
The role of trace Ag in the synthesis of Au nanorods†
Liane M.
Moreau
ab,
Matthew R.
Jones
ab,
Eric W.
Roth
ab,
Jinsong
Wu
ab,
Sumit
Kewalramani
ab,
Matthew N.
O'Brien
 cb,
Bor-Rong
Chen
a,
Chad A.
Mirkin
*acb and
Michael J.
Bedzyk
*adb
cb,
Bor-Rong
Chen
a,
Chad A.
Mirkin
*acb and
Michael J.
Bedzyk
*adb
aDepartment of Materials Science and Engineering, Northwestern University, Evanston, IL 60208, USA. E-mail: bedzyk@northwestern.edu
bInternational Institute for Nanotechnology, Northwestern University, Evanston, IL 60208, USA
cDepartment of Chemistry, Northwestern University, Evanston, IL 60208, USA
dDepartment of Physics and Astronomy, Northwestern University, Evanston, IL 60208, USA
First published on 5th June 2019
Abstract
One of the more useful syntheses of single crystalline, uniform Au nanorods from Au spherical seeds relies on the addition of trace Ag ions, yet the role that Ag+ plays has remained both elusive and controversial, due in part to lack of knowledge of how the Ag distribution in the nanorod evolves over time. In this work, we fill in this knowledge gap by correlating the spatial distribution of Ag within Au nanorods with nanorod anisotropic growth through time-course X-ray absorption spectroscopy (XAFS)-derived atomic-level elemental coordination paired with electron microscopy for nanoscale morphological analysis. Using this method, a plausible pathway for the conversion of spherical seeds into Au nanorods is proposed. Evidence shows that the nanorod anisotropic growth is directly related to the Ag surface coverage. Anisotropy is induced early in the reaction when Ag first deposits onto the nanoparticle surface, but growth occurs more isotropically as the reaction progresses and Ag diffuses into the nanorod bulk. The results of this investigation and methods employed should be extendable to many anisotropic nanoparticle syntheses that make use of trace elemental species as shape-control additives.
Introduction
Control over nanoparticle morphology has led to optimized nanoparticles for use in catalysis due to their preferential faceting,1 in optics due to their size and shape-dependent local surface plasmon resonance (LSPR),2–4 and in programmable assembly due to the introduction of valency.5,6 One particularly successful synthetic approach for producing uniform anisotropic Au nanoparticles begins with colloidal growth from small Au nanoparticles (<2 nm in diameter) that act as “seeds” to template growth in an Au salt solution with trace Ag+. Although the trace Ag+ has been determined to be a necessary reactant in controlling the morphology and aspect ratio of the resulting product,7,8 the role of Ag in the synthesis and how these particles form remains elusive and controversial.9Au nanorods are the most utilized and studied morphology synthesized via this approach and thus are an ideal particle to investigate the pathway behind the synthesis of anisotropic Au nanoparticles using trace Ag+.10 Historically, nanorods synthesized in growth media containing cetyl trimethylammonium bromide (CTAB) as a surfactant were observed in low yield, with non-uniform size and shape.11,12 When trace amounts of Ag+ were added into this reactant solution, however, the uniformity was improved and nanorod aspect ratio could be controlled based on the amount of Ag+ introduced.10 These results sparked an interest into the role that Ag plays in controlling the reaction product, in addition to the overall pathway behind anisotropic growth in colloidal nanoparticle systems.
Although there are many ideas surrounding nanorod anisotropic growth, two general hypotheses dominate current literature (Fig. 1): (1) the surfactant CTAB adheres with a greater affinity or a greater packing density to specific hkl surface facets, which induces a slower rate of Au reduction onto these facets relative to others and leads to an anisotropic shape.13–15 In these arguments, solution Ag+ is entirely ignored from a mechanistic perspective, and (2) trace Ag deposits preferentially on specific surface facets, which similarly induces a slower relative rate of growth and leads to anisotropy.9 It should be noted that even when anisotropy is attributed to Ag, a source of bromide is also deemed necessary to achieve the desired nanorod product.7,14,16,17 The argument for CTAB's involvement in anisotropic growth originates, in part from ligand exchange experiments, where CTAB is difficult to remove from the sides of the nanorods, but can be easily exchanged with thiolated ligands at the tips. This suggests that CTAB is bound more strongly to the sides of the rods than the tips, which may result in anisotropic growth.18,19 For the case of Ag, the argument has depended on the determined oxidation state of deposited Ag, as it remains controversial whether the deposited Ag is Ag0 (ref. 20–22) or Ag+.22,23 For the case of Ag+, it is believed that Ag may form a complex with bromine on particular surface facets, preventing growth on these facets.16 The preference of Ag for particular surface facets for the Ag0 case is explained by the underpotential deposition (UPD) hypothesis,20 based on the technique commonly used to deposit a metallic monolayer onto the surface of a more noble metal due to a decrease in required reduction potential.24,25 In bulk systems, the decrease in reduction potential has been determined to be greater for higher-energy surface facets.25,26 The dependence of nanoparticle morphology on solution Ag+ concentration has also been attributed to this phenomenon.7,27,28 This has led to the hypothesis that with Au nanoparticles, Ag+ is preferentially reduced onto higher energy surface facets, leading to anisotropic growth. In this study, we specifically investigate the Ag hypothesis and consider the role that Ag plays in the anisotropic growth of Au nanorods.
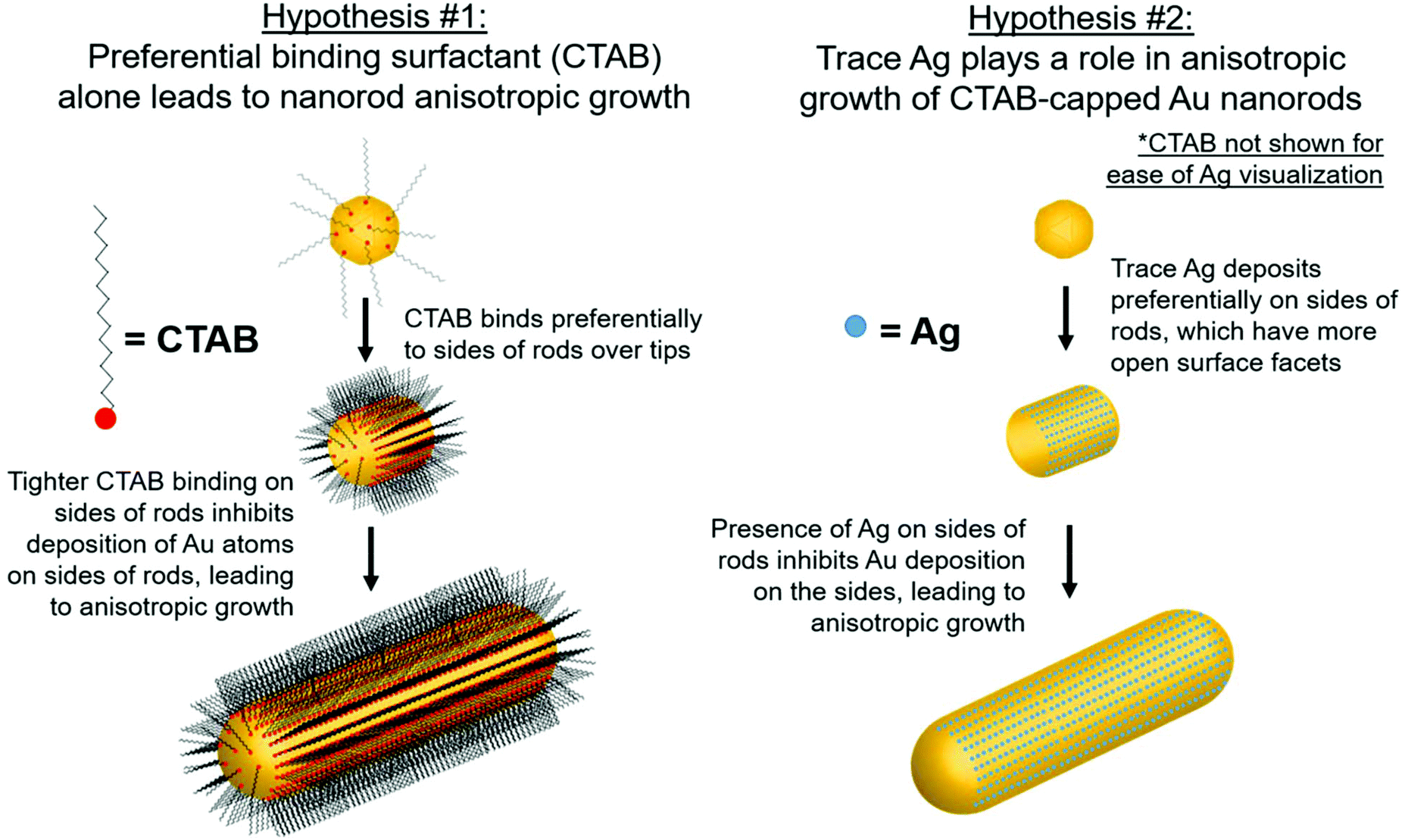 | ||
| Fig. 1 Schematic of proposed anisotropic growth pathways in the synthesis of Au nanorods. | ||
Previous investigations into the nanorod growth pathway using electron microscopy (EM),10,17,20,21 EDX,29 XPS,28 and inductively coupled plasma-atomic emission spectroscopy (ICP-AES)22 attempted to extrapolate the synthesis pathway from only the final nanorod product. With the exception of a time-course TEM study30 and HRTEM studies of initial growth off of Au seeds,31 the earlier time points in the reaction have not been addressed experimentally. More importantly, there is little knowledge of how the Ag distribution in the nanorod evolves over time. The present study aims to fill in this knowledge gap and determine the structure of Ag within the nanorods and its role, or lack thereof, in anisotropic growth.
