
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceWhy nature chose the Mn4CaO5 cluster as water-splitting catalyst in photosystem II: a new hypothesis for the mechanism of O–O bond formation
Biaobiao
Zhang
 a and
Licheng
Sun
*ab
a and
Licheng
Sun
*ab
aDepartment of Chemistry, KTH Royal Institute of Technology, 10044 Stockholm, Sweden. E-mail: lichengs@kth.se
bState Key Laboratory of Fine Chemicals, Institute of Artificial Photosynthesis, DUT-KTH Joint Education and Research Center on Molecular Devices, Dalian University of Technology (DUT), 116024 Dalian, China
First published on 21st August 2018
Abstract
Resolving the questions, namely, the selection of Mn by nature to build the oxygen-evolving complex (OEC) and the presence of a cubic Mn3CaO4 structure in OEC coupled with an additional dangling Mn (Mn4) via μ-O atom are not only important to uncover the secret of water oxidation in nature, but also essential to achieve a blueprint for developing advanced water-oxidation catalysts for artificial photosynthesis. Based on the important experimental results reported so far in the literature and on our own findings, we propose a new hypothesis for the water oxidation mechanism in OEC. In this new hypothesis, we propose for the first time, a complete catalytic cycle involving a charge-rearrangement-induced MnVII–dioxo species on the dangling Mn4 during the S3 → S4 transition. Moreover, the O–O bond is formed within this MnVII–dioxo site, which is totally different from that discussed in other existing proposals.
Introduction
The oxygen-evolving complex (OEC) of photosystem II (PSII), which is the most important natural complex for maintaining aerobic life, has existed in photosynthetic organisms on Earth for almost three billion years.1,2 Understanding the structure and catalytic mechanism of OEC is motivated not only by humans’ desire to discover the secret of natural photosynthesis. It can also provide a blueprint to advance the development of water-oxidation catalysts (WOCs) for artificial photosynthesis for renewable fuel production using solar energy.1The general structure of OEC has been determined by a series of X-ray techniques.3–7Fig. 1A shows that the core of OEC comprises a cubic Mn3CaO4 structure coupled with an additional dangling Mn (Mn4) via μ-O atom. The entire Mn4CaO5 cluster is surrounded by amino acid residues (YZ, D1–D61, D1–H190, etc.), two Cl− ions, and many H2O molecules. However, the pathway of water-oxidation mechanism at the OEC, which proceeds through the Kok cycle via five intermediates (called Si (i = 0–4) states),8 remains unknown. Although researchers studying PSII are generally in agreement on the oxidation states9 and structures of the metastable S0, S1, and S2 intermediates (Fig. 1B),3,4,10,11 details of O–O bond formation are still unclear. This is due to lack of experimental evidence for the most important S3 → S4 and S4 → S0 steps, which directly involve the highest oxidized S4 state that produces oxygen.12–14 Currently, two O–O bond formation pathways are widely discussed: water nucleophilic attack mechanism14,15 and oxo–oxyl radical coupling mechanism,16–18 which involve a MnV–oxo electrophile and a MnIV–oxyl radical as the active S4 intermediate (Fig. 1B), respectively. However, the existence of a MnV–oxo electrophile or MnIV–oxyl radical in OEC catalysis has not been experimentally proved. Hence, mechanism of water oxidation by the Mn4CaO5 cluster is still an open question, requiring scientists to propose more appropriate answers.
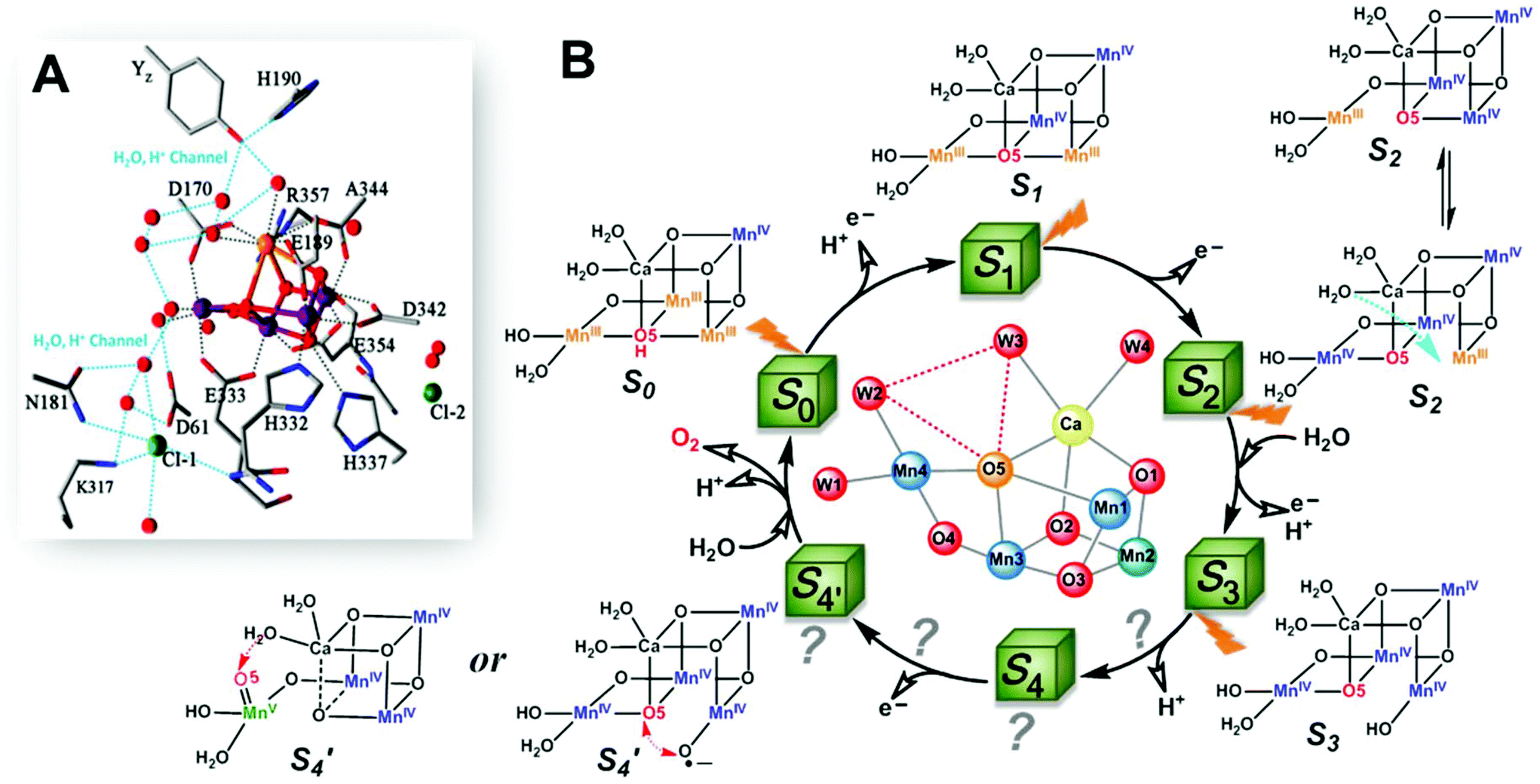 | ||
