
Bright electrochemiluminescent films of efficient aggregation-induced emission luminogens for sensitive detection of dopamine†
Zihua
Li
a,
Wei
Qin
 *a,
Jialong
Wu
a,
Zhiyong
Yang
b,
Zhenguo
Chi
b and
Guodong
Liang
*a
*a,
Jialong
Wu
a,
Zhiyong
Yang
b,
Zhenguo
Chi
b and
Guodong
Liang
*a
aPCFM Lab, School of Materials Science and Engineering, Sun Yat-sen University, Guangzhou 510275, P. R. China. E-mail: qinw9@mail.sysu.edu.cn; lgdong@mail.sysu.edu.cn
bPCFM Lab, School of Chemistry, Sun Yat-sen University, Guangzhou 510275, P. R. China
First published on 30th July 2019
Abstract
The development of electrochemiluminescent (ECL) luminogens is of great importance for sensitive detection of biomolecules in various applications. Herein, two efficient red luminogens bearing benzothiadiazole and arylamino moieties, namely BTD-TPA and BTD-NPA, have been synthesized through one-step Suzuki reaction. They show aggregation-induced emission (AIE) features with high fluorescence quantum efficiency and reversible redox pairs with high stability. Taking advantage of these merits, bright ECL non-doped films are achieved based on the AIE luminogens. Interestingly, the ECL intensity of the films is proportional to the film thickness, which enables the optimization of their brightness through the variation of luminogen loading. Furthermore, the bright ECL films are utilized for sensitive detection of dopamine (DA) with a broad linear range (0.05–350 μM) and a low detection limit of 17.0 nM. Such bright ECL films provide an ideal platform for sensitive analysis of important biomolecules with high selectivity.
Introduction
Electrochemiluminescence (ECL) has attracted enormous attention in immunoassay and analytical science due to its remarkable advantages, including simple instruments, low background, and high sensitivity.1–3 To date, different ECL systems based on metal complexes and quantum dots have been developed for in vitro diagnosis, DNA detection,4,5 and other biological applications.6–9 However, they encounter a few inherent problems such as high cost, heavy-metal toxicity, and poor processability. To circumvent the problems, organic fluorescent materials based on anthracene, fluorene, thienyltriazole, boron dipyrromethene (BODIPY), and pyridopyrimidine functional moieties have been developed for ECL applications due to their low cost, easy functionalization, and good biocompatibility.10–13 Most of the organic chromophores are constructed from hydrophobic aromatic moieties and used in solution.14 Compared with solution-based ECL systems, solid ECL films have overwhelming advantages, such as environmental friendliness, enhanced and stable ECL signal, and high cost-efficiency.15 However, conventional organic chromophores suffer from severe aggregation-caused quenching (ACQ) effect when used in solid films. Indeed, limited by the ACQ effect, efficient ECL films based on conventional organic chromophores are rarely reported.16 Although doping or labeling in polymers alleviates the ACQ effect, the chromophores are normally retained at low concentration (<3 wt%) in the host materials, which greatly hinders their practical applications.17The discovery of the aggregation-induced emission (AIE) phenomenon has opened a new avenue to solve the ACQ problem.18–20 Opposite to the ACQ effect, the fluorescence emission of AIE luminogens is enhanced by aggregate formation, which offers a new chance for the development of solid ECL films.21,22 A few of the efficient luminogens with AIE features have been developed and utilized in bioimaging and organic light-emitting diodes (OLEDs) due to their high brightness in the aggregated state.23–26 However, to our best knowledge, there is no report of efficient organic luminogens for bright ECL film applications, possibly due to their irreversible redox waves and poor stability.27–29
Dopamine (DA) is a significant neurotransmitter, which plays a key role in the function of the central nervous, hormonal, and cardiovascular systems. The abnormal concentration levels of DA are associated with some notorious diseases of the nervous system, such as Parkinson's, Alzheimer's and schizophrenia disease.30 Common methods for DA detection are based on high-performance liquid chromatography (HPLC) or mass spectrometry (MS).31,32 However, these methods are limited in practical applications due to expensive instruments, time-consuming analysis, and laborious sample pretreatment.33 Recently, electrochemical methods based on fullerene, ZnO nanowires, and carbon nanotubes, were employed to detect DA with a detection limit in the range of 0.1–60 nM.34–40
Herein, we develop a type of bright ECL film based on efficient AIE luminogens (BTD-TPA and BTD-NPA). The AIE luminogens are constructed with a benzothiadiazole (BTD) core as an electron acceptor and an arylamino unit as an electron donor,41,42 giving rise to red luminogens through intramolecular charge transfer effect. Freely rotating rotors from the functional arylamino units endow them with AIE features. They show stable and reversible redox pairs inherited from the functional arylamino moieties. Taking advantage of the AIE features and reversible redox pairs, bright and tunable ECL films are achieved. Moreover, the bright ECL films are further utilized for sensitive detection of DA with a high Stern–Volmer constant (5.5 × 105 M−1) and a low detection limit (17.0 nM). The interference experiment demonstrates the sensitive detection of DA with good selectivity. Such bright ECL films provide a promising platform for rapid and quantitative analysis of bioassays with high sensitivity and selectivity.
Experimental
Synthesis of BTD-Ph, BTD-TPA, and BTD-NPA
The compounds were synthesized according to the procedure reported in the literature.43 The detailed synthetic procedure is presented in the ESI.†Preparation of non-doped films
The films were facilely prepared by drop-casting 5 μL of DCM solution of luminogens on L-type GCE, followed by air drying. The film thickness is regulated by using luminogen solutions at various concentrations. The film thickness of BTD-TPA was measured using a fluorescence microscope (Leica M205 FA).Electrochemical and ECL measurements
All electrochemistry experiments were performed with a three-electrode cell on an electrochemical workstation (CHI 660D, Chenhua instrument, China). Glassy carbon electrodes (GCE, 3 mm diameter) and gold ultramicroelectrodes (UME, 12.5 μm diameter) were employed as the working electrodes. Platinum wire was used as the counter electrode and silver wire was used as the quasi-reference electrode. An L-type GCE (3 mm diameter) was used as the working electrode for ECL experiments. Before usage, all working electrodes were polished with 0.5, 0.3, and 0.05 μm alumina powder dispersed in water and sonicated in ethanol and deionized water. Before the end of each experiment, the Ag reference electrode was calibrated against ferrocene as an internal redox standard (0.464 V vs. SCE).44 Electrochemistry and ECL experiments were obtained in DCM and the ECL of BTD-TPA and BTD-NPA films were performed in CH3CN/H2O (v/v = 7![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) mixture with 0.1 M TBAPF6 as the supporting electrolyte. 20 mM TPrA was used as a co-reactant in the ECL experiments of BTD-TPA and BTD-NPA, while 5 mM BPO was employed as a co-reactant in the ECL experiment of BTD-Ph. A photomultiplier tube detector (PMT, Hamamatsu, Japan) with the voltage at 800 V was employed to collect the ECL signal. The ECL spectrum was obtained via pulsing 80 mV past the peak potentials of the oxidation or reduction waves for BTD-TPA, BTD-NPA, and BTD-Ph with a pulse width of 1 s by differential pulse amperometry. The ECL intensity-time profiles were obtained by pulsing between 80 mV past the oxidation peak and 0 V for BTD-TPA and BTD-NPA, and pulsing between 80 mV past the reduction peak and 0 V for BTD-Ph by means of multi-potential steps, with a pulse width of 1 s. The relative ECL efficiencies of BTD-Ph, BTD-TPA, and BTD-NPA in the solution state are calculated through integration of both ECL intensity and current value versus time with DPA (ΦECL is about 0.25% in DMF11) or [Ru(bpy)3]2+ (ΦECL is about 5.0% in acetonitrile12) as a standard (eqn (S1), ESI†).
