
Synthesis of indoles and quinazolines via additive-controlled selective C–H activation/annulation of N-arylamidines and sulfoxonium ylides†
Ruizhi
Lai
,
Xiaohua
Wu
,
Songyang
Lv
,
Chen
Zhang
,
Maoyao
He
,
Yuncan
Chen
,
Qiantao
Wang
,
Li
Hai
 * and
Yong
Wu
*
* and
Yong
Wu
*
Key Laboratory of Drug-Targeting of Education Ministry and Department of Medicinal Chemistry, West China School of Pharmacy, Sichuan University, Chengdu 610041, China. E-mail: wyong@scu.edu.cn; smile@scu.edu.cn
First published on 6th March 2019
Abstract
Selective synthesis of indole and quinazoline products was achieved through a precise control of the C–H activation/annulation by changing the additives from NaOAc to CuF2/CsOAc. This strategy constructs indole and quinazoline scaffolds efficiently, and hence is of great interest in pharmaceutical, agricultural and chemical industries.
Transition-metal-catalyzed C–H activation has been extensively explored as it usually avoids the multistep preactivation of the starting materials providing an atom- and step-economical strategy for organic synthesis.1 For instance, C–H activation/annulation, which has been one of the hottest topics in recent years,2 does not only specifically functionalize the inert C–H bonds, but also forms a variety of cyclic compounds by coupling and cyclization with the introduced functional groups.
Indoles and quinazolines are two well-known classes of nitrogen-containing heterocyclic compounds that pose a broad-spectrum of biological activities and have been widely used in pharmaceutical, agricultural and chemical industries (Fig. 1).3 Therefore, a more economical and environment-friendly synthetic strategy would be of great interest in the field. Among the published works, the formation of indoles was achieved mainly through the C–H activation of aniline derivatives with coupling reagents, such as