
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Recent advances in synthetic facets of immensely reactive azetidines
Vishu Mehra
a,
Isha Lumb
b,
Amit Anand
*c and
Vipan Kumar
 *d
*d
aDepartment of Chemistry, Hindu College, Amritsar-143005, India
bDepartment of Chemistry, Baring Union Christian College, Batala-143505, India
cDepartment of Chemistry, Khalsa College, Amritsar-143005, India
dDepartment of Chemistry, Guru Nanak Dev University, Amritsar-143005, Punjab, India. E-mail: vipan_org@yahoo.com; Fax: +91-183-2258819-20; Tel: +91-183-2258802 extn 3320
First published on 27th September 2017
Abstract
The synthetic chemistry of azetidine constitutes an important yet undeveloped research area, in spite of their ubiquity in natural products and importance in medicinal chemistry. Their aptness as amino acid surrogates along with their potential in peptidomimetic and nucleic acid chemistry is considered remarkable. Azetidines also possess important prospects in other settings such as catalytic processes including Henry, Suzuki, Sonogashira and Michael additions. They also represent an important class of strained compounds making them excellent candidates for ring-opening and expansion reactions. With this in mind, the present review article addresses the recent developments in synthetic strategies towards functionalized azetidines along with their versatility as heterocyclic synthons.
 Vishu Mehra | Dr Vishu Mehra obtained his Master's degree in Applied Chemistry with first division in 2007 from Guru Nanak Dev University, Amritsar. In 2007, he joined Jubilant Chemsys (R & D) at Noida and worked as a Trainee Research Associate in the field of organic synthesis. He joined the research group of Dr Vipan Kumar in 2009 in the Department of Chemistry, Guru Nanak Dev University, and worked extensively on the synthetic transformations of C-3 functionalized azetidin-2-ones leading towards the synthesis of functionally decorated heterocyclic compounds. He has also received the best poster presentation award in an international conference held at Punjab University, Chandigarh in 2011. He has published fourteen research papers in journals of international repute and is currently working as an Assistant Professor at Hindu College, Amritsar. His research interests include utilization of β-lactam synthon protocol for the preparation of functionally enriched heterocyclic scaffolds having biological relevance. He is also working on designing synthetic conjugates of medicinally active compounds to architect novel hybrid pharmacophores with higher potential and efficacy as compared to their analogues. |
 Isha Lumb | Dr Isha Lumb Ph.D. has been working as an Assistant Professor in Department of Chemistry, Baring Union Christian College, Batala since 2016. She obtained her Master's degree in Chemistry with first division in 2007 from Panjab University. She joined research group of Prof. Geeta Hundal in the Department of Chemistry, GNDU, Amritsar in 2009 and exclusively worked on synthesis, characterization and crystal structure determination of amide based polydentate ligands and their complexes with different transition metals. During her doctoral work, she has published four research papers in journals of international repute. Her research interest includes the designing and synthesis of polydentate ligands along their complexes with different metals and characterized them with different spectroscopic techniques including X-ray crystallography. She is also working on designing synthetically medicinally active complexes, having high potential and efficacy against antimicrobial and antifungal activities. |
 Amit Anand | Dr Amit Anand, Ph.D. has been working as an Assistant Professor in Department of Chemistry, Khalsa College, Amritsar since 2004. He completed his undergraduate and post graduate degree in Chemistry from Department of Chemistry, Guru Nanak Dev University, Amritsar, Punjab, He obtained his Ph.D degree with Prof. M. P. Mahajan in the Department of Applied Chemistry, GNDU, Amritsar in 2009. His research interest includes designing and synthesis of diverse heterocyclic compounds with biological and medicinal potential utilising the well documented β-lactam synthon strategy. He is also working on designing synthetic conjugates of medicinally active compounds to architect novel hybrid pharmacophores with higher potential and efficacy as compared to their analogues. |
 Vipan Kumar | Dr Vipan kumar Ph.D. has been working as an Assistant Professor in the Department of Chemistry, Guru Nanak Dev University, Amritsar since 2009. He obtained his Ph.D with Prof. Mohinder P Mahajan, in the Department of Applied Chemistry, GNDU. In 2007, he moved to the University of Cape Town (UCT), South Africa to pursue his postdoctoral studies with Prof. Kelly Chibale. In 2015, he was awarded research fellowship from Carl Tryggers research foundation, Sweden to carry out research at University of Umea, Sweden. His research interests include the development of diverse synthetic protocols for synthesis of novel molecular frameworks targeting tropical infections. He has also been engaged in the utilization of β-lactam synthon protocols for the synthesis of functionally decorated and biologically relevant heterocycles with medicinal potential. |
1. Introduction
Azetidines constitute a vital class of aza-heterocyclic scaffolds with prevalence in diverse natural and synthetic products exhibiting a wide range of biological activities.1 α-Amino azetidin-2-carboxylic acid is a proline antagonist in plant tissue cultures, exenta and melagatran act as thrombin inhibitors, while nicotianamine acts as a phytosiderophore, and mugineic acid and 2′′-hydroxynicotianamine are ACE-inhibitors2 (Fig. 1).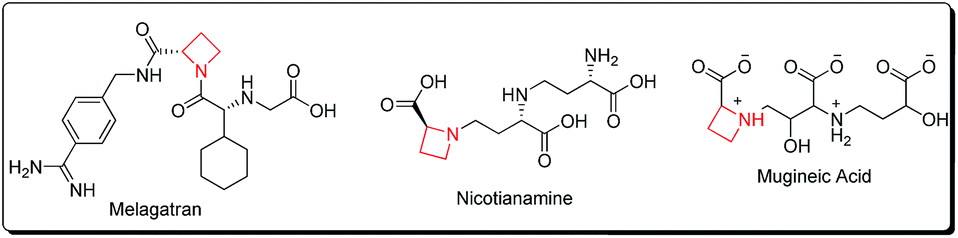 | ||
| Fig. 1 Azetidine based biologically active scaffolds. | ||
The modern mimicking approaches for recognition enhancement by biological receptors suffer from conformational constraints. Small size rings especially the azetidine framework, provide the necessary size for complementary receptor binding and achieving a high level of activity. The usefulness of azetidines as surrogates of natural amino-acids3a and as a tool in peptidomimetics3b is highly commendable.3c–e Their use as ligands in various catalytic processes including reductions, cycloadditions, cyclopropanation and C–C bond forming reactions is endowed with promising results.4 Moreover, azetidines belong to the class of strained compounds which makes them excellent candidates for nucleophilic ring-opening or ring-expansion reactions yielding highly substituted acyclic amines or expanded ring systems. As a result, substituted azetidines have proven to be excellent synthons for rearrangements towards pyrroles, pyrrolidines, pyrrolidinones, imidazolidinones, isoxazolidines, piperidines, 1,2-oxazines, piperidin-2-ones, 2-iminopiperidines, azepanes and azepan-2-ones.5 The purpose of the present review is therefore to address the audience of synthetic chemists about the recent developments (2011–2016) in strategies for the preparation of this four-membered ring along with its role as versatile synthon in heterocyclic chemistry. For the sake of convenience, the review article is divided into following sections and subsections:
• Intramolecular cyclization enroute to racemic-/enantio-/diastereoselective access to azetidines.
• C-3 functionalized azetidines via β-lactam synthon approach.
• Nucleophilic ring opening of aziridines.
• Metal-catalyzed synthesis of azetidines.
• Synthesis of azetidines via organocatalysis.
• Miscellaneous syntheses.
• Azetidines as versatile synthons.
2. Synthetic approaches to functionalized azetidines
2.1 Intramolecular cyclization enroute to racemic-/enantio-/diastereoselective access to azetidines
The selective introduction of fluorine, as a trifluoromethyl (CF3) group strongly alters the biological and pharmacological properties such as pKa, lipophilicity, toxicity and metabolic stability of bioactive compounds.6 The chemistry of 2-CF3-azetidines has largely remained unexplored both in terms of their synthesis and reactivity. De Kimpe et al.7 has developed protocol for the synthesis 1-alkyl-2-(trifluoromethyl)azetidines 5 starting from ethyl 4,4,4-trifluoroacetoacetate via imination, hydride reduction, chlorination and base-induced ring closure. Thus, the treatment of ethyl 4,4,4-trifluoroacetoacetate 1 with a primary amine in the presence of acetic acid yielded the enamine 2 which upon reduction with sodium borohydride afforded the corresponding 3-alkylamino-4,4,4-trifluorobutan-1-ol 3. Further, treatment of γ-amino alcohol 3 with thionyl chloride in dichloromethane resulted in isolation of N-alkyl-4-chloro-1,1,1-trifluorobutan-2-amine 4. The intramolecular cyclization of 4 in the presence of LiHMDS in THF under refluxing for 4 h, resulted in the formation of 1-alkyl-2-(trifluoromethyl)azetidines 5 as depicted in Scheme 1. The electron-withdrawing nature of trifluoromethyl substituent reduced the nucleophilicity of nitrogen atom, thus requiring a strong base (LiHMDS) to achieve this cyclization.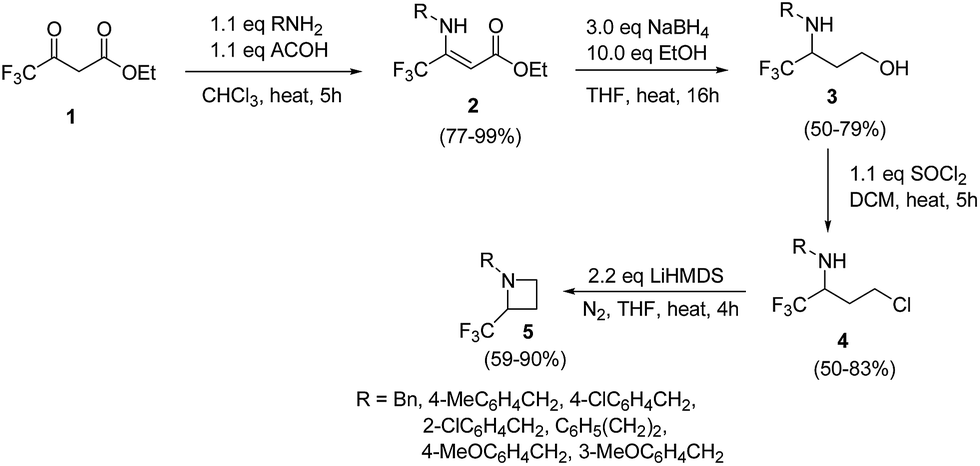 | ||
| Scheme 1 | ||
Fossey and co-workers8 have explored the iodocyclization of homoallyl amines enroute to functionalized 2-(iodomethyl)azetidine derivatives. The synthetic protocol involved the treatment of homoallyl amine 6 with three equivalents of iodine in acetonitrile in the presence of sodium hydrogen carbonate at room temperature resulting in the stereoselective synthesis of cis-azetidine 7 along with pyrrolidine 8 as depicted in Scheme 2. It was observed that the nature of substituent (electron releasing/withdrawing) affected the relative ratio of products viz. azetidine 7 and pyrrolidine 8 (Scheme 2).
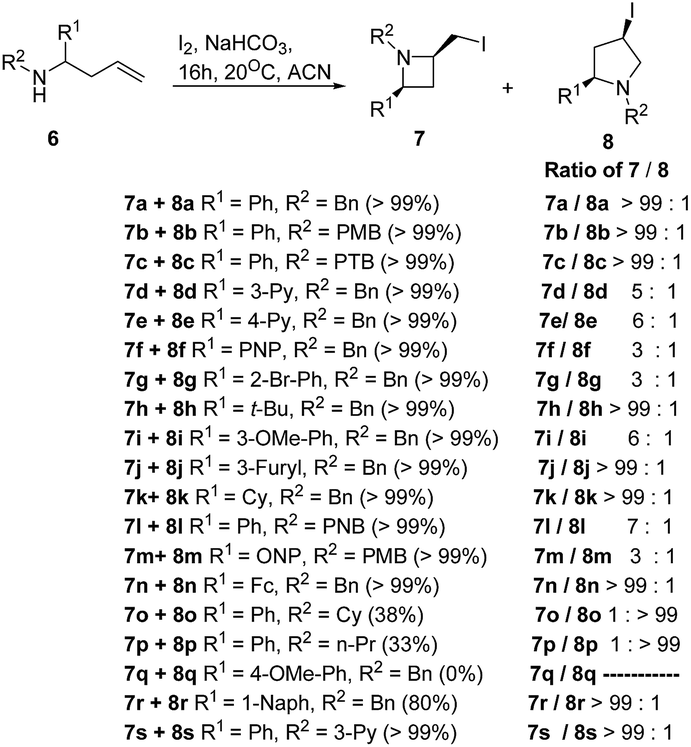 | ||
| Scheme 2 | ||
De Kimpe and co-workers9 have utilized α-chloro-β-aminosulfinyl imidates 9 for the synthesis of enantiopure trans-2-aryl-3-chloroazetidines 13. The key step involved the deprotection of α-chloro-β-aminosulfinyl imidates 9 with 4 N HCl in dioxane to afford the corresponding imidate hydrochloride 10 which underwent hydrolysis at 50 °C to yield ester 11. Reduction of ester 11 using LAH in dry THF at low temperature yielded β-chloro-γ-sulfonylamino alcohol 12 which underwent intramolecular cyclization under Mitsunobu conditions to afford trans-2-aryl-3-chloroazetidines 13 (Scheme 3).
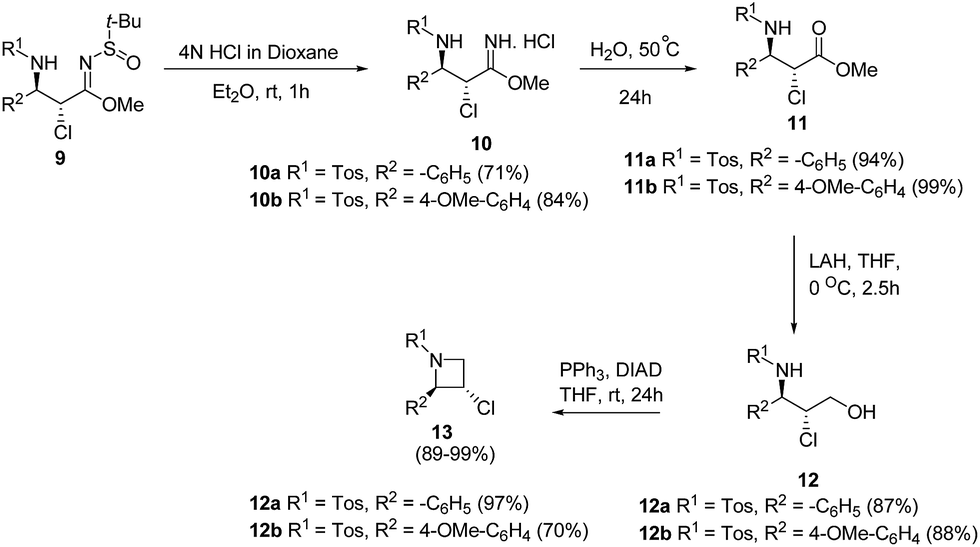 | ||
| Scheme 3 | ||
Smith and co-workers10 have recently reported the synthesis of (S)-2-methylazetidine(R)-(−)-camphorsulfonate (CSA) from 1,3-butanediol. The synthetic protocol involved an initial mesylation of (R)-(−)-1,3-butanediol 14 to yield bis-mesylated product 15 which on treatment with benzylamine afforded the desired azetidine 16 in 53% yield with 95% ee. Recrystallization of (R)-(−)-CSA salt of azetidine 16 in ethyl acetate![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :dichloromethane mixture improved the enantiopurity from 95% to >99%. Subsequent hydrogenolysis of 16 afforded (S)-2-methylazetidine 17 as shown in Scheme 4.
:dichloromethane mixture improved the enantiopurity from 95% to >99%. Subsequent hydrogenolysis of 16 afforded (S)-2-methylazetidine 17 as shown in Scheme 4.