| Fig. 1 (A) Structure of Mn4CaO5 cluster of the OEC in PSII.3 (B) Extended classic Kok cycle8,19 for water oxidation in PSII, involving electron transfer and proton release, and the widely proposed structures of intermediate “S” states. Si (i = 0–4) states represent oxidation states of the Mn4CaO5 cluster relative to the S0 state. S1 is the only dark-stable state. S2 state exists as two interconverting isomers: “open” and “closed” configurations.10 A significant structural change occurs in the S3 state.19–23 S4 and S′4 states are hypothetical states without experimental evidence, due to their ultra-short life-times14,19 and related reaction steps being unclear. | ||
The current proposed mechanism for water oxidation by the Mn4CaO5 cluster is far from complete and cannot effectively explain the selection of Mn by nature, the construction of OEC from multiple Mn ions, and the presence of a dangling Mn4 outside the cubic structure. Herein, we propose a MnVII–dioxo-based mechanism for O–O bond formation by the Mn4CaO5 cluster. This mechanism is distinct from the widely discussed water nucleophilic attack and oxo–oxyl radical coupling mechanisms. Our mechanism suggests that after charge accumulation in the first three steps, charge rearrangement occurs in the fourth step to form a MnVII–dioxo site on the dangling Mn4, where the O–O bond forms in the S4 state. This MnVII–dioxo-based mechanism may open new possibilities for revealing the actual mechanism of water oxidation in PSII, and consequently offer cogent guidance for developing more efficient synthetic WOCs for artificial photosynthesis.
Proposing the MnVII species involved mechanism
Aside from the abundance of Mn on Earth, a fundamental reason for nature to incorporate Mn in OEC could be the specificity of its redox chemistry.24,25 It is reasonable to consider mechanism of water oxidation by the Mn4CaO5 cluster in terms of the particular redox chemistry of Mn. For first-row transition metals in the periodic table, from Sc to Zn, the highest accessible valency and number of accessible valencies first increase up to Mn and then decrease toward Zn, which only has a valency of +2.24 Therefore, Mn has the highest oxidation state (MnVII), and the largest number of oxidation states, i.e., Mn is the only metal (at least among those involved in plants) that can carry five charges (from MnII to MnVII). Furthermore, Mn participates in many disproportionation reactions between two or multiple Mn ions. The unique redox chemistry of Mn makes it an ideal element for building the OEC, in which accumulation of four charges is needed to oxidize two water molecules to molecular oxygen. Based on these unique features of Mn and experimental evidences presented below, we propose a new mechanism for water oxidation by the Mn4CaO5 cluster in PSII, which involves charge-rearrangement-induced MnVII–dioxo species.In this new mechanism, we propose that first three steps, i.e., S0 → S1, S1 → S2, and S2 → S3, are the charge accumulation steps. As shown in Fig. 2, electron transfers, proton transfers, and structural changes during these three steps are consistent with the widely accepted processes with abundant experimental evidences.9,12–14 The dark-stable S1 [III, IV, IV, III] state, which evolves from the S0 [III, IV, III, III] state via a proton-coupled electron-transfer process, is transformed into the S2 state after loss of one electron (please note that the order of oxidation states follows the numbering of Mn atoms in Fig. 2). In the following crucial S2 → S3 step, W3 H2O on the Ca site inserts into the open position between Ca and O517,20,26 and forms a new O5 for the next catalytic cycle. The original O5 is pulled towards Mn4 as a π-donating ligand. Loss of one proton and another electron gives the S3 intermediate with oxidation states [IV, IV, IV, IV]. We believe that charge accumulation on the Mn4CaO5 cluster is the driving force for insertion of the W3 H2O, because one negatively charged ligand is needed to stabilize the S3 [IV, IV, IV, IV] state. The insertion of W3 H2O and subsequent deprotonation help to balance the positive charges accumulated on the Mn core structure after the first three steps of oxidation.