alkene, alkyne and diazo (Scheme 1a).4 On the other hand, quinazolines were mainly afforded via the C–H activation of benzimidates with amino reagents (dioxazolones and alkyl azides) or by the reactions of N-arylamidines with the “one-carbon coupling reagents” (C1 unit) like isonitrile and alkyne derivatives (Scheme 1b).5
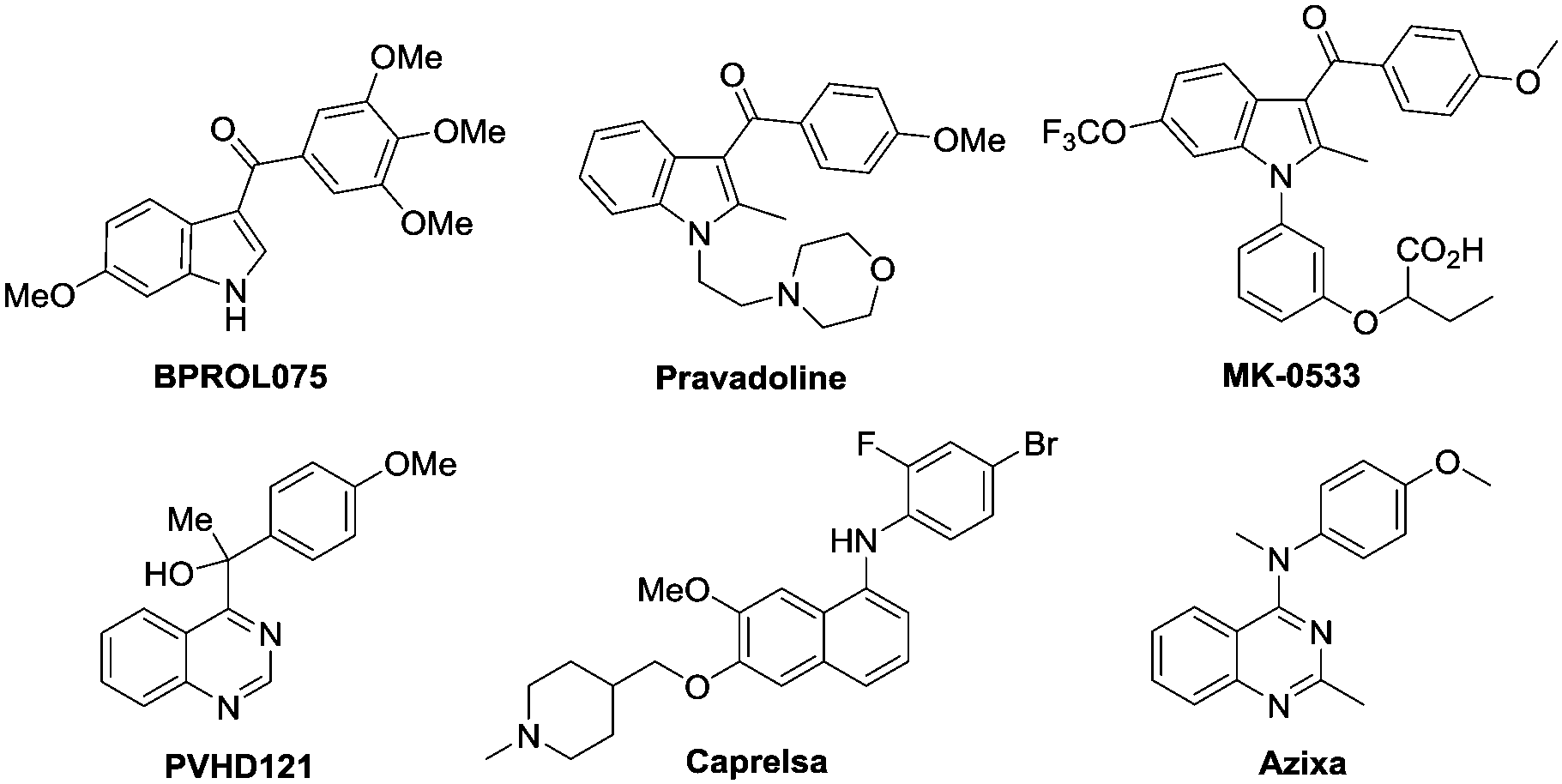 | ||
| Fig. 1 Selected examples of bioactive indoles and quinazolines. | ||
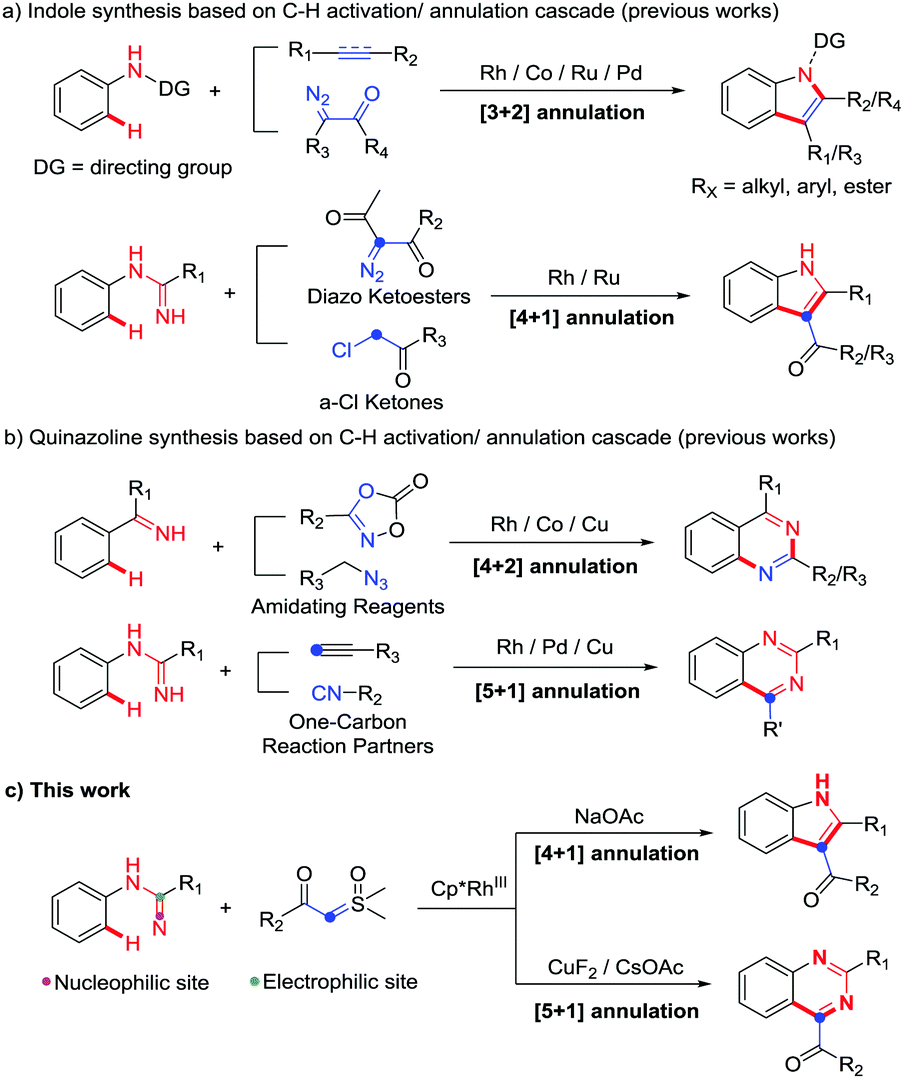 | ||
| Scheme 1 C–H activation/annulation for the synthesis of indoles and quinazolines. | ||
Although the synthesis of indoles and quinazolines has been widely studied, most of the methods still have some drawbacks, such as the use of toxic and dangerous reagents (isonitrile, diazo, azide, etc.) and the unsatisfactory yields. Recently, sulfoxonium ylides have been widely used as a convenient and safe carbene precursor reagent in transition-metal-catalyzed C–H activation/annulation.6 Many groups independently reported C–H activation/annulation cascade reactions of different directing groups (DGs) with sulfoxonium ylides as the C2 unit to obtain lactones, lactams, isoquinolines, azolopyrimidines, naphthols, etc.6a–i In addition, Kim's group recently used azobenzene with sulfoxonium ylide as the C1 unit to synthesize indazoles.6j Collectively, these results revealed that sulfoxonium ylide played an important role in C–H activation/annulation. As our group has been interested in C–H activation in recent years,7 we were wondering whether it is possible to use N-arylamidines and sulfoxonium ylides to synthesize N-heterocycles through C–H activation/annulation. Herein, to answer this question, we report our most recent work on additive-controlled selective synthesis of indoles and quinazolines by using N-arylamidines and sulfoxonium ylides as the starting materials (Scheme 1c).