In this study, the local atomic-scale structure of Ag within the nanorods is probed using a combination of X-ray absorption fine structure (XAFS), X-ray fluorescence (XRF) and EDX-mapping to correlate the Ag distribution in the nanorods to morphology evolution traced using EM. The emphasis on X-ray techniques provides the capability to probe nanoparticles in situ in their native solution and in a statistically meaningful sample population. Use of this structural toolbox enables us to evaluate the aforementioned hypotheses with new detail at the atomic scale, and ultimately establish support for the Ag underpotential deposition hypothesis of Au nanorod growth (hypothesis 2, Fig. 1). Most notably, Ag adsorbs onto the nanoparticle surface early in the reaction, which correlates with anisotropic growth. As the reaction progresses, the incorporation rate of Ag slows, such that by ∼30 minutes into the 120 minutes reaction, very little Ag remains on the nanoparticle surface. In other words, by 30 minutes, Ag has diffused into the nanoparticle bulk. The key observation is that while the diameter growth rate of the nanorods does not depend on the amount of Ag incorporated into the nanoparticle surface, the length growth rate of the nanorods is directly correlated with the amount of surface Ag.
Experimental
Synthesis
Au nanorods were synthesized via the procedure established by El-Sayed and co-workers.10 Briefly, ∼2 nm Au seed nanoparticles were synthesized by addition of 0.6 mL of 0.01 M ice cold NaBH4 (Sigma-Aldrich) to a stirring solution containing 5 mL 0.2 M CTAB (bioWORLD), 0.25 mL 0.01 M HAuCl4 (Sigma-Aldrich) and 4.75 mL NANOpure™ water (18.2 MΩ cm resistivity). In a separate vial, 5 mL 0.2 M CTAB, 0.3 mL 0.004 M AgNO3 (Sigma-Aldrich), 0.5 mL 0.01 M HAuCl4 and 4.5 mL NANOpure™ water were combined and 0.07 mL of 0.078 M ascorbic acid (Sigma-Aldrich) added to reduce Au3+ to Au+. 0.012 mL of the as-synthesized seeds were added to this solution and reacted for 120 minutes to form the final nanorod product. In order to quench the reaction at different timepoints, an aliquot from the reaction solution was brought to 1 mM bis(p-sulfonatophenyl)phenylphosphine (BSPP, Sigma Aldrich), which exchanged with CTAB on the surface of the nanorods and halted further nanorod growth. UV-vis spectroscopy (Fig. S5†) and small angle X-ray scattering results (Fig. S6†) confirm that the nanoparticle morphological evolution is the same under in situ and BSPP-quenched conditions, thus validating this approach.UV-Vis spectroscopy
UV-Vis spectroscopy scans of samples in 1 ml NANOpure™ water were taken using a Cary 5000 UV-vis spectrophotometer across the range of 200–1000 nm at a 1 cm path length.Electron microscopy
Using the NU EPIC Facility, samples were dropcasted on a carbon coated grid for electron microscopy characterization. Scanning transmission electron microscopy (STEM) images were collected using a JEOL JEM-2100 FasTEM at 200 keV. Early time point nanorod samples were plunge frozen at different time points on glow discharged 200 mesh lacy carbon grids with an FEI Vitrobot Mark III and loaded into a Gatan Cryo Transfer Holder held at −165 °C. Image data was gathered in a Hitachi HD-2300 STEM at 200 kV utilizing phase contrast transmission and high angle annular dark field detectors. EDX mapping of 12-minute nanorod samples was performed on an aberration-corrected Hitachi HD-2700 STEM operated at 200 kV. The image shown in Fig. 6 is a STEM image collected by the high angle annular dark field detector showing Z-contrast, while the X-ray energy dispersive spectra (EDS) used for mapping is collected using an Oxford X-ray detector and processed by AZtecEnergy software.Small-angle X-ray scattering
(SAXS) patterns were collected using 10.00 keV X-rays at DND-CAT station 5ID-D located at the Advanced Photon Source (APS) at Argonne National Laboratory (ANL). The aqueous nanoparticle dispersions were placed in quartz capillary tubes (inner diameter ∼1.5 mm, Charles Supper) for measurement. The scattering patterns from ex situ (BSPP-quenched) and in situ samples were collected and compared at various stages throughout the timecourse reaction.X-ray fluorescence
(XRF) spectra were obtained at APS stations 10BM-B and 5BM-D using a Vortex four element silicon drift diode detector (SDD). Spectra were collected at 26.014 keV (above the Ag K edge energy, 25.514 keV) to determine the Ag-to-Au atomic ratio from the areas under the Au Lα and Ag Kα fluorescence lines. Elemental XRF cross sections,32 detector efficiency, and attenuation due to solvent media were taken into account in determining the Ag/Au ratio.X-ray absorption fine-structure
(XAFS) spectra at the Au L3 edge and Ag K edge (11.919 keV and 25.514 keV, respectively) were collected at MR-CAT station 10BM-B located at the APS. Energy scans were taken over a range from −150 eV to 600 eV with respect to the Au or Ag absorption edge using a Si(111) monochromator. XAFS spectra of the samples were collected in XRF-mode using a four-element Vortex SDD, and calibrated with an Au or Ag metal foil standard in transmission-mode. Samples were concentrated via centrifugation to micromolar concentrations of Au/Ag atoms and placed in 3 mm inner diameter quartz capillary tubes.XAFS data was processed using ATHENA and ARTEMIS software, part of the IFEFFIT package.33 EXAFS spectra were modeled according to the EXAFS equation:34–37
