
An alternative route to single ion conductivity using multi-ionic salts†
Sumanth
Chereddy
a,
Parameswara Rao
Chinnam
*a,
Vijay
Chatare
a,
Stephen Patrick
diLuzio
a,
Mallory P.
Gobet
 b,
Steven G.
Greenbaum
b and
Stephanie L.
Wunder
*b
b,
Steven G.
Greenbaum
b and
Stephanie L.
Wunder
*b
aDepartment of Chemistry, Temple University, Philadelphia, PA 19122, USA. E-mail: slwunder@temple.edu; chprbhu@temple.edu
bDepartment of Physics, Hunter College, CUNY, New York, NY 10065, USA
First published on 20th February 2018
Abstract
Multi-ionic lithium salts comprised of polyoligomeric silsesquioxanes (POSS) functionalized with eight – (LiNSO2CF3) groups, referred to as POSS-(LiNSO2CF3)8, can be dissolved at very high loadings into tetraglyme (G4), where they can be considered solvent-in-salt electrolytes. With increasing dilution, colloidal solutions are formed. Two systems were investigated, neat POSS-(LiNSO2CF3)8 in G4 and mixtures of POSS-(LiNSO2CF3)8 with LiPF6 or lithium bis(trifluoromethanesulfonyl)imide (LiTFSI). PFG-NMR indicates that Li can be un-dissociated, completely dissociated and surrounded by G4 molecules, or as contact ion pairs (in which there are 3–4 ether oxygen contacts and one contact with the oxygen from the anion). Equilibria exist between the species of POSS-(LiNSO2CF3)8 and if there is rapid equilibration between the Li states, and close enough proximity between the POSS-(LiNSO2CF3), then the Li+ ions can migrate by a Grotthus-type coordinated hopping mechanism, as well as by a purely diffusive motion. Unlike polymer single ion conductors, where the backbone flexibility permits cluster/aggregate formation, which inhibits escape and mobility of the Li+ ions, the rigid POSS cube and its colloidal structure in G4 prevents formation of POSS-(NSO2CF3−)⋯Li+⋯(−CF3NSO2)-POSS triplets. Instead, the solvated Li+ in POSS-(NSO2CF3−)⋯Li+⋯G4 can be more easily removed to form conductive G4⋯Li+−⋯G4. Good ionic conductivities (∼10−4 S cm−1) and lithium ion transference numbers of tPP+ = 0.65 can be achieved in these systems. Mixtures of 80 wt% LiTFSI and 20 wt% POSS-(LiNSO2CF3)8 in G4 at an O/Li ratio of 20/1, yield both high conductivity (σ = 3.3 × 10−3 S cm−1) and high (tPP+ = 0.65) transference number. Stable cycling between C/2 and 2C with high capacity retention was achieved using Li/[G4/80 wt% LiTFSI/20 wt% POSS-(LiNSO2CF3)8]/LiFePO4 half-cells.
Conceptual insightsCritical goals for the achievement of safe, nonvolatile solid organic electrolytes in lithium ion batteries are the development of materials with both high ionic conductivity (σ) and lithium ion transference numbers, tLi+. Liquids offer advantages over solids because they can permeate porous cathodes and access the active components more effectively. This article shows that the use of multi-ionic lithium salts based on polyoligomeric silsesquioxanes (POSS) with pendant anions, POSS-(LiNSO2CF3)8, can result in both high ionic conductivity (σ > 10−4 S cm−1) and high lithium ion transference numbers (tLi+ = 0.65) when dissolved in low volatility tetraglyme (G4) solvents. Solvent in salt electrolytes are formed at high POSS-(LiNSO2CF3)8 concentrations and colloids are formed with increasing dilution. The absence of aggregation in the partially charged, rigid, symmetric POSS-(LiNSO2CF3)8 in tetraglyme prevents trapping of Li+ ions that can slow down Li+ ion migration in polymer single ion conductors. Li+ ion migration can occur by both diffusive and coordinated hopping mechanisms. The addition of mono-ionic lithium salts increases conductivity (σ > 10−3 S cm−1) while maintaining tLi+ = 0.65. |
Introduction
One of the major problems in the development of separators for lithium-ion or metal batteries (LIBs, LMBs) is to optimize ionic conductivity (σ) and reduce concentration polarization. Concentration gradients adversely affect cell performance by contributing to the growth of dendrites, which are branched or needle-like structures that form instead of desirable flat, uniform Li0 deposition. Dendrites can break off, resulting in a decrease in energy density, or span the cell separator causing internal shorting, heating, thermal runaway and catastrophic cell failure. Dendrites are particularly a concern for lithium metal batteries (LMBs), but also occur in lithium-ion batteries (LIBs), since Li0 metal rather than intercalated Li+ can deposit on the anode in LIBs during fast charging or at low temperatures.1 The transition from graphitic to metallic lithium anodes would enhance energy density 10 fold, and metallic lithium anodes would enable the use of un-lithiated materials such as sulfur or air to replace intercalation cathodes for lithium/sulfur or lithium/air batteries2,3 with improved energy density. In these cases, the safety issues associated with dendrite growth when using metallic Li0 are a major concern.There has been extensive theoretical and experimental research on the prevention of dendritic growth, including mechanical inhibition4 and limiting concentration gradients that result in anion depletion near the anode.5 The latter can be avoided by the use of electrolytes with high ionic conductivities and low anion mobility, i.e. high Li+ ion high transference numbers (tLi+). The transference number is by definition the contribution of a single charged species to transport under the influence of an electric field. Since tLi+ + tX− = 1, if the anion is immobile tX− = 0, tLi+ = 1, and concentration polarization does not develop. A tLi+ = 1 was shown to enhance power and energy density, particularly at high discharge rates, over electrolytes with tLi+ = 0.2, even with an order of magnitude decrease in conductivity.6 Recent modelling studies suggest that tLi+ ≈ 0.7 would also allow a higher attainable state of charge at high charge rates.7
However, most electrolytes are not ideal and lithium can be transported in the form of neutral contact ion pairs, and neutral or charged aggregates. When the electrolyte is non-ideal, an equilibrium exists between Li+, X−, LiX, Li2X+, LiX2− (and even larger aggregates), and tLi+ depends on the concentration (activity), mobility and charge of each charged species.8 The measurement of tLi+ is therefore not straightforward and an accurate determination is not always possible.9 The two most common methods used to measure transport properties of lithium are spin echo pulse-field gradient (PFG)-NMR and electrochemical techniques,10 particularly potentiostatic polarization, developed by Bruce and Vincent.11,12 In spin echo PFG-NMR,13–16 the average static self-diffusion coefficients of all species containing a nucleus with a unique chemical shift is monitored, whereas only charged species are measured using potentiostatic polarization. Both methods can overestimate the effective tLi+, and they often do not agree with each other.
Concentration gradients are avoided in polymer single ion conductors (SICs) by covalent attachment of anions to the polymer backbone so that tLi+ → 1. However, conductivities of polymer SICs have remained low (σ < 10−5 S cm−1).17 In polymer electrolytes the cation motion is believed to be coupled to the backbone dynamics, so attempts to increase conductivity have included incorporating flexible chains with low glass transition temperatures (Tgs) such as polydimethylsiloxanes. One of the reasons for the low conductivity in polymer SICs is extensive ion aggregation. As in the case of bi-ionic conductors, there are completely dissociated ions, neutral contact ion pairs (CIPs) and poorly conductive ion triplets and aggregates (AGG) (Fig. 1A). However, for polymer SICs larger ion aggregates also form, as shown schematically in Fig. 1B and there are few dissociated ions.18,19 In this case, polymer flexibility adversely affects conductivity by contributing to the ion clustering, since the pendant ion groups have the mobility necessary to phase separate from the more hydrophobic backbone. Confirmation that the formation of these aggregates causes a decrease in ionic conductivity comes from the unexpected finding that addition of a tetraglyme plasticizer to an ionomer resulted in a reduction of ionic conductivity by 4–5 orders of magnitude, as the result of recoupling of the ionic conductivity with the polymer segmental dynamics.20 Single ion conductors can be crosslinked and swollen with liquid solvents to achieve tLi+ = 1 and higher ionic conductivity than dry systems21,22,23 and as in the dry systems, highly dissociative ionic species reduce the formation of aggregates and increase conductivity. Thus, replacement of carboxylate, phosphate and sulfonate anions with the LiTFSI-like anion in lithium[(4-styrenesulfonyl)trifluoromethane-sulfonyl)imide]24,25 and poly[(4-styrenesulfonyl)(trifluoromethyl(S-trifluoromethylsulfonylimino)sulfonyl)imide]26 have resulted in the best ion conductivities to date.
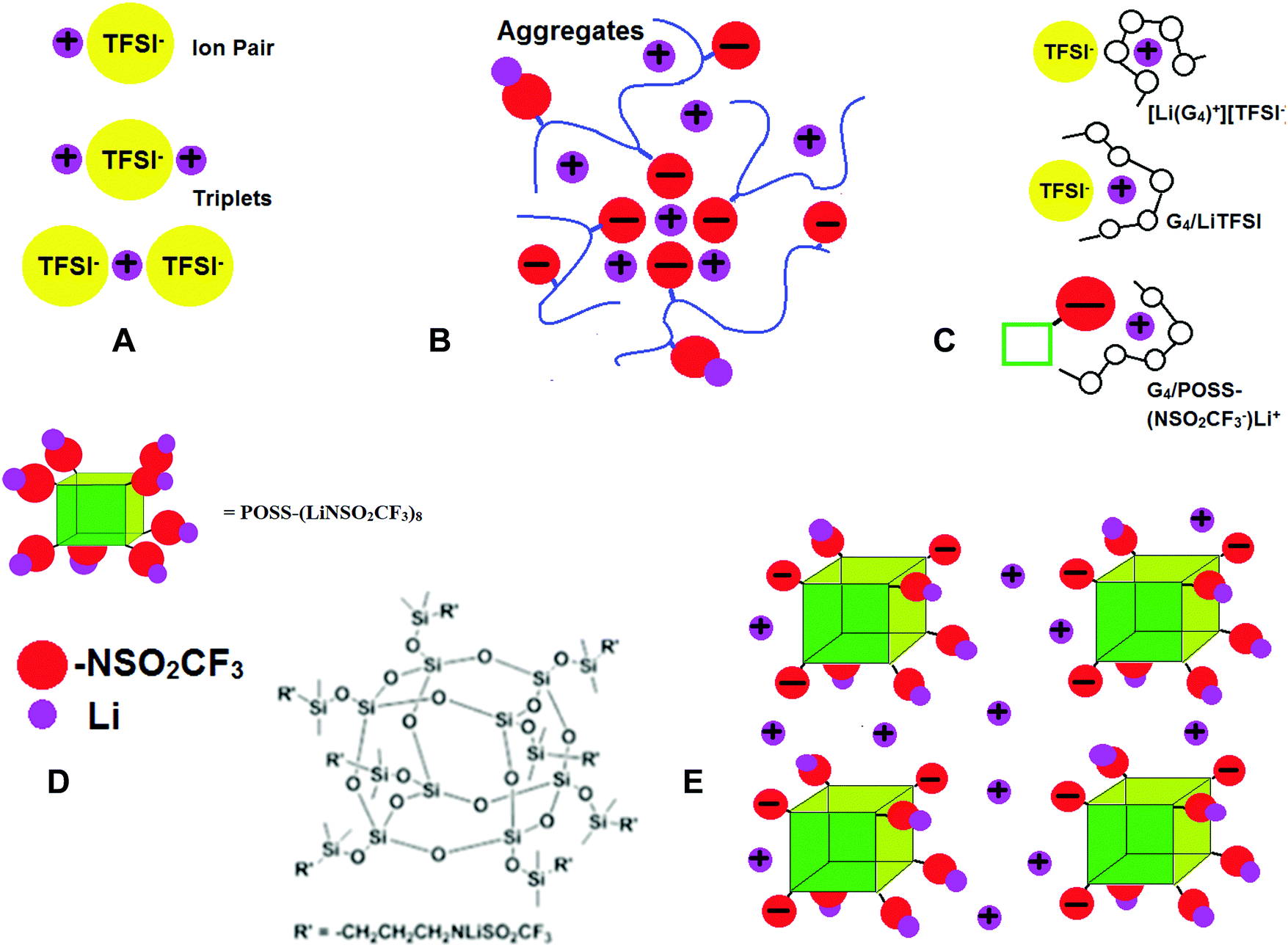 | ||
| Fig. 1 Schematic of (A) ion pairs and ion triplets found in LiX salts and polymer SICs; (B) higher order aggregates that can occur in polymer single ion conductors (SICs); (C) solvate ionic liquid [Li(G4)+]/[TFSI−], mixtures of G4 and LiTFSI and mixtures of G4 and POSS-(NSO2CF3−)Li+; (D) structure of POSS-(LiNSO2CF3)8; and (E) POSS-(NSO2CF3−)8 anions dispersed in G4; G4 not shown. | ||
Nonflammable liquids have some advantages over solids when used as battery electrolytes. In particular, they can permeate the space between the composite electrodes so that more of the active material is accessible. In order to increase tLi+ in liquids, the strategy has been to use large, slow moving anions. Anions tethered to SiO2 nanoparticles27 and polyanions dissolved in liquid solvents28–30 have been investigated, and can have high room temperature conductivities (σ > 10−4 S cm−1) and tLi+ > 0.7. Recently investigated solvent-in-salt electrolytes also exhibit tLi+ ∼ 0.7 and high conductivity.31 Tetraglyme (G4), CH3–O–(CH2CH2O)4–CH3, is an alternative to expensive nonflammable ionic liquids (ILs), since it forms a solvate (or chelate) IL, where a third component (here G4) strongly coordinates Li+, thus forming a complex cation. This has been shown to occur for a 1/1 molar mixture of G4 and LiTFSI, [Li(G4)+][TFSI−], with brackets used to indicate the molar ratios, while for other compositions or salts, coordination of Li+ can occur between both G4 and the anion (Fig. 1C).32–38
Here we propose the use of functionalized symmetric, multi-ionic polyhedral oligomeric silsesquioxane (POSS) with dissociative lithium salts (Fig. 1D) dissolved in tetraglyme (G4), CH3–O–(CH2CH2O)4–CH3, to increase lithium ion transference numbers (tLi+) and avoid the formation of large ion clusters that trap the Li+ ions. The salts are composed of (SiO1.5)8 cubes, where the eight corners have been functionalized with LiTFSI-type salts (see Experimental details, synthesis and Scheme S1 in ESI†), and will be referred to as POSS-(LiNSO2CF3)8. The POSS-(LiNSO2CF3)8 can be considered solvent-in-salt electrolytes, since salt weights = 3× solvent weight can be achieved.31 When dissolved in G4, some of the Li ions dissociate, resulting in partially charged species, and form a colloidal solution. These large negatively charged anions repel each other, preventing aggregation Fig. 1E. We have investigated POSS-(LiNSO2CF3)8 in G4, and compared the properties of G4/POSS-(LiNSO2CF3)8 with those of G4/LiX, X = TFSI−, PF6−, and lithium bis(perfluoroethanesulfonyl)imide (BETI−) = [N(SO2C2F5)2]−, and their mixtures, G4/POSS-(LiNSO2CF3)8/LiX. Mixtures of POSS-(LiNSO2CF3)8 and LiTFSI or LiPF6 dissolved in G4 have tLi+ ∼ 0.65 in the liquid state, with conductivities σ > 10−3.