Initially, we selected N-phenylacetimidamide 1a as a substrate to react with dimethyloxosulfonium benzoylmethylide 2a using [Cp*RhCl2]2 (5 mol%)/AgSbF6 (20 mol%) as the catalyst. To our delight, (2-methyl-1H-indol-3-yl)(phenyl)methanone 3a and 2-methyl-4-benzoylquinazoline 5a were obtained in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio with 62% total yield (Table 1, entry 1). Then, the relationship between additives and reaction results was explored (entries 2–11). It was shown that NaOAc as a base was obviously favored to generate the indole product 3a (entry 3), while Cu salt, especially CuF2, afforded quinazoline product 5a mostly (entry 11). The effect of the solvent was also examined, and it turned out that DCE was the best (see the ESI†). Increasing the reaction temperature would cause serious side-reactions of ylide hence lowering the yield (see the ESI†). Increasing the amount of 2a only improved the yield of 3a (entries 12 and 13), and the formation of 5a was favored by the oxygen environment (entry 14). In addition, as we found that CsOAc had a mild preference on 5a (entry 5), we combined it with CuF2 to see if it can further boost the yield. Fortunately, 5a was obtained in 74% yield (entry 15).
:1 ratio with 62% total yield (Table 1, entry 1). Then, the relationship between additives and reaction results was explored (entries 2–11). It was shown that NaOAc as a base was obviously favored to generate the indole product 3a (entry 3), while Cu salt, especially CuF2, afforded quinazoline product 5a mostly (entry 11). The effect of the solvent was also examined, and it turned out that DCE was the best (see the ESI†). Increasing the reaction temperature would cause serious side-reactions of ylide hence lowering the yield (see the ESI†). Increasing the amount of 2a only improved the yield of 3a (entries 12 and 13), and the formation of 5a was favored by the oxygen environment (entry 14). In addition, as we found that CsOAc had a mild preference on 5a (entry 5), we combined it with CuF2 to see if it can further boost the yield. Fortunately, 5a was obtained in 74% yield (entry 15).
|
|
|||
|---|---|---|---|
| Entry | Additive | Yieldb (%) | |
| 3a | 5a | ||
| a Unless otherwise noted, all the reactions were carried out using N-phenylacetimidamide 1a (0.20 mmol) and dimethyloxosulfonium benzoylmethylide 2a (0.40 mmol) in the presence of [Cp*RhCl2]2 (0.01 mmol), AgSbF6 (0.04 mmol), additive (0.40 mmol) and DCE (1.0 ml) in a Schlenk tube, and the mixture was stirred at 80 °C for 24 h under Ar. b Isolated yield by chromatography on a silica gel. c Dimethyloxosulfonium benzoylmethylide 2a = 0.60 mmol. d O2 atmosphere. e CsOAc = 0.20 mmol. | |||
| 1 | — | 33 | 29 |