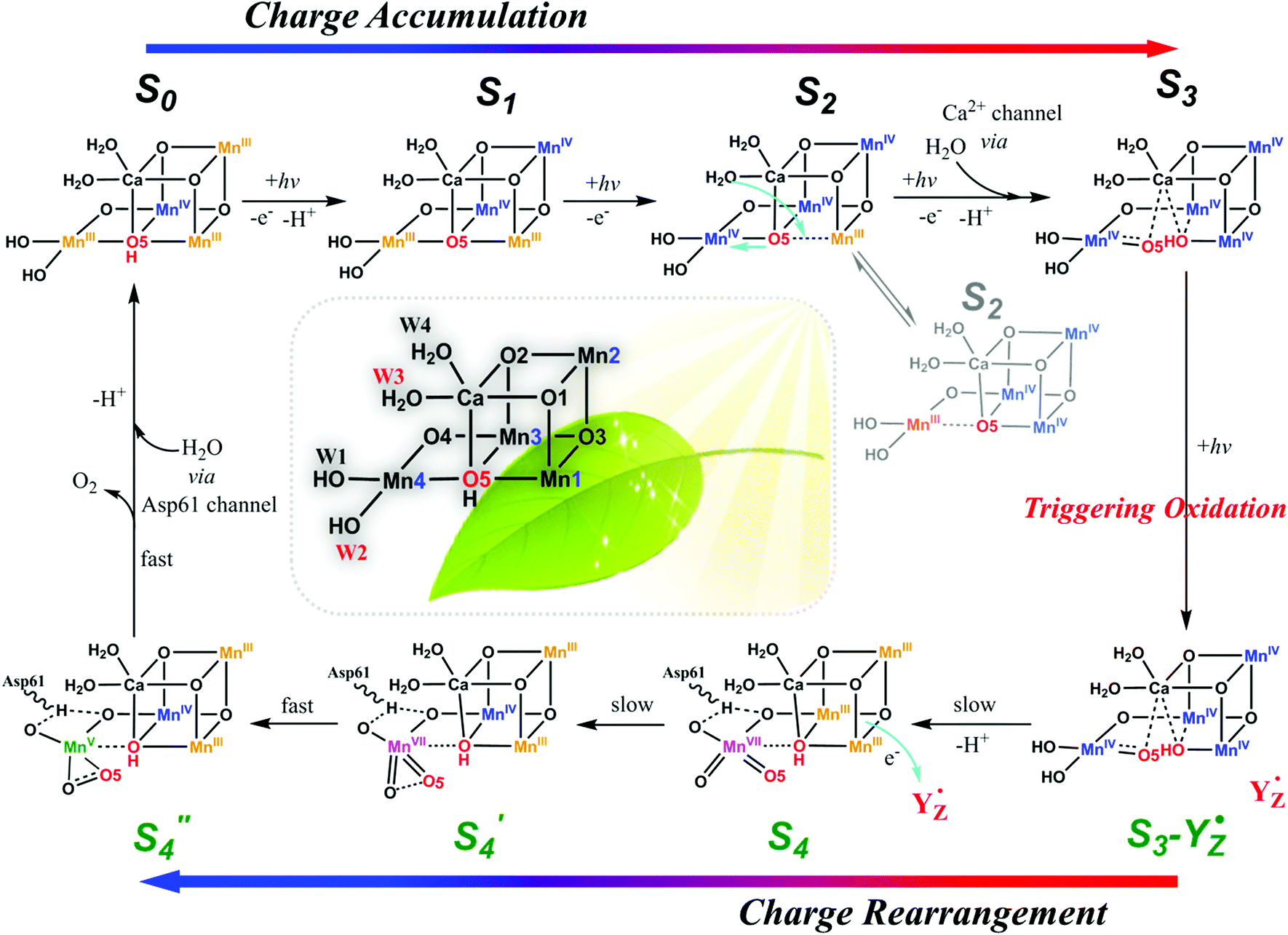 | ||
| Fig. 2 Proposed catalytic mechanism for water oxidation by Mn4CaO5 cluster in PSII involving MnVII–dioxo species. Charge disproportionation of Mn4CaO5 cluster at the S3-YZ˙ state after accumulation of three charges, i.e., charge rearrangement, leads to the formation of a super-active S′4 [III, III, IV, VII] state after the S3 → S4 step. An O–O bond forms within the MnVII–dioxo site (between W2 and O5) during the S4 → S′4 step. W2, O5, and W3 all participate in O2 evolution. | ||
After accumulation of three charges on the Mn4CaO5 cluster at the S3 state, the oxidation state of Mn ions in the Mn4CaO5 cluster is [IV, IV, IV, and IV]. According to the Latimer diagram of Mn, this is the highest state that can be reached through a one-electron oxidation (Scheme 1). Thus, in contrast to the first three oxidation steps, during the S3 → S4 step, the fourth charge obtained from P680+ is retained for a longer period on YZ˙ than that in the first three steps. This results in the observation of a S3-YZ˙ state19,21 because the direct oxidation of [IV, IV, IV, IV] to [V, IV, IV, IV] by YZ˙ may not be the lowest energy pathway. To accept the fourth charge from YZ˙, i.e., to further oxidize the Mn4CaO5 cluster, a charge rearrangement (i.e. disproportionation) occurs within the four MnIV ions in the S3 state. This is accompanied with release of a proton, resulting in an S4 [III, III, III, VII] resting state with a MnVII–dioxo site. The fourth oxidation by the P680+ can be a driving force that triggers the acceleration of charge rearrangement because it introduces the YZ˙, which changes the positively charged environment around the Mn4CaO5 cluster and the pKa balance of proton transfer channels.20,27,28
 | ||
| Scheme 1 The Latimer diagram for Mn illustrates its standard reduction potentials (in 1 M acid) at oxidation states from +7 to 0. | ||
These multiple processes involved in the S3 → S4 step, including storage of one charge on YZ˙, release of one proton, charge rearrangement, and formation of the MnVII–dioxo site, are crucial for restoring the severely charged Mn cluster and producing a standby state to accept a fourth charge and form the O–O bond. Indeed, significant structural rearrangements have been experimentally observed during the S3-YZ˙ → S4 → S0 steps.19,21 After such charge rearrangement, the S4 transition state [III, III, III, VII] is easily oxidized by its paired YZ˙ to the active S′4 [III, III, IV, VII] state. This highly active S′4 species immediately releases a molecular oxygen via a peroxo transition state S′′4 [III, III, IV, V] and is transformed back to the S0 [III, IV, III, III] state by binding one ready H2O, supplied from the ASP61/Cl− water channel, to vacant sites on the dangling Mn4 and losing one proton, thus completing one catalytic cycle.
In this new mechanism, two unique and attractive points are to be necessarily discussed in detail: (1) charge rearrangement involving disproportionation of 4 MnIV into 3 MnIII and 1 MnVII (i.e., MnIV–MnIV–MnIV–MnIV → MnVII–MnIII–MnIII–MnIII), and (2) O–O bond formation at the MnVII–dioxo site. Although we did not perform special verifiable experimental studies for this article, there are sufficient published experimental results to support these two essential proposals.
