
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDirected nickel-catalyzed 1,2-dialkylation of alkenyl carbonyl compounds†
Joseph
Derosa
,
Vincent A.
van der Puyl
,
Van T.
Tran
,
Mingyu
Liu
 and
Keary M.
Engle
*
and
Keary M.
Engle
*
Department of Chemistry, The Scripps Research Institute, 10550 North Torrey Pines Road, La Jolla, California 92037, USA. E-mail: keary@scripps.edu
First published on 16th May 2018
Abstract
A nickel-catalyzed conjunctive cross-coupling of non-conjugated alkenes, alkyl halides, and alkylzinc reagents is reported. Regioselectivity is controlled by chelation of a removable bidentate 8-aminoquinoline directing group. Under optimized conditions, a wide range of 1,2-dialkylated products can be accessed in moderate to excellent yields. To the best of our knowledge, this report represents the first example of three-component 1,2-dialkylation of non-conjugated alkenes to introduce differentiated alkyl fragments.
Introduction
Synthetic methods that enable expeditious access to carbogenic skeletons with high sp3 character are a vital component of the synthetic toolkit, given the prevalence of such structures in natural products and their importance in modern drug discovery.1 In the past several years, cross-coupling reactions involving alkenes as conjunctive reagents have emerged as a powerful platform for forging two C(sp3) stereocenters in a single stroke.2 There are several challenging aspects to developing such reactions, such as (1) achieving appropriate reaction rates of the constituent elementary steps to favor the desired pathway among other competitive side reactions, and (2) controlling the regioselectivity, stereoselectivity, and 1,1- versus 1,2-selectivity. To address these issues, several approaches have been pursued, including intramolecular tethering of the alkene to one reaction partner,3 use of a conjugated alkene (e.g., styrenes, acrylates, and allenes),4 metalate rearrangements,5 and chelation control.6 These developments notwithstanding, existing methods typically require at least one C(sp2) reaction partner. A generally applicable, three-component “all-alkyl” conjunctive cross-coupling, capable of delivering differentiated alkyl fragments across a non-conjugated alkene, would be synthetically enabling but has not been developed to date.7 This is due not only to the aforementioned challenges with conjunctive cross-coupling but also to the inherent difficulties associated with C(sp3)–C(sp3) cross-coupling processes.8,9 In the present study we describe our efforts to bridge this gap through the development of a nickel-catalyzed three-component conjunctive cross-coupling of an alkyl organometallic nucleophile, an alkyl halide electrophile, and a non-conjugated alkene using a chelation control strategy.Several literature precedents of alkene 1,2-dialkylation spoke to the viability of such a reaction but also illustrated challenges to be anticipated (Scheme 1A).