| 2 | HOAc | 28 | 31 |
| 3 | NaOAc | 62 | 10 |
| 4 | AgOAc | 58 | 10 |
| 5 | CsOAc | 21 | 43 |
| 6 | CsCO3 | 27 | 35 |
| 7 | Cu(OAc)2 | <5 | 51 |
| 8 | Cu(TFA)2 | <5 | 42 |
| 9 | Cu(OTf)2 | <5 | 45 |
| 10 | CuCl2 | <5 | 31 |
| 11 | CuF2 | <5 | 54 |
| 12 | NaOAc | 78 | <5 |
| 13c | CuF2 | <5 | 52 |
| 14d | CuF2 | <5 | 68 |
| 15 , | CuF 2 /CsOAc | <5 | 74 |
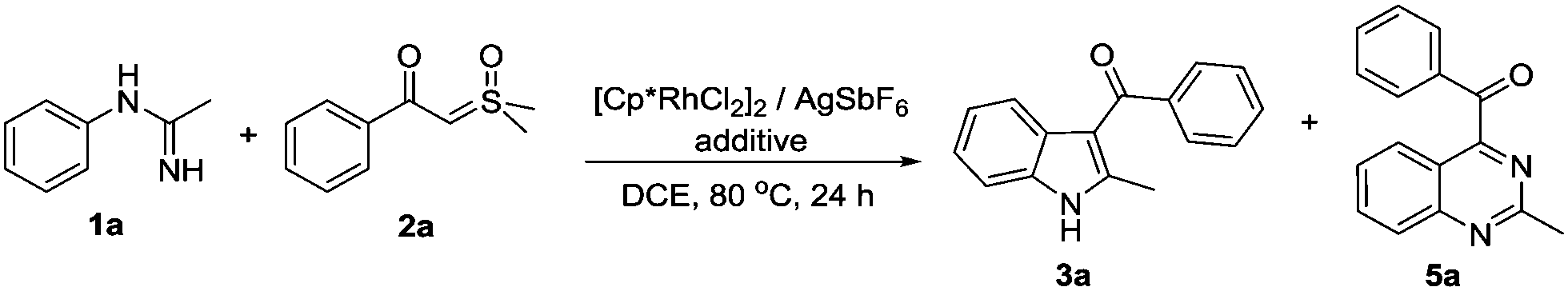
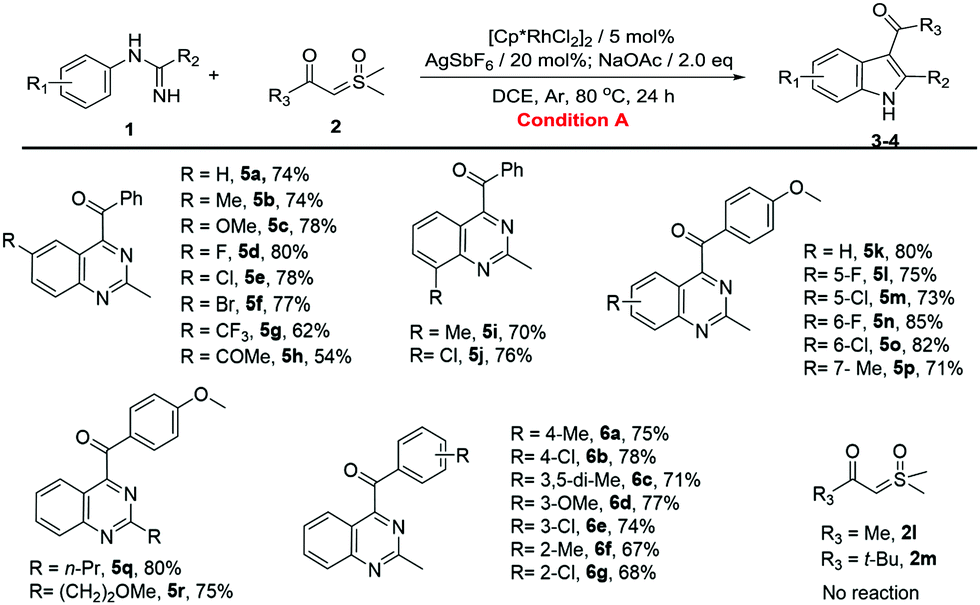
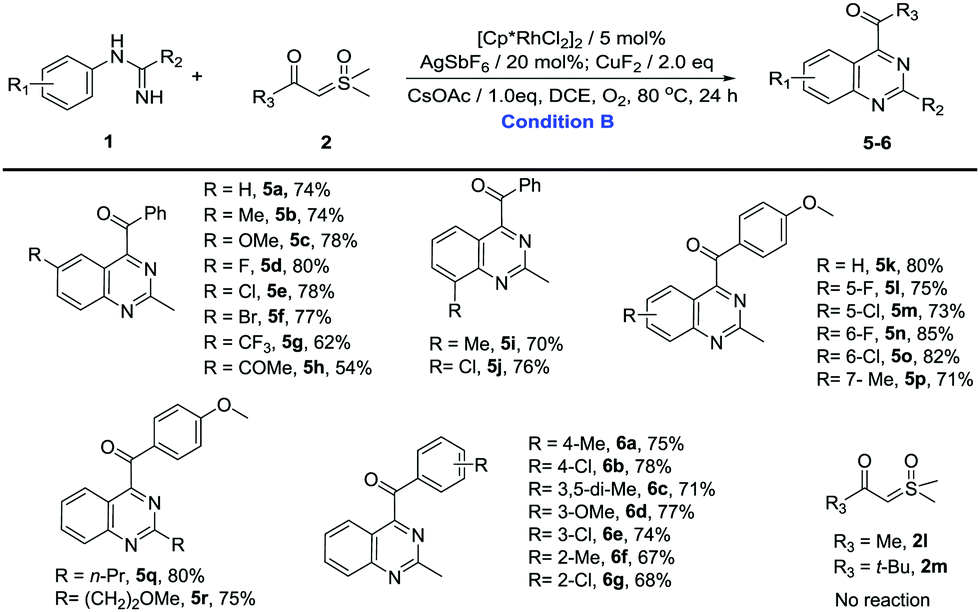
With the optimized reaction conditions in hand, the C–H activation of imidamides was examined. Firstly, the substrate generality of indole products was explored, and the results are shown in Table 2. Unsubstituted N-phenylacetimidamide afforded 3a with 78% isolated yield. The electron-donating, electron-withdrawing and halogen groups introduced into different positions of the benzene ring were fully tolerated (3b–3q), giving good to excellent yields (60–85%) for the indole products. Notably, when isopropyl, isobutyl, methoxyethyl and benzyl groups were introduced into C-alkyl imidamides, the reaction also successfully afforded desired products (3r–3u). Given the above results, it was found that the electron-donating groups, such as Me and OMe, and halogens on the phenyl were more favorable for this transformation than the electron-withdrawing groups including CF3, nitro, acetyl and ester (3g–3j). In addition, the different substituting positions of the benzene ring affected the yields as well. The para-substitution was slightly better than the meta-, and the meta- was better than the ortho-. It was also found that when the imidamide was meta-monosubstituted by Me, both 3k and 3′k were produced in a ratio of 5:1. However, when Me was changed to F or Cl, only one regioisomer could be obtained (3l and 3m). The difference in regioisomer formation may be the result of a combination of the steric effects and the electrical effects. Then, the change of sulfoxonium ylides was also investigated. When Me, MeO and halogen groups were attached to different positions on the benzene ring of sulfoxonium ylides, the reactions were fully tolerated (4a–4i), furnishing the desired products in good to excellent yields (66–82%). Notably, sulfoxonium ylides with MeO ortho-bisubstitution on the benzene ring had a yield of 66% only (4i). This may be due to the greatly increasing steric hindrance. When the benzene ring of ylide was changed to a naphthalene ring or a 1,3-benzodioxole ring, the reaction still proceeded smoothly and gave good yields (4j and 4k). Once the benzene ring of the ylide was changed to Me (2l) or t-Bu (2m), no product was formed. It seemed that the aromaticity of the sulfoxonium ylide could be necessary in this reaction.