Disproportionation of MnIV–MnIV–MnIV–MnIV to MnVII–MnIII–MnIII–MnIII
The mechanism of water oxidation by MnIV sulphate was investigated by Shilov et al. ca. 40 years ago.29 Their experimental results showed that the intermediate involved in oxygen evolution is a MnVII species generated by disproportionation of MnIV ions.29,30 The detailed kinetic study of this reaction indicated that charge disproportionation of 4 MnIV to 1 MnVII and 3 MnIII regularly happens under MnIV-rich conditions. Furthermore, a kinetic study by Dzhabiev et al. showed that a tetranuclear MnIV intermediate may be involved in the charge disproportionation of 4 MnIV to 1 MnVII and 3 MnIII (eqn (1)).31| 4 MnIV → [MnIV4] tetramer → MnVII + 3 MnIII | (1) |
Moreover, formation of MnO4− species has been observed during water oxidation using several Mn based catalytic systems.32–36 For example, Kaneko et al. reported that MnO4− was formed from the oxidation of [(bpy)2Mn(μ-O)2Mn(bpy)2] (Mn-bpy, bpy = 2,2′-bipyridine).33 As shown in eqn (2), they suggested that two molecules of Mn-bpy dimer, i.e., four Mn cores, are involved in MnO4− formation.
| 2[MnIV–MnIV] → MnVII + MnIII + MnIII + MnIII | (2) |
| [MnV–MnV] → MnVII + MnIII | (3) |
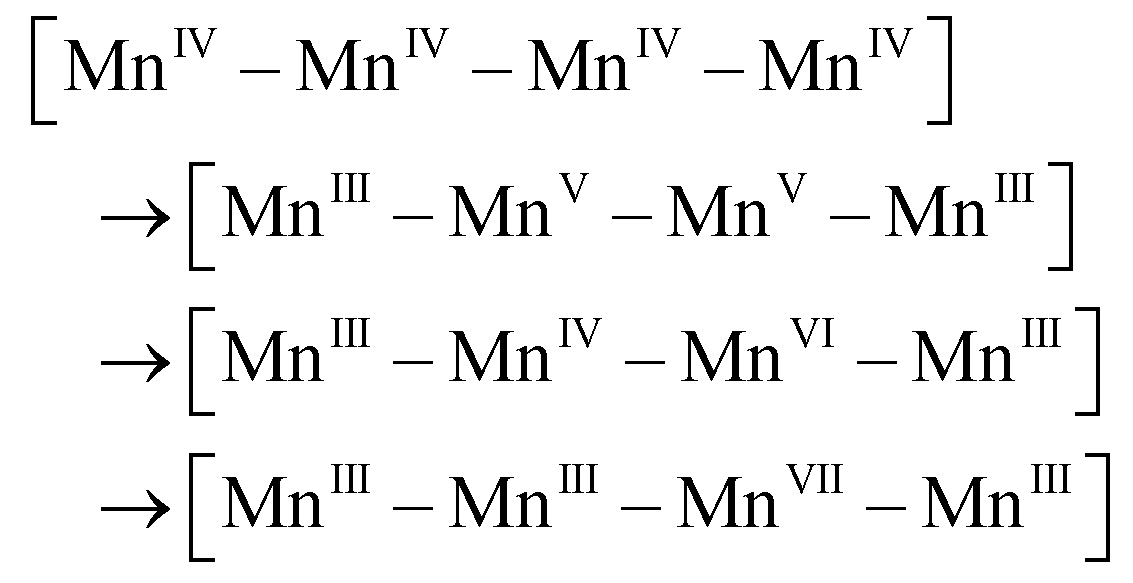 | (4) |
Similarly, Brudvig et al. observed the formation of MnO4− while studying water oxidation by [(H2O)(tpy)Mn(μ-O)2Mn(tpy)(H2O)] (Mn-tpy, tpy = 2,2′:6′,2′′-terpyridine) dimer catalysts.32,34 They proposed that MnO4− formed via disproportionation of [(tpy)(O)MnV(μ-O)2MnV(tpy)(H2O)] (eqn (3)). Furthermore, Yagi et al. found that two molecules of Mn-tpy dimer were involved in the rate-determining step of MnO4− formation by kinetic analysis. This is consistent with proposal by Kaneko et al. These experimental observations strongly support that one possible evolution pathway for a [IV, IV, IV, IV] state is to disproportionate into a [VII, III, III, III] state. A detailed process of charge rearrangement is proposed as shown in eqn (4), in which the transformation from MnIV–MnVI to MnVII–MnIII is a fast step.30
Although theoretical studies are not sufficient to explain this complicated process, a Latimer diagram of Mn can preliminarily help us understand why multiple Mn ions are involved in the generation of MnVII species instead of a step by step one-electron oxidation (Scheme 1). The Latimer diagram of Mn shows that oxidation potential from MnIV to MnV is 4.27 V, which is much higher than all other redox potentials of Mn. This implies that the stepwise one-electron oxidation of MnIV to MnVII may need very high energy. This also explains why MnV is the only species missing in the Pourbaix diagram of Mn and why MnO4− lies in close proximity to MnO2.
However, the oxidation potentials of PSII are sufficient to generate a high-valent MnVII site in the Mn4CaO5 cluster. In the Pourbaix diagram for Mn, theoretical potential for the formation of MnO4− at pH 7 is 1.05 V.36 The estimated redox potentials for P680/P680+ and YZ/YZ˙ are approximately +1.26 and +1.21 V, respectively,37 and are thermodynamically adequate for initiating the formation of MnVII species. Indeed, MnII can be oxidized to MnVII by moderate one-electron oxidants29 such as Ru(bipyridine)33+ with an oxidation potential of +1.26 V, which is close to the potentials of P680+ and YZ˙.
These analyses together with the consideration of the facts of PSII, i.e., the unique dangling Mn4 site and existence of the S3-YZ˙ state, reasonably support the disproportionation of the [IV, IV, IV, IV] state to a [III, III, III, VII] state during the S3 → S4 transition.
