
CTAB-assisted growth of self-supported Zn2GeO4 nanosheet network on a conductive foam as a binder-free electrode for long-life lithium-ion batteries†
Guoxin
Gao‡
a,
Yang
Xiang‡
a,
Shiyao
Lu
a,
Bitao
Dong
a,
Sheng
Chen
a,
Lei
Shi
a,
Yuankun
Wang
a,
Hu
Wu
a,
Zhaoyang
Li
a,
Amr
Abdelkader
 bc,
Kai
Xi
*d and
Shujiang
Ding
*a
bc,
Kai
Xi
*d and
Shujiang
Ding
*a
aDepartment of Applied Chemistry, School of Science, Xi'an Jiaotong University, Xi'an 710049, P. R. China. E-mail: dingsj@xjtu.edu.cn
bNational Graphene Institute (NGI), University of Manchester, Booth Street East, Manchester, M13 9QS, UK
cFaculty of Science and Technology, Bournemouth University, Poole House, Talbot Campus, Poole, Dorset BH12 5BB
dDepartment of Materials Science and Metallurgy, University of Cambridge, Cambridge CB3 0FS, UK. E-mail: kx210@cam.ac.uk
First published on 7th November 2017
Abstract
The Ge-based compounds show great potential as replacements for traditional graphite anode in lithium-ion batteries (LIBs). However, large volume changes and low conductivity of such materials result in a poor electrochemical cycling and rate performance. Herein, we fabricate a self-supported and three-dimensional (3D) sponge-like structure of interlinked Zn2GeO4 ultrathin nanosheets anchored vertically on a nickel foam (ZGO NSs@NF) via a simple hydrothermal process assisted by cetyltrimethyl ammonium bromide (CTAB). Such robust self-supported hybrid structures greatly improve the structural tolerance of the active materials and accommodate the volume variation that occurs during repeated electrochemical cycling. As expected, the self-supported ZGO NSs@NF composites demonstrate an excellent lithium storage with a high discharge capacity, a long cycling life, and a good rate capability when used as binder-free anodes for LIBs. A high reversible discharge capacity of 794 mA h g−1 is maintained after 500 cycles at 200 mA g−1, corresponding to 81% capacity retention of the second cycle. Further evaluation at a higher current density (2 A g−1) also delivers a reversible discharge capacity (537 mA h g−1) for this binder-free anode. This novel 3D structure of the self-supported ultrathin nanosheets on a conductive substrate, with its volume buffer effect and good interfacial contacts, can stimulate the progress of other energy-efficient technologies.
Introduction
Lithium-ion batteries (LIBs) are considered to be one of the most promising power sources for portable electronic devices, plug-in hybrid electric vehicles (HEVs) and stationary grid applications due to their high energy densities, long-term cycling stability, no memory effects and eco-friendliness.1–4 One challenge that is faced by the further development of high-performance LIBs is the limited capacity of graphite negative electrodes (372 mA h g−1).2,5,6 Tremendous efforts have been made to develop new anode materials that could replace the traditional graphite anodes, including using group IV elements (silicon, germanium and tin)7–10 and transition metal oxides/sulfides.11–18 Among these, special attention has been paid to the Ge-based anodes due to their high theoretical specific capacities (ca. 1600 mA h g−1) by forming the alloy Li4.4Ge, fast Li+ diffusivity (400 times faster than Si) and good electric conductivity (almost 100 times higher than Si).19–25 However, like most metal-based electrodes, the pure Ge anodes are expensive and suffer from rapid capacity decays caused by the large volume changes (about 370%) that occur between the lithium alloying and de-alloying process.23–25Stable and high-capacity ternary metal germinate compounds have a great potential to improve the electrochemical cycling performances and reduce the fabrication costs of the Ge-based anodes. Partially substituting expensive Ge with cheaper metal elements such as Zn, Cu, Fe, Cd, Ca and Co in the ternary oxide structure could generate a moderate buffer zone to accommodate the volume variation of electrode materials by means of the favorable synergistic effect and simultaneously improve the electrode electrochemical performance.26–39 The ternary Zn2GeO4-based anodes have been well studied in literature and can deliver a high theoretical capacity of 1443 mA h g−1 based on the conversion and lithium alloying reactions (Zn2GeO4 + 8 Li+ + 8 e− → 2 Zn + Ge + 4 Li2O, Ge + 4.4 Li+ + 4.4 e− ↔ Li4.4Ge, Zn + Li+ + e− ↔ LiZn).40 Despite significant efforts, the practical application of such Zn2GeO4-based anodes is still hampered by their fast capacity fading and poor rate capability, which can be ascribed to their intrinsic poor electronic conductivity and large volumetric variation upon cycling.41–43 Moreover, severe pulverization and structural collapse of the electrodes, significant aggregation of the intermediates, and subsequent detachment of the active materials from the current collector are also responsible for limiting the electrochemical cycling performance of the Zn2GeO4-based anodes.44,45