:1) mixture with 0.1 M TBAPF6 as the supporting electrolyte. 20 mM TPrA was used as a co-reactant in the ECL experiments of BTD-TPA and BTD-NPA, while 5 mM BPO was employed as a co-reactant in the ECL experiment of BTD-Ph. A photomultiplier tube detector (PMT, Hamamatsu, Japan) with the voltage at 800 V was employed to collect the ECL signal. The ECL spectrum was obtained via pulsing 80 mV past the peak potentials of the oxidation or reduction waves for BTD-TPA, BTD-NPA, and BTD-Ph with a pulse width of 1 s by differential pulse amperometry. The ECL intensity-time profiles were obtained by pulsing between 80 mV past the oxidation peak and 0 V for BTD-TPA and BTD-NPA, and pulsing between 80 mV past the reduction peak and 0 V for BTD-Ph by means of multi-potential steps, with a pulse width of 1 s. The relative ECL efficiencies of BTD-Ph, BTD-TPA, and BTD-NPA in the solution state are calculated through integration of both ECL intensity and current value versus time with DPA (ΦECL is about 0.25% in DMF11) or [Ru(bpy)3]2+ (ΦECL is about 5.0% in acetonitrile12) as a standard (eqn (S1), ESI†).
Results and discussion
BTD-TPA, BTD-NPA, and their counterpart (BTD-Ph) for comparison were facilely synthesized through one-step Suzuki coupling with satisfactory yields as shown in Scheme 1 (Fig. S1–S6, ESI†). They possess good solubility in common organic solvents, such as toluene (Tol), chloroform, dichloromethane (DCM), 1,4-dioxane (DO), and tetrahydrofuran (THF), but are insoluble in acetonitrile or water. The UV spectra of BTD-TPA and BTD-NPA in THF are peaked at 458 nm with a large red-shift of 78 nm compared to BTD-Ph (380 nm) (Fig. S7, ESI†). Compared to that of BTD-Ph, the fluorescence emission of BTD-TPA and BTD-NPA is more sensitive to solvent polarity (Fig. 1 and Fig. S8, Table S1, ESI†). All the results indicate that BTD-TPA and BTD-NPA demonstrate strong charge transfer effects from the electron-donating arylamino units to the electron-accepting benzothiadiazole core.45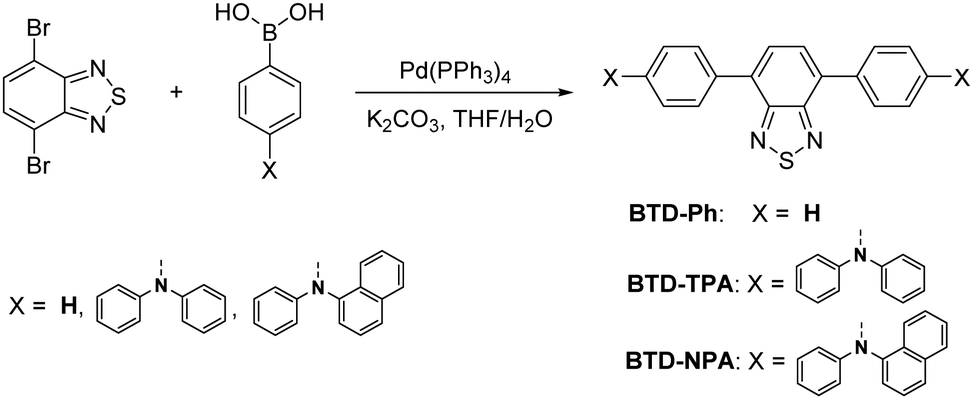 | ||
| Scheme 1 Synthetic routes and chemical structures of BTD-Ph, BTD-TPA and BTD-NPA. | ||
 | ||
| Fig. 1 Normalized PL emission spectra of (A) BTD-TPA, and (B) BTD-NPA in solvents with different polarities. | ||
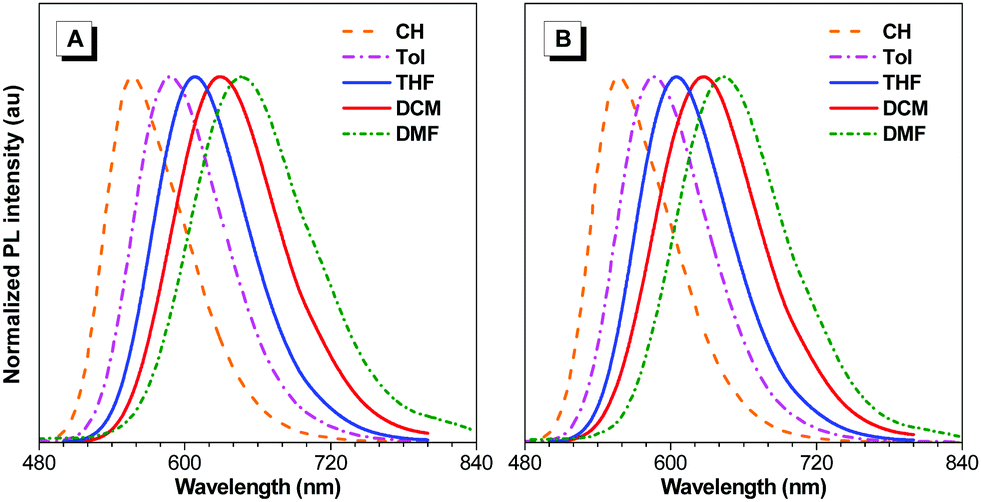
To study the effects of aggregation and solvent polarity on the emission process, the photoluminescence (PL) spectra of BTD-Ph, BTD-TPA, and BTD-NPA were explored in THF/water mixtures with different water fractions (fw). As shown in Fig. 2 and Fig. S9 (ESI†), BTD-Ph emits green fluorescence with an emission maximum at 481 nm in pure THF. After the addition of water into THF, its PL intensity is decreased gradually. At fw = 90%, the PL intensity is merely 15% of that in pure THF solution accompanied by a 25 nm red-shift, indicative of the typical ACQ feature. In contrast, BTD-TPA and BTD-NPA in pure THF emit orange-red fluorescence with an emission maximum at 609 and 605 nm, respectively. With the gradual addition of water into THF (fw ≤ 60%), the emission is weakened and red-shifted to ca. 625 nm due to transformation to the twisted intramolecular charge transfer (TICT) state in mixed solvents.46 Afterwards (fw > 60%), the PL intensity starts to increase due to the formation of luminogen aggregates and the AIE effect from the freely rotating rotors of the arylamino units. The higher the water fraction, the stronger the emission intensity. At fw = 90%, the PL intensity recovers remarkably, and the emission maximum moves to 609 and 606 nm, respectively. BTD-TPA and BTD-NPA solids possess high fluorescence quantum yields (ΦF) of 44% and 56%, respectively, which favors their application in solid films.