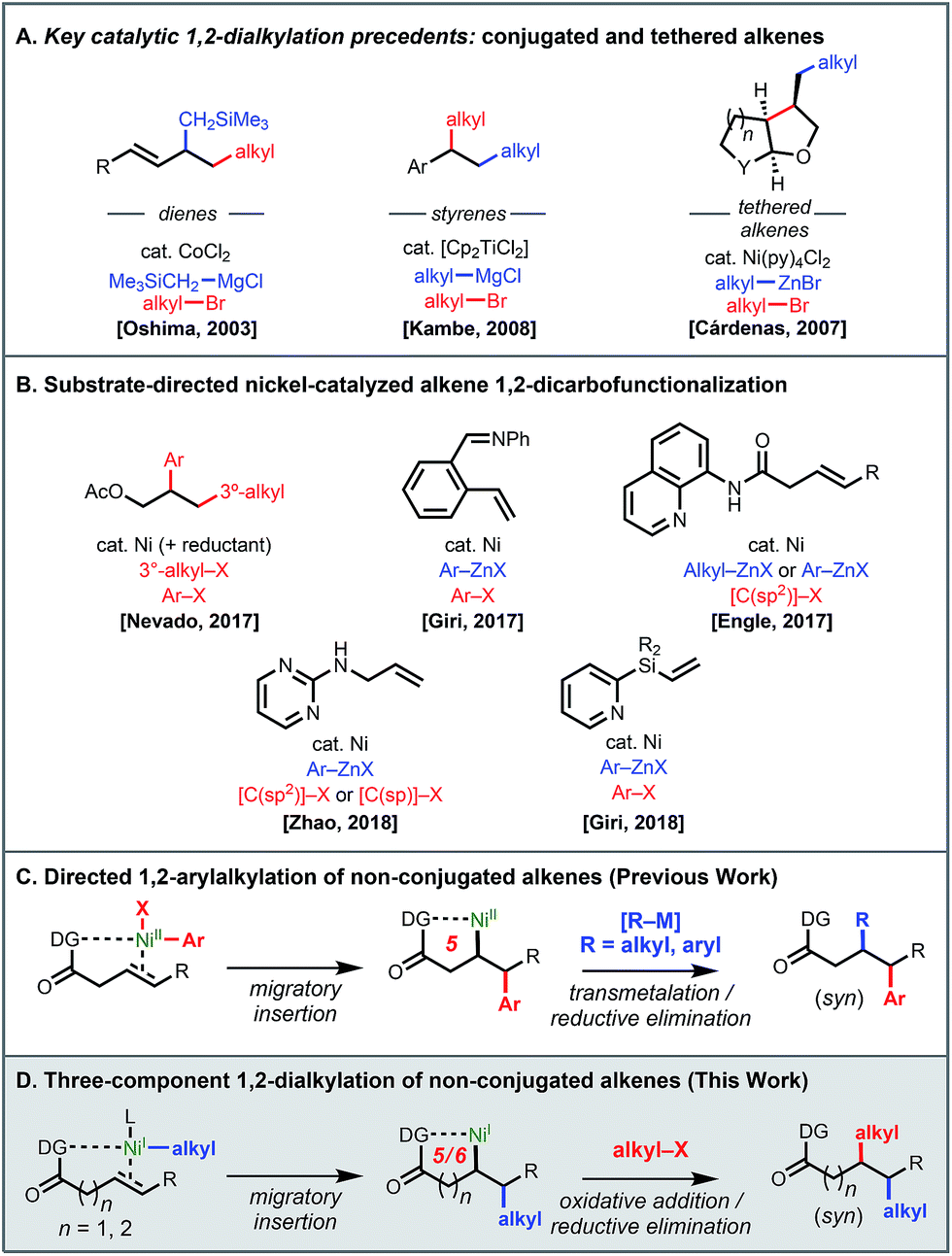 | ||
| Scheme 1 Three-component 1,2-dicarbofunctionalization with C(sp3) reaction partners: precedents and current work. | ||
Particularly, key precedents by Oshima and Kambe showcased the possibility of introducing two differentiable alkyl fragments across conjugated alkenes with alkyl bromides and alkyl Grignard reagents, presumably enabled by the stabilization of resulting π-allyl and π-benzyl nickel species after initial insertion.4e,f Early work by Cárdenas and coworkers demonstrated nickel-catalyzed intramolecular dialkylation.3 In the past year, there have been several reports on three-component 1,2-dicarbofunctionalization using substrate directivity strategies (Scheme 1B).6,10 In terms of examples with alkyl coupling partners, Nevado and coworkers demonstrated that allyl acetate and related alkenes could react with tertiary alkyl iodides and aryl iodides in a nickel-catalyzed reductive conjunctive cross-coupling.6a Our group developed a 1,2-arylalkylation reaction using aryl iodides and organozinc reagents under nickel catalysis.6c This reaction employed a removable bidentate directing group11 to control the regiochemical outcome through putative nickelacycle stabilization and to prevent otherwise rapid β-hydride (β-H) elimination (Scheme 1C).
The efficiency of our 1,2-arylalkylation reaction prompted us to consider whether it would be possible to use alkyl halides and alkyl organometallic reagents with non-conjugated terminal and internal alkenes via chelation control (Scheme 1D). At the outset we recognized that successful realization of the proposed transformation would necessitate overcoming a series of obstacles, most notably the fact that alkylmetal species capable of undergoing β-H elimination are present in every step of the catalytic cycle. Additionally, with alkyl halide electrophiles it is known that single-electron oxidative addition pathways are sometimes preferred, particularly with first-row metals,8,9 which meant that there was potential for mechanistic divergence compared to our earlier work (NiI/NiIIIvs. Ni0/NiII).
Results and discussion
To reduce this idea to practice, we began by exposing an alkenyl carbonyl compound 1a to reaction conditions using iodoethane as the electrophile and dimethyl zinc as the nucleophile under nickel catalysis (Table 1). To suppress β-H elimination, we hypothesized that a strong, bidentate directing group would be beneficial. Given our group's previous success in employing Daugulis's 8-aminoquinoline (AQ) directing group10 in 1,2-difunctionalization reactions,6c,11 we focused our efforts on this directing group.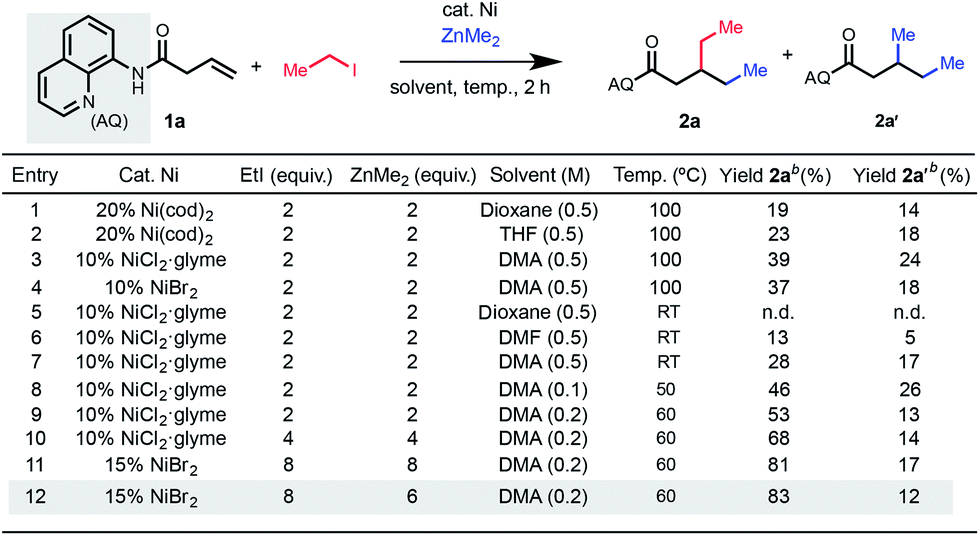
