
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSalen supported Al–O–C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/h2_char_e002.gif) P and Ga–P
P and Ga–P![[double bond, length as m-dash]](https://www.rsc.org/images/entities/h2_char_e001.gif) CO complexes†
CO complexes†
Yanbo
Mei
a,
Jaap E.
Borger
 *a,
Dong-Jun
Wu
ab and
Hansjörg
Grützmacher
*ab
*a,
Dong-Jun
Wu
ab and
Hansjörg
Grützmacher
*ab
aDepartment of Chemistry and Applied Biosciences, ETH Zürich, Vladimir-Prelog Weg 1, Hönggerberg, 8093 Zürich, Switzerland. E-mail: hgruetzmacher@ethz.ch; borger@inorg.chem.ethz.ch
bLehn Institute of Functional Materials (LIFM), School of Chemistry, Sun Yat-sen University, 510275 Guangzhou, China
First published on 12th March 2019
Abstract
The first OCP adducts of aluminium and gallium are reported. The complexes are supported by sterically encumbered salen ligands and reveal a selective binding to O and P, respectively. Their reactivity with diazaphosphenium Lewis acids and N-heterocyclic carbene Lewis bases is described, in addition to cycloaddition reactions with s-tetrazines.
The development of versatile phosphorus building blocks allows new avenues to be explored in organophosphorus chemistry. A prominent example is the 2-phosphaethynolate anion, OCP−,1 which represents the P-analogue of cyanate (OCN−) and can be prepared on multi-gram scale as sodium salt.2 In recent years, its reactivity has been studied extensively revealing a plethora of transformations to be feasible leading to a broad spectrum of novel phosphorus compounds.3 A prominent reaction step involves salt metathesis which leads to O- or, more frequently, to P-substituted products (Scheme 1). These facile syntheses of functionalized oxyphosphaalkynes (A) or phosphaketenes (B) allows the access to valuable starting materials which can be applied, for example, in P-heterocycle synthesis.3
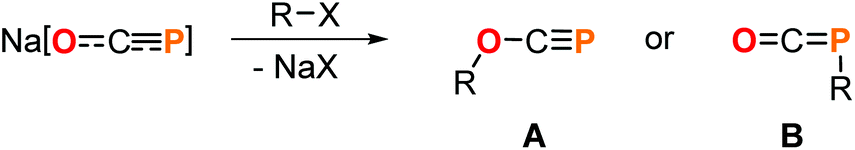 | ||
| Scheme 1 Generic salt metathesis reaction of Na[OCP] to give O- or P-substituted products. | ||
Compounds of the type A can be generated in reactions of Na[OCP] with highly oxophilic s-block and actinide metal halide complexes (M = Ca,4 Mg,5 U,6,7 Th6), as well as with those containing rare-earth metals (M = Sc,8 Y,9 Nd,9 Sm9). Compounds of type B are more common and form when p-block element (E = P,10 C,2b,11 Ge,12 Sn12a,13) or d-block metal (M = Re,14 Co,15 Ir,15 Au,15 Ti16) halide precursors are employed.17,18 In case of oxophilic organyl-substituted chlorosilanes13d,19 or -boranes,20 a mixture of both isomers A and B is obtained. The ratio is highly dependent on the reaction conditions. Notably, a diazaboryl chloride was recently reported to permit the synthesis of the first, and so far only, stable example of a main group element-coordinated phosphaethynolato adduct of the type A, that is (N(dipp)CH)2B–O–C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) P (dipp = 2,6-diisopropylphenyl).21,22
P (dipp = 2,6-diisopropylphenyl).21,22
The coordination behaviour of Na[OCP] toward the heavier elements of the group 13 has not been studied to date, yet should provide further insight into the ambident nature of the phosphaethynolate salt and access to novel rare O-substituted compounds. Herein, the synthesis and isolation of the first OCP adducts of aluminium and gallium are reported. The complexes contain sterically encumbered salen ligands and revealed a selective binding preference to oxygen or phosphorus, respectively. The reactivity of the products toward Lewis acids and bases was probed, and their engagement in cycloaddition reactions with tetrazines assessed.
