
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTandem catalysis in multicomponent solvent-free biofluids†
Dylan Luke
Atkins
 *abc,
José Augusto
Berrocal
cd,
Alexander Francesco
Mason
ce and
Ilja Karina
Voets
*ac
*abc,
José Augusto
Berrocal
cd,
Alexander Francesco
Mason
ce and
Ilja Karina
Voets
*ac
aLaboratory of Self-Organizing Soft Matter, Department of Chemical Engineering and Chemistry, Eindhoven University of Technology, 5600 MB Eindhoven, The Netherlands. E-mail: d.atkins@tue.nl; i.voets@tue.nl
bLaboratory of Physical Chemistry, Department of Chemical Engineering and Chemistry, Eindhoven University of Technology, 5600 MB Eindhoven, The Netherlands
cInstitute for Complex Molecular Systems, Department of Chemical Engineering and Chemistry, Eindhoven University of Technology, 5600 MB Eindhoven, The Netherlands
dLaboratory of Macromolecular and Organic Chemistry, Department of Chemical Engineering and Chemistry, Eindhoven University of Technology, 5600 MB Eindhoven, The Netherlands
eLaboratory of Bio-Organic Chemistry, Department of Chemical Engineering and Chemistry, Eindhoven University of Technology, 5600 MB Eindhoven, The Netherlands
First published on 11th October 2019
Abstract
Enzymes are widely employed to reduce the environmental impact of chemical industries as biocatalysts improve productivity and offer high selectively under mild reaction conditions in a diverse range of chemical transformations. The poor stability of biomacromolecules under reaction conditions is often a critical bottleneck to their application. Protein engineering or immobilization onto solid substrates may remedy this limitation but, unfortunately, this is often at the expense of catalytic potency or substrate specificity. In this work, we show that the combinatorial approach of chemical modification and supramolecular nanoencapsulation can endow mechanistically diverse enzymes with apparent extremophilic behavior. A protein–polymer surfactant core–shell architecture facilitates construction of increasingly complex biofluids from individual biosynthetic components, each of which retain biological activity at hydration levels almost two orders of magnitude below solvation. The herein constructed multifunctional biofluids operate in tandem up to 150 °C and in the total absence of solvent under apparent diffusional mass-transport limitation. The biosynthetic promotion of extremophilic traits for enzymes with diverse catalytic motions and chemical functions highlights the extraordinary capacity for a viscous surfactant milieu to replace both hydration and bulk waters.
Introduction
The application of enzymes in biotechnology is dependent upon their performance in often harsh processing conditions such as high temperature and pressure, extreme pH, or a non-aqueous reaction environment.1 Whilst biocatalysts offer an extremely diverse range of accessible reactions with extensive substrate specificities, limiting factors to their commercial implementation are often poor stability and activity under industrial processing conditions.1,2 Various strategies have been developed to address these critical bottlenecks, including protein engineering,3 chemical modifications,4 and immobilization, entrapment or encapsulation.5 However, increased stability and tolerance to adverse reaction environments via post-translational engineering strategies often comes at the cost of decreased catalytic potency or substrate specificity.Extremozymes (enzymes derived from extremophiles) showcase an evolutionary honed tolerance to conditions which are considered hostile to life.6,7 Consequently, extremozymes have been touted as ideal candidates for use as industrial biocatalysts.8,9 This motivated considerable interest in extremophile discovery, especially for the isolation and characterization of extremophilic enzyme variants for potential industrial application.10,11 Further, cultivation methodologies for the large-scale expression of extremozymes have been developed.9,11 To date however, these are still less developed than conventional expression systems, and typically require conditions which mimic the natural environment of the extremophile. An alternative approach aims to understand in detail the specific structure–function adaptations of extremozymes to rationally re-design selected enzymes to enhance their tolerance to harsh environments.12–14 The use of homology modelling between TLP-ste and thermolysin to engineer an enzyme resistant to boiling14 is a prominent example showcasing the translational success of this route, which has also served to advance our understanding of the stabilizing mechanisms of extremozymes. Disadvantageously however, this strategy is protein-specific, suffers significant lag-time and consequently is expensive.
Protein–polymer surfactant biohybrids in the form of self-contained solvent-free biofluids have been evidenced to greatly enhance the stability and activity of mesophile-derived biocatalysts.15,16 Pioneering work by Perriman et al. (2010) revealed that solvent-free myoglobin biohybrids exhibited activity at extremely low hydration levels up to unusually high temperatures.16 Elastic incoherent neutron scattering experiments evidenced that this is because a polymer surfactant corona can act as a substitute for hydration waters, mediating motions which closely resemble those observed in the hydrated state.17 Similarly, substrate solubility, mass transport and catalytic activity were reported for solvent-free biofluids in a broad temperature range from the onset of melting at approximately Tm ∼ 30 °C up to 150 °C.18
Herein we report on the construction of a quasi-ternary biofluid, composed of three mechanistically diverse and mesophile-derived enzymes, which displays tandem catalytic behavior under solvent-free conditions (Fig. 1). We created the mixed self-contained biofluids from horseradish peroxidase (HRP), glucose oxidase (GOx), and Thermomyces lanuginosus lipase (TLL), each of which independently retained biological activity at hydration levels roughly two orders of magnitude below protein solvation. Significantly, the protein–polymer surfactant core–shell architecture promotes biohybrid miscibility and extremophilic behavior enabling the cascade to operate at temperatures far above room temperature. Surprisingly, we find an apparent thermal activation temperature of 80 °C and achieve tandem catalysis up to 150 °C under seemingly diffusional mass transport limitations within the viscous biofluid. The retention of biological activity for enzymes with distinctly different catalytic motions and chemical functions highlights the extraordinary capacity for a viscous surfactant milieu to replace both hydration and bulk waters.