As shown in Fig. 1A–C, directly growing the electroactive materials with well-designed nanostructures (0D, 1D, 2D and 3D) on conductive substrates as binder-free electrodes has become an effective strategy for enhancing the battery cycling stability and rate capability.46–49 The improvement in cycling stability is due to the enhanced electronic conductivity and interfacial contacts between the current collector and the active materials compared with those in the simple physical mixing. For example, Wang and co-workers synthesized carbon coated coaxial Zn2GeO4 nanowires on a copper foil as the anode material, which exhibits a remarkably stable discharge capacity of 790 mA h g−1 after 100 cycles.50 Subsequently, they grew a carbon-coated Zn2GeO4 particle film on a nickel foam as a binder-free anode for LIBs.51 Shen and co-workers also reported a good cycling stability for the anodes engineered from metal germanate nanowires such as Ca2Ge7O16, SrGe4O9, BaGe4O9, and Zn2GeO4 grown on carbon textiles.39,52 To the best of our knowledge, the reported morphologies of the metal germanate compounds in the literature mainly include nanoparticles, nanorods and nanowires.37–45,53–59 Compared with the reported morphologies, the free-standing ultrathin nanosheets with an interlinked self-supported network aligning on a conductive substrate usually offer a more robust mechanical structure as well as a better electrolyte diffusion within the electrode, a larger exposed specific surface area and more abundant active sites to carry out the electrochemical reactions, thus delivering a good electrochemical performance.13,14,60
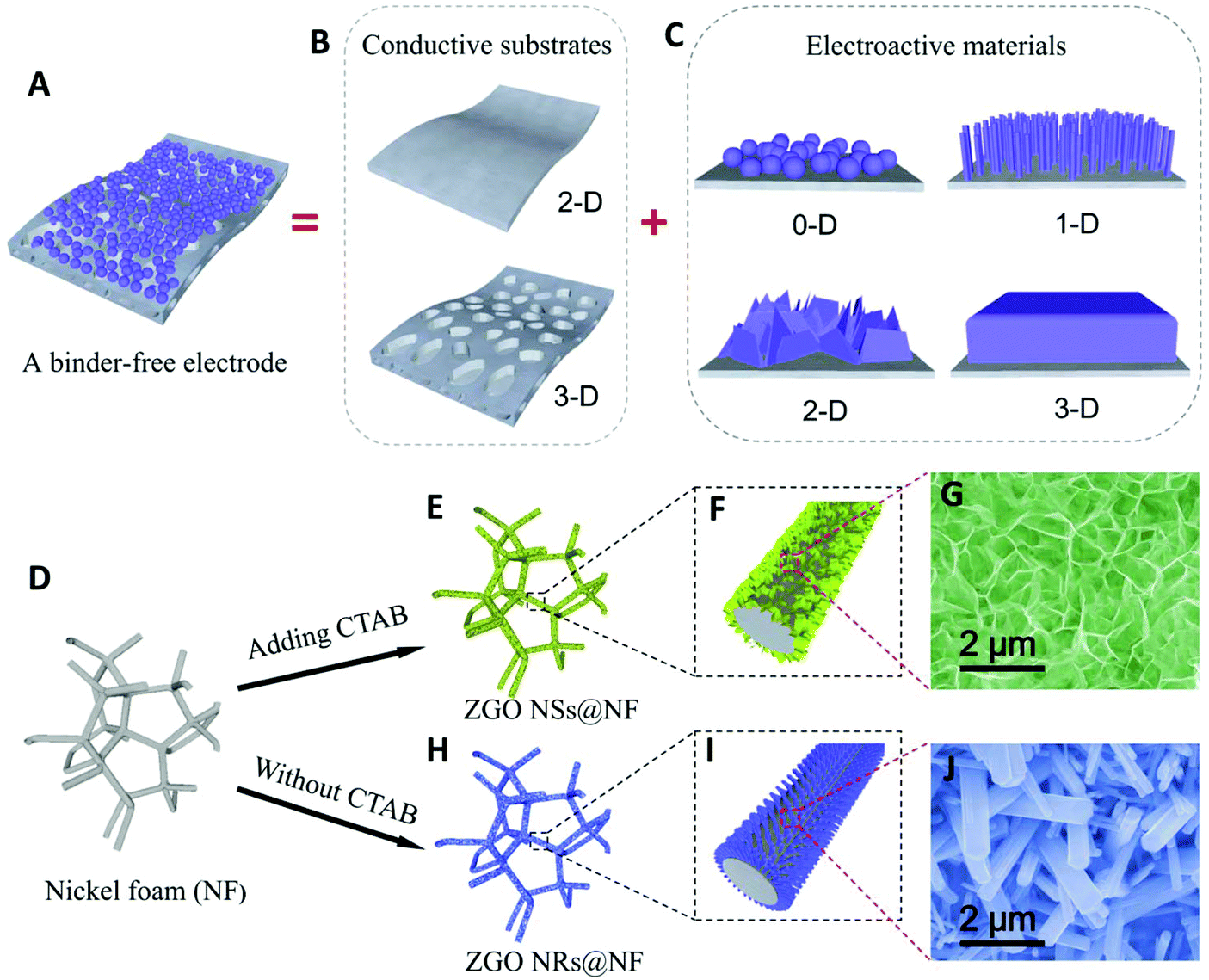 | ||
| Fig. 1 Schematic of a binder-free electrode and the influence of CTAB on the morphology of the hierarchical hybrid ZGO NSs@NF and ZGO NRs@NF. | ||
In this work, we successfully develop a novel hierarchical hybrid nanostructure of free-standing ultrathin Zn2GeO4 nanosheets anchored on the nickel foam (NF) substrates (denoted as ZGO NSs@NF) via a simple hydrothermal process assisted by cetyltrimethyl ammonium bromide (CTAB), as illustrated in Fig. 1D–J. The free-standing ultrathin ZGO NSs are interconnected to demonstrate a 3D self-supported sponge-like structure, which uniformly covers the conductive NF substrate. When used as the binder-free anodes for LIBs directly, the self-supported ZGO NFs@NF composites demonstrate a highly enhanced electrochemical performance due to their large surface area, high porosity, and robust structural tolerance. A high reversible discharge capacity of about 793 mA h g−1 has been successfully maintained after 500 cycles at a current density of 200 mAg−1, corresponding to about an 81% reversible discharge capacity retention of the second cycle. Even when cycled at a higher current density of 2 A g−1, the self-supported ZGO NSs@NF anodes still deliver a stable discharge capacity of about 537 mA h g−1, exhibiting an excellent rate capability.
Results and discussion
Fig. 2A shows the digital picture of the NF substrate and the as-prepared 3D self-supported ZGO NSs@NF composites. Clearly, after the simple CTAB-assisted hydrothermal process, the surface of the NF substrate becomes darker, confirming the successful growth of ZGO layer on the NF substrate. Remarkably, binding, scratching, and twisting the composite electrode does not result in any visible changes to the black powder peeling off from the substrate as seen in Fig. S1 (see the ESI†). The X-ray diffraction (XRD) patterns of the as-prepared self-supported ZGO NSs@NF are shown in Fig. S2 (see the ESI†) to distinguish their crystallographic structure and the phase purity. Except for three characteristic diffraction peaks derived from the NF substrate (JCPDS no. 04-0850), all of other diffraction peaks are indexed to the rhombohedral Zn2GeO4 crystalline structure (JCPDS no. 11-0687).43,61 No additional peaks can be found, which strongly suggests that the self-supported ZGO NSs@NF composites have a high purity. More importantly, the energy dispersive X-ray (EDX) analysis as shown in Fig. S3 (see the ESI†) further confirms that the self-supported hybrid composites are mainly composed of Ni, Zn, Ge and O elements, and the atomic ratio (At%) of Zn![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Ge is close to 2:1. The Ni elements in the EDX plot are from the NF substrate.
:Ge is close to 2:1. The Ni elements in the EDX plot are from the NF substrate.