| a Reaction conditions: N-arylethanimidamides 1 (0.20 mmol), dimethyloxosulfonium benzoylmethylide 2a (0.60 mmol), [Cp*RhCl2]2 (0.01 mmol), AgSbF6 (0.04 mmol), NaOAc (0.40 mmol) and DCE (1.0 ml) in a Schlenk tube. The mixture was stirred at 80 °C for 24 h under Ar. Then, without any post processing, the reaction mixture was purified by column chromatography on a silica gel (eluent: PE/DCM = 1/1) to afford the desired product. |
|---|
|
Then, we turned our attention to the synthesis of quinazolines (Table 3). Unsubstituted N-phenylacetimidamide gave 74% isolated yield of 5a. The electron-donating, electron-withdrawing and halogen groups introduced into different positions of the benzene ring were fully tolerated (5a–5p), and the extension of the terminal carbon chain seemed to have no effect on the reaction (5q and 5r), giving moderate to excellent yields (54–85%). Notably, when the benzene ring was meta-monosubstituted with Me, C–H activation took place in the C(6) site producing 5p. However, when Me was changed to F or Cl, C–H activation took place in the C(2) site producing 5l or 5m. The results above may be the interaction of the steric effects and the electrical effects. Then, sulfoxonium ylides were investigated to further explore the substrate generality of quinazoline products. When Me, MeO and halogen groups were attached to different positions on the benzene ring of sulfoxonium ylides, the reactions were fully tolerated (6a–6g), furnishing the desired products in good yields (67–78%). Similarly, once the benzene ring of the ylide was changed to Me (2l) or t-Bu (2m), no product was found.
| a Reaction conditions: N-arylethanimidamides 1 (0.20 mmol), dimethyloxosulfonium benzoylmethylide 2a or 2c (0.40 mmol), [Cp*RhCl2]2 (0.01 mmol), AgSbF6 (0.04 mmol), CuF2 (0.40 mmol), CsOAc (0.20 mmol) and DCE (1.0 ml) in a Schlenk tube. The mixture was stirred at 80 °C for 24 h under O2. Then, without any post processing, the reaction mixture was purified by column chromatography on a silica gel (eluent: PE/acetone = 50/1) to afford the desired product. |
|---|
|
To gain mechanistic insight into the reaction, a series of experiments have been conducted (see the ESI†).8 Based on the above results and previous related studies,6g,9 a possible mechanism for this protocol is shown in Scheme 2. Cyclometalation of N-phenylacetimidamide 1a and the active Rh(III) catalyst gives a rhodacyclic intermediate A. Coordination of dimethyloxosulfonium benzoylmethylide 2a generates a Rh(III) alkyl species B, and the subsequent α-elimination of DMSO from B affords a reactive rhodium α-oxo carbene species C. Following the formation of C, two pathways may be possible. In pathway A, C is proposed to undergo migratory insertion of the Rh–C bond to generate a seven-membered rhodacyclic intermediate D. Intermediate E is then formed from unstable intermediate D by Rh–C(alkyl) migratory insertion into the C–N bond. Eventually, the final product 3a is released from E by elimination of the active Rh(III) catalyst and one molecule of ammonia from E upon protonolysis and intramolecular protonolysis. In pathway B, the seven-membered rhodacyclic intermediate D is still generated. Then, the reductive elimination from intermediate D forms partially reduced quinazoline F. Subsequently, the oxidation of F affords the quinazoline product 5a. On the other hand, the resulting Cp*Rh(I) can be reoxidized to the starting Rh(III) species by the action of Cu(II) and/or O2 to complete the catalytic cycle.
