
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Exploring short strong hydrogen bonds engineered in organic acid molecular crystals for temperature dependent proton migration behaviour using single crystal synchrotron X-ray diffraction (SCSXRD)†
Lucy K.
Saunders
 *a,
Harriott
Nowell
a,
Lauren E.
Hatcher
b,
Helena J.
Shepherd
c,
Simon J.
Teat
d,
David R.
Allan
a,
Paul R.
Raithby
b and
Chick C.
Wilson
*b
*a,
Harriott
Nowell
a,
Lauren E.
Hatcher
b,
Helena J.
Shepherd
c,
Simon J.
Teat
d,
David R.
Allan
a,
Paul R.
Raithby
b and
Chick C.
Wilson
*b
aDiamond Light Source, Harwell Science and Innovation Campus, Didcot OX11 0DE, UK. E-mail: lucy.saunders@diamond.ac.uk
bDepartment of Chemistry, University of Bath, Bath, BA2 7AY, UK
cSchool of Physical Sciences, Ingram Building, University of Kent, Canterbury, CT2 7NH, UK
dAdvanced Light Source, Lawrence Berkeley National Laboratory, 1 Cyclotron Road, Berkeley, CA 94720-8229, USA
First published on 12th August 2019
Abstract
Seven multi-component molecular crystals containing O–H⋯O/O+–H⋯O− and N+–H⋯O− short strong hydrogen bonds (SSHBs) have been engineered by combining substituted organic acids with hydrogen bond acceptor molecules N,N-dimethylurea and isonicotinamide. In these materials, the shortest of the SSHBs are formed in the N,N-dimethylurea set for the ortho/para nitro-substituted organic acids whilst a twisted molecular approach favours the shorter SSHBs N+–H⋯O− in the isonicotinamide set. Temperature dependent proton migration behaviour has been explored in these systems using single crystal synchrotron X-ray diffraction (SCSXRD). By using a protocol which considers a combination of structural information when assessing the hydrogen atom (H-atom) behaviour, including refined H-atom positions alongside heavy atom geometry and Fourier difference maps, temperature dependent proton migration is indicated in two complexes (2: N,N-dimethylurea 2,4-dinitrobenzoic acid 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 and 5: isonicotinamide phthalic acid 2:1). We also implement Hirshfeld atom refinement for further confidence in this observation; this highlights the importance of having corroborating trends when applying the SCSXRD technique in these studies. Further insights into the SSHB donor–acceptor distance limit for temperature dependent proton migration are also revealed. For the O–H⋯O/O+–H⋯O− SSHBs, the systems here support the previously proposed maximum limit of 2.45 Å whilst for the charge assisted N+–H⋯O− SSHBs, a limit in the region of 2.55 Å may be suggested.
:1 and 5: isonicotinamide phthalic acid 2:1). We also implement Hirshfeld atom refinement for further confidence in this observation; this highlights the importance of having corroborating trends when applying the SCSXRD technique in these studies. Further insights into the SSHB donor–acceptor distance limit for temperature dependent proton migration are also revealed. For the O–H⋯O/O+–H⋯O− SSHBs, the systems here support the previously proposed maximum limit of 2.45 Å whilst for the charge assisted N+–H⋯O− SSHBs, a limit in the region of 2.55 Å may be suggested.
Introduction
Short strong hydrogen bonds (SSHBs) are those with very short donor–acceptor distances, typically less than 2.5 Å for O–H⋯O HBs1,2 and less than 2.6 Å for N–H⋯O HBs.3 These interactions may be partly covalent in character4,5 and have a potential energy surface (PES) which approaches the shape of a symmetric single well potential with a diminished barrier to hydrogen atom (H-atom) motion.6 Proton transfer, in the form of migration, may occur in the presence of this ‘low’ barrier, involving a gradual shift/movement in H-atom position across the HB; this effect often occurs under an external stimulus including the application of an electric field,7,8 or changes in pressure9 or temperature.10Proton migration across SSHBs can lead to a number of potentially useful properties in solid-state materials including ferroelectric11 or colour changing12 behaviour. Its study can reveal insight into the occurrence of such properties and have implications in functional materials design. A temperature dependency of this behaviour has been observed across SSHBs in a number of solid-state organic materials. These include across O–H⋯O SSHBs in substituted urea:organic acid molecular complexes10,13 and across charge assisted N+–H⋯O− SSHBs in N-heterocycle:organic acid molecular complexes.14–16 For the urea:organic acid systems, the SSHB donor–acceptor distance is found to ‘tune’ the migration behaviour; an O–H⋯O donor–acceptor distance of 2.45 Å has been proposed as a limit for migration, above which proton migration behaviour would not be expected.10,13 No such limit has been proposed for charge assisted N+–H⋯O− SSHBs. However, temperature dependent proton migration is reported only for molecular complexes where the donor–acceptor distance is less than 2.55 Å.14–16
Solid-state proton transfer processes across HBs, including temperature dependent proton migration, are typically followed using neutron diffraction; a method which allows the unambiguous determination of H-atom positions and, often important in such studies, also reveals H-atom anisotropic displacement parameters (ADPs).17 Other experimental methods such as 14NQR (nuclear quadrupole resonance) spectroscopy,18 X-ray photoelectron spectroscopy19 and solid-state NMR20,21 combined with computational methods22,23 can offer alternatives to diffraction methods in determining H-atom positions. Recently, X-ray diffraction has been shown to be useful in such studies, notwithstanding the fact that the precision of the determination of H-atom parameters is lower.24–27 Hirshfeld atom refinement (HAR)27 can contribute to overcoming such deficiencies allowing the precise and accurate location of H-atoms, comparable to neutron determinations,26,28 from X-ray diffraction data.
The present work explores SSHBs engineered in multi-component organic acid:N,N-dimethylurea/isonicotinamide molecular crystals for temperature dependent proton migration behaviour. These systems have arisen from a wider study into the SSHB propensity of organic co-formers.29N,N-Dimethylurea and isonicotinamide molecules readily recognise carboxylic acid groups to form robust heterodimeric hydrogen bonded motifs30,31 across which migration behaviour has been observed previously.10,13–16 The selected organic acid co-formers (Fig. 1) in this study include ortho or nitro-substituted benzoic acids targeted for their increased acidity32 with the potential to form HBs with a charge-assisted component that can impart additional strength to the interaction,33 increasing the likelihood of exhibiting migration behaviour.
 | ||
| Fig. 1 The N,N-dimethylurea/isonicotinamide components and organic acid co-formers. | ||
Single crystal synchrotron X-ray diffraction (SCSXRD) has been implemented here to explore the temperature dependent proton migration behaviour of the SSHBs in the prepared systems; nearly all of these form with SSHB donor–acceptor distances approaching the region where this behaviour has been previously reported.13,15,16 This technique is shown to be valid in identifying such behaviour by using a protocol which assesses potential migration behaviour across a number of parameters and which additionally implements HAR. This combined approach allows greater confidence in any behaviour observed. The use of SCSXRD in this context is advantageous because the high intensity of the synchrotron X-rays allows for rapid data collection enabling multiple measurements under different conditions or of different complexes to be carried out faster than a single laboratory XRD or neutron experiment.
Experimental
Complex preparation
All single crystal samples were prepared by evaporation from solution. Single crystals of 1 (N,N-dimethylurea phthalic acid 2:1) and 5 (isonicotinamide phthalic acid 2:1) were grown from a 2:1 stoichiometric ratio of the two components, respectively, whilst 2 (N,N-dimethylurea 2,4-dinitrobenzoic acid 1:1), 3 (N,N-dimethylurea 3,5-dinitrobenzoic acid 1:1), 4 (N,N-dimethylurea 3,5-dinitrobenzoic acid 2:2), 6 (isonicotinamide 2,4-dinitrobenzoic acid 1:1), 7 (isonicotinamide 2,4-dinitrobenzoic acid 2:2) and 8 (isonicotinamide 3,5-dinitrobenzoic acid 1:1) were grown from a 1:1 stoichiometric ratio of the two components. Crystallisations were carried out in methanol solvent for 2 (at 40 °C), 5 (30 °C), 7 (at room temperature) or in ethanol for 1, 3, 6 (at room temperature) and 8 (at 35 °C).