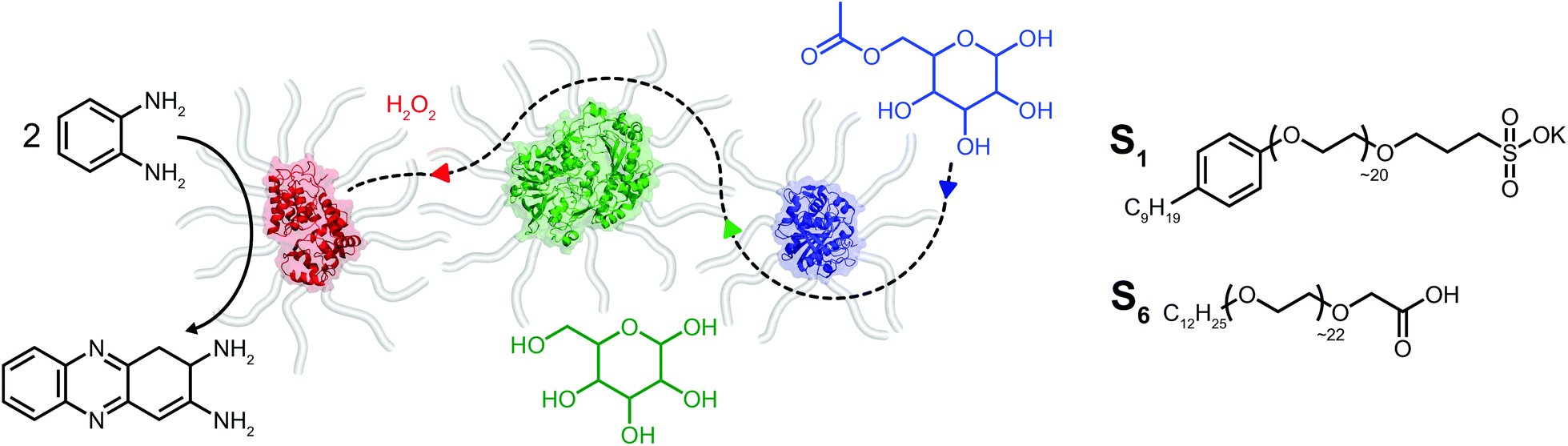 | ||
| Fig. 1 Chemical structures of polymer surfactants poly(ethylene glycol)-4-nonylphenyl-3-sulfopropyl ether (S1) and carboxylated Brij-L23 (S6) which are used in the production of solvent-free protein polymer hybrid nanoconjugates. Biohybrids produced from the three enzymes horseradish peroxidase (red), glucose oxidase (green), and Thermomyces lanuginosus lipase (blue) are utilized for constructing enzymatic cascades in the solvent-free phase. The enzyme-specific substrates acetylated glucose, glucose, and hydrogen peroxide are indicated in blue, green and red, respectively. Horseradish peroxidase biohybrid-mediated formation of 2,3-diaminophenazine from the o-phenylenediamine substrate is shown in black. The efficient diffusion, turnover and release of species is represented schematically by the dotted line. | ||
Results and discussion
Biohybrid preparation and characterization
Solvent-free protein–polymer surfactant biofluids were prepared using a modified two-step protocol (Fig. S1†).19 Briefly, cationic supercharged variants of horseradish peroxidase (cHRP), glucose oxidase (cGOx), and Thermomyces lanuginosus lipase (cTLL) were produced via EDC-mediated coupling of 3-(dimethylamino)-1-propylamine (DMAPA) to solvent accessible Asp and Glu residues. Coupling efficiencies of 78% (cHRP), 97% (cGOx), and 80% (cTLL) were estimated which correspond to the successful modification of 7 of 9, 37 of 38, and 20 of 25 solvent accessible acidic residues, respectively (Fig. S2†). An anionic polymer surfactant poly(ethylene glycol)-4-nonylphenyl-3-sulfopropyl ether (S1) was electrostatically coupled to the supercharged proteins (producing cHRP-S1, cGOx-S1, and cTLL-S1). Extensive dialysis was performed to remove unbound surfactant, and the resulting protein–polymer conjugates were then lyophilized extensively and annealed at 70 °C to produce free-flowing solvent-free biofluids.Thermogravimetric analysis (TGA) revealed half-decomposition temperatures ranging from 385 °C to 395 °C for the biohybrids. Further, mass losses upon thermal annealing at 110 °C for 60 min corresponded to 2–10 specifically bound waters per nanoconjugate (Fig. S4† and Table 1). For comparison, we computed the number of waters required for complete coverage of the solvent accessible surface area using the HyPred20 online tool, and associated PDB files (1hch, 1cf3, and 1ein21–23 (ESI† and Table 1). Our estimated water contents by TGA are at least two orders of magnitude less than those computed using HyPred (1400 for HRP, 5200 for the GOx dimer, and 1200 for TLL), thus confirming the preparation of solvent-free biohybrids. In the context of enzyme activity, these values correspond to extremely small hydration levels (h < 0.002), which are also roughly two orders of magnitude less than the apparent hydration limit for activity of h ∼ 0.2. Differential scanning calorimetry (DSC) showed reversible endothermic melting transitions at 24 °C, 29 °C and 22 °C for cHRP-S1, cGOx-S1, and cTLL-S1 respectively (Fig. S5† and Table 1). Taken together, TGA and DSC analysis confirmed the preparation of solvent-free biohybrids with liquid-like behaviors which persisted in the absence of hydration. These characteristics are highly consistent with previous reports in the literature.16,18,19
| Species | n bw | h | n theo | T m/°C |
|---|---|---|---|---|
| Neat S1 | 3.2 | — | — | 10 |
| cHRP-S1 | 9.4 | 0.0019 | 1398 | 24 |
| cGOx-S1 | 6.2 | 0.0015 | 5183 | 29 |
| cTLL-S1 | 2.2 | 0.0007 | 1194 | 22 |
Given the successful preparation of solvent-free melts, circular dichroism (CD) spectroscopy was performed in solution on re-suspended hybrids to elucidate the impact of supercharging and subsequent electrostatic coupling of a polymer surfactant on the respective protein secondary structures (Fig. 2a–c). Due to high absorbance of the nonylphenyl moiety in S1, complementary CD experiments were performed using biohybrids prepared with a carboxylated24 Brij-L23 (S6) surfactant (ESI†). Despite the increased surface charge due to extensive covalent modification of the solvent accessible acidic sidechains, we observed near-native folding of each protein, corresponding to approximately 93.7% (cHRP), 94.4% (cGOx), and 100% (cTLL) retained secondary structures (Table 2). These were reduced to 84.0%, 83.7%, and 77.0% upon polymer surfactant conjugation for cHRP-S6, cGOx-S6, and cTLL-S6, respectively. Since S1 and S6 are structurally similar, we expect similarly modest misfolding for the S1-containing hybrids.