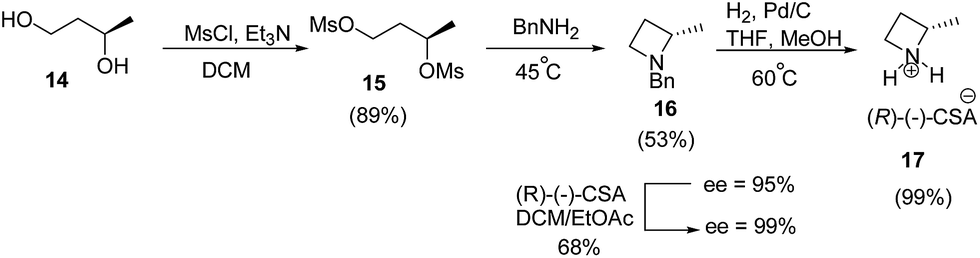 | ||
| Scheme 4 | ||
Further, enantioenriched (S)-2-methylazetidine 17 was prepared through one-pot generation and cyclization of a bis-triflate under different conditions. Best results in terms of yield was obtained via dropwise addition of Tf2O to a premixed solution of (R)-1,3-butanediol 14 and Hunig's base at −35 °C followed by treatment with benzhydrylamine resulted in the formation of 18 which upon hydrogenolysis in the presence of palladium hydroxide to afford (S)-2-methylazetidine(R)-(−)-camphorsulfonate (CSA) 17 (Scheme 5).10
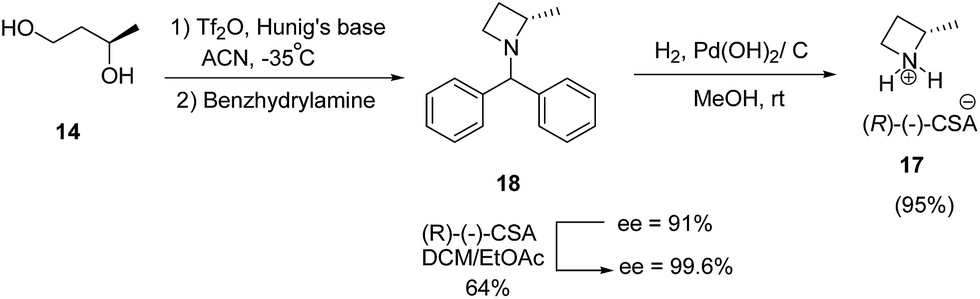 | ||
| Scheme 5 | ||
De Kimpe and co-workers11 have developed thermal isomerization of aziridines for the synthesis of 3-bromoazetidine-3-carboxylic acid derivatives. The key step involved the base promoted cyclization of dibromo amino esters 21 to yield the desired aziridines 22 and/or azetidines 23. Thermal isomerization of kinetically controlled product aziridine to azetidine was carried out by refluxing in DMSO at 70 °C. Further, treatment of 3-bromoazetidines 23 with different nucleophiles viz. potassium cyanide, potassium thiocyanate, sodium azide and potassium phenoxide in DMSO at 60 °C yielded the corresponding 3-cyano, 3-thiocyano, 3-azido and 3-phenoxy azetidine-3-carboxylic acid esters 24. These compounds are imperative in the preparation of libraries of bioactive compounds like bronchodilating and anti-inflammatory drugs12 and antibacterial agents.13 Hydrolysis of ethyl 3-azidoazetidine-3-carboxylate 24 (R1 = Et, R2 = t-Bu, t-pentyl, Nu = N3) with 2 N NaOH solution at room temperature yielded the corresponding acid 25 which upon hydrogenolysis afforded 3-aminoazetidine-3-carboxylic acid 27. The hydrogenolysis of ethyl 3-azidoazetidine-3-carboxylates 24 (R1 = Et, R2 = t-Bu, Nu = N3) with Pd/C yielded the corresponding 3-aminoazetidine-3-carboxylate 26 (Scheme 6).11
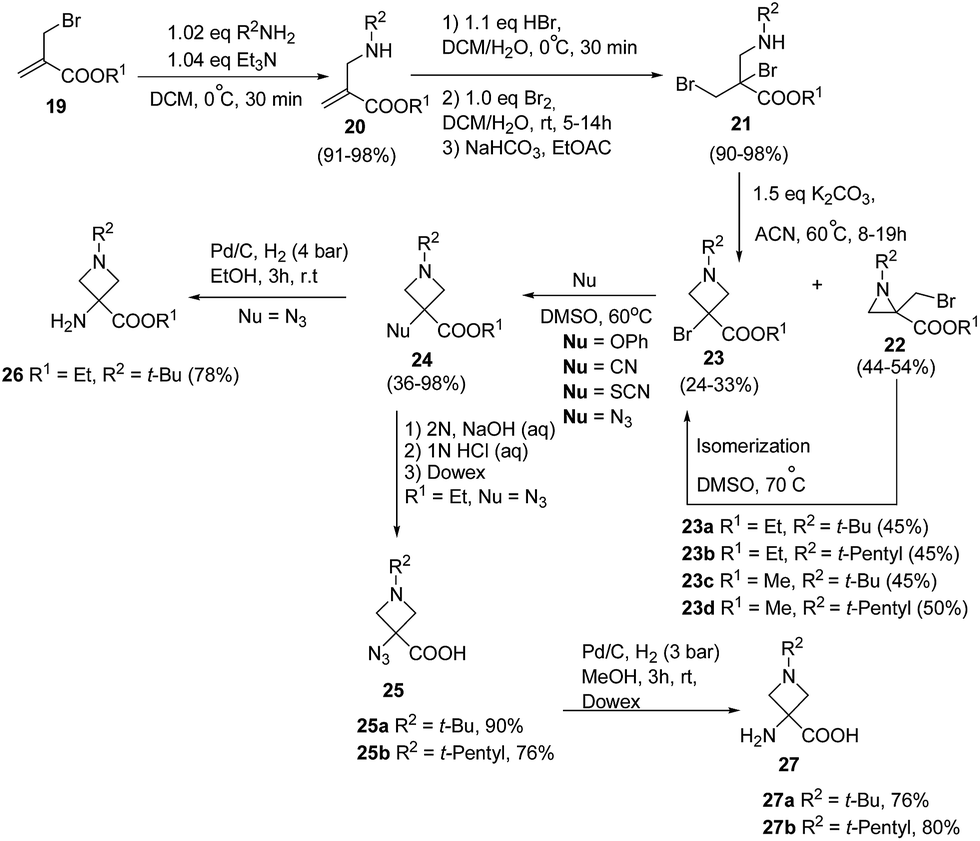 | ||
| Scheme 6 | ||
The above methodology was further extended by De Kimpe and co-workers14 to afford a facile access to 1-t-butyl- and 1-(4-methylbenzyl)-3-bromo-3-methylazetidines as shown in Scheme 7. The synthetic approach followed a sequence of synthetic events with the key step involving the thermal isomerisation of aziridine 30 in acetonitrile to afford 3-bromoazetidine 31 (Path A). Further, room temperature stirring of N-t-butylimine 32 with NaBH4 selectively gave N-(t-butyl)-2,3-dibromo-2-methylpropanamine 33 instead of azetidine derivative (Scheme 7, Path B). Refluxing of 33 in ethanol or methanol for 12 h yielded a mixture of thermodynamically preferred azetidines 34 and 35. The refluxing of 33 in a less nucleophilic solvent such as isopropanol afforded 3-bromoazetidine 34 as the sole product. Further treatment of imine 32 with LAH in diethyl ether gave 3-bromoazetidine 34 as the major product without the traces of corresponding aziridine (Scheme 7, Path C).
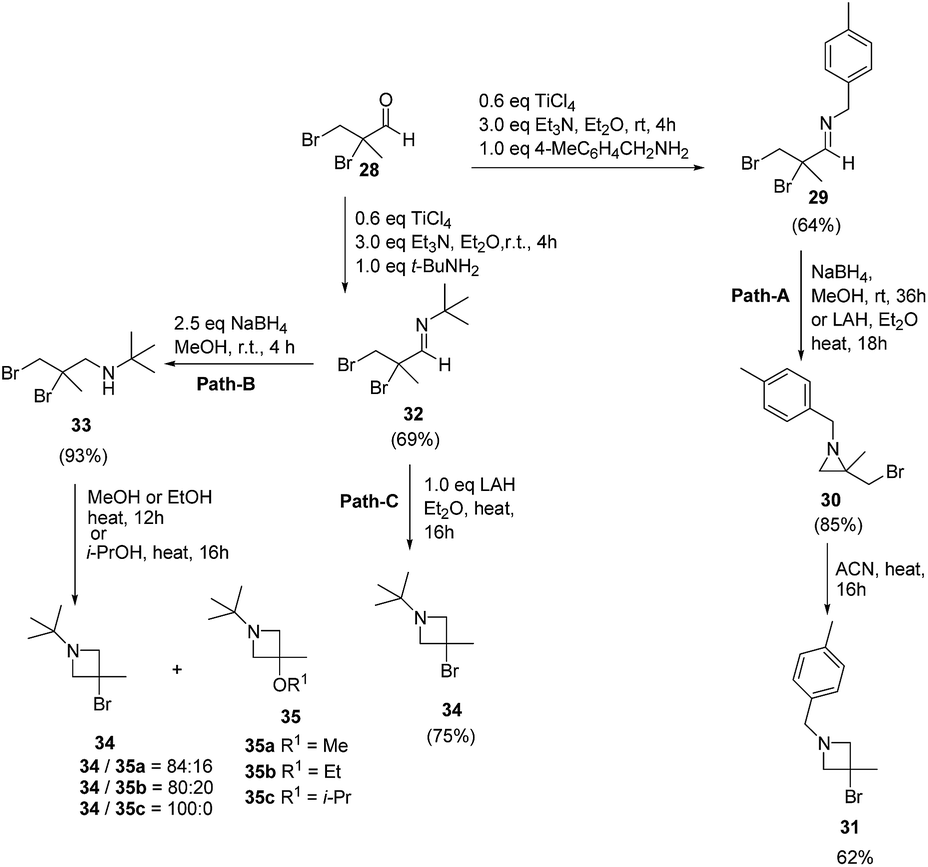 | ||
| Scheme 7 | ||
The plausible mechanism for the formation of side product 3-alkoxyazetidines 35 might involve an initial intramolecular nucleophilic displacement in 34 to yield bicyclic aziridinium ion 36, which underwent alcoholysis (methanolysis or ethanolysis) (Scheme 8).
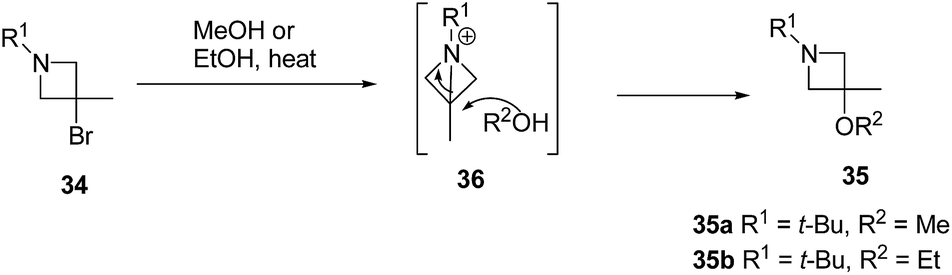 | ||
| Scheme 8 | ||
The synthetic potential of 3-bromo-substituted azetidines was further explored by its reaction with different nucleophiles resulting in the formation of C-3-substituted azetidines as depicted in Scheme 9.14
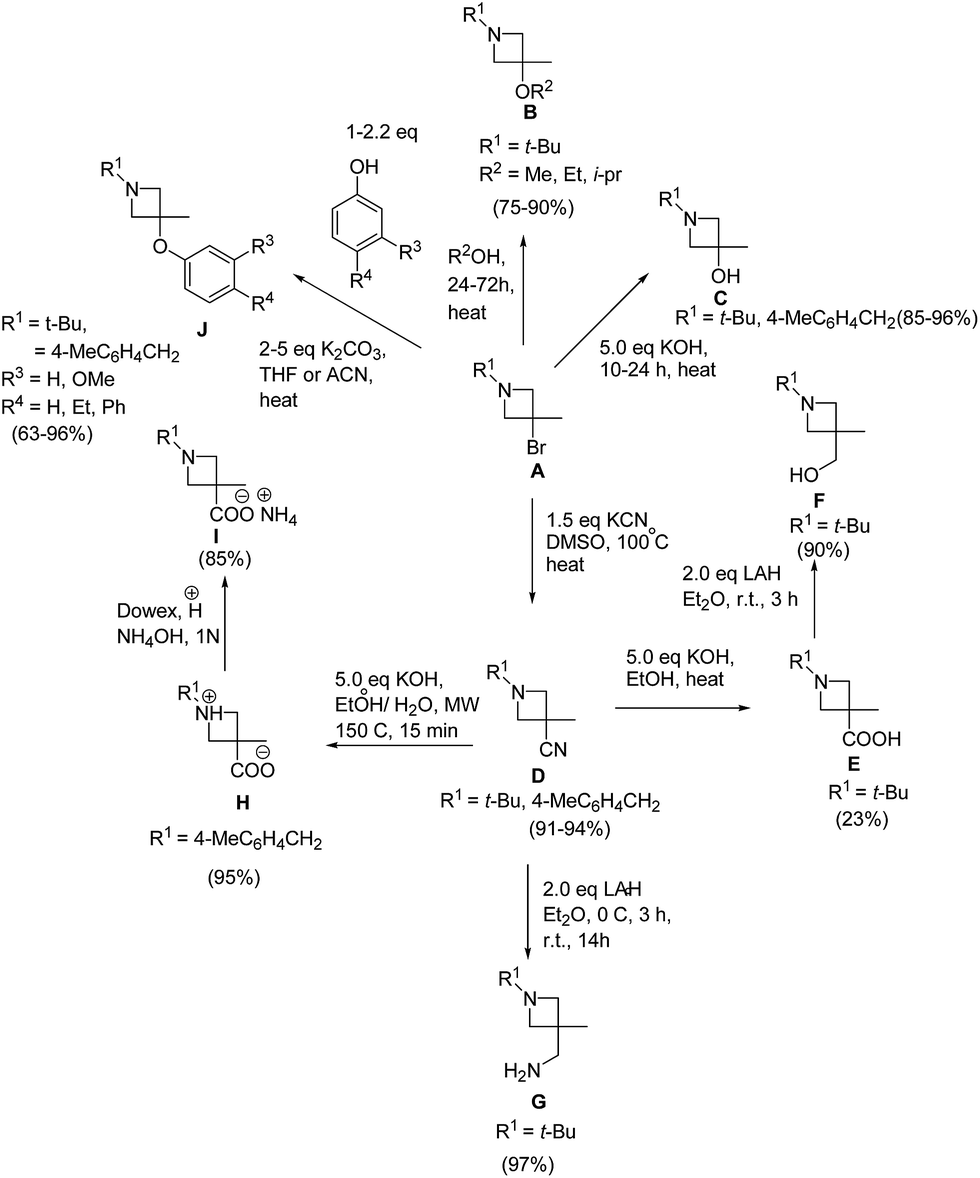 | ||
| Scheme 9 | ||
Szymoniak et al.15 have recently explored the stereoselective synthesis of cis-2,3-disubstituted azetidines through diastereoselective hydrozirconation. The synthetic approach involved the treatment of 37 with Schwartz reagent to yield the hydrozirconated intermediate which on reaction with iodine afforded iodocarbamate intermediate 38. The base promoted intramolecular nucleophilic ring closure of intermediate 38 afforded enantiopure azetidine 39 as elucidated in Scheme 10.
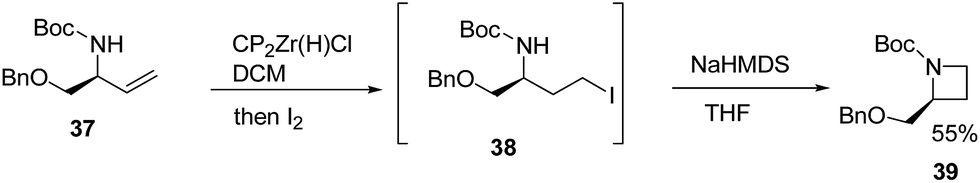 | ||
| Scheme 10 | ||
A range of substituents were explored leading to diastereoselective access of cis-2,3-disubstituted azetidine 42 in as depicted in Scheme 11.15
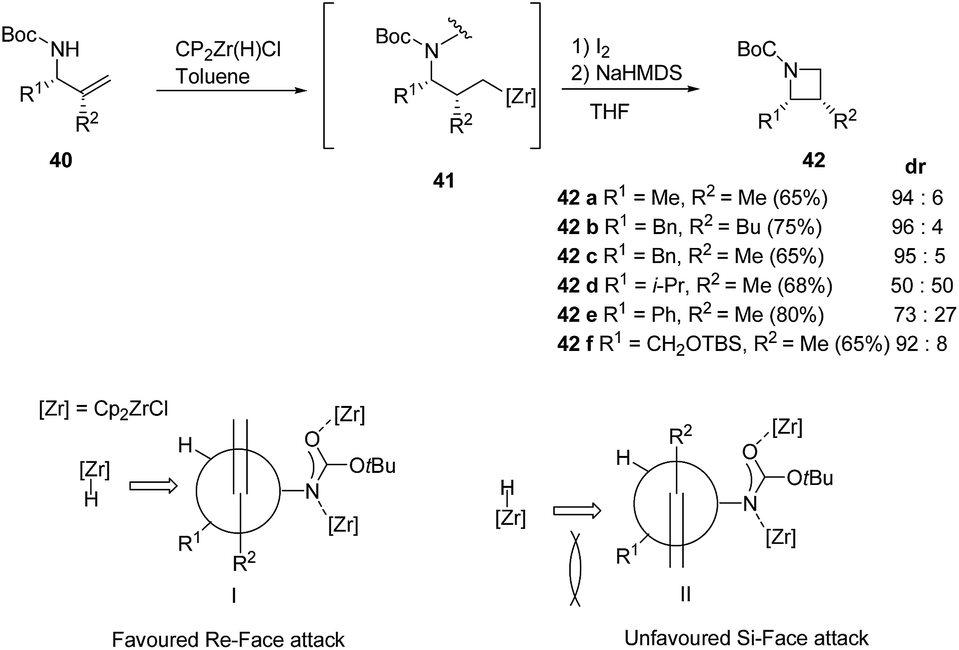 | ||
| Scheme 11 | ||
However the lack of diastereoselectivity was observed in case of 42d and 42e (R1 = i-Pr, Ph and R2 = Me) could be attributed due to steric hinderance created by the bulky substituents. When (R1 = allylamine, R2 = Ph) dehydrozirconation turned out to be more favorable pathway and the reaction did not lead to iodonation/cyclisation product (Scheme 11).