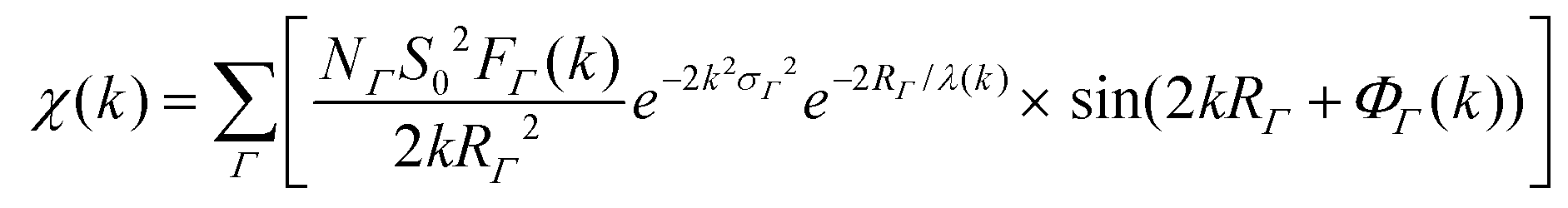
Results
STEM images collected at various time-points throughout the 120 minutes nanorod synthesis reaction are shown in Fig. 2 (see Methods section for a description of the synthesis process). While time-dependent dimensional parameters for Au nanorods have been previously reported,22,30,38 we herein repeated this work such that dimensional parameters could be connected to the Ag structural environment within the nanorods at varying time points from the same batch of nanorods. Most notably, from the spherical seed particles, anisotropy is induced early in the reaction (between 6 and 8 minutes). This anisotropy event is also observed from UV-vis absorption spectra (Fig. 3b), which show within the same 6–8 minutes time frame, the emergence of a longitudinal localized surface plasmon resonance (LSPR) around 800 nm and transverse LSPR around 550 nm that are characteristic of a nanorod morphology.10 These results agree with previous HRTEM studies of symmetry breaking of Au seeds, which occurred only after the seeds reached a critical size (>5 minutes).31,39,40 By 8 minutes into the reaction, the seeds evolve into anisotropic rods with an aspect ratio of 1.5. Over the next few minutes, the nanorods exhibit a rapid elongation, reaching an aspect ratio of ∼4 (Table S1†). While the UV-vis longitudinal LSPR blue shifts slightly between 20 and 120 minutes (Fig. 3b), suggesting a slight decrease in aspect ratio, values extracted from STEM remain the same within error (Table S1†) for the remainder of the reaction as the nanorods continue to grow until the reaction is complete. | ||
| Fig. 2 Timecourse STEM images. Starting from 2 nm Au seeds, STEM images taken from colloidally synthesized Au nanorods quenched with BSPP at time points from 2 minutes into growth (top-left) to final product nanorods (bottom-right). The first three images are collected using cryo-STEM (see ESI† for details). | ||
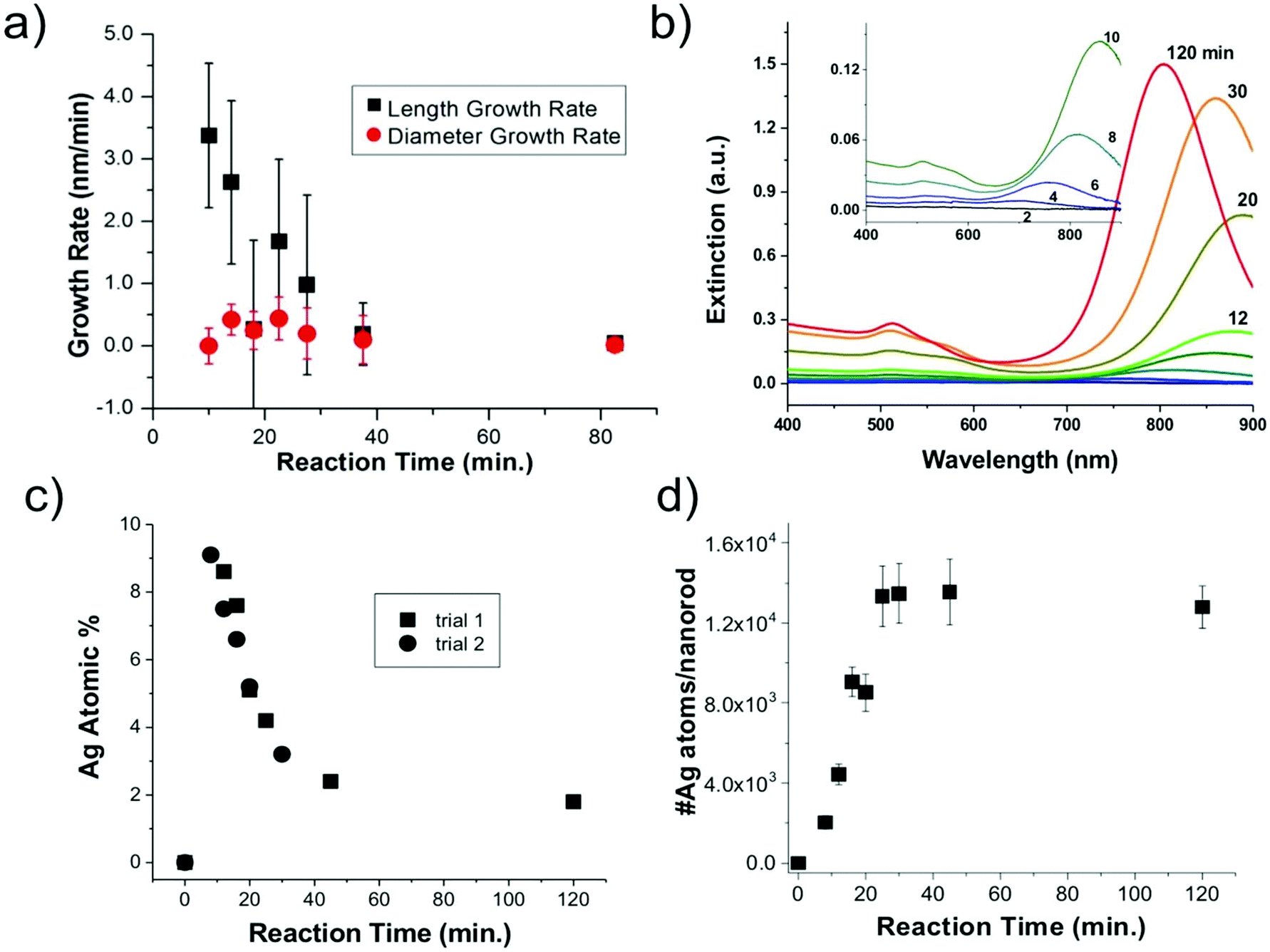 | ||
| Fig. 3 Nanoparticle growth and composition. (a) EM determined length and diameter growth rates as a function of reaction time, (b) UV-vis spectra as a function of reaction time; inset shows a magnified view of early time point spectra, (c) XRF-determined nanoparticle composition and (d) XRF and EM determined number of Ag atoms per nanorod. | ||
Statistics from STEM-determined particle dimensions were used to track the length and diameter growth rates of the nanorods as a function of reaction time (Fig. 3a). While the diameter growth rate appears relatively constant throughout the reaction, the length growth rate exceeds the diameter growth rate during the 8–30 minutes time frame. After 30 minutes, the length and width growth rates are very low, and minimal particle growth is observed from 45 to 120 minutes. It should be noted that this slowing of growth rate is not the result of depletion of Ag+ or Au+ ions, which both our own and previous work22 found to be in excess at reaction completion. Excess solution ions would be expected given that the concentration of ascorbic acid is insufficient to reduce all solution Au+. These results reveal that the anisotropic growth steps leading to nanorod formation occur early in the reaction, while the late reaction stages do not contribute to particle anisotropy.
From XRF intensities under element-specific fluorescence lines, relative percentages of Ag vs. Au can be determined. XRF analysis (Fig. 3c) show that the Ag incorporates early in the Au nanorods and the Ag atomic % decreases with reaction time. This finding was reproducible through two separate trials. Further analysis that combines XRF and STEM results (Fig. 3d) shows that the absolute number of Ag atoms per nanorod increases in the first 45 min. This shows that the decrease in the atomic% Ag as a function of reaction time does not result simply from a decrease in the number of Ag atoms incorporated onto or into the nanorods.
To consider the oxidation state of Ag throughout the reaction, XANES spectra were compared to relevant Ag+ standards, which include AgBr (a species previously proposed to cap the elongated nanorod surface facets)17 and Ag2O, as well as Ag0 standards, which include an Ag metal foil and the as-synthesized Au nanorods overgrown with an Au shell, which should encapsulate any surface Ag5 (Fig. 4). Each of these standards possesses a different fingerprint signature that is affected by differences in the 3d unoccupied densities of states, which can be used to identify the state of the Ag. The Au nanorod spectrum near edge features mimic those of the Ag0 standards (Fig. 4, right). In particular, the spectrum appears the same for the Ag in the final-product nanorods (green) as when they are overgrown with an Au shell (blue), when all Ag should be Ag0 due to encapsulation of any surface species into the FCC interior. When Au nanorods are not isolated from solution (magenta), the signature is primarily Ag+ due to excess AgNO3 in solution. This confirms that a majority of the Ag ions present in solution do not incorporate into the nanorods.22 We not only observe these comparisons for the final product nanorods (Fig. 4, green), but also throughout each step of the time-course reaction, even early on when Ag is the dominant surface species (Fig. S7†). Ag therefore incorporates into the nanorod as Ag0.