O–O bond formation within the MnVII–dioxo site
Oxygen evolution at MnVII–dioxo site is thermodynamically possible, but kinetically hampered. For example, it is well known that MnO4− can evolve O2, but the rate is very slow.38However, the rate of oxygen evolution from MnVII–dioxo site can be greatly enhanced by various promoters. For example, MnO2 has been known as a catalyst for oxygen evolution from MnO4− by Skrabal since 1910.38 Shafirovich et al. studied the mechanism of oxygen formation during oxidation of water by MnIV sulfate. They suggested a mechanism (Fig. 3A) where a MnIV–MnVII surface complex is the active intermediate for O–O bond formation.29–31 Interestingly, a crystalline MnIV–MnVII complex, (H3O)2–[MnIV(MnVIIO4)6] 11H2O, immediately evolves oxygen when kept at or over −4 °C (Fig. 3B).39 When MnO4− is activated by a strong Lewis acid such as BF3, O2 is rapidly evolved via intramolecular coupling of MnVII–dioxo site (Fig. 3C).40 During photo-induced oxygen evolution from MnO4−, Lee et al. investigated the presence of an excited state of MnO4−, which is a much more active oxidant than permanganate itself and the subsequent formation of a MnV–dioxo intermediate prior to oxygen evolution (Fig. 3D).41 A MnVII![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O species has also been proposed as an essential intermediate for a MnV–nitrido complex during CeIV-driven water oxidation (Fig. 3E).42 In 2017, a highly reactive pendant MnVIIO moiety on a cubic Mn–nitride complex has been reported as a synthetic structural model of the proposed S4 state in PSII (Fig. 3F).43
O species has also been proposed as an essential intermediate for a MnV–nitrido complex during CeIV-driven water oxidation (Fig. 3E).42 In 2017, a highly reactive pendant MnVIIO moiety on a cubic Mn–nitride complex has been reported as a synthetic structural model of the proposed S4 state in PSII (Fig. 3F).43
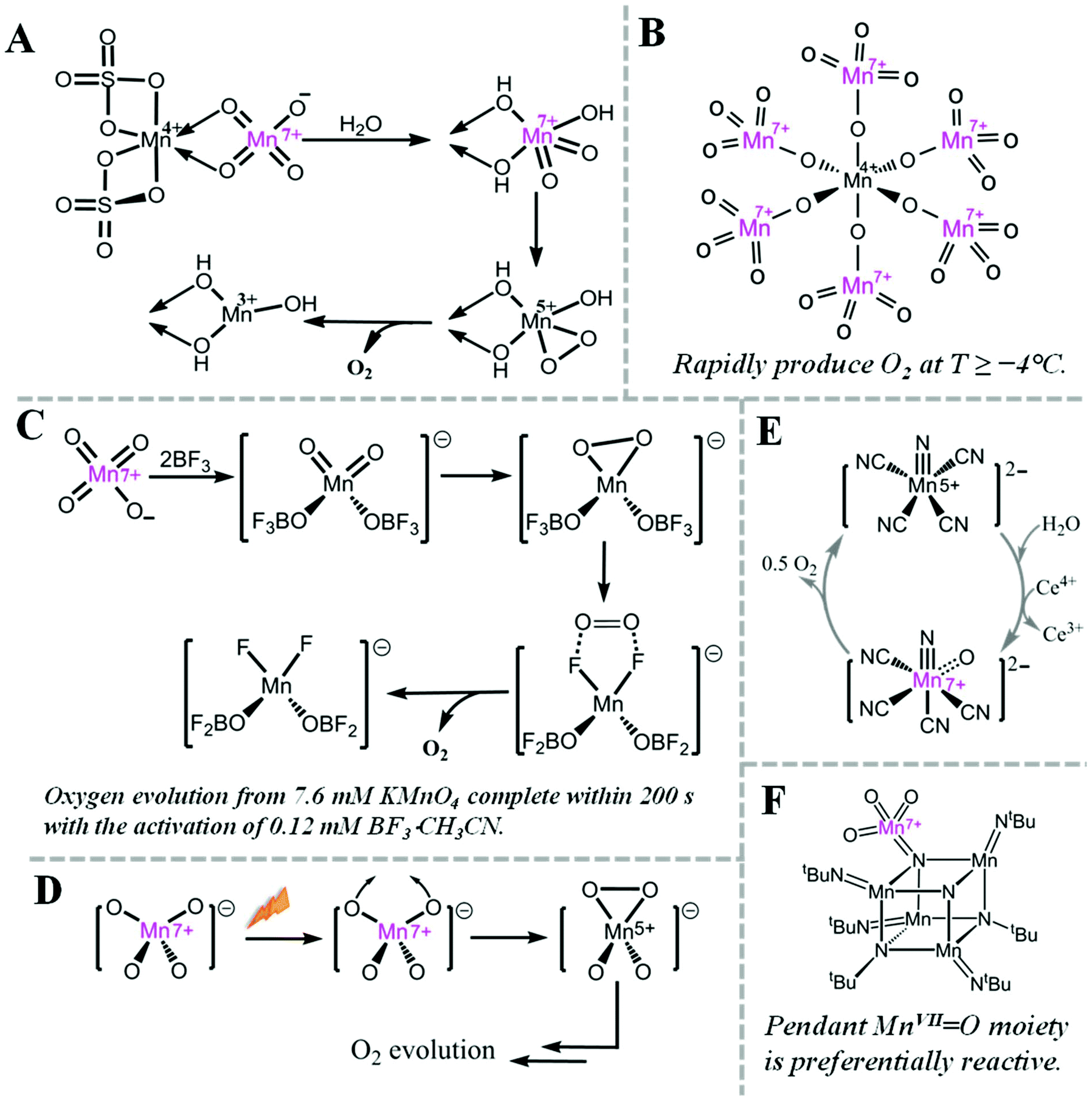 | ||
| Fig. 3 Highly reactive MnVII species and related O2 evolution reactions. (A) Pathway for oxygen evolution from MnO4−, catalysed by MnIV ion.29 (B) Structure of [MnIV(MnVIIO4)6]2− complex.39 (C) Mechanism for fast oxygen evolution from MnO4−, activated by BF3, a strong Lewis acid.40 (D) Proposed structures of intermediates for O2 evolution from photodecomposition of MnO4−.41 (E) Mechanism for water oxidation by MnV–nitrido molecular catalyst.42 (F) Structure of cubic Mn–nitride complex with pendant MnVIIO moiety.43 | ||
Very recently, we reported the presence of an essential MnVII–dioxo intermediate in a synthetic c-disordered δ-MnOx-based water oxidation catalyst (i.e., MnOx-300),45 where a MnIV–O–MnVIIO was proposed as the active species for O–O bond formation.44 As shown in Fig. 4, one active Mn site and three related Mn atoms of the manganese oxides are suggested to participate actively in the catalysis. After multiple electrochemical oxidations of the active site, the initial [MnIIIMnIIIMnIV(HO–MnIII–OH2)] state is oxidized to [MnIVMnIVMnIV(HO–MnIVO)] accompanied by the loss of three electrons and two protons. [MnIVMnIVMnIV(HO–MnIVO)] is assumed to undergo charge rearrangement with release of one proton, resulting in a resting [MnIIIMnIIIMnIIIMnVII(O)2] state. A structural moiety of this intermediate has been proved by a characteristic CV reduction peak at 0.93 V and an IR absorption frequency of 912 cm−1, which strictly corresponds to the occurrence of water-oxidation reaction. Moreover, several experimental evidences demonstrated that this surface-bonded MnVII–dioxo intermediate is more prone to oxidation than the free MnO4−. However, we consider it as a resting state instead of a final active species for O–O bond formation since this MnVII–dioxo intermediate has a long life time. Subsequently, the MnIII ion directly bonded to the dangling MnVIIO site is thought to be further oxidized to MnIV, which drastically increases reactivity of the MnVIIO site, forming the active state [MnIIIMnIIIMnIVMnVII(O)2] state for oxygen evolution. Results of this study can be experimental evidences to support the assumption that an activated permanganate-like MnVIIO moiety is highly reactive and it may be present during the catalytic cycle of water oxidation in PSII.