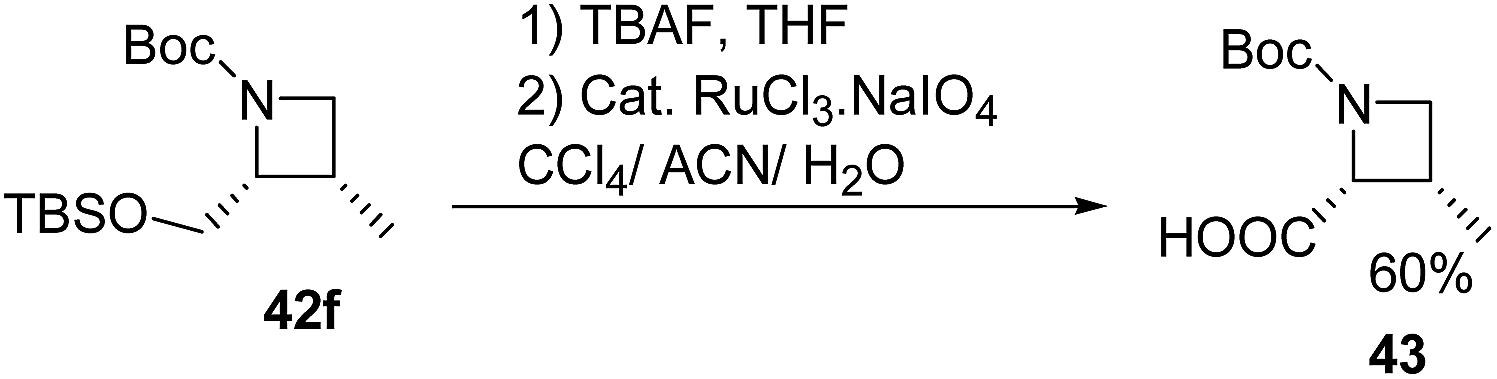
Deprotection of 42f with TBAF and subsequent ruthenium promoted oxidation led to cis-azetidin-2-carboxylic acid 43 which is a key constituent of a number of natural products and their derivatives (Scheme 12).2,16
 | ||
| Scheme 12 | ||
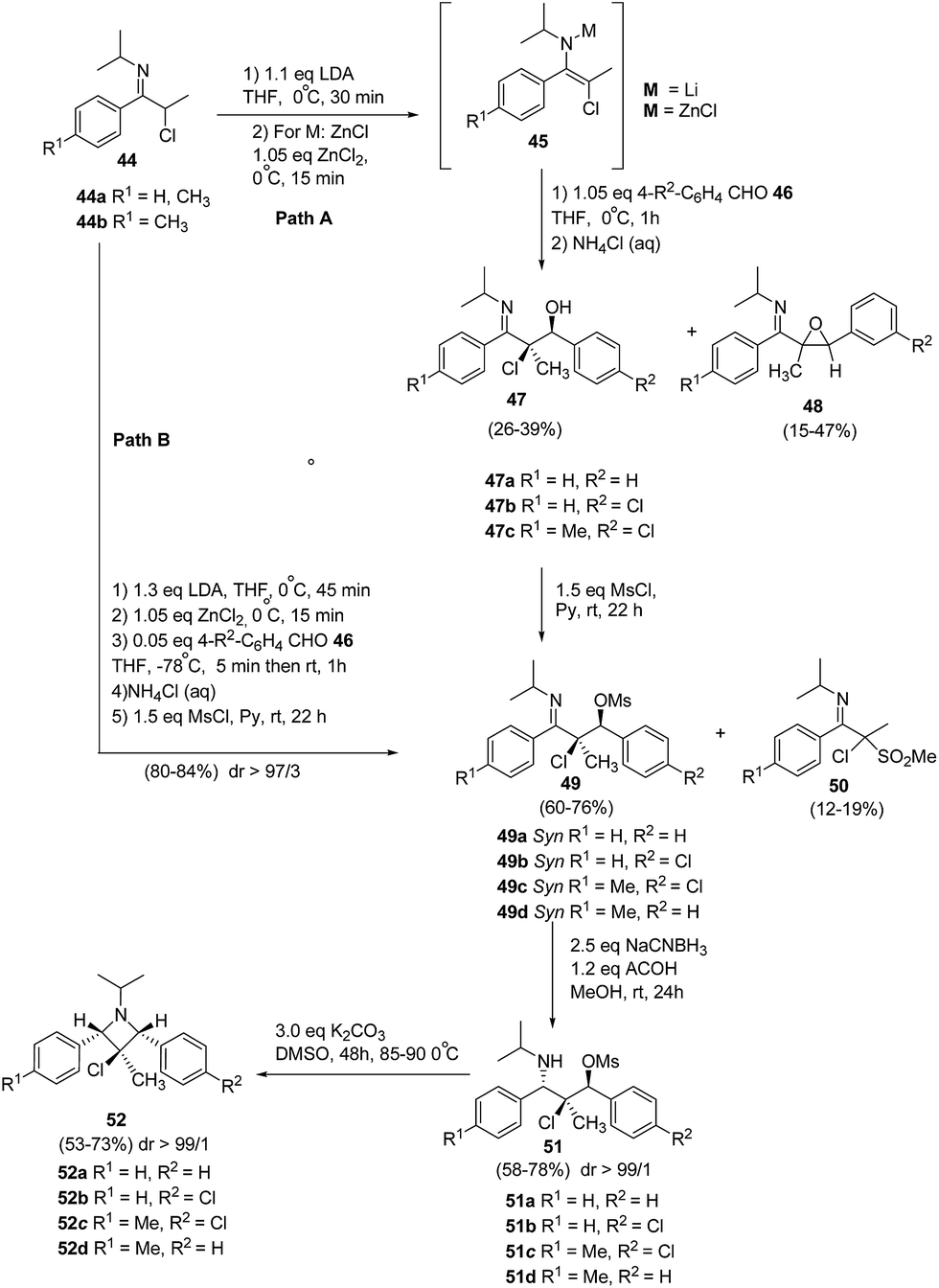
De Kimpe and co-workers17 have utilized the zincated 3-chloro-3-methyl-1-azaallylic anions 45 in stereoselective aldol condensation with aromatic aldehydes and subsequent mesylation to produce syn-α-chloro-β-mesyloxyketimines 49. This upon stereoselective reduction and cyclisation afforded 1,2,3,4-tetrasubstituted 3-chloroazetidines 52 having three stereogenic centers as depicted in Scheme 13. The synthetic protocol involved the treatment of N-isopropyl-α-chloro imine 44a with lithium diisopropylamide in tetrahydrofuran at 0 °C to yield lithiated 3-chloro-3-methyl-1-azaallylic anion 45a, which on aldol condensation with arylaldehyde 46 at 0 °C resulted in a mixture of syn-α-chloro-β-hydroxyketimine 47a as major product along with the corresponding cis- and trans-imidoylepoxides 48a as the side products. The transmetalation of the lithiated anion with ZnCl2 at 0 °C, resulted in zincated 3-chloro-3-methyl-1-azaallylic anion 45a, which proved to be beneficial as it led to slight improvement in the diastereoselectivity of the aldol reaction with benzaldehyde and afforded only smaller amounts of the epoxide 48a. Further lowering the reaction temperature to −78 °C led to the vast improvement in syn-diastereoselectivity of aldol reaction. Nevertheless, it was not possible to effectively separate the syn-α-chloro-β-hydroxyketimine 47a from its anti-adduct via crystallization due to the occurrence of concomitant retro-aldol reaction. However during workup, almost all of the zincated anti-adduct got converted into syn isomer. Subsequent treatment of 47a with mesyl chloride in pyridine yielded the corresponding syn-α-chloro-β-mesyloxyketimines 49a in 80% yield along with α-chloro-α-methanesulfonylketimines 50a isolated as a by product in low yield (Path B). The stereoselective reduction of β-mesyloxyketimines 49 with NaCNBH3 resulted in the formation of β-chloro-γ-mesyloxypropylamines 51 which upon heating in DMSO followed by base-induced cyclization, stereoselectively generated 1,2,3,4-tetrasubstituted 3-chloroazetidines 52.
 | ||
| Scheme 13 | ||
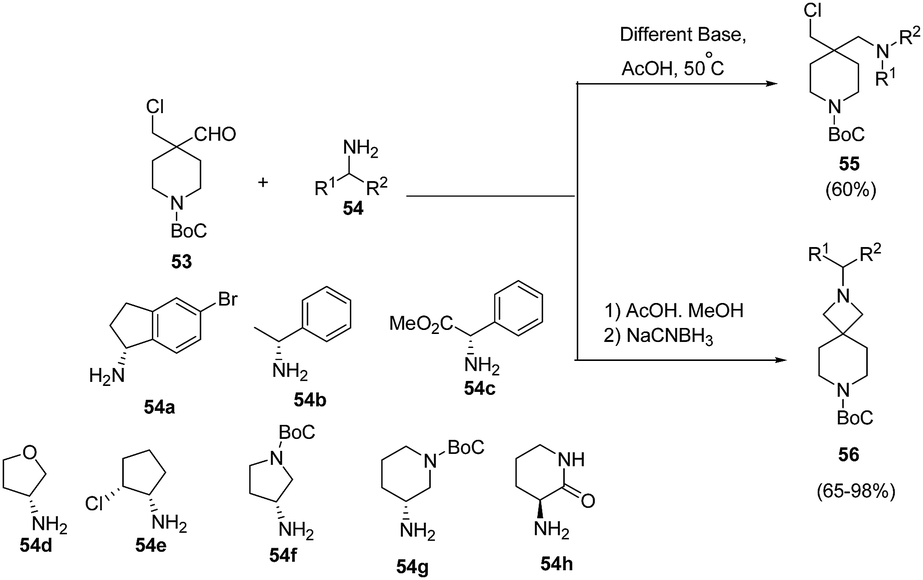
Makowski et al.18 have developed a convenient access to spiro-azetidine-piperidine ring system via reaction of piperidine chloroaldehyde with chiral amines. Initial attempts involving heating of 53 with chiral amines in the presence of base afforded the uncyclized reductive amination chloroamine product 55. The treatment of 53 with chiral amine 54 in AcOH and methanol at 50 °C, however yielded the corresponding imine which subsequently was cyclized in the presence of NaCNBH3 affording chiral azetidine-piperidines 56 as depicted in Scheme 14.
 | ||
| Scheme 14 | ||
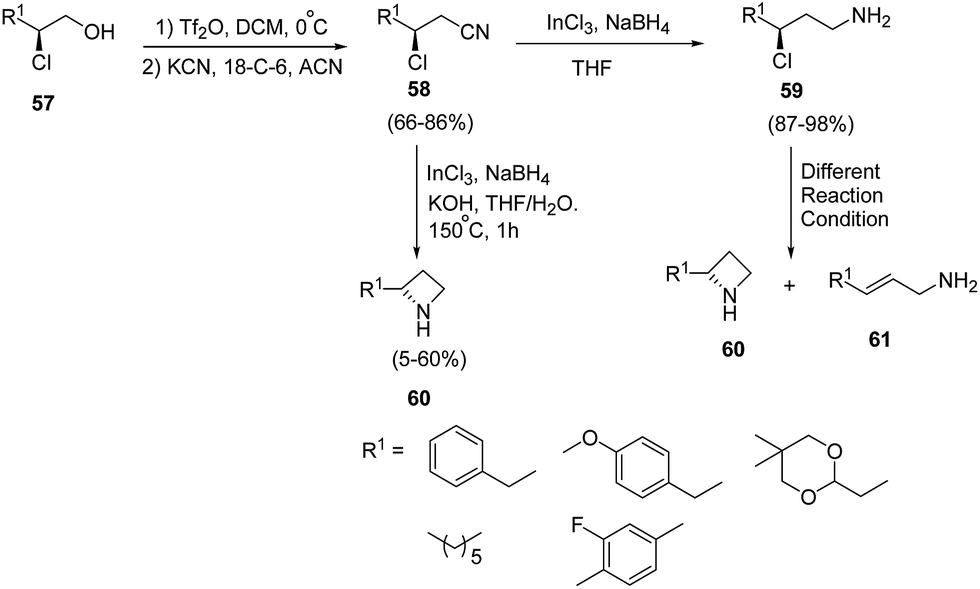
Lindsley and co-workers19 have devised an enantioselective synthesis of C2-functionalized azetidines from β-chloroalcohol. The methodology involved the initial activation of β-chloro alcohol 57 as triflate with subsequent displacement of triflate ion with cyanide in presence of 18-crown-6 to afford β-chloronitrile 58. The synthesized β-chloronitrile 58 was reduced with indium(III)chloride–sodium borohydride with simultaneously achievement of 4-exo-tet cyclization in the presence of KOH/THF to afford the desired azetidine 60 without the isolation of γ-chloroamine 59. It has been found that the reduction of β-chloronitrile 58 to γ-chloroamine 59 was achieved using indium(III)chloride–sodium borohydride. Different reaction conditions (K2CO3 in NMP, NaH in DMF, LHMDS in DMF and K2CO3 in DMF) were attempted for 4-exo-tet cyclization of γ-chloroamine 59 but it led to olefin 61 along with small quantities of desired azetidine 60 as depicted in Scheme 15.
 | ||
| Scheme 15 | ||
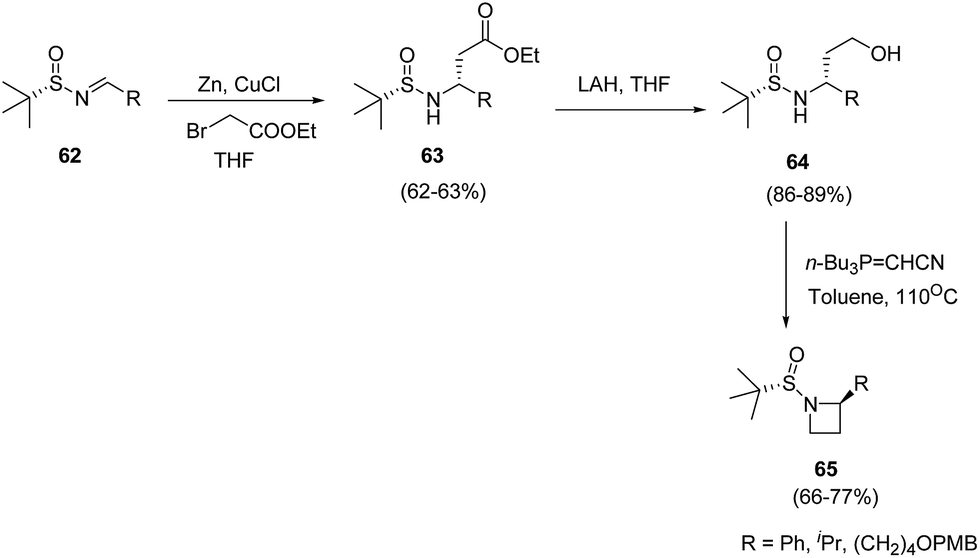
Voss and co-workers20 described the synthesis of C-2-substituted azetidines by using chiral tert-butanesulfinamide as auxiliary to achieve high levels of stereoselectivity. The synthetic methodology involved Reformatsky reaction of sulfinimines 62 yielding 63 followed by LAH promoted reduction to yield corresponding alcohol 64. The treatment of 64 with Tsunoda reagent in toluene at 110 °C resulted in the formation of C-2-substituted azetidines 65 as depicted in Scheme 16.
 | ||
| Scheme 16 | ||
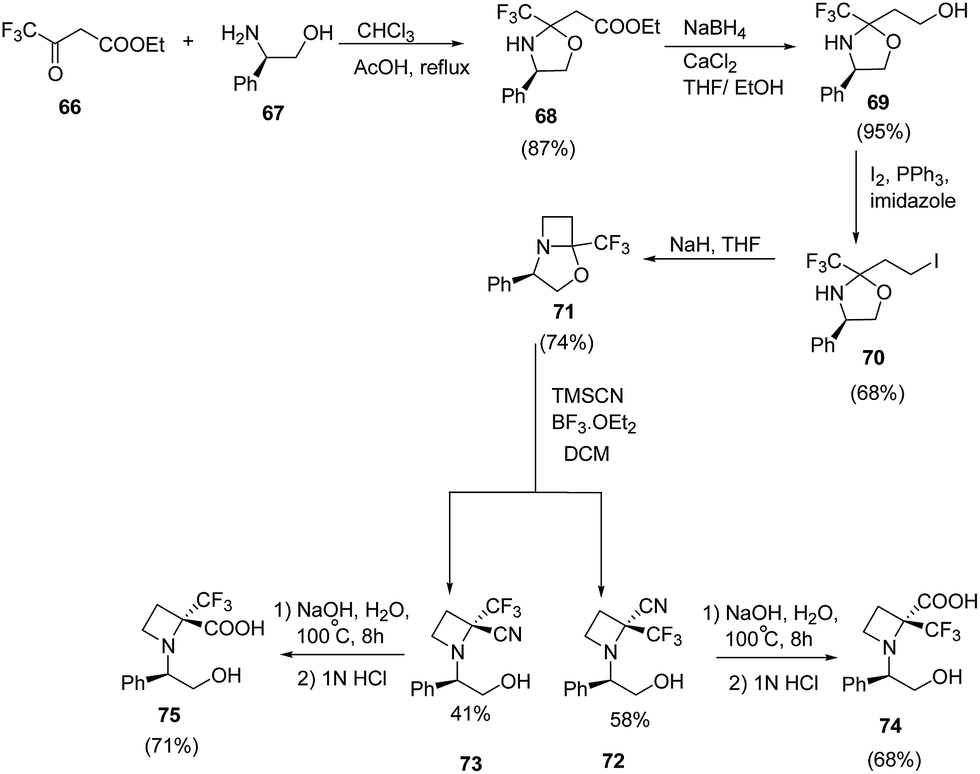
Brigaud et al.21 have recently developed a route for the synthesis of enantiopure α-trifluoromethylated azetidine-2-carboxylic acid from chiral oxazolidine. The synthetic methodology involved the condensation reaction of ethyl trifluoroacetoacetate 66 with (R)-phenylglycinol 67 to yield the oxazolidine 68 as a single diastereomer. This on selective reduction with NaBH4/CaCl2 (ref. 22) in a freshly distilled ethanol/THF (2:1) solution resulted in desired alcohol 69 without any degradation of the oxazolidine moiety (Scheme 17). The alcohol 69 was converted to corresponding iodo derivative 70 which upon NaH promoted cyclization in refluxing THF afforded bicyclic trifluoromethylated oxazolidine 71. The synthesized oxazolidine 71 was subjected to Strecker-type reaction using TMSCN to afford C-2 substituted azetidine as a diastereomeric mixture of 72 and 73. Sodium hydroxide promoted hydrolysis of 72/73 yielded the α-trifluoromethylated azetidine-2-carboxylic acids viz. 74 and 75 repectively (Scheme 17).
 | ||
| Scheme 17 | ||
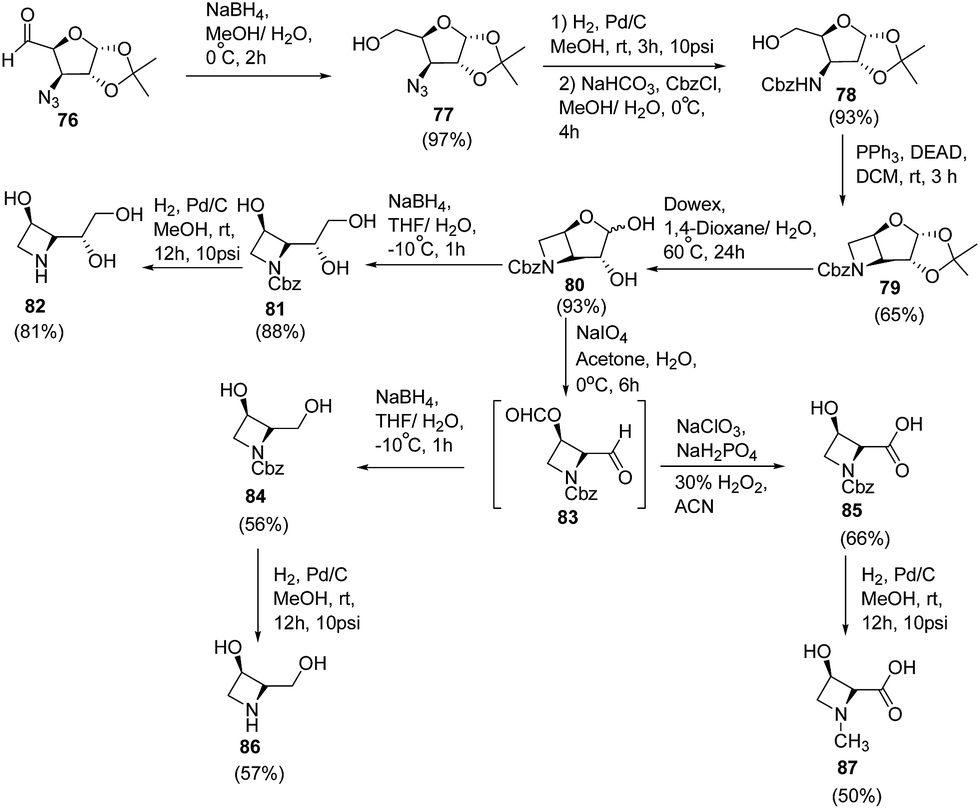
Polyhydroxylated nitrogen containing compounds popularly known as iminosugars are well known glycosidase inhibitors.23,24 Shinde et al.25 have developed a convenient methodology for the synthesis of azetidine iminosugars and 3-hydroxy-N-methylazetidine-2-carboxylic acid from D-glucose using intramolecular Mitsunobu reaction. The synthetic methodology involved the treatment of azido aldehyde 76 with sodium borohydride in aqueous methanol at 0 °C to afford the corresponding azido alcohol 77. The reduction of C3-azido functionality was achieved by using 10% Pd/C in methanol to afford the corresponding amine, which on protection with benzyl chloroformate (CbzCl) resulted in carbamate derivative 78. Further, reaction of 78 with triphenylphosphine and diethyl azodicarboxylate (DEAD) under Mitsunobu condition yielded cyclic azetidine intermediate 79. Deprotection of 1,2-acetonide group of 79 with the Dowex (H+) resin gave anomeric mixture of hemiacetals 80 which on treatment with sodium borohydride in THF/H2O generated N-Cbz protected triol 81. Hydrogenolysis of 81 using 10% Pd/C in methanol afforded (2S,3R)-2-((R)-1,2-dihydroxyethyl)azetidin-3-ol 82. Synthesized hemiacetal 80 also underwent oxidative cleavage with sodium metaperiodate, to yield the corresponding dialdehyde 83. This upon reduction with sodium borohydride in methanol afforded N-Cbz protected diol 84 which upon hydrogenolysis using 10% Pd/C in methanol yielded (2R,3R)-2-(hydroxymethyl)azetidin-3-ol 86. The hemiacetal 80 upon treatment with sodium metaperiodate, followed by the Pinnick oxidation26 using sodium chlorite and hydrogen peroxide afforded N-Cbz protected acid 85 which on subsequent hydrogenolysis yielded (2S,3R)-3-hydroxy-N-methylated azetidine-2-carboxylic acid 87 (Scheme 18).