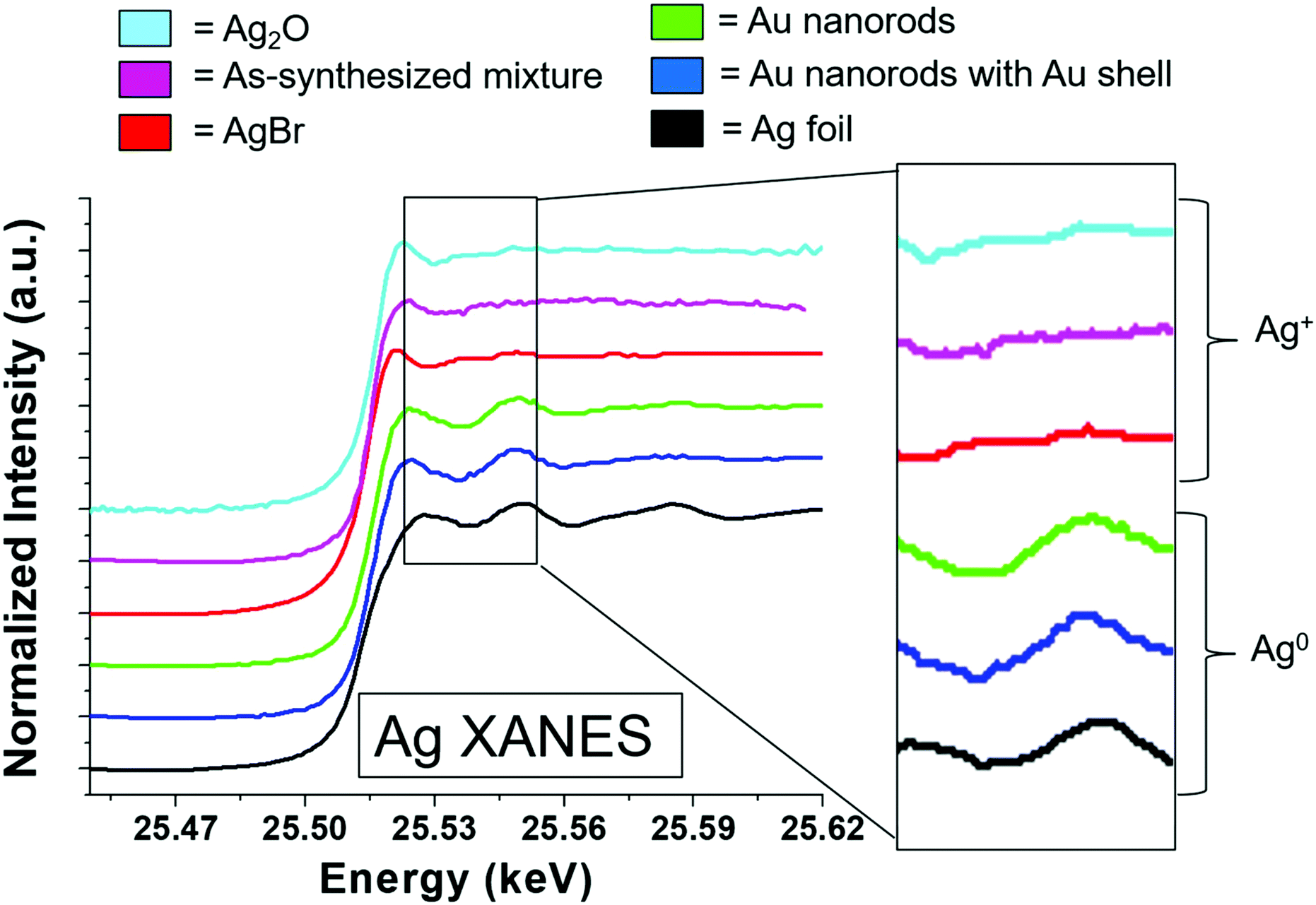 | ||
| Fig. 4 Ag K-edge XANES data from 120 min final product nanorods (middle, green) compared to standards with Ag+ (top 3) and Ag0 (bottom 2). A blowup of the near edge region is shown on the right-hand-side for each spectrum. | ||
EXAFS-derived coordination numbers (CNs) show whether or not atoms in the nanoparticle are on the nanorod surface (CN < 12) or in FCC bulk (CN = 12) (Fig. 5a). These coordination numbers are representative of only Ag and Au atoms that are affiliated with the quenched timecourse nanorod samples (the excess AgNO3 and HAuCl4 are removed from solution). While the Au CN throughout the reaction is 12, the Ag CN increases from <8 at 8 minutes into the reaction, to a full shell of 12 by 40 min. This suggests that Ag starts on the nanoparticle surface and incorporates into the bulk as the reaction progresses. Ag incorporation into the nanorod bulk, provides, in conjunction with the increasing total number of Ag atoms incorporated into the nanorod (Fig. 3d) evidence that the decreasing atomic% Ag in the nanorods over time is inconsistent with a Galvanic exchange process wherein excess solution Au+ would react with surface Ag.41,42
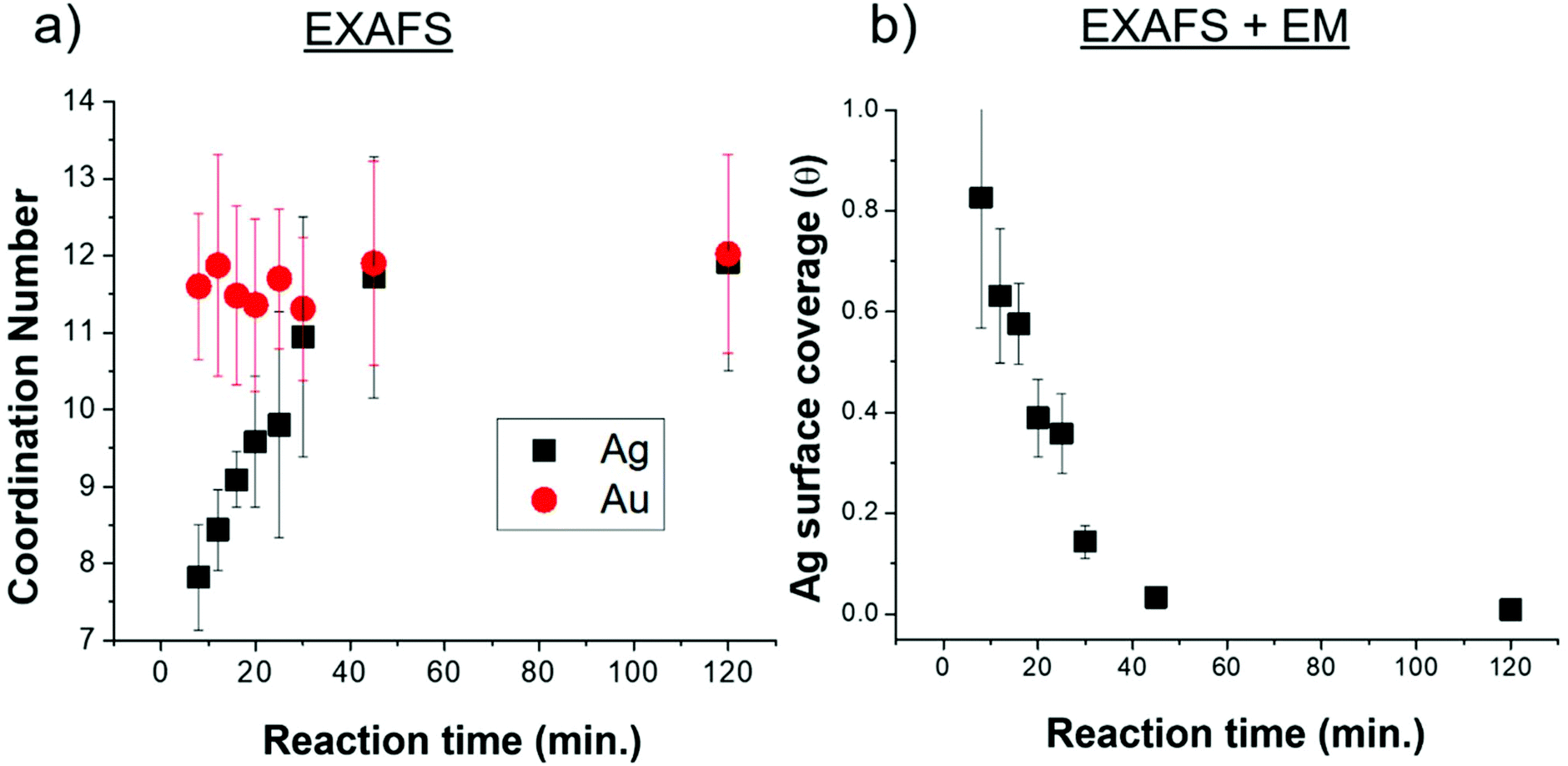 | ||
| Fig. 5 Ag coordination number and surface coverage. (a) Coordination numbers extracted from Ag K and Au L3 edge XAFS spectra. (b) Ag surface coverage, extracted from a combination of XAFS-extracted coordination numbers, EM-determined dimensions and the assumption that Ag on the surface has a CN of 7. | ||
The fraction of the Ag atoms in the nanorod that are on the surface is determined from the Ag CN (Fig. 5a), assuming a surface CN of 7 (as would be the case for {110} facets), which has been reported as the identity of the elongated nanorod facets10,20 and a bulk CN of 12 (see ESI† for details). This fraction is converted into an Ag surface coverage (Fig. 5b) when combined with the overall Ag atomic% and nanoparticle dimensions (Fig. 3). Using this method, nanorod surface coverage is determined to be 84 atomic% Ag at 8 minutes into the reaction and reduces to only a few percent after 45 minutes into the reaction. This trend persists even if we consider the identity of the elongated facets to be {520} rather than {110} as has been more recently proposed16,30,43 (Fig. S2†). This analysis shows that the increase in coordination number is not only due to the reduction in surface area to volume ratio during nanorod growth. Rather, the Ag surface coverage is high at the start of the reaction, and deceases as the reaction progresses. The nanorod surface coverage has been hypothesized to strongly influence the deposition rate of the Au and Ag atoms during nanorod evolution,7,20,28 but to our knowledge, has not been previously measured.