Results and discussion
The preparation and compositions of all the samples are given in Table S1 (ESI†). G4/POSS-(LiNSO2CF3)8 and G4/LiX, X = TFSI−, PF6− and BETI−, were characterized based on the ratio of ether oxygens in G4 to Li+ ions (O/Li ratio), as is common in PEO based electrolytes, and/or their molar ratios. In the case of the 1/1 molar ratio for G4 and LiX, this is O/Li = 5/1. In the case of G4/POSS-(LiNSO2CF3)8, a 1/1 molar ratio is O/Li = 5/8, while to provide one G4 for all of the eight Li on the POSS-(LiNSO2CF3)8, requires O/Li = 40/1. In mixtures of POSS-(LiNSO2CF3)8 with LiX, the weight ratio of POSS-(LiNSO2CF3)8 to LiX varied from 100/0 to 0/100.Temperature dependent ionic conductivities (see ESI† for experimental details) of G4/POSS-(LiNSO2CF3)8 and G4/LiTFSI as a function of O/Li, (Fig. 2 and Table S2, ESI†) show about an order of magnitude decrease in conductivity of G4/POSS-(LiNSO2CF3)8 compared with that of G4/LiTFSI. This is encouraging since if the pendant anion were on a polymer SIC, a 2–4 order of magnitude decrease in conductivity is expected. The conductivity of G4/LiTFSI agrees well with previously reported values at 30 °C (6.56 × 10−3 S cm−1).39 Of further interest is the temperature dependent viscosity of both electrolytes (Table S3, ESI†), where the viscosity of the G4/LiTFSI is only ∼21% less than that of the G4/POSS-(LiNSO2CF3)8 at 20 °C. This suggests that large scale aggregation does not occur in the G4/POSS-(LiNSO2CF3)8 (which would otherwise result in large viscosity increases). These results suggest that the difference in conductivity is related to a decreased contribution from the large POSS-(NSO2CF3−)8 anions (2520 g mol−1) compared with the TFSI− anion (215 g mol−1) and/or to the better dissociative properties of the LiTFSI (CF3SO2NLiSO2CF3) compared with –(LiNSO2CF3)8, which has only half the number of electron withdrawing groups. As in the case of bi-ionic conductors and polymer SICs, the conductivity increases as the electron withdrawing groups of the anion increase, and as the negative charge becomes more delocalized. Addition of LiTFSI to G4/POSS-(LiNSO2CF3)8 increases the conductivity, as expected (Table S2, ESI†).
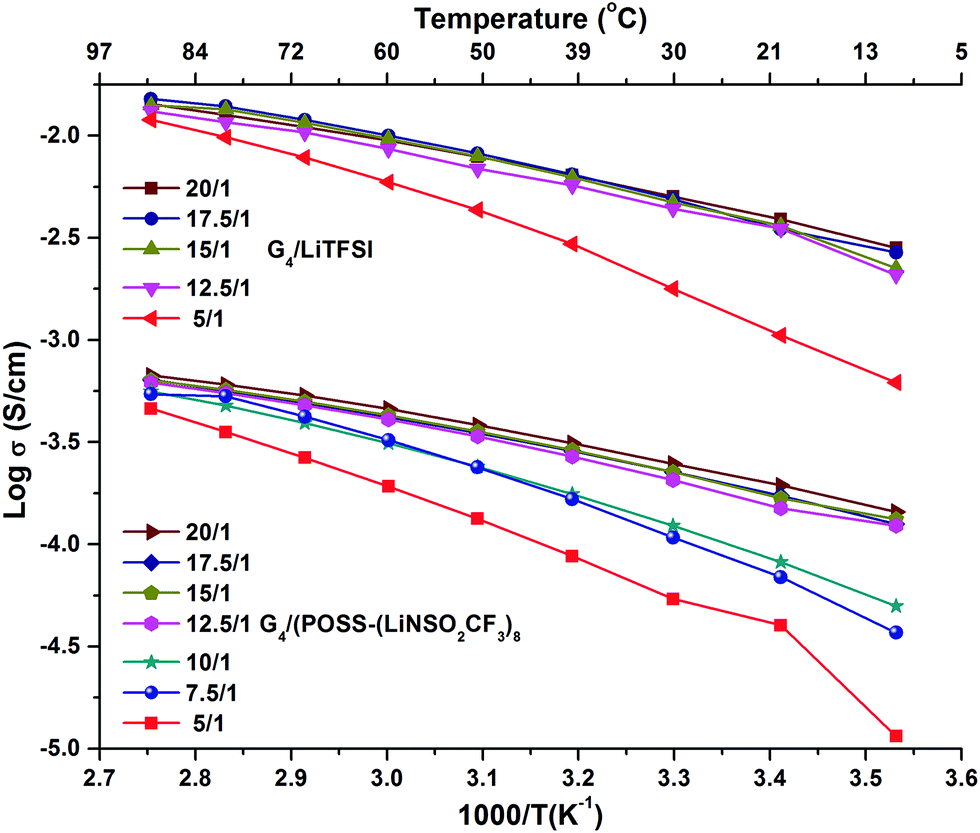 | ||
| Fig. 2 Temperature dependent ionic conductivity as a function of O/Li ratio for G4/LiTFSI and G4/POSS-(LiNSO2CF3)8. | ||
The absence of aggregation and the formation of a stable colloid for G4/POSS-(LiNSO2CF3)8 are indicated by the lack of phase separation or precipitation over months, even though the weight of POSS-(LiNSO2CF3)8 is up to 3× greater than the weight of G4 (see Table S1, ESI†). TEM images (Fig. 3) also show no aggregates, and EDAX data (Fig. S1, ESI†) confirm that these images contain POSS-(LiNSO2CF3)8.
 | ||
| Fig. 3 TEM image of G4/POSS-(LiNSO2CF3)8 with O/Li = 7.5/1. Sample was prepared by dissolution of POSS-(LiNSO2CF3)8 in polyethylene glycol monomethyl ether acrylate (Mn = 350 g mol−1), 0.1% AIBN, and dilution with acetonitrile (ACN). After dropping the solution on a TEM grid, and evaporation of the ACN, the sample polymerized. | ||
Differential scanning calorimetry (DSC) data (Table S4 and Fig. S2, ESI†) of the neat G4 indicated it melts at Tm = −28.3 °C in agreement with literature values (Tm = −3035), with no glass transition temperature (Tg). Attempts to crystallize POSS-(LiNSO2CF3)8 were unsuccessful, and it decomposed before Tg was reached. At comparable O/Li ratios, G4/LiTFSI more effectively suppresses crystallization than does POSS-(LiNSO2CF3)8. For G4/LiTFSI the melt is completely suppressed for O/Li = 15/1 to 5/1, and there are only very small (crystallization and remelt) peaks for the 20/1 and 17.5/1 samples. For G4/POSS-(LiNSO2CF3)8, the melt temperature Tm only completely disappear at O/Li = 7.5/1 and 5/1 and there is a very small crystallization and remelt at O/Li = 10/1. Since Li+ ions are typically solvated by a combination of 4–5 ether oxygens and/or anion contacts, in the case of G4/POSS-(LiNSO2CF3)8, more of these contacts may come from contact ion pairs than from the ether oxygens of G4 than in the case of [Li(G4)+]/[TFSI−], where the Li+ ions can be completely solvated by G4 (Fig. 1C). This would leave more G4 molecules available for crystallization in the case of G4/POSS-(LiNSO2CF3)8 compared with G4/LiTFSI, as observed.
When both G4/POSS-(LiNSO2CF3)8 and G4/LiTFSI are amorphous, Tg of G4/LiTFSI is 10 °C higher than G4/POSS-(LiNSO2CF3)8 at the same O/Li ratio. This also suggests that there is more Li+ dissociation in G4/LiTFSI raising its Tg, while in G4/POSS-(LiNSO2CF3)8 the Li+ ion dissociates less and forms more contact ion pairs (–NSO2CF3−⋯Li+⋯G4). Both G4/LiTFSI and G4/POSS-(LiNSO2CF3)8 show the expected increase in Tg with increasing salt concentration, which can be been attributed to the effects of increased viscosity and –CH2CH2O⋯Li+⋯OCH2CH2– crosslinks with increased salt concentration.
Thermogravimetric analysis (TGA) data (Fig. 4) also provide information on the complex formed between the Li+ cation and G4. Unlike the case of [Li(G4)+][TFSI−] (1/1 mole ratio, O/Li = 5/1), where removal of G4 is delayed until ∼200 °C36 compared with G4 removal at ∼100 °C in compositions with excess G4, delayed removal of G4 does not occur for any composition of G4/POSS-(LiNSO2CF3)8 (Fig. S3, ESI†) except for the 4/1 mole ratio (O/Li = 2.5/1) (Fig. 4). The TGA data therefore indicates that for G4/POSS-(LiNSO2CF3)8 (O/Li ≥ 5/1) the Li+/G4 complex is not formed. Instead, the Li+ ion is more loosely complexed with the G4, and possibly also interacting with the NSO2CF3− anions of POSS-(NSO2CF3−)8. For G4/POSS-(LiNSO2CF3)8 (4/1 mole ratio, O/Li = 2.5/1), G4 comes off predominantly at 300 °C, at an even higher temperature than the pure POSS-(LiNSO2CF3)8. In this case, the G4 is even more tightly complexed than for the [Li(G4)+][TFSI−], which is removed at 200 °C. This arises since one G4 must be shared between two Li+ of G4/POSS-(LiNSO2CF3)8 so that the G4 is trapped between the cubes, as shown in a 2D representation (Fig. 4). It should be noted that even for this composition, where the weight of the POSS-(LiNSO2CF3)8 is 3× that of the G4, the mixture is stable against any signs of phase separation or precipitation for months.