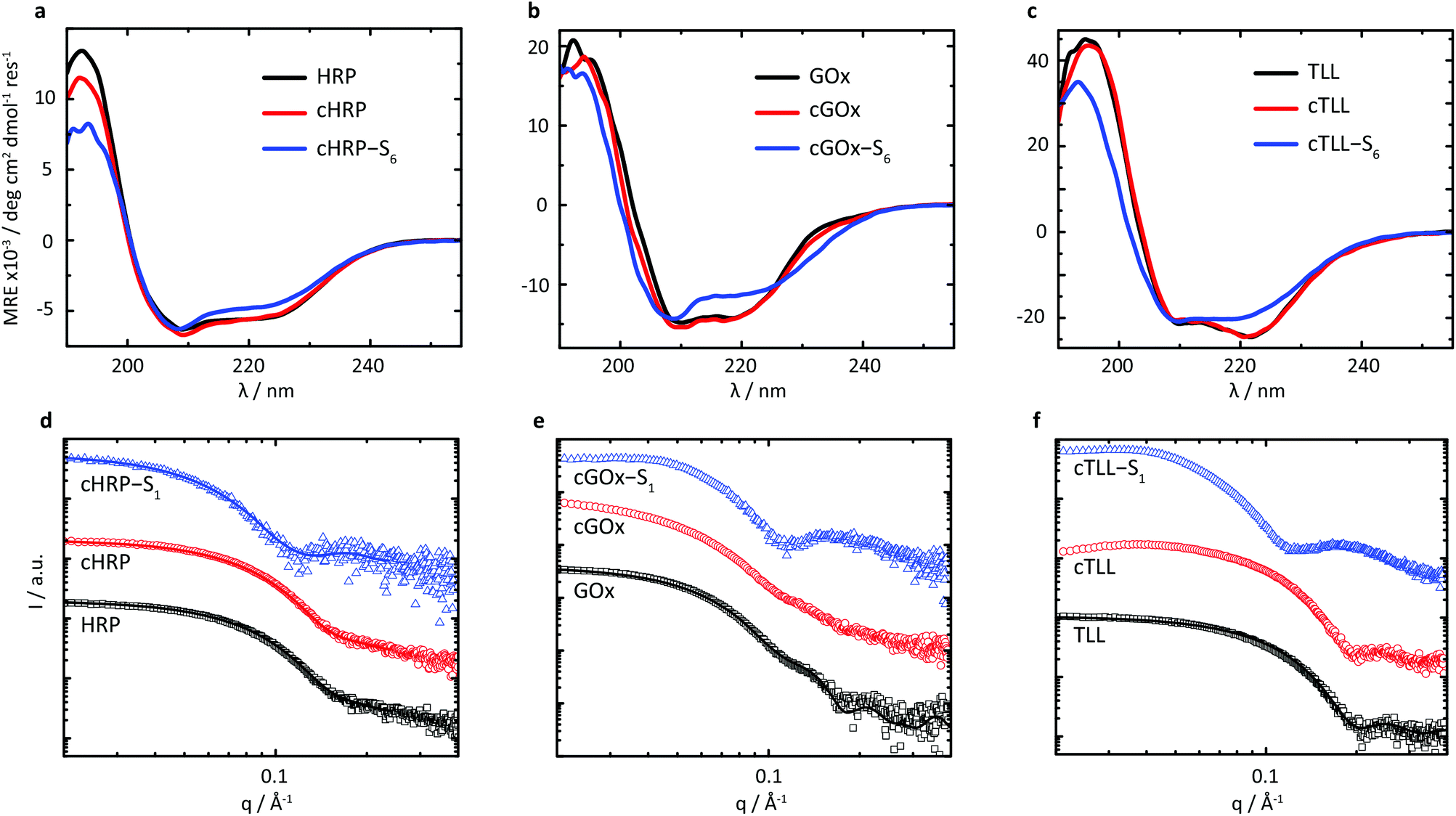 | ||
| Fig. 2 (a–c) Circular dichroism spectroscopy traces of native (black), supercharged (red), and S6-coupled (blue) variants for (a) horseradish peroxidase, (b) glucose oxidase, and (c) thermomyces lanuginosus lipase. CD traces show high degrees of secondary structure retention upon cationization, and small losses of native folds upon electrostatic conjugation of polymer surfactant. (d–f) Solution-state SAXS profiles of native (black), supercharged (red), and S1-coupled (blue) variants for (d) horseradish peroxidase, (e) glucose oxidase, and (f) Thermomyces lanuginosus lipase. Scattering profiles indicate high degrees of retention of native morphologies upon cationization, and the successful construction of polymeric coronas. | ||
| Variant | Secondary structure retained/% |
|---|---|
| cHRP | 93.7 |
| cHRP-S6 | 84.0 |
| cGOx | 94.4 |
| cGOx-S6 | 83.7 |
| cTLL | 100 |
| cTLL-S6 | 77.0 |
We performed small-angle X-ray scattering (SAXS) experiments in the solution-state to characterize morphological changes of the globular protein structure, and to validate the encapsulation of the proteins to produce biosynthetic nanoconjugates (Fig. 2d–f). The SAXS profiles of native HRP and cHRP were fitted with an ellipsoid form factor25 of comparable dimensions (HRP: rmin = 18.2 Å, rmax = 32.9 Å; cHRP: rmin = 18.6 Å, rmax = 33.1 Å), which were roughly consistent with the crystallographic dimensions determined from the associated PDB file 1hch. The slightly larger radii used to fit the cHRP SAXS curves (Table S1†) are in line with the small loss of native globular fold observed by CD. Significantly, the construction of nanoencapsulated biosynthetic enzymes was confirmed for cHRP-S1 by fitting the SAXS profile with fixed cHRP protein dimensions while accounting for a thin polymeric corona using a core–shell ellipsoid26,27 form factor. This revealed a compressed polymeric corona of approximately 3.1 nm primarily located along the minor axis of cHRP, with a very minimal contribution (∼0.5 nm) at the ellipsoidal extremities (Table S2†). For comparison we show a scattering profile using a homogenous shell thickness (3.1 nm) encapsulating cHRP which resulted in a significantly poorer fit (Fig. S10†).
The compacted and patchy coverage of a supercharged enzyme in aqueous solution is in line with previous reports on polymer-tethered lysozyme and myoglobin studied by a combination of small-angle neutron scattering and molecular dynamics simulations.28 We further confirmed its correspondence with the cationized protein's surface charge distribution by performing continuum electrostatics calculations via the PDB2PQR web server29,30 to map the electrostatic potential of the supercharged protein surface (Fig. S3†). Convincingly, this revealed considerably high positive charge densities on the cHRP minor axis, with a relatively neutral charge remaining at the major axis extremities.