Having determined the evolution of the overall nanorod surface coverage, we next explored the composition of particular nanorod faces, since an inequivalence in Ag surface composition has been hypothesized to lead to an inequivalence in nanorod growth rates along different crystal directions.20 Even though EDX mapping of final product nanorods has been previously explored,29 it is important to consider the distribution early in the reaction when a majority of Ag atoms are on the nanorod surface. Additionally, EM images of nanorods from this previous study showed core–shell structure based on the electron density, which may be a result of subtle differences in the synthesis procedure (most notably a lower CTA+ concentration). This is not observed in our EM images (Fig. 2 and 6, top). EDX mapping (Fig. 6) was effective for studying the Au and Ag distribution in a nanorod 12 minutes into the reaction, when the majority of surface atoms in the nanoparticle are Ag (Fig. 5), making the Ag distribution found at this time point relevant to Ag deposition conditions. Here, it can be seen that Ag is present on the sides of the nanorods, as illustrated in Fig. 6 (bottom, center) rather than only on the ends (Fig. 6 bottom, right). The resolution of the measurement, in part due to low Ag signal in comparison to Au (Fig. S13†), is not sufficient to determine whether the Ag is on both the sides and the ends of the nanorods (Fig. 6 bottom, left). The Ag distribution within early time point nanorods and its implication on anisotropic growth will be further discussed in the following paragraphs.
 | ||
| Fig. 6 Ag distribution after 12 minutes during nanorod growth. STEM (top), and STEM EDX maps of Ag and Au at 12 minutes into the reaction where Ag is predominantly at the surface (Fig. 5). The bottom schematic shows the three types of Ag (blue) surface distributions considered. EDX eliminates the case for Ag only on the ends of the nanorods (bottom right). | ||
Discussion
The above structural characterization results can be used to determine whether or not the role of trace Ag+ in the synthesis of Au nanorods aligns with those that have been previously proposed (Fig. 7). It is first important to consider whether or not Ag incorporates into the particles at the time that symmetry is broken. From STEM images and UV-Vis spectra, we find that anisotropy is induced between 6 and 8 minutes into the reaction. By 8 minutes into the reaction, Ag has already deposited onto the nanorod surface such that the rods are ∼10% Ag, and 84% of the Ag in the nanorod is on the surface. This proves that Ag is incorporated early on in the reaction, which is also when we observe induction of anisotropy. This observation agrees with previous studies that suggest that Ag stabilizes the elongated nanorod facets prior to 8 minutes into the reaction,31 leading to anisotropic growth at early reaction stages.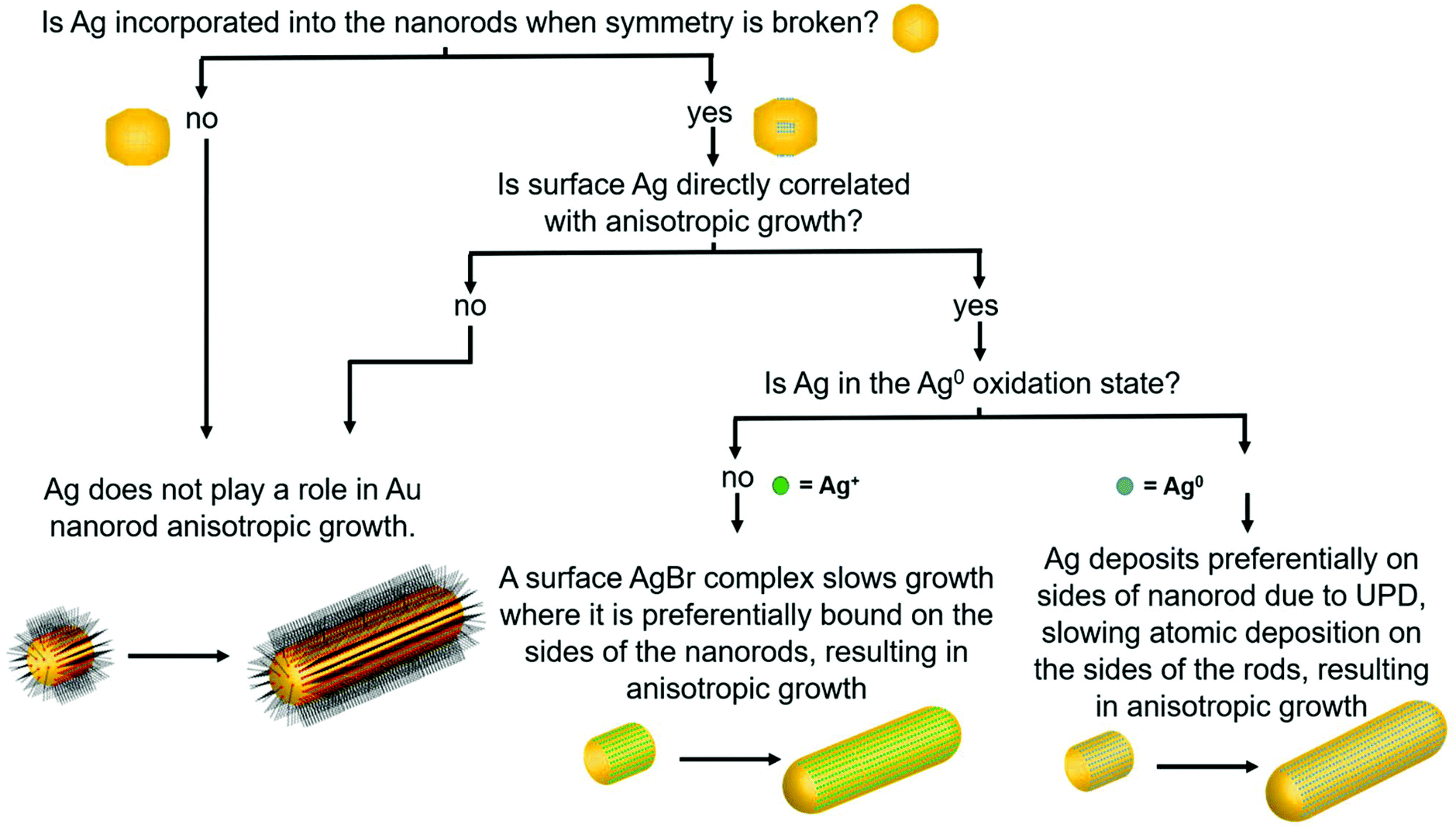 | ||
| Fig. 7 Flow chart considering the role of Ag in nanorod synthesis. The flow chart relates nanorod structural attributes to the appropriate hypothesis for the role of Ag in nanorod anisotropic growth. This study provides answers to the questions posed, leading to identification of the UPD hypothesis as the most consistent with the pathway behind anisotropic growth. | ||
To determine whether this surface incorporated Ag plays a role in nanorod anisotropic growth, XRF, EXAFS, and STEM results are combined to derive the nanorod length and diameter growth rates as a function of Ag surface coverage (Fig. 8). Interestingly, the length growth rate of the nanorods is directly correlated with the amount of surface Ag, whereas the diameter growth rate shows no correlation. This indicates that towards the beginning of the reaction when the majority of the surface is Ag, the length growth rate exceeds the diameter growth rate, resulting in an anisotropic nanoparticle. As the reaction progresses and Ag becomes increasingly incorporated into the bulk of the nanorod and its surface coverage drops, the length growth rate slows for the remainder of the reaction. At later reaction stages when a majority of surface atoms are Au, nanorod length and width growth rates are very low and similar within experimental error.