 | ||
| Scheme 18 | ||
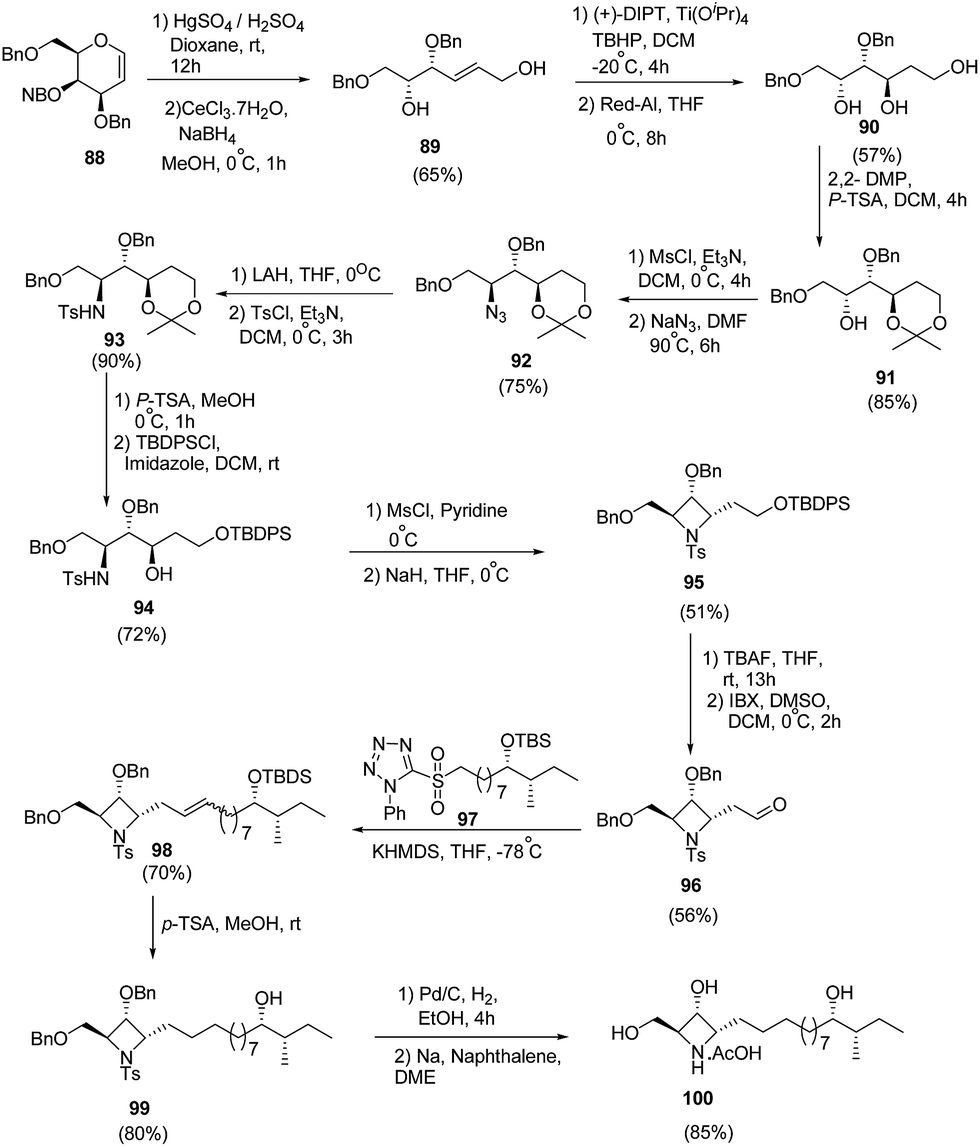
Reddy et al.27 have developed a synthetic methodology for the stereoselective synthesis of azetidine analogue of natural product penaresidin A. Key step involved the treatment of 3,4,6-tri-O-benzyl-D-galactal 88 with HgSO4 in the presence of H2SO4 afforded the hydroxy-trans-enal which upon chemoselective reduction with CeCl3·7H2O/NaBH4 yielded the allyl alcohol 89. Sharpless epoxidation of 89 with subsequent ring opening using Red-Al generated triol 90. The triol 90 was protected with 2,2-DMP to yield 1,3-acetonide 91. Mesylation of 91 with subsequent treatment with NaN3 led to azide 92. The reduction of azide 92 with LAH and subsequent tosylation yielded tosyl intermediate 93. Removal of acetonide group of 93 was carried out with p-TSA in methanol while subsequent protection of primary hydroxyl group with tert-butyldiphenylsilyl chloride afforded the TBDPS ether intermediate 94. This upon a sequence of mesylation and cyclization afforded 95. Desilylation of 95 with TBAF followed by oxidation of the hydroxyl group with IBX in DMSO yielded the corresponding aldehyde 96. Julia–Kocienski olefination of 96 with sulfone fragment 97 in presence of KHDMS afforded olefin 98. Further, desiylation of 98 with p-TSA led to the formation of 99. The utilization of 99 in the formation of penaresidin A 100 was achieved via its reduction with 10% Pd/C followed by treatment with Na/naphthalene (Scheme 19).
 | ||
| Scheme 19 | ||
Fleet and co-workers28 have developed a route for the synthesis of azetidine analogues of mannosidase inhibitor swainsonine and 1,4-dideoxy-1,4-imino-D-mannitol (DIM) through bicyclic azetidine. The methodology involved the protection of D-altrose 101 with acetone in the presence of sulfuric acid and anhydrous copper(II)sulfate to yield an inseparable mixture of pyranose 102 and furanose 103 diacetonides. Further, selective hydrolysis of the side chain acetonide of 103 was achieved with acetic acid resulting in monoacetonide 104 along with pure pyranose diacetonide 102. Further, treatment of 104 with TBDMS chloride in the presence of imidazole yielded the silyl ether 105 while the remaining two hydroxyl groups underwent esterification with triflic anhydride to afford the ditriflate 106. Reaction of 106 with benzylamine resulted in the formation of bicyclic azetidine intermediate 107. Further, hydrolysis of 107 with aqueous trifluoroacetic acid resulted in the 108 which upon reduction with sodium borohydride in water, yielded N-benzylazetidine 109. Hydrogenation of 109 with ammonium formate in the presence of palladium on carbon yielded the desired azetidine analogue of DIM (1,4-dideoxy-1,4-imino-D-mannitol) 110 as depicted in Scheme 20.
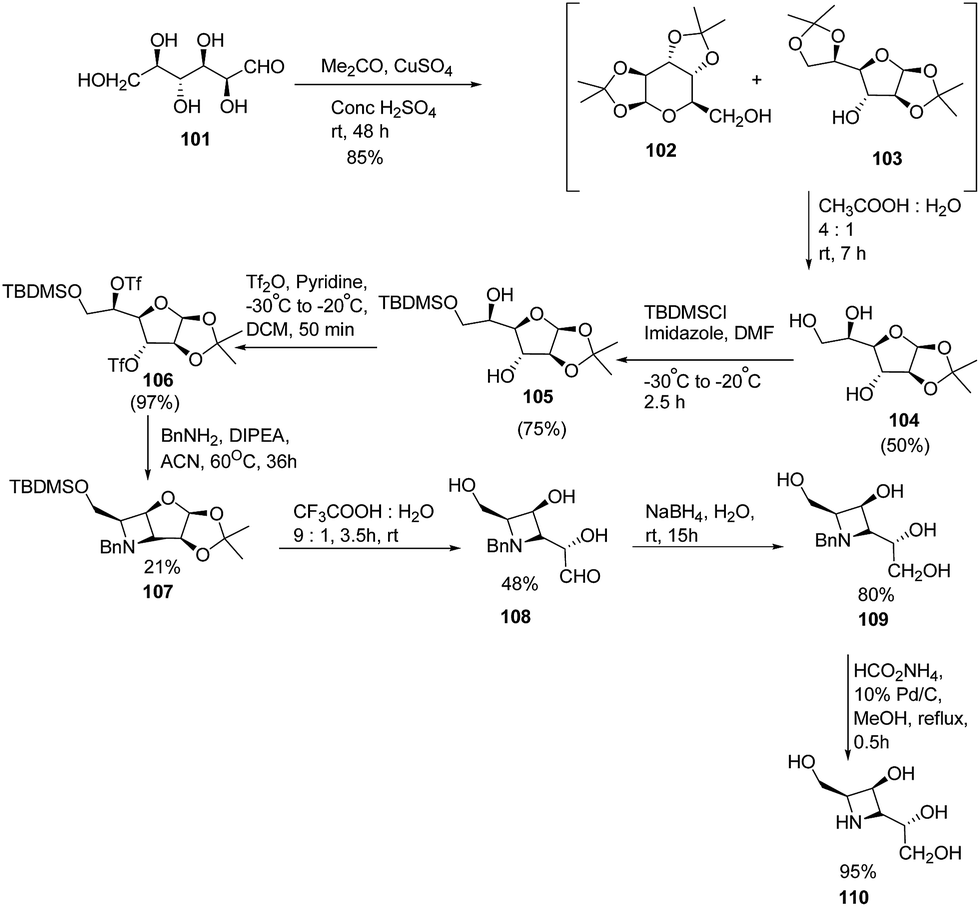 | ||
| Scheme 20 | ||
2.2 C-3 functionalized azetidines via β-lactam synthon approach
β-Lactam antibiotics is generally recognized as a cornerstone of human health care due to the unparalleled clinical efficacy and safety of this type of antibacterials.29 Besides their biological relevance, the stereocontrolled transformation of β-lactam ring provides efficient access to a variety of non-protein amino acids, functionalized piperazines, 1,4-diazepanes, quinolones, isoquinolones, isoquinolines, enantiopure succinimides, oligopeptides, peptidomimetics and biologically relevant heterocycles, such as indolizidines, paclitaxel, docetaxel, taxoids, cryptophycins, lankacidins. Such transformation is popularly known as β-lactam-synthon protocol.30,31 Reduction of β-lactams (azetidin-2-ones) with DIBAL-H and chloroalanes is considered as one of the most convenient approach for the chemoselective synthesis of azetidines.32 However, the presence of a Lewis acid and alanes could result in the ring opening of the strained four membered ring. The effect is generally more pronounced with electron rich phenyl group substituents on azetidine nucleus. Kumar et al.33 have recently developed a protocol for the diastereoselective synthesis of 2,3-disubstituted 1-arylazetidines via NaBH4 promoted reduction of C-3 functionalized azetidin-2-ones. The synthetic protocol involved the treatment of azetidin-2-ones 111 with sodium borohydride in isopropanol to afford the diastereoselective formation of trans-azetidines 112 (Scheme 21).33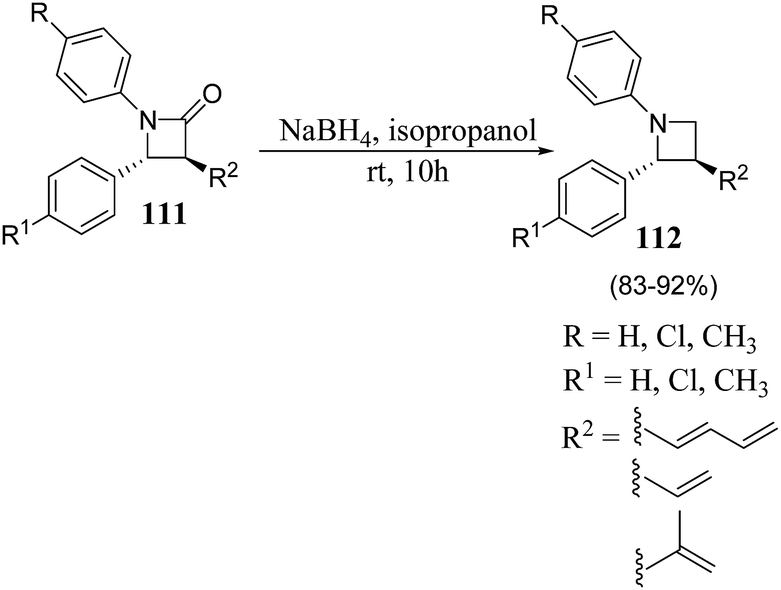 | ||
| Scheme 21 | ||
The developed protocol was further extended towards the synthesis of a series of C-3 azido/amino azetidines as depicted in Scheme 22.33
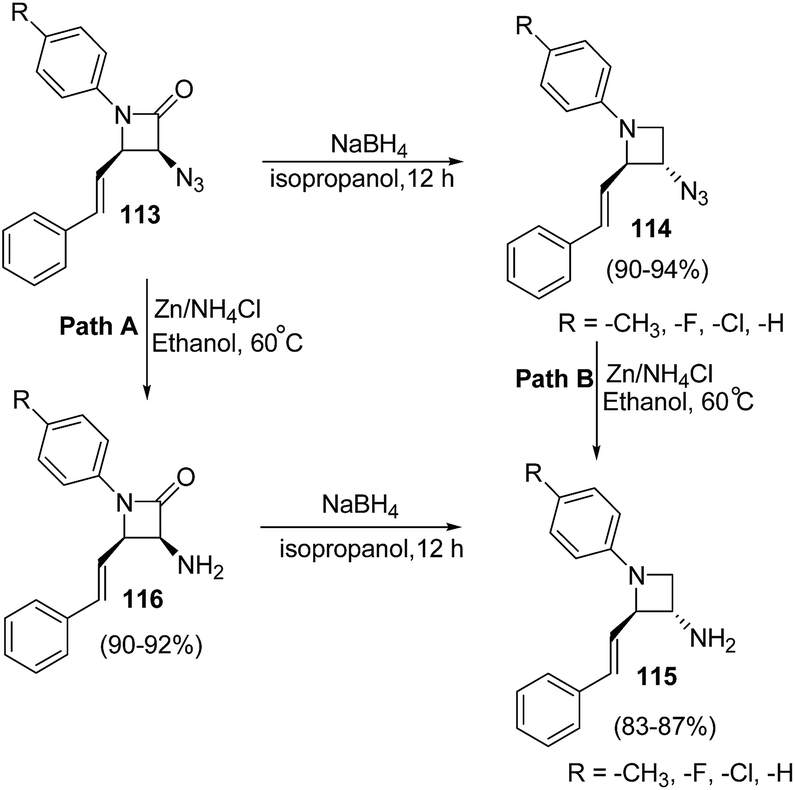 | ||
| Scheme 22 | ||
2.3 Nucleophilic ring opening of aziridines
The thermodynamically controlled rearrangement of aziridine to azetidine has very few precedents in the chemical literature and hence provides a window of opportunities for the selective synthesis of 3-substituted azetidines. De Kimpe et al.34 explored the intrinsic reactivity of 2-bromomethyl-2-methylaziridines 117 to ring enlargement in different solvents with a variety of nucleophiles such as thiocyanate, cyanide, phenoxide and acetate in order to assess the scope of the developed transformation (Scheme 23).34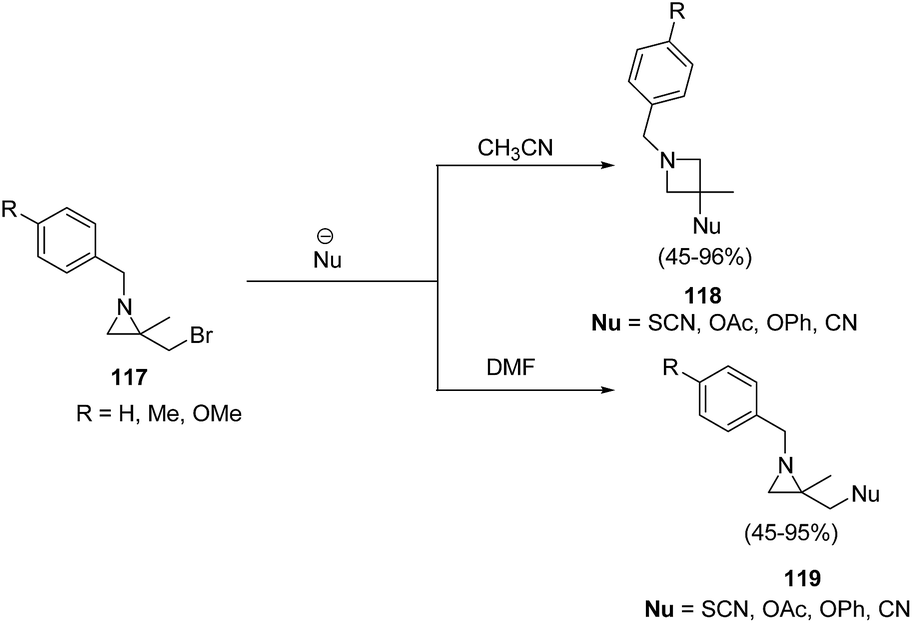 | ||
| Scheme 23 | ||
Ukaji and co-workers35 have recently developed the route for the synthesis of four-membered heterocyclic azetidines via [3 + 1] cycloaddition reaction of azomethine ylide with aromatic isocyanides. The key step of reaction involved the treatment of azridine 120 having dicarboxylate moiety with aromatic isocyanide 121 in the presence of Y(OTf)3 to afford azetidine derivatives 122/123 in good yields as depicted in Scheme 24. It has been found that the aliphatic isocyanides could not initiate the reaction due to its low reactivity. Aromatic isocyanides, irrespective of electron-withdrawing or donating substituents led to the formation of desired four membered heterocycle in reasonably good yields.