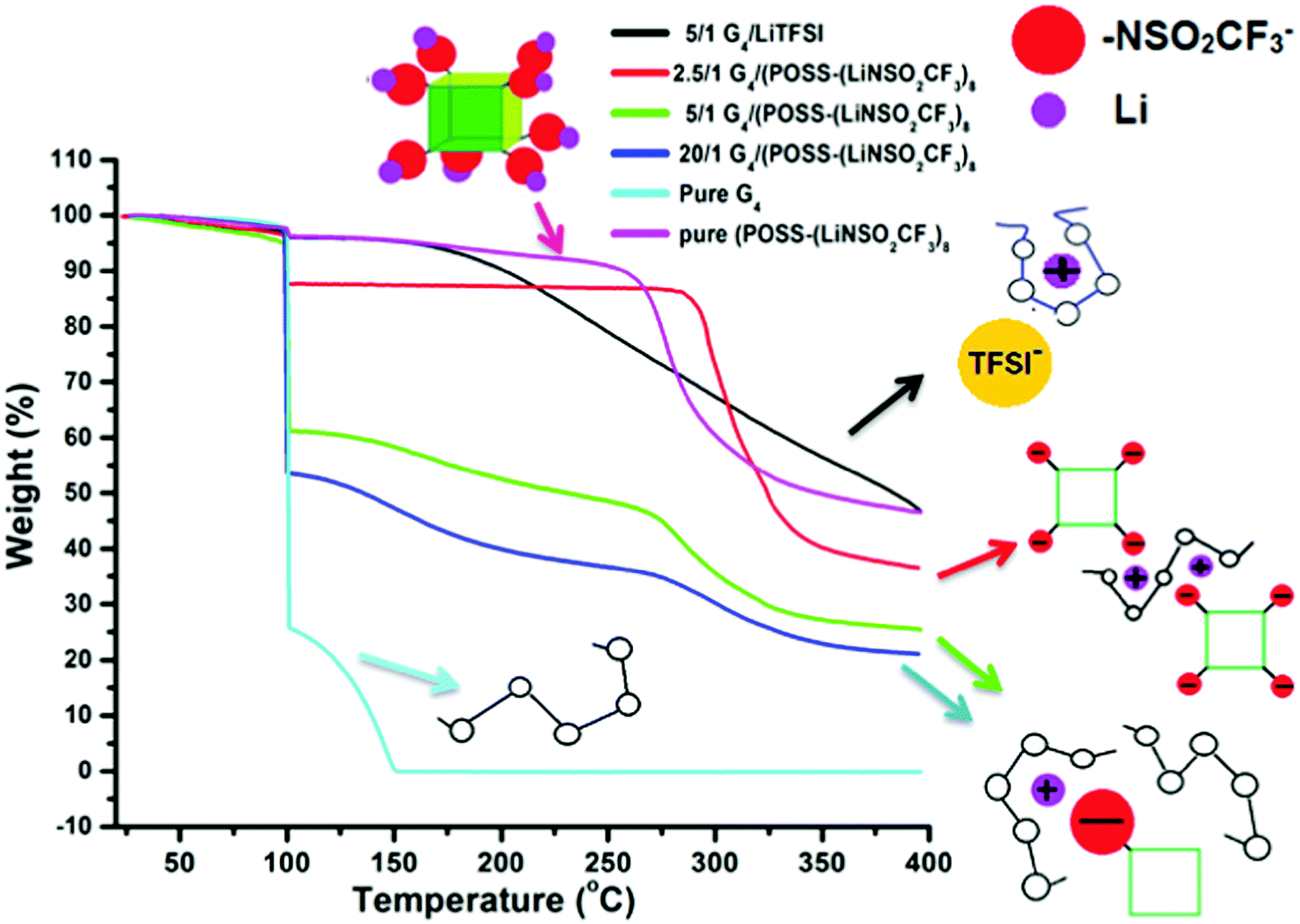 | ||
| Fig. 4 TGA weight loss data: The samples were heated to 100 °C (at 10 °C min−1), held at that temperature for 1 h, and then heated to 400 °C at 10 °C min−1. | ||
Structure of the liquids by X-ray scattering
Wide angle X-ray scattering (WAXS) data obtained at room temperature for neat G4 and neat POSS-(LiNSO2CF3)8 are shown in Fig. 5A. G4 crystallized at low temperatures (Tm = −30 °C; shown at −173 °C), but POSS-(LiNSO2CF3)8, which is a glass, had similar WAXS at all temperatures (not shown). At room temperature, G4 shows a single broad peak centered at 2θ = 21–22° (q ∼ 15 nm−1; d ∼ 0.45 nm) and is a feature typical of molecular liquids that can distinguish between intra- and intermolecular distances.40 POSS-(LiNSO2CF3)8 exhibits a shoulder peak at 2θ ∼ 22° (q ∼ 15.5 nm−1; d ∼ 0.40 nm), and two broad features at 2θ ∼ 18.5° (q ∼ 13.1 nm−1; 0.48 nm) and 2θ ∼ 6.9° (q = 4.9 nm−1, d = 1.28 nm). Assignments of the peaks can be made in analogy with LiTFSI in the (simulated) liquid state (530 K). For LiTFSI, deconvolution of S(q) obtained from MD simulations41 in the 4 < q/nm−1 < 20 region show three Gaussian functions: a shoulder peak at 2θ = 21.94° (q ∼ 15.5 nm−1; d = 0.405 nm), and peaks centered at 2θ = 16.8° (q = 11.9 nm−1; d = 0.53 nm) and 2θ = 8.7° (q = 6.2 nm−1; d = 1.01 nm) and. The 2θ = 21.94° (q ∼ 15.5 nm−1; d = 0.405 nm) shoulder peak in LiTFSI, known as the contact peak, corresponds to distances between neighboring atoms of different ions, defining the boundary between intra- and inter-molecular features,41 and can be similarly assigned for amorphous POSS-(LiNSO2CF3)8. The 2θ = 16.8° feature in LiTFSI has been attributed to a charge-ordering peak (COP), often referred to as the polar network, and is a feature observed in ionic liquids and molten salts, since ions need to be surrounded by counter-ions to establish local electroneutrality.42 In LiTFSI, this assignment is corroborated by a more ordered COP at d = 0.52 nm in the crystalline state.43 The 2θ =18.5° (q ∼ 13.1 nm−1; d = 0.48 nm) feature in POSS-(LiNSO2CF3)8 can also possibly be assigned to the COP peak, but crystalline POSS-(LiNSO2CF3)8 is not available to make a similar comparison. Finally, the 2θ = 8.7° (6.2 nm−1, 1.01 nm) peak in LiTFSI was attributed to distances between sulfur atoms belonging to two TFSI− ions, oriented side by side without an intervening Li, and occurs at 0.86 nm in crystalline LiTFSI.42 By analogy, the 2θ = 6.9° (q ∼ 4.9 nm−1; 1.28 nm) peak in POSS-(LiNSO2CF3)8 may correspond to some larger distance between the sulfur containing pendant anions. Small angle X-ray scattering (SAXS) data (not shown) showed no structure at small q for either G4 or POSS-(LiNSO2CF3)8. | ||
Fig. 5 (A) WAXS data for neat G4 (25 and −173 °C), neat POSS-(LiNSO2CF3)8, and mole ratios of [G4]/[POSS-(LiNSO2CF3)8] also shown as (O/Li ratio); and (B) SAXS data at 25 °C for [G4]/[POSS-(LiNSO2CF3)8], O/Li = 7.5/1 obtained in 2 regions ( , ,  ); and the (C) S(q) and (D) P(R) data. ); and the (C) S(q) and (D) P(R) data. | ||
WAXS data for mixtures of G4/POSS-(LiNSO2CF3)8 at room temperature with O/Li = 2.5/1, 5/1, 7.5/1 are shown in Fig. 5A. Temperature dependent data (not shown) were identical, and none of the samples showed crystalline peaks of G4 at −100 °C. With the addition of G4 to POSS-(LiNSO2CF3)8 the (shoulder) contact peak of POSS-(LiNSO2CF3)8 at 2θ ∼ 22° (q ∼ 15.5 nm−1; 0.40 nm) shifts to lower angles, i.e. larger distances, and merges with that of G4, and the intense charge ordering peak at 2θ ∼ 18.5° (q ∼ 13.1 nm−1; 0.48 nm) disappears. In the case of G4/LiTFSI (1/1 molar ratio), the contact peak (shoulder) at 2θ = 21.94° (q ∼ 15.5 nm−1; d = 0.405 nm) also shifts to lower values of 2θ = 21.2° (q ∼ 15.0 nm−1; d = 0.418 nm), i.e. larger distances, and the intense charge ordering peak also disappears. The addition of G4 to POSS-(LiNSO2CF3)8 for O/Li = 2.5/1 and 5/1 results in a shift of the feature at 2θ ∼ 6.9° (q = 4.9 nm−1, d = 1.28 nm), tentatively assigned to separation between the anionic side-groups, to lower angles 2θ = 5.48° (q = 3.9 nm−1; d = 1.61 nm), consistent with an increase in the separation distance between POSS-(LiNSO2CF3)8 due to intervening G4 molecules. In the case of G4/LiTFSI a new peak appears that shifts from higher angles to 2θ = 13.38° (q ∼ 9.5 nm−1, d = 0.66 nm) for the 1/1 molar ratio, and is attributed to a characteristic separation distance between anions consistent with glyme-solvated Li ions.40,41
The peak associated with anion separation in G4/POSS-(LiNSO2CF3)8 at 2θ ∼ 6.9° (q = 4.9 nm−1, d = 1.28 nm) can only be observed for G4/POSS-(LiNSO2CF3)8 with O/Li = 2.5/1 and 5/1, which correspond to the mole ratios [4G4][1 POSS-(LiNSO2CF3)8] and [8G4][1 POSS-(LiNSO2CF3)8], where 1 G4 is shared between two Li or there is 1 G4 for each Li, respectively. Since it was not possible to investigate values of 2θ < 5° by WAXS (due to instrumental limitations), SAXS was used to determine whether aggregation/clustering or ordering could be observed for G4/POSS-(LiNSO2CF3)8 for O/Li = 7.5/1. This sample was chosen since it had the highest concentration of POSS-(LiNSO2CF3)8 whose viscosity allowed it to be put into narrow quartz capillaries. SAXS data were obtained in two q regions, which were spliced together (Fig. 5B). Subtraction of G4 from G4/POSS-(LiNSO2CF3)8 in the 3 to 20 q (nm−1) region showed that the 2θ = 18.5 peak of POSS-(LiNSO2CF3)8 persisted in the mixture and was distinct from the close 2θ = 21–22 peak (q ∼ 15 nm−1; d = 0.45 nm) of G4; subtraction of G4 was not done in WAXs data, Fig. 5A. SAXS data from q = 1 nm−1 to q = 5 nm−1 obtained for G4/POSS-(LiNSO2CF3)8 with O/Li = 7.5/1 was fit to a structure factor, assuming a model where the primary particles were hard spheres (Fig. 5C). The form factor (Fig. 5D) gave “particles” of 1.57 nm radius. The SiO1.5 core has an approximate radius of 0.45 nm.44 The large radius observed by SAXS suggests that the POSS-(LiNSO2CF3)8 cubes do not aggregate and instead form solvated structures, with the space between adjacent SiO1.5 cores of POSS-(LiNSO2CF3)8 occupied by free G4, G4 participating in contact ion pairs (one anion –NSO2CF3−⋯Li+ contact and 3–4 ether oxygen –Li+ contacts from G4), and solvated Li+G4. If the “particles” were in a cubic array, the center to center distance would be approximately 1.64 nm apart, smaller than the center to center distance of the “particles” measured by SAXS. However, it is likely that the structure factor should consider interpenetrating particles.