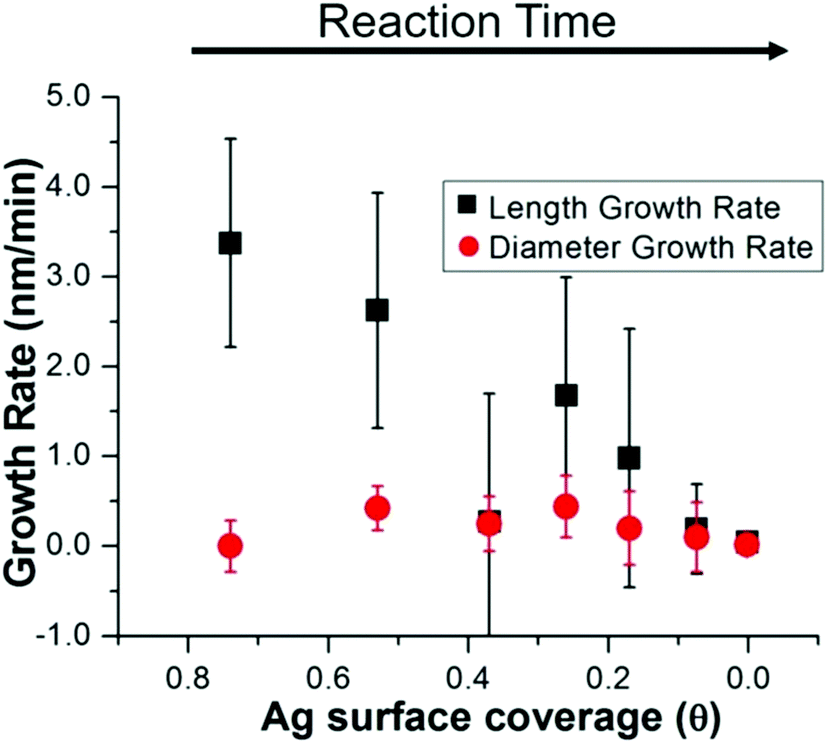 | ||
| Fig. 8 Nanorod growth rates vs. Ag surface coverage. Combining the results of XRF, EXAFS and STEM, the diameter growth rate is shown to be unaffected by surface Ag, while the length growth rate is directly correlated. | ||
Results show that surface Ag is directly correlated with nanorod anisotropic growth. Yet to provide an explanation for how surface Ag may contribute to anisotropic growth, results must be evaluated to see if the previously proposed UPD hypothesis, or if a different or new hypothesis better explains the observed phenomenon. In bulk UPD, Ag deposits as Ag0 rather than as an Ag+ adsorbate complex.25,26 From XANES, in the nanorod case, Ag also deposits as Ag0. The spectrum features match those of Ag0 standards and not other proposed species including AgBr and Ag2O.9
The UPD hypothesis also relies on the preferential deposition of Ag0 onto higher surface energy facets (i.e., those with lower coordination number) compared to other facets.20,28 EXAFS-derived coordination numbers give insight into which surface Ag is deposited on, as atoms incorporated into different surface facets have a different associated coordination number. A coordination number of CN = 12 corresponds to bulk, while for unreconstructed fcc surfaces CN = 9 for {111}, 8 for {100} and 7 for {110}. Given that the aforementioned are the proposed surface facets for single-crystal Au nanorods synthesized via the same synthetic procedure,10,20 we will refer to them moving forward, although it should be noted that the Ag distribution and resulting Ag surface coverage are still consistent when {520} facets are assumed for the sides of the nanorods instead of {110} (Fig. S2†). The EXAFS measured coordination number at the 8 minutes time point (7.8 ± 0.8) indicates that the Ag likely deposits on the {110} facets, since the measured CN should not be lower than the facet to which the Ag is coordinated due to atoms in the bulk that have a full coordination shell of 12. This agrees with the previous hypothesis that Ag is on the {110} facets.20,43 If we assume that the Ag is indeed on the {110} facets at the 8 minutes time point, then 84% Ag on the surface is on {110} facets with the remaining 16% in the bulk (i.e., 0.84 × 7 + 0.16 × 12 = 7.8). EDX mapping provides further evidence that Ag deposits onto the {110} facets (sides of the nanorods). The low CN eliminates the possibility of deposition onto lower energy {111} facets, which directly supports the UPD hypothesis that Ag has preference for higher surface energy facets on the nanorod.
In summary, the structural characterization herein is consistent with what would be expected for Ag UPD. Time course local and global structural details lead to the proposal of a plausible pathway for the anisotropic growth of Ag nanorods. In particular, quantitative details about the Ag content and distribution within the nanorods as a function of reaction time are provided. Fig. 9 outlines pictorially the following stages in the proposed reaction pathway highlighting the role of trace Ag in nanorod synthesis: (1) from quasi-spherical ∼2 nm Au seeds, Ag deposits as Ag0 between 6–8 minutes as a result of UPD, stabilizing {110} facets, and inducing anisotropic growth. (2) By 8 minutes into the reaction, the rods are anisotropic with an aspect ratio of ∼1.6, with an Ag surface coverage of 0.8. (3) By 12 minutes into the reaction, the nanorod aspect ratio reaches ∼3.6 (which is the same as at reaction completion within error), while a majority of surface atoms are still Ag. (4) As the reaction progresses, the rate of Ag deposition slows, such that the Ag surface coverage reduces and the length growth rate of the nanorod decreases. This may be due, in part, to a decrease in solution ascorbic acid concentration (Fig. S14†). The length growth rate of the nanorods is correlated to the amount of surface Ag, whereas the diameter growth rate is not. The most plausible explanation for why surface Ag inhibits Au deposition in comparison to surfaces that are Au rich is the higher bond-strength of Au–Au vs. Au–Ag, resulting in preferential Au deposition onto surfaces where Ag is not present. It is also the case that reduction potential is lowered for a metal onto a more noble metal (Au) than a less noble metal (Ag).25 Alternatively, there may be some synergistic interaction with CTAB and surface Ag that inhibits Au deposition, given that a bromide source is also essential to monodisperse nanorod growth.7,14,16,17 This is also supported by a previous study of Ag UPD-based nanoparticle synthesis that has found evidence for surface facet dependent characteristics in Ag–halogen binding using a similar surfactant (CTAC).44 (5) Ag diffuses into the layers below the nanorod surface, as additional Au atoms are deposited, such that the Ag coverage is reduced. Thus, the length growth rate slows, such that the ∼3.6 aspect ratio is maintained within error during growth. (6) By 45 minutes into the reaction, the Ag surface coverage approaches zero, and the length and diameter growth rates are diminishingly small until 120 minutes, when the nanorods cease growing, likely due to depletion of solution ascorbic acid.