 | ||
| Fig. 2 (A) PL spectra of BTD-TPA in THF/water mixtures with different water fractions (fw). (B) Plots of the relative PL intensity (I/I0) versus the composition of the THF/water mixtures of BTD-Ph, BTD-TPA, and BTD-NPA. I0 denotes emission intensity in pure THF. Concentration: 10 μM. (C) Optimized geometries and molecular orbitals of HOMO and LUMO levels of the molecules. (D) CVs of BTD-Ph, BTD-TPA, and BTD-NPA in anhydrous DCM. | ||
To study the photophysical properties of the compounds at the molecular level, density functional theory (DFT) calculations were carried out (Fig. 2C). The highest occupied molecular orbitals (HOMOs) of the studied molecules are dispersed on the whole molecules. While the electron clouds of the lowest unoccupied molecular orbitals (LUMO) are dominated on the BTD core and the adjacent benzene rings. The remarkable difference in electron distributions of the HOMO and LUMO levels of BTD-TPA and BTD-NPA indicates an intrinsic intramolecular charge transfer effect. Compared to the bandgap of BTD-Ph, the narrow band gaps of BTD-TPA and BTD-NPA are indicative of stronger electron donor–acceptor systems, which is consistent with the experimental data (Fig. S7–S9, ESI†).
Cyclic voltammetry (CV) measurements were carried out to investigate the electrochemical behaviours of BTD-Ph, BTD-TPA, and BTD-NPA (Fig. 2D). BTD-TPA and BTD-NPA exhibit a stable and reversible redox pair at about +1.1 V due to the presence of arylamine moieties. The anodic peak current (ipa) and cathodic peak current (ipc) are linearly dependent on the square root of the scan rate (ν1/2) (Fig. S10–S12, ESI†), demonstrating diffusion control in these redox processes. The peak current ratio (ipa/ipc) of the redox pair of BTD-TPA and BTD-NPA is close to unity, revealing that radical cations generated during anodic potential scanning are stable.47 CVs of BTD-TPA and BTD-NPA using gold ultramicroelectrodes (UME) obtain similar results (Fig. S13, S14 (ESI†), and Table 1). Such stable and reversible redox properties of BTD-TPA and BTD-NPA are essential for their further ECL applications.
| Compound | E 1/2 (V vs. SCE)a | 10−6Db [cm2 s−1] | E g [eV] | E HOMO [eV] | E LUMO [eV] | |
|---|---|---|---|---|---|---|
| A/A+a |
A/A−a |
|||||
| a The values of E1/2 are obtained by averaging between the cathodic and anodic peak potentials. b D = πa2/16S2, a is the radius of gold UME, S is the slope of i–t−1/2 in chronoamperometry. c HOMO–LUMO band gap (Eg) is calculated from the onset of the absorption spectrum. d HOMO is the highest occupied molecular orbital derived from the onset oxidation potential. e LUMO is the lowest unoccupied molecular orbital estimated by using the equation ELUMO = EHOMO + Eg. f nd: not detected due to without oxidation wave of BTD-Ph. | ||||||
| BTD-Ph | ndf | −0.74 | ndf | 2.87 | −5.74 | −2.87 |
| BTD-TPA | 1.09 | −0.76 | 4.37 | 2.33 | −5.24 | −2.91 |
| BTD-NPA | 1.11 | −0.77 | 14.3 | 2.33 | −5.27 | −2.94 |
The ECL properties of BTD-Ph, BTD-TPA, and BTD-NPA in DCM solution were investigated. In a co-reactant system, the ECL and corresponding CVs of all compounds were performed in DCM solution in the presence of benzoyl peroxide (BPO) or tripropylamine (TPrA) (Fig. S15, ESI†). The bright ECL of BTD-Ph, BTD-TPA, and BTD-NPA can be observed by the naked eye with emission peaks at 493, 633, and 632 nm, respectively (Fig. S16, ESI†). The ECL spectra resemble their PL spectra, which confirms that the ECL is from the luminogens. The ECL efficiencies of BTD-Ph, BTD-TPA, and BTD-NPA in solution are calculated to be 0.02%, 2.5%, and 2.4%, respectively. The ECL stabilities of BTD-Ph, BTD-TPA, and BTD-NPA were investigated by the pulsing technique (Fig. S17, ESI†). BTD-TPA and BTD-NPA show good ECL stabilities and retain at an almost constant value during the consecutive cyclic potential scanning with a relative standard derivation (RSD) of 1.0% and 2.3%, respectively. In contrast, the ECL signal of BTD-Ph is decreased with increasing scanning cycles. The decreased ECL signal of BTD-Ph is ascribed to the unstable radical anion, which greatly hinders the generation of the excited state of BTD-Ph.10