To start, commercially available diisobutylaluminum chloride (DIBAC) was reacted with Na[OCP] in C6D6 at room temperature. The reaction produced a single species according to the multinuclear NMR spectra of the black mixture, with a singlet 31P resonance signal at −331.5 ppm (see the ESI† for further details). In situ infrared spectroscopy revealed a strong absorption at ν = 1676 cm−1, which is indicative of O-bound phosphaethynolato complex 1 (Scheme 2). For comparison, the respective OCP stretching frequency in (N(dipp)CH)2B–O–CP is found in the same range at ν = 1649 cm−1,21 while that in Ph3Sn–P![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CO is located at ν = 1946 cm−1.13d Indeed, the calculated 31P NMR chemical shift of the (iBu)2Al–O–CP isomer is δ = −333.0 ppm, which corresponds well with the experimentally observed one and is significantly different from the calculated 31P chemical shift of the phosphaketene isomer (iBu)2Al–PCO at δ = −353.1 ppm (DFT: B3LYP/6-311+G(2d,p)).23 Remarkably, while the latter is not observed in solution, it is predicted to be 4.4 kcal mol−1 (ΔE) lower in energy than the former which implies 1 to be a kinetic product. Its selective formation may be explained by a preference for oxygen binding of Na[OCP] to (iBu)2AlCl giving the anionic Al–O adduct Na[(iBu)2Al(OCP)Cl] as intermediate prior to salt elimination, which is calculated to be 3.4 kcal mol−1 lower in energy than the corresponding P-coordinated salt Na[(iBu)2Al(PCO)Cl].
CO is located at ν = 1946 cm−1.13d Indeed, the calculated 31P NMR chemical shift of the (iBu)2Al–O–CP isomer is δ = −333.0 ppm, which corresponds well with the experimentally observed one and is significantly different from the calculated 31P chemical shift of the phosphaketene isomer (iBu)2Al–PCO at δ = −353.1 ppm (DFT: B3LYP/6-311+G(2d,p)).23 Remarkably, while the latter is not observed in solution, it is predicted to be 4.4 kcal mol−1 (ΔE) lower in energy than the former which implies 1 to be a kinetic product. Its selective formation may be explained by a preference for oxygen binding of Na[OCP] to (iBu)2AlCl giving the anionic Al–O adduct Na[(iBu)2Al(OCP)Cl] as intermediate prior to salt elimination, which is calculated to be 3.4 kcal mol−1 lower in energy than the corresponding P-coordinated salt Na[(iBu)2Al(PCO)Cl].
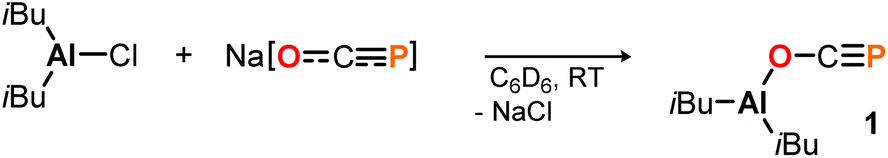 | ||
| Scheme 2 Synthesis of 1 by reaction of DIBAC with Na[OCP]. | ||