In a series of initial experiments, we were delighted to observe formation of the desired 1,2-dialkylated product 2a in 19% yield along with 14% of dimethylated byproduct 2a′ based on conditions from our previous work using 20 mol% Ni(cod)2.6c Interestingly, the product was determined to have electrophile incorporation at the β-position, opposite to the results in our previously reported 1,2-arylalkylation methodology, suggestive of a different underlying mechanism that is induced by use of an alkyl halide electrophile (vide infra). We then moved on to examine nickel(II) precatalysts, which are inexpensive and bench-stable alternatives to air-sensitive nickel(0) precatalysts. With NiCl2·glyme as catalyst, the procedure could reliably be reproduced without the use of an inert atmosphere glovebox, delivering the desired 1,2-dialkylation product in 39% yield (entry 3). We were also encouraged by the efficaciousness of a range of nickel(II) sources, including NiBr2 which performed comparably to NiCl2·glyme (entry 4). The reaction only proceeded in appreciable yield when polar aprotic solvents were used, so we opted to use DMA as the solvent for subsequent optimization. Through systematic screening, we were pleased to observe that the yield increased when the reaction was performed at concentrations lower than 0.5 M. With this change, the temperature could be decreased to 60 °C, and under these conditions, the desired product was formed in 53% yield. Next, we reasoned that the nucleophile and electrophile were being consumed through competitive two-component cross-coupling or homodimerization. Indeed, we observed a dramatic increase in yield proportional to the increase in equivalents of both electrophile and nucleophile that were used (entry 8). The reaction provided 68% yield using 4 equivalents of each (entry 10), and the yield could be further improved to 83% using 6 equivalents of nucleophile and 8 equivalents of electrophile (entry 12).12 Because the reaction partners in this study were generally inexpensive and commercially available, we performed most of the ensuing experiments using the conditions shown in entry 12, though we also carried out several examples under the conditions shown in entry 10 for comparison.