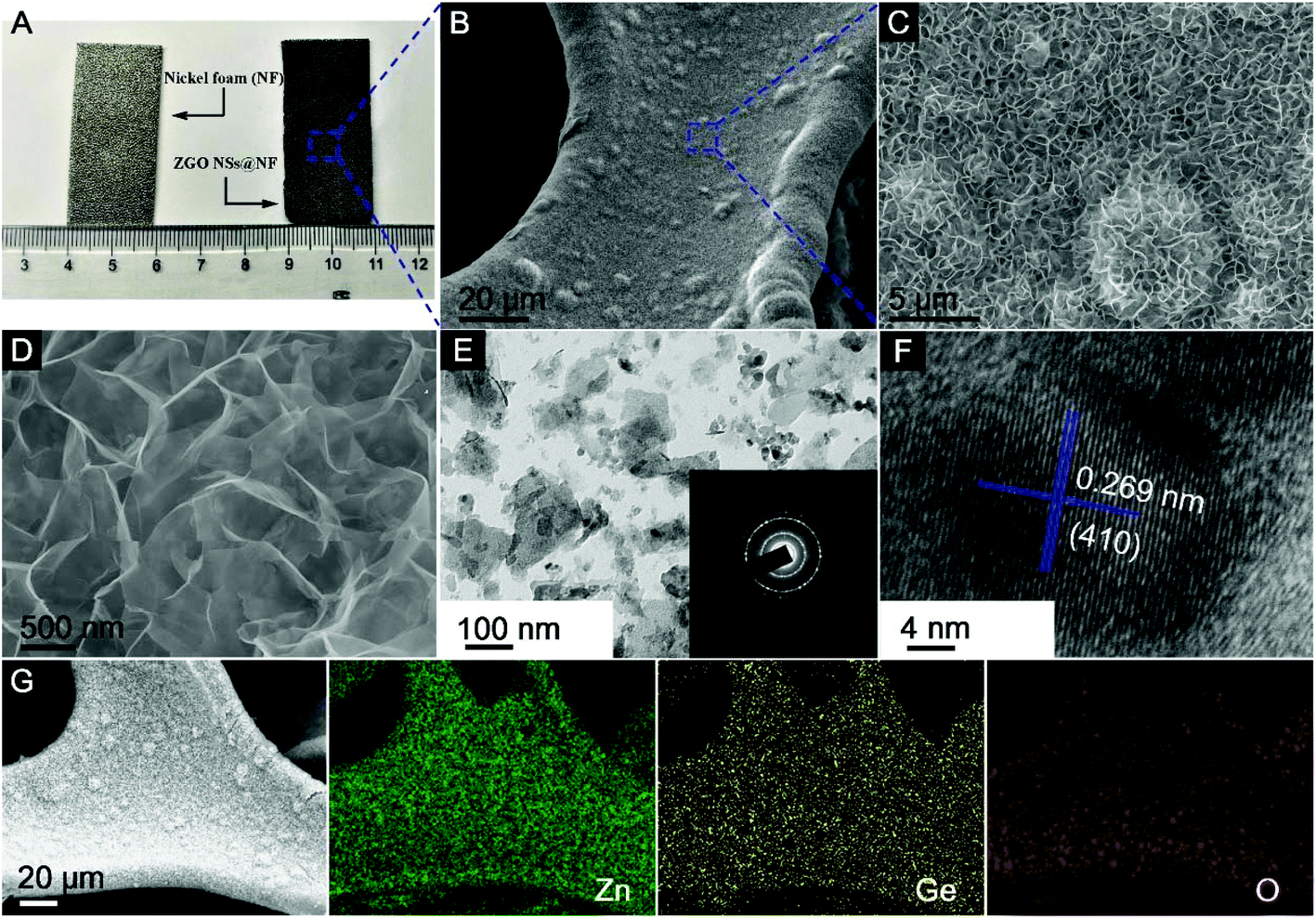 | ||
| Fig. 2 (A) Digital photo, (B–D) FESEM, (E) TEM, (F) HRTEM images, and (G) EDX – elemental mapping of the ZGO NSs@NF obtained through the CTAB-assisted hydrothermal reaction at 160 °C for 180 min. The inset image in E shows the SAED pattern. | ||
The morphologies of the as-prepared ZGO NSs@NF are examined by field-emission scanning electron microscopy (FESEM), transmission electron microscopy (TEM) and high-resolution TEM (HRTEM) as shown in Fig. 2B–F. Fig. 2B shows the morphology of the novel ZGO NSs@NF at a low magnification. The coating seems to be homogeneous with no clear aggregations of ZGO NSs on the NF substrate. At a higher magnification, Fig. 2C and D distinctly reveal that those ultrathin ZGO NSs are interconnected to each other and arranged in a 3D sponge-like network. Such interlinked porous structure is self-supported and firmly anchored on the conductive substrate. However, when we repeated the experiment without placing the NF substrate in the hydrothermal system, only a few pristine ZGO nanoflakes are obtained as shown in Fig. S4A and S4B (see the ESI†). In addition, a close observation (Fig. S4B†) reveals that most ZGO nanoflakes stack together to form the clusters, which means that one of the roles of NF is to prevent the aggregation of the ultrathin ZGO flakes. To obtain more detailed microstructural information, the self-supported ZGO interlinked NSs are exfoliated from the NF by ultrasonication in ethanol and then dropped directly onto a copper grid to carry out TEM analysis. Fig. 2E shows that the flake size of the exfoliated ZGO NSs is around 200 nm. We believe that the actual flake size on the NF substrate is much greater than 200 nm. Due to the strong adhesion between ZGO NSs and the NF substrate, an extensive sonication was required, resulting in breaking the ZGO flakes into smaller fragments. The polycrystalline nature of the rhombohedral Zn2GeO4 phase is also confirmed by the intermittent diffraction rings shown in the inset of Fig. 2E. In addition, the structural feature of a typical ZGO NS fragment with visible lattice fringes has been observed in an HRTEM image (Fig. 2F). The marked interplanar distance of 0.269 nm corresponds to the (410) crystal planes of the rhombohedral Zn2GeO4 phase. The elemental mapping further confirms the chemical components of the as-prepared ZGO NSs@NF composites (Fig. 2G). All of the key elements, including Zn, Ge and O, are detected distinctly with a homogeneous distribution throughout the measurement area.
We find that the CTAB surfactant plays a crucial role in the successful formation of the 3D self-supported ZGO NSs on the NF substrate. We have conducted several experiments with different amounts of CTAB added during the hydrothermal reaction. The FESEM images of the as-synthesized materials are shown in Fig. S5 (see the ESI†). Fig. S5A† shows the products of the hydrothermal process without CTAB addition. Clearly, only short ZGO nanorods (NRs) with a hexagonal cross-section can be detected on the NF (denoted as ZGO NRs@NF), but these free-standing ZGO NRs anchored on the NF surface are isolated from each other. When a little amount (0.3 mmol) of CTAB is added to this hydrothermal system, most free-standing ZGO NRs disappear and are transferred into small sheet-like protuberances covering the NF surface as shown in Fig. S5B.† Further increasing the amount of CTAB to 0.7 mmol causes the growth of sheet-like tiny protuberances onto the interconnected ZGO NSs, which are free-standing, anchored on the NF support (Fig. S5C†). Moreover, less and less rod-like structure of ZGO NRs could be observed, suggesting that CTAB has an important influence on the formation of ultrathin ZGO NSs with the self-supported structure in this work. We believe that the adsorption of the CTAB molecules on some facets of the crystalline ZGO subunits significantly lowers their surface free energy and drives the formation of the sheet-like nanostructure on the NF.62 Similar phenomenon was observed in the fabrication of silver nanoplates.63 However, as seen from Fig. S5D,† even when the amount of CTAB is increased to 2 mmol, we still only obtain the ultrathin ZGO NSs anchored on the NF substrate. Therefore, we chose 1 mmol as the optimal amount of CTAB in this research.