Complex 1 proved to be persistent in solution for at least two days, however, decomposed to unidentified products upon concentration of the reaction mixture under vacuum. By contrast, employing the five coordinate Schiff-base precursors salen(tBu)AlCl24 and Salophen(tBu)AlCl24 in the reaction with Na[OCP] yielded stable aluminum oxyphosphaalkyne complexes 2a and 2b (Scheme 3), which could be isolated from toluene in 68% and 40% yield, respectively. Both, the IR OCP stretching frequencies (2a: ν = 1692 cm−1; 2b: ν = 1690 cm−1), as well as the 31P NMR chemical shifts (2a: δ = −336.8 ppm in C6D6; 2b: δ = −335.2 ppm in toluene) are comparable to those found for 1, and similar to the values reported for related Th–,6 Sc–8 and Y–O–CP9 complexes (δ31P = −334.0, −343.5 and −346.9 ppm, respectively), yet significantly shifted from M[OCP] salts (M = Li, Na, K; δ31P = −384 to −397 ppm).1,2
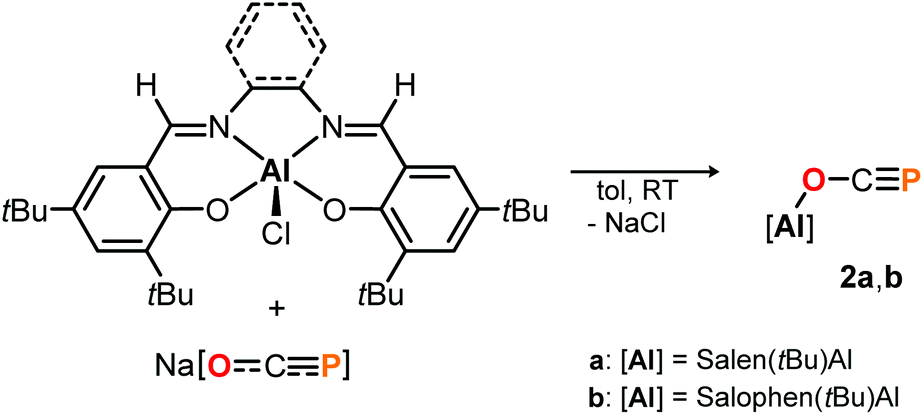 | ||
| Scheme 3 Synthesis of salen supported [Al]–O–CP complexes 2. | ||
Light yellow crystals of 2a were grown from a saturated toluene solution layered with n-hexane and used for X-ray diffraction to determine the structure in solid state; a plot is presented in Fig. 1. As expected, the oxygen center of the OCP unit binds to the aluminum center. Compared to Na[OCP] (d(O–C)avg. = 1.208 Å; d(C–P)avg. = 1.575 Å),2a the O1–C1 bond of 1.2538(10) Å is elongated while the C1–P1 bond of 1.5646(9) Å is slightly shortened due to the electron withdrawing aluminium substituent at O1, giving rise to more pronounced single and triple bond character, respectively. This phenomenon is likewise observed in the only other related main group element example of A (Scheme 1), that is (N(dipp)CH)2B–O–CP (d(O–C) = 1.269(2) Å; d(C–P) = 1.545(2) Å).21
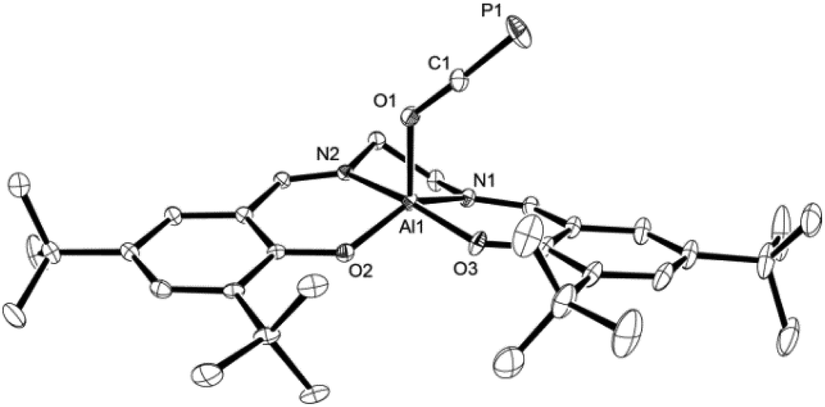 | ||
| Fig. 1 Molecular structure of 2a in the crystal25 (ellipsoids are shown at 50% probability; hydrogen atoms are omitted for clarity). Selected bond distances [Å] and angles [°]: C1–P1 1.5646(9), O1–C1 1.2538(10), Al1–O1 1.8206(7), Al1–O2 1.7670(6), Al1–O3 1.7901(6), C1–O1–Al1 131.38(6), O1–C1–P1 177.61(8). | ||
DFT calculations on the truncated model of 2a and the corresponding phosphaketene isomer, that is 2a′ and 2a′-I (tBu = H; B3LYP/6-311G(2d,p)), were performed to gain insight into their relative energies.23 In contrast to what was found for the respective isomers of 1, the O-bound structure is computed to be 5.0 kcal mol−1 (ΔE) lower in energy than the P-bound one and thereby the thermodynamically favoured product in this case. Presumably, the reverse in order of stability is caused by the more electron deficient O-bound aryloxy groups in 2a compared to the alkyl substituents in 1, which make the Al-atom in the former even harder and consequently more oxophilic. When the aluminum center was exchanged for a softer gallium center, the P-coordinated isomer of the type B is calculated to be 8.5 kcal mol−1 lower in energy compared to the corresponding gallium-oxyphosphaalkyne type A. Indeed, mixing salen(tBu)GaCl26 with Na[OCP] in toluene at room temperature gave only phosphaketene 3 which could be isolated in 81% yield (Scheme 4).