Lithium ion transference numbers (tPP+) by potentiostatic polarization
Lithium ion transference numbers (Table 1) were obtained by potentiostatic polarization experiments (tPP+), where they were calculated (eqn (1)) after corrections for the solid electrolyte interphase (SEI) that forms on the electrodes (eqn (2)).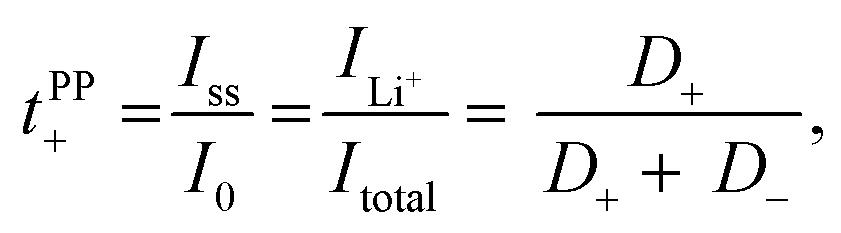 | (1) |
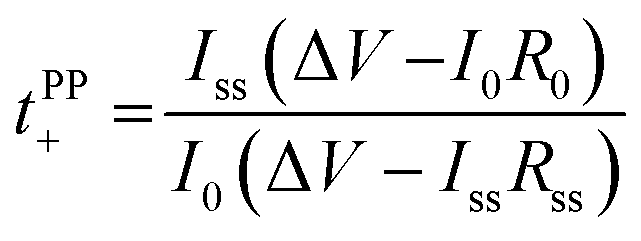 | (2) |
| Sample O/Li = 20/1 | Diffusion constant, D, m2 s−1 × 1011 | Lithium ion transference numbers | Radii (nm) or normalized radii | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| R i s from ηb |
|
||||||||||||
| D G4 | D PF6 | D 8mer | D Li | t NMR+ | t PP+ | t NMR− | R Lis |
|
R POSSs | Li | PF6 | POSS | |
a 8mer = POSS-(LiNSO2CF3).
b
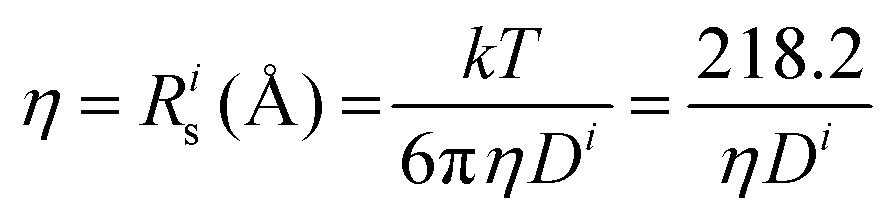 (@25 °C, where η (mPa s), D (m2 s−1 × 10−11).
c If equal moles of two species are assumed.
d O/Li = 18.3/1. (@25 °C, where η (mPa s), D (m2 s−1 × 10−11).
c If equal moles of two species are assumed.
d O/Li = 18.3/1.
|
|||||||||||||
| G4/LiPF6 | 7.70 | 5.83 | 4.41 | 0.43 | 0.51 | 0.57 | 0.33 | 0.25 | 1.75 | 1.32 | |||
| G4/(LiPF6 + 20 wt% 8mera)d | 6.41 | 4.97 | 1.14 | 3.53 | 0.42 | 0.65 | PF6− 0.583 | 0.33 | 0.24 | 1.03 | 1.81 | 1.29 | 5.6 |
| POSS− 0.002 | |||||||||||||
| G4/8mera | 11.3 | 2.05 | 1.6 | 0.44 | 0.65 | 0.60c | 0.92 | 0.63 | 7.1 | 5.5 | |||
| 1.1 | 0.35 | 1.34 | 10.3 | ||||||||||
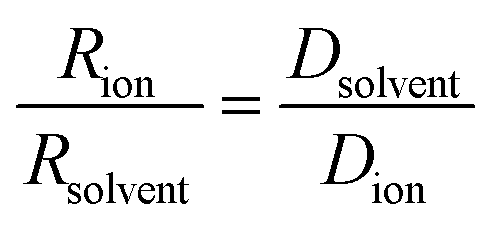
What is interesting is that for all the G4/LiX (X = TFSI−, PF6− and BETI−), tPP+ = 0.5 ± 0.01, but for G4/POSS-(LiNSO2CF3)8, and all the mixtures, i.e. G4/LiX/POSS-(LiNSO2CF3)8, tPP+ = 0.65 ± 0.01. Any system with greater than 10 wt% POSS-(LiNSO2CF3)8 also has tPP+ > 0.65 (Table S2, ESI†).
Evidence that we do not have significant amounts of neutral species (although we could have other charged species) of POSS-(LiNSO2CF3)8 or LiX in G4 comes from measurements of σeff (σeff = Iss/ΔV, σ0 = I0/ΔV). σeff is expected to be independent of the applied voltage when ΔV: (i) ≤10 mV for a fully dissociated ideal electrolyte;11 (ii) <10 V for high concentrations of LiX (since the solution acts like an insulator).46 Therefore measurement of the steady state current itself can indicate when significant ion pairs are present.47 For all of the systems we investigated, values of ΔV = 10, 20 and 30 mV were tested, and the current increased as ΔV increased, indicating the absence of significant neutral species.
Although measurements of σeff/σ0 (σeff = Iss/ΔV, σ0 = I0/ΔV) do not yield t+ (eqn (1)) when there are ion pairs or multiply charged species (since t+ is defined for a single species, typically Li+), it is important to stress that in practical applications, what is of equal importance is the steady state DC current, which is a measure of the net flux of the electroactive constituent (lithium) in the cell. Although the initial DC current (I0) under the same applied potential (∼20 mV) is about 3 times higher for LiX (X = TFSI−, PF6−, BETI−) than for POSS-(LiNSO2CF3)8, the steady state current (Iss) is always higher for the POSS-(LiNSO2CF3)8 or mixtures of POSS-(LiNSO2CF3)8/LiX than for LiX. A high value of σeff is desirable, since in battery applications, during discharge, lithium can be carried by Li+, aggregates and LiX. It is less important which species carries the current, provided that the total flux of Li from anode to cathode is high, which is the case here.
Diffusion measurements and lithium ion transference (tNMR+) from PFG-NMR
Diffusion coefficients obtained by PFG-NMR are summarized in Table 1. In order to also measure the mixed diffusion of POSS-(LiNSO2CF3)8 and LiX in G4, LiPF6 was selected since it has a distinct 19F NMR signal compared with –CF3 from POSS-(LiNSO2CF3)8. Three systems were investigated: G4/POSS-(LiNSO2CF3)8, G4/LiPF6 and G4/[80 wt% LiPF6 + 20 wt% POSS-(LiNSO2CF3)8].1H-NMR (Fig. S5, ESI†) was only used to measure the diffusion coefficient of the G4, using chemical shifts in the 3.5–5 ppm range. The diffusivity of the PF6− was measured by 19F PFG-NMR at its characteristic signal, a doublet resonating at around −72 ppm, and the diffusivity of the POSS anion was followed using the CF3 resonance at −76 ppm (Fig. 6). As expected, the mixed G4/(LiPF6 + 20 wt% POSS-(LiNSO2CF3)8) samples had features from both the PF6− and the –CF3 species. Of interest is that in the 7Li spectra of neat G4/POSS-(LiNSO2CF3)8, two broader but separate peaks are observed.
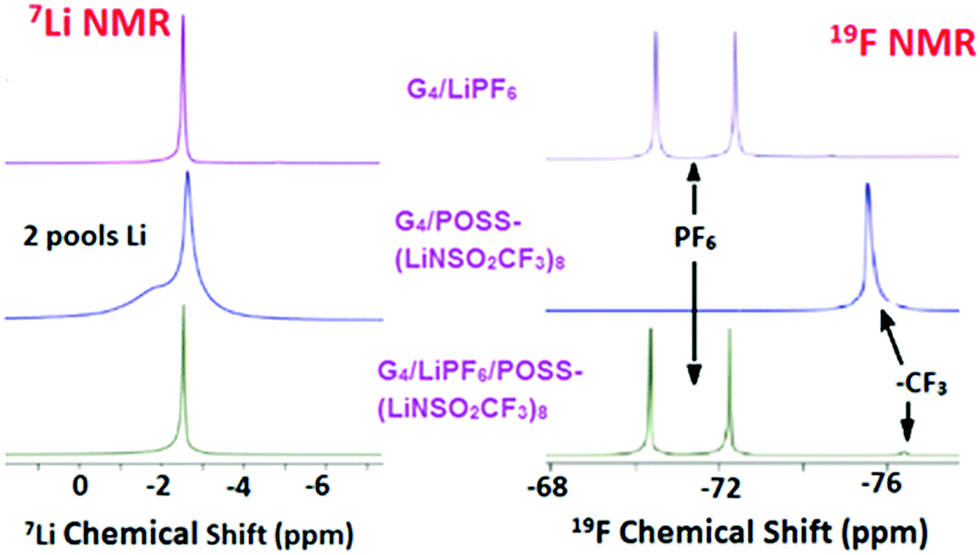 | ||
| Fig. 6 7Li and 19F PFG-NMR data for G4/LiPF6, G4/POSS-(LiNSO2CF3)8, and G4/(LiPF6+ POSS-(LiNSO2CF3)8). | ||
In all cases, the self-diffusion constants increase in the order DG4 > Danion > DLi+, the same order observed previously for dilute LiX solutions of glyme/LiX36,39 and other aprotic solvents. The diffusion constant of neat G4 decreased with the addition of G4/LiPF6, G4/POSS-(LiNSO2CF3)8, and G4/(LiPF6+ POSS-(LiNSO2CF3)8), consistent with previous reports for addition of LiX salts to glymes.36,39 Since DG4 has the fastest diffusion constant, it indicates that there is free glyme in all the solutions. Further, DG4 is fastest for G4/POSS-(LiNSO2CF3)8, indicating that there is more free G4 and less G4 participating in solvation shells than for G4/LiPF6, consistent with the DSC results. The diffusion constants of Li+ and PF6− in G4/LiPF6 are slower by a factor of ∼2 compared with Li+ and TFSI− in G4/LiTFSI (O/Li = 20/1) (DTFSI− = 11 × 10−7, and DLi+ = 9 × 10−7 cm2 s−1) at 30 °C36) consistent with the known low lattice energy of LiTFSI compared with other LiX salts (such as LiPF6) and its tendency for less ion pair formation in solution.
In the case of POSS-(LiNSO2CF3)8, the most salient feature is that for G4/POSS-(LiNSO2CF3)8, the anion shows a single peak in the 19F NMR spectra, while Li exhibits two peaks in the 7Li NMR spectra. For 19F, this may indicate a single diffusing species or a variety of diffusing species with rapid chemical exchange between them. However, the existence of 2 peaks in the 7Li NMR spectrum indicates that there are two lithium “pools”, and that each lithium pool consists of lithium containing species that exchange with each other and/or have similar chemical shifts. The broadness of the 7Li lines is indicative of a lower mobility for the Li+ ions.
While we cannot definitely assign the two Li+ peaks, the narrower (“fast-diffusing” species) peak is likely to involve Li in a more symmetric environment. It is similar in position and shape (although slightly broader) than that of Li in LiPF6 and other LiX salts and it may be assigned to solvated Li+. The solvated Li+ can be “free”, Li+(G4)n, but not necessarily in a 1/1 molar ratio with G4, in the form of a contact ion pair (–NSO2CF3−⋯Li+⋯G4), or even a “free” Li that moves with the rest of the cage (Fig. 7). The broader (“slow-diffusing” species) peak is then assigned to Li that has not disassociated from the POSS cage. Their relative populations based on the integrated intensities of the static 7Li NMR peaks is 33/67 (fast/slow), whereas the ratio in a two component fit of the decay in the 7Li NMR PFG-NMR experiment is 48/52 (fast/slow). The two ratios are different because the broad peak (“slow-diffusing” species) has a significantly faster relaxation.