Having identified optimal conditions, we first explored potential alkyl electrophiles and nucleophiles that were compatible in this 1,2-dialkylation reaction (Table 2). It is important to note that competitive 1,2-dialkylation with the nucleophile was also observed in 5–20% by crude 1H NMR in most cases. To our satisfaction, a wide range of primary alkyl electrophiles and nucleophiles were tolerated. By simply changing the identity of the nucleophilic and electrophilic components (i.e., iodoethane/dimethylzinc to iodomethane/diethylzinc), both regioisomers could be synthesized in high yields (2a and 2h). In general, secondary and tertiary alkyl reaction partners did not perform well under the optimized conditions, likely due to steric constraints of the resulting chelation-stabilized nickelacycle and/or rapid β-hydride elimination for partners with 4 or more accessible β-hydrogen atoms. Cyclobutyl coupling partners were an exception, however, and the corresponding difunctionalized products 2f and 2n could be obtained in high yields. Several synthetically useful functional groups were tolerated in this reaction, allowing for the installation of acetals, esters, and free alcohols in moderate to good yields (2d, 2e, 2l, and 2m). Heterocycle-containing reaction partners were generally incompatible, which we hypothesize is due to catalyst coordination. Interestingly, 1,2-arylalkylation products could be accessed using diphenyl zinc (2k). Terminal alkene-tethered electrophiles were compatible but delivered the desired product in modest yield (2g).
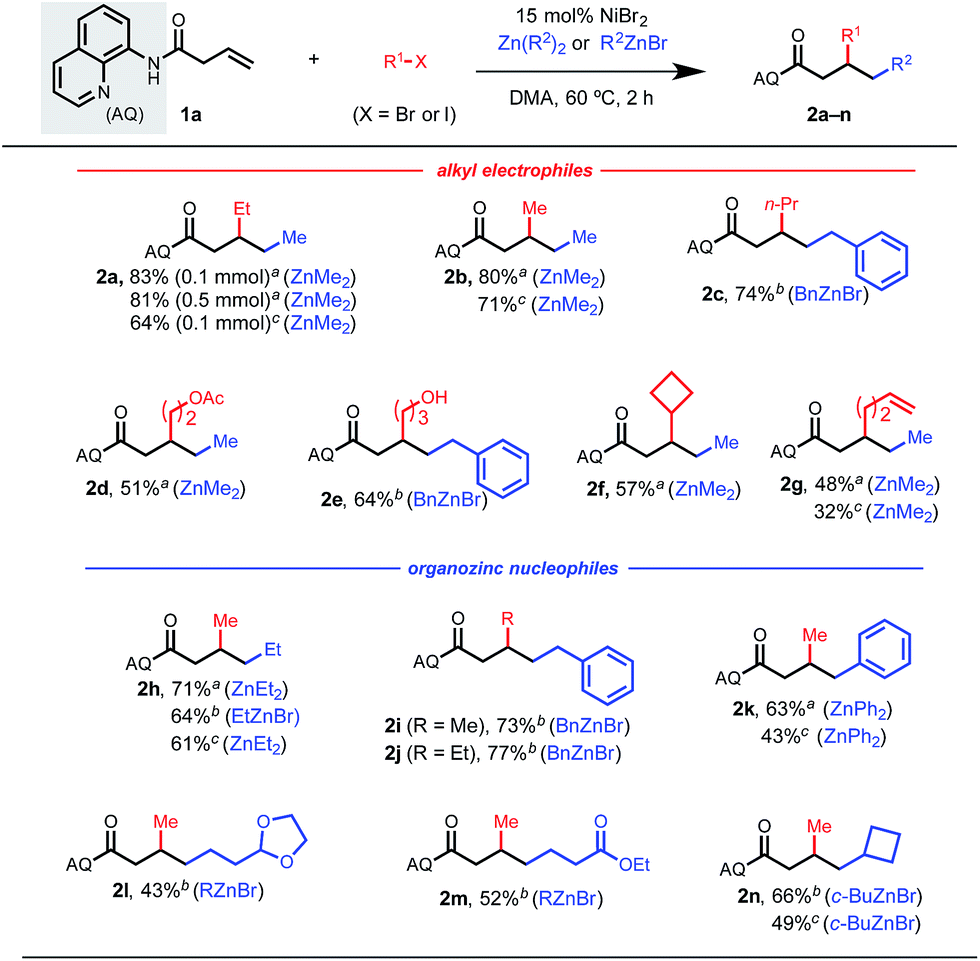
To provide a representative survey of the effects of coupling component equivalents, we conducted several examples using 4 equivalents of both electrophile and nucleophile. The yields were typically within 20% of the optimized yields, providing potential end-users with the opportunity to prioritize yield over reagent equivalents, or vice versa. We found that the AQ auxiliary of a representative product could be unmasked to the corresponding carboxylic acid or methyl ester under standard hydrolysis or methanolysis13 conditions, respectively (Scheme 2).
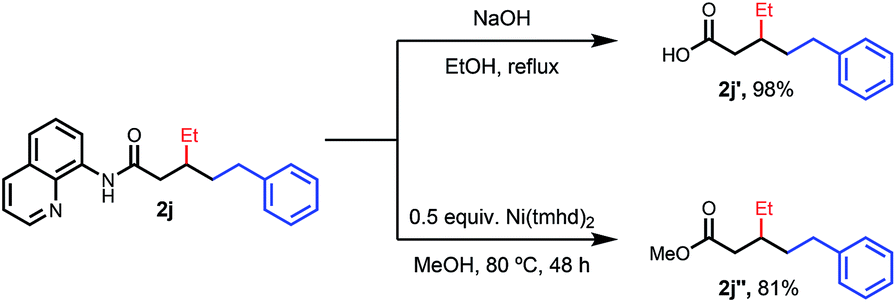 | ||
| Scheme 2 Removal of the AQ directing group. | ||
Next, we examined internal and α-substituted terminal β,γ-unsaturated alkenes (Table 3). For these substrates, we elected to use iodoethane as the electrophile and dimethyl zinc or benzylzinc bromide as the nucleophile. Internal alkenes provided the desired syn-1,2-difunctionalized products in good yields and with high diastereomeric ratios (3a–3g). The relative stereochemistry was determined by reacting representative internal alkene 1b with diphenyl zinc and methyl iodide, yielding a 1,2-arylalkylation product reported in our previous publication (see ESI†). Upon subjecting a trisubstituted alkene to the reactions conditions, we were pleased to discover that quaternary centers could be formed at the γ-position (3h). We questioned whether a 1,1-disubstituted alkene could react; however, the reaction did not proceed at all with this substrate (3i). We hypothesize that the putative chelation-stabilized alkylnickel intermediate is very sensitive to the proximal steric environment.