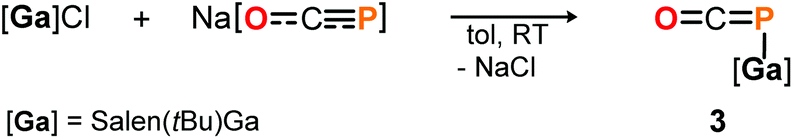 | ||
| Scheme 4 Synthesis of salen supported [Ga]–PCO complex 3. | ||
The identity of 3 was established in solution by multinuclear NMR spectroscopy and in the solid state by IR spectroscopy and single crystal X-ray diffraction. The 31P NMR chemical shift was observed at δ = −376.9 ppm (in THF-d8) and is in the same range as the ones found for related R–PCO species where R = Ph3Sn or iPr3Si (δ31P = −378.0 or −370.1 ppm, respectively), yet upfield to those arising from examples where R = Ph3Si or Ph3Ge (δ31P = −340.5 or −344.0 ppm, respectively).13d The asymmetric IR PCO stretching frequency at ν = 1910 cm−1 is characteristic for phosphaketenes (cf. Mes*-PCO:27ν = 1953 cm−1; Ph3E–PCO (E = Si, Ge, Sn, Pb):13dν = 1923–1962 cm−1), as are the crystallographically determined P1–C1 and C1–O1 bond lengths of 1.625(4) Å and 1.189(4) Å, respectively (Fig. 2). These are, however, significantly contracted in comparison to the predicted lengths for PC and CO double bonds (∑rcov(PC) = 1.69 Å; ∑rcov(CO) = 1.24 Å).28
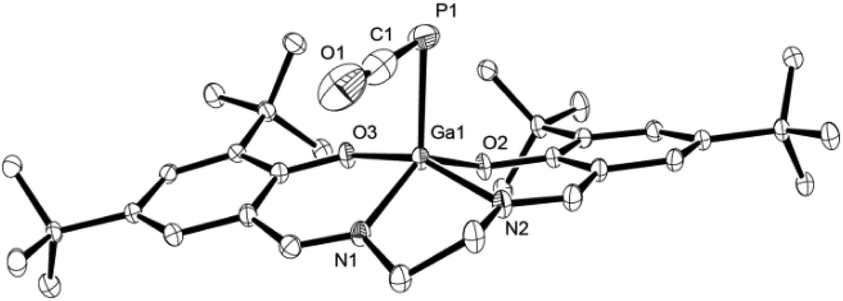 | ||
| Fig. 2 Molecular structure of 3 in the crystal25 (ellipsoids are shown at 50% probability; hydrogen atoms are omitted for clarity). Selected bond distances [Å] and angles [°]: C1–P1 1.625(4), O1–C1 1.189(4), Ga1–P1 2.3761(8), Ga1–O2 1.8748(15), Ga1–O3 1.9073(16), C1–P1–Ga1 88.43(11), O1–C1–P1 177.3(3). | ||
Some insight into the reactivity of the new adducts 2 and 3 could be acquired by closer inspection of their 31P NMR spectra in THF-d8 and C6D6. In case of the Ga–PCO compound 3, spectra in both solvents showed sharp signals at the same chemical shift indicating no significant interaction with the solvents. On the other hand, those recorded of the oxyphosphaalkyne Al adduct 2a displayed a 17.1 ppm upfield shift and broadening of the 31P resonance in THF-d8 compared to the one in C6D6 (δ31P = −353.9 vs. −336.8 ppm, respectively; see the ESI† for a variable temperature NMR spectroscopy study). These observations may be explained by the reversible coordination of a THF molecule to the Lewis acidic aluminium center in 2a to form 2a-THF, which does not occur in case of 3 (Scheme 5). This is corroborated by DFT calculations using the model compounds 2a′ and 3′ (tBu = H),23 which revealed the formation of 2a′-THF to be 6.0 kcal mol−1 downhill in energy (ΔE), while no minimum for 3′-THF was found. Indeed, crystallization of 2a from a THF solution layered with toluene/n-hexane yielded X-ray quality crystals of 2a-THF, which allowed elucidation of its solid state structure and revealed a THF bound six-coordinate aluminium core with an elongated Al1–O1 bond (1.920(3) Å) compared to solvent-free complex (Fig. 3; cf. Fig. 1). Consistently, the C1–O1 bond (1.228(5) Å) is found to be slightly shorter and the C1–P1 bond (1.587(5) Å) longer, reflecting a more pronounced delocalization of electron density from O1 into the π*-orbital of the C1P1 bond.