Scattering profiles were also computed using Crysol31 and respective crystal structures for GOx (symmetrical dimer) and TLL. These results first confirmed that the associated homodimeric state of GOx was largely lost upon modification of solvent accessible acidic residues. This is evidenced by the loss of subtle undulations associated with the quaternary structure of dimeric assembly at values of q > 0.1 Å. Further, the globular state of TLL is distinctly well-preserved upon covalent modification to produce cTLL, as demonstrated by the excellent correspondence between the respective scattering profiles. Finally, the subsequent charge-driven self-assembly of supercharged protein and polymer surfactant is signalled by distinct transformations in scattering profiles of cGOx-S1 and cTLL-S1 compared to their respective supercharged variants. Convincingly, these transformations closely match that of cHRP-S1, and are qualitatively similar to previously published scattering profiles of core–shell biohybrid enzymes.32 Given these insights, we reasonably conclude the successful construction of core–shell structured biosynthetic enzymes.
Multi-enzyme biofluids for solvent-free tandem catalysis
Given the successful preparation of discrete protein nanocompartments which displayed near room temperature melting and conserved globular protein states, we decided to probe enzyme activity in the solvent-free environment. The formation of 2,3-diaminophenazine (DAP, ε418 = 16.7 mM−1 cm−1)32 from o-phenylenediamine (OPD) by the biosynthetic cHRP-S1 was followed by absorbance using a Linkam-modified UV-vis spectrophotometer.33 An initial experiment was performed at 90 °C, whereby H2O2 was supplied to the reaction environment by soaking a quartz plate in a 1% H2O2 solution for 5 min and allowing it to air-dry (Fig. S11†). Remarkably, residual H2O2 was able to diffuse into the solvent-free melted phase to be scavenged for the production of DAP at an estimated initial rate of 3.28 ± 0.82 μM s−1. Negative control experiments performed at 110 °C confirm that cHRP is essential for catalysis and OPD auto-oxidation is negligible as no DAP was produced for cHRP-S1 without H2O2, or in the neat surfactant S1 with H2O2 but without cHRP (Fig. S11†). Notably, the catalytic turnover of OPD by cHRP-S1 confirms that the heme redox cofactor remained specifically bound. Importantly, the heme group of HRP is considerably more buried than that of other heme-coordinated proteins such as myoglobin. In solution the heme access pathway for H2O2 is afforded by a dynamic Phe68/Phe142 gate, and the penetrative access of oxygen likely proceeds via this fluctuating entry point (Fig. 3a).34 Critically, the retention of enzyme activity confirms that the small internal pore of the heme pocket remained open, and these findings suggest that conformational motions of the fluctuating gate were unhindered at 90 °C.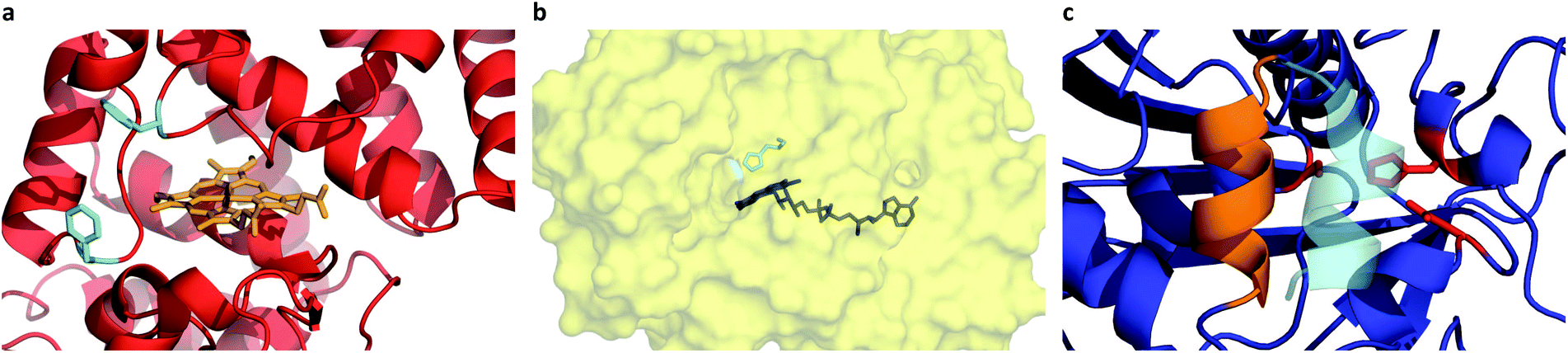 | ||
| Fig. 3 (a) Coordinated heme redox cofactor (orange) of horseradish peroxidase. The fluctuating entry point formed by the Phe68/Phe142 gate (cyan) mediates access of H2O2 and O2. (b) Deep internalized binding pocket of glucose oxidase showing FAD cofactor (blue) and the flexible site of oxygen activation His516 (cyan). (c) Open (orange) and closed (cyan) conformations of the helical lid motif of the Thermomyces lanuginosus lipase. The catalytic triad formed by residues Ser146-His258-Asp201 is shown in red. | ||
We were now confident that the amphiphilic environment afforded by the surfactant should effectively mediate small molecule transport. Aiming to interrogate the chemical and thermal limits of enzymatic catalysis in solvent-free liquids, and to supply H2O2 in a well-controlled manner, we devised a 2-step catalytic pathway producing H2O2in situ via the cGOx-S1 catalyzed conversion of glucose into glucono-1,5-lactone and H2O2. We anticipated that this would be followed by the cHRP-S1 catalyzed and H2O2 mediated conversion of ODP into DAP given that H2O2 diffusion appeared to be possible despite a bulk surfactant milieu.35,36 Preliminary microscopy experiments confirmed efficient solubilization of glucose at the solid–liquid interface, and independent liquid–liquid miscibility of the substrate OPD above 100 °C (Fig. S7 and S8†).
A series of high temperature assays from 40–140 °C were subsequently performed for cGOx-S1/cHRP-S1 by first annealing neat desiccated OPD with the two-component biofluid, followed by the addition of solid glucose substrate (Fig. 4a and b). We found no appreciable DAP production below 80 °C (Fig. S12†) despite temperatures far exceeding the melting point of the mixed fluid (Fig. S6†). By contrast, DAP was efficiently produced above 80 °C. An impressive initial rate of 3.46 ± 1.81 μM s−1 was measured at 80 °C, which was followed by an apparent exponential enhancement in enzymatic turnover with respect to temperature (Table 3). For example, at 140 °C we found an almost 22-fold enhancement in turnover up to 74.50 ± 1.58 μM s−1. The enhanced solvent-free turnovers clearly demonstrate that glucose can access the deep internalized binding pocket of cGOx-S1 despite the absence of solvent (Fig. 3b). The unhindered access of the deep pocket is striking considering the hybrid undergoes structural reordering upon polymer conjugation. This reordering does not appear to have greatly perturbed the deep pocket opening or collapsed the internal cavity. Further, glucose binding necessitates a degree of flexibility of the oxygen activation site (His516) in order to accommodate hydrogen bonding with neighboring residues within the rigid, tight cavity.22,37 Catalysis confirms a polymer surfactant corona effectively replaced hydration water in order to mediate the small-scale motion required for residue-specific flexibility. However, we cannot rule out that the 6 waters per cGOx-S1 nanoconjugate observed by TGA are located at the entrance, or within the deep pocket itself. Residual waters may assist in small molecule transport or aid in stabilization of the deep pocket.