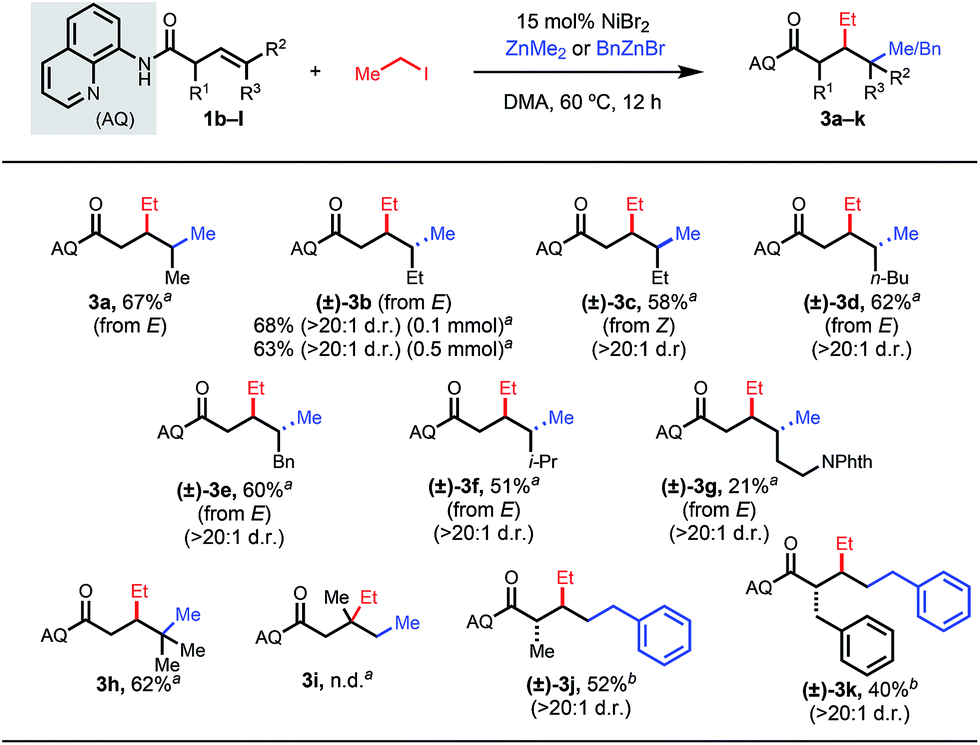
To investigate the steric constraints of our optimized system, we subjected α-substituted substrates to the reaction conditions (3j and 3k).14 More sterically bulky groups led to lower yields. In both successful cases, only a single diastereomer was detected, which was assigned as trans in analogy to our previous work.15
It is typically difficult to extend catalytic directed alkene functionalizations to substrates containing more distal functional groups due to the instability of the metalacycles that are six-membered or larger. Recently our group published a tridentate directing group strategy for the hydrofunctionalization of γ,δ-unsaturated alkenes; the tridentate directing group is thought to suppress β-H elimination in six-membered palladacycles.16
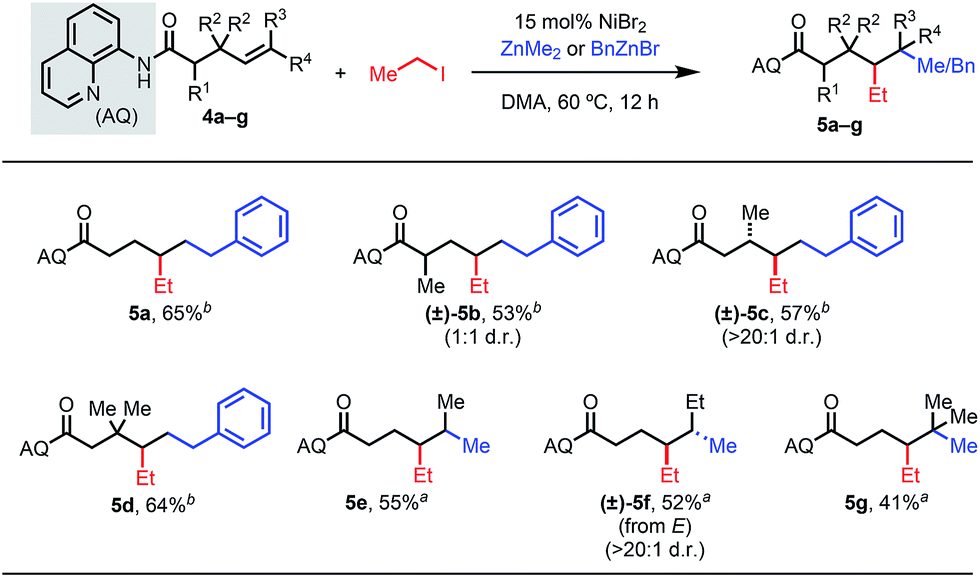
An alternative strategy is to intercept the metalacycle in a rapid subsequent step, such that this downstream reaction can outcompete undesired β-H elimination. Given the unique reactivity patterns in this 1,2-dialkylation reaction, we thus questioned whether γ,δ-dialkylation would be feasible via a six-membered AQ-bound nickelacycle (Table 4). To this end, we employed a terminal γ,δ-unsaturated alkene substrate with iodoethane and benzylzinc bromide. Our standard reaction conditions delivered the desired product in 65% yield (5a), showcasing the ability of this transformation to introduce alkyl fragments at the γ- and δ-positions, which are challenging to functionalize using existing chemistry. We then introduced substituents at various positions to investigate the effects on diastereoselectivity. Interestingly, α-substituted starting materials gave the desired product, but with significantly diminished diastereoselectivity (5b). When the analogous β-substituted starting material was used, the desired product was delivered as a single diastereomer (5c).15 These results shed light on the influence of proximal substituents on competing nickelacycle formation pathways. Internal alkenes within this substrate class also reacted well to give dialkylated products (5e and 5f). To our delight, the corresponding trisubstituted alkene also proved to be reactive, allowing formation of a quaternary center (5g). We further probed a representative δ,ε-unsaturated substrate but in this case could only detect isomerization byproducts.