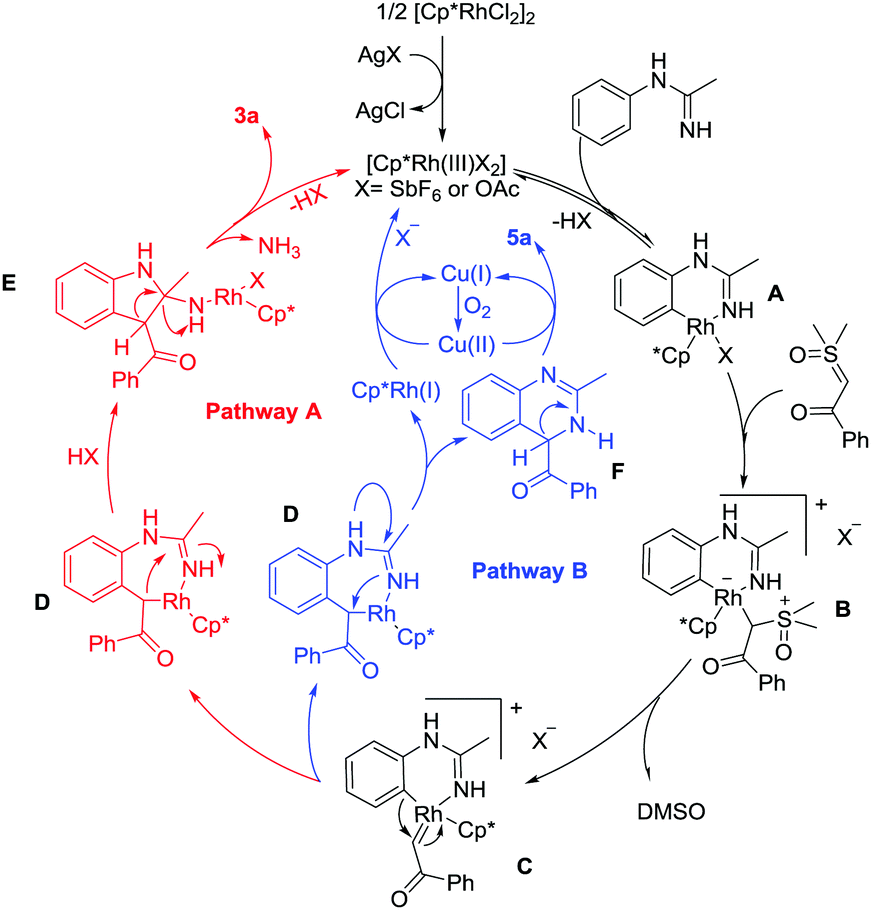 | ||
| Scheme 2 Possible pathways of the C–H activation/cyclization. | ||
It is noteworthy that this protocol really has many practical applications. For example, Pravadoline, a phase II drug, was recognized as a cannabinoid CB1 receptor agonist and has a strong analgesic effect (IC50 = 4.9 μM).10N-methylacetimidamide 1a and dimethyloxosulfonium 4-methoxybenzoylmethylide 2c were used to synthesize Pravadoline 9 in a shorter route yet with a higher yield, i.e. a total yield of 70%, in two steps (Scheme 3a). On the other hand, indole and quinazoline scaffolds have a broad-spectrum of biological activities. Based on a previous work,11 we synthesized several indole compounds using our protocol (Scheme 3b), and we also choose several quinazoline compounds we synthesized (Scheme 3c). The anti-tumor activity of these indole and quinazoline compounds was evaluated. 3-Aroylindoles (11a–11d) all had excellent anti-tumor activity, while quinazoline products (5n, 5o, 5q, 5r) put up a poor show. In comparison with BPR-0L-075, compounds 11b, 11c and 11d displayed similar or greater growth inhibitory activities (see the ESI†). It is worth pinpointing that our highly efficient and scalable methodology for the synthesis of 3-aroylindoles and 4-aroylquinazolines will be extremely useful in discovering the novel anti-tumor compounds, and it may provide new ideas for drug design and synthesis.
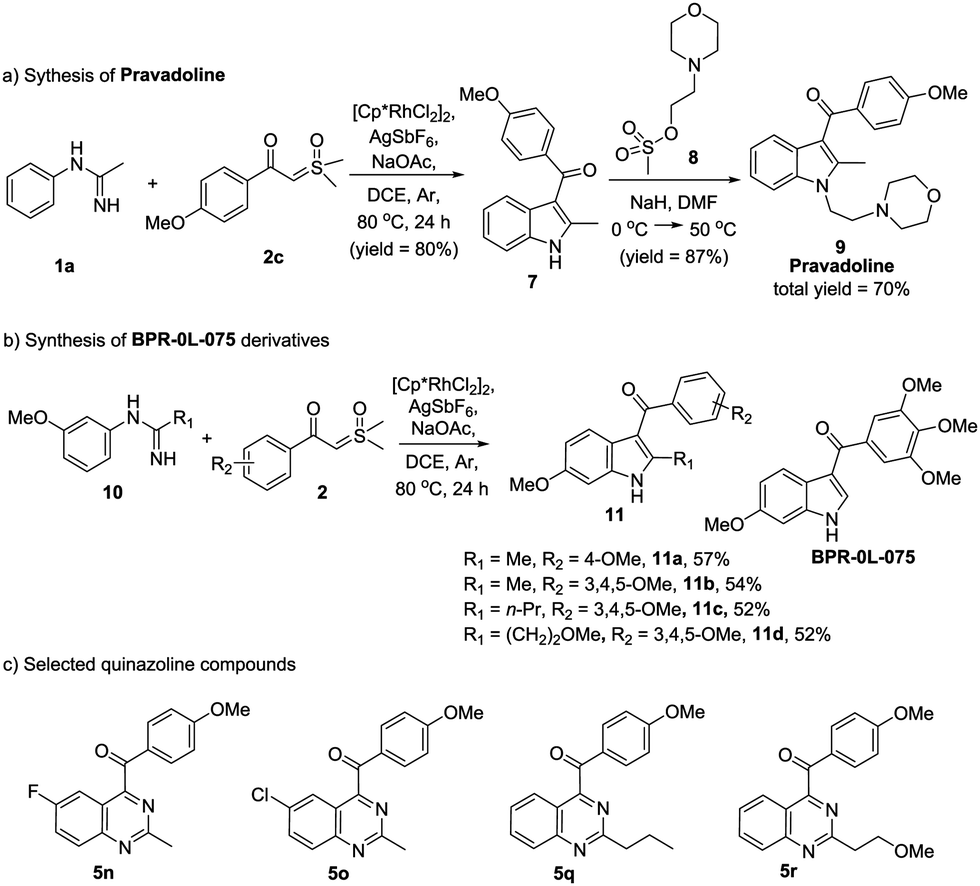 | ||
| Scheme 3 The practical applications of this protocol. | ||
In summary, we report the first example, to the best of our knowledge, of an additive-controlled selective C–H activation/annulation reaction of N-arylamidines and sulfoxonium ylides to selectively synthesize indoles and quinazolines. In this process, the additives were shown to play a key role in selectively controlling the [4+1] and [5+1] annulation. With the additive NaOAc, the reaction predominantly gave the indoles because of the [4+1] annulation. Changing the additives to CuF2/CsOAc, the preference of the annulation was switched to [5+1], hence selectively affording the quinazoline products. Furthermore, this simple, rapid and efficient strategy may provide a new tool for the synthesis of N-heterocycles, hence facilitating chemical synthesis and drug design.