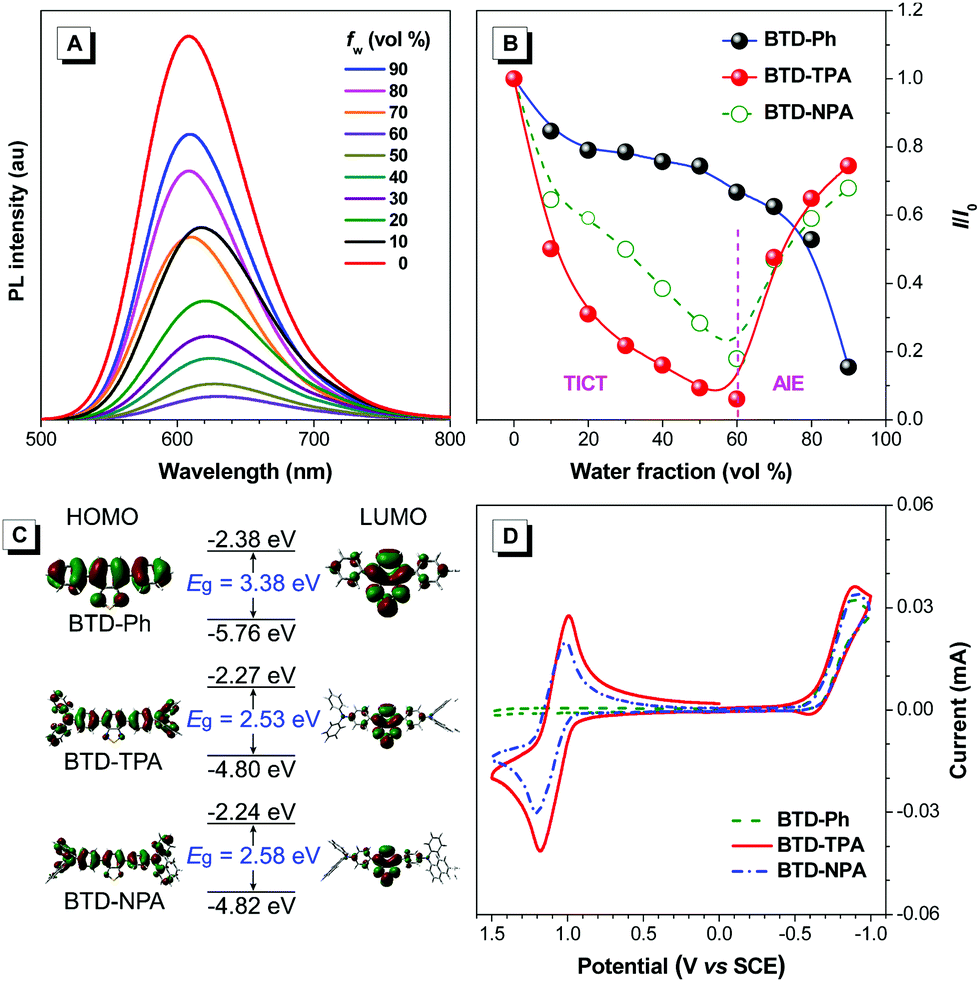
Encouraged by their promising solution ECL results and AIE features, we developed solid ECL films based on BTD-TPA and BTD-NPA (Fig. 3A). The films were facilely prepared by drop-casting DCM solution of the luminogens on a glassy carbon electrode (GCE). The solid films of BTD-TPA and BTD-NPA turn on at low voltages of +0.80 V and +0.65 V, respectively, which are much lower than that of Ru(bpy)32+ (+1.2 V).48 The ECL intensities of BTD-TPA and BTD-NPA reach the maximum values at +1.05 V and +1.14 V, respectively (Fig. S18–S20, ESI†). The ECL spectrum of the BTD-TPA film resembles its PL spectrum (Fig. 3B). The ECL quantum efficiency of BTD-TPA films is calculated to be 4.0%, using Ru(bpy)32+ as a reference with the ECL quantum efficiency of 5.0% (eqn (S1), ESI†). The ECL signal of BTD-TPA films remains almost at a constant value during consecutive cyclic potential scanning with an RSD of 1.4% (Fig. 3C). In contrast, the ECL intensity of BTD-NPA films gradually decreases to 36.5% of its maximum value after 27 cycles (Fig. S21, ESI†) due to the formation of a thin polymer film on the electrode surface.13 The corresponding Commission Internationale de l’Eclairage (CIE) coordinates of BTD-Ph (I), BTD-TPA (II), and BTD-NPA (III) in DCM solution are calculated to be (0.17, 0.35), (0.62, 0.36), and (0.62, 0.36), respectively. While the CIE coordinates of the spectrum of BTD-TPA solid film are calculated to be (0.56, 0.41), as shown in Fig. 3D. The ECL of BTD-TPA films is explained through an oxidative-reduction co-reactant mechanism (Fig. 3E). Both BTD-TPA and co-reactant TPrA are first oxidized to the corresponding radical cation BTD-TPA˙+ and intermediate radical cation TPrA˙+. TPrA˙+ loses a proton to generate the neutral radical TPrA˙ on the electrode surface. Then BTD-TPA˙+ is reduced by TPrA˙ to form excited-state BTD-TPA*, which finally emits light and returns to its ground state (eqn (S2)–(S6), ESI†).49,50
 | ||
| Fig. 3 (A) Digital photos of the experimental setup after the ECL experiment under daylight and UV illumination. (B) Normalized ECL and PL spectra of the BTD-TPA films. Excitation: 460 nm. (C) ECL stability of the BTD-TPA films. (D) ECL photos of BTD-Ph (I), BTD-TPA (II), and BTD-NPA (III) in DCM solution, the BTD-TPA film (IV), and CIE chromaticity coordinates of their ECL spectra. (E) Proposed ECL mechanism of BTD-TPA films. | ||
The brightness of the ECL films is tunable. The ECL intensity of the BTD-TPA films is remarkably enhanced with the increase in the luminogen loading from 25 ng mm−2 to 1 μg mm−2 (Fig. 4). The thickness of the solid films was further measured using a fluorescence microscope. Interestingly, the ECL intensity of the BTD-TPA films with increasing thickness demonstrates a linear relationship (R = 0.9973). The ECL intensity of the films is enhanced by 58-fold when the film thickness is increased from 26 μm to 130 μm. Evidently, the ECL intensity of BTD-TPA films is tunable through facilely regulating luminogen loading or film thickness, which is useful for their practical applications. Such tunable ECL intensity of BTD-TPA films benefits from AIE features of the luminogen. The current during ECL of BTD-TPA films was monitored. The current increases linearly with increasing luminogen loading (Fig. S22, ESI†), suggesting that BTD-TPA molecules in the films involve ECL reactions.