Data collection and treatment
SCSXRD data were collected from 2, 4, 6 and 8 on beamline I19 (EH1) at Diamond Light Source, U.K. (NR18193-1) at λ = 0.6889 Å using a Fluid Film Devices Ltd diffractometer equipped with a PILATUS 2M detector. The sample temperature was controlled (between 100 and 350 K) using an Oxford Cryosystems Cryostream Plus. Data were collected using the in-house General Data Acquisition (GDA) software and processed using in-house xia2 software.34 For 8, data were additionally scaled using the Bruker software SADABS.35 SCSXRD data were collected from 1, 3, 5 and 7 on beamline 11.3.1 at the Advanced Light Source, U.S.A. using Si (111) monochromated radiation at λ = 0.7749 Å (1) and λ = 0.8856 Å (3, 5 and 7). SCSXRD data were collected from 1 using a Bruker AXS D8 three-circle diffractometer equipped with a Bruker AXS APEX2 CCD detector. SCSXRD data were collected from 3, 5 and 7 using a Bruker AXS D8 three-circle diffractometer equipped with a Bruker AXS PHOTON 100 CMOS detector. The sample temperature was controlled for collections between 100 and 350 K using an Oxford Cryosystems Cryostream Plus. Data were collected and processed using Bruker AXS APEX236 and Bruker software SADABs35 versions 2014/2 and 2014/15.Structure solution and refinement details
All 100 K structures of 1–8 were solved using SHELXS-201837 and refined using SHELXL-201838 within the WinGX package.39 For successive temperatures, the 100 K structure was used as the starting point for the SHELXL refinements. H-Atoms were nearly all located from Fourier difference maps and their positions and isotropic thermal parameters allowed to refine freely. Exceptions to this are in 1–4 where disorder is indicated in the N,N-dimethylurea methyl groups at elevated temperatures and so the methyl H-atoms were placed geometrically using HFIX 137 at these temperatures. A SHEL command was used for each system set to the maximum diffraction resolution of the highest temperature data set (cut-off criteria where I/σ ≥ 1–2, Rint ≤ 50%) allowing comparability of multi-temperature data sets. Crystallographic data for 1–8 are given in ESI† Tables S1 to S8. HAR was performed for systems 2, 5 and 7 that exhibited the most interesting proton migration behaviour from the SHELXL refinements. Data were first merged in Bruker software XPREP40 followed by crystal structure refinement using SHELXL. On finalising the structure, a LIST 4 FVAR2 command was used and the output Fobs structure factors and finalised CIF were used as the input to HAR. The programme HAR (16.05.31 v. c4e213d) was run on the Linux platform (Linux-4.13.0-41-generic) on the Diamond Light Source (U.K.) cluster using the selected refinement options: shelx-f2 (reflection file F2 format), dispersion details for X-ray wavelength, basis-set using def2-TZVP level of theory, anisotropic or isotropic refinement of H-atom ADPs (depending on refinement stability) and the use of a restricted Hartree–Fock self-consistent field. The crystal fragment used was the asymmetric unit. The refinements took between 5 to 48 hours depending on complexity of asymmetric unit. The refinement was deemed to be successful when run to convergence with values of Chi2 < 10 and maximum shift values refined to 3 decimal places. HAR refinement data are included in the ESI† Tables S2, S5 and S7.Results and discussion
SSHB formation and proton transfer
:1 system of N,N-dimethylurea and phthalic acid (1) respectively, a 1:1 adduct of N,N-dimethylurea and 2,4-dinitrobenzoic acid (2), a 1:1 adduct of N,N-dimethylurea 3,5-dinitrobenzoic acid (3) and a 2:2 system of N,N-dimethylurea and 3,5-dinitrobenzoic acid (4). 3 and 4 are non-polymorphic forms crystallising with the same component stoichiometry (1:1) but differing values of Z′ (3Z′ = 1 versus4Z′ = 2). In the crystal structures of 1–4, molecular association of the two components occurs via a carboxylic acid:amide R22(8) HB synthon (Fig. 2) in which the O–H⋯O HB dominates synthon formation.41 This synthon is commonly formed between substituted urea and organic acid components;42,43 its frequency of formation is consistent with complex stoichiometry, as noted by Alhalaweh et al. (2010).44
 | ||
| Fig. 2 The carboxylic acid:amide R22(8) hydrogen bonded synthons formed in the crystal structures of 1–4. | ||
The donor–acceptor distances of the O–H⋯O HBs in 1–4 at 100 K (Table 1) are below 2.5 Å and can therefore be classed as short strong hydrogen bonds.45 In most cases, the donor–acceptor distances are within the range of the previously proposed limit for temperature dependent proton migration across O–H⋯O SSHBs (∼2.45 Å)13 and are good candidates for multi-temperature studies. The shortest HBs are formed in 2 and 4 having donor–acceptor distances in the region of 2.44 Å, just below the proposed migration limit. The complementary N–H⋯O HB in the R22(8) HB synthon mirrors this interaction and tends to be shorter if the O–H⋯O HB is shorter.
:amide R22(8) HB synthon in 1–4: D is the HB donor atom and A is the HB acceptor atom
| System | Components | Interaction | dD–H (Å) | H⋯A (Å) | dD⋯A (Å) | <DHA (°) |
|---|---|---|---|---|---|---|
| 1 | 2:1 N,N-dimethylurea phthalic acid |
O2–H2⋯O5 | 1.01(3) | 1.46(3) | 2.4520(16) | 167(2) |
| N2–H14⋯O1 | 0.95(2) | 2.03(2) | 2.9743(19) | 170.1(18) | ||
| O3–H1⋯O6 | 1.09(3) | 1.44(3) | 2.4925(16) | 160(3) | ||
| N4–H21⋯O4 | 0.93(2) | 2.06(2) | 2.9778(19) | 174(2) | ||
| 2 | 1:1 N,N-dimethylurea 2,4-dinitrobenzoic acid |
O1–H1⋯O2 | 1.10(2) | 1.36(2) | 2.4435(10) | 168.6(18) |
| N1–H2⋯O3 | 0.884(16) | 2.030(16) | 2.9100(12) | 173.1(14) | ||
| 3 | 1:1 N,N-dimethylurea 3,5-dinitrobenzoic acid |
O1–H1⋯O2 | 1.22(7) | 1.26(7) | 2.469(3) | 172(6) |
| N4–H12⋯O3 | 0.88(4) | 2.11(5) | 2.964(3) | 162(3) | ||
| 4 | 2:2 N,N-dimethylurea 3,5-dinitrobenzoic acid |
O1–H1⋯O7 | 1.06(2) | 1.39(2) | 2.4445(12) | 170(2) |
| N3–H5⋯O2 | 0.890(19) | 1.99(2) | 2.8795(14) | 173.9(17) | ||
| O8–H13⋯O14 | 1.03(2) | 1.45(2) | 2.4640(11) | 167(2) | ||
| N7–H17⋯O9 | 0.905(18) | 2.041(18) | 2.9272(14) | 166.0(16) |
The O–H⋯O SSHBs form with varying degrees of proton transfer in the 100 K structures. In 1 and 4, no proton transfer is evident as the carboxylic acid proton resides on the acid molecule; 1 and 4 are therefore neutral complexes at 100 K and considered co-crystals.46 For 2 and 3, the O–H⋯O SSHBs are charge assisted (O+–H⋯O−) as a result of the H-atom being fully (2) or partially (3) transferred between the organic acid and N,N-dimethylurea molecule. 2 is therefore a salt at this temperature46 whilst the refined H-atom positions at 100 K for 3 indicate a centralised position (the dO–H 1.23(7) Å and dH⋯O 1.26(7) Å distances are not significantly different given the esds) meaning it is an intermediate between a salt and co-crystal.47
Generally, where the interaction has a charge assisted component (O+–H⋯O−), as in 2 and 3, the donor–acceptor distances are shorter. This can be explained where the charges on the donor–acceptor atoms significantly increase the strength of the interaction33 by reinforcing its electrostatic character;48,49 leading to the shorter donor–acceptor distances. Furthermore, where the charge assisted component of the interaction is enhanced by the organic acid having substituents that are either electron withdrawing50 (2–4) or in the ortho/para position (2),32,51,52 the donor–acceptor distances are shorter; these substituents increase the acidity of the carboxylic acid group and impart a greater charge assisted component to the SSHB. These observations are useful in targeting short O–H⋯O SSHBs in the future. The shorter O–H⋯O donor–acceptor distances also form where the DĤA angle approaches ca. 170° suggesting this may be an optimum angle to facilitate SSHB formation for this set of materials.