In an attempt to gain insight regarding the mechanism of the oxidation addition step of this 1,2-dialkylation reaction, we conducted a radical clock experiment using (bromomethyl)cyclopropane as an electrophile (Scheme 3). Upon treating standard substrate 1a with 2 equivalents of electrophile and 2 equivalents of dimethyl zinc, we observed the formation of ring-opened product 2g in 27% yield. This result is consistent with a radical oxidation step involving single-electron transfer (SET). Such SET processes have been previously reported to occur from organonickel(I) species that are formed via transmetalation.3b,8
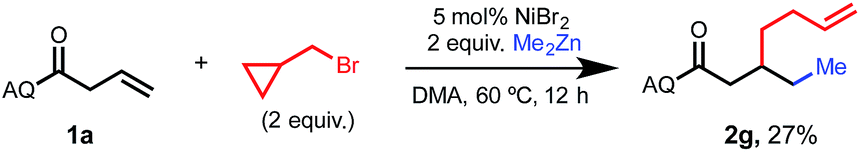 | ||
| Scheme 3 Mechanistic experiment involving (bromomethyl)cyclopropane as an electrophile. | ||
Though a detailed mechanistic investigation of this transformation remains outside of the scope of the present manuscript, a plausible catalytic cycle consistent with experimental data is shown in Scheme 4. The cycle follows a NiI/NiIII redox manifold, in line with other alkyl–alkyl cross-coupling processes.3b,8d,e,9h Initially, a NiI catalyst (presumably bound as shown in A) undergoes a transmetalation/syn-1,2-migratory insertion sequence to yield putative nickelacycle B. This species then performs SET with the alkyl halide electrophile to generate NiII-bound species C and the corresponding alkyl radical. After radical recombination with the resulting alkyl radical, NiIII intermediate D undergoes reductive elimination to yield the desired product and regenerate the active catalyst.
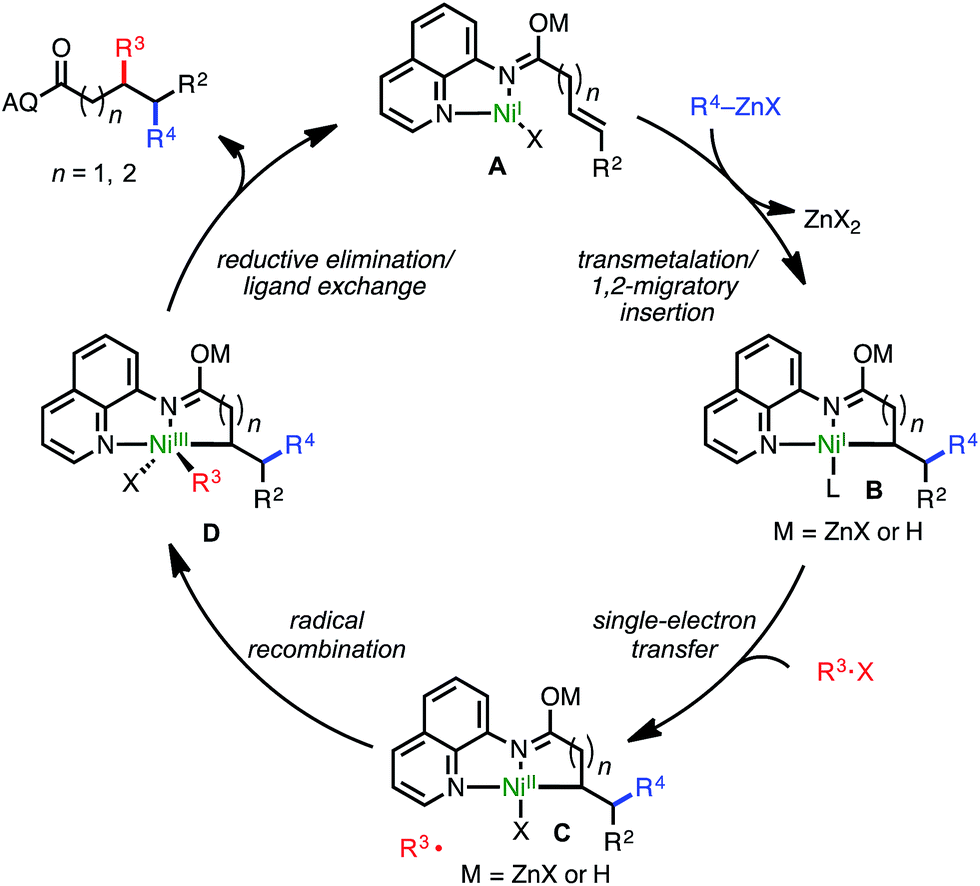 | ||
| Scheme 4 Proposed catalytic cycle. | ||
An alternative mechanism could also be envisioned in which oxidative addition of the alkyl halide occurs first, followed by syn-insertion to deliver the electrophile alkyl fragment proximal to the directing group and generate the 6- or 7-membered nickelacycle for substrates classes 1 and 4, respectively. Though we have not conclusively ruled out this pathway at this stage, several observations stand counter to this mechanism. First, assuming this alternative pathway were operative, the observed regiochemical outcome when utilizing γ,δ-unsaturated alkenes (4) would indicate that the catalyst selectively forms a 7-membered alkyl nickelacycle in preference to a 6-membered alkyl nickelacycle, which is inconsistent with established trends in alkyl metalacycle formation.16 Second, assuming the former point were true, it would stand to reason that δ,ε-unsaturated carbonyl compounds would also be compatible; however, in practice this substrate class was not tolerated. Third, formation of products such as 5g following this alternative mechanism would require the intermediacy of a 7-membered tertiary-alkyl nickelacycle, a species that would be expected to be highly unstable.