To further understand the growth mechanism of the self-supported ZGO NSs@NF, we collected a series of samples at different intervals in the hydrothermal process and investigated their morphologies using FESEM, and the results are shown in Fig. 3. The ZGO flakes start to appear on the NF substrate in the initial few minutes of the hydrothermal treatment (Fig. 3A). These original flakes work as seeds for the further growth and it takes only 30 min of processing to fully cover the NF substrate with thin ZGO flakes (Fig. 3B). The second layer, grown on the top of the first ZGO layer, seems to be composed of many thinner flakes. The new flakes appear to be oriented vertically on the substrate as seen from the SEM image of the sample after 60 min (Fig. 3C). Longer processing times, 120 min (Fig. 3D) and 180 min (Fig. 3E), lead to a more ordered arrangement of these ultrathin flakes, creating the honeycomb-like structures with the ZGO flakes interlinked at the edges and aligned vertically on the NF support. However, a prolonged treatment (such as 240 min) leads to an excessive growth of the thin ZGO flakes in a direction parallel to the substrate, thus showing increasing flake thickness (Fig. 3F). Therefore, we settled on an ideal hydrothermal period of 180 min at 160 °C to obtain the self-supported ZGO NSs@NF composites. The gradual evolution process is illustrated in Fig. 3G–K.
 | ||
| Fig. 3 FESEM images of the ZGO@NF products obtained at 160 °C for various hydrothermal times: (A) 10 min, (B) 30 min, (C) 60 min, (D) 120 min, (E) 180 min, and (F) 240 min. | ||
Next, we carried out the CV measurements at a scanning rate of 0.5 mV s−1 in the voltage window of 0.01–3 V vs. Li/Li+ to identify the electrochemical reactions of the self-supported ZGO NSs@NF composites by directly using them as the binder-free anodes for LIBs. As shown in Fig. 4A, the initial CV scan is very different from the subsequent ones, but there are no apparent differences from the second cycle onwards, particularly in the charging branch. In the initial cathodic sweep, two reduction peaks can be observed on the CV plots. The predominant peak located at about 0.63 V may be ascribed to the decomposition of Zn2GeO4 into Zn, Ge, Li2O and the formation of a solid electrode interface (SEI) film, as well as the partial degradation of the organic electrolyte.38,64 Another relatively peak at around 0.31 V can be attributed to the alloying reactions of Zn–Li and Ge–Li.50,55 Compared with the reduction peak in the first cycle, the strong reduction peak at 0.63 V shifts to a higher voltage (about 0.84 V) in the second cycle and then moves back slightly to a lower voltage again in the third cycle.51 In the anodic sweep process, a distinct oxidation peak can be detected at about 1.39 V corresponding to the reoxidation of metallic Zn and Ge into ZnO and GeO2, respectively.42 The weak and broad peak between 0.4 and 0.9 V is assigned to the delithiation reactions of the Li-metal alloys.50 Therefore, according to the CV analysis and the previously reported lithium-storage mechanisms of ZnO and GeO2, the corresponding electrochemical reactions for the ZGO NSs@NF anode might be expressed as follows.38,40,65
| Zn2GeO4 + 8 Li+ + 8 e− → 2 Zn + Ge + 4 Li2O | (1) |
| Zn + Li+ + e− ↔ LiZn | (2) |
| Ge + 4.4 Li+ + 4.4 e− ↔ Li4.4Ge | (3) |
| Zn + Li2O ↔ ZnO + 2 Li+ + 2 e− | (4) |
| Ge + 2 Li2O ↔ GeO2 + 4 Li+ + 4 e− | (5) |
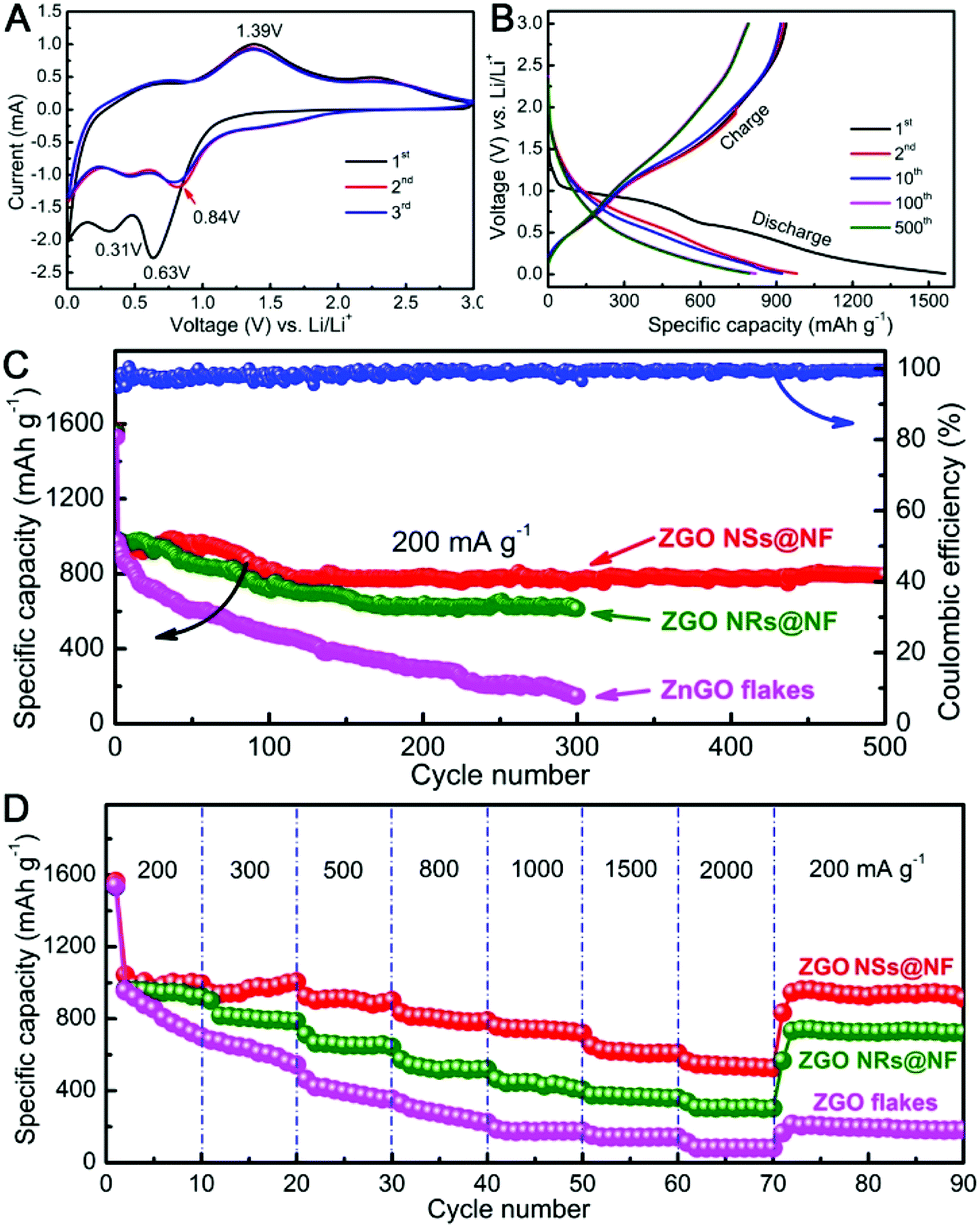 | ||
| Fig. 4 Electrochemical characterization: (A) CVs of ZGO NSs@NF tested at a scan rate of 0.5 mV s−1 between 0.01 and 3 V; (B) discharge–charge voltage profiles of ZGO NSs@NF at a current density of 200 mA g−1 in the cut-off voltage of 0.01–3 V; (C) cycling performance; and (D) rate capability of ZGO NSs@NF, ZGO NRs@NF, and ZGO flakes evaluated at different current densities between 0.01 and 3 V. | ||
The lithium storage properties were further investigated in a single half-cell using galvanostatic discharge/charge measurements. Fig. 4B reveals the corresponding discharge/charge voltage profiles of the 1st, 2nd, 10th, 100th and 500th cycles of the binder-free ZGO NSs@NF electrode at a current density of 200 mA g−1 within the cut-off voltage of 0.01–3.0 V vs. Li/Li+. For the first cycle, the self-supported ZGO NSs@NF electrode delivers high discharge and charge capacities of 1564 and 928 mA h g−1, respectively, presenting an irreversible loss of about 40.7%. Such irreversible capacity loss in the first cycle is usually due to the possible decomposition of the electrolyte, the formation of an SEI layer on the surface of the electrodes, and an incomplete conversion reaction or irreversible lithium uptake.64 The discharge plateaus at 0.2–1.0 V and the charge plateaus at 0.5–1.4 V observed in the discharge/charge curves can be attributed to the lithium alloying and de-alloying reactions.55 All of the data are in good agreement with the above-mentioned CV results and the