:1 system of isonicotinamide and phthalic acid respectively (5), a 1:1 system of isonicotinamide and 2,4-dinitrobenzoic acid (6), a 2:2 system of isonicotinamide and 2,4-dinitrobenzoic acid (7) and a 1:1 system of isonicotinamide and 3,5-dinitrobenzoic acid (8). 6 and 7 are non-polymorphic forms crystallising with the same component stoichiometry (1:1) but differing values of Z′ (6Z′ = 1 versus7Z′= 2). The crystal structure of 8 has been reported previously and is included for comparison with this related set of structures.53 In the crystal structures of 5–8, molecular association of the two components occurs via charge assisted N+–H⋯O− HBs. For 6 and 7, the N+–H⋯O− interaction is assisted by a weak C–H⋯O HB to give a pyridinium:carboxylate R22(7) hydrogen bond synthon (Fig. 3). This synthon is commonly reported in pyridine carboxylic acid molecular complexes54 and can contribute to favourable mechanical properties or plasticity in materials.41 Two symmetry independent pyridinium:carboxylate R22(7) HB synthons form in 7 consistent with complex stoichiometry (2:2).
 | ||
| Fig. 3 The charge assisted N+–H⋯O− HBs in 5–8 formed singularly (5 and 8) or as part of a pyridinium:carboxylate R22(7) hydrogen bond synthon (6, 7). | ||
The refined H-atom positions indicate that 5–8 are all salts at 100 K; an acidic proton has been transferred to the isonicotinamide molecule in each case. In 5, both phthalic acid carboxylic acid groups form HBs to a different isonicotinamide molecule; only one of these interactions is charge assisted, the other is a neutral O–H⋯N HB which is moderate in strength (dN⋯O 2.696(1) Å). This interaction is assisted by a weak C–H⋯O hydrogen bond and is a pyridine:carboxylic acid R22(7) HB synthon.
The charge assisted N+–H⋯O− HBs in 5–8 have donor–acceptor distances (Table 2) equivalent to or shorter than 2.6 Å and are therefore classed as strong HBs. For all but 6, these distances are close to the region where temperature dependent proton migration across N+–H⋯O− SSHBs has previously been observed, near 2.55 Å;3,14–16 the shortest is formed in 8. The shorter N+–H⋯O− HBs are formed as a discrete link D11(3) between isonicotinamide and organic acid co-former, as in 5 and 8. In this scenario, the carboxylate group is significantly twisted out of co-planarity of the isonicotinamide N-heterocyclic ring (CONC torsions: 5 −85.41(9)°, 8 29.3(2)°). This arrangement may allow a closer approach of the carboxylate group to the protonated isonicotinamide molecule.
| System | Components | Interaction | D–H (Å) | H⋯A (Å) | dD…A (Å) | <DHA (°) |
|---|---|---|---|---|---|---|
| 5 | 2:1 isonicotinamide phthalic acid |
N3–H3⋯O3 | 1.102(18) | 1.449(18) | 2.5499(10) | 176.7(2) |
| O1–H1⋯N1 | 1.04(2) | 1.66(2) | 2.6955(10) | 179(2) | ||
| 6 | 1:1 isonicotinamide 2,4-dinitrobenzoic acid |
N1–H1⋯O1 | 1.046(18) | 1.572(18) | 2.6000(8) | 166.2(2) |
| 7 | 2:2 isonicotinamide 2,4-dinitrobenzoic acid |
N2–H1⋯O9 | 0.985(18) | 1.660(19) | 2.6347(12) | 169.5(2) |
| N1–H2⋯O5 | 1.05(2) | 1.50(2) | 2.5542(12) | 178(2) | ||
| 8 | 1:1 isonicotinamide 3,5-dinitrobenzoic acid |
N1–H1⋯O1 | 1.084(18) | 1.484(18) | 2.5350(13) | 161.4(2) |
Beyond the charge assisted N+–H⋯O− interactions, the isonicotinamide molecules (protonated and neutral) associate via centrosymmetric amide:amide R22(8) HB dimers formed of N–H⋯O HBs. Common structural features are found in these dimers where the charge assisted N+–H⋯O− HBs are shorter; the amide:amide HB distances are longer (ESI† Table S9, dD⋯A IN:IN) whilst the isonicotinamide amide group is increasingly out of co-planarity of the N-heterocycle ring (ESI† Table S9, <OCCC). It may be that these features work to facilitate or compensate for the formation of the shorter charge assisted N+–H⋯O− HBs in these types of materials.
In terms of crystal engineering approaches, there is no clear distinction between one acid co-former over the other, in terms of best choice to access the shorter of the charge assisted N+–H⋯O− HBs. However, it is clear that from this set, the combination of isonicotinamide with either ortho-substituted or dinitrobenzoic acids is a good route to forming shorter hydrogen bonding interactions. As is the case for the O–H⋯O SSHBs, this may be a result of the increased acidity of the carboxylic acid group32,50–52 and thus an enhanced charge-assisted component of the HB55,56 for these types (nitro) or positions (ortho) of substituent group.
Multi temperature measurements reveal migratory behaviour in 2 and 5
Multi temperature SCSXRD measurements were performed on 1 to 8 between 100 and 350 K to explore the SSHBs for temperature dependent proton migration behaviour. The high throughput nature of the SCSXRD technique was exploited in collecting a significant number of data points for each system whilst attenuating the synchrotron beam to ensure the sample continued to diffract across the whole temperature range. Following these measurements, migratory behaviour is indicated in 2 and 5 only.For systems 2 and 5, in both the SHELXL57 and HAR27 crystal structures, a lengthening of the D–H distance and a shortening of the H⋯A distance occurs in the SSHB as a function of temperature (Fig. 4, ESI† Tables S10 and S11) suggestive of a migrating H-atom. In 2, the H-atom migrates away from the urea group up to 250 K where it occupies a more central position. Between 300 and 350 K, almost complete transfer to the organic acid is indicated. In 5, the H-atom migrates away from the HB donor group (N+) to a more central position in the charge assisted N+–H⋯O− SSHB as a function of temperature. The D–H/H⋯A distances change gradually as a function of temperature and in a consistent manner for 2 (up to 300 K) and 5 (up to 350 K) whilst at the extremes of the migration temperature range they are appreciably different (2dO–H 1.10(2) Å at 100 K vs. 1.34(3) Å at 350 K, 5dN+–H 1.102(18) Å at 100 K vs. 1.19(2) Å at 350 K). The fact there is agreement between the SHELXL57 and HAR27 crystal structures is significant and adds further confidence to the observation of the migration behaviour. The reliability of the SHELXL57 refinements is aided by low values for the standard uncertainties on the D–H and H⋯A distances (of 0.02–0.03 Å) over the temperature range. On implementing the HAR27 method, the precision of the H-atom positions is improved at low temperature, as is typically seen;26 the observed migration trend is also more consistent. As is usually observed between X-rays and neutrons,24 the changes in H-atom position observed as a function of temperature between 100 and 350 K are generally larger from the SHELXL57 refinements for 2 and 5 (0.24(4) and 0.09(3) Å, respectively) than they are from the HAR27 refinements (2, 0.18(1) and 5, 0.038(12) Å), giving values closest to those found by neutron diffraction.
 | ||
| Fig. 4 The D–H and H⋯A distances in 2 and 5 as refined using SHELXL57 and HAR27 over the 100 to 350 K temperature range. | ||
The extent of temperature dependent proton migration occurring in 2 is similar to that in 1:1 adduct urea phosphoric acid,10 where the H-atom migrates across the charge assisted O+–H⋯O− hydrogen bond away from the protonated donor (urea) to a central position at 300 K. For 5, there are similarities in the nature of the migration occurring with 4,4-bipyridine 1,2,3,4-tetracarboxylic acid16 and pyridine-3,5-dicarboxylic acid16 where the H-atom moves away from the nitrogen donor towards the centre of the N+–H⋯O− CAHB as a function of temperature. In contrast to 5, in these literature systems, the proton movement occurs to a far greater extent, located closer to the organic acid at room temperature.
Over the migration temperature range, the donor–acceptor distances of the SSHBs of interest are essentially invariant in both 2 and 5 (Fig. 5). This indicates that the observed H-atom migration behaviours are not simply a result of a lengthening SSHB due to lattice expansion as a function of temperature. In 2, the most significant change is between 300 and 350 K and may explain the slight inconsistency in the O+–H and H⋯O− distances in this region that suggest a sudden large jump in H-atom position, as can be seen in Fig. 4.