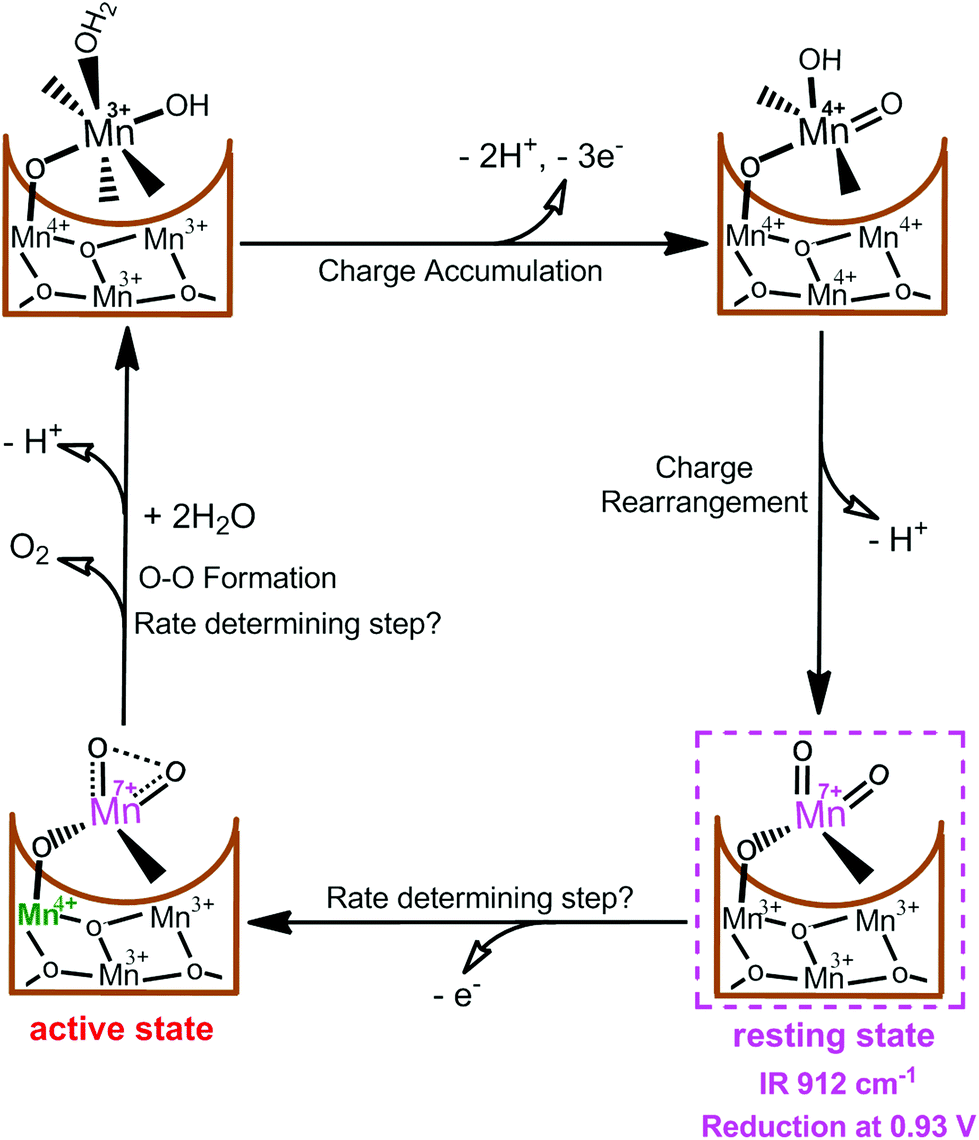 | ||
| Fig. 4 Proposed catalytic cycle in MnOx-300-catalyzed water-oxidation reaction.44 | ||
The above examples clearly show that O–O bond formation from species with activated permanganate-like MnVIIO moiety is a fast and efficient pathway for oxygen evolution. In contrast, a fast O–O bond formation mechanism from MnIVO or MnVO species is not commonly available in the published studies on oxygen evolution reaction. Since the turnover frequency of PSII is about 100–400 s−1,36 it is necessary to consider a fast pathway for the O–O bond formation, involved in the mechanism of water oxidation by PSII.