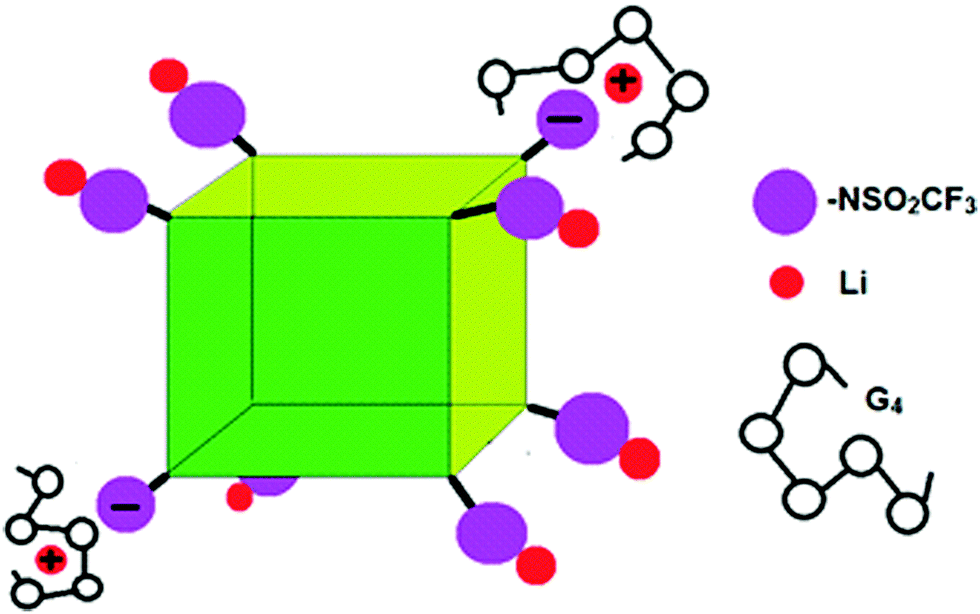 | ||
| Fig. 7 Schematic of G4/POSS-(LiNSO2CF3)8 showing contact ion pairs, solvent separated ion pairs and un-dissociated Li. | ||
Further insight can be gained by analyzing the radii of the solvated ions, by: (i) the Stokes–Einstein equation originally defined for hard spheres:
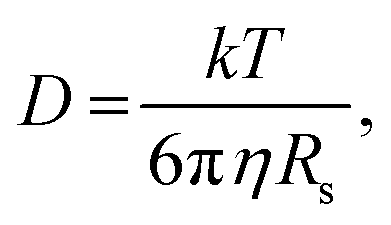 | (3) |
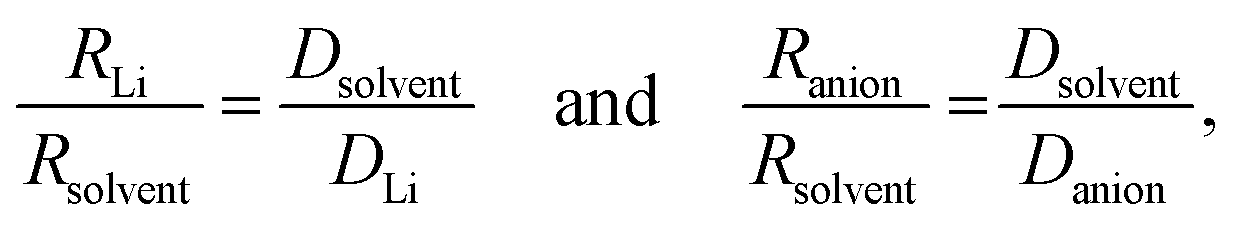 | (4) |
Radii obtained by both methods are presented in Table 1, using measured solution viscosities (Table S3, ESI†) for the Stoke–Einstein equation.
The transference numbers were calculated from self-diffusion coefficients (Table 1) using eqn (5) and (6):
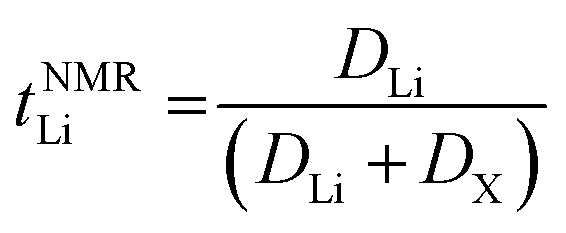 | (5) |
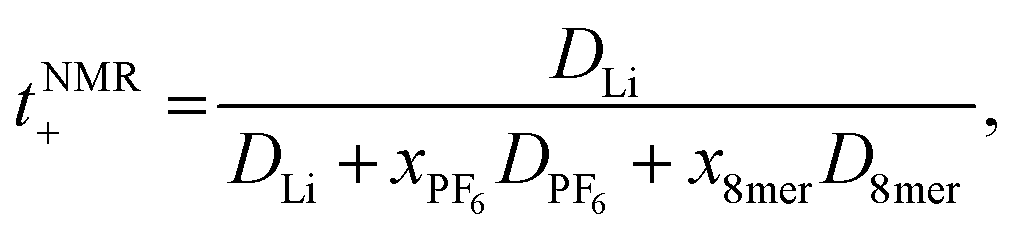 | (6) |
In order to better understand the measured diffusion coefficients, associated radii and transference numbers, the actual species present in solution must be considered. In particular, we want to elucidate why tPP+ > tNMR+ and why there is only one 19F NMR signal and two 7Li NMR signals. In general, lithium ion transference numbers depend on the possible species and their relative amounts in solution. For simple LiX salts in aprotic polar solvents, LiX ↔ Li+ + X− equilibria exist, so that free (solvated) ions, contact ion pairs (CIPs) and higher order multiplets can be present. In most treatments, only free solvated ions and CIPs are considered.
For LiPF6, the smaller value of Rs (eqn (3)) for PF6− than for Li+ indicates that there is less interaction between the anion and G4 than between Li+ and G4. That is, G4 preferentially solvates Li+ ions compared with the PF6− anions, as also reported for G4/Li triflate (Tf = LiCF3SO3) [RLi ∼ 0.317–0.383 nm and RTf ∼ 0.289–0.326 nm]. The same trends for LiPF6 are observed using eqn (4), where the ionic radii are normalized against the solvent radius.
In the case of POSS-(LiNSO2CF3)8, there are many possible multiply charged as well as neutral species.
For POSS-(LiNSO2CF3)8, abbreviated here as PLi8:
| PLi8 + nXG4 ↔ x1PLi71− + x2PLi62− + x3PLi53− + x4PLi44− + x5PLi35− + x6PLi26− + x7PLi17− + x8PLi08− + XLi+(G4)n | (7) |
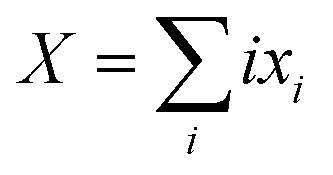 and n is the number of solvent molecules.
and n is the number of solvent molecules.
If there is rapid exchange between all the Li containing species, the diffusion coefficient measured by PFG-NMR is:
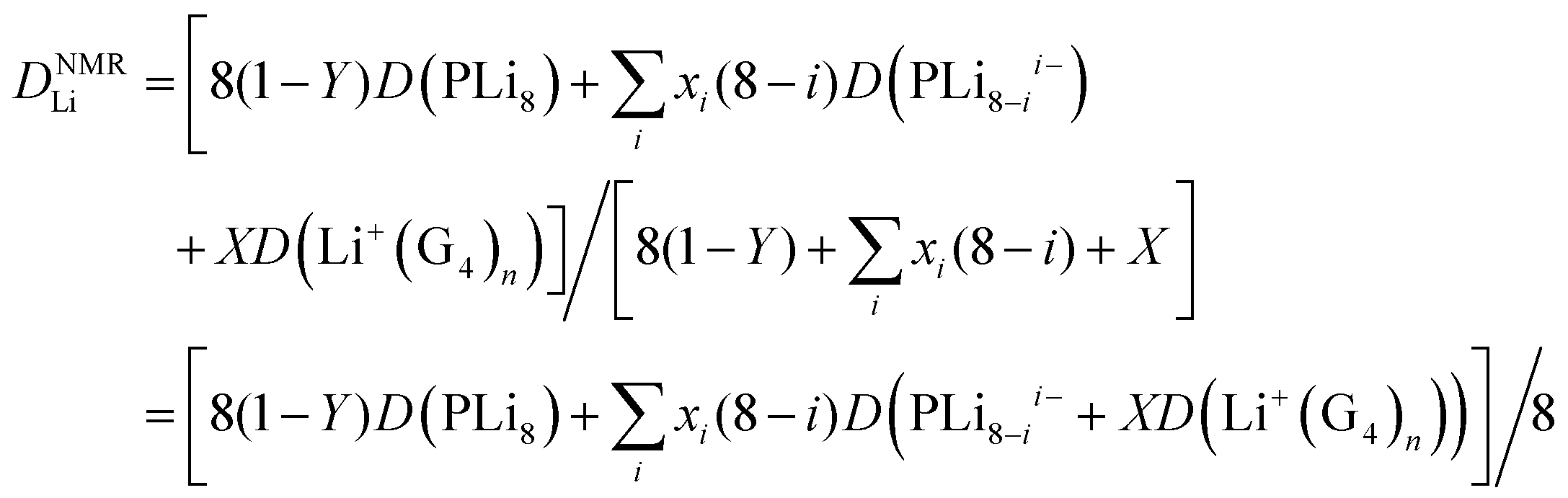 | (8) |
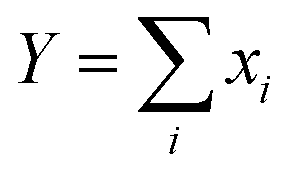 .
.
In the case of complete dissociation,
| DNMRLi = D(Li+(G4)n) | (9) |
The corresponding conductivities are:
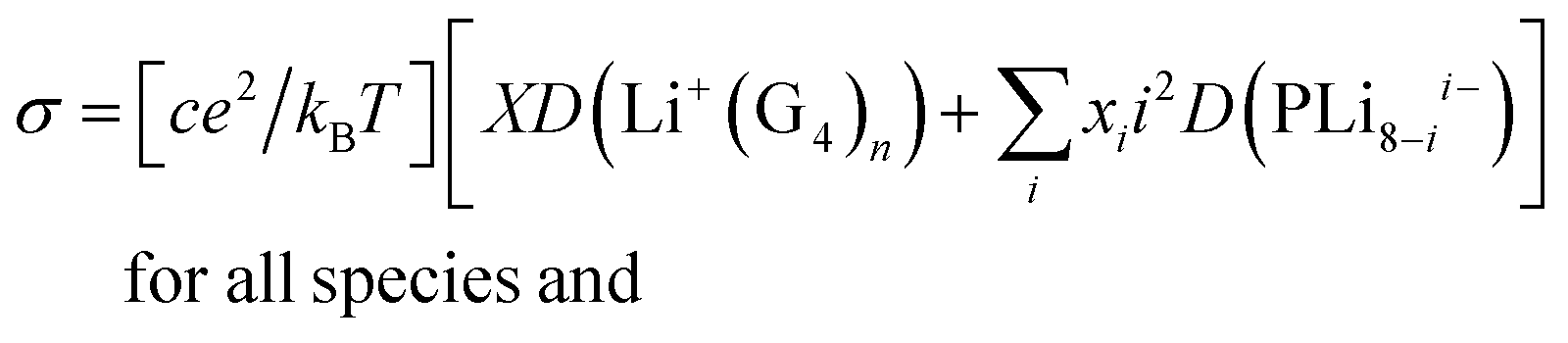
| σ = [ce2/kBT][8D(Li+(G4)n) + 64D(PLi08−)] for complete dissociation |
The Li+ ion transference numbers are then:
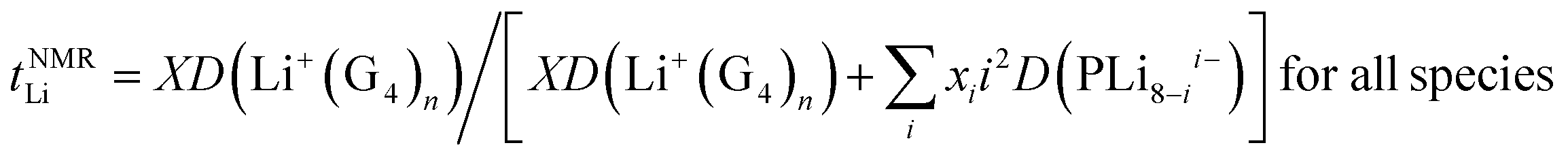 | (10) |
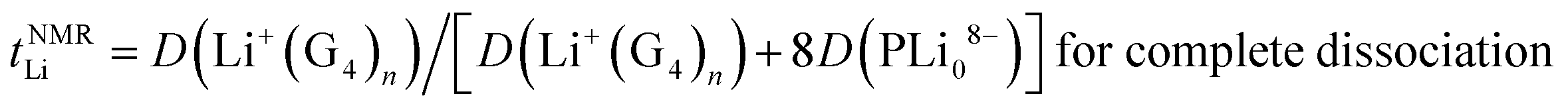 | (11) |
The value of the radius obtained from the 19F PFG-NMR diffusion coefficient (eqn (3)) indicates that RPOSS is small (0.63 nm), in the range of sizes found for POSS compounds (where radii of ∼0.5–0.6 nm are expected from the geometry of the cubes). Since a trimer composed of POSS-(LiNSO2CF3)8⋯Li⋯POSS-(LiNSO2CF3)8 would have a size much larger than those measured in the 19F PFG-NMR experiment, aggregates of this type are rare and/or not long lived.