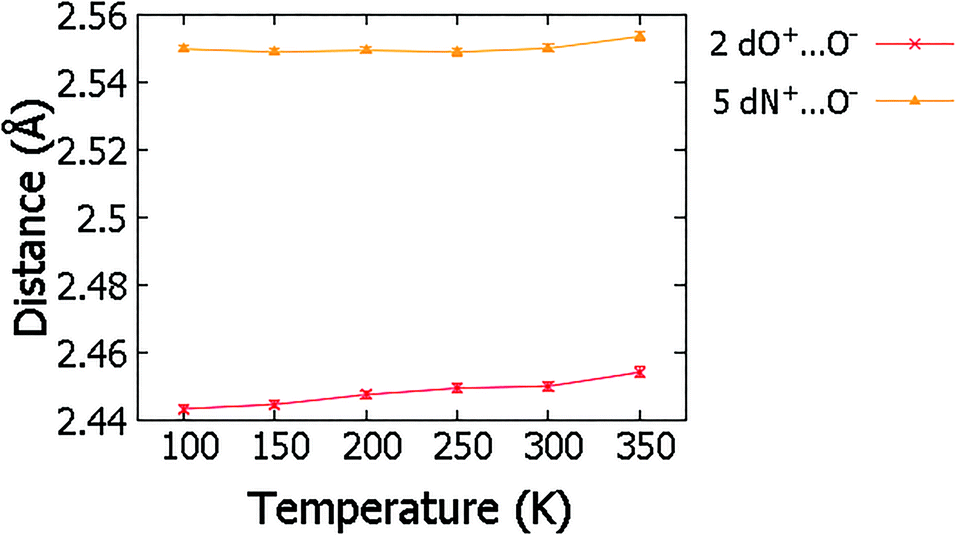 | ||
| Fig. 5 The SSHB donor–acceptor distance in complex 2 and 5 obtained from refinement in SHELXL57 over the 100 to 350 K temperature range. | ||
Direct visualisation of the H-atom electron density in Fourier difference electron density maps can yield additional insight into proton transfer behaviour.58 These maps are a valuable tool, giving a clearer and independent picture from X-ray diffraction data,24 as they are not dependent on the refined parameters of the H atom involved in the SSHB, which is removed in the calculation of the map. Two dimensional Fourier difference electron density maps (FMAPs) have been generated as a function of temperature for SSHBs O1+–H1⋯O5− (2) (Fig. 6) and N3+–H3⋯O3− (5) (Fig. 7) from the SCSXRD data using the LIST 3 command in the SHELX37 instruction file. These maps have been generated with the H-atom of interest removed from the model such that its density is visible (contours in the red to white region) when visualised using the MAPVIEW option in WinGX.39 In the 100 K FMAPs for 2 and 5, the H-atom density is partially smeared along the HB, asymmetric in shape with a tail end that points towards the acceptor atom; this is characteristic of a H-atom in a short strong interaction.13 In 2, this smearing increases with increasing temperature up to 250 K where it becomes more elongated in shape. At 300 K, the peak maxima has moved to a central position and the H-atom density is evenly smeared across the centre of the SSHB, indicating a more centrally located H-atom in agreement with the refined H-atom positions at this temperature. At 350 K, the H-atom density peak maxima is located closer to the acceptor O2+ and the extended tail points more towards the opposite end of the SSHB, indicating its transfer. This switch in asymmetry of the H-atom density as a function of temperature is similar to that seen in the FMAPs of an equivalent temperature dependent proton migration material, the 1:1 adduct of N,N′-dimethylurea oxalic acid.59 Similarly in 5, ramping the temperature to 250 K causes the H-atom density to change shape and become more elongated along the SSHB. At 300 K the H-atom density peak maxima broadens suggesting increased thermal motion that could correspond to its movement towards the O3− acceptor atom. At 350 K, the H-atom density is more symmetrically smeared along the interaction and the peak maximum occupies a more central position. It is less clear in this case (5) whether the changes occurring in the H-atom density as a function of temperature are consistent with proton migration or simply an effect of increased thermal motion of the H-atom with increasing temperature of the system;13 the changes are less significant than seen for 2.
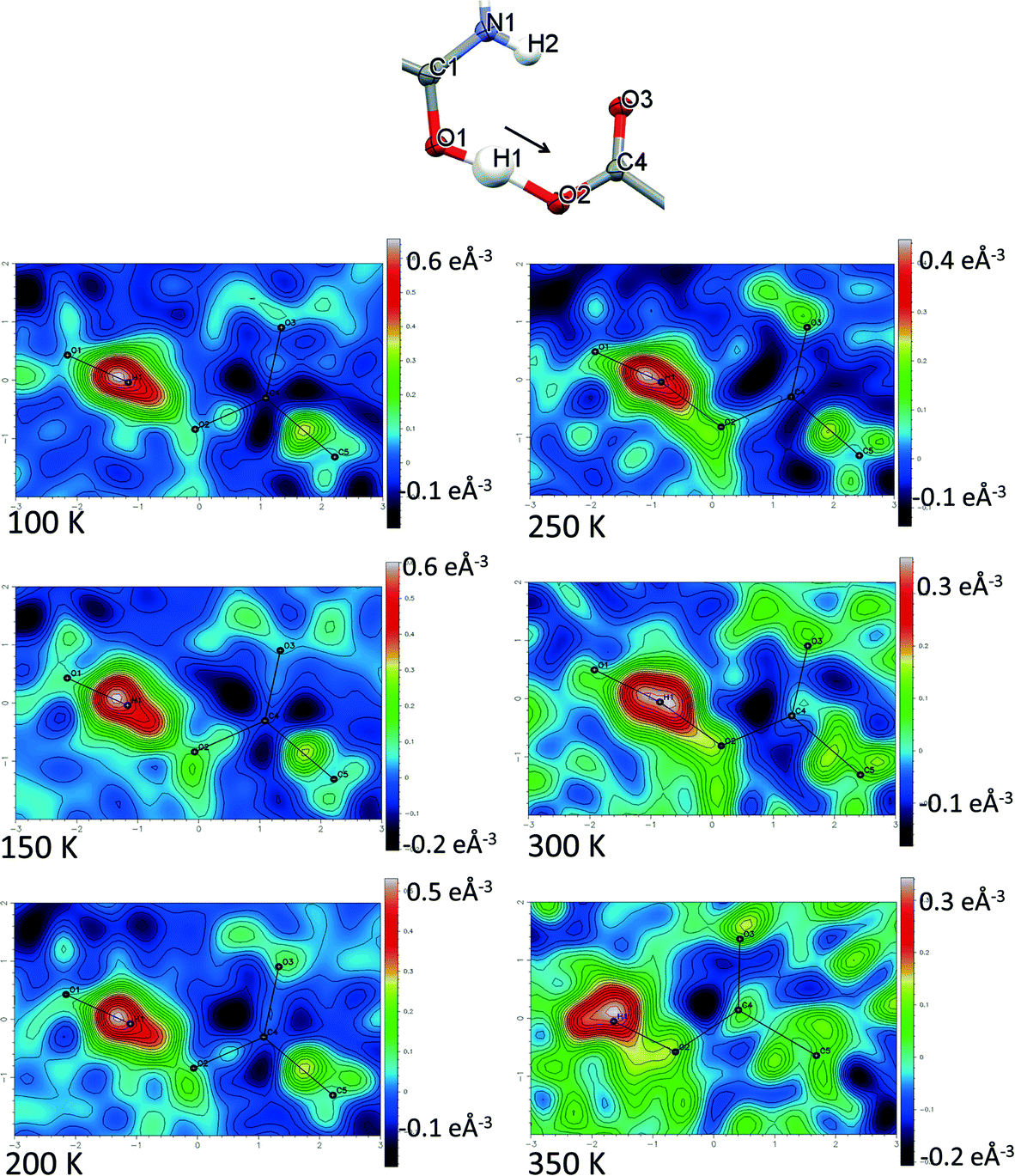 | ||
| Fig. 6 Fourier difference electron density maps showing the electron density associated with the H-atom in the O+–H⋯O− SSHB in 2 over the 100 to 350 K temperature range. The 350 K FMAP is missing atom O1 where it was not possible to generate a 2D plane containing all three atoms of the SSHB. | ||
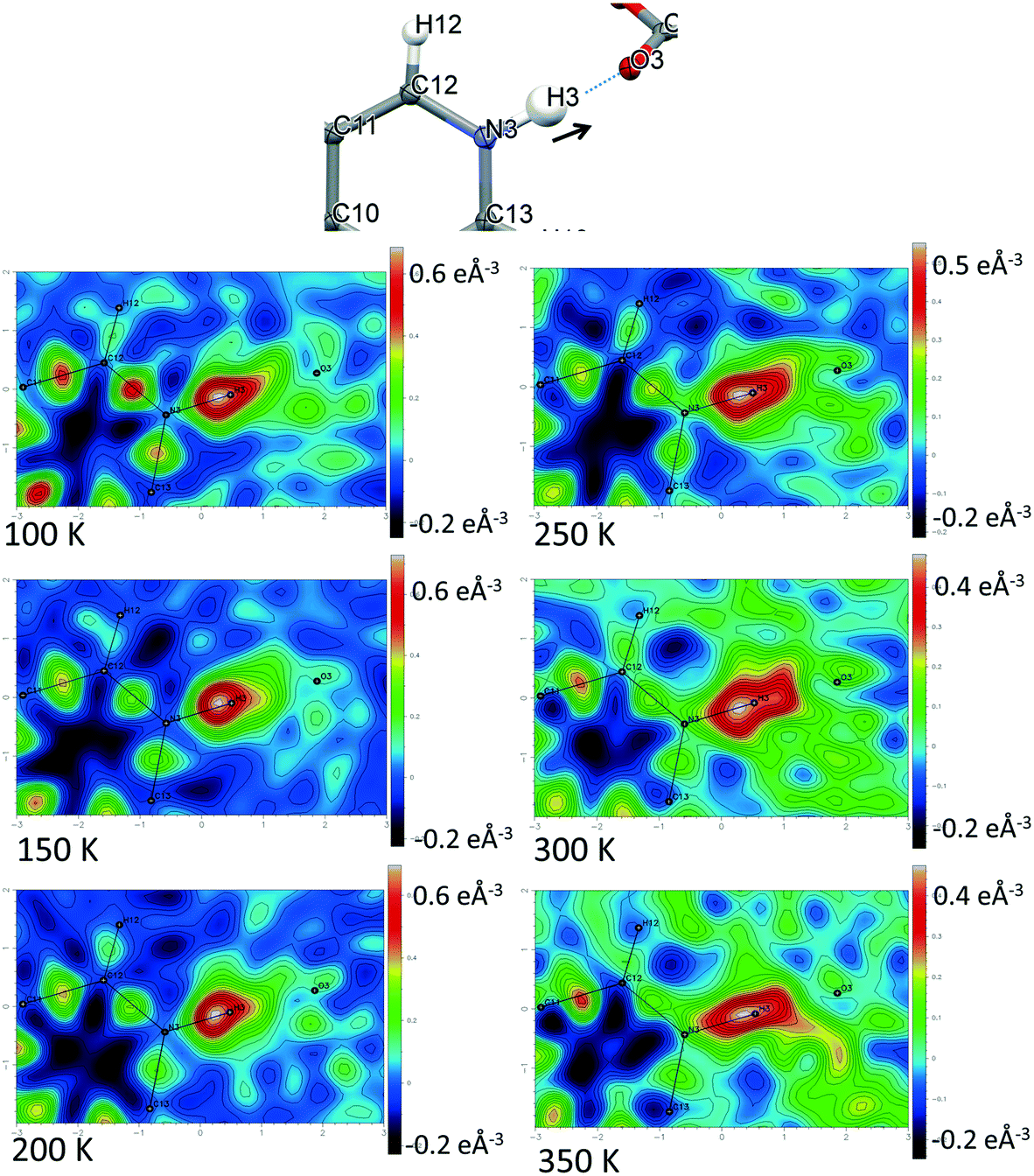 | ||
| Fig. 7 Fourier difference electron density maps showing the electron density associated with the H-atom in the N+–H⋯O− SSHB in 5 over the 100 to 350 K temperature range. | ||
The generated FMAPs for 2 and 5 are of good quality in each case featuring low and consistent noise over the temperature range aiding the determination of the H-atom behaviour. In general, there is a slight difference in the FMAPs in the refined H-atom position and the peak maximum. This is likely due to the SSHB H-atom being refined with an isotropic thermal parameter which, characteristically in an SSHB, has a more anisotropic shape; this leads to an apparent shift of the atom away from its true position during the refinement.24 The difference is, however, small and consistent over the temperature range meaning the refined H-atom positions are made more reliable for deducing proton migration trends.