literature.38,50,51 From the second cycle onwards, the voltage profiles of the self-supported ZGO NSs@NF anodes are nearly overlapping, indicating the reversibility of the electrochemical reactions. Fig. 4C further compares the cycling properties of ZGO NSs@NF, ZGO NRs@NF and ZGO flakes at a current density of 200 mA g−1. The self-supported ZGO NSs@NF anodes exhibit the best specific capacity and cycling stability among three electrodes. The discharge capacities of this binder-free electrode consistently maintain a high and stable value of about 794 mA h g−1 even after 500 cycles at 200 mA g−1, with the exception of a slight increase before the initial 30 cycles. The gentle increase in the capacity in the initial cycles implies a slow activation process, which might be related to the inadequate surface contact at the beginning of the measurement between the electroactive ZGO NSs@NF and the electrolyte.55 Moreover, the build-up of a stable SEI layer on the active materials also needs a certain number of cycles, which leads to a slight increase in the discharge capacity in the initial cycles.66 It is worth noting from Fig. 4C that the Coulombic efficiencies of the self-supported ZGO NSs@NF anodes are more than 98% after the initial cycles and remain almost unchanged in the subsequent cycles. The high Coulombic efficiencies highlight the excellent energy-conversion efficiency of the self-supported ZGO NSs@NF. Like the self-supported ZGO NSs@NF anode, the ZGO NRs@NF anode also presents the good electrochemical cycling stability since the ZGO NRs are also anchored to a conductive NF substrate. However, the discharge capacities decrease slowly from 983 mA h g−1 for the second cycle and maintain a low value of about 613 mA h g−1 over 300 cycles. We believe that the relatively small specific surface area of the hybrid ZGO NRs@NF is responsible for making the discharge capacities lower than those of the self-supported ZGO NSs@NF.67 However, without the support of the conductive NF, the pristine ZGO flakes demonstrate the poorest cycling stability compared with other two binder-free electrodes. After the second cycle, the discharge capacity decreased over 300 cycles from 971 mA h g−1 to 147 mA h g−1 at the 300th cycle. To compare the rate capabilities of three samples, the test cells were further evaluated at different current densities ranging from 200 to 2000 mA g−1, and the results are shown in Fig. 4D. The discharge capacities of ZGO NSs@NF and ZGO NRs@NF slowly decrease with increasing current densities. The ZGO NSs@NF and ZGO NRs@NF electrodes still deliver reversible discharge capacities of 537 and 302 mA h g−1, respectively, at a current density as high as 2000 mA g−1. Both electrodes are able to recover high discharge capacities: 938 mA h g−1 for ZGO NSs@NF and 722 mA h g−1 for ZGO NRs@NF when the current densities fall back to 200 mA g−1. However, the pristine ZGO flake electrode demonstrates a rather inferior rate capability compared to that of other binder-free electrodes. It delivers a much lower discharge capacity of about 82 mA h g−1 at 2000 mA g−1. Then, the recovered discharge capacity is as low as 180 mA h g−1 with the current density returning to 200 mA g−1.
In order to explore the reaction kinetics of the as-prepared ZGO NSs@NF electrodes, the CV measurements of the ZGO NSs@NF anode at different scan rates were recorded and are shown in Fig. S6A (see the ESI†). It is well-known that the relation between the current (I) and the scanning rate (v) can be described by the following equations:68–70
| i = avb | (6) |
| log(i) = blog(v) + log(a) | (7) |
To mimic the practical application of the ZGO NSs@NF electrode, we further assembled them into full-cells using the commercial LiFePO4 as a cathode and the electrochemically pre-lithiated ZGO NSs@NF as an anode. The loading mass ratio of the LiFePO4 cathode and the pre-lithiated ZGO NSs@NF anode was controlled to about 4:1 in order to ensure that the ZGO NSs@NF anode capacity exceeded a little. The full-cells were charged and discharged at 0.2 C (1 C = 170 mA h g−1, respect to LiFePO4) within the cut-off voltage of 2.0–3.8 V. Fig. S7A (see the ESI†) shows the charge–discharge curves at 1, 2, and 10 cycles. It can be seen that the initial discharge capacity was 160 mA h g−1, while the initial Coulombic efficiency was as high as 96.7%, suggesting a superb reversible performance. Even after 80 cycles, the discharge capacity could remain at 104 mA h g−1 (Fig. S7B, see the ESI†). Clearly, the LiFePO4/ZGO NSs@NF full-cells exhibit a good capacity and cycling stability and a high Coulombic efficiency. The electrochemical results of the full-cells verified the potential of ZGO NSs@NF as a promising candidate for the anode of LIBs. In addition, to present the superior electrochemical performance of our well-designed self-supported ZGO NSs@NF composites, other reported ZGO-based anodes are compared in Table S1 (see the ESI†).