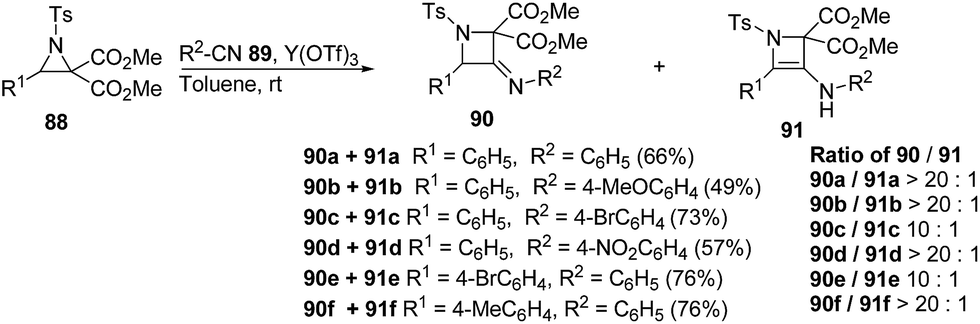 | ||
| Scheme 24 | ||
Mechanistically, it has been found that Y(OTf)3, coordinated with the oxygen atom of dicarboxylate moiety 120, to generate complexed intermediate I which undergoes C–C bond cleavage to afford the corresponding azomethine ylide II. The Ugi-type nucleophilic attack of the isocyanide 121 generated the nitrilium intermediate III which subsequently underwent internal trapping by α-anion of the azomethine ylide resulting in the formation of azetidine derivatives 122 as shown in Scheme 25.
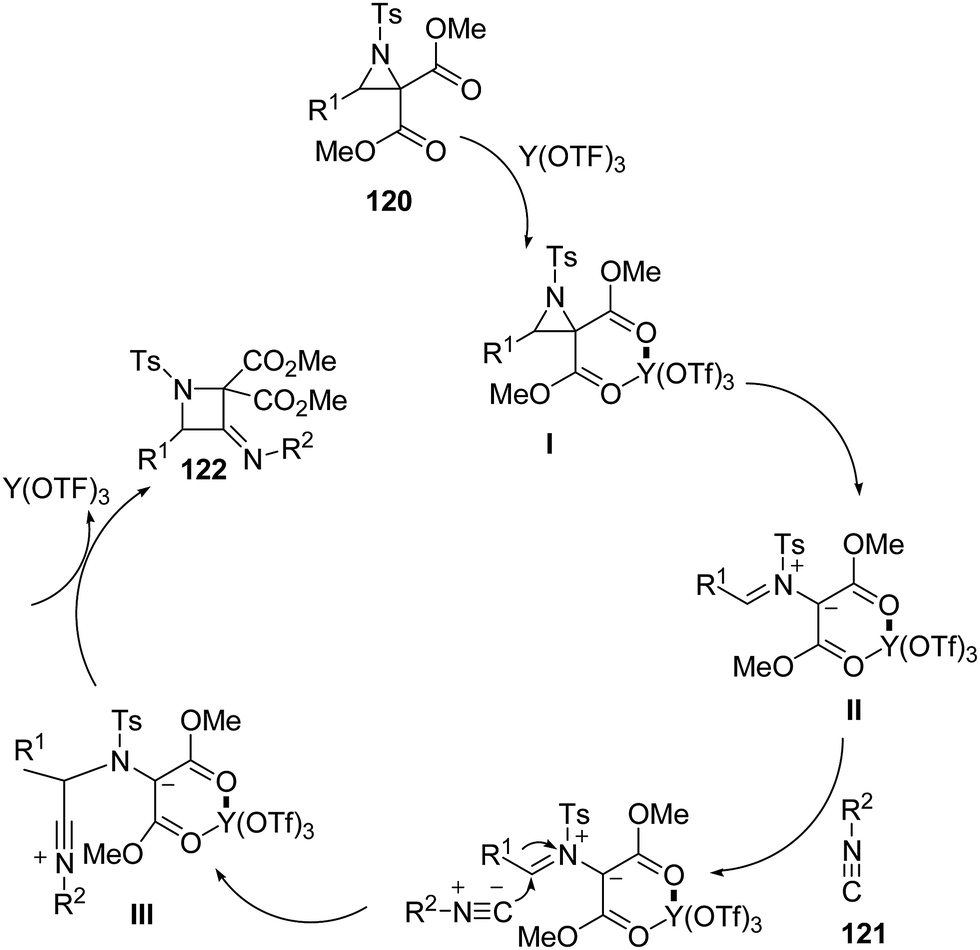 | ||
| Scheme 25 | ||
2.4 Metal-catalyzed synthesis of azetidines
Zhang et al.36 have recently reported the synthesis of chiral azetidine using Pd-catalyzed asymmetric allylation of azalactones 124 with 4-styryl-1,3-dioxolan-2-one 125 via 127 as intermediate. Different ligands, additives and solvents were attempted to increase the yield and enantioselectivity of 127 but the best results were obtained when reaction was carried out in presence of ligand 126 and NaBArF as additive. The acetylation of 127 with Ac2O afforded 128 which underwent Pd-catalyzed allylic amination resulting in the enantioselective formation of azetidine 129 as depicted in Scheme 26.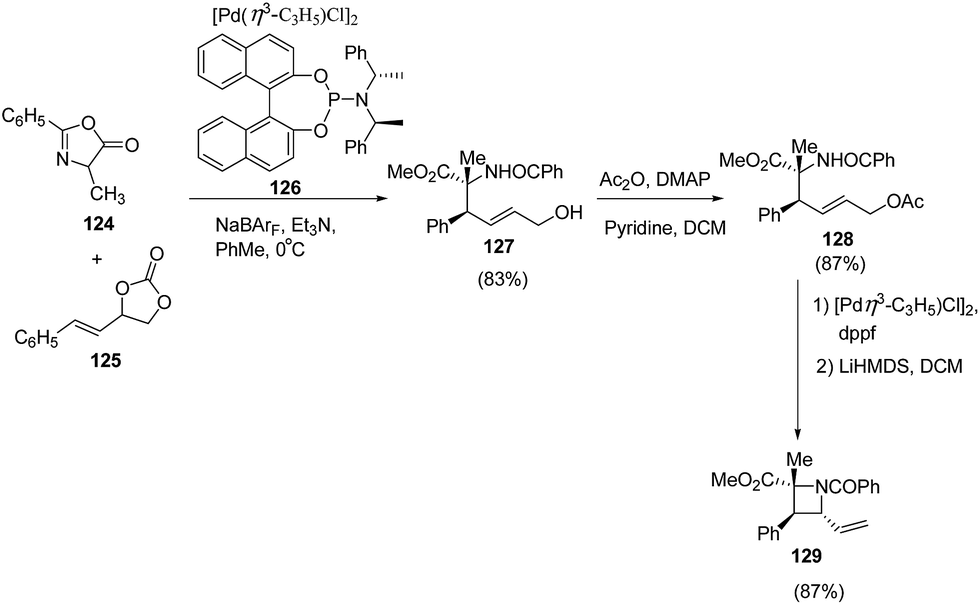 | ||
| Scheme 26 | ||
Schafer et al.37 have utilized tantalum catalyzed hydroaminoalkylation reaction for the synthesis of azetidines. The protocol involved hydroaminoalkylation reaction of between 130 with 131 in presence of tantalum catalyst 132 to form an intermediate intermediate 133 which underwent cyclization in presence of TsF and DBU at 130 °C to afford the corresponding azetidine 134 as shown in Scheme 27.
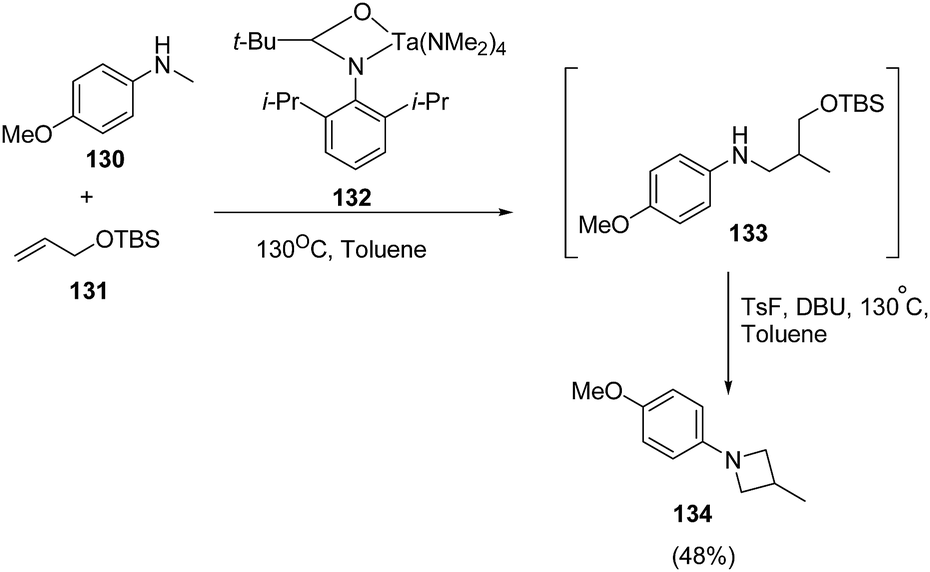 | ||
| Scheme 27 | ||
Ferrocene has drawn the attention of chemists due to its various applications in organic synthesis38,39 and asymmetric catalysis.40 Mandal and co-workers41 have developed a protocol for the diastereoselective synthesis of ferrocenyl azetidinimines via copper catalyzed alkyne–azide cascade reaction as shown in Scheme 28. The synthetic approach involved the cascade reaction of ferrocenylimine 135, p-toluenesulfonyl azide 136 and substituted acetylene 137 yielding the ferrocenyl azetidinimines 138. Best result in terms of yields was obtained using 10% CuI (catalyst) or with 1.5 equiv. of Et3N at ambient temperatures.
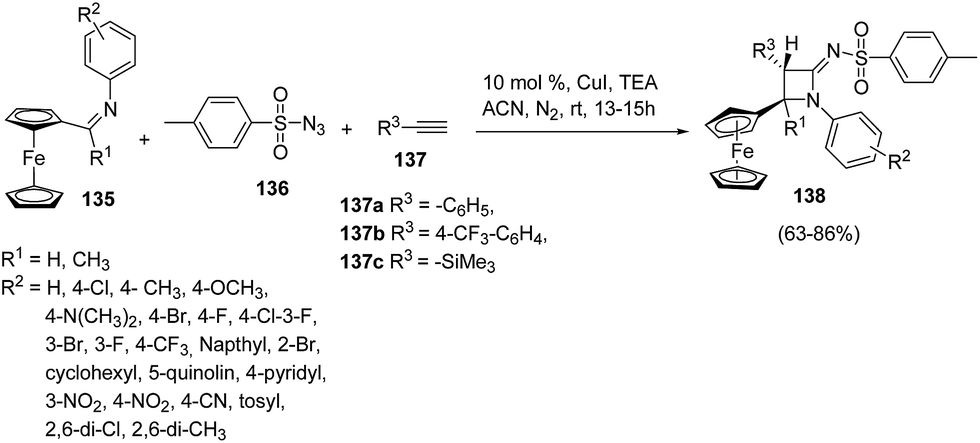 | ||
| Scheme 28 | ||
Mechanistically the alkyne 137 reacted with sulfonyl azide 136 to afford intermediate 139 which could follow two different pathways leading to azetidinimine 138. Nitrogen extrusion of intermediate 139 generated ring opened intermediate 140 which upon protonation afforded ketenimine intermediate 142 (Path A). Alternatively intermediate 139 could result in the intermediate 141 which on protonation and nitrogen extrusion led to the formation of ketenimine 142 (Path B). [2 + 2]-Cyclo-addition of 142 with ferrocenylimine 135 led to the diastereoselective synthesis of ferrocenyl substituted azetidinimine 106 (Scheme 29).41
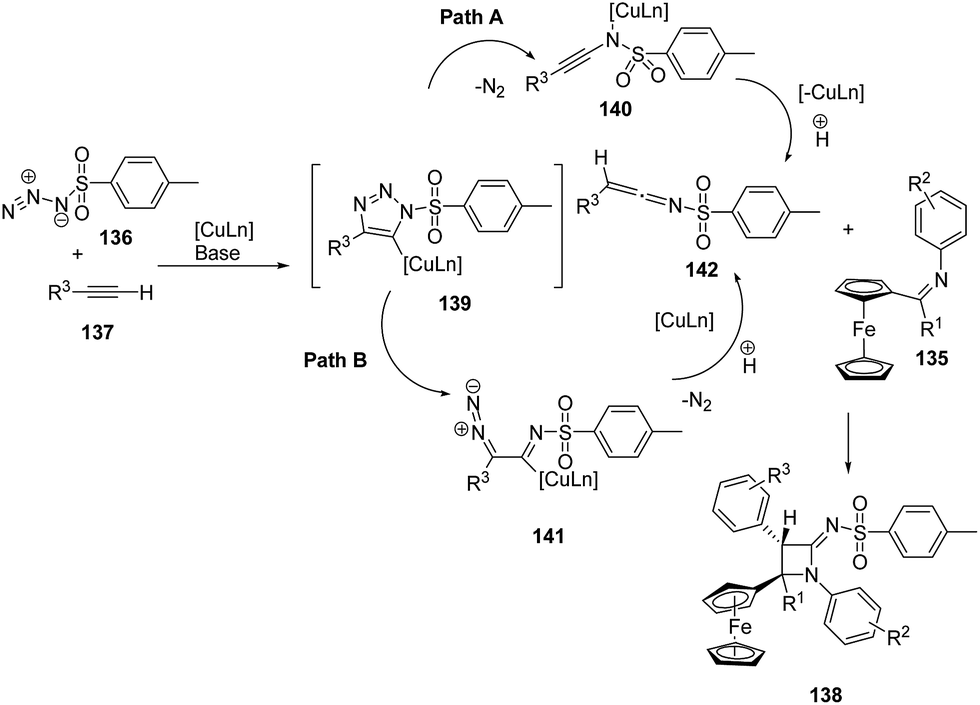 | ||
| Scheme 29 | ||
Hu et al.42 has developed an efficient method for the synthesis of disulfide-linked N-sulfonylazetidin-2-imines. The synthetic methodology involved the multicomponent reaction between different sulfonyl azides 143, phenylacetylenes 144 and benzenethiol Schiff bases 145 in presence of CuI and Et3N at 0 °C as depicted in Scheme 30. The reaction was optimised in different bases and solvents but best results were obtained using Et3N as base and THF as solvent. The presence of electron donating substituents, both on sulfonyl azides 143 and Schiff bases 145 led to the formation of corresponding azetidines 146 in good yields.