For 2 and 5, changes in molecular geometries of the heavy atom groups forming the SSHBs as a function of temperature provide corroborating evidence for a migrating H-atom (Fig. 8, ESI† Tables S12 and S13). In both 2 and 5, the bond distances of the protonated groups (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) OUREA+ and C–NIN+) forming the SSHBs undergo some shortening as a function of temperature indicating a slight increase in their bond orders and supporting the migration of the H-atom away from this end of the HB. At 350 K, the bond distance for the COUREA+ group in 2 is longer than might be expected for a fully deprotonated N,N-dimethylurea (WIFKEB02 1.245(2) Å at 273 K).60 This post migration distance is, however, consistent with related N,N-dimethylurea systems exhibiting similar migration behaviour.13 At the opposite end of the SSHB, the C–O− bond distances for both 2 and 5 also shorten as a function of temperature. This is less consistent with migration behaviour; an increase in this bond length would be expected where the H-atom migrates towards this group. This effect is, however, very small and could be caused by a redistribution of charge in the carboxylate group as a function of temperature.
OUREA+ and C–NIN+) forming the SSHBs undergo some shortening as a function of temperature indicating a slight increase in their bond orders and supporting the migration of the H-atom away from this end of the HB. At 350 K, the bond distance for the COUREA+ group in 2 is longer than might be expected for a fully deprotonated N,N-dimethylurea (WIFKEB02 1.245(2) Å at 273 K).60 This post migration distance is, however, consistent with related N,N-dimethylurea systems exhibiting similar migration behaviour.13 At the opposite end of the SSHB, the C–O− bond distances for both 2 and 5 also shorten as a function of temperature. This is less consistent with migration behaviour; an increase in this bond length would be expected where the H-atom migrates towards this group. This effect is, however, very small and could be caused by a redistribution of charge in the carboxylate group as a function of temperature.
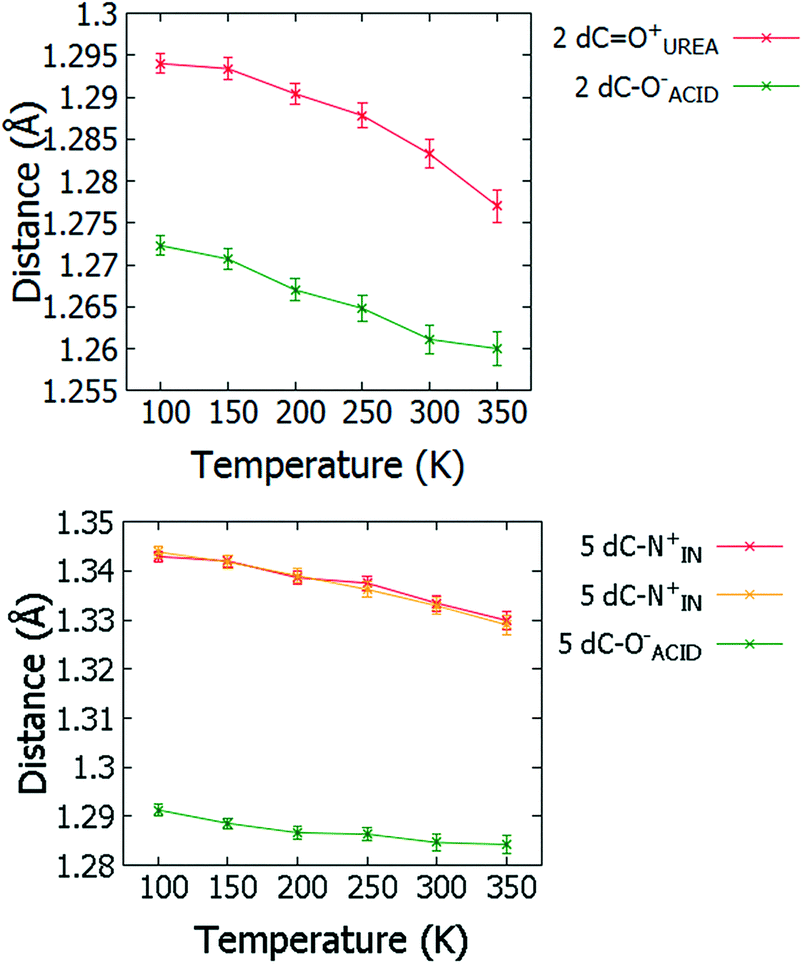 | ||
| Fig. 8 Bond distances of the groups forming the SSHBs in 2 (top) and 5 (bottom) over the 100 to 350 K temperature range. | ||
Inconclusive or absent behaviour
Temperature dependent proton migration behaviour in contrast cannot be confirmed in 1, 3, 4, 6–8. For 7 and 8, the behaviour of the SSHB H-atom appears on the edge of migratory behaviour. Changes in the N+–H and H⋯O− distances as a function of temperature correspond to only a slight migration of the H-atom towards the centre of the N+–H⋯O− SSHBs (Fig. 9, ESI† Tables S14 and S15). Investigating these migrations in more detail in 7, it is clear that these changes are within error in the SHELXL57 refinement. Also, the agreement between the SHELXL57 and HAR27 methods is not good; this is likely due to the HAR27 method affording longer D–H distances closer in value to those found from neutron diffraction which are typically longer probing nuclear position rather than electron density.24 A significant lengthening of the SSHB donor–acceptor distance also occurs for 7 (Fig. 10) such that any changes in the SSHB H-atom are more likely attributed to HB expansion. The supporting FMAPs in the region of the SSHBs show inconsistent behaviour (Fig. 11) meaning it is not possible to confidently identify migration behaviour in this material. They do not support the changes in the N+–H and H⋯O− distances as a function of temperature, instead showing no identifiable trend. An elongation of the hydrogen atom electron density occurs along the hydrogen bond but this does not increase consistently across the temperature range. From 250 K, this density appears as two peaks suggesting that proton disorder is more likely to be occurring and by 350 K, the H-atom peak is becoming lost in the increased thermal motion of the entire system; its signal cannot be deciphered from background noise. In 8, the SSHB donor–acceptor distances in contrast show minor variations as a function of temperature (Fig. 10) meaning any observed migratory behaviour would be ruled out from being attributed to lattice expansions. However, in the FMAPs (Fig. 12) the H-atom density remains close to the N+ donor atom indicating a static H-atom over the temperature range. Only significant structural changes in the HB groups (Fig. 13, ESI† Tables S16 and S17) that would support migratory behaviour as a function of temperature occur in 7; shortening of the isonicotinamide HB donor group C–N bond distances occur as a function of temperature, similar to those in 5. System 7 would benefit from additional experimental studies to fully elucidate the proton transfer behaviour; it has however not been possible to grow further single crystals of 7 suitable for diffraction measurements since these SCSXRD measurements were undertaken, only the 1:1 system 6 is currently accessible.
