
Synthesis of low- and high-index faceted metal (Pt, Pd, Ru, Ir, Rh) nanoparticles for improved activity and stability in electrocatalysis
Agus R.
Poerwoprajitno
a,
Lucy
Gloag
ab,
Soshan
Cheong
 c,
J. Justin
Gooding
ab and
Richard D.
Tilley
*abc
c,
J. Justin
Gooding
ab and
Richard D.
Tilley
*abc
aSchool of Chemistry, The University of New South Wales, Sydney, NSW 2052, Australia. E-mail: r.tilley@unsw.edu.au
bAustralian Centre for NanoMedicine, The University of New South Wales, Sydney, NSW 2052, Australia
cMark Wainwright Analytical Centre, The University of New South Wales, Sydney, NSW 2052, Australia
First published on 7th August 2019
Abstract
Driven by the quest for future energy solution, faceted metal nanoparticles are being pursued as the next generation electrocatalysts for renewable energy applications. Thanks to recent advancement in solution phase synthesis, different low- and high-index facets on metal nanocrystals become accessible and are tested for specific electrocatalytic reactions. This minireview summarises the key approaches to prepare nanocrystals containing the most catalytically active platinum group metals (Pt, Pd, Ru, Ir and Rh) exposed with low- and high-index facets using solution phase synthesis. Electrocatalytic studies related to the different facets are highlighted to emphasise the importance of exposing facets for catalysing these reactions, namely oxygen reduction reaction (ORR), hydrogen oxidation reaction (HOR), alcohol oxidation including methanol (MOR) and ethanol oxidation reactions (EOR), formic acid oxidation reaction (FAOR), oxygen evolution reaction (OER), and hydrogen evolution reaction (HER). The future outlook discusses the challenges and opportunities for making electrocatalysts that are even more active and stable.
 Agus R. Poerwoprajitno | Agus R. Poerwoprajitno is a Scientia Ph.D. candidate in the School of Chemistry at UNSW. He graduated a B.Eng. from Diponegoro University Indonesia in 2014 and M.Sc. from NTUST Taiwan in 2015. Then he worked as a research assistant in Institute of Chemistry, Academia Sinica, Taiwan. His research interest is mainly about solution-phase synthesis to make metallic nanocrystals with structural precision including control of shape, size and composition for catalytic applications. |
 Lucy Gloag | Lucy Gloag is a postdoctoral research fellow in the School of Chemistry at UNSW. She graduated with a B.Sc. (Hons) from Victoria University of Wellington and a PhD from UNSW. Her current research focuses on shape control of metallic nanocrystals for catalysis and biomedicine applications. |
 Soshan Cheong | Soshan Cheong is a research fellow in the Solid State & Elemental Analysis Unit (SSEAU) and the Electron Microscope Unit (EMU) at UNSW. She received her PhD in Chemistry (2010) from Victoria University of Wellington, New Zealand. She studied solution-phase synthesis and growth of nanocrystals for applications in catalysis and biomedicine. |
 J. Justin Gooding | Scientia Professor J. Justin Gooding graduated with a B.Sc. (Hons) from Melbourne University, completed a PhD from the University of Oxford and postdoctoral training at the Institute of Biotechnology in Cambridge University. After starting as a Research Fellow at the University of New South Wales, he gained Professorship in 2006. He is an ARC Australian Laureate Fellow and co-director of the Australian Centre for NanoMedicine. He leads a research team in surface modification and nanotechnology for biosensors and biomaterials. |
 Richard D. Tilley | Professor Richard Tilley is the Director of Electron Microscope Unit and a Professor in the School of Chemistry at the University of New South Wales. He graduated with a Master of Chemistry from Oxford University and then completed his PhD at the University of Cambridge, after which he was a Postdoctoral Fellow at the Toshiba basic R&D Center, Tokyo. He leads a research team focused on the solution synthesis of nanocrystals for catalysis and biomedical imaging. |
1. Introduction
Electrochemical energy conversion and storage technologies, such as water splitting electrolysers and fuel cells, have come to the forefront of chemical research due to the ever increasing global demand for energy and the depletion of fossil fuels.1–3 The high cost and scarcity of the most catalytically active platinum group metals means that structures that maximise the intrinsic activity and stability of these catalysts while minimising the mass of material used is the quintessential target for catalyst design.2,4,5 Nanoparticle electrocatalysts that have high surface areas and precisely tuned structures provide the means to achieve this goal.6–9Specifically, the arrangements of atoms at the surface of the nanoparticles where catalysis occurs is the most important feature affecting the activity and stability of a catalyst. However, at present, commercial nanoparticles are typically spherical without precise surface facets.10 In many cases, the performance of the catalyst can be improved, while lowering the amount of scarce metal needed, by exposing specific crystallographic facets on the nanocrystals.6,11–13 The key questions for researchers are then: (1) What facets provide the most active, stable nanocatalysts? and (2) How are these facets synthesised? Crystal facets of nanocrystals are denoted by the Miller indices {hkl} In this paper we define nanocrystals in two categories; (i) nanocrystals with low-index facets with Miller indices of 0 and 1, such as {100} and {111} facets; and (ii) nanocrystals with high-index facets that have at least one Miller indices greater than 1, such as {210} and {311}.
The exposed facets determine the final shape of the nanocrystals.14 For example, a cube is enclosed by six {100} facets, an octahedron is enclosed by eight {111} facets, a rhombic dodecahedron is enclosed by eight {110} facets, and a tetrahexahedron is enclosed by 24 high-index {310} facets.
In the last two decades, high-index faceted nanocrystals have been targeted for many catalytic applications due to their higher energy surfaces that increase specific activity. This high activity comes from the large number of low and high coordinated atoms at atomic steps, edges and kinks, which are often highly active sites.15–18 This has driven synthesis research to achieve nanocrystals with very high-index facets, even as high as {730}.19 However, recent advances have shown that low-index facets are more suited for some catalytic reactions, as they provide additional stability as well as improving activity.20–22 This means that to achieve the highest performing nanocatalysts, it is crucial to understand the relationships between faceting and catalytic performance.
This review presents the most effective synthetic approaches to form well-defined faceted nanocrystals and shows how these surface structures influence their catalytic properties. The fundamental knowledge in this review should direct the design and synthesis of faceted nanocrystals for the next generation of electrocatalysts.
2. Synthesis of well-defined facets
Solution phase synthesis is a powerful approach to form faceted nanocrystals.23–27 Shape controlled synthesis became a focus in the mid-1990s at a time when field emission transmission electron microscopes were developed with the resolution to accurately characterise and image the shape of nanocrystals.28Generally, the formation of nanocrystals involves two stages: (i) Nucleation and (ii) growth of nuclei into nanocrystals.6,24,29 The final shape of the nanocrystals can be controlled by manipulating the nucleation and the growth.24,30–32 The nucleation is the first stage of any crystallisation process which plays important role on the final nanocrystal shape.33 As the environment keeps supplying the atoms, the nuclei can serve as a seed for further nanocrystal growth. For face centred cubic (fcc) metals, the nuclei can be either be single crystals, or contain defects such as twin planes. The nuclei shape can be precisely controlled by experimental conditions such as the concentration of precursor and the temperature.24 In the case of seed-mediated growth, the nucleation process is separated as pre-synthesized seeds are added to solution for growth process.
The growth of the seed into nanocrystals can occur either in a thermodynamic or kinetic regime.34 The interplay between thermodynamic and kinetic regimes has a strong role in dictating the final shape of nanocrystals. In the thermodynamic regime, the most thermodynamically stable products are dominant, leading to the formation of low-index facets with the lowest surface energies. Essentially growth is directed towards the formation of surfaces terminated with low-index facets due to their low surface energy, following the order of {111} < {100} < {110} < high-index facets.35 As a result, the thermodynamic equilibrium product of fcc metal is often octahedral (only {111}) or cuboctahedral ({111} and {100}) shapes.36 The kinetic growth regime, in contrast, promotes anisotropic or directional growth along specific crystal facets, leading to the formation of thermodynamically less favourable, high-index facets.
Tremendous advances have been reported in the facet-controlled syntheses of mono and multimetallic nanocrystals which has been thoroughly reviewed in recent articles.35,37,38 In the following section, we summarise the key approaches to synthesise low- and high-index faceted nanocrystals.
2.1 Synthesis of low-index faceted nanocrystals
In this section, we consider the key aspects of forming nanocrystals with low-index facets including; (i) facet-selective stabilising agent, (ii) mild reduction conditions and (iii) seed-mediated facet replication.| Facet | Metal | Facet-selective stabilising agent | Ref |
|---|---|---|---|
| CTAB = cetyltrimethylammonium bromide, TMAB = tetramethylammonium bromide, TOAB = tetraoctylammonium bromide, TTAB = trimethyl(tetradecyl) ammonium bromide, OAm = oleylamine, OLA = oleic acid, PAH = polyallylamine, HDBAC = hexadecyldimethylbenzyl ammonium chloride, PVP = polyvinylpyrrolidone, CTAC = cetyltrimethylammonium chloride. | |||
| 100 | Pd | Br− (KBr) | 44 |
| Pd | Br− (CTAB) | 45 | |
| Pt | Br− (TMAB) | 61 | |
| PtCu | Br− (TOAB) | 62,63 | |
| PtRu | Br− (CTAB) | 8 | |
| PtNi | Br− (KBr) | 46 | |
| Rh | Br− (TTAB) | 64 | |
| PdPt | Br− (KBr), I− (KI) | 47 | |
| PdPt | I− (NaI) | 65,66 | |
| Pt | NH2(OAm), COOH(OLA), CO | 49,50,51 | |
| Pt3Co | NH2(OAm), COOH(OLA), CO | 67 | |
| PtM (M = Co, Fe, Ni) | NH2(OAm), COOH(OLA), CO | 52,53 | |
| PtMn | NH2(OAm), COOH (OLA), CO | 54 | |
| PtRh | NH2(OAm), CO | 55 | |
| 111 | PdPt | C2O42− (Na2C2O4) | 47 |
| Pd | COO− (citric acid) | 44,68 | |
| Pd | NH2(OAm), HCHO | 69 | |
| Pd | NH2(PAH) | 56 | |
| PdNi | NH2(OAm) | 57 | |
| Pd | CO | 58 | |
| PdAg | CO | 59 | |
| PdPtAg | CO | 60 | |
| PdPt | Cl− (NaCl) | 65 | |
| PtRu | Cl− (HDBAC) | 8 | |
| 110 | Pd | Cl− (CTAC) | 73 |
| AuPd | Cl− (CTAC) | 72,74 | |
Stabilising {100} facets. Halide ions sourced from metal salts (e.g. potassium bromide) and ionic surfactants (e.g. cetyltrimethylammonium bromide or CTAB) are among the most commonly used stabilising agents for synthesising {100}-bound faceted metal nanocrystals.44,45 In particular, bromide is known to preferentially bind to the {100} facets of Pt and Pd nanocrystals, and so is often used in the synthesis of Pt- and Pd-based mono- and bimetallic nanocubes enclosed by {100} facets (Fig. 1a and b).44,46,47
 | ||
| Fig. 1 Mono- and bi-metallic nanocrystals synthesised with different stabilising agents. (a) Pt and (b) PtNi alloy nanocubes stabilised by bromide ions. (c) Pt and (d) PtCo alloy nanocubes stabilised by a mix of amine, acid and CO. (e and f) Pd and (d) PdNi alloy icosahedral nanocrystals stabilised by amine. (g) Pd and (h) PdAg alloy nanosheet stabilised by CO. Image (a) was adapted with permission from ref. 61. Copyright 2009, American Chemical Society. Image (b) was adapted with permission from ref. 46. Copyright 2012, American Chemical Society. Image (c) was adapted with permission from ref. 51. Copyright 2011 The Royal Society of Chemistry. Image (d) was adapted with permission from ref. 53, Copyright 2011, American Chemical Society. Image (e) was adapted with permission from ref. 56. Copyright 2013, American Chemical Society. Image (f) was adapted with permission from ref. 57. Copyright 2018, AAAS. Image (g) was adapted with permission from ref. 58. Copyright 2011, Nature Publishing Group. Image (h) was adapted with permission from ref. 59. Copyright 2015, Wiley-VCH. | ||
Small molecules such as carbonyl and dissolved gas (e.g. H2 and CO) are effective stabilising agents for achieving {100} facets of metal nanocrystals.48 Although these small molecules play a critical role in the formation of {100} facets, they are only effective in the presence of additional larger surfactant molecules such as oleylamine and oleic acid that stabilise the dispersion of nanocrystals in solution.49–51 For instance, Wu et al. demonstrated that the co-adsorption of dissolved CO gas and oleylamine onto Pt {100} was essential for the formation of Pt nanocubes (Fig. 1c).51 The stabilisation of Pt{100} by similar CO-surfactant molecule combination is also broadly applicable to prepare various Pt–M (M = Rh, Co, Mn, Fe, Ni) nanocubes (Fig. 1d).52–55
It is noteworthy that although oleylamine and oleic acid are widely used for shape control in organic solution synthesis, there is no indication that either of these molecules alone is effective in stabilising {100} facets. To facilitate the formation and stabilisation of {100} facets, a combination of both oleylamine and oleic acid is required as well as tuning of the ratio and concentration, and in many cases coupled with the presence of CO.49–51
Stabilising {111} facets. Nanocrystals with exposed {111} facets take on shapes of octahedra, decahedra, icosahedra, or plate-like structures depending on their nucleation and growth processes. Amine-based stabilisers such as oleylamine and polyallylamine are commonly used in the synthesis of both Pd mono- and bimetallic nanocrystals enclosed by {111} facets. Amine binds strongly to Pd, which promotes the preferential adsorption of amine-based stabilisers on Pd {111} facets, resulting in the formation of icosahedral nanocrystals (Fig. 1e and f).56,57
Strong adsorption of CO onto Pd{111} has been used to synthesise mono- and multimetallic Pd nanosheets with extended {111} facets (Fig. 1g and h).58–60 In these syntheses, polymer molecules such as polyvinylpyrrolidone (PVP) is required to facilitate nanocrystal dispersion but do not participate in preferential binding. This application is unlike the stabilisation of Pt{100} by CO, where co-adsorption of surfactant molecules is essential.51,58,60
Stabilising {110} facets. Nanoparticles exclusively enclosed by {110} facets adopt the shape of a rhombic dodecahedron. CTAC is an effective stabilising agent to synthesize Pd based rhombic dodecahedral nanoparticles.70–72 In these syntheses, fast growth rate is important for the preferential formation of {110} over {100} and {111} facets that have lower total surface energy.70 Therefore, the combination of stabilising agent and growth rate is a key factor to achieve rhombic dodecahedra. Compared to those of {100} and {111} facets, reports on nanoparticles enclosed by {110} facets are still limited.