Lastly, we emphasize that in this new proposed mechanism, the essential charge rearrangement involved in the steps of S3-YZ˙ → S4 → S0 is consistent with the kinetic studies where a slow kinetic phase involving a structural rearrangement has been strongly suggested,19,21–23,27,28 giving a possible explanation to this slow kinetic phase. Two molecules of water are transferred into the Mn4CaO5 cluster during each catalytic cycle: one from the Ca water channel (as the successor of O5 for the next catalytic cycle) and one from the Asp61 water channel (for the new W2) consumed for forming O2. This is consistent with the fact that the proton/water channels in PSII are closely related to the Ca site and the Mn4 site.46 This finding also agrees with the fact that W2, W3 and O5 are usually considered as the O substrates involved in O–O bond formation.46
Based on our findings, we prefer this mechanism that involves MnVIIO moiety and O–O bond formation compared to the coupling of O5 and O of W2 for water oxidation by the OEC. However, without experimental evidence from direct studies on the OEC, particularly for the S3 → S4 step, other potential O–O bond formation pathways may be possible, for example, O–O bond formation from O5 and the new O (O6) on Mn117,18,47 or from O4 and O of W1.48
Conclusions
Involvement of MnVII in the O–O bond formation step is a highly appropriate alternative mechanism for water-oxidation catalysis by the Mn4CaO5 cluster. This mechanism involves a complete catalytic cycle with reasonable valency changes, structural transformations, and O–O bond formation steps. In this new mechanism, we propose that Mn4 is the active site for the O–O bond formation in the S4 state by forming an essential MnVII–dioxo species. This Mn4 site also functions as a gate for releasing protons from the Mn4CaO5 cluster. Mn1, Mn2, and Mn3 sites, within the cubane, function as a battery for charge storage in the first three steps. During the S3 → S4 step, all stored charges accumulate on Mn4 to form the appropriate MnVII–dioxo state, which subsequently triggers O–O bond formation and oxygen evolution. Hence, OEC is constructed as a Mn3CaO4 cubane coupled with a dangling Mn4. In addition, OEC has an extremely open coordination sphere that is located in the water and proton channels and approaches the redox mediator YZ.Therefore, we suggest that synthetic multinuclear Mn complexes could be promising candidates for efficient artificial WOCs. Different coordination environments should be considered in the ligand structure design for different types of Mn cores with specific roles, i.e., multiple Mn cores for charge storage and Mn with open coordination sites for handling substrate water, proton release, and O–O bond formation. Furthermore, special attention should be paid to the ligands that are needed to stabilize the MnVII site.
Finally, on the basis of our new mechanistic proposal, we can answer the question in the title: why did nature choose the Mn4CaO5 cluster as the catalyst for water oxidation in PSII?
- Mn is abundant in the Earth, has rich redox chemistry, and can bear four charges by varying its valencies between MnIII and MnVII.
- Four Mn can coordinate and create the MnVII–dioxo site via charge disproportionation of four MnIV, which is unique, i.e., distinguishable from other 3 Mn ions by being placed outside the cubane structure.
- Five μ-oxo bridges are essential, with one (O5) available for O–O bond formation and the other four for balancing the positive charges. These bridges can also prevent the integral Mn4CaO5 cluster structure from falling apart (maintaining cooperation among four Mn and one Ca).
- Ca is needed as a “taxi stand” for the essential substrate water molecules before they are transferred to the open site during the S2 → S3 step to regenerate a new O5 for the next catalytic cycle.
Therefore, as an economic and perfect “team-playing” catalyst, the Mn4CaO5 cluster was chosen by nature for water oxidation in PSII three billion years ago. Since investigations and conclusions on the mechanism of oxygen evolution in PSII are still far from clear, we hope our new hypothesis opens new possibilities for uncovering the secrets of water oxidation in PSII.
Conflicts of interest
The authors declare no competing interests.Acknowledgements
We acknowledge financial support of this work by the Swedish Research Council (2017-00935), Swedish Energy Agency, Knut and Alice Wallenberg Foundation, and the National Basic Research Program of China (973 program, 2014CB239402). We thank Professor Christina Moberg at KTH for reviewing the manuscript and the valuable discussions.References
- J. P. McEvoy and G. W. Brudvig, Chem. Rev., 2006, 106, 4455–4483 CrossRef PubMed.
- S. A. Crowe, L. N. Dossing, N. J. Beukes, M. Bau, S. J. Kruger, R. Frei and D. E. Canfield, Nature, 2013, 501, 535–538 CrossRef PubMed.
- M. Suga, F. Akita, K. Hirata, G. Ueno, H. Murakami, Y. Nakajima, T. Shimizu, K. Yamashita, M. Yamamoto, H. Ago and J. R. Shen, Nature, 2015, 517, 99–103 CrossRef PubMed.
- Y. Umena, K. Kawakami, J. R. Shen and N. Kamiya, Nature, 2011, 473, 55–60 CrossRef PubMed.
- K. N. Ferreira, T. M. Iverson, K. Maghlaoui, J. Barber and S. Iwata, Science, 2004, 303, 1831–1838 CrossRef PubMed.
- A. Zouni, H.-T. Witt, J. Kern, P. Fromme, N. Krauβ, W. Saenger and P. Orth, Nature, 2001, 409, 739–743 CrossRef PubMed.
- B. Loll, J. Kern, W. Saenger, A. Zouni and J. Biesiadka, Nature, 2005, 438, 1040–1044 CrossRef PubMed.
- B. Kok, B. Forbush and M. McGloin, Photochem. Photobiol., 1970, 11, 457–475 CrossRef PubMed.
- V. Krewald, M. Retegan, N. Cox, J. Messinger, W. Lubitz, S. DeBeer, F. Neese and D. A. Pantazis, Chem. Sci., 2015, 6, 1676–1695 RSC.
- D. A. Pantazis, W. Ames, N. Cox, W. Lubitz and F. Neese, Angew. Chem., Int. Ed., 2012, 51, 9935–9940 CrossRef PubMed.
- T. Lohmiller, V. Krewald, A. Sedoud, A. W. Rutherford, F. Neese, W. Lubitz, D. A. Pantazis and N. Cox, J. Am. Chem. Soc., 2017, 139, 14412–14424 CrossRef PubMed.
- J. Yano and V. Yachandra, Chem. Rev., 2014, 114, 4175–4205 CrossRef PubMed.
- M. Perez-Navarro, F. Neese, W. Lubitz, D. A. Pantazis and N. Cox, Curr. Opin. Chem. Biol., 2016, 31, 113–119 CrossRef PubMed.