 | ||
| Fig. 9 Bond distances of the groups forming the SSHBs in 7 (top) and 8 (bottom) over the 100 to 350 K temperature range. | ||
 | ||
| Fig. 10 The SSHB donor–acceptor distance in complex 3, 7 and 8 over the 100 to 350 K temperature range. | ||
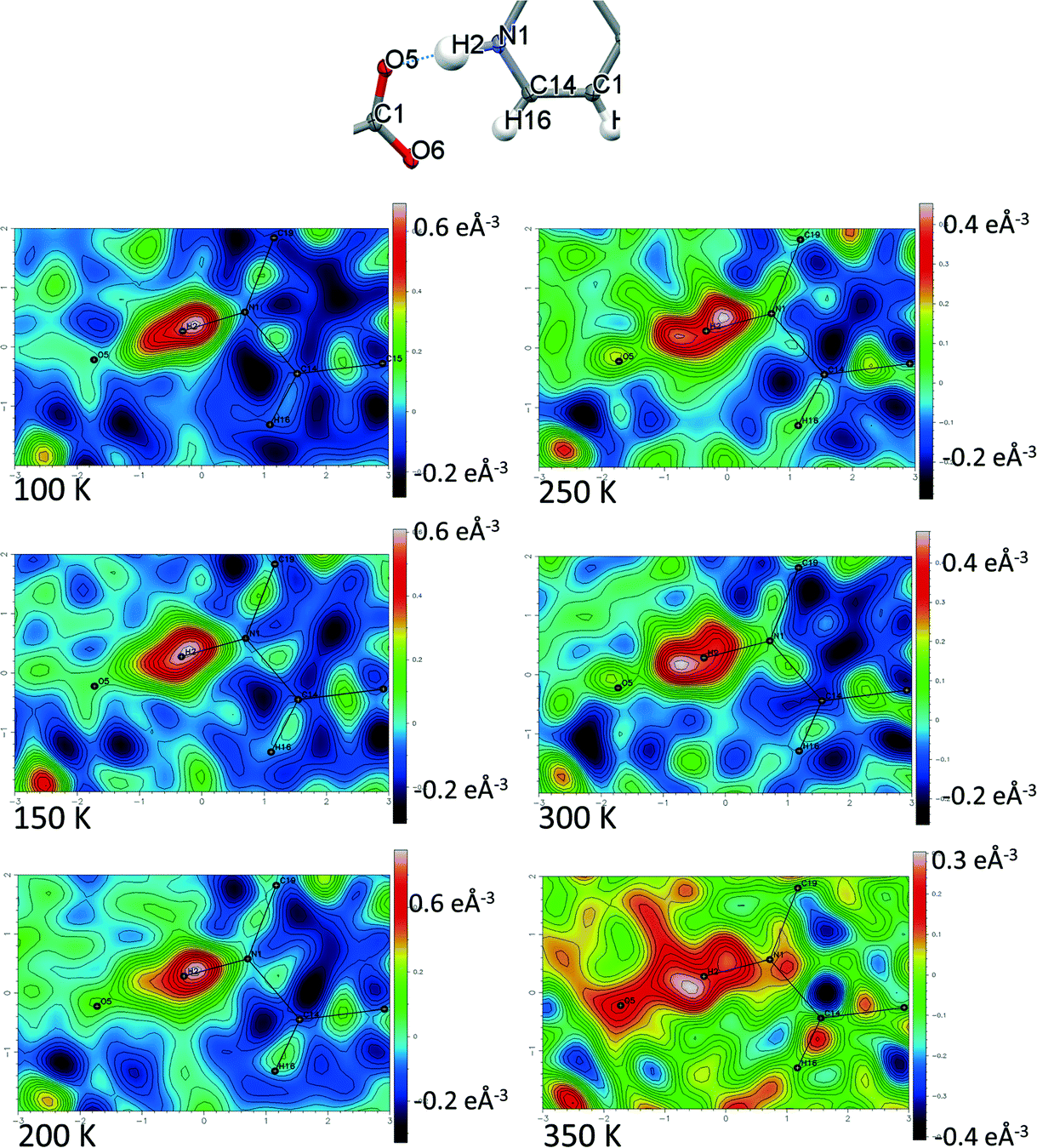 | ||
| Fig. 11 Fourier difference electron density maps showing the electron density associated with the H-atom in the N+–H⋯O− SSHB in 7 over the 100 to 350 K temperature range. | ||
 | ||
| Fig. 12 Fourier difference electron density maps showing the electron density associated with the H-atom in the N+–H⋯O− SSHB in 8 over the 100 to 350 K temperature range. | ||
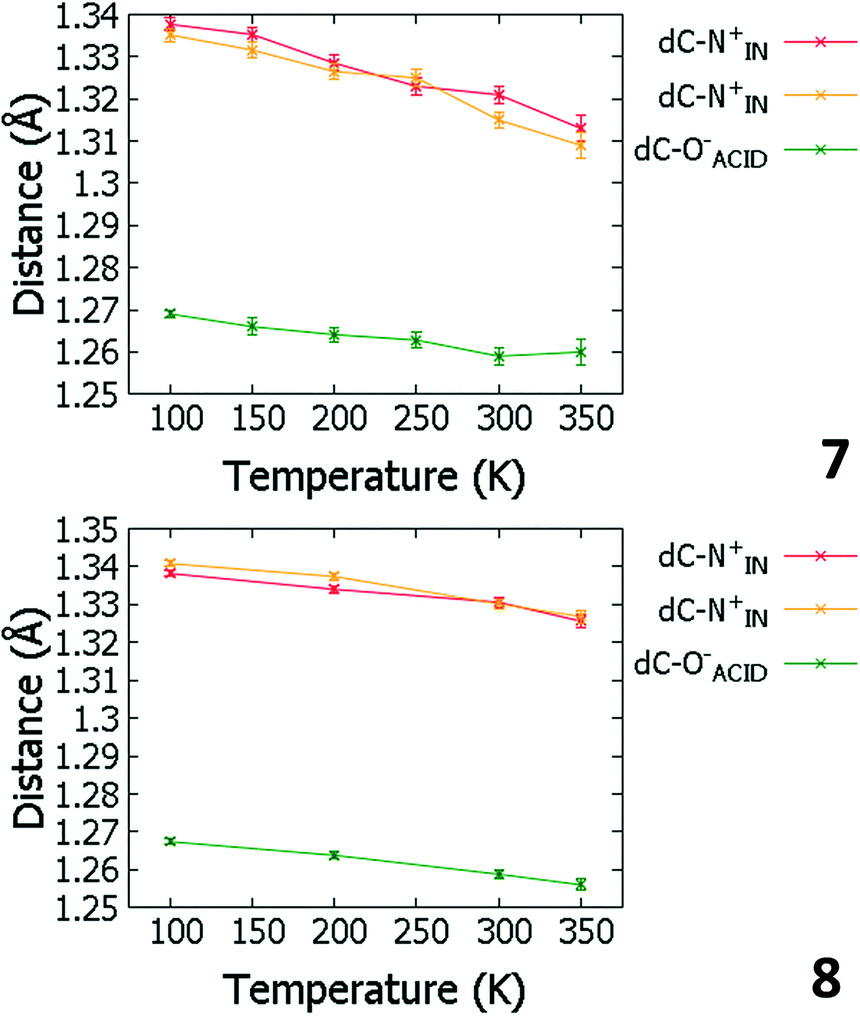 | ||
| Fig. 13 Bond distances of the groups forming the SSHBs in 7 (top) and 8 (bottom) over the 100 to 350 K temperature range. | ||
The migration behaviour for 3 is unclear when considering the refined H-atom positions (ESI† Fig. S1, Table S18) and FMAPs (ESI† Fig. S2) over the 100 to 300 K temperature range. The H-atom position appears to fluctuate as a function of temperature whilst the determination of behaviour is made difficult by the significant noise present in the H-atom positions and FMAPs. The SSHB donor–acceptor distance also fluctuates significantly (Fig. 10) with temperature, meaning any migration behaviour is more likely to be attributed to an expanding HB. This system could benefit from multi-temperature neutron diffraction measurements (requiring the growth of larger single crystals) as there may be interesting proton disorder behaviour above 100 K; though this suggestion is based on FMAPs with high levels of noise and is not suggested in the acid CO/C–O bond lengths. In 1, 4 and 6 the evidence points to systems where temperature dependent proton migration behaviour is absent. Little variation in the D–H and H⋯A distances occur as a function of temperature (ESI† Table S19–S21, Fig. S1) and they are not significantly different at the temperature extremes. The FMAPs corroborate a static H-atom whose density remains close to the SSHB donor atom as a function of temperature (ESI† Fig. S3–S5).