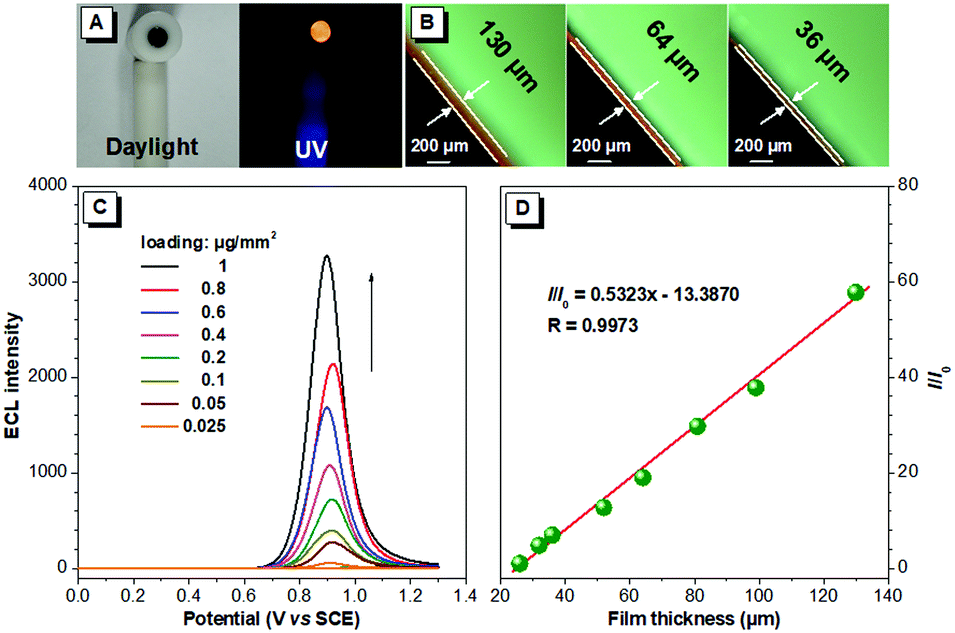 | ||
| Fig. 4 (A) Digital photos of BTD-TPA film under daylight and UV illumination. (B) Optical microscope images of BTD-TPA films with different thicknesses. (C) ECL intensity of BTD-TPA films with various loadings. (D) Plot of relative ECL intensity (I/I0) versus the different thickness of BTD-TPA films; I0 denotes the ECL emission intensity of BTD-TPA film with a thickness of 26 μm. | ||
To investigate the morphologies and microstructure of BTD-TPA films, scanning electron microscopy (SEM) and X-ray diffraction (XRD) were performed. As shown in Fig. S23 (ESI†), continous and dense films are observed. BTD-TPA films have a smooth surface. No ordered sub-structures are recognized in the films. Moreover, the XRD spectrum displays that the diffraction peaks of BTD-TPA film are in good agreement with the simulated peak from single crystals,45 confirming the formation of a crystal structure in BTD-TPA films (Fig. S24, ESI†). Furthermore, electrochemical impedance spectroscopy (EIS) was employed to monitor the electrochemical properties of BTD-TPA films at various loadings. The semicircle diameter at higher frequencies reflects the electron-transfer resistance (Rct), which is related to the electron-transfer kinetics. The bare GCE displays a small Rct value (225 Ω, Fig. S25, ESI†). Modification of the GCE with BTD-TPA at 0.1 μg mm−2 leads to a 5-fold Rct value (1022 Ω). With increasing BTD-TPA loading, the Rct value increases. The GCE modified with 1 μg mm−2 of BTD-TPA shows an Rct value of 2199 Ω. The moderate Rct values of the BTD-TPA modified GCE in the order of kΩ show an acceptable electrical conductivity of BTD-TPA films and efficient electrical communication among the GCE, BTD-TPA films, and supporting electrolyte.
In ECL films, a small amount of luminogens (typically 50 ng to 14 μg) is used.16 In contrast, much higher amounts of luminogens by two orders of magnitude are required in solution ECL systems (typically 2.0 mg).10,51 Thus, the present ECL films greatly reduce the consumption of luminogens. Moreover, in ECL films, luminogens are loaded on the electrode surface. Poor solvents of luminogens such as water and acetonitrile are used as supporting liquid. A large volume of toxic and volatile solvents such as DCM is required in solution ECL systems. Evaporation of the toxic solvents during ECL measurements causes not only unstable ECL but also environmental issues. Thus, the ECL films are a promising platform for further applications. In recent years, there have been a few reports of immobilization of metal complexes on the electrode surface for ECL through the Langmiur–Blodegtt method or self-assembly.15 However, the immobilization methods are far from satisfactory due to the problems from unstable biased potentials and easy desorption of luminophores from solid electrodes. Although a series of approaches for ECL enhancement through the preparation of modified electrodes are reported,52–54 they involve complicated modification processes on electrodes. This greatly hinders their practical applications. Thus, the present work using inexpensive organic AIE luminogens demonstrates a simple and direct approach for the development of bright and tunable ECL devices without involving any ancillary methods.