The highly enhanced electrochemical performance including high discharge capacity, long-term cycle life and excellent rate capability of the novel self-supported ZGO NSs@NF electrodes can be ascribed to the well-designed 3D self-supported porous network structure. First, using the free-standing ultrathin ZGO NSs as the building blocks and arranging them in a sponge-like structure increase the available electrode–electrolyte contact area, which shortens the Li+ ion diffusion pathway and facilitates the electrochemical process.13,65 Second, the 3D interlinked ultrathin ZGO NSs with the self-supported structure can efficiently accommodate the stress induced by a drastic volume variation and avoid the structural collapse of the active electrode materials on the repeated discharge/charge process as shown in Fig. 5. The intumescent ZGO NSs resulting from the lithiation process were observed in Fig. 5E after the initial five discharge/charge cycles at 200 mA g−1. Even after 100 cycles, the interlinked 3D porous network structure can be well-maintained in the self-supported ZGO NSs@NF composites (Fig. 5F). For the same current density and cycle number, however, a clear distortion, even severe fracture, of the ZGO NRs arrays from the NF substrate can be observed on the ZGO NRs@NF electrodes (Fig. 5K and L, and Fig. S8Aand S8B, see the ESI†) due to the limited tolerance to the volume changes of the ZGO NRs. Third, the conductive NF skeleton not only prevents severe aggregation of the original ZGO NSs and the subsequent intermediate, which can improve the reversibility of the conversion reactions and facilitate the Li+ ion accessibility to the electrode,49,51 but also improves the electronic conductivity of the active materials in the total electrode even without other conductive additives. The Nyquist plots (Fig. S9, see the ESI†) show that the charge transfer resistance of the ZGO NSs@NF is lower than that of the pristine ZGO flakes and ZGO NRs@NF. Benefiting from these advantages, the novel self-supported ZGO NSs@NF anodes could demonstrate an enhanced electrochemical cycling stability.
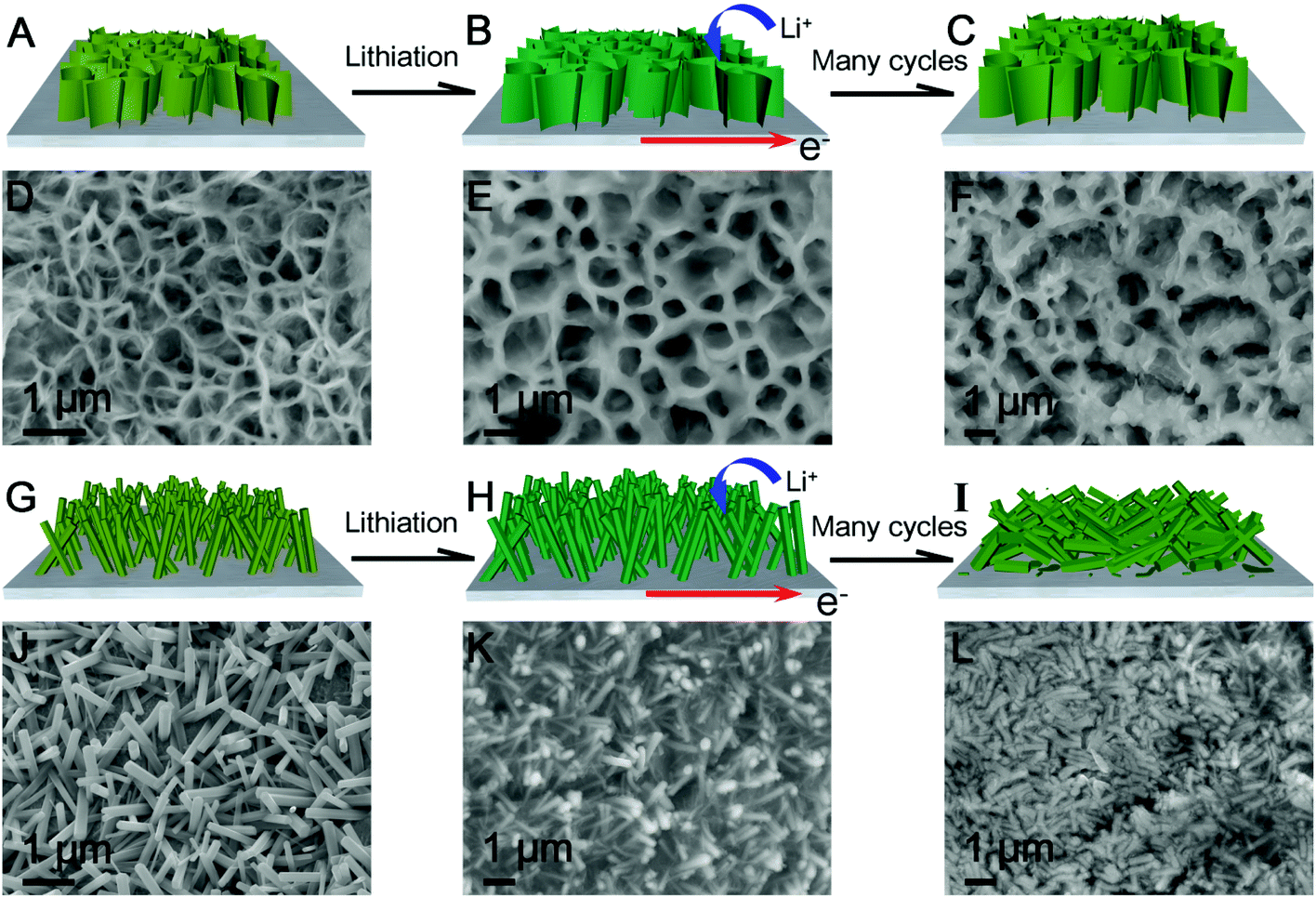 | ||
| Fig. 5 Schematic of the buffer function of the interlinked ultrathin ZGO NSs (A, B, C) and ZGO NRs (G, H, I) on the nickel foam during the lithiation/delithiation process. Initial FESEM (D, J) and post-mortem FESEM images (E, F, K, L) of ZGO NSs@NF and ZGO NRs after 5 cycles (E, K) and 100 cycles (F, L) at 200 mA g−1. | ||
Conclusions
In this study, we successfully developed a novel hierarchical hybrid 3D self-supported structure composed of free-standing ultrathin ZGO NSs on a conductive nickel foam (NF) through a simple CTAB-assisted hydrothermal process at 160 °C for 3 h. With the help of CTAB, the ultrathin ZGO NSs were interconnected and aligned vertically on the NF substrate to form a self-supported sponge-like network structure. This novel structure facilitated the electronic/ionic transport and stabilized the total electrode structure upon the repeated cycling. The ZGO NSs@NF exhibited an excellent electrochemical performance with a high discharge capacity, a long-term cycling stability, and a good rate capability. A high reversible discharge capacity of about 794 mA h g−1 was readily maintained after 500 cycles at a current density of 200 mA g−1, corresponding to an 81% capacity retention of the 2nd cycle. Even after cycling at a higher current density of 2000 mA g−1, the novel ZGO NSs@NF delivered a stable discharge capacity of 537 mA h g−1. Furthermore, the LiFePO4/ZGO NSs@NF full-cells also delivered a stable discharge capacity of 104 mA h g−1 after 80 cycles. Such self-supported structural construction of a 3D free-standing ultrathin NSs network on a conductive substrate could offer an excellent volume buffer effect and improve the interfacial contacts, which may stimulate the progress of other energy-efficient technologies.Author contributions
G. G. and Y. X. fabricated the electrode materials and analysed the electrochemical performance. S. L., B. D. and C. S. contributed to SEM and TEM measurements. S. L. carried out XRD measurements. L. S. performed TGA, and Y. W. performed BET tests. G. G and K. X. wrote the manuscript. All authors discussed the results. S. D. and K. X. supervised the project.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was supported partially by the Fundamental Research Funds for the Central Universities (xjj2015119), the National Natural Science Foundation of China (No. 51273158, 21303131), the China Postdoctoral Science Foundation (No. 2016M592776), and the Natural Science Foundation of Shaanxi Province (No. 2016JM5021).References
- N. Mahmood, T. Y. Tang and Y. L. Hou, Adv. Energy Mater., 2016, 6, 1600374 CrossRef.