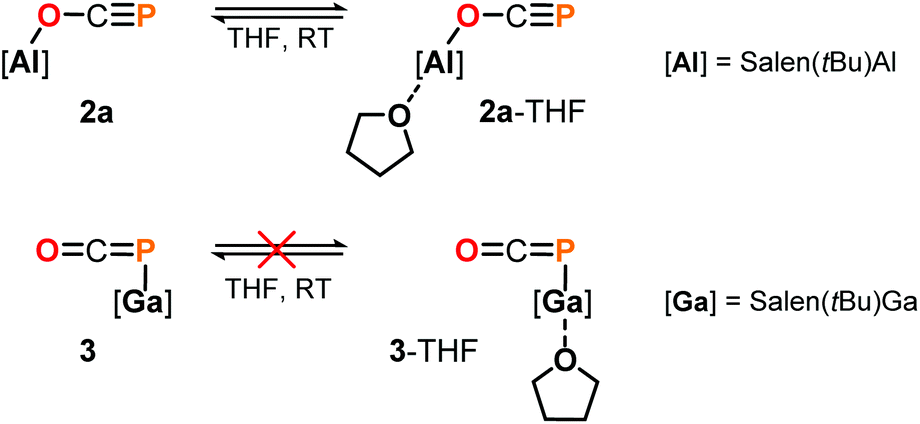 | ||
| Scheme 5 Reactivity of 2a and 3 in THF. | ||
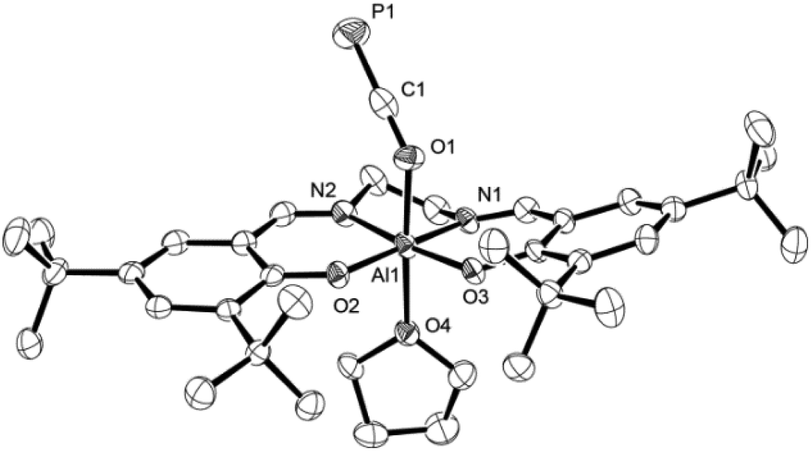 | ||
| Fig. 3 Molecular structure of 2a-THF in the crystal25 (ellipsoids are shown at 50% probability; hydrogen atoms and three toluene molecules are omitted for clarity). Selected bond distances [Å] and angles [°]: C1–P1 1.587(5), C1–O1 1.228(5), Al1–O1 1.920(3), Al1–O2 1.802(3), Al1–O3 1.796(2), Al1–O4 2.040(3), C1–O1–Al1 137.5(2), O1–C1–P1 178.5(3). | ||
To further explore the Lewis acidic properties of 2a and 3, stoichiometric reactions with N-heterocyclic carbene I were scrutinized in toluene (Scheme 6, top). Instead of forming an adduct similar to 2a-THF, however, compound 4 was produced in both reactions according to single crystal X-ray diffraction studies for which crystals were used that precipitated directly from the reaction solutions (see the ESI†).25 These results imply acid–base reactions to have occurred, in which presumably the salen ligands in 2a and 3 are deprotonated by the NHC and subsequently [OCP]− is liberated to form the [NHC-H][OCP] ion pair. Note that 4 is a rare example of a phosphaethynolate anion with a weakly coordinating cation,15,29 a related example of which could be accessed from the reaction of Na[OCP] with the non-methylated 1,3-diisopropyl imidazolium chloride (δ31P = −388.0 ppm; see the ESI† for synthetic details).