With the merits of bright ECL and superior stability, BTD-TPA films were used for the detection of dopamine (DA). The blank BTD-TPA film emits strong ECL with an emission maximum at 607 nm. The ECL signal of BTD-TPA films is stable and reproducible. ECL intensity changes rarely during consecutive scanning (Fig. S26, ESI†). The ECL intensity of BTD-TPA films is gradually quenched with increasing concentration of DA. When the concentration of DA is increased to 350 μM, the ECL intensity of BTD-TPA thin film is almost quenched with only <4% of its initial ECL intensity (Fig. 5A). ECL quenching of BTD-TPA films follows the Stern–Volmer equation at low DA concentration.55
| I0/I = K[Q] + 1 | (1) |
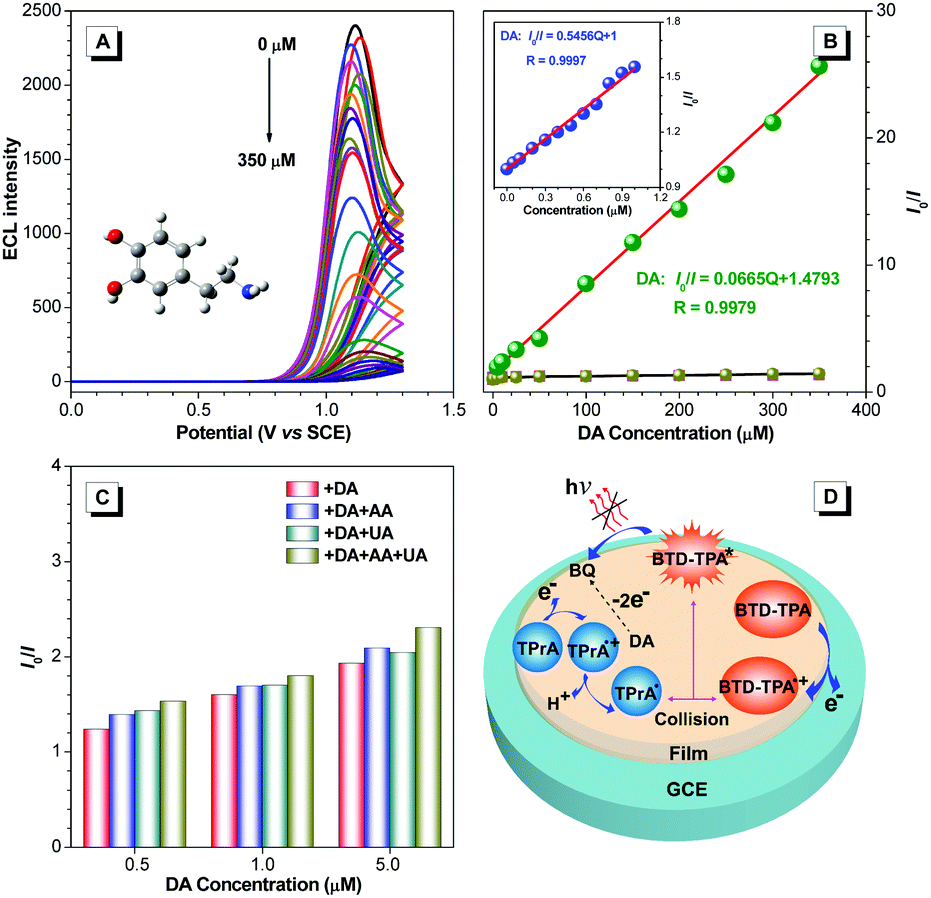 | ||
| Fig. 5 (A) ECL intensity versus potential profiles of BTD-TPA film with different concentrations of DA. (B) Stern–Volmer plots of I0/I of BTD-TPA film versus the concentration of DA, AA, and UA. Inset: Stern–Volmer plot at low concentration of DA (0–1 μM). (C) I0/I of BTD-TPA film with different concentrations of DA containing 100-fold concentrations of AA and UA. (D) Proposed ECL quenching mechanism of the BTD-TPA film modified GCE. | ||
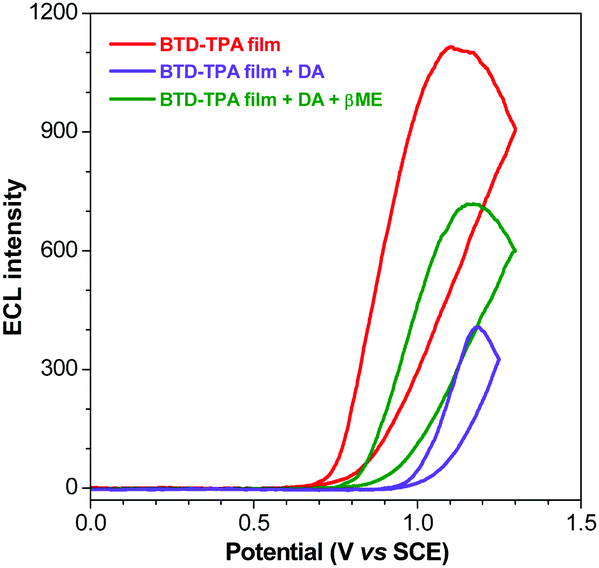 | ||
| Fig. 6 ECL curves of the BTD-TPA film (red line), +5 μM DA (violet line) and +100 μM βME (green line). | ||
Thus, the ECL films in this work offer a promising platform for specific detection of DA. Recently, there have been a few reports of DA detection using different modified electrodes (Fig. 7 and Table S2, ESI†).58–73 Most of the reported methods have detection limits from 33 nM to 1400 nM. Moreover, relevant quenching constants are rarely reported.58,60 Compared to previous reports, the present results are interesting with a high quenching constant (5.5 × 105 M−1), a wider linear range (0.05–350 μM), and a lower detection limit (17.0 nM).
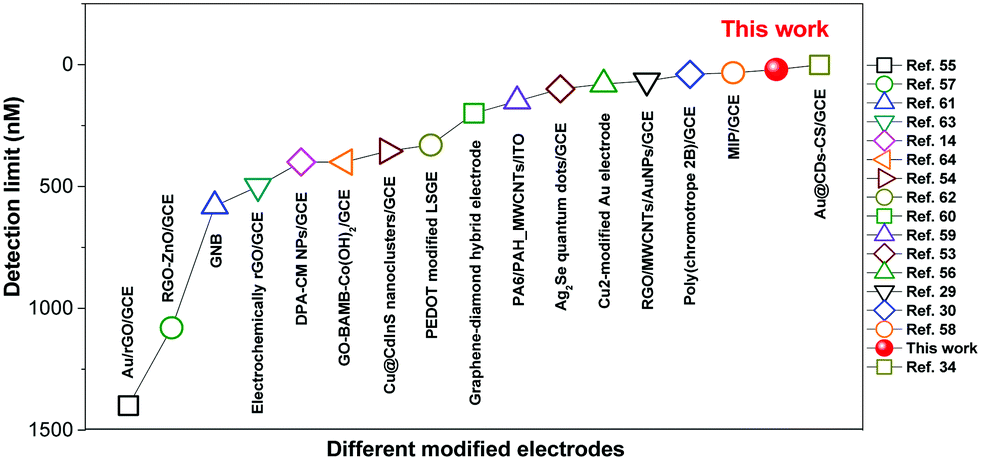 | ||
| Fig. 7 Summary of detection limits for DA using different modified electrodes in the literature. | ||
Conclusions
In summary, we have constructed two red luminogens from benzothiadiazole (BTD) and arylamino units, abbreviated as BTD-TPA and BTD-NPA, through one-step Suzuki reaction. The resultant molecules show AIE effects with high fluorescence efficiency and reversible redox pairs with high stability. Bright ECL solid films are fabricated based on the efficient AIE luminogens. Interestingly, the ECL intensity of the non-doped films is proportional to the film thickness, which allows tuning their brightness through facile variation of luminogen loading. Moreover, the bright ECL films are utilized for sensitive detection of dopamine (DA). A high Stern–Volmer constant (5.5 × 105 M−1) and a low detection limit of 17.0 nM are achieved with good selectivity. Such bright ECL films open a promising avenue for rapid and quantitative analysis of bioassays with high sensitivity and selectivity.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The financial support is partially from NSFC (21374136), and the Fundamental Research Funds for the Central Universities (17lgjc03 and 18lgpy04).Notes and references
- M. M. Richter, Chem. Rev., 2004, 104, 3003–3036 CrossRef CAS PubMed
.