Insights into temperature dependent proton migration across SSHBs
The study of SSHBs in molecular crystals for temperature dependent proton migration using the SCSXRD technique adds to an understanding of the migration process in the solid-state. The high throughput nature of the SCSXRD technique means the number of systems explored for this migration behaviour can be greatly increased. In particular, this study has identified two new temperature dependent proton migration materials, 2 and 5, two in which this behaviour is less clear (7 and 8) and four (1, 3, 4 and 6) in which this behaviour is absent.A comparison with proton migration systems found previously3,10,13–16,61 reveals insights related to SSHB donor–acceptor distance. For O–H⋯O SSHBs, in substituted urea:organic acid systems, a limiting donor–acceptor distance of 2.45 Å has been proposed,13 above which temperature-dependent proton migration is not expected. The related multi-component molecular crystals studied here containing O–H⋯O/O+–H⋯O− SSHBs are consistent with this proposal;59 proton migration is not indicated in any of the molecular complexes with O⋯O donor–acceptor distances above 2.45 Å, only in 2 where the O⋯O distance is 2.441(1) Å at 100 K. For N+–H⋯O− SSHBs in related N-heterocycle:organic acid molecular complexes, temperature dependent proton migration has not been identified in systems with donor–acceptor distances longer than 2.55 Å, having been reported previously in isoniazid 4-aminosalicylic acid (dN⋯O 2.548(2) Å),14 pyridine-3,5-dicarboxylic acid (dN⋯O 2.523(2) Å)15 and 4,4′-bipyridine benzene-1,2,4,5-tetracarboxylic acid (dN⋯O 2.5220(17) Å).165 has the longest N+–H⋯O− hydrogen bond across which temperature dependent proton migration is suggested (dN⋯O 2.551(1) Å). It is not possible to confidently identify the presence of temperature dependent proton migration in 7, with a slightly longer SSHB donor–acceptor distance (dN⋯O 2.555(1) Å). It may be, for N+–H⋯O− SSHBs, that the donor–acceptor limiting distance is in the region of 2.55 Å. This is, however, a preliminary assessment and more cases are needed before a firm conclusion about N+–H⋯O− donor–acceptor distances can be made. It is interesting that for 8, despite the very short HB donor–acceptor distance of 2.5350(13) Å at 100 K, temperature dependent migration behaviour appeared to be absent. This may be related to the SSHB angle of this interaction which has reduced directionality (ca. 161°) compared to in systems 5 and 7 (ca. 180°). It may be that enhanced directionality in charge assisted N+–H⋯O− HBs favour migratory behaviour; pyridine-3,5-dicarboxylic acid (CSD refcode DINICA12),15 isoniazid 4-aminosalicylic acid (URUDER)14 and 4,4′-bipyridine benzene-1,2,4,5-tetracarboxylic acid (WISNAN)62 all have HB angles greater than ca. 168°.
In the complexes reported here, the donor–acceptor distance is therefore shown to ‘tune’ the occurrence of temperature dependent proton migration across SSHBs, occurring in some instances but not in others. This is in part known where donor–acceptor distances affect the shape of the HB PES6 and can be correlated to HB strength and PES symmetry.63 A shorter donor–acceptor distance favours an increasingly symmetric HB PES with a low barrier to proton transfer; in this situation, migration behaviour is more likely.
Conclusions
In this work we have engineered seven multi-component molecular crystals containing SSHBs by combining isonicotinamide and N,N-dimethylurea components with organic acid co-formers. Analysis of the SSHBs across the systems revealed that the strategy of selecting nitro-substituted organic acids favoured the formation of shorter hydrogen bonds in general whilst for the N,N-dimethylurea set, the presence of ortho/para substituents was additionally favourable. These findings contribute to the predictable design of these short interactions in the solid-state. If donor–acceptor distances in SSHBs can be predictably tuned, this would be a step forward in accessing proton migration materials more predictably in future.SCSXRD has been used to investigate the SSHBs for temperature dependent proton migration behaviour in eight multi component molecular crystals. This technique has potential in identifying such behaviour by implementing a protocol which considers the refined H-atom positions alongside those from Hirshfeld atom refinement in conjunction with other information including Fourier difference maps (FMAPs) and selected heavy atom structural parameters. Following this protocol, two new temperature dependent proton migration materials have been identified (2 and 5) in which migration of the H-atom occurs from the donor atom across the SSHB either towards the centre (5) or to the opposite side of the HB (2). The evaluation of such behaviour is aided by the availability of high quality, multi-condition data using the SCSXRD technique. These findings contribute to the development of the SCSXRD method to allow it to be used routinely and with more confidence in future studies of proton transfer behaviour. Further insight into the dependence of temperature dependent proton migration on SSHB donor–acceptor distances is also revealed which can help to identify potential proton migration materials in future.
Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
We acknowledge the support from the University of Bath and the Diamond Light Source Ltd for a studentship to L. K. S. C. C. W. and P. R. R. are grateful to the EPSRC for continued support (EP/K004956/1). We thank Florian Kleemis from the University of Bremen for his assistance in implementing the HAR software. We thank Diamond Light Source for access to beamline I19 (NR18193-1) for data for samples 2, 4, 6, 8. We thank the Advanced Light Source, LBNL for beamtime to collect on samples 1, 3, 5 and 7 and S. J. Teat and K. J. Gagnon for assistance. The Advanced Light Source is supported by the Director, Office of Science, Office of Basic Energy Sciences, of the U.S. Department of Energy under contract no. DE-AC02-05CH11231.References
- L. H. R. Dos Santos, B. L. Rodrigues, Y. M. Idemori and N. G. Fernandes, J. Mol. Struct., 2012, 1014, 102–109 CrossRef CAS.
- P. Gilli, V. Bertolasi, V. Ferretti and G. Gilli, J. Am. Chem. Soc., 1994, 116, 909–915 CrossRef CAS.
- T. Steiner, I. Majerz and C. C. Wilson, Angew. Chem., Int. Ed., 2001, 40, 2651–2654 CrossRef CAS PubMed.
- M. Schmidtmann, M. J. Gutmann, D. S. Middlemiss and C. C. Wilson, CrystEngComm, 2007, 9, 743–745 RSC.
- M. V. Vener, E. O. Levina, A. A. Astakhov and V. G. Tsirelson, Chem. Phys. Lett., 2015, 638, 233–236 CrossRef CAS.
- C. L. Perrin, Acc. Chem. Res., 2010, 43, 1550–1557 CrossRef CAS PubMed.
- I. A. Abronin, I. A. Koval'chuk and V. P. Sakun, Russ. J. Phys. Chem. B, 2016, 10, 357–359 CrossRef CAS.
- Z.-S. Yao, K. Yamamoto, H.-L. Cai, K. Takahashi and O. Sato, J. Am. Chem. Soc., 2016, 138, 12005–12008 CrossRef CAS PubMed.
- D. M. S. Martins, D. S. Middlemiss, C. R. Pulham, C. C. Wilson, M. T. Weller, P. F. Henry, N. Shankland, K. Shankland, W. G. Marshall, R. M. Ibberson, K. Knight, S. Moggach, M. Brunelli and C. A. Morrison, J. Am. Chem. Soc., 2009, 131, 3884–3893 CrossRef CAS PubMed.
- C. C. Wilson, K. Shankland and N. Shankland, Z. Kristallogr., 2001, 216, 303–306 CAS.
- K. Lee, B. Kolb, T. Thonhauser, D. Vanderbilt and D. C. Langreth, Phys. Rev. B: Condens. Matter Mater. Phys., 2012, 86, 104102 CrossRef.
- M. T. Reetz, S. Höger and K. Harms, Angew. Chem., Int. Ed. Engl., 1994, 33, 181–183 CrossRef.