It is not possible to obtain lithium ion transference >0.5 using eqn (10) or (11) with any combination of PLi8−ii− and free Li+(G4)n. Eqn (11) indicates that for fully dissociated PLi8, i.e. PLi08− + 8Li+(G4)n, the diffusion coefficient of Li+(G4)n must be very much greater than 8D(PLi08−) to achieve a high Li+ transference number, and there would be only one peak in the 7Li NMR spectrum. The radius of this solvated Li+ ion would also be expected to be approximately the same size as that observed in G4/LiPF6, ∼0.33 nm, and in G4/Li triflate (Tf = LiCF3SO3) ∼ 0.317–0.383 nm,49 rather than 0.92–1.34 nm measured. Since these effects are not observed, the PLi8 must be only partially dissociated. Even in this case, i.e. for a distribution of PLi8−ii−, since the two Li peaks have similar areas, this interpretation indicates that the average charge 〈i〉 of the PLi8−ii− must be significantly greater than unity, assuming that the PLi8−ii− all have the same average diffusion coefficient, 〈DP〉. The diffusion coefficient of Li+(G4)n, D(Li+(G4)n), must then be greater than about 5 times 〈DP〉 to achieve a high Li+ ion transference number. However, the diffusion coefficients obtained from 7Li PFG-NMR are smaller for the two Li pools than the diffusion coefficient obtained by 19F PFG-NMR for the PLi8−ii−, so that the Li+ ion transference number would be substantially less than 0.5.
In order to obtain lithium ion transference numbers >0.5, the peaks in the Li PFG-NMR must be assigned to solvated and un-solvated Li+ ions. The peak at 1.34 nm is assigned to PLi8−ii− in which the Li is un-dissociated. The solvated Li+ peak (0.92 nm) is composed of free Li+(G4)n and PLi8−ii−·Li(G4)n, i.e. PLi8−ii− solvated with Li+. The PLi8−ii−·Li(G4)n can be contact ion pairs or Li+(G4)n moving with the anion; the latter can arise if the anion and cation fluxes are coupled (Fig. 7). Since PFG-NMR measures the average diffusion coefficient of the nuclei in each pool, the diffusion coefficient of the PLi8−ii−·Li+(G4)n (which do not contribute to Li+ ion conductivity since they are charge neutral) may be substantially smaller than the diffusion coefficient of the “free” solvated Li+ ions (which are the only lithium ions that contribute to the Li+ ion conductivity) and result in the (average) small diffusion coefficients measured.
In addition, the solvated lithium ions that are linked to the PLi8−ii− ions reduce their effective charges, and hence their contribution to the anion conductivity. In other words, the dissolved PLi8 exists as {PLi8−ii−·(LiG4)j}(i−j)−, and the effective degree of dissociation is small, thus reducing the anion conductivity. This makes PLi8 act very much like a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 salt in solution from a conductivity point of view. A small effective degree of dissociation enables the diffusion coefficient of the free solvated Li+ ions to be substantially larger than the average diffusion coefficient of all the solvated Li+ ions (that also include the slower moving Li+ associated with the large anion). If the diffusion coefficient of the free solvated Li+ ions is substantially larger than the average diffusion coefficient of all the solvated Li+ ions, a Li+ ion transference number greater than 0.5 can be achieved. A small degree of effective dissociation can also account for the much lower conductivity of POSSLi8 relative to simple lithium salts despite the high PFG NMR diffusion coefficients.
:1 salt in solution from a conductivity point of view. A small effective degree of dissociation enables the diffusion coefficient of the free solvated Li+ ions to be substantially larger than the average diffusion coefficient of all the solvated Li+ ions (that also include the slower moving Li+ associated with the large anion). If the diffusion coefficient of the free solvated Li+ ions is substantially larger than the average diffusion coefficient of all the solvated Li+ ions, a Li+ ion transference number greater than 0.5 can be achieved. A small degree of effective dissociation can also account for the much lower conductivity of POSSLi8 relative to simple lithium salts despite the high PFG NMR diffusion coefficients.
In the 19F NMR, there is broadening of the CF3 resonance compared with the narrower PF6− of the LiX salt, indicating a wider distribution of anion populations or environments that could arise from a range of solvated and non-solvated Li+ ions associated with the anion (but still with only a single 19F NMR peak). As in the case of other LiX salts, when contact ion pairs exist, the effect of un-dissociated dimers on the average diffusion constant (〈X− + LiX〉) of the anion (i.e. addition to a small Li to a larger X−) is smaller than the effect of un-dissociated dimers on the average diffusion constant (〈Li+ + LiX〉) of Li (i.e. addition of large X− to small Li)].49 This explains why there can be one 19F NMR signal but two 7Li NMR signals.
The association of Li+ ions with G4 and the anion are dynamic processes that are somewhat reminiscent of the Grotthuss mechanism in water. However, here charge does not move from one Li atom to another, so it is better described as coordinated hopping of Li+. The Li+ ion is associated dynamically with an average of n solvent molecules and/or anions. Thus D(Li+(G4)n) in the above equations should be interpreted as the diffusion coefficient of Li+ ions that are instantaneously but not permanently solvated by an average of n solvent molecules, and where one solvent molecule can displace another. In a contact ion pair, the –NSO2CF3−⋯Li+⋯G4 contact can be replaced with an oxygen from G4(G4⋯Li+) forming Li+(G4)n. The diffusion of Li+ ions can then occur either by a Grotthuss type (coordinated hopping) mechanism (rapid exchange between species) or translation (diffusion) of a complex of Li+(G4)n (if species long-lived). Grotthuss type mechanisms of Li+ ion transfer, where charge transport has been decoupled from mass transport, have been modelled using molecular dynamics simulations (MDS) of concentrated solutions of acrylonitrile (AN)–LiX, consisting of dissociated ions, neutral contact ion pairs (CIPs) or neutral aggregates (AGG).50 The coordinated hopping of Li+ between ion pairs or higher order clusters has been predicted by MDS,51 where hopping between aggregates is unlikely unless they are closely spaced.52 Therefore, this mechanism can occur between two POSS-(LiNSO2CF3)8 cubes only if they are close enough together. For G4/POSS-(LiNSO2CF3)8 O/Li = 20/1, the center-to-center distance is estimated as 2.27 nm (cubic array)/2.41 nm (FCC) so that the largest size (1.13 nm radius) measured for the Li species by 7Li PFG-NMR is the closest possible approach for two contacting spheres. The SAXS data for the more concentrated G4/POSS-(LiNSO2CF3)8 O/Li = 7.5 sample indicates that the spheres can be overlapping.
In the mixed system, the radii of Li+ ions are almost the same in G4/LiPF6 and mixed G4/LiPF6/POSS-(LiNSO2CF3)8 samples, and the radii of the PF6− ions are also almost the same in G4/LiPF6 and mixed G4/LiPF6/POSS-(LiNSO2CF3)8 samples, suggesting that each ion is similarly solvated in both systems. Since the POSS-(LiNSO2CF3)8 is expected to become more dissociated with dilution, more dissociated Li+ ions solvated by a single G4 are expected. Further, in mole ratios, the [LiPF6] = 85 [POSS-(LiNSO2CF3)8], so the dominant species is LiPF6. However, the radius of the POSS-(LiNSO2CF3)8 anion measured by 19F PFG-NMR increases, but not to twice the size expected if triplets were formed. We thus suggest that the POSS anions form complexes with the PF6− anion, i.e. –NSO2CF3−⋯Li+⋯PF6−. This effectively slows its diffusion constant and permits a higher transference number measured by potentiostatic polarization.
Electrochemical characterization