- L. Hu and G. Xu, Chem. Soc. Rev., 2010, 39, 3275–3304 RSC
- L. Li, Y. Chen and J. J. Zhu, Anal. Chem., 2017, 89, 358–371 CrossRef CAS
- W. Cao, J. P. Ferrance, J. Demas and J. P. Landers, J. Am. Chem. Soc., 2006, 128, 7572–7578 CrossRef CAS PubMed
- G. Jie, H. Huang, X. Sun and J. J. Zhu, Biosens. Bioelectron., 2008, 23, 1896–1899 CrossRef CAS PubMed
- S. Liu, H. Yuan, H. Bai, P. Zhang, F. Lv, L. Liu, Z. Dai, J. Bao and S. Wang, J. Am. Chem. Soc., 2018, 140, 2284–2291 CrossRef CAS PubMed
- G. Valenti, S. Scarabino, B. Goudeau, A. Lesch, M. Jović, E. Villani, M. Sentic, S. Rapino, S. Arbault, F. Paolucci and N. Sojic, J. Am. Chem. Soc., 2017, 139, 16830–16837 CrossRef CAS PubMed
- S. Voci, B. Goudeau, G. Valenti, A. Lesch, M. Jović, S. Rapino, F. Paolucci, S. Arbault and N. Sojic, J. Am. Chem. Soc., 2018, 140, 14753–14760 CrossRef CAS PubMed
- P. Bertoncello and R. J. Forster, Biosens. Bioelectron., 2009, 24, 3191–3200 CrossRef CAS PubMed
- R. Zhang, F. Tong, L. Yang, J. R. Adsetts, T. Yan, R. Wang, Z. Ding and H. B. Wang, Chem. Commun., 2018, 54, 9897–9900 RSC
- F. Rizzo, F. Polo, G. Bottaro, S. Fantacci, S. Antonello, L. Armelao, S. Quici and F. Maran, J. Am. Chem. Soc., 2017, 139, 2060–2069 CrossRef CAS PubMed
- H. Qi, J. J. Teesdale, R. C. Pupillo, J. Rosenthal and A. J. Bard, J. Am. Chem. Soc., 2013, 135, 13558–13566 CrossRef CAS PubMed
- F. Polo, F. Rizzo, M. Veiga Gutierrez, L. De Cola and S. Quici, J. Am. Chem. Soc., 2012, 134, 15402–15409 CrossRef CAS PubMed
- H. Qi, Y. H. Chen, C. H. Cheng and A. J. Bard, J. Am. Chem. Soc., 2013, 135, 9041–9049 CrossRef CAS PubMed
- Z. Guo and S. Dong, Anal. Chem., 2004, 76, 2683–2688 CrossRef CAS PubMed
- M. Shen, X. H. Zhu and A. J. Bard, J. Am. Chem. Soc., 2013, 135, 8868–8873 CrossRef CAS PubMed
- Z. Wang, S. Chen, J. W. Y. Lam, W. Qin, R. T. K. Kwok, N. Xie, Q. Hu and B. Z. Tang, J. Am. Chem. Soc., 2013, 135, 8238–8245 CrossRef CAS PubMed
- J. Mei, N. L. C. Leung, R. T. K. Kwok, J. W. Y. Lam and B. Z. Tang, Chem. Rev., 2015, 115, 11718–11940 CrossRef CAS PubMed
- J. Mei, Y. Huang and H. Tian, ACS Appl. Mater. Interfaces, 2018, 10, 12217–12261 CrossRef CAS PubMed
- C. Wang and Z. Li, Mater. Chem. Front., 2017, 1, 2174–2194 RSC
- X. Wei, M. J. Zhu, Z. Cheng, M. Lee, H. Yan, C. Lu and J. J. Xu, Angew. Chem., 2019, 131, 3194–3198 CrossRef
- Z. Han, Z. Yang, H. Sun, Y. Xu, X. Ma, D. Shan, J. Chen, S. Huo, Z. Zhang, P. Du and X. Lu, Angew. Chem., 2019, 131, 5976–5980 CrossRef
- W. Qin, P. Zhang, H. Li, J. W. Y. Lam, Y. Cai, R. T. K. Kwok, J. Qian, W. Zheng and B. Z. Tang, Chem. Sci., 2018, 9, 2705–2710 RSC
- W. Qin, Z. Yang, Y. Jiang, J. W. Y. Lam, G. Liang, H. S. Kwok and B. Z. Tang, Chem. Mater., 2015, 27, 3892–3901 CrossRef CAS
- X. Han, B. Zhang, J. Chen, S. H. Liu, C. Tan, H. Liu, M. J. Lang, Y. Tan, X. Liu and J. Yin, J. Mater. Chem. B, 2017, 5, 5096–5100 RSC
- X. Y. Wang, J. Zhang, Y. B. Dong, Y. Zhang, J. Yin and S. H. Liu, Dyes Pigm., 2018, 156, 74–81 CrossRef CAS
- L. Chen, Y. Jiang, H. Nie, P. Lu, H. H. Y. Sung, I. D. Williams, H. S. Kwok, F. Huang, A. Qin, Z. Zhao and B. Z. Tang, Adv. Funct. Mater., 2014, 24, 3621–3630 CrossRef CAS
- Y. Liu, S. Chen, J. W. Y. Lam, P. Lu, R. T. K. Kwok, F. Mahtab, H. S. Kwok and B. Z. Tang, Chem. Mater., 2011, 23, 2536–2544 CrossRef CAS
- Y. Liu, Y. Lv, H. Xi, X. Zhang, S. Chen, J. W. Y. Lam, R. T. K. Kwok, F. Mahtab, H. S. Kwok, X. Tao and B. Z. Tang, Chem. Commun., 2013, 49, 7216–7218 RSC
- S. R. Ali, Y. Ma, R. R. Parajuli, Y. Balogun, W. Y. C. Lai and H. He, Anal. Chem., 2007, 79, 2583–2587 CrossRef CAS PubMed
- C. Muzzi, E. Bertocci, L. Terzuoli, B. Porcelli, I. Ciari, R. Pagani and R. Guerranti, Biomed. Pharmacother., 2008, 62, 253–258 CrossRef CAS PubMed
- V. Carrera, E. Sabater, E. Vilanova and M. A. Sogorb, J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2007, 847, 88–94 CrossRef CAS PubMed
- F. Shang, L. Zhou, K. A. Mahmoud, S. Hrapovic, Y. Liu, H. A. Moynihan, J. D. Glennon and J. H. T. Luong, Anal. Chem., 2009, 81, 4089–4098 CrossRef CAS PubMed
- Z. Su, Y. Liu, Q. Xie, L. Chen, Y. Zhang, Y. Meng, Y. Li, Y. Fu, M. Ma and S. Yao, Biosens. Bioelectron., 2012, 36, 154–160 CrossRef CAS PubMed
- L. Wei, Y. Lei, H. Fu and J. Yao, ACS Appl. Mater. Interfaces, 2012, 4, 1594–1600 CrossRef CAS PubMed
- H. Y. Yue, S. Huang, J. Chang, C. Heo, F. Yao, S. Adhikari, F. Gunes, L.
