. Hu, Y. C. Huang, J. L. Chen, L. Tao, D. Yan, D. Chen, S. Shen, S. Chou and S. Wang, Adv. Funct. Mater., 2017, 27, 1702546 CrossRef.
- L. Xu, Q. Jiang, Z. Xiao, X. Li, J. Huo, S. Wang and L. Dai, Angew. Chem., Int. Ed., 2016, 55, 5277–5281 CrossRef CAS PubMed.
- X. Zhang, H. Li, X. Lv, J. Xu, Y. Wang, C. He, N. Liu, Y. Yang and Y. Wang, Chem.–Eur. J., 2018, 24, 8822–8832 CrossRef CAS PubMed.
- X. Zhang, F. Hou, H. Li, Y. Yang, Y. Wang, N. Liu and Y. Yang, Microporous Mesoporous Mater., 2018, 259, 211–219 CrossRef CAS.
- M. Liu, J. Liu, Z. Li and F. Wang, ACS Appl. Mater. Interfaces, 2018, 10, 7052–7060 CrossRef CAS PubMed.
- S. Gadipelli and Z. X. Guo, ChemSusChem, 2015, 8, 2123–2132 CrossRef CAS PubMed.
- M. K. Debe, Nature, 2012, 486, 43–51 CrossRef CAS PubMed.
- B. Lim, M. Jiang, P. H. C. Camargo, E. C. Cho, J. Tao, X. Lu, Y. Zhu and Y. Xia, Science, 2009, 324, 1302–1306 CrossRef CAS PubMed.
- J. Wu, J. Zhang, Z. Peng, S. Yang, F. T. Wagner and H. Yang, J. Am. Chem. Soc., 2010, 132, 4984–4985 CrossRef CAS PubMed.
- T. Zhao, S. Gadipelli, G. He, M. J. Ward, D. Do, P. Zhang and Z. Guo, ChemSusChem, 2018, 11, 1295–1304 CrossRef CAS PubMed.
- J. Meng, C. Niu, L. Xu, J. Li, X. Liu, X. Wang, Y. Wu, X. Xu, W. Chen, Q. Li, Z. Zhu, D. Zhao and L. Mai, J. Am. Chem. Soc., 2017, 139, 8212–8221 CrossRef CAS PubMed.
- X. Liu and L. Dai, Nat. Rev. Mater., 2016, 1, 16064 CrossRef CAS.
- C. Hu and L. Dai, Angew. Chem., Int. Ed., 2016, 55, 11736–11758 CrossRef CAS PubMed.
- S. Guo, S. Zhang, L. Wu and S. Sun, Angew. Chem., Int. Ed., 2012, 51, 11770–11773 CrossRef CAS PubMed.
- X. Ge, A. Sumboja, D. Wuu, T. An, B. Li, F. W. T. Goh, T. S. A. Hor, Y. Zong and Z. Liu, ACS Catal., 2015, 5, 4643–4667 CrossRef CAS.
- T. S. Olson, S. Pylypenko, P. Atanassov, K. Asazawa, K. Yamada and H. Tanaka, J. Phys. Chem. C, 2010, 114, 5049–5059 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ta10925g |
| ‡ Now at the University of Hong Kong, E-mail: zxguo@hku.hk. |
| This journal is © The Royal Society of Chemistry 2019 |