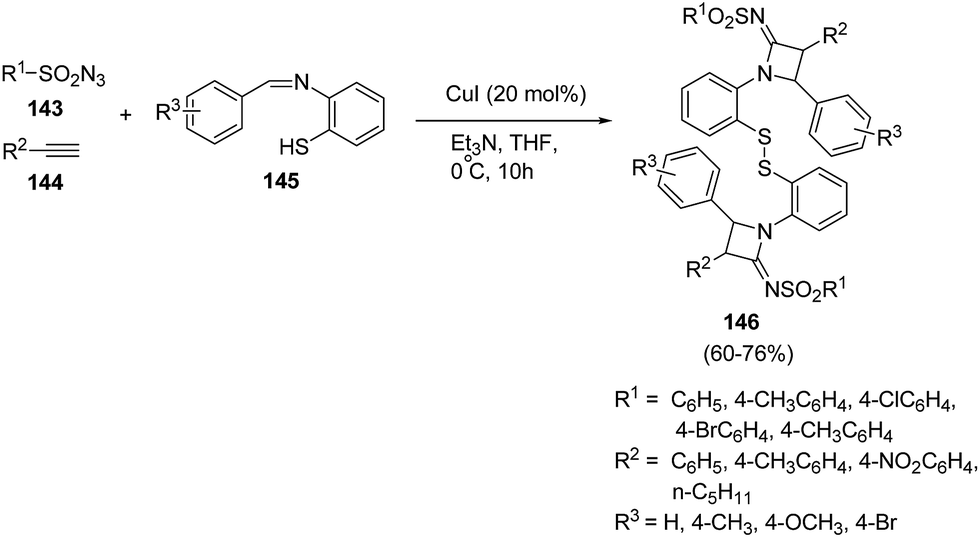 | ||
| Scheme 30 | ||
Mechanistically, sulfonyl azide 143 reacted with the alkyne 144 in the presence of CuI to afforded intermediate IV which on protonation gave the ketenimine V. Meanwhile, in presence of trace amounts of I2, the intermolecular dimerization of benzenethiol Schiff base 145 was achieved to generate intermediate VI. Further, [2 + 2] cycloaddition reaction of V with iminic component VI afforded the desired azetidinimine 146 (Scheme 31).42
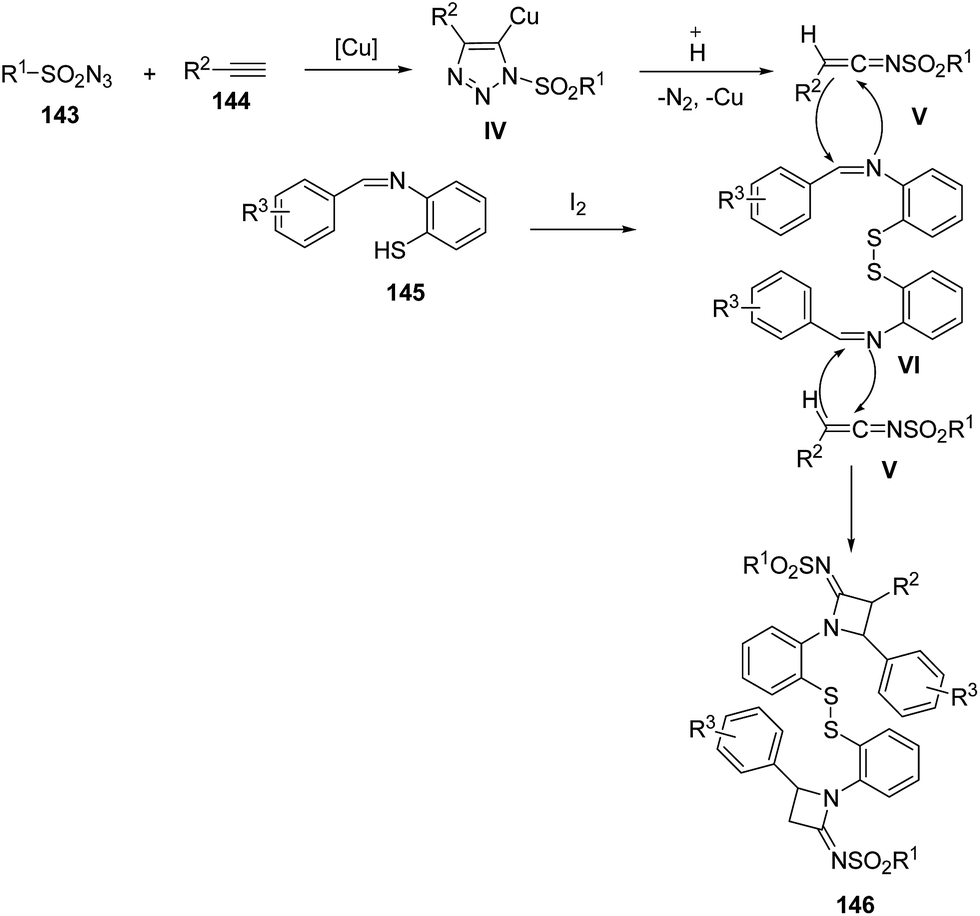 | ||
| Scheme 31 | ||
2.5 Synthesis of azetidines via organocatalysis
Yadav and co-workers43 have utilized [2 + 2] annulation of aldehydes with aldimines for stereoselective synthesis of azetidin-2-ols. The key step in the synthesis involved the treatment of aldehyde 147 with pyrrolidine based catalysts 148 to afford chiral enamine intermediate 149 which upon [2 + 2] annulation reaction with aldimines 150 in presence of K2CO3 resulted in the diastereoselective synthesis of corresponding azetidin-2-ols 151 as depicted in Scheme 32. The reaction was optimized with substituted pyrrolidine based catalysts and bases viz. DBU, DABCO, Et3N and NaHCO3. Best results in terms of yields were obtained using 116 as the catalyst and K2CO3 as base.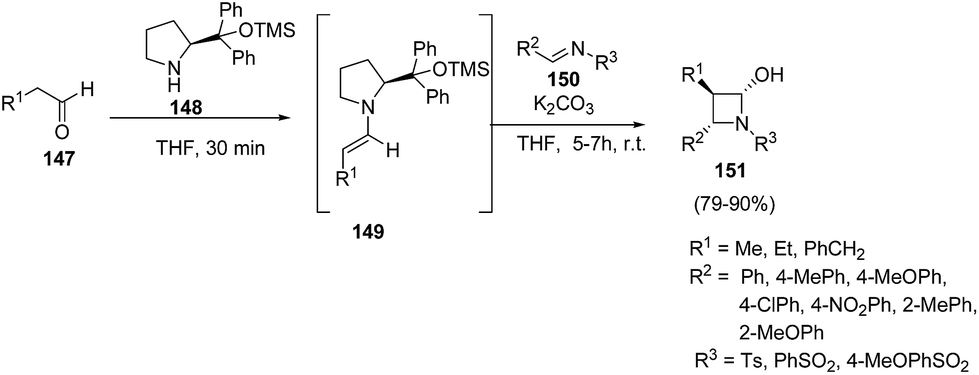 | ||
| Scheme 32 | ||
Mechanistically it was found that the catalyst diphenylprolinol trimethylsilyl ether 148 activated the aldehyde 147, generating the corresponding enamine 149. Stereoselectively reaction of 149 with aldimine 150 yielded the intermediate 152. Hydration of 152 afforded another intermediate 153, which underwent intramolecular cyclization to give the desired azetidin-2-ols 151 depicted in Scheme 33.43
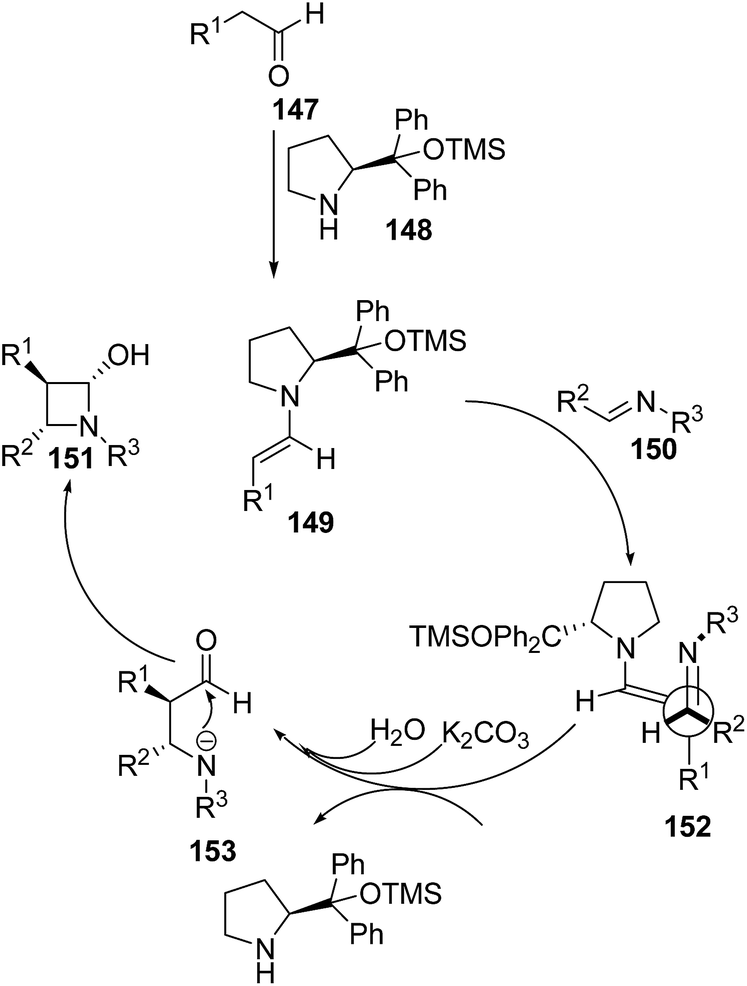 | ||
| Scheme 33 | ||
Sasai and co-workers44 have developed an enantioselective synthesis of azetidines 156 using aza-Morita–Baylis–Hillman (MBH) reaction of ketimines 154 and allenoate 155 in the presence of organocatalyst. The reaction was optimized using range of chiral amines as organocatalysts with the best results in terms of yields were obtained using β-isocupreidine (β-ICD) as catalyst in the presence of MS 3 Å. Azetidine 156 was reduced using DIBAL-H affording allylalcohol 157 while Suzuki Miyaura cross-coupling of 156 with phenylboronic acid afforded the corresponding biphenyl azetidine 158 as depicted in Scheme 34.
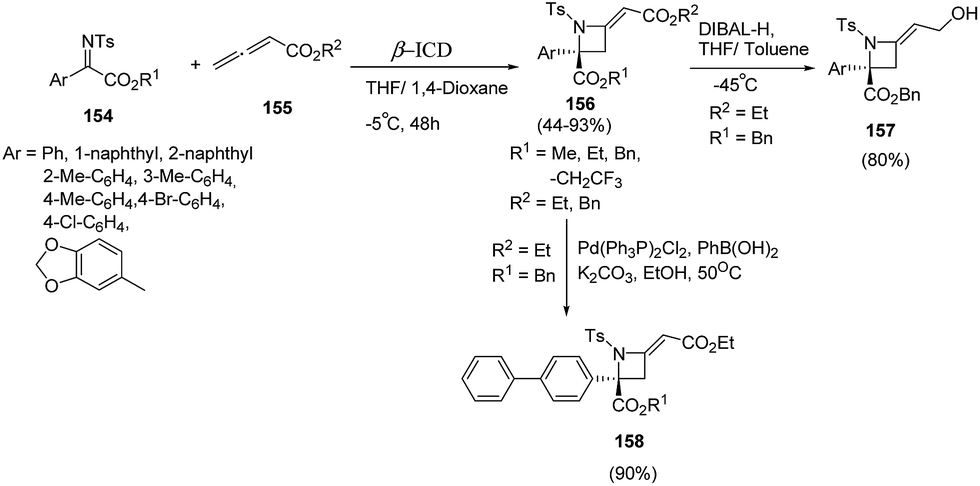 | ||
| Scheme 34 | ||
Synthetic approaches towards optically active polysubstituted azetidines demands greater exploration as compared to aziridines, pyrrolidines and piperidines. Kaufman et al.45 have developed on organocatalytic approach for the synthesis of optically pure 1,2,3-trisubstituted azetidines. The synthetic protocol involved sequence of synthetic steps as shown in Scheme 35, the key step being L-proline catalysed condensation reaction of substituted aldehydes 159 with substituted anilines 160 to yield Schiff bases 161. The reaction of 161 with aldehyde 162 in presence of NMP at −20 °C afforded intermediate 163 which upon reduction with NaBH4 in MeOH led to the corresponding γ-aminoalcohols 164. Microwave promoted intramolecular cyclization of 164 in presence of tosyl chloride led to the enantioselective synthesis of 1,2,3-trisubstituted azetidines 165. The steric as well as electronic nature of substituents both on aldehyde and amine has a profound role on the outcome of the reaction. The presence of substituent ortho to the formyl moiety resulted in intermediate imine 161 but failed to form the desired azetidine 165. 2-Phenylaniline failed to react while the reaction with α-naphthylamine and 2-methoxyaniline furnished the corresponding Schiff bases, but failed to form the desired product (Scheme 35).
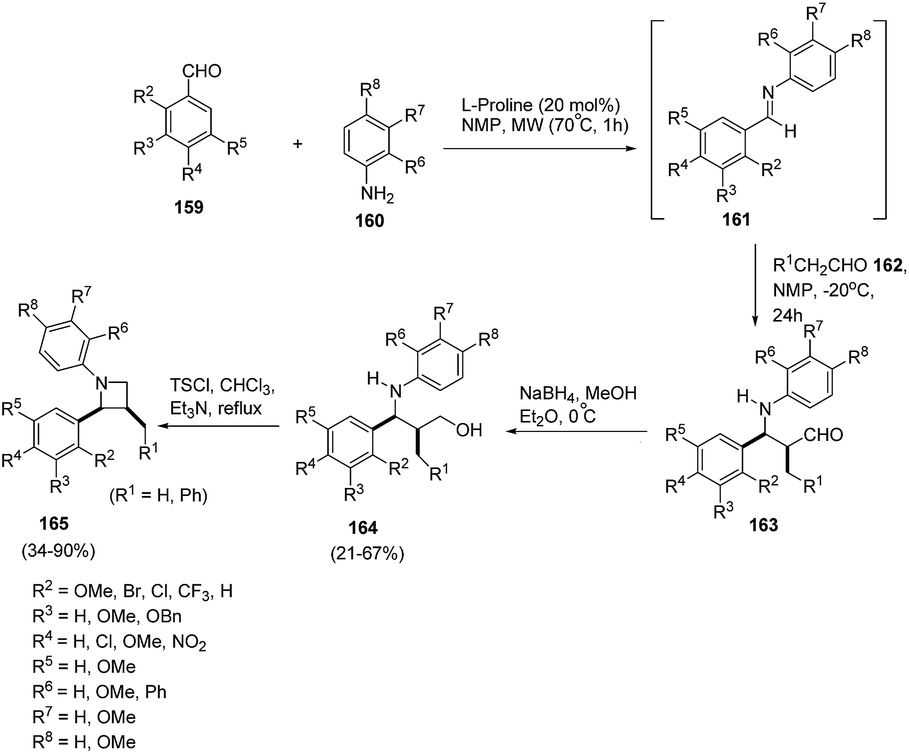 | ||
| Scheme 35 | ||
2.6 Miscellaneous syntheses
Blanc and co-workers46 have reported the synthesis of α-carbonylated N-sulfonylazetidines via ring contraction of α-bromo N-sulfonylpyrrolidinones (Scheme 36). The synthetic methodology involved the ring contraction of α-bromo N-sulfonylpyrrolidinone 166 with K2CO3 in presence of acetonitrile:methanol (9:1) to yield the α-carbonylated N-sulfonylazetidine 167 exclusively. A variety of bases and solvents were tried and the best results in terms of yields were obtained using K2CO3 as base. Interestingly, no conversion was observed in the absence of base (Scheme 37).
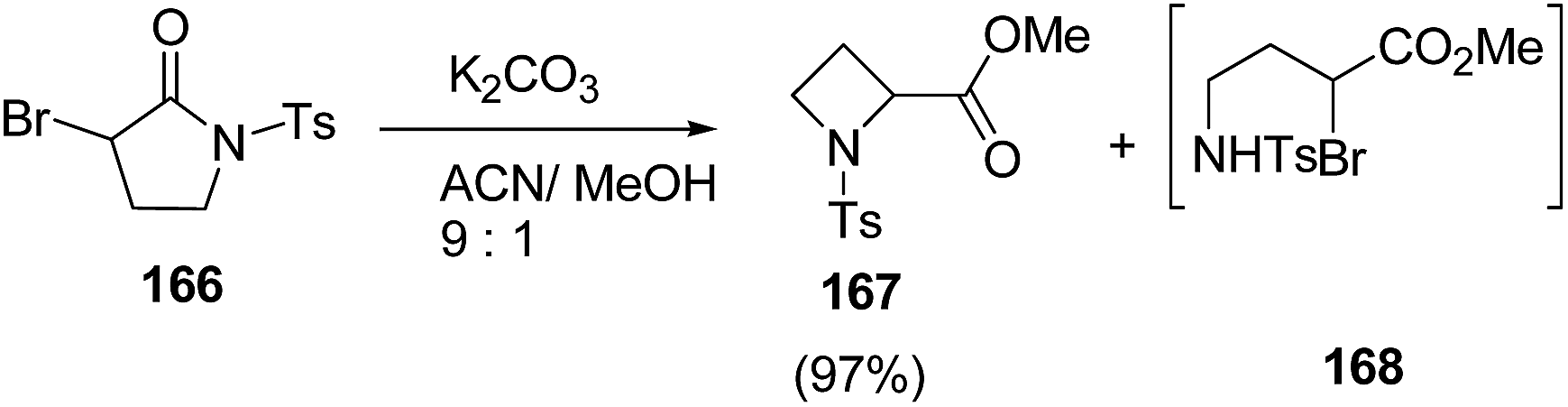 | ||
| Scheme 36 | ||
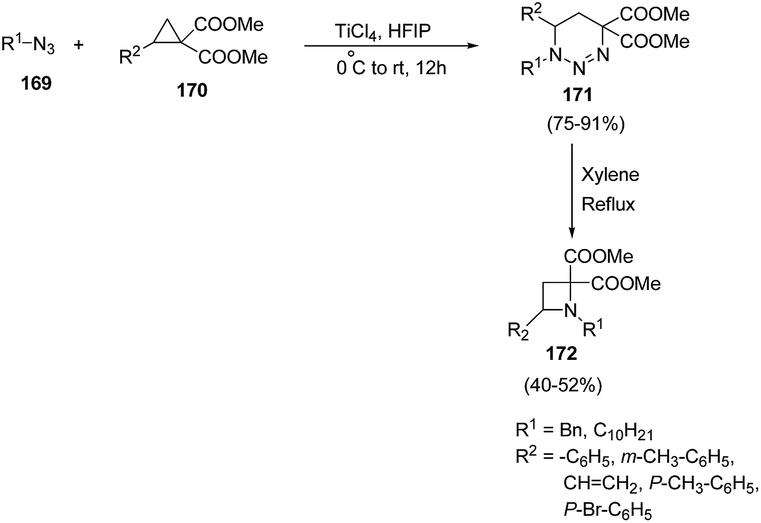 | ||
| Scheme 37 | ||
Xu and co-workers47 have described the synthesis of C-4 substituted azetidines via TiCl4 promoted [3 + 3] cycloaddition reaction. The synthetic methodology involved the treatment of cyclopropane 1,1-diesters 170 with alkyl azide 169 in presence of catalytic amount of TiCl4 in hexafluoro-2-propanol (HFIP), resulting in the synthesis of substituted triazines 171. The presence of electron withdrawing as well as electron donating substituent in cyclopropane 1,1-diesters 170 gave corresponding triazines 171 in good yields. The synthesized triazine 171 was subsequently refluxed in xylene to afford the corresponding C-4 substituted azetidines 172 in good yields (Scheme 37).
Mechanistically, it was found that TiCl4, promoted the ring-opening of cyclopropane 170 to afford 1,3-zwitterionic intermediate VII which underwent nucleophilic addition of an azide 169 to yield another zwitterionic intermediate VIII with subsequent intramolecular ring closure to afford the triazine 171. Refluxing of 171 in xylene generated intermediate IX which upon intramolecular cyclization and nitrogen extrusion yielded desired azetidine 172 as depicted in Scheme 38.47
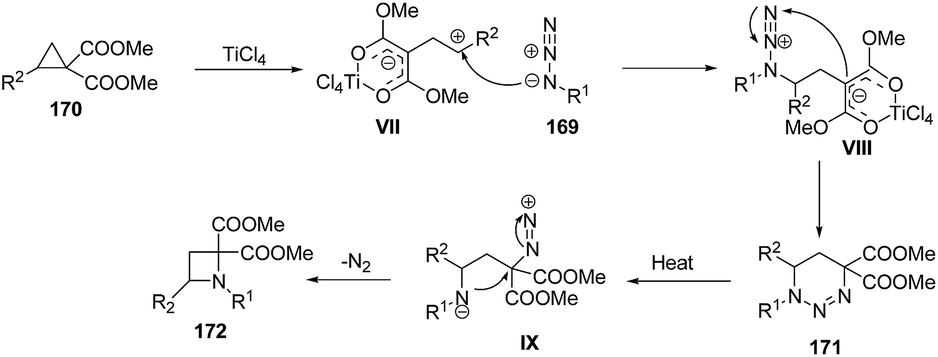 | ||
| Scheme 38 | ||
Santini and co-workers48 reduced the 3-oxo-azetidine 176 into corresponding alcohol in the presence of Zn/Cu couple49 which was subsequently utilized in spirocyclization. The synthetic methodology involved the reduction of 3-oxo-azetidine 173 with propargyl bromide in the presence of Zn/Cu couple resulting into azetidine 174. The treatment of azetidine 174 with catalytic amount of Au(PPh3)(NTf2) in presence of 3,5-dichloropyridine N-oxide and methanesulfonic acid resulted in the synthesis of spiro-3-furanone 175 which underwent LDA-promoted enolization to afford enol triflate 176 (Scheme 39).