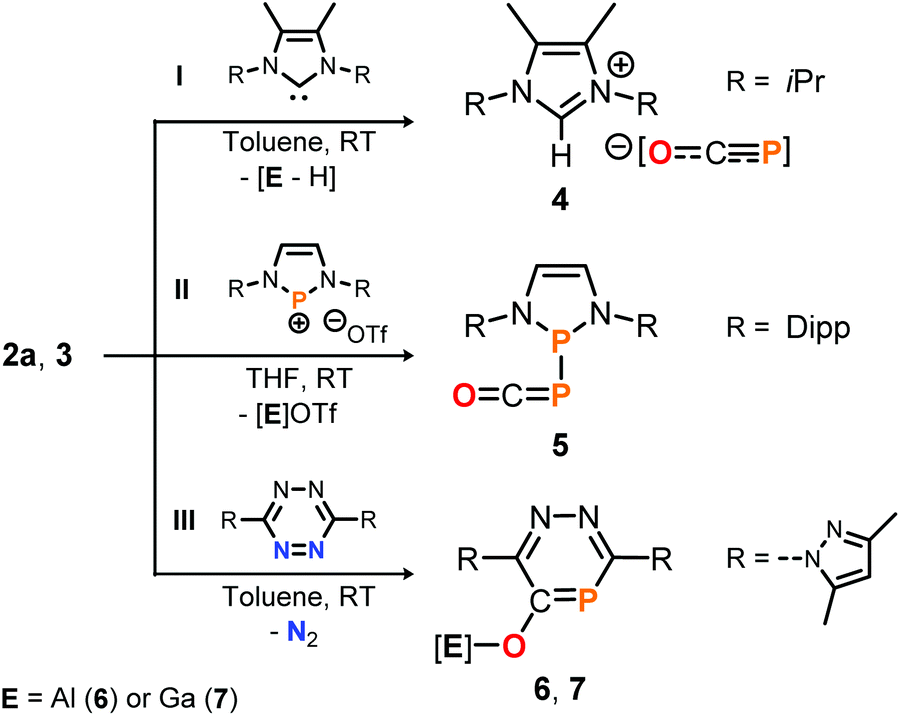 | ||
| Scheme 6 Reactivity of 2a and 3 toward NHC I, diazaphosphenium triflate II and s-tetrazine III. OTf = CF3SO3. [Al] = salen(tBu)Al, [Ga] = salen(tBu)Ga. | ||
The Lewis basic properties of 2a and 3 were investigated by monitoring their reaction with the diazaphosphenium salt II by 31P NMR spectroscopy (Scheme 6, middle). It was found that the [OCP]− ligand in both the Al- and Ga-adduct transfers instantly and irreversibly to the stronger Lewis acid to yield phosphanylphosphaketene 5 quantitatively, as confirmed by comparison of its 31P NMR resonance signal (δ = 166.1 and −231.8 ppm, 1JP, P = 253 Hz) with literature values. Compound 5 was previously prepared from II by salt metathesis using Na[OCP] instead.10f Finally, the reaction of 2a with s-tetrazine III in toluene afforded the O-salen(tBu)Al-1,2,4-diazaphosphinine-5-olate 6, which could be isolated in 46% yield and fully characterized including a structure determination by an X-ray diffraction study using a single crystal (Scheme 6, bottom; δ31P = 132.0 ppm; see ESI† for synthetic details25). The P-heterocycle is formed through Diels Alder type reactivity of the [Al]–O–CP triple bond in 2a with the tetrazine framework under elimination of dinitrogen. This reaction was recently reported to occur similarly with Na[OCP] as reagent.30 Remarkably, employing phosphaketene 3 under the same reaction conditions afforded the gallium derivative 7 (75% isolated yield; δ31P = 133.1 ppm), suggesting a related [4 + 2]-cycloaddition between the CP double bond in [Ga]–PCO 3 and the tetrazine followed by elimination of N2 and subsequent 1,3-P,O-migration of the salen gallium fragment.
In summary, the first aluminium and gallium adducts of the phosphaethynolate anion have been isolated and fully characterized. The complexes are supported by bulky salen ligands and reveal a selective binding to O and P, respectively. Preliminary reactivity studies showed that the OCP units in both derivatives are readily liberated from the group 13 element centers by an N-heterocyclic carbene Lewis base, or transferred to a stronger diazaphosphenium Lewis acid. In addition, cycloaddition reactions with s-tetrazines under elimination of N2 were found to give Al- or Ga-complexed 1,2,4-diazaphosphinine-5-olate heterocycles. The studied transformations mimic those previously reported for Na[OCP], which suggests that the Al–O–CP and Ga–PCO adducts 2 and 3 can serve as neutral and more soluble [OCP]− precursors. Moreover, the presence of the bulky Lewis acidic salen(tBu)E centers could facilitate the stabilization and subsequent derivatization of otherwise fleeting phosphaethynolate-derived products.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the ETH Zürich (project 0-20214-169), and the Lehn Institute of Functional Materials (LIFM), School of Chemistry at the Sun Yat-sen university, the National Natural Science Foundation of China (NSFC) project (21720102007).Notes and references
- The first preparation of the phosphaethynolate anion [OCP]− was reported by Becker et al. in 1992 as Li+ salt, see: G. Becker, W. Schwarz, N. Seidler and M. Westerhausen, Z. Anorg. Allg. Chem., 1992, 612, 72–82 CrossRef CAS.
- (a) F. F. Puschmann, D. Stein, D. Heift, C. Hendriksen, Z. A. Gal, H.-F. Grützmacher and H. Grützmacher, Angew. Chem., Int. Ed., 2011, 50, 8420–8423 CrossRef CAS PubMed; (b) D. Heift, Z. Benkő and H. Grützmacher, Dalton Trans., 2014, 831–840 RSC; (c) R. Suter, Z. Benkő, M. Bispinghoff and H. Grützmacher, Angew. Chem., Int. Ed., 2017, 56, 11226–11231 CrossRef CAS PubMed; (d) I. Krummenacher and C. C. Cummins, Polyhedron, 2012, 32, 10–13 CrossRef CAS; (e) A. R. Jupp and J. M. Goicoechea, Angew. Chem., Int. Ed., 2013, 52, 10064–10067 CrossRef CAS PubMed.