Financial support from the National Natural Science Foundation of China (NSFC; grant numbers 81573286, 81773577 and 81602954) and the Fundamental Research Funds for the Central Universities (YJ201561) is gratefully acknowledged.
Conflicts of interest
There are no conflicts to declare.Notes and references
- Selected reviews for C–H activation: (a) D. A. Colby, R. G. Bergman and J. A. Ellman, Chem. Rev., 2010, 110, 624 CrossRef CAS PubMed; (b) N. Kuhl, N. Schröder and F. Glorius, Adv. Synth. Catal., 2014, 356, 1443 CrossRef CAS; (c) X.-S. Zhang, K. Chen and Z.-J. Shi, Chem. Sci., 2014, 5, 2146 RSC; (d) P. L. Arnold, M. W. McMullon, J. Rieb and F. E. Kühn, Angew. Chem., Int. Ed., 2015, 54, 82 CrossRef CAS PubMed; (e) F. Wang, S. Yu and X. Li, Chem. Soc. Rev., 2016, 45, 6462 RSC.
- Selected reviews for C–H activation/annulation: (a) T. Satoh and M. Miura, Chem. – Eur. J., 2010, 16, 11212 CrossRef CAS PubMed; (b) G. Song, F. Wang and X. Li, Chem. Soc. Rev., 2012, 41, 3651 RSC.
- (a) M. Somei and F. Yamada, Nat. Prod. Rep., 2004, 21, 278 RSC; (b) A. J. Kochanowska-Karamyan and M. T. Hamann, Chem. Rev., 2010, 110, 4489 CrossRef CAS PubMed; (c) G. Marzaro, A. Guiotto and A. Chilin, Expert Opin. Ther. Pat., 2012, 22, 223 CrossRef CAS PubMed.
- Selected examples: (a) J. Chen, G. Song, C. Pan and X. Li, Org. Lett., 2010, 12, 5426 CrossRef CAS PubMed; (b) M. P. Huestis, L. R. Chan, D. R. Stuart and K. Fagnou, Angew. Chem., Int. Ed., 2011, 50, 1338 CrossRef CAS PubMed; (c) C. Wang, H. Sun, Y. Fang and Y. Huang, Angew. Chem., Int. Ed., 2013, 52, 5795 CrossRef CAS PubMed; (d) Y. Liang, K. Yu, B. Li, S. Xu, H. Song and B. Wang, Chem. Commun., 2014, 50, 6130 RSC; (e) D. Zhao, S. Vasquez-Cespedes and F. Glorius, Angew. Chem., Int. Ed., 2015, 54, 1657 CrossRef CAS PubMed; (f) Z. Qi, S. Yu and X. Li, Org. Lett., 2016, 18, 700 CrossRef CAS PubMed; (g) K. S. Halskov, H. S. Roth and J. A. Ellman, Angew. Chem., Int. Ed., 2017, 56, 9183 CrossRef CAS PubMed; (h) F. Xu, W.-F. Kang, Y. Wang, C.-S. Liu, J.-Y. Tian, R.-R. Zhao and M. Du, Org. Lett., 2018, 20, 3245 CrossRef CAS PubMed; (i) J. Zhou, J. Li, Y. Li, C. Wu, G. He, Q. Yang, Y. Zhou and H. Liu, Org. Lett., 2018, 20, 7645 CrossRef CAS PubMed.