As noted above, the present reaction delivers the opposite regiochemical outcome compared with previously reported conditions using an aryl iodide electrophile,6c potentially indicating mechanistic divergence in the two cases. Notably, Zhao and coworkers have also recently reported that the regiochemical outcome of nickel-catalyzed alkene 1,2-dicarbofunctionalization reactions can be switched based on the identity of the electrophilic coupling partner.6d
Conclusions
In conclusion, we have developed a three-component conjunctive coupling involving only alkyl components for β,γ- and γ,δ-difunctionalization of alkenyl carbonyl compounds. Using a removable directing group strategy, high regioselectivity and diastereoselectivity can be achieved, rendering this method a potentially powerful tool for rapidly assembling molecular complexity. In-depth studies are currently underway to determine the precise mechanism of nickel-catalyzed dicarbofunctionalization and to expand the scope of compatible electrophilic and nucleophilic coupling partners. These results will be reported in due course.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by The Scripps Research Institute (TSRI), Pfizer, Inc., Bristol-Myers Squibb (Unrestricted Grant), and the National Institutes of Health (1R35GM125052). We gratefully acknowledge the NSF for a Graduate Research Fellowship (NSF/DGE-1346837, J. D.) and a Summer Undergraduate Research Fellowship (NSF/DBI-1359160, V. A. V.). We thank Dr Dee-Hua Huang and Dr Laura Pasternack for assistance with NMR spectroscopy. We appreciate Prof. Nathan D. Schley (Vanderbilt) and Prof. Ryan A. Shenvi (TSRI) for helpful discussion.Notes and references
- F. Lovering, J. Bikker and C. Humblet, J. Med. Chem., 2009, 52, 6752–6756 CrossRef PubMed.
- For reviews on transition-metal-catalyzed two-component cross-coupling reactions, see: (a) C. C. C. Johansson Seechurn, M. O. Kitching, T. J. Colacot and V. Snieckus, Angew. Chem., Int. Ed., 2012, 51, 5062–5085 CrossRef PubMed; (b) S. Z. Tasker, E. A. Standley and T. F. Jamison, Nature, 2014, 509, 299–309 CrossRef PubMed. For reviews on transition-metal-catalyzed three-component cross-coupling reactions, see: (c) R. Giri and S. KC, J. Org. Chem., 2018, 83, 3013–3022 CrossRef PubMed; (d) R. K. Dhungana, S. KC, P. Basnet and R. Giri, Chem. Rec., 2018 DOI:10.1002/tcr.201700098; (e) J. Derosa, V. T. Tran, V. A. van der Puyl and K. M. Engle, Aldrichimica Acta, 2018, 51, 21–32 Search PubMed.
- (a) V. B. Phapale, E. Buñuel, M. García-Iglesias and D. J. Cárdenas, Angew. Chem., Int. Ed., 2007, 46, 8790–8795 CrossRef PubMed; (b) S. KC, P. Basnet, S. Thapa, B. Shrestha and R. Giri, J. Org. Chem., 2018, 83, 2920–2936 CrossRef PubMed.
- For representative examples of conjunctive coupling using allenes, 1,3-dienes, styrenes, or acrylates, see: (a) T.-H. Huang, H.-M. Chang, M.-Y. Wu and C.-H. Cheng, J. Org. Chem., 2002, 67, 99–105 CrossRef PubMed; (b) L. Liao, R. Jana, K. B. Urkalan and M. S. Sigman, J. Am. Chem. Soc., 2011, 133, 5784–5787 CrossRef PubMed; (c) T. Qin, J. Cornella, C. Li, L. R. Malins, J. T. Edwards, S. Kawamura, B. D. Maxwell, M. D. Eastgate and P. S. Baran, Science, 2016, 352, 801–805 CrossRef PubMed; (d) K. M. Logan, K. B. Smith and M. K. Brown, Angew. Chem., Int. Ed., 2015, 54, 5228–5231 CrossRef PubMed. For early work on 1,2-alkylation of styrenes and dienes with organometallic nucleophiles and alkyl electrophiles, see: (e) J. Terao, Y. Kato and N. Kambe, Chem.–Asian J., 2008, 3, 1472–1478 CrossRef PubMed; (f) K. Mizutani, H. Shinokubo and K. Oshima, Org. Lett., 2003, 5, 3959–3961 CrossRef PubMed.
- (a) L. Zhang, G. J. Lovinger, E. K. Edelstein, A. A. Szymaniak, M. P. Chierchia and J. P. Morken, Science, 2016, 351, 70–74 CrossRef PubMed. For a recent example of Ni-catalyzed 1,2-dicarbofunctionalization involving alkyl electrophiles via a metalate rearrangement, see: (b) G. J. Lovinger and J. P. Morken, J. Am. Chem. Soc., 2017, 139, 17293–17296 CrossRef PubMed.
- (a) A. García-Domínguez, Z. Li and C. Nevado, J. Am. Chem. Soc., 2017, 139, 6835–6838 CrossRef PubMed; (b) B. Shrestha, P. Basnet, R. K. Dhungana, S. KC, S. Thapa, J. M. Sears and R. Giri, J. Am. Chem. Soc., 2017, 139, 10653–10656 CrossRef PubMed; (c) J. Derosa, V. T. Tran, M. N. Boulous, J. S. Chen and K. M. Engle, J. Am. Chem. Soc., 2017, 139, 10657–10660 CrossRef PubMed; (d) W. Li, J. K. Boon and Y. Zhao, Chem. Sci., 2018, 9, 600–607 RSC; (e) S. Thapa, R. K. Dhungana, R. T. Magar, B. Shrestha, S. KC and R. Giri, Chem. Sci., 2018, 9, 904–909 RSC.