Electrochemical stability and reversibility
The anodic stability of G4/POSS-(LiNSO2CF3)8, G4/(LiTFSI + 20% POSS-(LiNSO2CF3)8), and G4/LiTFSI was measured with linear sweep voltammetry at 25 °C from open circuit voltage (OCV) to 5 V at a scan rate of 2 mV s−1, using stainless steel working electrodes and lithium counter/reference electrodes (Fig. S7, ESI†). The stability window was about the same (2 V to 4.1–4.3 V) for the three electrolytes, using a threshold current of 50 μA cm−2 typical for glyme electrolytes,38 and thus the electrolytes were stable in the voltage range (2.6 to 4 V) used for half-cell testing with the LiFePO4 cathode. The time dependence of the resistance under open circuit voltage at 25 °C (Fig. S8, ESI†) was obtained using a Li0/electrolyte/Li0 cell for G4/POSS-(LiNSO2CF3)8 and G4/(80 wt% LiTFSI/20 wt% POSS-(LiNSO2CF3)8) with electrochemical impedance spectroscopy. The bulk resistance was constant for both electrolytes, with the bulk resistance of G4/POSS-(LiNSO2CF3)8 greater than that of G4/[80 wt% LiTFSI/20 wt% POSS-(LiNSO2CF3)8], as expected from the conductivity data. There was a gradual but slight decrease in the interfacial resistance for [G4/80 wt% LiTFSI/20 wt% POSS-(LiNSO2CF3)8], but for G4/POSS-(LiNSO2CF3)8 the interfacial resistance dropped steeply for several days, which may be due to improved wetting of the electrodes with time or to decreased diffusion in the more viscous G4/POSS-(LiNSO2CF3)8, and then leveled off to the same value as for the mixed system. This suggests that POSS-(LiNSO2CF3)8 affects the formation of the interfacial layer on Li0 metal and its resistance similarly for both systems, and may be due to the formation of a particle rich coating on the Li0 metal, as previously suggested for hairy nanoparticles.53Lithium plating and stripping
Lithium plating and stripping for Li0/[G4/LiTFSI]/Li0, Li0/[G4/POSS-(LiNSO2CF3)8/Li0 and Li0/[G4/80 wt% LiTFSI/20 wt% POSS-(LiNSO2CF3)8]/Li0 were studied at different current densities (0.01 to 1 mA cm−2) for 2 hour charge and discharge at 25 °C. Cell failure occurred in the order G4/80% LiTFSI/20 wt% POSS-(LiNSO2CF3)8 < G4/LiTFSI < G4/POSS-(LiNSO2CF3)8, (although not all cells were cycled until failure). At 0.01 mA cm−2, G4/80 wt% LiTFSI/20 wt% POSS-(LiNSO2CF3)8 and G4/LiTFSI exhibited stable cycling, but for G4/POSS-(LiNSO2CF3)8, the polarization increased with cycle number (Fig. S9, ESI†). At 0.1 mA cm−2, both G4/80 wt% LiTFSI/20 wt% POSS-(LiNSO2CF3)8 and G4/LiTFSI exhibited cyclic stability and stable voltage profiles for 11 days (when cycling was stopped) (Fig. 8A), and the overvoltage was less in the case of the G4/80 wt% LiTFSI/20 wt% POSS-(LiNSO2CF3)8. At 1 mA cm−2 (Fig. S9, ESI†), cell failure occurred at <1 cycle, ∼1 day and was stopped at 5 days, for G4/POSS-(LiNSO2CF3)8, G4/LiTFSI and G4/80 wt% LiTFSI/20 wt% POSS-(LiNSO2CF3)8, respectively, although polarization increased with cycle number for G4/80 wt% LiTFSI/20 wt% POSS-(LiNSO2CF3)8.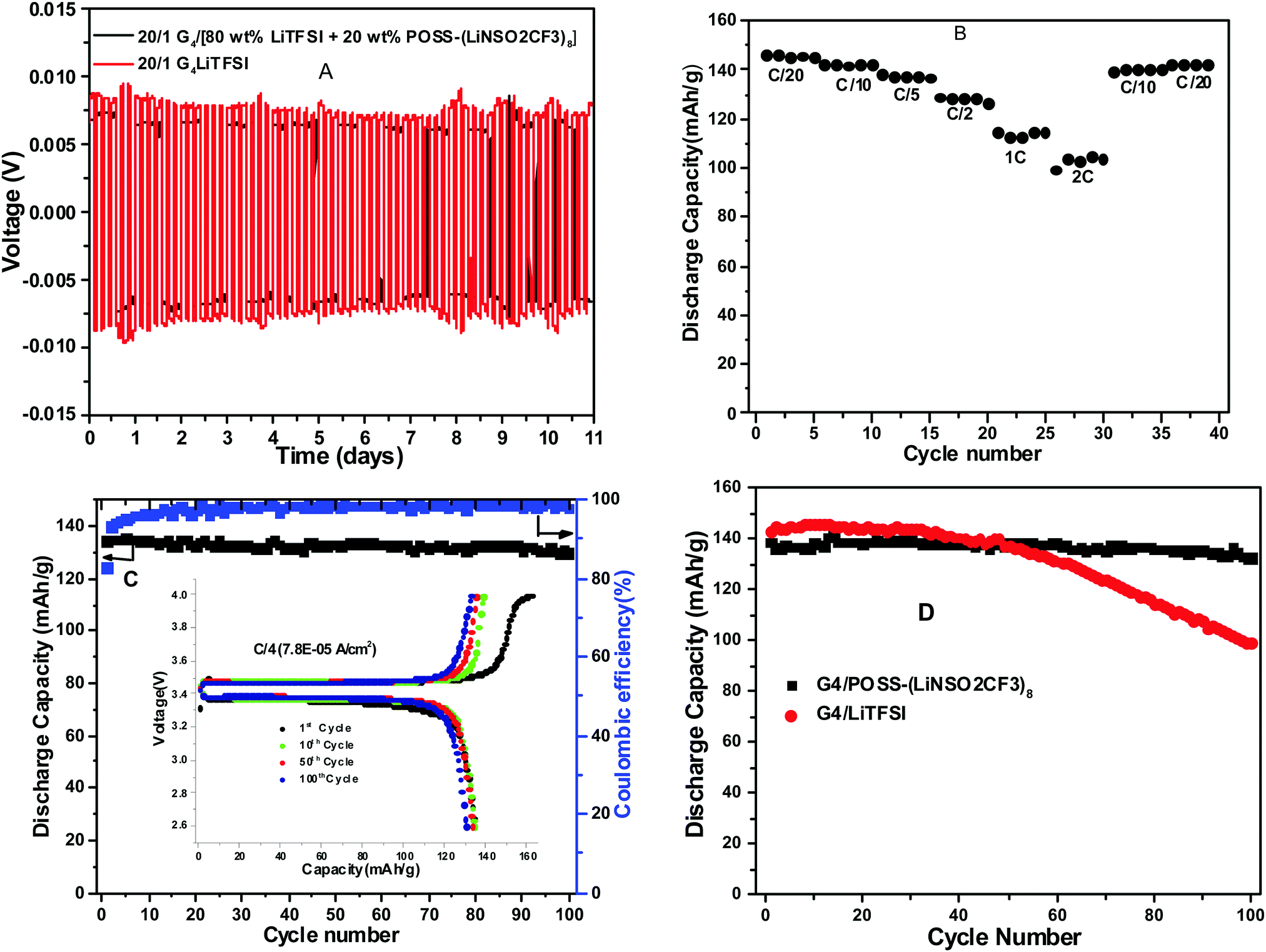 | ||
| Fig. 8 Electrochemical results (all cells O/Li = 20/1): (A) comparison of cycling data using 0.1 mA cm−2 and 2 h charge/2 h discharge for Li0/[G4/80 wt% LiTFSI/20 wt% POSS-(LiNSO2CF3)8]/Li0 and Li0/(G4/LiTFSI)/Li0; (B) rate capability Li0/[G4/80 wt% LiTFSI/20 wt% POSS-(LiNSO2CF3)8]/LiFePO4 at C/20, C/10, C/5, C/2, 1C, 2C, C/10 and C/20; (C) discharge capacity and Coulombic efficiency as a function of cycle number for Li0/[G4/80 wt% LiTFSI/20 wt% POSS-(LiNSO2CF3)8]/LiFePO4 cell at C/4 rate and 25 °C; inset shows cell voltage vs. specific capacity at 1, 10, 50 and 100 cycles; (D) comparison of discharge capacity for Li0/[G4/POSS-(LiNSO2CF3)8]/LiFePO4 and Li0/(G4/LiTFSI)/LiFePO4 at C/5 rate. | ||
Half-cell testing
Cyclic voltammetry (CV) was used to study the reversibility of the Li/(G4/POSS-(LiNSO2CF3)8 O/Li = 20/1)/LiFePO4 system. (Fig. S10, ESI†); the preparation of the LiFePO4 electrode is provided in ESI.† For practical applications, the battery should be able to run at different C-rates with high capacity. The galvanostatic performance of half cells using Li0 metal as the anode and LiFePO4 as the cathode were investigated for the G4/80 wt% LiTFSI/20 wt% POSS-(LiNSO2CF3)8, G4/LiTFSI and G4/POSS-(LiNSO2CF3)8 electrolytes as a function of C-rate. The high ionic conductivity of electrolytes (0.25 mS cm−1 to 3 mS cm−1) allows the batteries to be tested at RT. The expected decrease in capacity with C-rate for Li0/[G4/80 wt% LiTFSI/20 wt% POSS-(LiNSO2CF3)8]/LiFePO4 (Fig. 8B), which recovers when returned to low C-rates, demonstrates the stability of the electrolyte throughout the testing. Stable performance was demonstrated at C/4, with retention of 130 mA h g−1 until the 100th cycle, when cycling was stopped (Fig. 8C). Better cycling behavior was demonstrated for the G4/POSS-(LiNSO2CF3)8 than for the G4/LiTFSI electrolyte at C/5 (Fig. 8D): the former exhibited stable cycling until the 100th cycle, while the latter showed capacity-fade after the 50th cycle. At 3C, capacity fade began at ∼30 cycles (Fig. S11, ESI†), and decreased slightly less for the G4/80 wt% LiTFSI/20 wt% POSS-(LiNSO2CF3)8 than for the G4/LiTFSI electrolyte. By comparison, for 1 M LiPF6 in a mixture of ethylene carbonate (EC) and ethyl methyl carbonate (EMC) (EC:EMC 4:6 by weight) and a Li0/NCA cell (NCA = LiNi0.80Co0.15Al0.05O2), the specific capacity started decreasing after 15 cycles at 1C, 2C and 3C rates.54
The cause of cell failure or capacity fade was not directly ascertained. However, it is interesting to note that recent work on dendritic growth in liquid electrolytes has shown that this growth is inhibited by electrolytes with low viscosities and large anions (with little effect of conductivity).55 In the current investigation, cells lasted longest and had the best half-cell cycling using the electrolyte that had large anions as well as low viscosity (G4/80 wt% LiTFSI/20 wt% POSS-(LiNSO2CF3)8). The role of nanoparticles in stabilizing the interface and reducing interfacial resistance of Li0 metal has also been demonstrated.53
Conclusions
In summary, we have shown that multi-ionic lithium salts (POSS-(LiNSO2CF3)8, comprised of rigid polyoligomeric silsesquioxanes (POSS) cubes functionalized with eight lithium[(4-styrenesulfonyl)trifluoromethane-sulfonyl)imide] groups (LiNSO2CF3), can be added to solutions of LiTFSI or LiPF6 in tetraglyme (G4), increasing the lithium ion transference number but maintaining high ionic conductivity. The (POSS-(LiNSO2CF3)8 are only partially dissociated, and better ionic conductivity is expected with more dissociative pendant anions. Unlike single ion conducting (SIC) polymers added to electrolyte solutions, addition of the compact POSS-(LiNSO2CF3)8 provides a source of less mobile anions without the same large increase in viscosity as in the flexible SIC polymers, and avoids the formation of clusters/aggregates that trap Li+ ions and prevent high conductivity in these SIC systems. In neat G4/POSS-(LiNSO2CF3)8, solvent-in-salt electrolytes are formed at high POSS-(LiNSO2CF3)8 concentration and stable colloids are formed with increasing solvent. When the POSS-(LiNSO2CF3)8 are in close proximity to each other in G4 solutions, there are many possible rapidly equilibrating Li species such as contact ion pairs and solvent separated ion pairs that can result in the transport of Li+ ions by Grotthuss type coordinated hopping mechanisms as well as by diffusion.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Financial support of this work from NSF CBET award 1437814 is gratefully acknowledged. We thank Dr Gerd Langenbucher of Anton-Paar, Inc. for help in obtaining the SAXS data.References
- M. Armand and J. M. Tarascon, Building better batteries, Nature, 2008, 451(7179), 652–657 CrossRef CAS PubMed
.