Weak reducing agents. The use of reducing agents with weak reducing power can promote nanocrystal growth under thermodynamically controlled regime. Slow addition to the nanocrystal surface provides sufficient time for ad-atoms to rearrange to find high energy sites, resulting in lower energy surface facets. Hydrogen gas, ascorbic acid and polyol are commonly used as a weak reducing agent.37
Hydrogen gas. Tilley and co-workers have successfully synthesised many types of intricate metal nanocrystals using hydrogen gas as a weak reducing agent.20–22,75 For example, Au–Ru and Pd–Ru nanocrystals with well-defined three-dimensional branches were formed in a Fischer–Porter bottle containing hydrogen gas.21,75 The thermodynamically driven growth causes the branches to be bound by hexagonal close packed (hcp) low-index {0001} and {10–11} facets (Fig. 2). The formation of energetically stable, low-index facets from high surface energy intermediate products demonstrated that hydrogen gas is highly effective in promoting slow growth.20,75
 | ||
| Fig. 2 (a) Low resolution and (b–c) high resolution TEM images of Au–Ru branched nanoparticles enclosed by low-index facets formed using hydrogen gas as a reducing agent. (d) Growth mechanism. Adapted with permission from ref. 20. Copyright 2018, Wiley-VCH. | ||
Ascorbic acid. Ascorbic acid is another weak reducing agent that can be used in aqueous syntheses. As it is an easy-to-handle solid, ascorbic acid is more commonly used than hydrogen gas, despite being a slightly stronger reducing agent.37,45 Low concentrations of ascorbic acid are used to ensure slow growth, as demonstrated by Niu et al. in the synthesis of Pd nanocrystals.45 At low concentration, the final facet was a mixed {100} and {111} facets. Nanocubes bound by higher energy {100} facets were formed at higher ascorbic acid concentration, suggesting that increasing the amount of weak reducing agent allows access to shapes that are less thermodynamically stable but still have low-index facets.
Polyalcohols (polyols). The reducing power of polyols depends on their type, which follows the order of tetraethylene glycol (TEG) < triethylene glycol (TREG) < diethylene glycol (DEG) < ethylene glycol (EG).37,76 Similarly to changing the concentration of the reducing agent, the change in reducing power enables the rate of growth to be simply controlled. In the synthesis of Pd nanocrystals, the use of DEG resulted in icosahedra enclosed by {111} facets.76 When more strongly reducing EG was used instead, truncated octahedron covered by a mix of {100} and {111} facets were formed, which could be attributed to a faster deposition rate promoting the formation of higher surface energy facets.
Matching lattice between seed and shell metals. The seed and the shell metals ideally need to have a crystal structures with small lattice mismatch, usually <5%. Beyond 5% mismatch, island growth tends to dominate to lower the contact between the mismatched metals.79 With a lattice mismatch of merely 0.8%,80 Pd and Pt are considered ideally matched for forming core–shell nanocrystals via the seed-mediated facet replication approach. This has been demonstrated by the many reports of Pd–Pt core–shell nanocubes bound by {100},81,82 and icosahedral83 and octahedral84 nanocrystals terminated with {111} facets. Other examples of core–shell nanocrystals synthesised with seed facet replication include Pd–Ir,85,86 PdCu3–Ru87 and Ag–Pt,88 in which the core–shell metal pairs have lattice mismatch of 1.3%, 2.1% and 4.0% respectively.
Diffusion rate > deposition rate. A rate of diffusion that is faster than deposition is essential for faceting. It allows the shell metal atoms to diffuse over the seed surfaces and deposit uniformly before subsequent layer deposition occurs (Fig. 3a).89 Experimentally, a slow supply of shell metal atoms combined with an adequately high reaction temperature has been successful to form {100} and {111} faceted core–shell nanocrystals of different metals including Pd,89 Pt81–84 and PtNi.90
 | ||
| Fig. 3 (a) Role of deposition and diffusion rate to ensure the atomic layer epitaxial growth. Adapted with permission from ref. 85. Copyright 2014, American Chemical Society. (b–j) HAADF-STEM images and EDX line scan analyses of Pd–Pt core–shell nanocubes with different Pt atomic layers. Adapted with permission from ref. 81. Copyright 2014, American Chemical Society. | ||
Xia and co-workers showed that a slow supply of Pt atoms could be achieved via a controlled precursor injection. Slow injection ensured a slow deposition of Pt on the Pd surfaces, resulting in core–shell structures with fine control of the shell thickness down to single atomic layers (Fig. 3b).81,83 Elevated reaction temperatures promote a high diffusion rate to ensure atoms deposited on corners/vertices (high energy sites) can readily diffuse and spread evenly across the seed surfaces. This approach is so effective that metals that do not naturally crystallise with these facets exposed, such as Ir85,86 and fcc Ru,91 have formed core–shell nanocrystals with {100} and {111}.
Weak reducing agents are also effective for achieving slow deposition rates. Hydrogen gas has been shown to enable Pd atoms to diffuse to the most stable positions on faceted Au seed surfaces.89 This facilitated a layer-by-layer atomic deposition of Pd onto Au icosahedral seed to form Au–Pd core–shell nanocrystals retaining the seed morphology with controlled shell thickness.
2.2 Synthesis of high-index faceted nanocrystals
In a seminal study, Wang and co-workers demonstrated the high activity of high-index faceted Pt tetrahexahedral nanocrystals.92 Since this study in 2007, research on synthesising high-index faceted nanocrystals has boomed. In solution, high-index facets often rapidly disappear during crystal growth as monomers add to become more stable low-index facets. This is because the high-index facets are energetically unfavourable thus are preferentially eliminated to minimise the total surface energies.35In this section, we cover key aspects of forming nanocrystals with high-index facets including; (i) fast reduction in the presence of stabilising agents, (ii) selective etching and (iii) seed-mediated facet replication.
Strong reducing agent. The type and the amount of strong reducing agent is vital to ensure fast kinetically controlled growth.93–95 Glycine has been reported to be a strong reducing agent,37 which has promoted the formation of Pt concave nanocubes enclosed by {hk0} facets in a Teflon-lined stainless steel.96 The fast reduction means that monomers are available to rapidly deposit on the nanocrystal, without sufficient time to diffuse to form lower energy surfaces. Growth occurs at the corners of cubes, resulting in surface atoms that have high coordination numbers in the concave side and low coordination numbers at the corners. The same approach was also used to successfully synthesise high-index faceted Pt–Ni, PtPb and Pt3Mn concave nanocrystals from cubic seeds.97–99 It is important to control the temperature and the reaction time to ensure surface diffusion is slow relative to deposition.
Concentration of reducing agent. As with the formation of low-index facets, the concentration of reducing agent can control the rate of deposition of monomers. In the formation of Pt concave nanocubes, the concentration of glucose reducing agent highly influenced the facets on the final nanocrystals (Fig. 4).100 At low concentration of glucose, growth was slow which provided time for diffusion to occur and result in low-index facets. In contrast, the high concentration of glucose allowed fast reducing condition resulting in {720} facets. In a similar example demonstrated by Han and co-workers, a high concentration of the reducing agent ascorbic acid was used to induce a kinetically controlled regime, resulting in high-index {541} facets on Au–Pd hexoctahedral nanoparticles.70,101
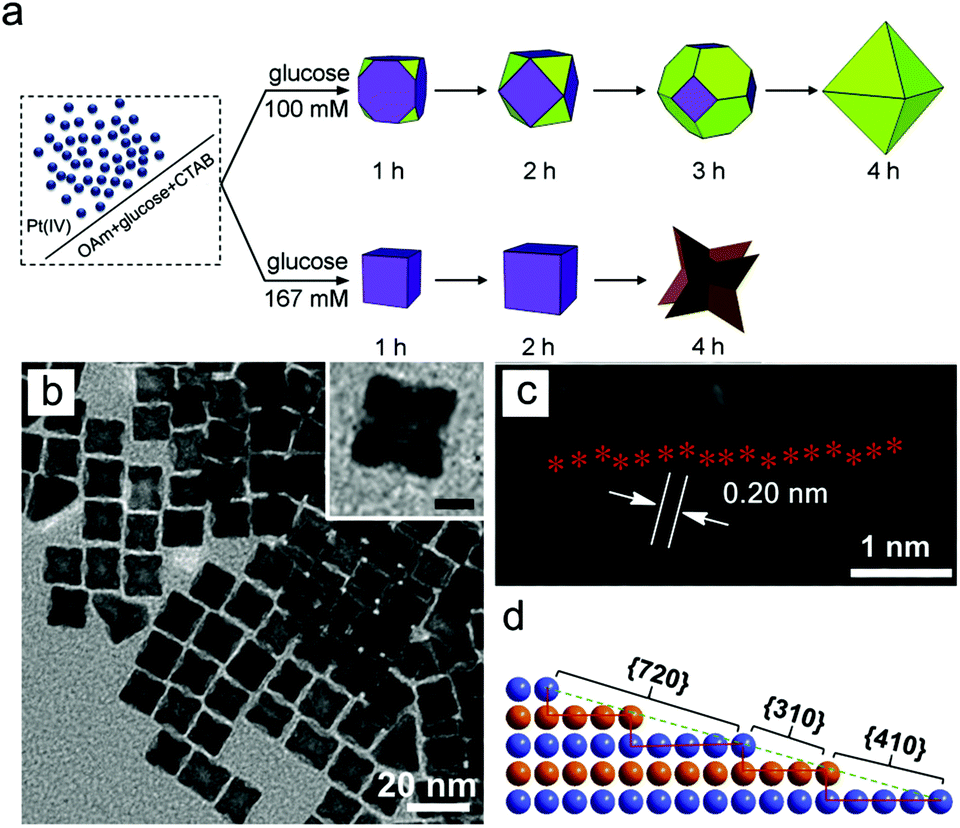 | ||
| Fig. 4 (a) Effect of reducing agent concentration for the formation of high-index faceted nanoparticles. (b–d) TEM images and the atomic model of high-index faceted Pt nanoparticles. Adapted with permission from ref. 100. Copyright 2018, Elsevier. | ||
Addition of stabilising agent. Stabilising agents are needed in the formation of high-index facets to ensure the nanocrystals remain dispersed in solution, but without preferentially stabilising high-index facets. Amine-based stabilising agents are commonly used to form high-index facets of Pt nanocrystals.102–105 For example, Huang et al. demonstrated that the role of methylamine was important in creating Pt concave nanocrystals enclosed by {411} facets.102 In the absence of methylamine (only polymer), low-index facets were formed as deposition and diffusion can occur readily. The use of methylamine or other amines (i.e., ethylamine, butylamine, 4-methylpiperidine, trimethylamine) resulted in concave structures, likely due to blocking of the sides of the cube seeds causing growth to only be possible from the corners. Similar to low-index faceted nanoparticles, the concentration of stabilising agent is also an important factor in facet control. For instance, Zheng and co-workers showed that Au–Pd {221} facets were achieved by doubling the concentration of octadecyltrimethyl ammonium chloride; whereas the {110} facets were preferred at a lower stabilising agent concentration.106
The interplay between reduction rate and stabilising agent has led to a wide variety of mono and multimetallic nanocrystals with high-index facets.107,108 Unlike the case of low-index faceted nanocrystals, the choice of stabilising agent to synthesise high-index faceted is challenging, which has resulted in other methods being used to access high-index facets.