- Z. Y. Wang, L. Zhou and X. W. Lou, Adv. Mater., 2012, 24, 1903–1911 CrossRef CAS PubMed.
- H. B. Wu, J. S. Chen, H. H. Hng and X. W. Lou, Nanoscale, 2012, 4, 2526–2542 RSC.
- F. Zhang and L. M. Qi, Adv. Sci., 2016, 3, 1600049 CrossRef PubMed.
- Z. S. Wu, W. C. Ren, L. Wen, L. B. Gao, J. P. Zhao, Z. P. Chen, G. M. Zhou, F. Li and H. M. Cheng, ACS Nano, 2010, 4, 3187–3194 CrossRef CAS PubMed.
- H. L. Wang, L. F. Cui, Y. A. Yang, H. S. Casalongue, J. T. Robinson, Y. Y. Liang, Y. Cui and H. J. Dai, J. Am. Chem. Soc., 2010, 132, 13978–13980 CrossRef CAS PubMed.
- Y. S. Hu, R. Demir-Cakan, M. M. Titirici, J. O. Muller, R. Schlogl, M. Antonietti and J. Maier, Angew. Chem., Int. Ed., 2008, 47, 1645–1649 CrossRef CAS PubMed.
- X. L. Wu, Y. G. Guo and L. J. Wan, Chem. – Asian J., 2013, 8, 1948–1958 CrossRef CAS PubMed.
- D. T. Ngo, R. S. Kalubarme, H. T. T. Le, J. G. Fisher, C. N. Park, I. D. Kim and C. J. Park, Adv. Funct. Mater., 2014, 24, 5291–5298 CrossRef CAS.
- J. Hwang, S. H. Woo, J. Shim, C. Jo, K. T. Lee and J. Lee, ACS Nano, 2013, 7, 1036–1044 CrossRef CAS PubMed.
- P. Poizot, S. Laruelle, S. Grugeon, L. Dupont and J. M. Tarascon, Nature, 2000, 407, 496–499 CrossRef CAS PubMed.
- Y. Z. Su, S. Li, D. Q. Wu, F. Zhang, H. W. Liang, P. F. Gao, C. Cheng and X. L. Feng, ACS Nano, 2012, 6, 8349–8356 CrossRef CAS PubMed.
- G. X. Gao, S. Y. Lu, Y. Xiang, B. T. Dong, W. Yan and S. J. Ding, Dalton Trans., 2015, 44, 18737–18742 RSC.
- G. X. Gao, S. Y. Lu, B. T. Dong, W. Yan, W. Wang, T. Zhao, C. Y. Lao, K. Xi, V. Kumar and S. J. Ding, J. Mater. Chem. A, 2016, 4, 10419–10424 CAS.
- G. Q. Zhang, B. Y. Xia, C. Xiao, L. Yu, X. Wang, Y. Xie and X. W. Lou, Angew. Chem., Int. Ed., 2013, 52, 8643–8647 CrossRef CAS PubMed.
- X. L. Jia, Z. Chen, X. Cui, Y. T. Peng, X. L. Wang, G. Wang, F. Wei and Y. F. Lu, ACS Nano, 2012, 6, 9911–9919 CrossRef CAS PubMed.
- X. Xu, W. Liu, Y. Kim and J. Cho, Nano Today, 2014, 9, 604–630 CrossRef CAS.
- S. H. Choi and Y. C. Kang, Small, 2014, 10, 474–478 CrossRef CAS PubMed.
- S. P. Wu, C. P. Han, J. Iocozzia, M. J. Lu, R. Y. Ge, R. Xu and Z. Q. Lin, Angew. Chem., Int. Ed., 2016, 55, 7898–7922 CrossRef CAS PubMed.
- Z. L. Hu, S. Zhang, C. J. Zhang and G. L. Cui, Coord. Chem. Rev., 2016, 326, 34–85 CrossRef CAS.
- C. J. Zhang, F. L. Chai, L. Fu, P. Hu, S. P. Pang and G. L. Cui, J. Mater. Chem. A, 2015, 3, 22552–22556 CAS.
- K. H. Seng, M. H. Park, Z. P. Guo, H. K. Liu and J. Cho, Nano Lett., 2013, 13, 1230–1236 CrossRef CAS PubMed.
- H. Y. Qiu, L. X. Zeng, T. B. Lan, X. K. Ding and M. D. Wei, J. Mater. Chem. A, 2015, 3, 1619–1623 CAS.
- Y. Wang and G. X. Wang, Chem. – Asian J., 2013, 8, 3142–3146 CrossRef CAS PubMed.
- Z. Fang, T. T. Qiang, J. X. Fang, Y. X. Song, Q. Y. Ma, M. Ye, F. Q. Qiang and B. Y. Geng, Electrochim. Acta, 2015, 151, 453–458 CrossRef CAS.
- H. H. Li, L. L. Zhang, C. Y. Fan, X. L. Wu, H. F. Wang, X. Y. Li, K. Wang, H. Z. Sun and J. P. Zhang, J. Mater. Chem. A, 2016, 4, 2055–2059 CAS.
- Z. Chen, Y. Yan, S. Xin, W. Li, J. Qu, Y. G. Guo and W. G. Song, J. Mater. Chem. A, 2013, 1, 11404–11409 CAS.
- J. K. Feng, M. O. Lai and L. Lu, Mater. Res. Bull., 2012, 47, 1693–1696 CrossRef CAS.
- S. P. Wu, R. Wang, Z. L. Wang and Z. Q. Lin, Nanoscale, 2014, 6, 8350–8358 RSC.
- S. X. Jin and C. X. Wang, Nano Energy, 2014, 7, 63–71 CrossRef CAS.
- J. Feng, C. Wang and Y. Qian, Mater. Lett., 2014, 122, 327–330 CrossRef CAS.
- W. W. Li, D. Chen and G. Z. Shen, J. Mater. Chem. A, 2015, 3, 20673–20680 CAS.
- T. Lv, X. Li and J. M. Ma, RSC Adv., 2014, 4, 49942–49945 RSC.