- For reviews, see: (a) H. Grützmacher and J. Goicoechea, Angew. Chem., Int. Ed., 2018, 57, 16968–16994 CrossRef PubMed; (b) L. Weber, Eur. J. Inorg. Chem., 2018, 2175–2227 CrossRef CAS.
- M. Westerhausen, S. Schneiderbauer, H. Piotrowski, M. Suter and H. Nöth, J. Organomet. Chem., 2002, 643–644, 189–193 CrossRef CAS.
- R. J. Gilliard, D. Heift, Z. Benkő, J. M. Keiser, A. L. Rheingold, H. Grützmacher and J. D. Protasiewicz, Dalton Trans., 2018, 666–669 RSC.
- C. Camp, N. Settineri, J. Lefèvre, A. R. Jupp, J. M. Goicoechea, L. Maron and J. Arnold, Chem. Sci., 2015, 6, 6379–6384 RSC.
- C. J. Hoerger, F. W. Heinemann, E. Louyriac, L. Maron, H. Grützmacher and K. Meyer, Organometallics, 2017, 36, 4351–4354 CrossRef CAS.
- L. N. Grant, B. Pinter, B. C. Manor, H. Grützmacher and D. J. Mindiola, Angew. Chem., Int. Ed., 2018, 57, 1049–1052 CrossRef CAS PubMed.
- S. Bestgen, Q. Chen, N. H. Rees and J. M. Goicoechea, Dalton Trans., 2018, 13016–13024 RSC.
- (a) M. M. Hansmann, D. A. Ruiz, L. Liu, R. Jazzar and G. Bertrand, Chem. Sci., 2017, 8, 3720–3725 RSC; (b) L. Liu, D. A. Ruiz, D. Munz and G. Bertrand, Chem, 2016, 1, 147–153 CrossRef CAS; (c) Z. Li, X. Chen, Z. Benkő, L. Liu, D. A. Ruiz, J. L. Peltier, G. Bertrand, C.-Y. Su and H. Grützmacher, Angew. Chem., Int. Ed., 2016, 55, 6018–6022 CrossRef CAS PubMed; (d) M. M. Hansmann, R. Jazzar and G. Bertrand, J. Am. Chem. Soc., 2016, 138, 8356–8359 CrossRef CAS PubMed; (e) M. M. Hansmann and G. Bertrand, J. Am. Chem. Soc., 2016, 138, 15885–15888 CrossRef CAS PubMed; (f) Z. Li, X. Chen, M. Bergeler, M. Reiher, C.-Y. Su and H. Grützmacher, Dalton Trans., 2015, 6431–6438 RSC.
- T. Krachko, A. W. Ehlers, M. Nieger, M. Lutz and J. C. Slootweg, Angew. Chem., Int. Ed., 2018, 57, 1683–1687 CrossRef CAS PubMed.
- (a) A. Hinz and J. M. Goicoechea, Chem. – Eur. J., 2018, 24, 7358–7363 CrossRef CAS PubMed; (b) Y. Xiong, S. Yao, T. Szilvási, E. Ballestero-Martínez, H. Grützmacher and M. Driess, Angew. Chem., Int. Ed., 2017, 56, 4333–4336 CrossRef CAS PubMed; (c) S. Yao, Y. Xiong, T. Szilvási, H. Grützmacher and M. Driess, Angew. Chem., Int. Ed., 2016, 55, 4781–4785 CrossRef CAS PubMed; (d) N. Del Rio, A. Baceiredo, N. Saffon-Merceron, D. Hashizume, D. Lutters, T. Müller and T. Kato, Angew. Chem., Int. Ed., 2016, 55, 4753–4758 CrossRef CAS PubMed; (e) Y. Wu, L. Liu, J. Su, J. Zhu, Z. Ji and Y. Zhao, Organometallics, 2016, 35, 1593–1596 CrossRef CAS.
- (a) A. Hinz and J. M. Goicoechea, Dalton Trans., 2018, 8879–8883 RSC; (b) Z. Li, X. Chen, Y. Li, C.-Y. Su and H. Grützmacher, Chem. Commun., 2016, 52, 11343–11346 RSC; (c) D. Heift, Z. Benkő and H. Grützmacher, Chem. – Eur. J., 2014, 20, 11326–11330 CrossRef CAS PubMed; (d) D. Heift, Z. Benkő and H. Grützmacher, Dalton Trans., 2014, 5920–5928 RSC.