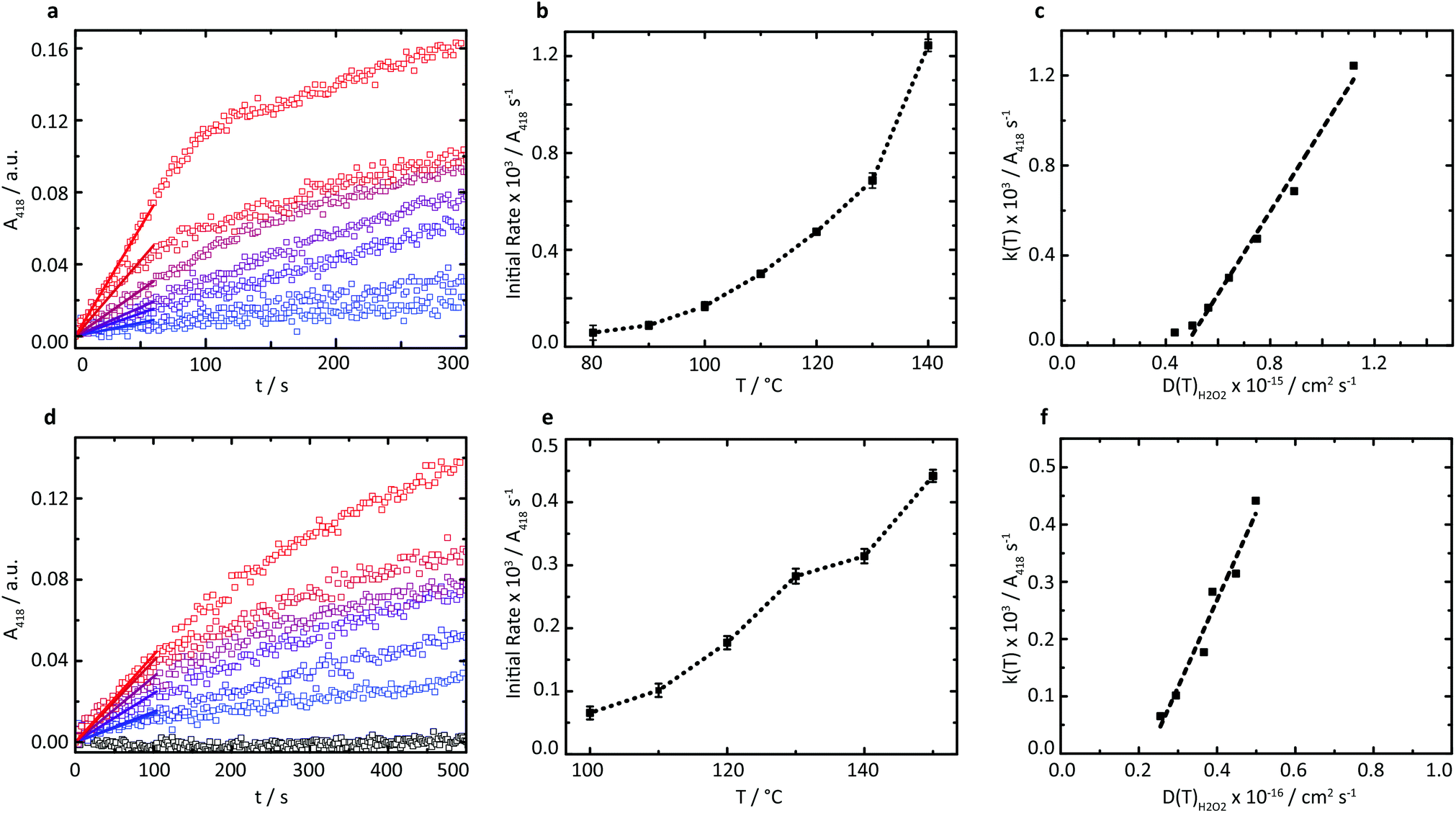 | ||
| Fig. 4 (a) Plots of absorbance at 418 nm versus time indicating the production of DAP from 80 °C to 140 °C at 10 °C intervals (blue to red) in the cGOx-S1/cHRP-S1 cascade. (b) Linear regression analysis of the two-enzyme cascade profiles (over 60 s) indicates a strong temperature dependence of initial reaction velocity. (c) Initial reaction velocity k(T) versus H2O2 diffusion coefficient D(T) is linear above 80 °C, indicating the production of DAP is diffusion-limited. (d) Plots of absorbance at 418 nm versus time indicating the production of DAP at 110 °C is inhibited in the cGOx-S1/cHRP-S1 pair upon addition of acetyl-protected glucose (open black squares). Addition of cTLL-S1 to the solvent-free biofluid re-initiates a three-enzyme cascade, as shown by the production of DAP from 100 °C to 150 °C at 10 °C intervals (blue to red). (e) Linear regression analysis of the three-enzyme cascade profiles (over 100 s) indicates a strong temperature dependence of initial reaction velocity. (f) Initial three-enzyme reaction velocity k(T) versus H2O2 diffusion coefficient D(T) is linear, indicating the production of DAP is diffusion-limited. | ||
| T/°C | |||||||
|---|---|---|---|---|---|---|---|
| 80 | 90 | 100 | 110 | 120 | 130 | 140 | |
| DAP production/μM s−1 | 3.46 | 5.30 | 10.04 | 18.03 | 28.41 | 41.09 | 74.50 |
| St. Dev./μM s−1 | 1.81 | 1.37 | 1.52 | 1.31 | 1.29 | 1.88 | 1.58 |
The rapid conversion of glucose in the solvent-free cGOx-S1/cHRPS1 biofluid confirms reactant solubilization, conversion, mass transport, and active site access despite the absence of hydration waters and bulk solvent at h < 0.002. Furthermore, the observed catalytic behavior of biosynthetic enzymes was strongly temperature-dependent above 80 °C. However, protection of the glucose substrate via acetylation36 (AcGl, ESI†) resulted in total loss of enzymatic recognition of the substrate at 110 °C (Fig. 4d). Control over substrate turnover and specificity further suggest a remarkably well-preserved active-site environment, with small-scale catalytic motions which should closely resemble aqueous GOx. Finally, we were able to reinitiate the cascade by introduction of a third biosynthetic enzyme component to yield the cTLL-S1/cGOx-S1/cHRP-S1 mixed biofluid.