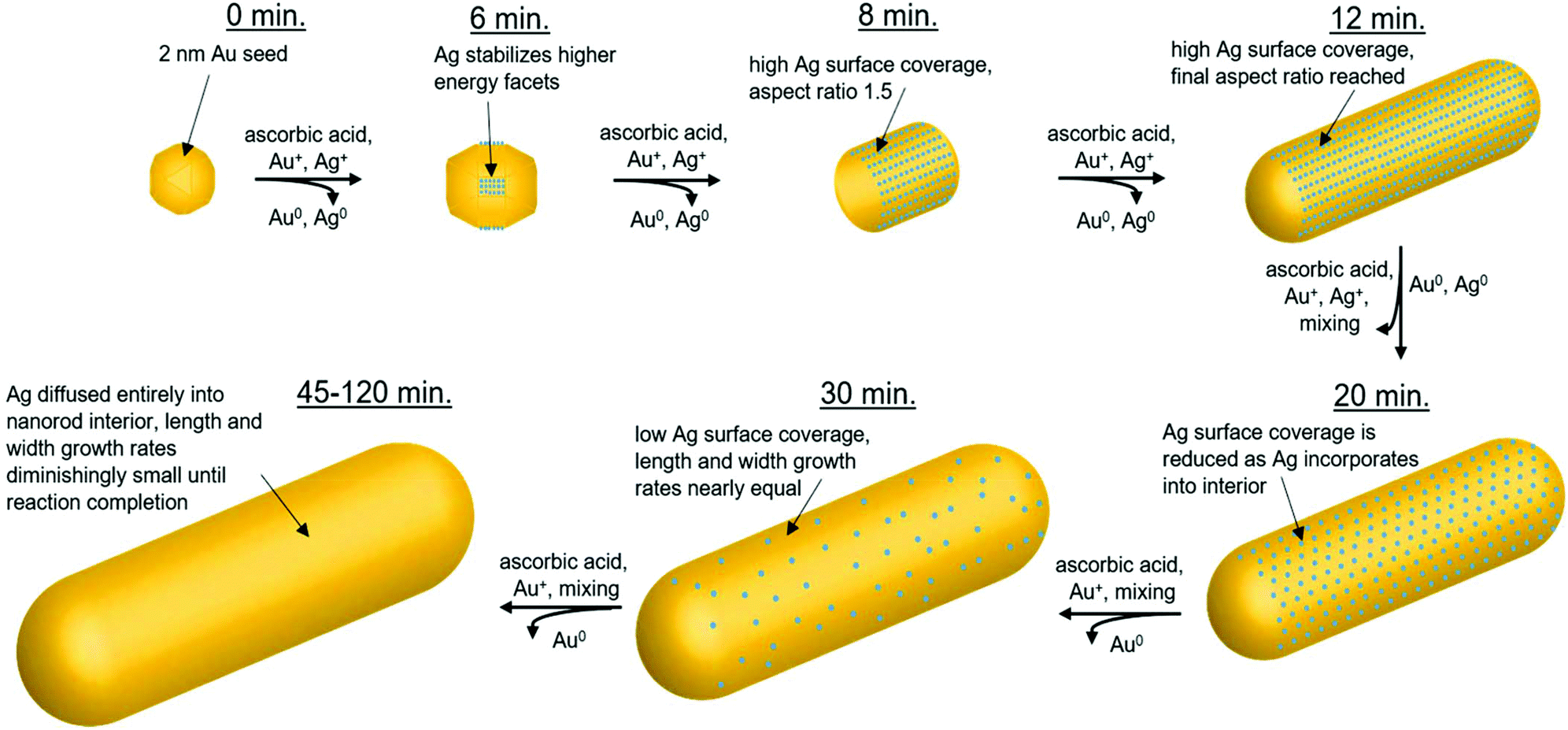 | ||
| Fig. 9 Proposed role of trace Ag within the reaction pathway for Au nanorod growth. Starting with ∼2 nm Au seeds (0 minutes), Ag deposits between 6–8 minutes, stabilizing {110} facets and inducing anisotropic growth. By 8 minutes into the reaction, the nanorods are anisotropic with an aspect ratio of 1.5. An aspect ratio of 3.6 is reached by 12 minutes into the reaction. Over time, Ag deposition slows and incorporated Ag diffuses into the nanorod interior. By 45 minutes into the reaction, Ag surface coverage approaches zero, and the length and diameter nanorod growth rates become diminishingly small until reaction completion (120 minutes, final product). | ||
Conclusions
Through the use of EM, UV-vis, XRF and XAFS applied in a time-course strategy, along with EDX mapping of 12-minute time point particles, the Ag distribution within Au nanorods has been quantified. This bridges the gap between the morphological evolution that results in the formation of the Au nanorods from Au seeds and the role that Ag plays in this process. Structural characterization reveals the key insight that surface Ag directs anisotropic facet growth rates of the nanorods and enables the proposal of a plausible reaction pathway based on the Ag distribution in the nanorods over time. Nanorod length growth rate is directly proportional to the nanorod Ag surface coverage, whereas Ag incorporation is not correlated with nanorod diameter growth rate. While this study provides insight into the role of Ag in the anisotropic growth of Au nanorods, the mechanism by which Ag first deposits remains elusive and requires further investigation. Time course atomic and nanoscale measurements demonstrate that Ag deposits as Ag0 preferentially onto higher surface energy {110} facets early in the reaction, a pattern which supports the UPD hypothesis previously proposed.20 The nanorods reach an aspect ratio of ∼3.6 by 12 minutes into the reaction, indicating that anisotropic growth dominates early in the reaction, when a majority of {110} surface atoms are Ag. As the reaction progresses, Ag incorporates into the bulk of the nanorod, and by 45 minutes into the reaction, Ag surface coverage approaches zero, leading to no perceivable difference in the diminishingly small length and diameter growth rates until reaction completion. These results are not just important for the nanorod synthesis, but may be applicable to the many Au anisotropic nanoparticle synthesis reactions, which make use of trace Ag7,28 and the many other nanoparticle synthesis reactions involving trace external species.Conflicts of interest
There are no conflicts to declare.Acknowledgements
S. K., C. A. M., and M. J. B. acknowledge support by the AFSOR under Award FA9550-11-1-0275. C. A. M. and L. M. M. acknowledge support from the Vannevar Bush Faculty Fellowship program sponsored by the Basic Research Office of the Assistant Secretary of Defense for Research and Engineering, and funded by the Office of Naval Research through grant N00014-15-1-0043. L. M. M. was also funded by a National Defense Science and Engineering Graduate (NDSEG) fellowship. M. R. J. and M. N. O. acknowledge support from the National Science Foundation through a graduate research fellowship. M. R. J. also acknowledges Northwestern University's International Institute for Nanotechnology for support through a Ryan Fellowship. S. K. and M. J. B. acknowledge support by Department of Energy DOE-BES through grant DE-SC0018093 for X-ray measurements and analysis. B. R. C. acknowledges support by DOE-BES through grant DE-FG02-03ER15457. This work made use of Northwestern University's NUANCE Center, which has received support from the Soft and Hybrid Nanotechnology Experimental (SHyNE) Resource (NSF ECCS-1542205); the MRSEC program (NSF DMR-1720139) at the Materials Research Center; the International Institute for Nanotechnology (IIN); the Keck Foundation; and the State of Illinois, through the IIN. EDX mapping was performed at the Center for Nanoscale Systems (CNS), a member of the National Nanotechnology Coordinated Infrastructure Network (NNCI), which is supported by the National Science Foundation under NSF award no. 1541959. CNS is part of Harvard University. SAXS experiments were performed at the APS DND-CAT 5ID-D beamline, which is supported through E. I. duPont de Nemours & Co., Northwestern University (NU), The Dow Chemical Co., and the NSF funded MRSEC at NU. XAFS and XRF experiments were performed at the APS 10BM-B beamline (MR-CAT), which is supported by DOE and the MRCAT member institutions, the APS 5BM-D beamline and the APS 20ID-B beamline (PNC-CAT). Use of the APS was supported by DOE-BES (DE-AC02-06CH11357). We thank Steven Weigand of DND-CAT for assistance with the SAXS setup and data reduction, John Katsoudas of MR-CAT, Qing Ma of DND-CAT and Dale Brewe of PNC-CAT for assistance with the EXAFS setup.References
- N. Tian, Z.-Y. Zhou, S.-G. Sun, Y. Ding and Z. L. Wang, Science, 2007, 316, 732–735 CrossRef CAS PubMed.