Core–shell. The highest-index facets reported to date, Pd {730} facets, were formed using Cl−/O2 to etch away surface atoms during the formation of a Pd shell.116 The resulting high-index {730} facets are formed from steps, ledges, and kinks that are stabilised by CTAC.
Alloy. The success of this approach for core–shell nanocrystals has inspired researchers to synthesise high-index faceted alloy nanocrystals. For instance, the formation of high-index PtCo, PtSn, and PtMn nanocrystals has also been achieved using Cl−/O2 as oxidative etchant.118,119 In the synthesis of Pt3Co nanocubes, the introduction of deionised water (DI water) was vital for the formation of concave architecture (Fig. 5).118 DI water increased the amount of solubilised O2 and so increased the amount of Cl−/O2 pairs for oxidative etching. In the absence of DI water, nanocubes that were unetched resulted.
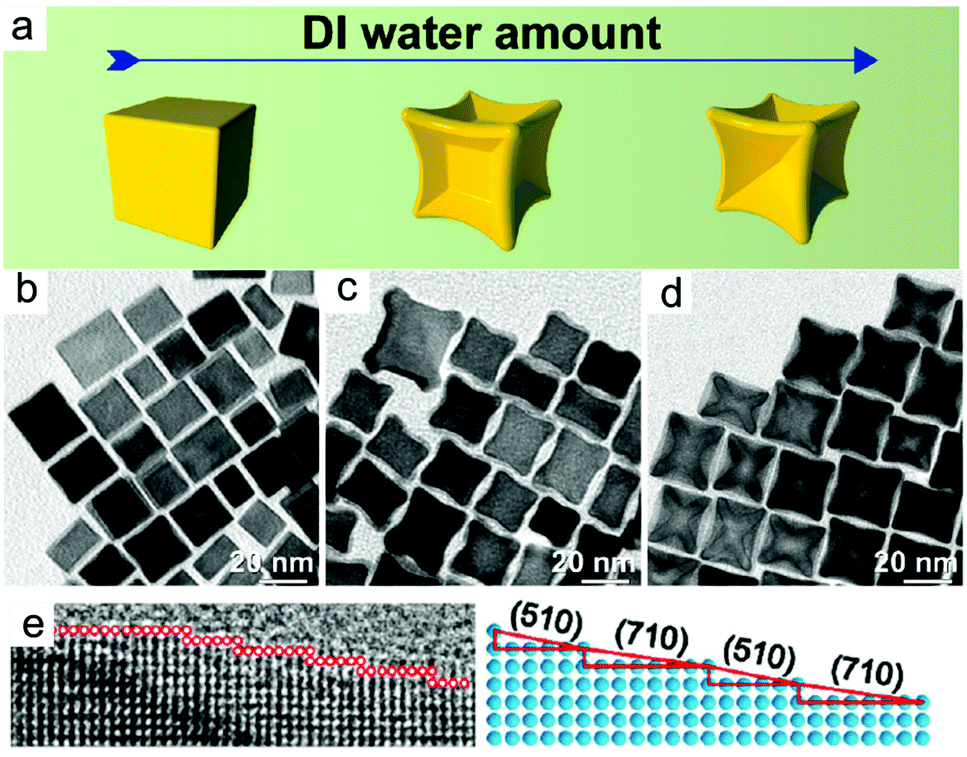 | ||
| Fig. 5 (a–d) Effect of DI water concentration in the shape of nanoparticles. (e and f) HRTEM image and atomic modelling of the edge of an individual excavated nanocubes synthesized by high amount of DI water. Adapted with permission from ref. 118. Copyright 2017, American Chemical Society. | ||
Low concentration of metal precursor. The controlled synthesis of Au nanocrystals with high-index facets is more well developed than other metals.123,124 To achieve nanocrystals of other metals with high-index facets, researchers have attempted to coat high-index Au nanocrystals. Yan and co-workers achieved some success with coating Pd shells on Au tetrahexahedral (THH) and trisoctahedral (TOH) nanocrystals, which are enclosed by {730} and {221} facets, respectively.121 Thin Pd shells were grown uniformly onto the Au seeds, while retaining the same facets.
In a different study, Yu et al. found that increasing the concentration of Pd precursor enabled access to different high-index faceted shapes, such as hexoctahedral (HOH) and tetrahexahedral (THH) Au–Pd core–shell nanocrystals.120 This was attributed to the fast rate of deposition that causes new high-index facets to form rather than diffusing onto the seed template. The trisoctahedral (TOH) Au–Pd nanocrystals was achieved by using low concentration of Pd precursor (0.25 molar equivalents), which ensures slow deposition and diffusion, replicating their Au TOH seeds.
Slow monomer addition. In the last few years, the synthesis of high-index faceted Pd nanocrystals has been achieved, which can also serve as a seed for epitaxial growth. Given close matching lattice between Pt and Pd, replicating high-index facets of Pd with Pt becomes readily achievable. Pd–Pt core–shell concave nanocubes were achieved by combining a slow injection rate with a mild reducing agent at high temperature. The surface of the concave could be indexed to a mixture of {210}, {100} and {110} facets with 3–5 Pt atomic layers (Fig. 6).122 A slow injection method was used to achieve thin Pt shell and avoid the formation of rough surface due to high energy barrier for surface diffusion on concave structure. As with seeded growth of low-index faceted nanocrystals, the rate of deposition and diffusion can be altered by changing the amount of ascorbic acid as reducing agent and the reaction temperature.
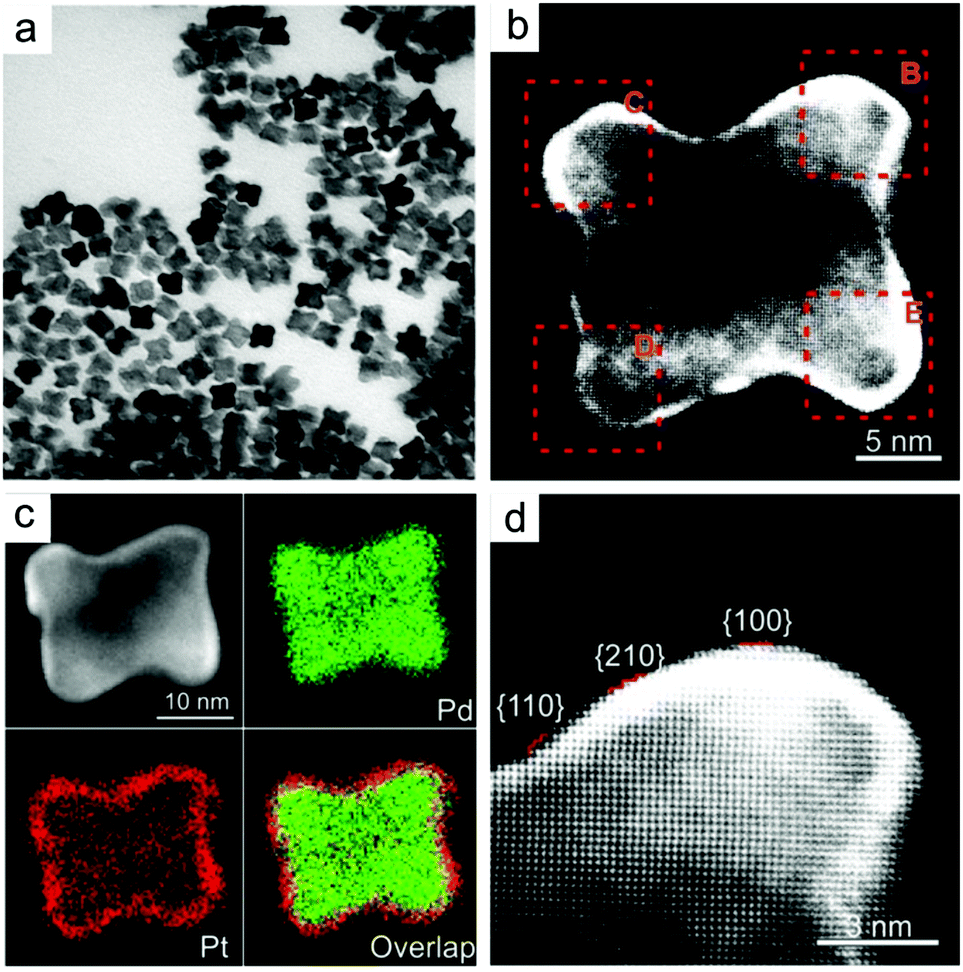 | ||
| Fig. 6 (a, b, d) TEM images and (c) EDS mapping of Pd–Pt concave nanocubes enclosed by high index facets using seed-mediated facet replication. Adapted with permission from ref. 122. Copyright 2016, Wiley-VCH. | ||
3. Electrocatalytic studies of faceted nanocrystals
As catalysis takes place on the nanocrystal surface, the arrangement of atoms at the surface determines the adsorption and desorption energies of reactants and intermediate species, as well as the number of active sites, all of which strongly affect the catalytic performance of the nanocrystal. Factors such as metallic composition, size and sub-surface crystal structure have been shown to influence interactions between catalytic species and the nanoparticles and have been reviewed in detail.125,126 With the latest advances in controlling surface atomic arrangement of nanoparticle electrocatalysts, it is important to understand specifically how nanocrystal facets generate the desired interactions for high activity and stability.Often the trends observed with nanocrystal facets are similar to observations for thin film single-crystal catalysts, which means that nanocatalysts can be designed by looking to high performing surfaces. In this section, we cover the electrocatalytic reactions that have been most influenced by surface facets, namely the oxygen reduction reaction, hydrogen oxidation reaction, alcohol oxidation reaction, formic acid oxidation reaction and oxygen evolution reaction. These reactions have clearly shown the effect of having specific facets on activity and stability. Hydrogen evolution and hydrogen oxidation are important reactions that are relatively unexplored for facet sensitivity, but are discussed to highlight the opportunity for further research.