A new series of high-temperature kinetic assays were performed by first annealing OPD with the three-enzyme complex biofluid, followed by addition of the protected AcGl. Initially, we confirmed that the absorbance profile for the multicomponent biofluid indeed plateaued over extensive time periods up to 2600 s (Fig. S13†). We then observed efficient production of DAP at elevated temperatures up to 150 °C (Fig. 4d and e) despite the requirement of a water molecule for the hydrolysis of the acetyl moiety. Further, pronounced thermophilic behavior was observed, corresponding to an almost 7-fold enhancement in turnover from 3.92 ± 0.63 μM s−1 at 100 °C up to 26.45 ± 0.58 μM s−1 at 150 °C (Table 4). The estimated turnover per mass of the solvent-free biofluid is roughly 5.3 μM s−1 mg−1 at 150 °C, which is similar to the catalytic performance of a reported three-component (HRP, GOx, and β-glucosidase) self-standing film operating in solution and at 37 °C for which we estimate a performance of 5.8 μM s−1 mg−1.38 Crucially, herein biocatalysis is achieved in the total absence of a solvent, whereby polymer surfactant replaces both hydration and bulk waters to stabilize enzymes and permits bioactivity up to at least 150 °C.
| T/°C | ||||||
|---|---|---|---|---|---|---|
| 100 | 110 | 120 | 130 | 140 | 150 | |
| DAP Production/μM s−1 | 3.92 | 6.07 | 10.60 | 16.92 | 18.83 | 26.45 |
| St. Dev./μM s−1 | 0.63 | 0.63 | 0.64 | 0.71 | 0.69 | 0.58 |
Evidently, the small loss of the native fold upon polymer surfactant conjugation for cTLL-S1 does not disrupt the catalytic-triad constructed by the Ser146-His258-Asp201 residues. This is in line with a previous study reporting on the hydrolysis of short- and long-chain fatty acid esters in cTLL-S1 melts at temperatures as low as 30 °C, corresponding roughly to the onset of melting.18 We anticipate that this remarkably low-temperature activation of the TLL helical lid-motif (Fig. 3c) is a result of the amphiphilic environment afforded by the polymer surfactant corona, which may facilitate the conformational transition from the closed-state (hydrophilic) to the open-state (hydrophobic).23 An alternative explanation that we cannot yet rule out is that the helical loop may be unstructured, which would provide a permanently exposed active-site.
Interestingly, while cTLL-S1 biofluids are active at low temperatures near the onset of melting,18 the temperatures required for the self-contained two- and three-enzyme cascades to function were almost 50 °C above their melting transition. Hence, we conclude that either GOx or HRP must limit the enzymatic cascade by the requirement for thermal activation. As the polymer surfactant appears to effectively replace hydration waters in the solvent-free environment,17 we anticipate that cGOx-S1 retains catalytic activity below 80 °C, given that the GOx internalized active site is extremely rigid and does not exhibit large-scale shape fluctuations.37 We speculate that the cascades are inactive at low temperatures, when the diffusive motions (a fluctuating-entry point) required for cHRP-S1 activity are impeded. Elevated temperatures are required to overcome this activation barrier affording sufficient conformational “flexibility” of the biomacromolecular machine. In future work, we plan to rigorously test this hypothesis in a systematic study involving scattering, spectroscopic and modelling tools. While the relative activities of the biohybrids are yet to be elucidated, our findings clearly reveal that whilst the capacity for substrate solubilization and mass transport is essential for enzyme activity, these alone are insufficient to ensure catalysis.
Due to the apparent high viscosity of the solvent-free biofluids we initially anticipated that the restricted diffusional mobility of substrates would play a critical role in limiting catalytic turnover. However, we find that enzyme activation is in fact the bottleneck at low temperatures. To examine the impact of substrate diffusion on catalysis, variable-temperature rheology experiments of the reaction mixtures were performed to probe the temperature-dependence of the viscosity of the solvent-free biofluids up to 150 °C (Fig. S9†). A comparison of the corresponding substrate diffusion coefficients with catalytic turnover above 80 °C reveals a linear relationship between initial reaction velocities and substrate diffusion coefficients (Fig. 4c and f), which is characteristic for diffusion-limited catalysis. These findings confirm that at sufficiently high temperatures when the expected catalytically vital conformations can be accessed efficiently, the polymer surfactant emulates both hydration and bulk solvent environments in the solvent-free biohybrids.
Conclusions
In conclusion, we have prepared solvent-free mixed enzyme-polymer hybrid biofluids to create efficient thermophilic enzymatic cascades. Our variable-temperature activity assays demonstrate that although substrate miscibility and mass transport are feasible within the biofluid, these alone will not ensure enzymatic activity. It is furthermore imperative that the polymer surfactant offers a suitable microenvironment substituting both hydration and bulk waters effectively. At temperatures above yet near the melting transition, the polymer corona does mediate small-scale protein fluctuations emulating efficiently the function of hydration waters. In stark contrast, catalytically vital, large-scale fluctuations as occurring in an aqueous bulk environment are seemingly inhibited. This results in a considerable energetic threshold for catalytic activity, which appears to be approximately 80 °C for HRP-containing biohybrids. Above this critical temperature, we find the solvent-free self-contained enzymatic cascade operates exclusively under diffusional constraints up to 150 °C. With the above results taken together, we find that a protein–polymer surfactant core–shell architecture can be exploited to promote biohybrid miscibility, facilitating the construction of complex multifunctional biofluids. The retention of biological activity for enzymes with distinctly different catalytic motions and chemical functions highlights the extraordinary capacity for a viscous surfactant milieu to replace both hydration and bulk waters. The capacity to produce and utilize these complex multicomponent biofluids will expand upon their potential application in bio-industries.Materials and methods
Protein–polymer surfactant biohybrid preparation