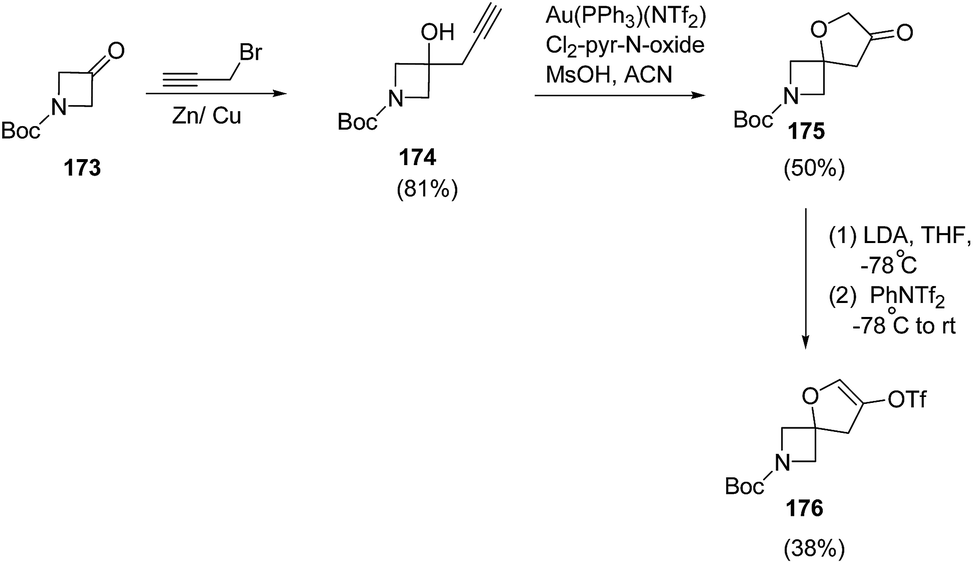 | ||
| Scheme 39 | ||
Lewis base catalyzed50 nucleophilic activation of allenoates has experienced a great expansion over the past few decades.51 Ma et al.52 introduced Lewis bases promoted [2 + 2] annulation reaction of allenoates and cyclic ketimines for the synthesis of fused azetidines in good yields. The synthetic protocol involved DABCO catalysed [2 + 2] annulation reaction of ethyl 2,3-butadienoate 177 with cyclic ketimine 178 in presence of 1,4-dioxane to afford the azetidine derivatives 179 in good yields (Scheme 40). It has been found that ketimine 178 having electron-withdrawing groups at 5th and/or 6th-position of the aromatic ring afforded the cycloadducts 179 in high yields whereas ketimine bearing electron-donating groups at 6th-position of the aromatic ring gave products 179 in moderate to good yields. A decrease in yield was observed with the introduction of a free NH group. The conversion of trifluoromethyl to difluoromethyl or phenyl group also resulted in the decrease in yields, confirming the role of CF3-group in the [2 + 2] annulation reaction (Scheme 40).
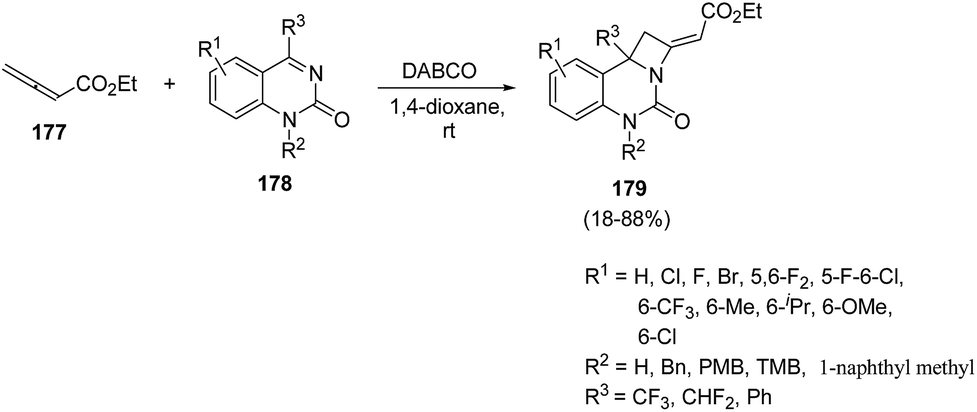 | ||
| Scheme 40 | ||
The plausible mechanism involved an intial addition of DABCO to the electrophilic β carbon of ethyl 2,3-butadienoate 177 to form the zwitterionic intermediate X which reacts with ketimine 178 through γ addition to give the intermediate XI. This upon intramolecular nucleophilic addition led to the formation of intermediate XII, which via subsequent catalyst elimination yielded fused azetidine 179 (Scheme 41).52
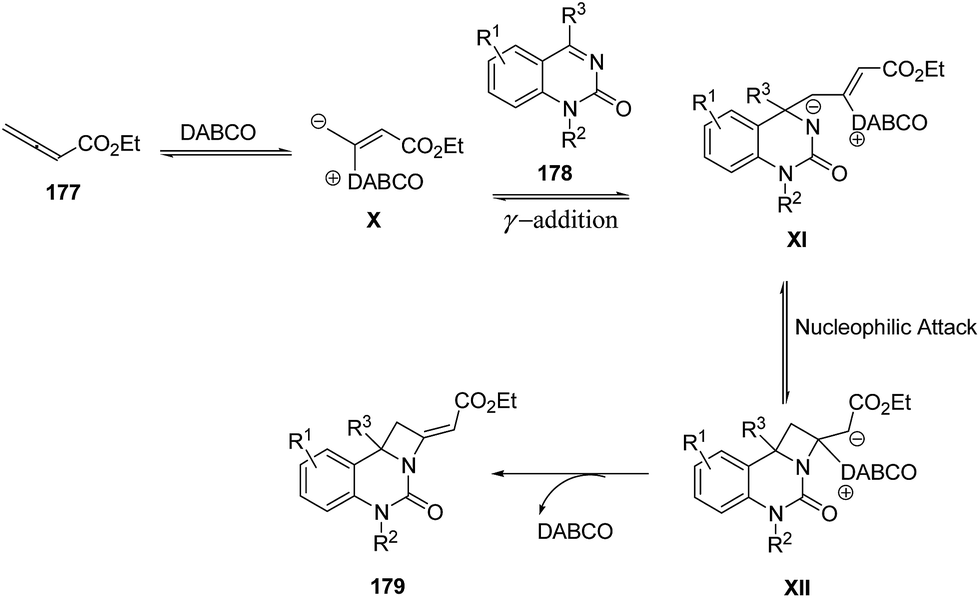 | ||
| Scheme 41 | ||
3. Azetidines as versatile synthons
Azetidines played an important role in medicinal chemistry programs.53 In recent years, ring transformations of azetidines has been exploited widely through nucleophilic ring-opening, cycloaddition, and rearrangement reactions which makes them highly valuable in organic synthesis.54Piperidin-4-ones represent an important class of bioactive heterocycles due to their observed biological and pharmaceutical properties along with their use as intermediates in the synthesis of a variety of biologically active compounds such as functionalized piperidines through the reduction of the carbonyl moiety. De Kimpe et al.55 have recently explored the synthesis of functionalized piperidin-4-ones via ring expansion-oxidation of corresponding azetidines. The key step in the synthesis involved the treatment of cis-2-(2-bromo-1,1-dimethylethyl)azetidines 180a–e with silver salt in DMSO at 100 °C for 18 h to afford the corresponding 5,5-dimethylpiperidin-4-one 181a–e. It has been found that same reaction when carried out with K2CO3 in DMSO did not result in the desired conversion and complex reaction mixtures were obtained. cis-2-(2-Mesyloxyethyl)azetidines 180f–h, however underwent similar ring expansion-oxidation in presence of K2CO3 and DMSO resulting in the synthesis of desired piperidin-4-ones 181f–h in good yields (Scheme 42).
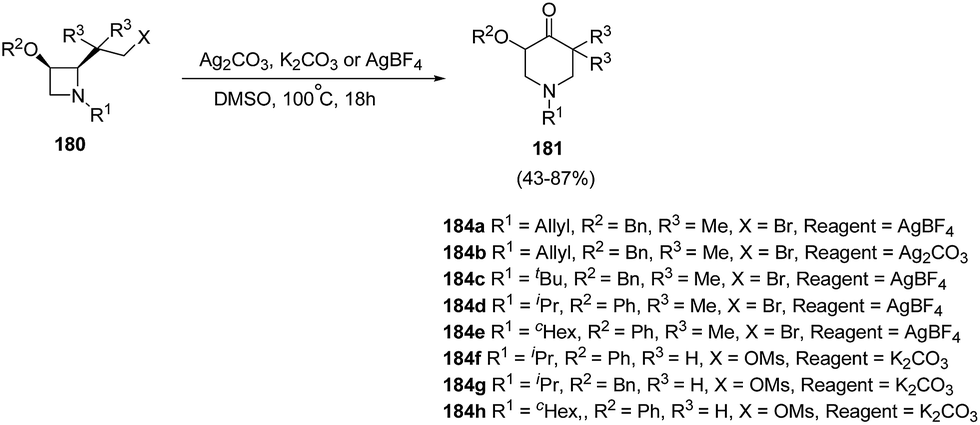 | ||
| Scheme 42 | ||
Mechanistically, it was suggested that azetidine 180 was transformed into reactive bicyclic azetidinium salts XIV through intramolecular nucleophilic displacement with subsequently conversion into piperidine intermediate XV via ring enlargement with dimethylsulfoxide. The abstraction of acidic proton at the oxygenated carbon atom resulted in the liberation of dimethylsulfide via α-elimination affording the formation of corresponding piperidin-4-ones 181 (Scheme 43).55
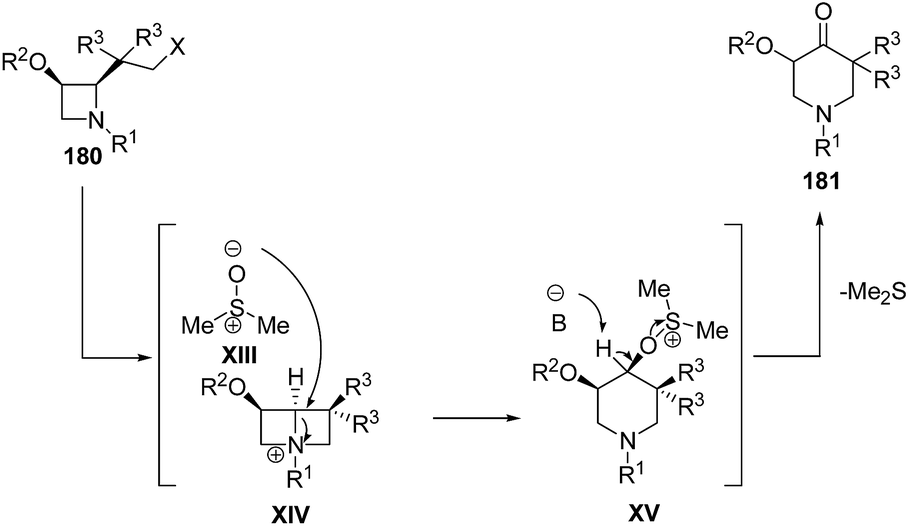 | ||
| Scheme 43 | ||
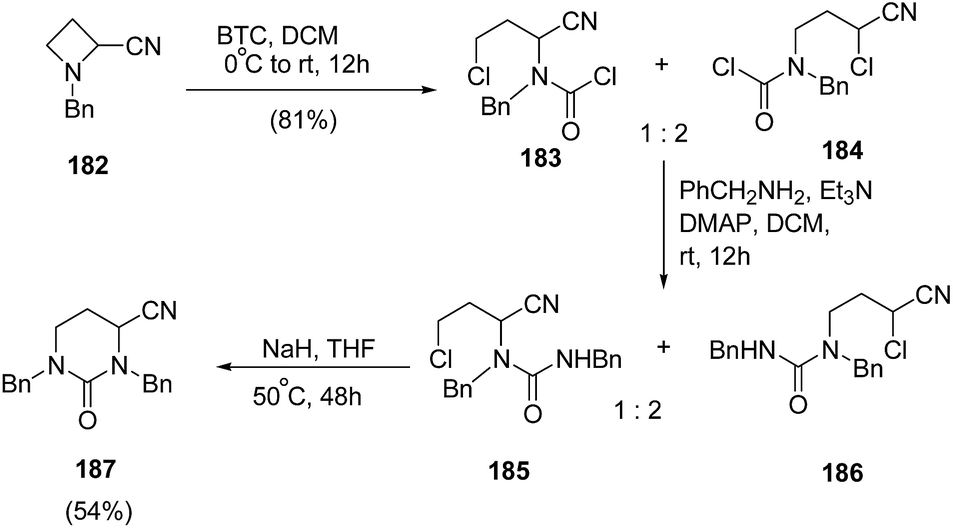
Couty and co-workers56 have reported BTC (bis-trichloromethylcarbonate, triphosgene) promoted selective bond cleavage of azetidine resulting in the formation of five or six membered urea. The synthetic protocol involved regioselective C-2 cleavage of azetidine 182 with BTC to afford 183 and 184 in ratio of 1:2. The reaction of 183 and 184 with benzylamine afforded the mixture of regioisomeric ureas 185 and 186, which underwent base promoted intramolecular ring closure to yield the pyrimidin-2-ones 187 (Scheme 44).
 | ||
| Scheme 44 | ||
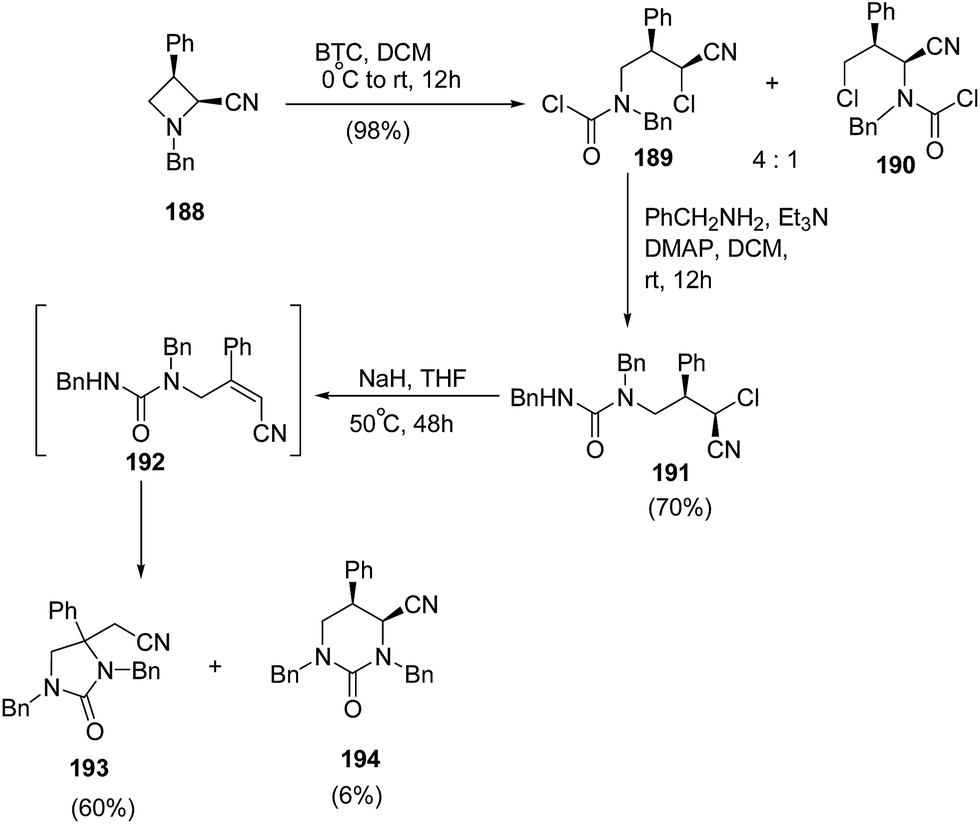
Further extension of the above protocol to azetidine 188 led to the formation of imidazolidin-2-one 193 as the major product along with formation of tetrahydropyrimidin-2-one 194, as the minor product as illustrated in Scheme 45.56
 | ||
| Scheme 45 | ||
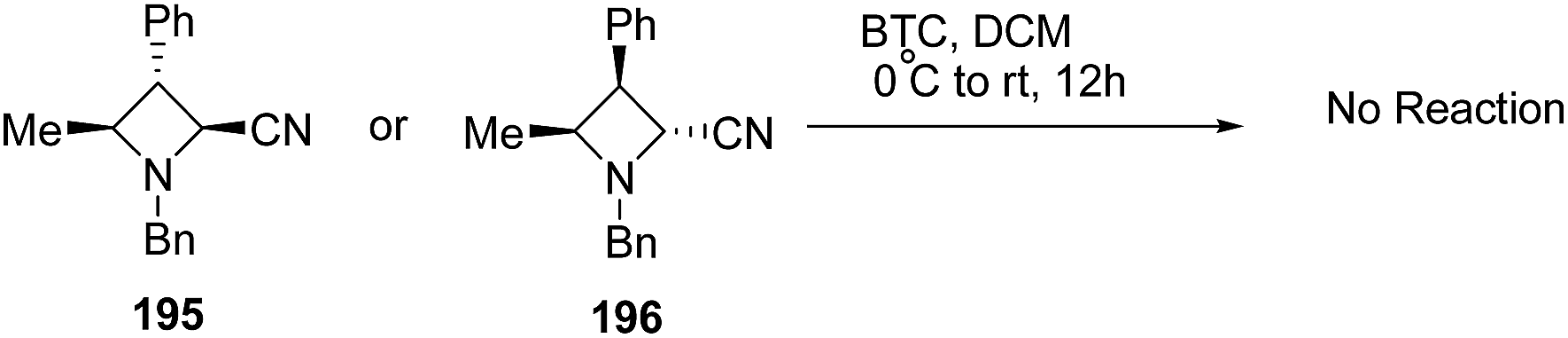
The above protocol, when attempted with trisubstituted azetidines viz. 195 or 196 did not result in any conversion due to its high sensitivity towards steric crowding around the nitrogen atom (Scheme 46).56
 | ||
| Scheme 46 | ||
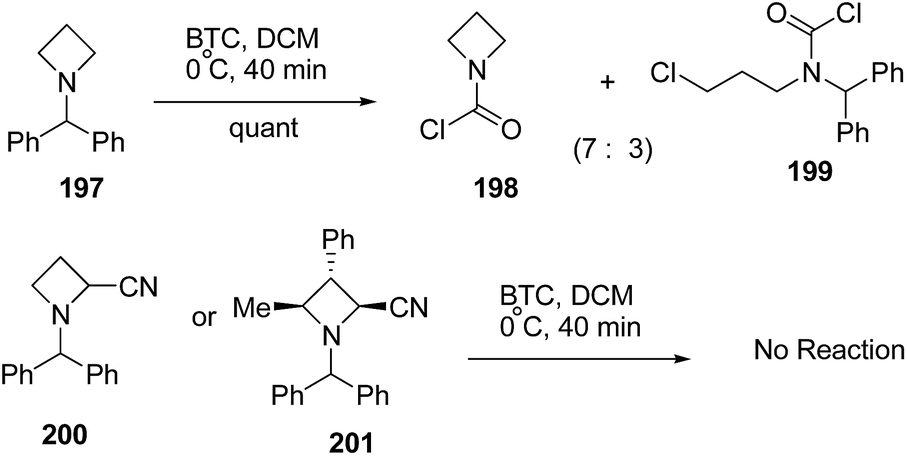
The treatment of N-benzhydryl azetidine 197 with BTC however, resulted in the isolation of a mixture of carbamoyl chloride 198 and ring opened product 199 in ratio 7:3. Further, N-benzhydryl 2-cyano azetidine 200 or trisubstituted N-benzhydrylazetidine 201 were found to be completely inert due to steric crowding around the nitrogen atom (Scheme 47).56
 | ||
| Scheme 47 | ||
Finally, the reaction of BTC with encumbered trisubstituted azetidines 202 and 207 having N-methyl substituent was carried out. Compound 202 having cis-configuration reacted rapidly to give good yield of ring-opened regioisomer 203 while the reaction with 207 having trans-configuration gave mixture of compounds, 208–210. Further, treatment of 204 with NaH in THF resulted in the synthesis of tetrahydropyrimidinones, 205 and 206 as a 6:4 epimeric mixture (Scheme 48).56
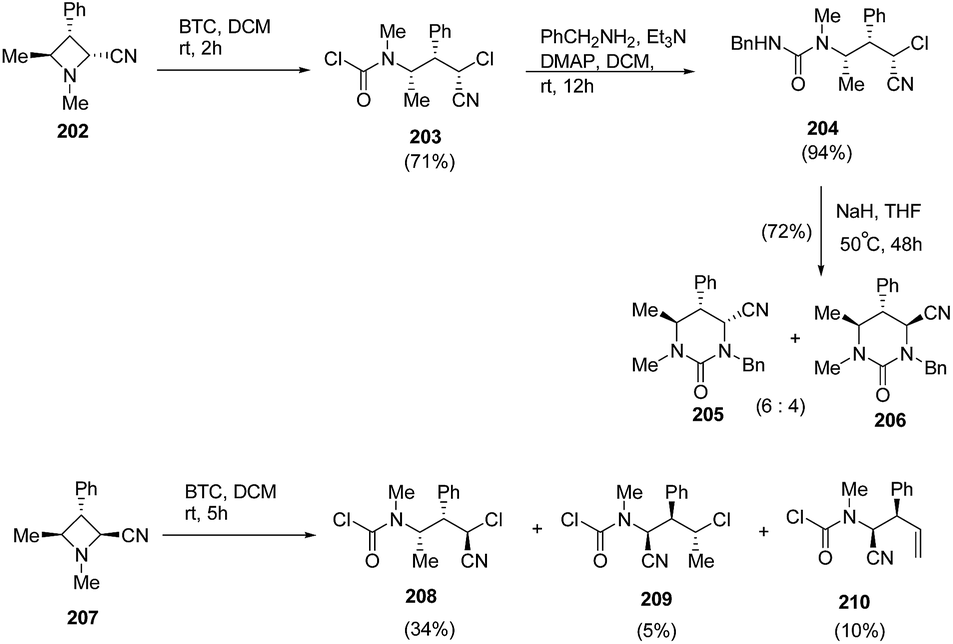 | ||
| Scheme 48 | ||
Compain and co-workers57 have developed the route for the synthesis of spirocyclopropyl γ-lactams via tandem intramolecular azetidine ring-opening/closing cascade reaction. The key step involved the SN2-type ring-opening of TMSOTf-activated azetidine ring by silyl ketene acetals. Thus, the treatment of azetidine 211 with 2 equiv. of TMSOTf in the presence of 2.5 equiv. of TEA in dichloromethane resulted in the formation of spirocyclopropyl-γ-lactam 212 in highly regio and stereoselective manner. Further reduction of the ester group with superhydride resulted into primary alcohol 213 while pyrrolidine analogue 214 was achieved by using LAH. The pyrrolidine analogue, 213 was efficiently converted to corresponding bromide 215 by using PPh3/Br2 and pyridine (Scheme 49).