3.1 Oxygen reduction reactions
Oxygen reduction reaction (ORR) is an important reaction at the cathode of proton-exchange membrane, direct alcohol and formic acid fuel cells, where Pt is the most active metallic catalyst.2 However, the slow kinetics of the present ORR catalysts means high amount of expensive Pt is required. The need to develop active cathode catalysts has driven research into controlling the surface facets of the electrocatalytic nanocrystals.Pt is the most active metal for ORR because the Pt–O binding energy (ΔG) is 1.57 eV which is near the peak on the volcano plot (∼1.80 eV).127 The main factor in designing an ORR catalyst is to provide a surface that binds OH groups strongly enough for effective adsorption of O2 and reaction intermediates to drive the reaction forward, but weakly enough for other oxygenated species in solution to not bind and block the catalyst surface.128 To achieve this, a binding energy (GOH–GOH(Pt111)) of M–OH of −0.1 eV is ‘ideal’ relative to Pt(111) (Fig. 7a).
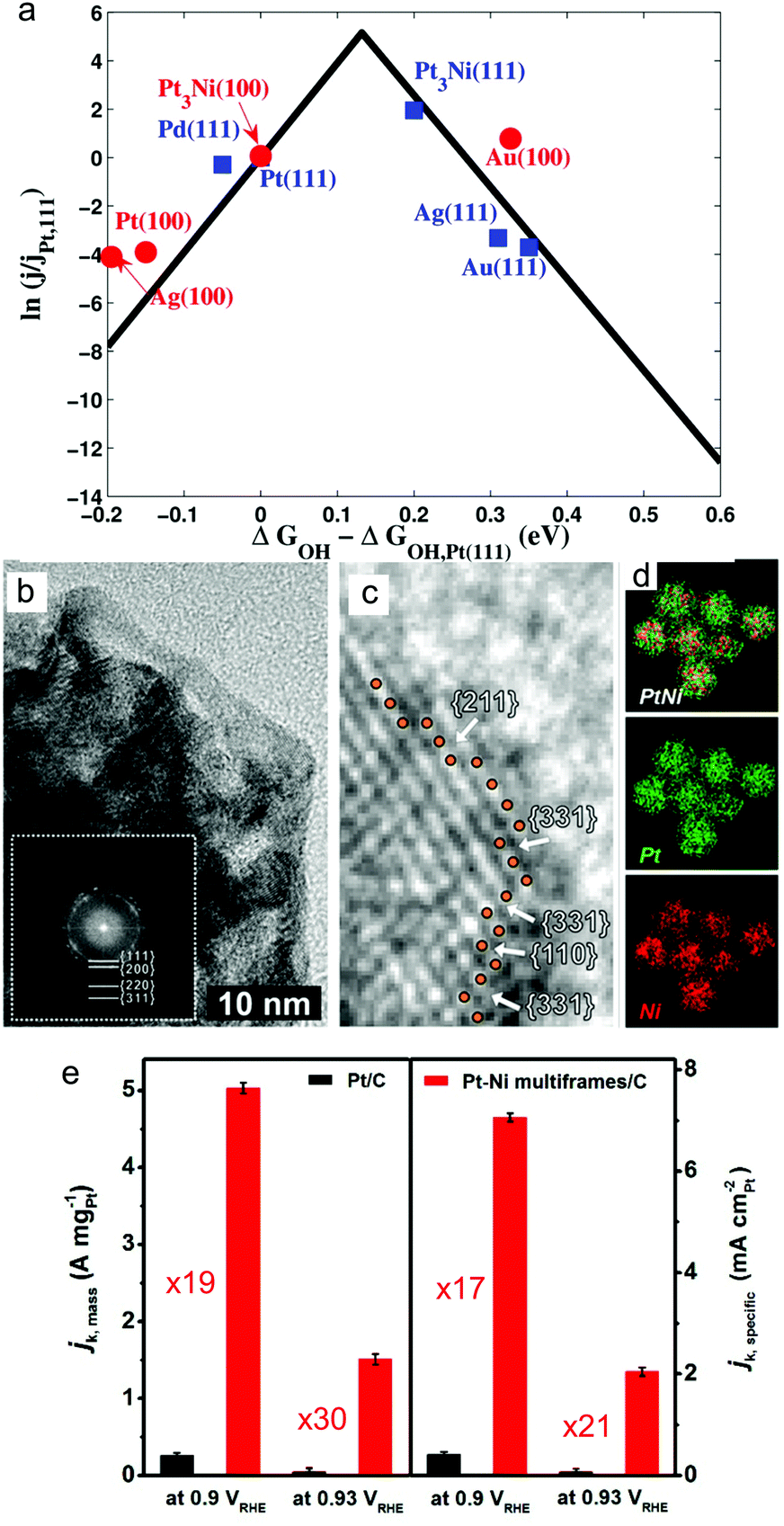 | ||
| Fig. 7 (a) Volcano plot showing the catalytic activity as a function of calculated free energy of OH bonding. Adapted from ref. 128. Copyright 2012, American Chemical Society. (b–e) Comparison of mass and specific activities of high index faceted PtNi alloy multiframes/C and the state-of-the-art Pt/C. Adapted from ref. 146. Copyright 2018, American Chemical Society. | ||
Pd is considered a good alternative for catalysing ORR as Pd–O has a binding energy (1.53 eV) that is also close to the ‘ideal’ (∼1.80 eV).127 However, Pd catalysts showed the opposite trend in terms of facet performance in ORR: (110) < (111) ≪ (100).133 This is because the coverage of oxide on Pd (100) is lower than on other facets yielding more available reaction sites.134 As a result, Pd nanocubes bound exclusively by {100} facets exhibited an order higher activity than Pd octahedra.134
Because of the significant enhancement in ORR activity by Pt3Ni(111) facets compared to other compositions and surface atom arrangements, research on the design and study of Pt3Ni alloy nanocrystals enclosed by {111} facets have received the most attention.110,136,138–141 Nanoparticles with diameters <10 nm with controlled facets have many high specific activity facets exposed to maximise the mass activity. For example, 10 nm Pt3Ni octahedra exhibited 5 times higher activity than Pt3Ni nanocube with a similar size and 7 times higher specific activity than non-faceted Pt nanoparticles.140
3.2 Hydrogen oxidation reaction
Hydrogen oxidation reaction (HOR) is the cathodic reaction of the hydrogen fuel cell which converts hydrogen gas to protons.148 HOR is not the kinetically limiting reaction in hydrogen fuel cell because it is a simple 2-electron process. As experimentally observed, Pt catalyst has already achieved high activities at low overpotentials due to the optimum free energy Hadsorption (ΔH ≈ 0).149 The catalytic activity of the facets increases in the order (111) < (100) < (110) for acid and alkaline environments due to the decrease in activation energy (ΔH).150,151 However, Pt-based bimetallic facets have not been studied as research has been focusing on less expensive, non-Pt catalysts. Perhaps studies to understand facet performance of earth abundant metal or alloy will open up new opportunities in the development of new active electrocatalysts that meets commercial requirements.3.3 Alcohol oxidation reactions
Methanol and ethanol have been considered alternative energy carriers to hydrogen gas because they are less hazardous and easier to handle in terms of transportation and storage.3 Pt is widely known as the most active metal for alcohol oxidation due to the facile dehydrogenation of alcohol.13,152 However, in both methanol and ethanol oxidation reactions, Pt nanocatalysts face different challenges, as discussed in the respective sections below.Although Pt is very active for MOR, Pt suffers from catalyst poisoning due to the strong binding of CO adsorbates that block the catalytic surface. The most efficient approach to avoid CO poisoning on Pt is to introduce oxophilic atoms such as Ru and Sn atoms. Ru can provide OH species at lower potentials that oxidises adjacent CO on Pt as well as weakens the Pt–CO bond.156,157 According to DFT calculation, the distance between Pt and Ru needs to be close to a ‘critical distance’ of 4.0 Å to enable the formation of a transition state (Pt–CO⋯OH–Ru).158 Wieckowski and co-workers reported an increasing MOR activity of PtRu single-crystal surface following the order (100) < (110) < (111).159 This trend can be a result of the ‘critical distance’ between Pt and Ru atoms and/or due to difference facet energies.8,158 This was translated into a high performing nanocatalyst recently reported by Dong and co-workers, who showed that PtRu alloy nanowires exposed with {111} facets being 2 times more active than PtRu alloy nanocubes bound by {100} (Fig. 8).8 In this case, the enhanced activity originates from the optimum adsorption energies of COads and OHads on {111} facets as well as the close proximity between Pt and Ru atoms. Alloying Pt with other less expensive metals such as Sn has also enhanced catalytic activity.160 Similar to Ru, Sn can promote the H2O dissociation to form Sn–OH and thus weakens the CO bond to Pt.161 For example, excavated PtSn enclosed by {110} facets showed higher activity than PtSn nanocubes, which could be attributed to the high energy {110} surfaces.162
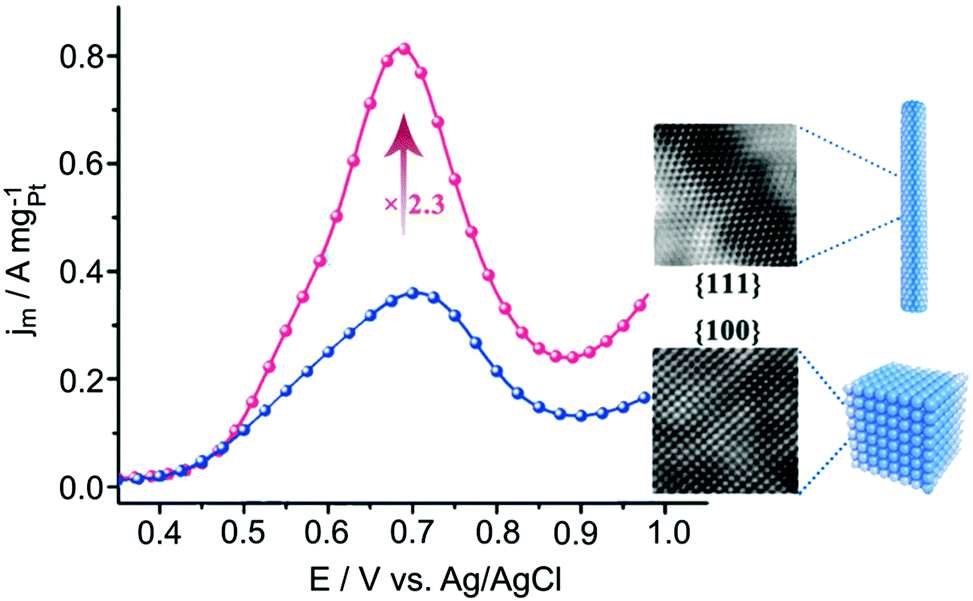 | ||
| Fig. 8 Comparison of methanol oxidation activities of PtRu nanowires enclosed by {111} facets and PtRu nanocubes enclosed by {100} facets. Adapted with permission from ref. 8. Copyright 2018, American Chemical Society. | ||
High-index faceted nanocrystals may also show higher activity than Pt commercial catalysts because the high energy Pt atoms on steps and kinks form weak bonds with CO and so are more resistant to poisoning.118,163,164 There are opportunities for research into the role of low- and high-index facets of many different Pt alloy nanoparticles that may decrease CO poisoning and increase MOR activities further.