Unless otherwise stated, all materials were purchased from Sigma-Aldrich, NL. Horseradish peroxidase (type-VI, Amoracia rusticana, Lot # SLBR4932 V) and glucose oxidase (type X-S, Aspergillus niger, Lot # BCBT3916) were used without further purification. Thermomyces lanuginosus lipase (Lot # SLBQ7422 V) was dialyzed against a weak (10 mM sodium phosphate, pH 6.5) protein buffer before use. In a typical synthesis, proteins were suspended in the protein buffer to achieve a final protein concentration of 2 mg mL−1. Under stirring, 3-(dimethylamino)-1-propylamine (DMAPA, 2.2 M and pH 6.2) was added in at least 500-fold excess to the number of solvent accessible acidic sidechains, followed by the immediate addition of solid N-(3-dimethylaminopropyl)-N′-ethylcarbodiimide hydrochloride (EDC) in a further 10-fold excess. After 4 h a secondary addition of EDC was performed, and the reaction was allowed to proceed overnight to produce a supercharged enzyme. Any precipitate was removed by centrifugation (4000g, 15 min), followed by extensive dialysis against protein buffer to remove excess reactants. The purified supercharged enzyme was added dropwise under stirring to neat poly(ethylene glycol)-4-nonylphenyl-3-sulfopropyl ether (S1), or carboxylated Brij® L23 (S6) dissolved in minimum protein buffer, to achieve a final ratio of 4 surfactants per solvent-accessible cationic site. The mixture was allowed to stir overnight, followed by centrifugation to remove any precipitate, and a secondary extensive dialysis was performed against decreasing buffer concentrations to remove excess salts and unbound surfactant. The resultant supercharged enzyme-polymer surfactant suspended in MilliQ quality water was freeze-dried for 48 h, and the soft-solid powder was finally annealed for 1 h at 70 °C to produce free-flowing solvent-free protein–polymer hybrids. To produce mixed enzyme solvent-free biofluids, supercharged enzyme-polymer surfactant hybrid solutions were first mixed in the desired enzyme ratios before freeze-drying and thermal annealing was performed.Circular dichroism spectroscopy
Far-UV circular dichroism spectroscopy experiments were performed on a Jasco J-815 instrument fitted with a Peltier PTC-423S/15 temperature controller. All samples were thermally controlled at 22 °C, and measurements were performed using a 0.1 cm quartz cuvette. Protein concentrations were adjusted to maximize signal-to-noise ratios and by ensuring HT values remained below 650 V. Experiments were performed from 260–190 nm at a scanning speed of 20 nm min−1, a data pitch of 0.5 nm, with 4 s accumulation and 2.0 nm bandwidth. Each CD trace is obtained from a minimum of 5 manually averaged and background subtracted spectra. All traces are plotted using a 3-point moving average. The degree of retention of native folding was estimated for supercharged and hybrid enzymes using the ratio of A222 and A208 for the modified and native enzymes.X-ray scattering data acquisition, reduction and analysis
All experiments were performed on a SAXSLAB GANESHA 300 XL system equipped with a GeniX-Cu ultra-low divergence micro focus sealed-tube source producing X-ray photons with a wavelength λ = 1.54 Å at a flux of 1 × 108 Ph s−1. The scattering intensity was measured as a function of momentum transfer vectorwhere λ is the radiation wavelength and 2θ is the scattering angle. The 2D scattering data were recorded on a Pilatus 300 K silicon pixel detector with 487 × 619 pixels of 172 μm × 172 μm in size. The beam-centre and q-range were calibrated using an AgBe standard. The calibrated detector response function, known sample-to-detector distances, and measured incident and transmitted beam intensities were using to scale the 2D patterns to absolute intensity. The corresponding 2D patterns were azimuthally averaged to produce 1D scattering profiles.

For solution-state investigations, small-angle X-ray scattering (SAXS) profiles were obtained for samples measured in 2.0 mm quartz capillaries (Hilgenberg), mounted with custom-built capillary holders. A known sample-to-detector distance of 713 mm gave an accessible q-range of 0.015 < q < 0.445 Å−1. The scattering profile of the protein buffer and quartz capillary were subtracted to produce a final SAXS profile. Background subtraction was performed using the SAXSutilities39 package (available for download http://www.sztucki.de/SAXSutilities), and the HRP series profiles were fitted using either ellipsoidal or core–shell ellipsoidal form factors25–27 in SASview (available for download http://www.sasview.org). For GOx and TLL, the online tool Crysol31 was used to compute scattering profiles based from crystallographic structure PDB files 1cf3 (symmetric dimer) and 1ein, respectively.22,23
Enzyme kinetic assays and analysis
All experiments were performed on a Linkam-modified Shimadzu UV-3102 PC UV-vis NIR scanning spectrophotometer (transmission-mode) using two-enzyme (cGOx-S1/cHRP-S1) or three-enzyme (cTLL-S1-cGOx-S1/cHRP-S1) biofluids and a 0.01 mm path length. In a typical assay, 5.0 ± 0.1 mg of a mixed protein–polymer hybrid was annealed at 80 °C on a quartz plate in the presence of 0.1 mg solid OPD substrate to enable solubilization. After 2 minutes, a further 0.1 mg of either solid glucose crystals or desiccated acetyl-protected glucose was added, and the second quartz plate was used to sandwich the reaction environment. The plates were then sealed at 80 °C with tape. The sealed sample was mounted and equilibrated at reaction temperature on the Linkam stage for a further 60 s, after which the production of DAP (ε418 = 16.7 mM−1 cm−1)32 was probed by measuring the increase in absorbance at 418 nm (A418). A constant 2 s interval and bandwidth of 2.0 nm was used for all assays for a time period of at least 15 min. For the cHRP-S1 assay the sample was prepared as above, except that no solid glucose or desiccated acetyl-protected glucose was added. Instead, H2O2 was delivered to the reaction environment by pre-soaking the second quartz plate in a 1.0 wt% H2O2 solution and allowed to air-dry at room-temperature. An equivalent control experiment performed without pre-soaking the second quartz plate showed no increase in A418.The temperature-dependent initial reaction velocities were estimated by linear regression analysis over 60 s (two-enzyme biofluid) or 100 s (three-enzyme biofluid) of each recorded profile. The concentration of DAP was then calculated from the initial slope using the molar extinction coefficient of 16.7 mM−1 cm−1 and a 0.01 mm pathlength. The standard deviation of regression analysis was then converted in the same way to obtain a confidence interval for DAP concentration. The calculated rates of DAP production were plotted with respect to temperature, revealing a distinct temperature-dependent increase in DAP production. The temperature-dependent parameters of DAP production and substrate diffusion coefficient were compared, where a diffusion-limited process is defined by linearity.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the European Union (ERC-2014-StG Contract No. 635928) and Dutch Ministry of Education, Culture and Science (Gravity Program 024.001.035).Notes and references
- C. Silva, M. Martins, S. Jing, J. Fu and A. Cavaco-Paulo, Crit. Rev. Biotechnol., 2018, 38, 335–350 CrossRef CAS PubMed
.