- J. E. Millstone, S. J. Hurst, G. S. Métraux, J. I. Cutler and C. A. Mirkin, Small, 2009, 5, 646–664 CrossRef CAS PubMed.
- F. Kim, J. H. Song and P. Yang, J. Am. Chem. Soc., 2002, 124, 14316–14317 CrossRef CAS PubMed.
- K. L. Kelly, E. Coronado, L. L. Zhao and G. C. Schatz, J. Phys. Chem. B, 2002, 107, 668–677 CrossRef.
- M. R. Jones, R. J. Macfarlane, B. Lee, J. Zhang, K. L. Young, A. J. Senesi and C. A. Mirkin, Nat. Mater., 2010, 9, 913–917 CrossRef CAS PubMed.
- R. J. Macfarlane, M. N. O'Brien, S. H. Petrosko and C. A. Mirkin, Angew. Chem., Int. Ed., 2013, 52, 5688–5698 CrossRef CAS PubMed.
- M. R. Langille, M. L. Personick, J. Zhang and C. A. Mirkin, J. Am. Chem. Soc., 2012, 134, 14542–14554 CrossRef CAS PubMed.
- M. R. Langille, J. Zhang, M. L. Personick, S. Li and C. A. Mirkin, Science, 2012, 337, 954–957 CrossRef CAS PubMed.
- C. J. Murphy, L. B. Thompson, A. M. Alkilany, P. N. Sisco, S. P. Boulos, S. T. Sivapalan, J. A. Yang, D. J. Chernak and J. Huang, J. Phys. Chem. Lett., 2010, 1, 2867–2875 CrossRef CAS.
- B. Nikoobakht and M. A. El-Sayed, Chem. Mater., 2003, 15, 1957–1962 CrossRef CAS.
- N. R. Jana, L. Gearheart and C. J. Murphy, J. Phys. Chem. B, 2001, 105, 4065–4067 CrossRef CAS.
- N. R. Jana, L. Gearheart and C. J. Murphy, Adv. Mater., 2001, 13, 1389–1393 CrossRef CAS.
- N. Garg, C. Scholl, A. Mohanty and R. Jin, Langmuir, 2010, 26, 10271–10276 CrossRef CAS PubMed.
- T. K. Sau and C. J. Murphy, Philos. Mag., 2007, 87, 2143–2158 CrossRef CAS.
- J. A. da Silva and M. R. Meneghetti, Langmuir, 2018, 34, 366–375 CrossRef CAS PubMed.
- N. Almora-Barrios, G. Novell-Leruth, P. Whiting, L. M. Liz-Marzán and N. López, Nano Lett., 2014, 14, 871–875 CrossRef CAS PubMed.
- Y. Niidome, Y. Nakamura, K. Honda, Y. Akiyama, K. Nishioka, H. Kawasaki and N. Nakashima, Chem. Commun., 2009, 1754–1756, 10.1039/B821402F.
- A. Petukhova, J. Greener, K. Liu, D. Nykypanchuk, R. Nicolaÿ, K. Matyjaszewski and E. Kumacheva, Small, 2012, 8, 731–737 CrossRef CAS PubMed.
- C. Kinnear, H. Dietsch, M. J. D. Clift, C. Endes, B. Rothen-Rutishauser and A. Petri-Fink, Angew. Chem., Int. Ed., 2013, 52, 1934–1938 CrossRef CAS PubMed.
- M. Z. Liu and P. Guyot-Sionnest, J. Phys. Chem. B, 2005, 109, 22192–22200 CrossRef CAS PubMed.
- F. Giannici, T. Placido, M. L. Curri, M. Striccoli, A. Agostiano and R. Comparelli, Dalton Trans., 2009, 10367–10374 RSC.
- C. J. Orendorff and C. J. Murphy, J. Phys. Chem. B, 2006, 110, 3990–3994 CrossRef CAS PubMed.
- Y. Chen, X. Gu, C.-G. Nie, Z.-Y. Jiang, Z.-X. Xie and C.-J. Lin, Chem. Commun., 2005, 4181–4183 RSC.
- D. M. Kolb, M. Przasnyski and H. Gerischer, J. Electroanal. Chem., 1974, 54, 25–38 CrossRef CAS.
- E. Herrero, L. J. Buller and H. D. Abruña, Chem. Rev., 2001, 101, 1897–1930 CrossRef CAS.
- D. M. Kolb, M. Przasnyski and H. Gerischer, J. Electroanal. Chem., 1974, 54, 25 CrossRef CAS.
- Q. Zhang, H. Jing, G. G. Li, Y. Lin, D. A. Blom and H. Wang, Chem. Mater., 2016, 28, 2728–2741 CrossRef CAS.
- M. L. Personick, M. R. Langille, J. Zhang and C. A. Mirkin, Nano Lett., 2011, 11, 3394–3398 CrossRef CAS PubMed.
- S. R. Jackson, J. R. McBride, S. J. Rosenthal and D. W. Wright, J. Am. Chem. Soc., 2014, 136, 5261–5263 CrossRef CAS PubMed.
- K. Park, L. F. Drummy, R. C. Wadams, H. Koerner, D. Nepal, L. Fabris and R. A. Vaia, Chem. Mater., 2013, 25, 555–563 CrossRef CAS.
- M. J. Walsh, S. J. Barrow, W. Tong, A. M. Funston and J. Etheridge, ACS Nano, 2015, 9, 715–724 CrossRef CAS PubMed.
- S. Puri, B. Chand, D. Mehta, M. L. Garg, N. Singh and P. N. Trehan, At. Data Nucl. Data Tables, 1995, 61, 289–311 CrossRef CAS.
- B. Ravel and M. Newville, J. Synchrotron Radiat., 2005, 12, 537–541 CrossRef CAS PubMed.
- E. A. Stern, Contemp. Phys., 1978, 19, 289–310 CrossRef.
- E. A. Stern, Phys. Rev. B: Solid State, 1974, 10, 3027–3037 CrossRef CAS.
- B. Ravel and S. D. Kelly, AIP Conf. Proc., 2007, 882, 150–152 CrossRef CAS.
- R. Scott, Physical Methods in Bioinorganic Chemistry: Spectroscopy and Magnetism, University Science Books, 2000 Search PubMed.
- Y. Hatakeyama, K. Sasaki, K. Judai, K. Nishikawa and K. Hino, J. Phys. Chem. C, 2018, 122, 7982–7991 CrossRef CAS.
- M. J. Walsh, W. Tong, H. Katz-Boon, P. Mulvaney, J. Etheridge and A. M. Funston, Acc. Chem. Res., 2017, 50, 2925–2935 CrossRef CAS PubMed.
- W. Tong, M. J. Walsh, P. Mulvaney, J. Etheridge and A. M. Funston, J. Phys. Chem. C, 2017, 121, 3549–3559 CrossRef CAS.
- Y. Sun and Y. Xia, J. Am. Chem. Soc., 2004, 126, 3892–3901 CrossRef CAS PubMed.
- J. Snyder, K. Livi and J. Erlebacher, J. Electrochem. Soc., 2008, 155, C464–C473 CrossRef CAS.
- E. Carbó-Argibay, B. Rodríguez-González, S. Gómez-Graña, A. Guerrero-Martínez, I. Pastoriza-Santos, J. Pérez-Juste and L. M. Liz-Marzán, Angew. Chem., Int. Ed., 2010, 49, 9397–9400 CrossRef PubMed.
- J. D. Padmos, M. L. Personick, Q. Tang, P. N. Duchesne, D.-e. Jiang, C. A. Mirkin and P. Zhang, Nat. Commun., 2015, 6, 7664 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Supporting materials include extended methods, XAFS data and associated models and derived parameters, sample calculations of nanorod growth parameters, UV-vis spectra, and SAXS patterns (PDF). See DOI: 10.1039/c9nr03246k |
| This journal is © The Royal Society of Chemistry 2019 |