3.4 Formic acid oxidation reaction
Direct formic acid fuel cells are another alternative energy conversion technology with a higher electromotive force (EMF = 1.45 V) as compared to hydrogen (EMF = 1.23 V) and methanol (EMF = 1.21 V) fuel cells.173 The oxidation process at the anode is a 2-electron process that converts formic acid to carbon dioxide and protons. This reaction is the limiting reaction due to high overpotentials and catalyst poisoning. Pd is considered the best metal catalyst for formic acid oxidation reaction (FAOR) because it is the only metal that can break the O–H bond at all potentials above 0 V where formic acid oxidation occurs.174 As a result, Pd promotes FAOR via a dehydrogenation pathway that is not limited by strongly adsorbing poisoning intermediates.173 In comparison, Pt has a propensity to break the C–O and/or C–H bond at low overpotentials, leading to the formation of CO that poisons the surface and decreases FAOR activity.174 The development of faceted Pd nanocatalysts has thus become the focus for improving FAOR.173,1753.5 Oxygen evolution reaction
The oxygen evolution reaction (OER) is the limiting reaction in proton exchange membrane water electrolyser which is important for energy storage. In this reaction water is broken down into protons and oxygen gas.4 The 4-electron process requires high overpotentials to drive the reaction forward. Ru and Ir-based catalyst are the most active for OER because these have standard enthalpies of lower to higher oxidation state transformation, which forms the catalytically active species, close to ∼1.48 eV with the lowest overpotential.1,4 However, the high oxidation state species that are active and form at potentials >1.23 V are easily dissolved into acid electrolyte, which causes stability to be the main challenge for OER catalysts.4,181 As a result, nanocrystals with facets that improve catalytic stability while retaining high activity are the main target for OER catalyst development.182Therefore, the synthesis of Ru nanocrystals with low-index facets are key targets for stable OER catalysts.20–22,184 Gloag et al. demonstrated that Ru nanoparticles that have low-index facets actually exhibited higher stability as well as higher activity compared to non-faceted nanoparticles in acidic electrolyte.20–22 This improved performance is due to the stability low-index {0001} and {10–11} facets against Ru dissolution (Fig. 9).26 Particularly important is that the improved stability actually enhances the activity by retaining the high number of active sites at the surface. As a result of this high stability, other low-index faceted Ru nanocrystals have achieved high activity (180 mV overpotential) and stability even up to 4 hours at 10 mA cm−2, which is 5 orders of magnitude higher than polycrystalline nanocrystals (Fig. 9).26 Importantly, post characterisation TEM after prolonged use showed the structure remains intact, which shows the durability of Ru nanoparticles with low index facets.
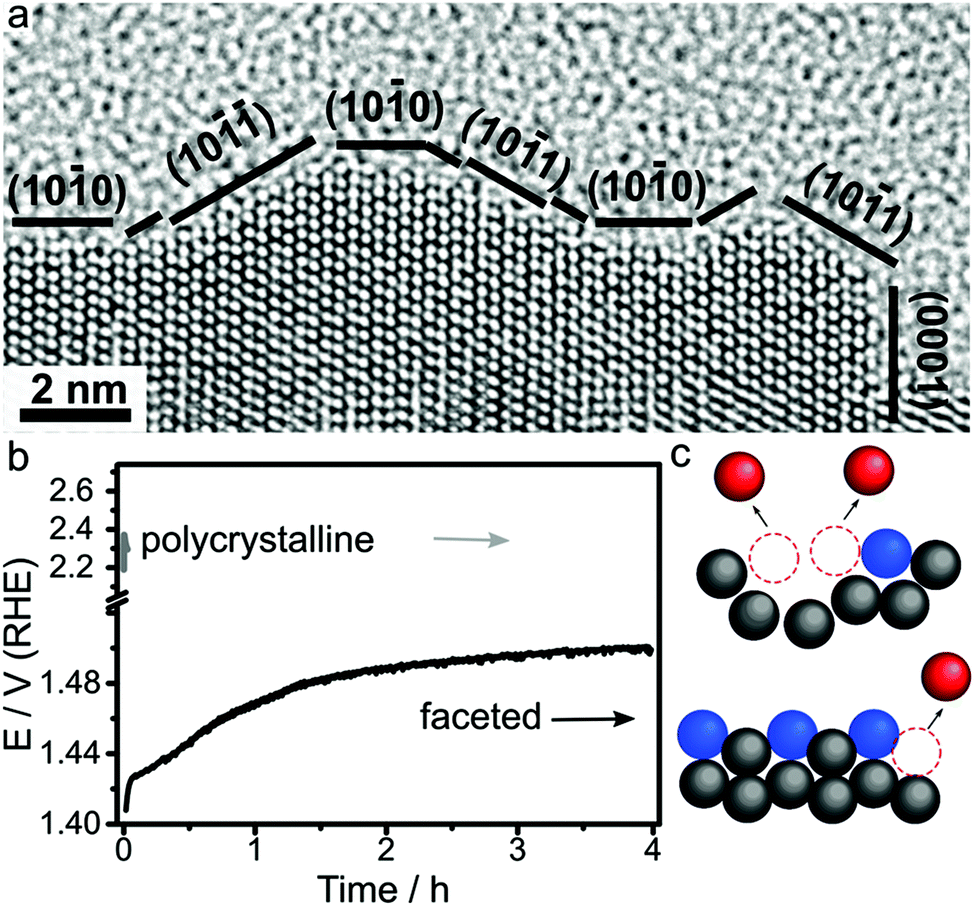 | ||
| Fig. 9 (a) HRTEM image of faceted Ru branched nanoparticles showing low index faceted surfaces. (b) Stability test of faceted Ru branched nanoparticles (black line) and polycrystalline Ru nanoparticles (grey line). (c) A representative model showing the stability of low index facets. Adapted with permission from ref. 26. Copyright 2019, Wiley-VCH. | ||
3.6 Hydrogen evolution reaction
Hydrogen evolution reaction (HER) occurs at the cathode of a water electrolyser, which converts protons to hydrogen gas.148 Because HER is 2-electron process, it is not kinetically limited as with OER.188 As a result, catalysts have been able to achieve high current densities 10 mA cm2geom at low overpotentials (0.05 V) without the need for extensive nanoparticle design.189 Even higher current densities at lower overpotentials are anticipated if the rate limiting Volmer step (second adsorption of hydrogen from the solution) is accelerated.190 Studies on the dependence of activity on facets has been limited to single-crystal surfaces. These revealed an activity trend of (111) < (100) < (110) for both acid and alkaline solution, which can be attributed to the decreases in activation energy (ΔH).150,151 As this is a relatively unexplored area, further study is likely to provide insight into the facet dependence on the nanoparticles. Further, as research moves away from expensive Pt metal, faceting of less noble metals or Pt alloys may be an important tool to reach high activities necessary for commercial application.1914. Summary and outlook
In solution phase synthesis, nanocrystals are formed in a complex reaction mixture comprising the precursor, solvent, stabiliser and reducing agent. Facet control not only needs to take into account the effects of these variables, but also the interplay between the thermodynamic and kinetic regimes during nanocrystal formation and growth. For platinum group metals Pt, Pd, Ru, Ir and Rh, forming and stabilising low-index {100} and {111} facets generally require the presence of facet-selective stabilising agents, sometimes combined with a slow reduction condition. In contrast, for the high-index facets {hkl} (where h, k or l >1), a fast reduction condition coupled with a suitable stabilising agent is indicated in the different studies reported. More creative approaches such as replicating the surface facets of seed crystals and selective etching have also been explored to overcome various facet control challenges.As different nanocrystal facets were achieved, their electrocatalytic performances were tested for targeted reactions. High-index facets are preferred for catalysing ORR, MOR and EOR as they exhibit much higher activities compared to low-index facets, and so have become the research focus of nanocatalyst synthesis in the past decade. However, recent catalyst development showed that OER, on the contrary, requires low-index facets to facilitate high electrocatalytic activities that are sustainable. For FAOR, although both low- and high-index facets show comparable activities, certain high-index facets are often preferred for being more cost-effective (requiring lower peak potential).
It is clear that there is no “one-size-fits-all” explanation to the relationship between surface facets and catalytic performance. This is because electrocatalytic performances are highly facet- and metal-dependent, which can be attributed to the different metal-reaction intermediate binding energies and mechanisms involved in the different reactions. Nevertheless, knowledge is being developed for identifying the right facets for specific reactions, which is vital for empowering nanocatalyst design.
Despite the tremendous progress in nanocrystal facet control and hence optimisation of electrocatalytic properties, there remain considerable challenges in the aspects of both synthesis and electrocatalysis: (1) There is still a lack of precise knowledge of the type of stabilising agents for each metal facet especially the high-index facets. (2) Growth mechanisms of many different facets are still not well understood, as knowledge is generally acquired via an experimental approach. This gap also hinders the further extension of the synthetic method for faceted multimetallic nanocrystals. (3) Understanding and control of surface facet changes during elecrocatalytic reactions are still lacking, yet so crucial for the development of stable catalysts.
In this regard, time-resolved or in situ studies are highly desired for probing nanocrystal growth mechanism and nanocatalyst behaviour. For instance, in situ liquid cell coupled with high-resolution transmission electron microscopy (HRTEM) can reveal the formation of facets in solution; exposing nanocatalyst to different atmosphere and temperature stimuli in an environmental TEM (ETEM) can disclose the surface atomic movement which can give an insight about the stability of the nanocatalyst during electrocatalysis reaction. Apart from further experimental studies, accurate theoretical modelling is required to aid in predicting facet formation and growth, as well as responses of the facets to electrocatalytic stimuli. There is still plenty to be accomplished for the development of the next generation electrocatalysts.
Conflicts of interest
The authors declare no competing interest.Acknowledgements
The authors acknowledge funding under the Australian Research Council Linkage grant (L. G., J. J. G. R. D. T., LP150101014), the Australian Laureate Fellowship (J. J. G., FT150100060) and the Discovery Project (R. D. T., DP190102659). A. R. P. is currently a Ph.D. candidate at School of Chemistry and is supported by an Australian Government Research Training Program Scholarship and the UNSW Scientia PhD Scholarship Scheme.Notes and references
- N.-T. Suen, S.-F. Hung, Q. Quan, N. Zhang, Y.-J. Xu and H. M. Chen, Chem. Soc. Rev., 2017, 46, 337–365 RSC.
- M. Shao, Q. Chang, J.-P. Dodelet and R. Chenitz, Chem. Rev., 2016, 116, 3594–3657 CrossRef CAS PubMed.
- N. Kakati, J. Maiti, S. H. Lee, S. H. Jee, B. Viswanathan and Y. S. Yoon, Chem. Rev., 2014, 114, 12397–12429 CrossRef CAS PubMed.
- T. Reier, H. N. Nong, D. Teschner, R. Schlögl and P. Strasser, Adv. Energy Mater., 2017, 7, 1601275 CrossRef.
- O. Pascu, S. Moisan, J.-D. Marty and C. Aymonier, J. Phys. Chem. C, 2014, 118, 14017–14025 CrossRef CAS.
- S. Cheong, J. D. Watt and R. D. Tilley, Nanoscale, 2010, 2, 2045–2053 RSC.
- Q. Li and S. Sun, Nano Energy, 2016, 29, 178–197 CrossRef CAS.
- L. Huang, X. Zhang, Q. Wang, Y. Han, Y. Fang and S. Dong, J. Am. Chem. Soc., 2018, 140, 1142–1147 CrossRef CAS PubMed.
- C. Wang, X. Sang, J. T. L. Gamler, D. P. Chen, R. R. Unocic and S. E. Skrabalak, Nano Lett., 2017, 17, 5526–5532 CrossRef CAS PubMed.
- T. Yoshida and K. Kojima, Electrochem. Soc. Interface, 2015, 24, 45–49 CrossRef CAS.
- M. T. M. Koper, Nanoscale, 2011, 3, 2054–2073 RSC.
- J. W. Hong, Y. Kim, Y. Kwon and S. W. Han, Chem. – Asian J., 2016, 11, 2224–2239 CrossRef CAS PubMed.
- R. Rizo and B. Roldan Cuenya, ACS Energy Lett., 2019, 4, 1484–1495 CrossRef CAS PubMed.
- Z.-Y. Zhou, N. Tian, Z.-Z. Huang, D.-J. Chen and S.-G. Sun, Faraday Discuss., 2009, 140, 81–92 RSC.
- T. Sheng, N. Tian, Z.-Y. Zhou, W.-F. Lin and S.-G. Sun, ACS Energy Lett., 2017, 2, 1892–1900 CrossRef CAS.