- Classical vicinal-difunctionalization reactions of α,β-unsaturated carbonyl compounds are a related class of reactions with well-established utility in organic synthesis. For selected reviews, see: (a) M. Suzuki and R. Noyori, in Organocopper Reagents: A Practical Approach, ed. R. J. K. Taylor, Oxford University Press, Oxford, 1994, ch. 9, pp. 185–216 Search PubMed; (b) R. J. K. Taylor, Synthesis, 1985, 364–392 CrossRef; (c) M. J. Chapdelaine and M. Hulce, Org. React., 1990, 38, 225–653 Search PubMed; (d) M. Ihara and K. Fukumoto, Angew. Chem., Int. Ed., 1993, 32, 1010–1022 CrossRef.
- For reviews on C(sp3)–C(sp3) cross-coupling, see: (a) M. R. Netherton and G. C. Fu, Adv. Synth. Catal., 2004, 346, 1525–1532 CrossRef; (b) A. C. Frisch and M. Beller, Angew. Chem., Int. Ed., 2005, 44, 674–688 CrossRef PubMed; (c) A. Rudolph and M. Lautens, Angew. Chem., Int. Ed., 2009, 48, 2656–2670 CrossRef PubMed; (d) R. Jana, T. P. Pathak and M. S. Sigman, Chem. Rev., 2011, 111, 1417–1492 CrossRef PubMed; (e) X. Hu, Chem. Sci., 2011, 2, 1867–1886 RSC.
- For early examples of utilizing ancillary ligands to suppress β-hydride elimination in C(sp3)–C(sp3) cross-coupling, see: (a) K. Tamao, Y. Kiso, K. Sumitani and M. Kumada, J. Am. Chem. Soc., 1972, 94, 9268–9269 CrossRef; (b) T. Hayashi, M. Konishi, Y. Kobori, M. Kumada, T. Higuchi and K. Hirotsu, J. Am. Chem. Soc., 1984, 106, 158–163 CrossRef; (c) T. Katayama and M. Umeno, Chem. Lett., 1991, 20, 2073–2076 CrossRef. For representative examples of nickel-catalyzed C(sp3)–C(sp3) cross-coupling, see: (d) A. E. Jensen and P. Knochel, J. Org. Chem., 2002, 67, 79–85 CrossRef PubMed; (e) J. Zhou and G. C. Fu, J. Am. Chem. Soc., 2003, 125, 14726–14727 CrossRef PubMed; (f) J. Terao, H. Todo, H. Watanabe, A. Ikumi and N. Kambe, Angew. Chem., Int. Ed., 2004, 43, 6180–6182 CrossRef PubMed; (g) H. Gong, R. Sinisi and M. R. Gagné, J. Am. Chem. Soc., 2007, 129, 1908–1909 CrossRef PubMed. For a representative example of directed C(sp3)–C(sp3) cross-coupling, see: (h) A. Wilsily, F. Tramutola, N. A. Owston and G. C. Fu, J. Am. Chem. Soc., 2012, 134, 5794–5795 CrossRef PubMed.
- Z. Liu, T. Zeng, K. S. Yang and K. M. Engle, J. Am. Chem. Soc., 2016, 138, 15122–15125 CrossRef PubMed.
- For pioneering work on bidentate auxiliaries in Pd(II)-catalyzed C–H activation, see: (a) V. G. Zaitsev, D. Shabashov and O. Daugulis, J. Am. Chem. Soc., 2005, 127, 13154–13155 CrossRef PubMed. For examples with Ni, see: (b) H. Shiota, Y. Ano, Y. Aihara, Y. Fukumoto and N. Chatani, J. Am. Chem. Soc., 2011, 133, 14952–14955 CrossRef PubMed; (c) Y. Aihara and N. Chatani, J. Am. Chem. Soc., 2014, 136, 898–901 CrossRef PubMed. For selected reviews, see: (d) G. Rouquet and N. Chatani, Angew. Chem., Int. Ed., 2013, 52, 11726–11743 CrossRef PubMed; (e) O. Daugulis, J. Roane and L. D. Tran, Acc. Chem. Res., 2015, 48, 1053–1064 CrossRef PubMed.
- We attempted slow addition of the nucleophilic component to improve the efficiency but did not observe a substantial increase in yield.
- T. Deguchi, H.-L. Xin, H. Morimoto and T. Ohshima, ACS Catal., 2017, 7, 3157–3161 CrossRef.
- To examine whether epimerizable α-stereocenters underwent racemization under the reaction conditions, we performed an additive study in which a protected amino acid derivatives was added to the reaction and reisolated after the standard time. In this experiment racemization was negligible (>99% ee to 97% ee) (see ESI† for details).
- The relative configuration was assigned as trans by analogy to compounds prepared in ref. 6c. The preference for trans product formation is believed to arise from more facile formation of the trans nickelacycle.
- M. L. O'Duill, R. Matsuura, Y. Wang, J. L. Turnbull, J. A. Gurak Jr, D.-W. Gao, G. Lu, P. Liu and K. M. Engle, J. Am. Chem. Soc., 2017, 139, 15576–15579 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8sc01735b |
| This journal is © The Royal Society of Chemistry 2018 |