- K. M. Polizzi, A. S. Bommarius, J. M. Broering and J. F. Chaparro-Riggers, Curr. Opin. Chem. Biol., 2007, 11, 220–225 CrossRef CAS PubMed
- S. Jemli, D. Ayadi-Zouari, H. Ben Hlima and S. Bejar, Crit. Rev. Biotechnol., 2016, 36, 246–258 CrossRef CAS PubMed
- B. G. Davis, Curr. Opin. Biotechnol., 2003, 14, 379–386 CrossRef CAS PubMed
- R. A. Sheldon and S. van Pelt, Chem. Soc. Rev., 2013, 42, 6223–6235 RSC
- M. W. W. Adams, F. B. Perler and R. M. Kelly, Nat. Biotechnol., 1995, 13, 662–668 CrossRef CAS
- D. W. Hough and M. J. Danson, Curr. Opin. Chem. Biol., 1999, 3, 39–46 CrossRef CAS PubMed
- K. Dumorné, D. C. Cordova, M. Astorga-Elo and P. Renganathan, J. Microbiol. Biotechnol., 2017, 27, 649–659 CrossRef PubMed
- F. Sarmiento, R. Peralta and J. M. Blamey, Front. Bioeng. Biotechnol., 2015, 3, 148 Search PubMed
- B. van den Burg, Curr. Opin. Microbiol., 2003, 6, 213–218 CrossRef CAS PubMed
- C. Schiraldi and M. De Rosa, Trends Biotechnol., 2002, 20, 515–521 CrossRef CAS PubMed
- F. Rigoldi, S. Donini, A. Redaelli, E. Parisini and A. Gautieri, APL Bioeng., 2018, 2, 11501 CrossRef PubMed
- T. Davids, M. Schmidt, D. Böttcher and U. T. Bornscheuer, Curr. Opin. Chem. Biol., 2013, 17, 215–220 CrossRef CAS PubMed
- B. Van den Burg, G. Vriend, O. R. Veltman, G. Venema and V. G. H. Eijsink, Proc. Natl. Acad. Sci. U. S. A., 1998, 95, 2056–2060 CrossRef CAS PubMed
- A. W. Perriman and S. Mann, ACS Nano, 2011, 5, 6085–6091 CrossRef CAS PubMed
- A. W. Perriman, A. P. S. Brogan, H. Cölfen, N. Tsoureas, G. R. Owen and S. Mann, Nat. Chem., 2010, 2, 622–626 CrossRef CAS PubMed
- F. X. Gallat, A. P. S. Brogan, Y. Fichou, N. McGrath, M. Moulin, M. Härtlein, J. Combet, J. Wuttke, S. Mann, G. Zaccai, C. J. Jackson, A. W. Perriman and M. Weik, J. Am. Chem. Soc., 2012, 134, 13168–13171 CrossRef CAS PubMed
- A. P. S. Brogan, K. P. Sharma, A. W. Perriman and S. Mann, Nat. Commun., 2014, 5, 5058 CrossRef CAS PubMed
- A. W. Perriman, H. Cölfen, R. W. Hughes, C. L. Barrie and S. Mann, Angew. Chem., Int. Ed., 2009, 48, 6242–6246 CrossRef CAS PubMed
- J. J. Virtanen, L. Makowski, T. R. Sosnick and K. F. Freed, Biophys. J., 2010, 99, 1611–1619 CrossRef CAS PubMed
- G. I. Berglund, G. H. Carlsson, A. T. Smith, H. Szöke, A. Henriksen and J. Hajdu, Nature, 2002, 417, 463 CrossRef CAS PubMed
- G. Wohlfahrt, S. Witt, J. Hendle, D. Schomburg, H. M. Kalisz and H. J. Hecht, Acta Crystallogr., Sect. D: Biol. Crystallogr., 1999, 55, 969–977 CrossRef CAS PubMed
- A. M. Brzozowski, H. Savage, C. S. Verma, J. P. Turkenburg, D. M. Lawson, A. Svendsen and P. Sham, Biochemistry, 2000, 39, 15071–15082 CrossRef CAS PubMed
- J. Araki, C. Zhao and K. Ito, Macromolecules, 2005, 38, 7524–7527 CrossRef CAS
-
L. A. Feigin and D. I. Svergun, Structure Analysis by Small-Angle X-Ray and Neutron Scattering, Springer US, Boston, MA, 1987 Search PubMed
- M. Kotlarchyk and S. Chen, J. Chem. Phys., 1983, 79, 2461–2469 CrossRef CAS
- S. S. Berr, J. Phys. Chem., 1987, 91, 4760–4765 CrossRef CAS
- A. P. S. Brogan, R. B. Sessions, A. W. Perriman and S. Mann, J. Am. Chem. Soc., 2014, 136, 16824–16831 CrossRef CAS PubMed
- N. A. Baker, D. Sept, S. Joseph, M. J. Holst and J. A. McCammon, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 10037–10041 CrossRef CAS
- T. J. Dolinsky, J. E. Nielsen, J. A. McCammon and N. A. Baker, Nucleic Acids Res., 2004, 32, W665–W667 CrossRef CAS PubMed
- D. Svergun, C. Barberato, M. H. J. Koch and IUCr, J. Appl. Crystallogr., 1995, 28, 768–773 CrossRef CAS
- K. C. Brown, J. F. Corbett and N. P. Loveless, Spectrochim. Acta, Part A, 1979, 35, 421–423 CrossRef
- S. Fornera and P. Walde, Anal. Biochem., 2010, 407, 293–295 CrossRef CAS
- M. Khajehpour, I. Rietveld, S. Vinogradov, N. V. Prabhu, K. A. Sharp and J. M. Vanderkooi, Proteins: Struct., Funct., Genet., 2003, 53, 656–666 CrossRef CAS PubMed
- M. Vriezema, P. M. L. Garcia, N. Sancho Oltra, N. S. Hatzakis, S. M. Kuiper, R. J. M. Nolte, A. E. Rowan and J. C. M. van Hest, Angew. Chem., 2007, 119, 7522–7526 CrossRef
- S. F. M. van Dongen, M. Nallani, J. J. L. M. Cornelissen, R. J. M. Nolte and J. C. M. van Hest, Chem. – Eur. J., 2009, 15, 1107–1114 CrossRef CAS PubMed
- S. S. Tatke, C. K. Loong, N. D'Souza, R. T. Schoephoerster and M. Prabhakaran, Biopolymers, 2008, 89, 582–594 CrossRef CAS PubMed
- T. Farrugia, A. W. Perriman, K. P. Sharma and S. Mann, Chem. Commun., 2017, 53, 2094–2097 RSC
- M. Sztucki and T. Narayanan, J. Appl. Crystallogr., 2007, 40, s459–s462 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9nr06045f |
| This journal is © The Royal Society of Chemistry 2019 |