- L. Zhang, W. Niu and G. Xu, Nano Today, 2012, 7, 586–605 CrossRef CAS.
- F. Calle-Vallejo, J. Tymoczko, V. Colic, Q. H. Vu, M. D. Pohl, K. Morgenstern, D. Loffreda, P. Sautet, W. Schuhmann and A. S. Bandarenka, Science, 2015, 350, 185–189 CrossRef CAS PubMed.
- G. Collins and J. D. Holmes, Adv. Mater., 2016, 28, 5689–5695 CrossRef CAS PubMed.
- M. Jin, H. Zhang, Z. Xie and Y. Xia, Angew. Chem., 2011, 123, 7996–8000 CrossRef.
- L. Gloag, T. M. Benedetti, S. Cheong, Y. Li, X.-H. Chan, L.-M. Lacroix, S. L. Y. Chang, R. Arenal, I. Florea, H. Barron, A. S. Barnard, A. M. Henning, C. Zhao, W. Schuhmann, J. J. Gooding and R. D. Tilley, Angew. Chem., Int. Ed., 2018, 57, 10241–10245 CrossRef CAS PubMed.
- L. Gloag, T. M. Benedetti, S. Cheong, C. E. Marjo, J. J. Gooding and R. D. Tilley, J. Am. Chem. Soc., 2018, 140, 12760–12764 CrossRef CAS PubMed.
- A. R. Poerwoprajitno, L. Gloag, T. M. Benedetti, S. Cheong, J. Watt, D. L. Huber, J. J. Gooding and R. D. Tilley, Small, 2019, 15, 1804577 CrossRef PubMed.
- J. Pal and T. Pal, Nanoscale, 2015, 7, 14159–14190 RSC.
- Y. Xia, Y. Xiong, B. Lim and S. E. Skrabalak, Angew. Chem., Int. Ed., 2009, 48, 60–103 CrossRef CAS PubMed.
- C. Garnero, M. Lepesant, C. Garcia-Marcelot, Y. Shin, C. Meny, P. Farger, B. Warot-Fonrose, R. Arenal, G. Viau, K. Soulantica, P. Fau, P. Poveda, L.-M. Lacroix and B. Chaudret, Nano Lett., 2019, 19, 1379–1386 CrossRef PubMed.
- X. Hao, A. Yoko, C. Chen, K. Inoue, M. Saito, G. Seong, S. Takami, T. Adschiri and Y. Ikuhara, Small, 2018, 14, 1802915 CrossRef PubMed.
- G. Collins, F. Davitt, C. O'Dwyer and J. D. Holmes, ACS Appl. Nano Mater., 2018, 1, 7129–7138 CrossRef CAS.
- E. Ruska, Rev. Mod. Phys., 1987, 59, 627–638 CrossRef CAS.
- D. Saha, E. D. Bøjesen, A. H. Mamakhel, M. Bremholm and B. B. Iversen, ChemNanoMat, 2017, 3, 472–478 CrossRef CAS.
- B. C. Camargo, B. Lassagne, R. Arenal, C. Gatel, T. Blon, G. Viau, L.-M. Lacroix and W. Escoffier, Nanoscale, 2017, 9, 14635–14640 RSC.
- A.-A. Litwinowicz, S. Takami, S. Asahina, X. Hao, A. Yoko, G. Seong, T. Tomai and T. Adschiri, CrystEngComm, 2019, 21, 3836–3843 RSC.
- J.-L. Mi, P. Nørby, M. Bremholm, J. Becker and B. B. Iversen, Nanoscale, 2015, 7, 16170–16174 RSC.
- E. D. Bøjesen and B. B. Iversen, CrystEngComm, 2016, 18, 8332–8353 RSC.
- Y. Wang, J. He, C. Liu, W. H. Chong and H. Chen, Angew. Chem., Int. Ed., 2015, 54, 2022–2051 CrossRef CAS PubMed.
- Y. Xia, X. Xia and H.-C. Peng, J. Am. Chem. Soc., 2015, 137, 7947–7966 CrossRef CAS PubMed.
- M. M. Kash, V. A. Sautenkov, A. S. Zibrov, L. Hollberg, G. R. Welch, M. D. Lukin, Y. Rostovtsev, E. S. Fry and M. O. Scully, Phys. Rev. Lett., 1999, 82, 5229–5232 CrossRef CAS.
- T. S. Rodrigues, M. Zhao, T.-H. Yang, K. D. Gilroy, A. G. M. da Silva, P. H. C. Camargo and Y. Xia, Chem. – Eur. J., 2018, 24, 16944–16963 CrossRef CAS PubMed.
- K. D. Gilroy, A. Ruditskiy, H.-C. Peng, D. Qin and Y. Xia, Chem. Rev., 2016, 116, 10414–10472 CrossRef CAS PubMed.
- X. Hao, C. Chen, M. Saito, D. Yin, K. Inoue, S. Takami, T. Adschiri and Y. Ikuhara, Small, 2018, 14, 1801093 CrossRef PubMed.
- T. Fujii, S.-I. Kawasaki, A. Suzuki and T. Adschiri, Cryst. Growth Des., 2016, 16, 1996–2001 CrossRef CAS.
- O. Pascu, L. Marciasini, S. Marre, M. Vaultier, M. Pucheault and C. Aymonier, Nanoscale, 2013, 5, 12425–12431 RSC.
- U. Schlotterbeck, C. Aymonier, R. Thomann, H. Hofmeister, M. Tromp, W. Richtering and S. Mecking, Adv. Funct. Mater., 2004, 14, 999–1004 CrossRef CAS.
- T. S. Ahmadi, Z. L. Wang, T. C. Green, A. Henglein and M. A. El-Sayed, Science, 1996, 272, 1924–1925 CrossRef CAS PubMed.
- B. Lim, M. Jiang, J. Tao, P. H. C. Camargo, Y. Zhu and Y. Xia, Adv. Funct. Mater., 2009, 19, 189–200 CrossRef CAS.
- W. Niu, L. Zhang and G. Xu, ACS Nano, 2010, 4, 1987–1996 CrossRef CAS PubMed.
- Y. Wu, S. Cai, D. Wang, W. He and Y. Li, J. Am. Chem. Soc., 2012, 134, 8975–8981 CrossRef CAS PubMed.
- A.-X. Yin, X.-Q. Min, Y.-W. Zhang and C.-H. Yan, J. Am. Chem. Soc., 2011, 133, 3816–3819 CrossRef CAS PubMed.
- A. P. LaGrow, B. Ingham, S. Cheong, G. V. M. Williams, C. Dotzler, M. F. Toney, D. A. Jefferson, E. C. Corbos, P. T. Bishop, J. Cookson and R. D. Tilley, J. Am. Chem. Soc., 2012, 134, 855–858 CrossRef CAS PubMed.
- C. Wang, H. Daimon, Y. Lee, J. Kim and S. Sun, J. Am. Chem. Soc., 2007, 129, 6974–6975 CrossRef CAS PubMed.
- Y. Kang, X. Ye and C. B. Murray, Angew. Chem., Int. Ed., 2010, 49, 6156–6159 CrossRef CAS PubMed.
- B. Wu, N. Zheng and G. Fu, Chem. Commun., 2011, 47, 1039–1041 RSC.
- J. Zhang and J. Fang, J. Am. Chem. Soc., 2009, 131, 18543–18547 CrossRef CAS PubMed.
- J. Wu, A. Gross and H. Yang, Nano Lett., 2011, 11, 798–802 CrossRef CAS PubMed.
- Y. Kang and C. B. Murray, J. Am. Chem. Soc., 2010, 132, 7568–7569 CrossRef CAS PubMed.
- F. Gao, Y. Zhang, P. Song, J. Wang, T. Song, C. Wang, L. Song, Y. Shiraishi and Y. Du, J. Mater. Chem. A, 2019, 7, 7891–7896 RSC.
- G. Fu, X. Jiang, L. Tao, Y. Chen, J. Lin, Y. Zhou, Y. Tang and T. Lu, Langmuir, 2013, 29, 4413–4420 CrossRef CAS PubMed.
- Y. Feng, Q. Shao, Y. Ji, X. Cui, Y. Li, X. Zhu and X. Huang, Sci. Adv., 2018, 4, eaap8817 CrossRef PubMed.
- X. Huang, S. Tang, X. Mu, Y. Dai, G. Chen, Z. Zhou, F. Ruan, Z. Yang and N. Zheng, Nat. Nanotechnol., 2010, 6, 28 CrossRef PubMed.
- C. Hu, X. Mu, J. Fan, H. Ma, X. Zhao, G. Chen, Z. Zhou and N. Zheng, ChemNanoMat, 2016, 2, 28–32 CrossRef CAS.
- J. W. Hong, Y. Kim, D. H. Wi, S. Lee, S.-U. Lee, Y. W. Lee, S.-I. Choi and S. W. Han, Angew. Chem., Int. Ed., 2016, 55, 2753–2758 CrossRef CAS PubMed.
- C.-K. Tsung, J. N. Kuhn, W. Huang, C. Aliaga, L.-I. Hung, G. A. Somorjai and P. Yang, J. Am. Chem. Soc., 2009, 131, 5816–5822 CrossRef CAS PubMed.
- D. Xu, Z. Liu, H. Yang, Q. Liu, J. Zhang, J. Fang, S. Zou and K. Sun, Angew. Chem., Int. Ed., 2009, 48, 4217–4221 CrossRef CAS PubMed.
- X. Zhao, B. Luo, R. Long, C. Wang and Y. Xiong, J. Mater. Chem. A, 2015, 3, 4134–4138 RSC.
- Y. Zhang, M. E. Grass, J. N. Kuhn, F. Tao, S. E. Habas, W. Huang, P. Yang and G. A. Somorjai, J. Am. Chem. Soc., 2008, 130, 5868–5869 CrossRef CAS PubMed.
- X. Huang, Y. Li, Y. Li, H. Zhou, X. Duan and Y. Huang, Nano Lett., 2012, 12, 4265–4270 CrossRef CAS PubMed.
- X. Huang, H. Zhang, C. Guo, Z. Zhou and N. Zheng, Angew. Chem., Int. Ed., 2009, 48, 4808–4812 CrossRef CAS PubMed.
- H. Yang, J. Zhang, K. Sun, S. Zou and J. Fang, Angew. Chem., Int. Ed., 2010, 49, 6848–6851 CrossRef CAS PubMed.
- Y. Xiong, J. M. McLellan, Y. Yin and Y. Xia, Angew. Chem., 2007, 119, 804–808 CrossRef.
- Z. Niu, Q. Peng, M. Gong, H. Rong and Y. Li, Angew. Chem., Int. Ed., 2011, 50, 6315–6319 CrossRef CAS PubMed.
- Y. W. Lee, D. Kim, J. W. Hong, S. W. Kang, S. B. Lee and S. W. Han, Small, 2013, 9, 660–665 CrossRef CAS PubMed.
- M. Kim, Y. Kim, J. W. Hong, S. Ahn, W. Y. Kim and S. W. Han, Chem. Commun., 2014, 50, 9454–9457 RSC.
- Y. W. Lee, M. Kim, S. W. Kang and S. W. Han, Angew. Chem., Int. Ed., 2011, 50, 3466–3470 CrossRef CAS PubMed.
- A. Tao, P. Sinsermsuksakul and P. Yang, Angew. Chem., Int. Ed., 2006, 45, 4597–4601 CrossRef CAS PubMed.
- H.-C. Peng, S. Xie, J. Park, X. Xia and Y. Xia, J. Am. Chem. Soc., 2013, 135, 3780–3783 CrossRef CAS.
- J. Watt, C. Yu, S. L. Y. Chang, S. Cheong and R. D. Tilley, J. Am. Chem. Soc., 2013, 135, 606–609 CrossRef CAS PubMed.
- Y. Wang, H.-C. Peng, J. Liu, C. Z. Huang and Y. Xia, Nano Lett., 2015, 15, 1445–1450 CrossRef CAS PubMed.