C. Liu, T. H. Lee, E. S. Oh, B. Li, J. J. Zhang, T. Q. Huy, N. V. Luan and Y. H. Lee, ACS Nano, 2014, 8, 1639–1646 CrossRef CAS PubMed
- H. Muguruma, Y. Inoue, H. Inoue and T. Ohsawa, J. Phys. Chem. C, 2016, 120, 12284–12292 CrossRef CAS
- L. Jiang, G. W. Nelson, J. Abda and J. S. Foord, ACS Appl. Mater. Interfaces, 2016, 8, 28338–28348 CrossRef CAS PubMed
- F. Wu, T. Huang, Y. Hu, X. Yang, Y. Ouyang and Q. Xie, Microchim. Acta, 2016, 183, 2539–2546 CrossRef CAS
- W. Deng, X. Yuan, Y. Tan, M. Ma and Q. Xie, Biosens. Bioelectron., 2016, 85, 618–624 CrossRef CAS PubMed
- J. Chen, F. Ye, Y. Lin, Z. Chen, S. Liu and J. Yin, Sci. China: Chem., 2019, 62, 440–450 CrossRef CAS
- J. Chen, D. Li, W. Chi, G. Liu, S. H. Liu, X. Liu, C. Zhang and J. Yin, Chem. – Eur. J., 2018, 24, 3671–3676 CrossRef CAS PubMed
- K. Shin Ichiro, M. Taisuke, S. Motoyuki, G. Hideki, M. Shuuichi, I. I. Tsutomu and M. Shuntaro, Chem. – Eur. J., 2010, 12, 2303–2317 Search PubMed
- S. Sahami and M. J. Weaver, J. Electroanal. Chem. Interfacial Electrochem., 1981, 122, 155–170 CrossRef CAS
- S. i. Kato, T. Matsumoto, T. Ishi, T. Thiemann, M. Shigeiwa, H. Gorohmaru, S. Maeda, Y. Yamashita and S. Mataka, Chem. Commun., 2004, 2342–2343 RSC
- W. Qin, D. Ding, J. Liu, W. Z. Yuan, Y. Hu, B. Liu and B. Z. Tang, Adv. Funct. Mater., 2012, 22, 771–779 CrossRef CAS
- J. Suk, Z. Wu, L. Wang and A. J. Bard, J. Am. Chem. Soc., 2011, 133, 14675–14685 CrossRef CAS PubMed
- M. Mayer, S. Takegami, M. Neumeier, S. Rink, A. Jacobi von Wangelin, S. Schulte, M. Vollmer, A. G. Griesbeck, A. Duerkop and A. J. Baeumner, Angew. Chem., Int. Ed., 2018, 57, 408–411 CrossRef CAS PubMed
- R. Y. Lai and A. J. Bard, J. Phys. Chem. A, 2003, 107, 3335–3340 CrossRef CAS
- W. Miao, J. P. Choi and A. J. Bard, J. Am. Chem. Soc., 2002, 124, 14478–14485 CrossRef CAS PubMed
- K. N. Swanick, S. Ladouceur, E. Zysman Colman and Z. Ding, Chem. Commun., 2012, 48, 3179–3181 RSC
- B. Wu, C. Miao, L. Yu, Z. Wang, C. Huang and N. Jia, Sens. Actuators, B, 2014, 195, 22–27 CrossRef CAS
- Y. P. Dong, G. Chen, Y. Zhou and J. J. Zhu, Anal. Chem., 2016, 88, 1922–1929 CrossRef CAS PubMed
- Y. Yu, C. Lu and M. Zhang, Anal. Chem., 2015, 87, 8026–8032 CrossRef CAS PubMed
- O. Stern and M. Volmer, Phys. Z., 1919, 20, 183–188 CAS
- J. McCall, C. Alexander and M. M. Richter, Anal. Chem., 1999, 71, 2523–2527 CrossRef CAS PubMed
- X. Liu, H. Jiang, J. Lei and H. Ju, Anal. Chem., 2007, 79, 8055–8060 CrossRef CAS PubMed
- H. Liu, L. Wang, H. Gao, H. Qi, Q. Gao and C. Zhang, ACS Appl. Mater. Interfaces, 2017, 9, 44324–44331 CrossRef CAS PubMed
- D. Yuan, S. Chen, R. Yuan, J. Zhang and X. Liu, Sens. Actuators, B, 2014, 191, 415–420 CrossRef CAS
- R. Cui, Y. P. Gu, L. Bao, J. Y. Zhao, B. P. Qi, Z. L. Zhang, Z. X. Xie and D. W. Pang, Anal. Chem., 2012, 84, 8932–8935 CrossRef CAS PubMed
- F. Wang, J. Lin, H. Wang, S. Yu, X. Cui, A. Ali, T. Wu and Y. Liu, Nanoscale, 2018, 10, 15932–15937 RSC
- X. B. Li, M. M. Rahman, G. R. Xu and J. J. Lee, Electrochim. Acta, 2015, 173, 440–447 CrossRef CAS
- C. Wang, J. Du, H. Wang, C. E. Zou, F. Jiang, P. Yang and Y. Du, Sens. Actuators, B, 2014, 204, 302–309 CrossRef CAS
- G. Jiang, X. Gu, G. Jiang, T. Chen, W. Zhan and S. Tian, Sens. Actuators, B, 2015, 209, 122–130 CrossRef CAS
- X. Zhang, Y. C. Zhang and L. X. Ma, Sens. Actuators, B, 2016, 227, 488–496 CrossRef CAS
- M. Zhong, Y. Teng, S. Pang, L. Yan and X. Kan, Biosens. Bioelectron., 2015, 64, 212–218 CrossRef CAS PubMed
- L. A. Mercante, A. Pavinatto, L. E. O. Iwaki, V. P. Scagion, V. Zucolotto, O. N. Oliveira, L. H. C. Mattoso and D. S. Correa, ACS Appl. Mater. Interfaces, 2015, 7, 4784–4790 CrossRef CAS PubMed
- Q. Yuan, Y. Liu, C. Ye, H. Sun, D. Dai, Q. Wei, G. Lai, T. Wu, A. Yu, L. Fu, K. W. A. Chee and C. T. Lin, Biosens. Bioelectron., 2018, 111, 117–123 CrossRef CAS PubMed
- P. K. Kannan, S. A. Moshkalev and C. S. Rout, Nanotechnology, 2016, 27, 075504 CrossRef PubMed
- Q. Huang, H. Zhang, S. Hu, F. Li, W. Weng, J. Chen, Q. Wang, Y. He, W. Zhang and X. Bao, Biosens. Bioelectron., 2014, 52, 277–280 CrossRef CAS PubMed
- G. Xu, Z. A. Jarjes, V. Desprez, P. A. Kilmartin and J. T. Sejdic, Biosens. Bioelectron., 2018, 107, 184–191 CrossRef CAS PubMed
- L. Yang, D. Liu, J. Huang and T. You, Sens. Actuators, B, 2014, 193, 166–172 CrossRef CAS
- A. Ejaz, Y. Joo and S. Jeon, Sens. Actuators, B, 2017, 240, 297–307 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available: Reagents, experimental details, apparatus and characterization. Movies, UV and PL spectra, electrochemical data, ECL measurements, and quantitative detection. See DOI: 10.1039/c9qm00401g |
| This journal is © the Partner Organisations 2019 |