- D. J. Vinyard, S. Khan and G. W. Brudvig, Faraday Discuss., 2015, 185, 37–50 RSC.
- J. Barber, Nat. Plants, 2017, 3, 17041 CrossRef PubMed.
- P. E. M. Siegbahn, Acc. Chem. Res., 2009, 42, 1871–1880 CrossRef PubMed.
- M. Suga, F. Akita, M. Sugahara, M. Kubo, Y. Nakajima, T. Nakane, K. Yamashita, Y. Umena, M. Nakabayashi, T. Yamane, T. Nakano, M. Suzuki, T. Masuda, S. Inoue, T. Kimura, T. Nomura, S. Yonekura, L.-J. Yu, T. Sakamoto, T. Motomura, J.-H. Chen, Y. Kato, T. Noguchi, K. Tono, Y. Joti, T. Kameshima, T. Hatsui, E. Nango, R. Tanaka, H. Naitow, Y. Matsuura, A. Yamashita, M. Yamamoto, O. Nureki, M. Yabashi, T. Ishikawa, S. Iwata and J.-R. Shen, Nature, 2017, 543, 131–135 CrossRef PubMed.
- P. E. M. Siegbahn, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 4966–4968 CrossRef PubMed.
- M. Haumann, P. Liebisch, C. Müller, M. Barra, M. Grabolle and H. Dau, Science, 2005, 310, 1019–1021 CrossRef PubMed.
- H. Sakamoto, T. Shimizu, R. Nagao and T. Noguchi, J. Am. Chem. Soc., 2017, 139, 2022–2029 CrossRef PubMed.
- H. Bao and R. L. Burnap, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, E6139–E6147 CrossRef PubMed.
- I. Zaharieva, H. Dau and M. Haumann, Biochemistry, 2016, 55, 6996–7004 CrossRef PubMed.
- M. Haumann, C. Müller, P. Liebisch, L. Iuzzolino, J. Dittmer, M. Grabolle, T. Neisius, W. Meyer-Klaucke and H. Dau, Biochemistry, 2005, 44, 1894–1908 CrossRef PubMed.
- F. A. Armstrong, Philos. Trans. R. Soc., B, 2008, 363, 1263–1270 CrossRef PubMed.
- R. J. Pace, R. Stranger and S. Petrie, Dalton Trans., 2012, 41, 7179–7189 RSC.
- M. Capone, D. Narzi, D. Bovi and L. Guidoni, J. Phys. Chem. Lett., 2016, 7, 592–596 CrossRef PubMed.
- M. R. Razeghifard and R. J. Pace, Biochemistry, 1999, 38, 1252–1257 CrossRef PubMed.
- G. Renger, Biochim. Biophys. Acta, 2001, 1503, 210–228 CrossRef.
- V. Y. Shafirovich, N. K. Khannanov and A. E. Shilov, J. Inorg. Biochem., 1981, 15, 113–129 CrossRef.
- V. Y. Shafirovich, Kinet. Katal., 1978, 19, 1502–1507 Search PubMed.
- T. S. Dzhabiev, Kinet. Katal., 1989, 30, 1219–1224 Search PubMed.
- J. Limburg, J. S. Vrettos, L. M. Liable-Sands, A. L. Rheingold, R. H. Crabtree and G. W. Brudvig, Science, 1999, 283, 1524–1527 CrossRef PubMed.
- R. Ramaraj, A. Kira and M. Kaneko, Chem. Lett., 1987, 16, 261–264 CrossRef.
- J. Limburg, G. W. Brudvig and R. H. Crabtree, J. Am. Chem. Soc., 1997, 119, 2761–2762 CrossRef.
- M. Yagi and K. Narita, J. Am. Chem. Soc., 2004, 126, 8084–8085 CrossRef PubMed.
- M. M. Najafpour, G. Renger, M. Holynska, A. N. Moghaddam, E. M. Aro, R. Carpentier, H. Nishihara, J. J. Eaton-Rye, J. R. Shen and S. I. Allakhverdiev, Chem. Rev., 2016, 116, 2886–2936 CrossRef PubMed.
- M. Grabolle and H. Dau, Biochim. Biophys. Acta, 2005, 1708, 209–218 CrossRef PubMed.
- A. Skrabal, Anorg. Allg. Chem., 1910, 68, 48–51 CrossRef.
- B. Krebs and K.-D. Hasse, Angew. Chem., Int. Ed. Engl., 1974, 13, 603 CrossRef.
- S. M. Yiu, W. L. Man, X. Wang, W. W. Lam, S. M. Ng, H. K. Kwong, K. C. Lau and T. C. Lau, Chem. Commun., 2011, 47, 4159–4161 RSC.
- D. G. Lee, C. R. Moylan, T. Hayashi and J. I. Braurnan, J. Am. Chem. Soc., 1987, 109, 3003–3010 CrossRef.
- L. Ma, Q. Wang, W. L. Man, H. K. Kwong, C. C. Ko and T. C. Lau, Angew. Chem., Int. Ed., 2015, 54, 5246–5249 CrossRef PubMed.
- S. Vaddypally, S. K. Kondaveeti, S. Karki, M. M. Van Vliet, R. J. Levis and M. J. Zdilla, J. Am. Chem. Soc., 2017, 139, 4675–4681 CrossRef PubMed.
- B. Zhang, Q. Daniel, L. Fan, T. Liu, Q. Meng and L. Sun, iScience, 2018, 4, 144–152 CrossRef.
- B. Zhang, H. Chen, Q. Daniel, B. Philippe, F. Yu, M. Valvo, Y. Li, R. B. Ambre, P. Zhang, F. Li, H. Rensmo and L. Sun, ACS Catal., 2017, 7, 6311–6322 CrossRef.
- J. R. Shen, Annu. Rev. Plant Biol., 2015, 66, 23–48 CrossRef PubMed.
- H. Isobe, M. Shoji, J. R. Shen and K. Yamaguchi, Inorg. Chem., 2016, 55, 502–511 CrossRef PubMed.
- K. Kawashima, T. Takaoka, H. Kimura, K. Saito and H. Ishikita, Nat. Commun., 2018, 9, 1247 CrossRef PubMed.
| This journal is © The Royal Society of Chemistry 2018 |