- D. Li, C. Q. Feng, H. K. Liu and Z. P. Guo, Sci. Rep., 2015, 5, 11326 CrossRef CAS PubMed.
- F. Zhang, R. H. Zhang, Z. Zhang, H. K. Wang, J. K. Feng, S. L. Xiong and Y. T. Qian, Electrochim. Acta, 2014, 150, 211–217 CrossRef CAS.
- Y. Subramanian, K. Kaliyappan and K. S. Ramakrishnan, J. Colloid Interface Sci., 2017, 498, 76–84 CrossRef CAS PubMed.
- W. Li, Y. X. Yin, S. Xin, W. G. Song and Y. G. Guo, Energy Environ. Sci., 2012, 5, 8007–8013 CAS.
- F. Zou, X. L. Hu, Y. M. Sun, W. Luo, F. F. Xia, L. Qie, Y. Jiang and Y. H. Huang, Chem. – Eur. J., 2013, 19, 6027–6033 CrossRef CAS PubMed.
- W. W. Li, X. F. Wang, B. Liu, J. Xu, B. Liang, T. Luo, S. J. Luo, D. Chen and G. Z. Shen, Nanoscale, 2013, 5, 10291–10299 RSC.
- Y. R. Lim, C. S. Jung, H. S. Im, K. Park, J. Park, W. I. Cho and E. H. Cha, J. Mater. Chem. A, 2016, 4, 10691–10699 CAS.
- X. Liu, J. Zai, B. Li, J. Zou, Z. Ma and X. Qian, J. Mater. Chem. A, 2016, 4, 10552–10557 CAS.
- R. Wang, S. P. Wu, Y. C. Lv and Z. Q. Lin, Langmuir, 2014, 30, 8215–8220 CrossRef CAS PubMed.
- W. Wang, J. W. Qin and M. H. Cao, ACS Appl. Mater. Interfaces, 2016, 8, 1388–1397 CAS.
- L. Yu, R. Zou, Z. Zhang, G. Song, Z. Chen, J. Yang and J. Hu, Chem. Commun., 2011, 47, 10719–10721 RSC.
- R. Yi, J. K. Feng, D. P. Lv, M. L. Gordin, S. R. Chen, D. W. Choi and D. H. Wang, Nano Energy, 2013, 2, 498–504 CrossRef CAS.
- Y. M. Sun, X. L. Hu, J. C. Yu, Q. Li, W. Luo, L. X. Yuan, W. X. Zhang and Y. H. Huang, Energy Environ. Sci., 2011, 4, 2870–2877 CAS.
- S. Yuan, X. L. Huang, D. L. Ma, H. G. Wang, F. Z. Meng and X. B. Zhang, Adv. Mater., 2014, 26, 2273–2279 CrossRef CAS PubMed.
- L. F. Shen, Q. Che, H. S. Li and X. G. Zhang, Adv. Funct. Mater., 2014, 24, 2630–2637 CrossRef CAS.
- G. X. Gao, H. B. Wu, S. J. Ding, L. M. Liu and X. W. Lou, Small, 2015, 11, 804–808 CrossRef CAS PubMed.
- W. M. Chen, L. Y. Lu, S. Maloney, Y. Yang and W. Y. Wang, Phys. Chem. Chem. Phys., 2015, 17, 5109–5114 RSC.
- W. M. Chen, S. Maloney and W. Y. Wang, Electrochim. Acta, 2015, 176, 96–102 CrossRef CAS.
- W. W. Li, X. F. Wang, B. Liu, S. J. Luo, Z. Liu, X. J. Hou, Q. Y. Xiang, D. Chen and G. Z. Shen, Chem. – Eur. J., 2013, 19, 8650–8656 CrossRef CAS PubMed.
- J. K. Feng, M. O. Lai and L. Lu, Electrochem. Commun., 2011, 13, 287–289 CrossRef CAS.
- J. S. Kim, A. Y. Kim, Y. W. Byeon, J. P. Ahn, D. Byun and J. K. Lee, Electrochim. Acta, 2016, 195, 43–50 CrossRef CAS.
- Y. Feng, X. D. Li, Z. P. Shao and H. T. Wang, J. Mater. Chem. A, 2015, 3, 15274–15279 CAS.
- K. Y. Lin, B. J. Ma, W. G. Su and W. Y. Liu, Appl. Surf. Sci., 2013, 286, 61–65 CrossRef CAS.
- N. Zhang, S. X. Ouyang, T. Kako and J. H. Ye, Chem. Commun., 2012, 48, 1269–1271 RSC.
- J. H. Huang, K. N. Ding, Y. D. Hou, X. C. Wang and X. Z. Fu, ChemSusChem, 2008, 1, 1011–1019 CrossRef CAS PubMed.
- S. C. Yan, J. J. Wang and Z. G. Zou, Dalton Trans., 2013, 42, 12975–12979 RSC.
- G. X. Gao, H. B. Wu, B. T. Dong, S. J. Ding and X. W. Lou, Adv. Sci., 2015, 2, 1400014 CrossRef PubMed.
- J. Liu, G. K. Zhang, J. C. Yu and Y. D. Guo, Dalton Trans., 2013, 42, 5092–5099 RSC.
- X. Ge, S. Y. Song and H. J. Zhang, CrystEngComm, 2012, 14, 7306–7311 RSC.
- S. H. Chen and D. L. Carroll, J. Phys. Chem. B, 2004, 108, 5500–5506 CrossRef CAS.
- F. Zou, X. L. Hu, L. Qie, Y. Jiang, X. Q. Xiong, Y. Qiao and Y. H. Huang, Nanoscale, 2014, 6, 924–930 RSC.
- Q. Li, X. G. Miao, C. X. Wang and L. W. Yin, J. Mater. Chem. A, 2015, 3, 21328–21336 CAS.
- L. Zhang, X. F. Cao, Y. L. Ma, X. T. Chen and Z. L. Xue, CrystEngComm, 2010, 12, 3201–3206 RSC.
- G. X. Gao, H. B. Wu and X. W. Lou, Adv. Energy Mater., 2014, 4, 1400422 CrossRef.
- J. Wang, J. Polleux, J. Lim and B. Dunn, J. Phys. Chem. C, 2007, 111, 14925–14931 CAS.
- T. Brezesinski, J. Wang, S. H. Tolbert and B. Dunn, Nat. Mater., 2010, 9, 146–151 CrossRef CAS PubMed.
- Z. Hu, L. Wang, K. Zhang, J. Wang, F. Cheng, Z. Tao and J. Chen, Angew. Chem., Int. Ed., 2014, 53, 12794–12798 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental sections; digital picture, XRD and EDX patterns of ZGO NSs@NF; FESEM images of ZGO NRs@NF, pristine ZGO flakes and ZGO NRs; Nyquist plots of ZGO NSs@NF, ZGO NRs@NF and pristine ZGO flakes; and post-mortem FESEM images of ZGO NRs@NF. See DOI: 10.1039/c7nr05407f |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2018 |