- R. G. Weiner, M. R. Kunz and S. E. Skrabalak, Acc. Chem. Res., 2015, 48, 2688–2695 CrossRef CAS PubMed.
- K. D. Gilroy, X. Yang, S. Xie, M. Zhao, D. Qin and Y. Xia, Adv. Mater., 2018, 30, 1706312 CrossRef PubMed.
- F.-R. Fan, D.-Y. Liu, Y.-F. Wu, S. Duan, Z.-X. Xie, Z.-Y. Jiang and Z.-Q. Tian, J. Am. Chem. Soc., 2008, 130, 6949–6951 CrossRef CAS PubMed.
- S. E. Habas, H. Lee, V. Radmilovic, G. A. Somorjai and P. Yang, Nat. Mater., 2007, 6, 692 CrossRef CAS PubMed.
- S. Xie, S.-I. Choi, N. Lu, L. T. Roling, J. A. Herron, L. Zhang, J. Park, J. Wang, M. J. Kim, Z. Xie, M. Mavrikakis and Y. Xia, Nano Lett., 2014, 14, 3570–3576 CrossRef CAS PubMed.
- S. Bai, L. Yang, C. Wang, Y. Lin, J. Lu, J. Jiang and Y. Xiong, Angew. Chem., Int. Ed., 2015, 54, 14810–14814 CrossRef CAS PubMed.
- X. Wang, S.-I. Choi, L. T. Roling, M. Luo, C. Ma, L. Zhang, M. Chi, J. Liu, Z. Xie, J. A. Herron, M. Mavrikakis and Y. Xia, Nat. Commun., 2015, 6, 7594 CrossRef PubMed.
- P. Zhang, Y. Hu, B. Li, Q. Zhang, C. Zhou, H. Yu, X. Zhang, L. Chen, B. Eichhorn and S. Zhou, ACS Catal., 2015, 5, 1335–1343 CrossRef CAS.
- X. Xia, L. Figueroa-Cosme, J. Tao, H.-C. Peng, G. Niu, Y. Zhu and Y. Xia, J. Am. Chem. Soc., 2014, 136, 10878–10881 CrossRef CAS PubMed.
- X. Xia, J. Zhang, N. Lu, M. J. Kim, K. Ghale, Y. Xu, E. McKenzie, J. Liu and H. Ye, ACS Nano, 2015, 9, 9994–10004 CrossRef CAS PubMed.
- Y. Yao, D. S. He, Y. Lin, X. Feng, X. Wang, P. Yin, X. Hong, G. Zhou, Y. Wu and Y. Li, Angew. Chem., 2016, 128, 5591–5595 CrossRef.
- U. Aslam, S. Chavez and S. Linic, Nat. Nanotechnol., 2017, 12, 1000 CrossRef CAS PubMed.
- A. M. Henning, J. Watt, P. J. Miedziak, S. Cheong, M. Santonastaso, M. Song, Y. Takeda, A. I. Kirkland, S. H. Taylor and R. D. Tilley, Angew. Chem., Int. Ed., 2013, 52, 1477–1480 CrossRef CAS PubMed.
- X. Zhao, S. Chen, Z. Fang, J. Ding, W. Sang, Y. Wang, J. Zhao, Z. Peng and J. Zeng, J. Am. Chem. Soc., 2015, 137, 2804–2807 CrossRef CAS PubMed.
- M. Zhao, L. Figueroa-Cosme, A. O. Elnabawy, M. Vara, X. Yang, L. T. Roling, M. Chi, M. Mavrikakis and Y. Xia, Nano Lett., 2016, 16, 5310–5317 CrossRef CAS PubMed.
- N. Tian, Z.-Y. Zhou, S.-G. Sun, Y. Ding and Z. L. Wang, Science, 2007, 316, 732–735 CrossRef CAS PubMed.
- L. Graham, G. Collins, J. D. Holmes and R. D. Tilley, Nanoscale, 2016, 8, 2867–2874 RSC.
- J. Watt, S. Cheong, M. F. Toney, B. Ingham, J. Cookson, P. T. Bishop and R. D. Tilley, ACS Nano, 2010, 4, 396–402 CrossRef CAS PubMed.
- X. Xia, S. Xie, M. Liu, H.-C. Peng, N. Lu, J. Wang, M. J. Kim and Y. Xia, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 6669–6673 CrossRef CAS PubMed.
- Z.-C. Zhang, J.-F. Hui, Z.-C. Liu, X. Zhang, J. Zhuang and X. Wang, Langmuir, 2012, 28, 14845–14848 CrossRef CAS PubMed.
- X. Xu, X. Zhang, H. Sun, Y. Yang, X. Dai, J. Gao, X. Li, P. Zhang, H.-H. Wang, N.-F. Yu and S.-G. Sun, Angew. Chem., 2014, 126, 12730–12735 CrossRef.
- L. Huang, X. Zhang, Y. Han, Q. Wang, Y. Fang and S. Dong, Chem. Mater., 2017, 29, 4557–4562 CrossRef CAS.
- Y. Wang, H. Zhuo, H. Sun, X. Zhang, X. Dai, C. Luan, C. Qin, H. Zhao, J. Li, M. Wang, J.-Y. Ye and S.-G. Sun, ACS Catal., 2019, 9, 442–455 CrossRef CAS.
- J. Qian, M. Shen, S. Zhou, C.-T. Lee, M. Zhao, Z. Lyu, Z. D. Hood, M. Vara, K. D. Gilroy, K. Wang and Y. Xia, Mater. Today, 2018, 21, 834–844 CrossRef CAS.
- D. Kim, Y. W. Lee, S. B. Lee and S. W. Han, Angew. Chem., Int. Ed., 2012, 51, 159–163 CrossRef CAS PubMed.
- X. Huang, Z. Zhao, J. Fan, Y. Tan and N. Zheng, J. Am. Chem. Soc., 2011, 133, 4718–4721 CrossRef CAS PubMed.
- B. Y. Xia, H. B. Wu, X. Wang and X. W. Lou, Angew. Chem., Int. Ed., 2013, 52, 12337–12340 CrossRef CAS PubMed.
- J. Lai, W. Niu, W. Qi, J. Zhao, S. Li, W. Gao, R. Luque and G. Xu, ChemCatChem, 2015, 7, 1064–1069 CrossRef CAS.
- L. Peres, D. Yi, S. Bustos-Rodriguez, C. Marcelot, A. Pierrot, P.-F. Fazzini, I. Florea, R. Arenal, L.-M. Lacroix, B. Warot-Fonrose, T. Blon and K. Soulantica, Nanoscale, 2018, 10, 22730–22736 RSC.
- J. Zhang, C. Hou, H. Huang, L. Zhang, Z. Jiang, G. Chen, Y. Jia, Q. Kuang, Z. Xie and L. Zheng, Small, 2013, 9, 538–544 CrossRef CAS PubMed.
- M. Luo, Y. Sun, X. Zhang, Y. Qin, M. Li, Y. Li, C. Li, Y. Yang, L. Wang, P. Gao, G. Lu and S. Guo, Adv. Mater., 2018, 30, 1705515 CrossRef PubMed.
- L. Bu, J. Ding, S. Guo, X. Zhang, D. Su, X. Zhu, J. Yao, J. Guo, G. Lu and X. Huang, Adv. Mater., 2015, 27, 7204–7212 CrossRef CAS PubMed.
- L. Zhang, L. T. Roling, X. Wang, M. Vara, M. Chi, J. Liu, S.-I. Choi, J. Park, J. A. Herron, Z. Xie, M. Mavrikakis and Y. Xia, Science, 2015, 349, 412–416 CrossRef CAS PubMed.
- C. Chen, Y. Kang, Z. Huo, Z. Zhu, W. Huang, H. L. Xin, J. D. Snyder, D. Li, J. A. Herron, M. Mavrikakis, M. Chi, K. L. More, Y. Li, N. M. Markovic, G. A. Somorjai, P. Yang and V. R. Stamenkovic, Science, 2014, 343, 1339–1343 CrossRef CAS PubMed.
- T. M. Benedetti, C. Andronescu, S. Cheong, P. Wilde, J. Wordsworth, M. Kientz, R. D. Tilley, W. Schuhmann and J. J. Gooding, J. Am. Chem. Soc., 2018, 140, 13449–13455 CrossRef CAS PubMed.
- R. Long, S. Zhou, B. J. Wiley and Y. Xiong, Chem. Soc. Rev., 2014, 43, 6288–6310 RSC.
- C. Qin, A. Fan, X. Zhang, X. Dai, H. Sun, D. Ren, Z. Dong, Y. Wang, C. Luan, J.-Y. Ye and S.-G. Sun, Nanoscale, 2019, 11, 9061–9075 RSC.
- S. Cheong, J. Watt, B. Ingham, M. F. Toney and R. D. Tilley, J. Am. Chem. Soc., 2009, 131, 14590–14595 CrossRef CAS PubMed.
- Q. Li, M. Rellán-Piñeiro, N. Almora-Barrios, M. Garcia-Ratés, I. N. Remediakis and N. López, Nanoscale, 2017, 9, 13089–13094 RSC.
- C.-L. Lu, K. S. Prasad, H.-L. Wu, J.-A. A. Ho and M. H. Huang, J. Am. Chem. Soc., 2010, 132, 14546–14553 CrossRef CAS PubMed.
- H. Jing, Q. Zhang, N. Large, C. Yu, D. A. Blom, P. Nordlander and H. Wang, Nano Lett., 2014, 14, 3674–3682 CrossRef CAS PubMed.
- H. Du, S. Luo, K. Wang, M. Tang, R. Sriphathoorat, Y. Jin and P. K. Shen, Chem. Mater., 2017, 29, 9613–9617 CrossRef CAS.
- H. Rong, J. Mao, P. Xin, D. He, Y. Chen, D. Wang, Z. Niu, Y. Wu and Y. Li, Adv. Mater., 2016, 28, 2540–2546 CrossRef CAS PubMed.
- Y. Yu, Q. Zhang, B. Liu and J. Y. Lee, J. Am. Chem. Soc., 2010, 132, 18258–18265 CrossRef CAS PubMed.
- F. Wang, C. Li, L.-D. Sun, H. Wu, T. Ming, J. Wang, J. C. Yu and C.-H. Yan, J. Am. Chem. Soc., 2011, 133, 1106–1111 CrossRef CAS PubMed.
- S. R. Lee, J. Park, K. D. Gilroy, X. Yang, L. Figueroa-Cosme, Y. Ding and Y. Xia, ChemCatChem, 2016, 8, 3082–3088 CrossRef CAS.
- T. Ming, W. Feng, Q. Tang, F. Wang, L. Sun, J. Wang and C. Yan, J. Am. Chem. Soc., 2009, 131, 16350–16351 CrossRef CAS PubMed.
- J. Zhang, M. R. Langille, M. L. Personick, K. Zhang, S. Li and C. A. Mirkin, J. Am. Chem. Soc., 2010, 132, 14012–14014 CrossRef CAS PubMed.
- Y. Yan, J. S. Du, K. D. Gilroy, D. Yang, Y. Xia and H. Zhang, Adv. Mater., 2017, 29, 1605997 CrossRef PubMed.
- Z. Fan and H. Zhang, Chem. Soc. Rev., 2016, 45, 63–82 RSC.
- J. K. Nørskov, J. Rossmeisl, A. Logadottir, L. Lindqvist, J. R. Kitchin, T. Bligaard and H. Jónsson, J. Phys. Chem. B, 2004, 108, 17886–17892 CrossRef.
- V. Viswanathan, H. A. Hansen, J. Rossmeisl and J. K. Nørskov, ACS Catal., 2012, 2, 1654–1660 CrossRef CAS.
- J.-C. Dong, X.-G. Zhang, V. Briega-Martos, X. Jin, J. Yang, S. Chen, Z.-L. Yang, D.-Y. Wu, J. M. Feliu, C. T. Williams, Z.-Q. Tian and J.-F. Li, Nat. Energy, 2019, 4, 60–67 CrossRef CAS.