- A. O. F. Jones, M.-H. Lemee-Cailleau, D. M. S. Martins, G. J. McIntyre, I. D. H. Oswald, C. R. Pulham, C. K. Spanswick, L. H. Thomas and C. C. Wilson, Phys. Chem. Chem. Phys., 2012, 14, 13273–13283 RSC.
- P. Grobelny, A. Mukherjee and G. R. Desiraju, CrystEngComm, 2011, 13, 4358–4364 RSC.
- J. A. Cowan, J. A. K. Howard, G. J. McIntyre, S. M. F. Lo and I. D. Williams, Acta Crystallogr., Sect. B: Struct. Sci., 2005, 61, 724–730 CrossRef PubMed.
- J. A. Cowan, J. A. K. Howard, G. J. McIntyre, S. M.-F. Lo and I. D. Williams, Acta Crystallogr., Sect. B: Struct. Sci., 2003, 59, 794–801 CrossRef PubMed.
- L. H. Thomas, N. Blagden, M. J. Gutmann, A. A. Kallay, A. Parkin, C. C. Seaton and C. C. Wilson, Cryst. Growth Des., 2010, 10, 2770–2774 CrossRef CAS.
- A. Gregorovič, T. Apih, V. Žagar and J. Seliger, Phys. Chem. Chem. Phys., 2019, 21, 306–313 RSC.
- J. S. Stevens, S. J. Byard, C. C. Seaton, G. Sadiq, R. J. Davey and S. L. M. Schroeder, Phys. Chem. Chem. Phys., 2014, 16, 1150–1160 RSC.
- L. Rajput, M. Banik, J. R. Yarava, S. Joseph, M. K. Pandey, Y. Nishiyama and G. R. Desiraju, IUCrJ, 2017, 4, 466–475 CrossRef CAS PubMed.
- P. Cerreia Vioglio, M. R. Chierotti and R. Gobetto, Adv. Drug Delivery Rev., 2017, 117, 86–110 CrossRef CAS PubMed.
- R. K. Harris, P. Y. Ghi, R. B. Hammond, C.-Y. Ma and K. J. Roberts, Chem. Commun., 2003, 2834–2835 RSC.
- M. Siskos, M. Choudhary and I. Gerothanassis, Molecules, 2017, 22, 415 CrossRef PubMed.
- A. Parkin, S. M. Harte, A. E. Goeta and C. C. Wilson, New J. Chem., 2004, 28, 718–721 RSC.
- A. O. F. Jones, N. Blagden, G. J. McIntyre, A. Parkin, C. C. Seaton, L. H. Thomas and C. C. Wilson, Cryst. Growth Des., 2013, 13, 497–509 CrossRef CAS.
- M. Woińska, S. Grabowsky, P. M. Dominiak, K. Woźniak and D. Jayatilaka, Sci. Adv., 2016, 2, e1600192 CrossRef PubMed.
- S. C. Capelli, H.-B. Burgi, B. Dittrich, S. Grabowsky and D. Jayatilaka, IUCrJ, 2014, 1, 361–379 CrossRef CAS PubMed.
- M. Fugel, D. Jayatilaka, E. Hupf, J. Overgaard, V. R. Hathwar, P. Macchi, M. J. Turner, J. A. K. Howard, O. V. Dolomanov, H. Puschmann, B. B. Iversen, H.-B. Burgi and S. Grabowsky, IUCrJ, 2018, 5, 32–44 CrossRef CAS PubMed.
- L. K. Saunders, PhD Thesis, University of Bath, U.K., 2016 Search PubMed.
- D. Braga and F. Grepioni, Making Crystals by Design: Methods, Techniques and Applications, Wiley-VCH, Weinheim, 2007 Search PubMed.
- M. Schmidtmann, L. J. Farrugia, D. S. Middlemiss, M. J. Gutmann, G. J. McIntyre and C. C. Wilson, J. Phys. Chem. A, 2009, 113, 13985–13997 CrossRef CAS PubMed.
- J. D. Hepworth, D. R. Waring and M. J. Waring, Aromatic Chemistry, Royal Society of Chemistry, Cambridge, 2002 Search PubMed.
- A. J. L. Jesus and J. S. Redinha, J. Phys. Chem. A, 2011, 115, 14069–14077 CrossRef CAS PubMed.
- G. Winter, J. Appl. Crystallogr., 2010, 43, 186–190 CrossRef CAS.
- SADABS, Bruker AXS Inc., Madison, Wisconsin, USA Search PubMed.
- Apex2, Bruker AXS Inc., Madison, Wisconsin, U.S.A Search PubMed.
- G. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed.
- P. Muller, R. Herbst-Irmer, A. L. Spek, T. R. Schneider and M. R. Sawaya, Crystal Structure Refinement: A Crystallographer's Guide to SHELXL, Oxford University Press, Oxford, 2006 Search PubMed.
- L. Farrugia, J. Appl. Crystallogr., 2012, 45, 849–854 CrossRef CAS.
- XPREP, Bruker AXS Inc., Madison, Wisconsin, USA Search PubMed.
- S. Saha and G. R. Desiraju, J. Am. Chem. Soc., 2018, 140, 6361–6373 CrossRef CAS PubMed.
- A. O. F. Jones, C. K. Leech, G. J. McIntyre, C. C. Wilson and L. H. Thomas, CrystEngComm, 2014, 16, 8177–8184 RSC.
- L. K. Saunders, H. Nowell, P. R. Raithby and C. C. Wilson, CrystEngComm, 2016, 18, 5916–5929 RSC.
- A. Alhalaweh, S. George, D. Boström and S. P. Velaga, Cryst. Growth Des., 2010, 10, 4847–4855 CrossRef CAS.
- G. A. Jeffrey, An Introduction to Hydrogen Bonding, Oxford University Press, New York, 1997 Search PubMed.
- C. B. Aakeröy, M. E. Fasulo and J. Desper, Mol. Pharmaceutics, 2007, 4, 317–322 CrossRef PubMed.
- A. R. Eberlin, M. D. Eddleston and C. S. Frampton, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2013, 69, 1260–1266 CrossRef CAS PubMed.
- C. B. Aakeroy and K. R. Seddon, Chem. Soc. Rev., 1993, 22, 397–407 RSC.
- M. D. Ward, Struct. Bonding, 2009, 132, 1–24 CrossRef CAS.
- M. A. Hernandez and A. Rathinavelu, Basic Pharmacology Understanding Drug Actions and Reactions, CRC Press, Boca Raton, 2006 Search PubMed.
- P. M. Dewick, Essentials of Organic Chemistry: For Students of Pharmacy, Medicinal Chemistry and Biological Chemistry, John Wiley & Sons, Ltd, Chichester, 2006 Search PubMed.
- T. L. Lemke, Review of Organic Functional Groups: Introduction to Medicinal Organic Chemistry, Lippincott Williams & Wilkins, Baltimore, 2003 Search PubMed.
- S. Tothadi and G. R. Desiraju, Philos. Trans. R. Soc., A, 2012, 370, 2900–2915 CrossRef CAS PubMed.
- N. Shan, A. D. Bond and W. Jones, Cryst. Eng., 2002, 5, 9–24 CrossRef CAS.
- L. H. Thomas, A. O. F. Jones, A. A. Kallay, G. J. McIntyre and C. C. Wilson, Cryst. Growth Des., 2016, 16, 2112–2122 CrossRef CAS.
- S. J. Grabowski, Annu. Rep. Prog. Chem., Sect. C: Phys. Chem., 2006, 102, 131–165 RSC.
- G. Sheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 Search PubMed.
- L. H. Thomas, A. J. Florence and C. C. Wilson, New J. Chem., 2009, 33, 2486–2490 RSC.
- A. O. F. Jones, PhD Thesis, University of Bath, 2012 Search PubMed.
- G. Song, F.-Y. Bai, N. Xing, C. Chen, H. Shan and Y.-H. Xing, Chem. Res. Chin. Univ., 2013, 29, 408–413 CrossRef CAS.
- S. J. Ford, G. J. McIntyre, M. R. Johnson and I. R. Evans, CrystEngComm, 2013, 15, 7576–7582 RSC.
- A. J. Lough, P. S. Wheatley, G. Ferguson and C. Glidewell, Acta Crystallogr., Sect. B: Struct. Sci., 2000, 56, 261–272 CrossRef PubMed.
- M. E. Tuckerman, D. Marx, M. L. Klein and M. Parrinello, Science, 1997, 275, 817–820 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Crystallographic data; selected molecular geometry tables; selected hydrogen bond tables/plots; selected Fourier difference electron density maps. CCDC 1921985–1922038. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c9ce00925f |
| This journal is © The Royal Society of Chemistry 2019 |