- S. Alidori, D. Heift, G. Santiso-Quinones, Z. Benkő, H. Grützmacher, M. Caporali, L. Gonsalvi, A. Rossin and M. Peruzzini, Chem. – Eur. J., 2012, 18, 14805–14811 CrossRef CAS PubMed.
- L. Liu, D. A. Ruiz, F. Dahcheh, G. Bertrand, R. Suter, A. M. Tondreau and H. Grützmacher, Chem. Sci., 2016, 7, 2335–2341 RSC.
- L. N. Grant, B. Pinter, B. C. Manor, R. Suter, H. Grützmacher and D. J. Mindiola, Chem. – Eur. J., 2017, 23, 6272–6276 CrossRef CAS PubMed.
- Protonation occurs likewise at the P-atom, see: A. Hinz, R. Labbow, C. Rennick, A. Schulz and J. M. Goicoechea, Angew. Chem., Int. Ed., 2017, 56, 3911–3915 CrossRef CAS PubMed.
- Carbene-coordinated Ni(C5H5), Ag(OSO2CF3) and Cu(OtBu) compounds were likewise found to engage in a salt metathesis reaction with Na[OCP], giving pi-coordinated phosphaketene complexes. See: (a) G. Hierlmeier, A. Hinz, R. Wolf and J. M. Goicoechea, Angew. Chem., Int. Ed., 2018, 57, 431–436 CrossRef CAS PubMed; (b) M. M. D. Roy, M. J. Ferguson, R. McDonald and E. Rivard, Chem. Commun., 2018, 54, 483–486 RSC; (c) Ref. 15 .
- K. Nakajima, W. Liang and Y. Nishibayashi, Org. Lett., 2016, 18, 5006–5009 CrossRef CAS PubMed.
- R. Suter, Y. Mei, M. Baker, Z. Benkő, Z. Li and H. Grützmacher, Angew. Chem., Int. Ed., 2017, 56, 1356–1360 CrossRef CAS PubMed.
- D. W. N. Wilson, A. Hinz and J. M. Goicoechea, Angew. Chem., Int. Ed., 2018, 57, 2188–2193 CrossRef CAS PubMed.
- The coordination chemistry of Na[OCP] toward pentaphenylborole and arylboranes was also studied and revealed the formation of P-heterocycles, see: (a) K. M. Szkop, A. R. Jupp, R. Suter, H. Grützmacher and D. W. Stephan, Angew. Chem., Int. Ed., 2017, 56, 14174–14177 CrossRef CAS PubMed; (b) Y. Li, R. K. Siwatch, T. Mondal, Y. Li, R. Ganguly, D. Koley and C.-W. So, Inorg. Chem., 2017, 56, 4112–4120 CrossRef CAS PubMed.
- See the ESI† for further details.
- Salen(tBu) = N,N′-ethylenebis(3,5-di-tert-butylsalicylideneimine; Salophen(tBu) = N,N′-benzenylidenebis(3,5-di-tert-butyl salicylideneamine). The [Al]Cl precursors were prepared according to literature procedures, see: D. Rutherford and D. A. Atwood, Organometallics, 1996, 15, 4417–4422 CrossRef CAS.
- CCDC 1860668 (2a), 1860669 (2a-THF), 1860673 (3), 1860732 (6) and 1860759 (4)† contain the supplementary crystallographic data for this paper.
- The [Ga]Cl precursor was prepared according to a literature procedure: D. J. Darensbourg and D. R. Billodeaux, C. R. Chim., 2004, 7, 755–761 CrossRef CAS.
- R. Appel and W. Paulen, Angew. Chem., 1983, 95, 807–808 CrossRef . Mes* = 2,4,6-tri-tert-butylphenyl.
- P. Pyykkö and M. Atsumi, Chem. – Eur. J., 2009, 15, 12770–12779 CrossRef PubMed.
- M. Jost, L. H. Finger, J. Sundermeyer and C. von Hänisch, Chem. Commun., 2016, 52, 11646–11648 RSC.
- (a) A. V. Polezhaev, D. M. Beagan, A. C. Cabelof, C.-H. Chen and K. G. Caulton, Dalton Trans., 2018, 5938–5942 RSC; (b) M. M. Hansmann, Chem. – Eur. J., 2018, 24, 11573–11577 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1860668, 1860669, 1860673, 1860732, 1860759 and 1876471. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9dt00485h |
| This journal is © The Royal Society of Chemistry 2019 |