- A. Kulkarni, S. Siahrostami, A. Patel and J. K. Nørskov, Chem. Rev., 2018, 118, 2302–2312 CrossRef CAS PubMed.
- M. E. Gamboa-Aldeco, E. Herrero, P. S. Zelenay and A. Wieckowski, J. Electroanal. Chem., 1993, 348, 451–457 CrossRef.
- W. Zhou, J. Wu and H. Yang, Nano Lett., 2013, 13, 2870–2874 CrossRef CAS PubMed.
- S. Kondo, M. Nakamura, N. Maki and N. Hoshi, J. Phys. Chem. C, 2009, 113, 12625–12628 CrossRef CAS.
- M. Shao, T. Yu, J. H. Odell, M. Jin and Y. Xia, Chem. Commun., 2011, 47, 6566–6568 RSC.
- V. R. Stamenkovic, B. Fowler, B. S. Mun, G. Wang, P. N. Ross, C. A. Lucas and N. M. Marković, Science, 2007, 315, 493–497 CrossRef CAS PubMed.
- C. Cui, L. Gan, M. Heggen, S. Rudi and P. Strasser, Nat. Mater., 2013, 12, 765 CrossRef CAS PubMed.
- Z. Duan and G. Wang, J. Phys. Chem. C, 2013, 117, 6284–6292 CrossRef CAS.
- X. Huang, Z. Zhao, L. Cao, Y. Chen, E. Zhu, Z. Lin, M. Li, A. Yan, A. Zettl, Y. M. Wang, X. Duan, T. Mueller and Y. Huang, Science, 2015, 348, 1230–1234 CrossRef CAS PubMed.
- X. Huang, Z. Zhao, Y. Chen, E. Zhu, M. Li, X. Duan and Y. Huang, Energy Environ. Sci., 2014, 7, 2957–2962 RSC.
- J. Zhang, H. Yang, J. Fang and S. Zou, Nano Lett., 2010, 10, 638–644 CrossRef CAS PubMed.
- J. Wu, L. Qi, H. You, A. Gross, J. Li and H. Yang, J. Am. Chem. Soc., 2012, 134, 11880–11883 CrossRef CAS PubMed.
- N. Hoshi, M. Nakamura and A. Hitotsuyanagi, Electrochim. Acta, 2013, 112, 899–904 CrossRef CAS.
- J. Yue, Z. Du and M. Shao, J. Phys. Chem. Lett., 2015, 6, 3346–3351 CrossRef CAS PubMed.
- T. Yu, D. Y. Kim, H. Zhang and Y. Xia, Angew. Chem., Int. Ed., 2011, 50, 2773–2777 CrossRef CAS PubMed.
- L. Mingchuan, S. Yingjun, Z. Xu, Q. Yingnan, L. Mingqiang, L. Yingjie, L. Chunji, Y. Yong, W. Lei, G. Peng, L. Gang and G. Shaojun, Adv. Mater., 2018, 30, 1705515 CrossRef PubMed.
- H. Kwon, M. K. Kabiraz, J. Park, A. Oh, H. Baik, S.-I. Choi and K. Lee, Nano Lett., 2018, 18, 2930–2936 CrossRef CAS PubMed.
- S. Luo, M. Tang, P. K. Shen and S. Ye, Adv. Mater., 2017, 29, 1601687 Search PubMed.
- Z. W. Seh, J. Kibsgaard, C. F. Dickens, I. Chorkendorff, J. K. Nørskov and T. F. Jaramillo, Science, 2017, 355, eaad4998 CrossRef PubMed.
- E. Skúlason, V. Tripkovic, M. E. Björketun, S. Gudmundsdóttir, G. Karlberg, J. Rossmeisl, T. Bligaard, H. Jónsson and J. K. Nørskov, J. Phys. Chem. C, 2010, 114, 18182–18197 CrossRef.
- N. M. Markovića, S. T. Sarraf, H. A. Gasteiger and P. N. Ross, J. Chem. Soc., Faraday Trans., 1996, 92, 3719–3725 RSC.
- N. M. Marković, B. N. Grgur and P. N. Ross, J. Phys. Chem. B, 1997, 101, 5405–5413 CrossRef.
- J. Kua and W. A. Goddard, J. Am. Chem. Soc., 1999, 121, 10928–10941 CrossRef CAS.
- T. D. Jarvi, S. Sriramulu and E. M. Stuve, Colloids Surf., A, 1998, 134, 145–153 CrossRef CAS.
- T. H. M. Housmans, A. H. Wonders and M. T. M. Koper, J. Phys. Chem. B, 2006, 110, 10021–10031 CrossRef CAS PubMed.
- P. Ferrin and M. Mavrikakis, J. Am. Chem. Soc., 2009, 131, 14381–14389 CrossRef CAS PubMed.
- S. Lu, K. Eid, D. Ge, J. Guo, L. Wang, H. Wang and H. Gu, Nanoscale, 2017, 9, 1033–1039 RSC.
- J. Park, H. J. Kim, A. Oh, T. Kwon, H. Baik, S.-I. Choi and K. Lee, Nanoscale, 2018, 10, 21178–21185 RSC.
- L. Zhuang, J. Jin and H. D. Abruña, J. Am. Chem. Soc., 2007, 129, 11033–11035 CrossRef CAS PubMed.
- W. Chrzanowski and A. Wieckowski, Langmuir, 1998, 14, 1967–1970 CrossRef CAS.
- Y. Liu, D. Li, V. R. Stamenkovic, S. Soled, J. D. Henao and S. Sun, ACS Catal., 2011, 1, 1719–1723 CrossRef CAS.
- X. Lu, Z. Deng, C. Guo, W. Wang, S. Wei, S.-P. Ng, X. Chen, N. Ding, W. Guo and C.-M. L. Wu, ACS Appl. Mater. Interfaces, 2016, 8, 12194–12204 CrossRef CAS PubMed.
- Q. Chen, Y. Yang, Z. Cao, Q. Kuang, G. Du, Y. Jiang, Z. Xie and L. Zheng, Angew. Chem., Int. Ed., 2016, 55, 9021–9025 CrossRef CAS PubMed.
- C. Wang, L. Zhang, H. Yang, J. Pan, J. Liu, C. Dotse, Y. Luan, R. Gao, C. Lin, J. Zhang, J. P. Kilcrease, X. Wen, S. Zou and J. Fang, Nano Lett., 2017, 17, 2204–2210 CrossRef CAS PubMed.
- N. Zhang, L. Bu, S. Guo, J. Guo and X. Huang, Nano Lett., 2016, 16, 5037–5043 CrossRef CAS PubMed.
- C. Busó-Rogero, V. Grozovski, F. J. Vidal-Iglesias, J. Solla-Gullón, E. Herrero and J. M. Feliu, J. Mater. Chem. A, 2013, 1, 7068–7076 RSC.
- H.-F. Wang and Z.-P. Liu, J. Am. Chem. Soc., 2008, 130, 10996–11004 CrossRef CAS PubMed.
- A. Chen and C. Ostrom, Chem. Rev., 2015, 115, 11999–12044 CrossRef CAS PubMed.
- C. Bianchini and P. K. Shen, Chem. Rev., 2009, 109, 4183–4206 CrossRef CAS PubMed.
- E. D. Wang, J. B. Xu and T. S. Zhao, J. Phys. Chem. C, 2010, 114, 10489–10497 CrossRef CAS.
- Z. Luo, J. Lu, C. Flox, R. Nafria, A. Genç, J. Arbiol, J. Llorca, M. Ibáñez, J. R. Morante and A. Cabot, J. Mater. Chem. A, 2016, 4, 16706–16713 RSC.
- S. J. Ye, D. Y. Kim, S. W. Kang, K. W. Choi, S. W. Han and O. O. Park, Nanoscale, 2014, 6, 4182–4187 RSC.
- N.-F. Yu, N. Tian, Z.-Y. Zhou, T. Sheng, W.-F. Lin, J.-Y. Ye, S. Liu, H.-B. Ma and S.-G. Sun, ACS Catal., 2019, 9, 3144–3152 CrossRef CAS.
- K. Jiang, H.-X. Zhang, S. Zou and W.-B. Cai, Phys. Chem. Chem. Phys., 2014, 16, 20360–20376 RSC.
- S. Uhm, H. J. Lee and J. Lee, Phys. Chem. Chem. Phys., 2009, 11, 9326–9336 RSC.
- V. Mazumder and S. Sun, J. Am. Chem. Soc., 2009, 131, 4588–4589 CrossRef CAS PubMed.
- S.-I. Choi, J. A. Herron, J. Scaranto, H. Huang, Y. Wang, X. Xia, T. Lv, J. Park, H.-C. Peng, M. Mavrikakis and Y. Xia, ChemCatChem, 2015, 7, 2077–2084 CrossRef CAS.
- N. Hoshi, K. Kida, M. Nakamura, M. Nakada and K. Osada, J. Phys. Chem. B, 2006, 110, 12480–12484 CrossRef CAS PubMed.
- M. Jin, H. Zhang, Z. Xie and Y. Xia, Energy Environ. Sci., 2012, 5, 6352–6357 RSC.
- X. Zhang, H. Yin, J. Wang, L. Chang, Y. Gao, W. Liu and Z. Tang, Nanoscale, 2013, 5, 8392–8397 RSC.
- Z. Xi, J. Li, D. Su, M. Muzzio, C. Yu, Q. Li and S. Sun, J. Am. Chem. Soc., 2017, 139, 15191–15196 CrossRef CAS PubMed.
- S. H. Chang, N. Danilovic, K.-C. Chang, R. Subbaraman, A. P. Paulikas, D. D. Fong, M. J. Highland, P. M. Baldo, V. R. Stamenkovic, J. W. Freeland, J. A. Eastman and N. M. Markovic, Nat. Commun., 2014, 5, 4191 CrossRef CAS PubMed.
- L. Gloag, T. M. Benedetti, S. Cheong, R. F. Webster, C. E. Marjo, J. J. Gooding and R. D. Tilley, Nanoscale, 2018, 10, 15173–15177 RSC.
- J. X. Wang, N. S. Marinković, H. Zajonz, B. M. Ocko and R. R. Adžić, J. Phys. Chem. B, 2001, 105, 2809–2814 CrossRef CAS.
- M. Zhao, Z. Chen, Z. Lyu, Z. D. Hood, M. Xie, M. Vara, M. Chi and Y. Xia, J. Am. Chem. Soc., 2019, 141, 7028–7036 CrossRef CAS PubMed.
- J. Zhu, Z. Chen, M. Xie, Z. Lyu, M. Chi, M. Mavrikakis, W. Jin and Y. Xia, Angew. Chem., Int. Ed., 2019, 58, 7244–7248 CrossRef CAS PubMed.
- B. Jiang, Y. Guo, J. Kim, A. E. Whitten, K. Wood, K. Kani, A. E. Rowan, J. Henzie and Y. Yamauchi, J. Am. Chem. Soc., 2018, 140, 12434–12441 CrossRef CAS PubMed.
- J. Ohyama, D. Kumada and A. Satsuma, J. Mater. Chem. A, 2016, 4, 15980–15985 RSC.
- Y. Jiao, Y. Zheng, M. Jaroniec and S. Z. Qiao, Chem. Soc. Rev., 2015, 44, 2060–2086 RSC.
- C. C. L. McCrory, S. Jung, I. M. Ferrer, S. M. Chatman, J. C. Peters and T. F. Jaramillo, J. Am. Chem. Soc., 2015, 137, 4347–4357 CrossRef CAS PubMed.
- Y. Zheng, Y. Jiao, A. Vasileff and S.-Z. Qiao, Angew. Chem., Int. Ed., 2018, 57, 7568–7579 CrossRef CAS PubMed.
- J. Mou, Y. Gao, J. Wang, J. Ma and H. Ren, RSC Adv., 2019, 9, 11755–11761 RSC.
| This journal is © The Royal Society of Chemistry 2019 |