- Selected examples: (a) Y. Wang, H. Wang, J. Peng and Q. Zhu, Org. Lett., 2011, 13, 4604 CrossRef CAS PubMed; (b) H. Wang, M. M. Lorion and L. Ackermann, Angew. Chem., Int. Ed., 2016, 55, 10386 CrossRef CAS PubMed; (c) F. Wang, H. Wang, Q. Wang, S. Yu and X. Li, Org. Lett., 2016, 18, 1306 CrossRef CAS PubMed; (d) X. Wang and N. Jiao, Org. Lett., 2016, 18, 2150 CrossRef CAS PubMed; (e) F. Xu, W.-F. Kang, Y. Wang, C.-S. Liu, J.-Y. Tian, R.-R. Zhao and M. Du, Org. Lett., 2018, 20, 3245 CrossRef CAS PubMed.
- (a) A. M. Phelps, V. S. Chan, J. G. Napolitano, S. W. Krabbe, J. M. Schomaker and S. Shekhar, J. Org. Chem., 2016, 81, 4158 CrossRef CAS PubMed; (b) J. Vaitla, A. Bayer and K. H. Hopmann, Angew. Chem., Int. Ed., 2017, 56, 4277 CrossRef CAS PubMed; (c) Y. Xu, G. Zheng, X. Yang and X. Li, Chem. Commun., 2018, 54, 670 RSC; (d) X. Wu, H. Xiong, S. Sun and J. Cheng, Org. Lett., 2018, 20, 1396 CrossRef CAS PubMed; (e) K. S. Halskov, M. R. Witten, G. L. Hoang, B. Q. Mercado and J. A. Ellman, Org. Lett., 2018, 20, 2464 CrossRef CAS PubMed; (f) G. L. Hoang and J. A. Ellman, Tetrahedron, 2018, 74, 3318 CrossRef CAS PubMed; (g) G. Zheng, M. Tian, Y. Xu, X. Chen and X. Li, Org. Chem. Front., 2018, 5, 998 RSC; (h) C. Zhou, F. Fang, Y. Cheng, Y. Li, H. Liu and Y. Zhou, Adv. Synth. Catal., 2018, 360, 2546 CrossRef CAS; (i) C.-F. Liu, M. Liu and L. Dong, J. Org. Chem., 2019, 84, 409 CrossRef CAS PubMed; (j) H. Oh, S. Han, A. K. Pandey, S. H. Han, N. K. Mishra, S. Kim, R. Chun, H. S. Kim, J. Park and I. S. Kim, J. Org. Chem., 2018, 83, 4070 CrossRef CAS PubMed.
- C. Zhang, X.-M. Chen, Y. Luo, J.-L. Li, M. Chen, L. Hai and Y. Wu, ACS Sustainable Chem. Eng., 2018, 6, 13473 CrossRef CAS.
- (a) E. M. Simmons and J. F. Hartwig, Angew. Chem., Int. Ed., 2012, 51, 3066 CrossRef CAS PubMed; (b) M. Tian, B. Liu, J. Sun and X. Li, Org. Lett., 2018, 20, 4946 CrossRef CAS PubMed.
- (a) M. Brasse, J. Cámpora, J. A. Ellman and R. G. Bergman, J. Am. Chem. Soc., 2013, 135, 6427 CrossRef CAS PubMed; (b) X. Zhou, Z. Qi, S. Yu, L. Kong, Y. Li, W. Tian and X. Li, Adv. Synth. Catal., 2017, 359, 1620 CrossRef CAS; (c) Y. Xu, G. Zheng, X. Yang and X. Li, Chem. Commun., 2018, 54, 670 RSC; (d) B. Alcaide, P. Almendros, I. Fernández, T. M. D. Campo and T. Naranjo, Adv. Synth. Catal., 2013, 355, 2681 CrossRef CAS; (e) J. Schießl, J. Schulmeister, A. Doppiu, E. Wörner, M. Rudolph, R. Karch and A. S. K. Hashmi, Adv. Synth. Catal., 2018, 360, 3949 CrossRef.
- J. M. Frost, M. J. Dart, K. R. Tietje, T. R. Garrison, G. K. Grayson, A. V. Daza, O. F. El-Kouhen, B. B. Yao, G. C. Hsieh, M. Pai, C. Z. Zhu, P. Chandran and M. D. Meyer, J. Med. Chem., 2010, 53, 295 CrossRef CAS PubMed.
- (a) J.-P. Liou, Y.-L. Chang, F.-M. Kuo, C.-W. Chang, H.-Y. Tseng, C.-C. Wang, Y.-N. Yang, J.-Y. Chang, S.-J. Lee and H.-P. Hsieh, J. Med. Chem., 2004, 47, 4247 CrossRef CAS PubMed; (b) M. A. Soussi, O. Provot, G. Bernadat, J. Bignon, D. Desravines, J. Dubois, J.-D. Brion, S. Messaoudi and M. Alami, ChemMedChem, 2015, 10, 1392 CrossRef CAS PubMed; (c) K. Kuroiwa, H. Ishii, K. Matsuno, A. Asai and Y. Suzuki, ACS Med. Chem. Lett., 2015, 6, 287 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c9cc01146c |
| This journal is © The Royal Society of Chemistry 2019 |