- P. G. Bruce, S. A. Freunberger, L. J. Hardwick and J. M. Tarascon, Li-O(2) and Li-S batteries with high energy storage, Nat. Mater., 2012, 11(1), 19–29 CrossRef CAS PubMed
- L. Grande, E. Paillard, J. Hassoun, J. B. Park, Y. J. Lee, Y. K. Sun, S. Passerini and B. Scrosati, The Lithium/Air Battery: Still an Emerging System or a Practical Reality?, Adv. Mater., 2015, 27(5), 784–800 CrossRef CAS PubMed
- C. Monroe and J. Newman, The impact of elastic deformation on deposition kinetics at lithium/polymer interfaces, J. Electrochem. Soc., 2005, 152(2), A396–A404 CrossRef CAS
- J. N. Chazalviel, Electrochemical aspects of the generation of ramified metallic electrodeposits, Phys. Rev. A: At., Mol., Opt. Phys., 1990, 42(12), 7355–7367 CrossRef CAS
- M. Doyle, T. F. Fuller and J. Newman, The importance of the lithium ion transference number in lithium/polymer cells, Electrochim. Acta, 1994, 39(13), 2073–2081 CrossRef CAS
- K. M. Diederichsen, E. J. McShane and B. D. McCloskey, Promising Routes to a High Li+ Transference Number Electrolyte for Lithium Ion Batteries, ACS Energy Lett., 2017, 2563–2575 CrossRef CAS
- P. G. Bruce, J. Evans and C. A. Vincent, Conductivity and transference number measurements on polymer electrolytes, Solid State Ionics, 1988, 28, 918–922 CrossRef
- N. P. Balsara and J. Newman, Relationship between Steady-State Current in Symmetric Cells and Transference Number of Electrolytes Comprising Univalent and Multivalent Ions, J. Electrochem. Soc., 2015, 162(14), A2720–A2722 CrossRef CAS
- S. Zugmann, M. Fleischmann, M. Amereller, R. M. Gschwind, H. D. Wiemhöfer and H. J. Gores, Measurement of transference numbers for lithium ion electrolytes via four different methods, a comparative study, Electrochim. Acta, 2011, 56(11), 3926–3933 CrossRef CAS
- P. G. Bruce and C. A. Vincent, Steady state current flow in solid binary electrolyte cells, J. Electroanal. Chem., 1987, 225(1–2), 1–17 CrossRef CAS
- J. Evans, C. A. Vincent and P. G. Bruce, Electrochemical measurement of transference numbers in polymer electrolytes, Polymer, 1987, 28(13), 2324–2328 CrossRef CAS
- E. L. Hahn, Spin echos, Phys. Rev., 1950, 80(4), 580–594 CrossRef
- E. O. Stejskal and J. E. Tanner, Spin diffusion measurements: spin echos in the presence of a time-dependent field gradient, J. Chem. Phys., 1965, 42(1), 288–292 CrossRef CAS
-
P. T. Callaghan, Principles of Nuclear Magnetic Resonance, Oxford University Press, Oxford, 1991 Search PubMed
- S. Abbrent and S. Greenbaum, Recent progress in NMR spectroscopy of polymer electrolytes for lithium batteries, Curr. Opin. Colloid Interface Sci., 2013, 18(3), 228–244 CrossRef CAS
- H. Zhang, C. M. Li, M. Piszcz, E. Coya, T. Rojo, L. M. Rodriguez-Martinez, M. Armand and Z. B. Zhou, Single lithium-ion conducting solid polymer electrolytes: advances and perspectives, Chem. Soc. Rev., 2017, 46(3), 797–815 RSC
-
S. Mogurampelly, O. Borodin and V. Ganesan, Computer Simulations of Ion Transport in Polymer Electrolyte Membranes, in Annual Review of Chemical and Biomolecular Engineering, ed. J. M. Prausnitz, Annual Reviews, Palo Alto, 2016, vol. 7, pp. 349–371 Search PubMed
- M. F. Lu, J. Runt and P. Painter, An Infrared Spectrocopic Study of a Polyester Copolymer Ionomer Based on Poly(ethylene oxide), Macromolecules, 2009, 42(17), 6581–6587 CrossRef CAS
- Y. V. Oza, D. R. MacFarlane, M. Forsyth and L. A. O'Dell, Unexpected effect of tetraglyme plasticizer on lithium ion dynamics in PAMPS based ionomers, Phys. Chem. Chem. Phys., 2016, 18(28), 19011–19019 RSC
- Y. Y. Lu, M. Tikekar, R. Mohanty, K. Hendrickson, L. Ma and L. A. Archer, Stable Cycling of Lithium Metal Batteries Using High Transference Number Electrolytes, Adv. Energy Mater., 2015, 5(9), 7 Search PubMed
- H. Oh, K. Xu, H. D. Yoo, D. S. Kim, C. Chanthad, G. Yang, J. Z. Jin, I. A. Ayhan, S. M. Oh and Q. Wang, Poly(arylene ether)-Based Single-Ion Conductors for Lithium-Ion Batteries, Chem. Mater., 2016, 28(1), 188–196 CrossRef CAS
- L. Porcarelli, A. S. Shaplov, F. Bella, J. R. Nair, D. Mecerreyes and C. Gerbaldi, Single-Ion Conducting Polymer Electrolytes for Lithium Metal Polymer Batteries that Operate at Ambient Temperature, ACS Energy Lett., 2016, 1(4), 678–682 CrossRef CAS
- R. Meziane, J.-P. Bonnet, M. Courty, K. Djellab and M. Armand, Single-ion polymer electrolytes based on a delocalized polyanion for lithium batteries, Electrochim. Acta, 2011, 57, 14–19 CrossRef CAS
- S. W. Feng, D. Y. Shi, F. Liu, L. P. Zheng, J. Nie, W. F. Feng, X. J. Huang, M. Armand and Z. B. Zhou, Single lithium-ion conducting polymer electrolytes based on poly(4-styrenesulfonyl)(trifluoromethanesulfonyl)imide anions, Electrochim. Acta, 2013, 93, 254–263 CrossRef CAS
- Q. Ma, H. Zhang, C. W. Zhou, L. P. Zheng, P. F. Cheng, J. Nie, W. F. Feng, Y. S. Hu, H. Li, X. J. Huang, L. Q. Chen, M. Armand and Z. B. Zhou, Single Lithium-Ion Conducting Polymer Electrolytes Based on a Super-Delocalized Polyanion, Angew. Chem., Int. Ed., 2016, 55(7), 2521–2525 CrossRef CAS PubMed
- J. L. Schaefer, D. A. Yanga and L. A. Archer, High Lithium Transference Number Electrolytes via Creation of 3-Dimensional, Charged, Nanoporous Networks from Dense Functionalized Nanoparticle Composites, Chem. Mater., 2013, 25(6), 834–839 CrossRef CAS
- K. D. Kreuer, A. Wohlfarth, C. C. de Araujo, A. Fuchs and J. Maier, Single Alkaline-Ion (Li+, Na+) Conductors by Ion Exchange of Proton-Conducting Ionomers and Polyelectrolytes, ChemPhysChem, 2011, 12(14), 2558–2560 CrossRef CAS PubMed
- M. Videa, W. Xu, B. Geil, R. Marzke and C. A. Angell, High Li+ self-diffusivity and transport number in novel electrolyte solutions, J. Electrochem. Soc., 2001, 148(12), A1352–A1356 CrossRef CAS
- H. G. Buss, S. Y. Chan, N. A. Lynd and B. D. McCloskey, Nonaqueous Polyelectrolyte Solutions as Liquid Electrolytes with High Lithium Ion Transference Number and Conductivity, ACS Energy Lett., 2017, 2(2), 481–487 CrossRef CAS
- L. Suo, Y.-S. Hu, H. Li, M. Armand and L. Chen, A new class of Solvent-in-Salt electrolyte for high-energy rechargeable metallic lithium batteries, Nat. Commun., 2013, 4, 1481 CrossRef PubMed
- P. Johansson, J. Tegenfeldt and J. Lindgren, Modelling amorphous lithium salt-PEO polymer electrolytes: ab initio calculations of lithium ion-tetra-, penta- and hexaglyme complexes, Polymer, 1999, 40(15), 4399–4406 CrossRef CAS
- S. Tsuzuki, W. Shinoda, M. Matsugami, Y. Umebayashi, K. Ueno, T. Mandai, S. Seki, K. Dokko and M. Watanabe, Structures of Li(glyme) (+) complexes and their interactions with anions in equimolar mixtures of glymes and Li TFSA: analysis by molecular dynamics simulations, Phys. Chem. Chem. Phys., 2015, 17(1), 126–129 RSC
- G. M. Mao, M. L. Saboungi, D. L. Price, M. B. Armand and W. S. Howells, Structure of liquid PEO-LiTFSI electrolyte, Phys. Rev. Lett., 2000, 84(24), 5536–5539 CrossRef CAS PubMed
- T. Tamura, K. Yoshida, T. Hachida, M. Tsuchiya, M. Nakamura, Y. Kazue, N. Tachikawa, K. Dokko and M. Watanabe, Physicochemical Properties of Glyme-Li Salt Complexes as a New Family of Room-temperature Ionic Liquids, Chem. Lett., 2010, 39(7), 753–755 CrossRef CAS
- K. Yoshida, M. Tsuchiya, N. Tachikawa, K. Dokko and M. Watanabe, Change from Glyme Solutions to Quasi-ionic Liquids for Binary Mixtures Consisting of Lithium Bis(trifluoromethanesulfonyl)amide and Glymes, J. Phys. Chem. C, 2011, 115(37), 18384–18394 CAS
- C. Zhang, A. Yamazaki, J. Murai, J. W. Park, T. Mandai, K. Ueno, K. Dokko and M. Watanabe, Chelate Effects in Glyme/Lithium Bis(trifluoromethanesulfonyl)amide Solvate Ionic Liquids, Part 2: Importance of Solvate-Structure Stability for Electrolytes of Lithium Batteries, J. Phys. Chem. C, 2014, 118(31), 17362–17373 CAS
- T. M. Pappenfus, W. A. Henderson, B. B. Owens, K. R. Mann and W. H. Smyrl, Complexes of lithium imide salts with tetraglyme and their polyelectrolyte composite materials, J. Electrochem. Soc., 2004, 151(2), A209–A215 CrossRef CAS
- K. Hayamizu, E. Akiba, T. Bando and Y. Aihara,
1H, 7Li and 19F nuclear magnetic resonance and ionic conductivity studies for liquid electrolytes composed of glymes and polyethyleneglycol dimethyl ethers of CH3O(CH2CH2O)nCH3 (n = 3–50) doped with LiN(SO2CF3)2, J. Phys. Chem., 2002, 117(12), 5929–5939 CrossRef CAS
- L. Aguilera, S. Z. Xiong, J. Scheers and A. Matic, A structural study of LiTFSI-tetraglyme mixtures: From diluted solutions to solvated ionic liquids, J. Mol. Liq., 2015, 210, 238–242 CrossRef CAS
- K. Shimizu, A. A. Freitas, R. Atkin, G. G. Warr, P. A. FitzGerald, H. Doi, S. Saito, K. Ueno, Y. Umebayashi, M. Watanabe and J. N. C. Lopes, Structural and aggregate analyses of (Li salt plus glyme) mixtures: the complex nature of solvate ionic liquids, Phys. Chem. Chem. Phys., 2015, 17(34), 22321–22335 RSC
- K. Shimizu, C. E. S. Bernardes, A. Triolo and J. N. C. Lopes, Nano-segregation in ionic liquids: scorpions and vanishing chains, Phys. Chem. Chem. Phys., 2013, 15(38), 16256–16262 RSC
- J. L. Nowinski, P. Lightfoot and P. G. Bruce, Structure of LiN(CF3SO2)2, a Novel Salt for Electrochemistry, J. Mater. Chem., 1994, 4(10), 1579–1580 RSC
- K. Larsson, The crystal structure of octa-(silsesquioxane)(HSiO1.5)8, Ark. Kemi, 1960, 16(3), 215–219 CAS
- F. Wohde, M. Balabajew and B. Roling, Li+ Transference Numbers in Liquid Electrolytes Obtained by Very-Low-Frequency Impedance Spectroscopy at Variable Electrode Distances, J. Electrochem. Soc., 2016, 163(5), A714–A721 CrossRef CAS
- P. G. Bruce, M. T. Hardgrave and C. A. Vincent, Steady-state current flow in solid binary electrolyte cells. 2. The effect of ion association, J. Electroanal. Chem., 1989, 271(1–2), 27–34 CrossRef CAS
- P. G. Bruce and C. A. Vincent, Effect of ion association on transport in polymer electrolytes, Faraday Discuss., 1989, 88, 43–54 RSC
- K. Hayamizu, Y. Aihara, S. Arai and W. S. Price, Self-diffusion coefficients of lithium, anion, polymer, and solvent in polymer gel electrolytes measured using Li-7, F-19, and H-1 pulsed-gradient spin-echo NMR, Electrochim. Acta, 2000, 45(8–9), 1313–1319 CrossRef CAS
- K. Hayamizu, Y. Aihara, S. Arai and C. G. Martinez, Pulse-gradient spin-echo H-1, Li-7, and F-19 NMR diffusion and ionic conductivity measurements of 14 organic electrolytes containing LiN(SO2CF3)2, J. Phys. Chem. B, 1999, 103(3), 519–524 CrossRef CAS PubMed
- D. M. Seo, O. Borodin, D. Balogh, M. O'Connell, Q. Ly, S. D. Han, S. Passerini and W. A. Henderson, Electrolyte Solvation and Ionic Association III. Acetonitrile-Lithium Salt Mixtures-Transport Properties, J. Electrochem. Soc., 2013, 160(8), A1061–A1070 CrossRef CAS
- K. R. Lu, J. K. Maranas and S. T. Milner, Ion-mediated charge transport in ionomeric electrolytes, Soft Matter, 2016, 12(17), 3943–3954 RSC
- M. Doyle, M. E. Lewittes, M. G. Roelofs and S. A. Perusich, Ionic conductivity of nonaqueous solvent–swollen ionomer membranes based on fluorosulfonate, fluorocarboxylate, and sulfonate fixed ion groups, J. Phys. Chem. B, 2001, 105(39), 9387–9394 CrossRef CAS
- S. Choudhury, A. Agrawal, S. Y. Wei, E. Jeng and L. A. Archer, Hybrid Hairy Nanoparticle Electrolytes Stabilizing Lithium Metal Batteries, Chem. Mater., 2016, 28(7), 2147–2157 CrossRef CAS
- D. P. Lv, Y. Y. Shao, T. Lozano, W. D. Bennett, G. L. Graff, B. Polzin, J. G. Zhang, M. H. Engelhard, N. T. Saenz, W. A. Henderson, P. Bhattacharya, J. Liu and J. Xiao, Failure Mechanism for Fast-Charged Lithium Metal Batteries with Liquid Electrolytes, Adv. Energy Mater., 2015, 5(3), 7 Search PubMed
- M. S. Park, S. B. Ma, D. J. Lee, D. Im, S. G. Doo and O. Yamamoto, A Highly Reversible Lithium Metal Anode, Sci. Rep., 2014, 4, 8 Search PubMed
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7mh01130j |
| This journal is © The Royal Society of Chemistry 2018 |