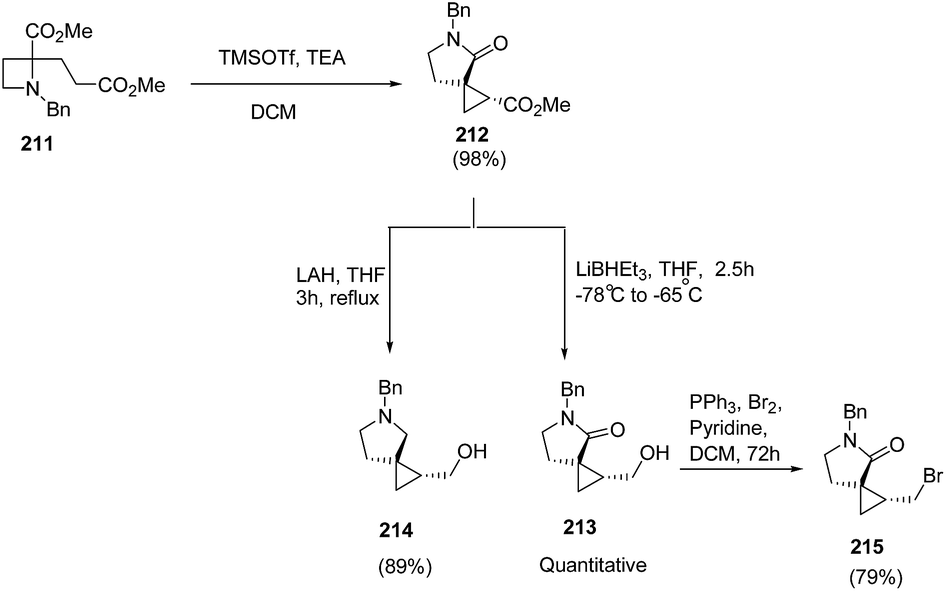 | ||
| Scheme 49 | ||
Mechanistically SN2-type ring-opening1f,58,59 of azetidine 211 with silyl ketene acetal, generated by treatment with TMSOTf and TEA,60 resulted in amino ester A which underwent intramolecular cyclization to afford the five membered lactam 212 as depicted in Scheme 50.
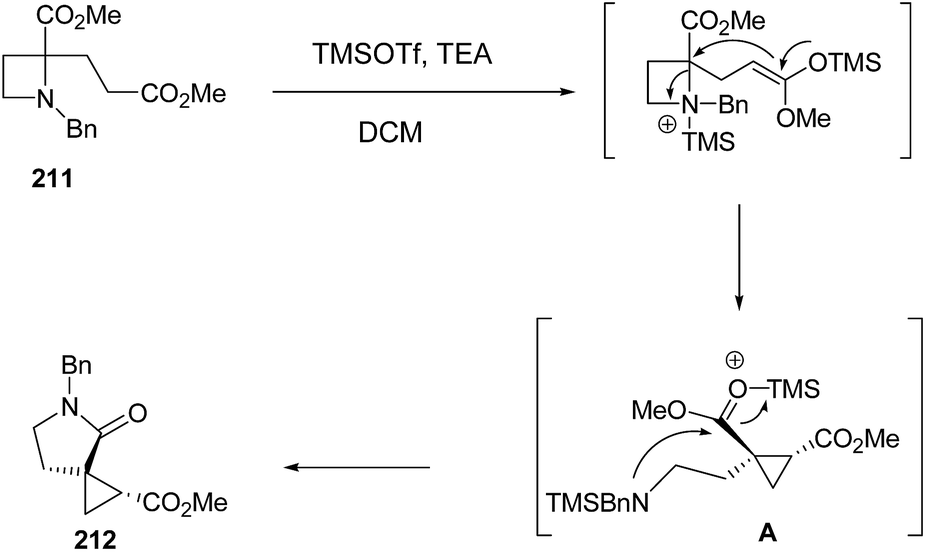 | ||
| Scheme 50 | ||
King et al.61 have recently explored triflic acid promoted conversion of N-cinnamoyl azetidine 216 into fused tricyclic product 217 having biological relevance (Scheme 51).
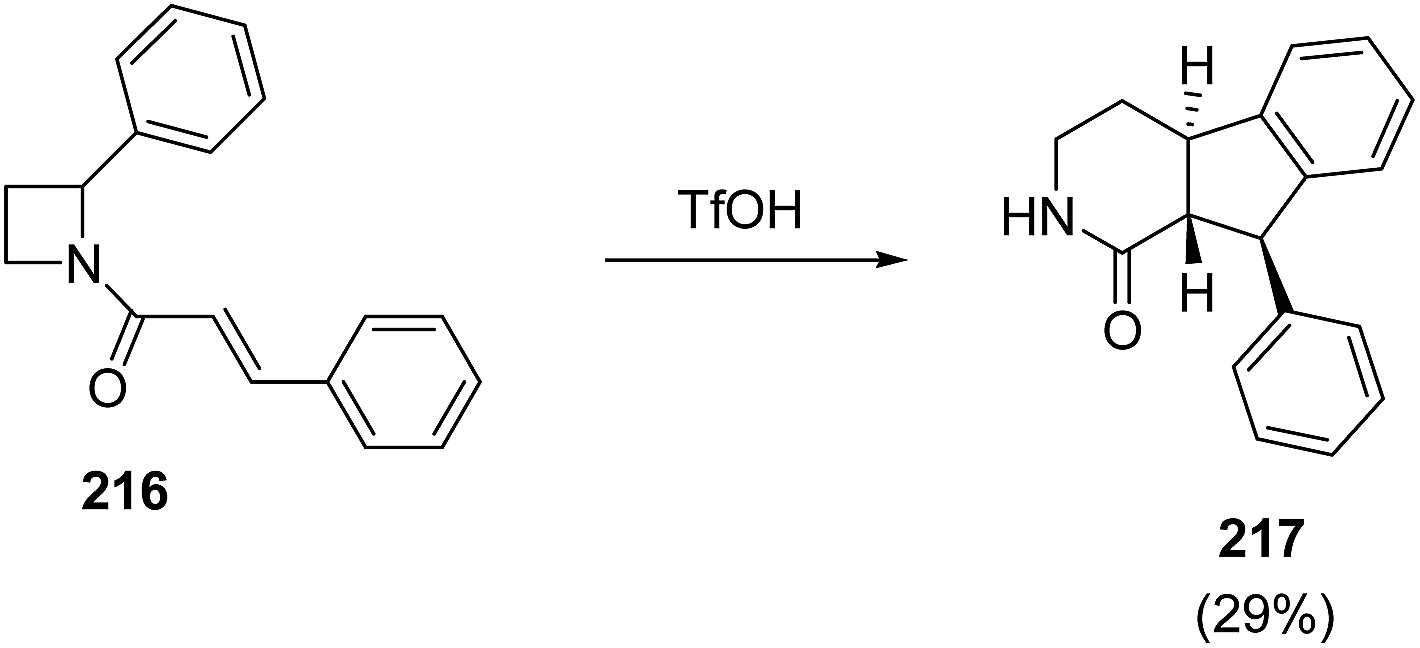 | ||
| Scheme 51 | ||
Couty and co-workers62 have utilized [1,2] Meisenheimer rearrangement of functionalized azetidines to isoxazolidines without trapping of the transient N-oxide. It was found that with an unsubstituted azetidine, the rearrangement took place very efficiently and the ring cleavage was highly regioselective when an ester or nitrile was present at C-2 position. The treatment of azetidine 218 with hydrogen peroxide in the presence of Na2WO4·2H2O resulted in the formation of N-benzhydryl isoxazolidine 219. The nitrile substituted azetidine 220 under basic condition led to the synthesis of isoxazolidinyl amide 221. Further, the reaction of azetidine 220 and 223 with m-CPBA in presence of Na2CO3 yielded 2-benzyl-isoxazolidine-5-carbonitrile 222 and 2-benzyl-isoxazolidine-5-carboxylic acid ethyl ester 224, respectively (Scheme 52).62
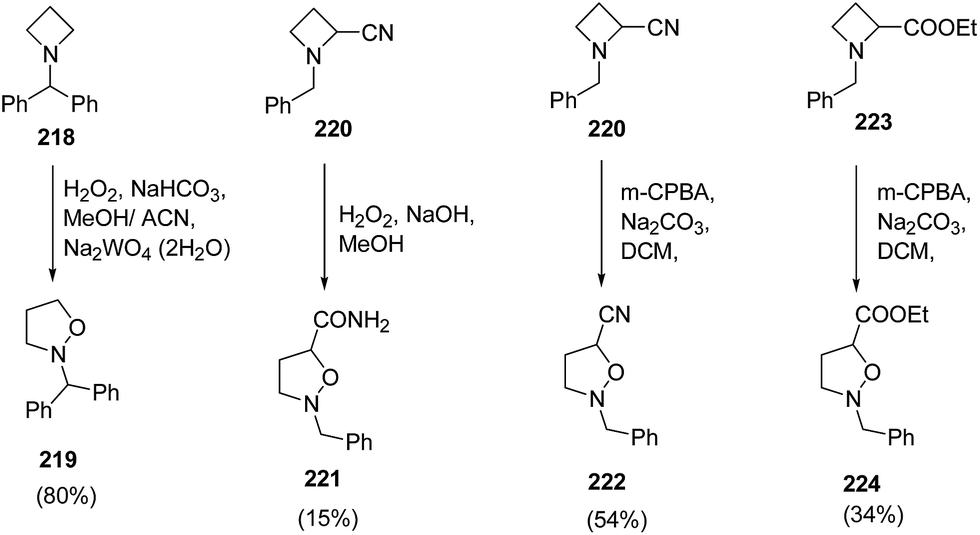 | ||
| Scheme 52 | ||
The exposure of diastereomeric azetidines 225 and 228 under similar conditions led to epimeric mixture of products. Thus, the treatment of azetidine 225 with m-CPBA in presence of Na2CO3 resulted in the synthesis of isoxazolidines 226 and 227 in ratio 3:1, while azetidine 228 gave isoxazolidines 229 and 230 in ratio 7:1 as depicted in Scheme 53.62
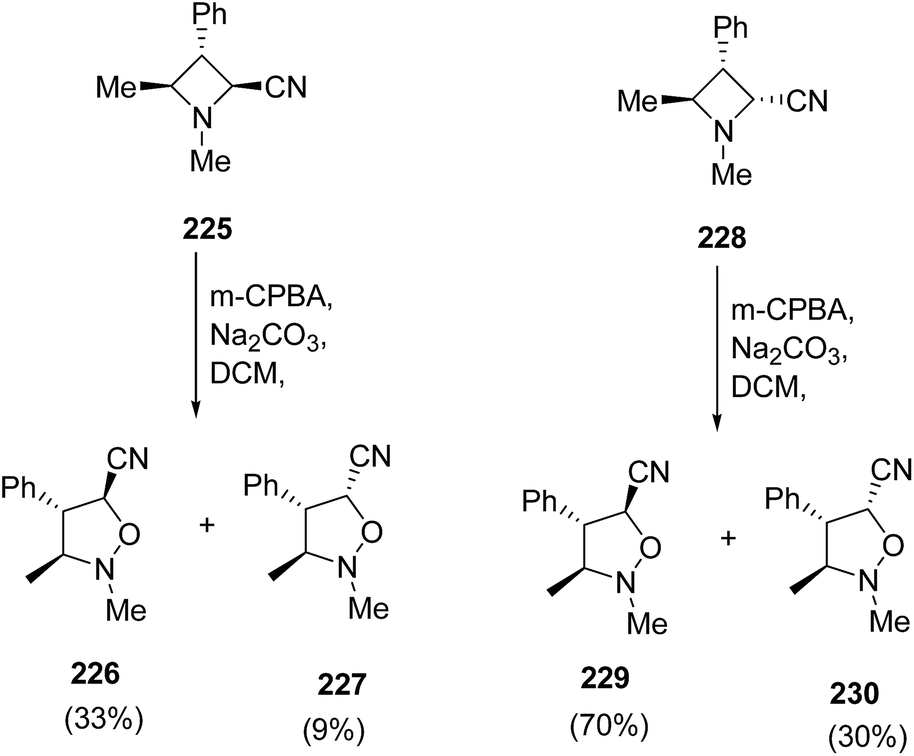 | ||
| Scheme 53 | ||
The degree of epimerization was found to depend on the nature of the substituent viz. nitrile or ester at C-2 position while no epimerization was observed with N-Bn analogue azetidine ester 231. Furthermore, the nature of substituent on nitrogen also play a crucial role, since extensive epimerization was observed in case of N-Me azetidine 234. Moreover, it was found that when N-oxide has cis relationship to the 4-methyl substituent, Cope elimination took place to yield hydroxylamine 233 and 236 along with isoxazolidines, 232 and 235 respectively (Scheme 54).62
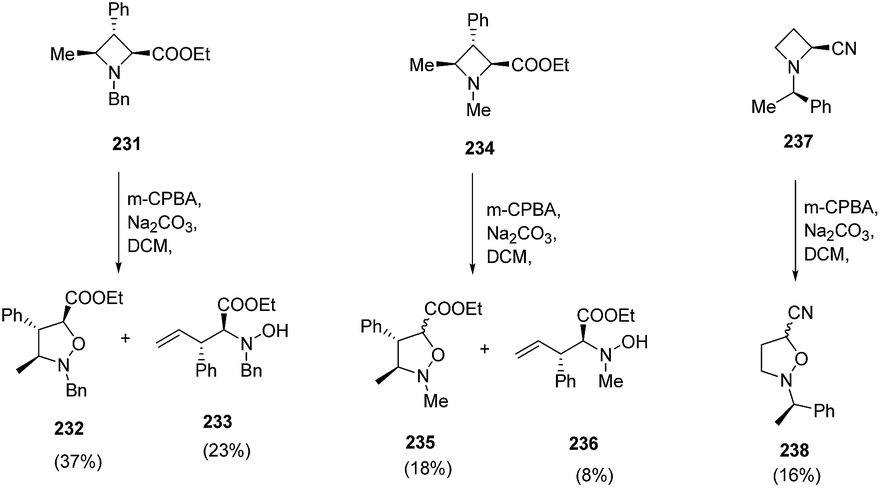 | ||
| Scheme 54 | ||
Zhang et al.63 have recently developed the route for the chiral synthesis of dihydrothiazoles and dihydro-oxazoles in a regio- and stereoselective manner via ring opening of azetidines. The synthetic protocol involved the treatment of 3-amino-4-aryl-azetidine 239 with isothiocyanates 240 in DCM at ambient temperature resulting in the synthesis of dihydrothiazoles 241 without the formation of corresponding thiourea. The reaction was carried out under different conditions but best results in term of yields were obtained in dichloromethane (Scheme 55).
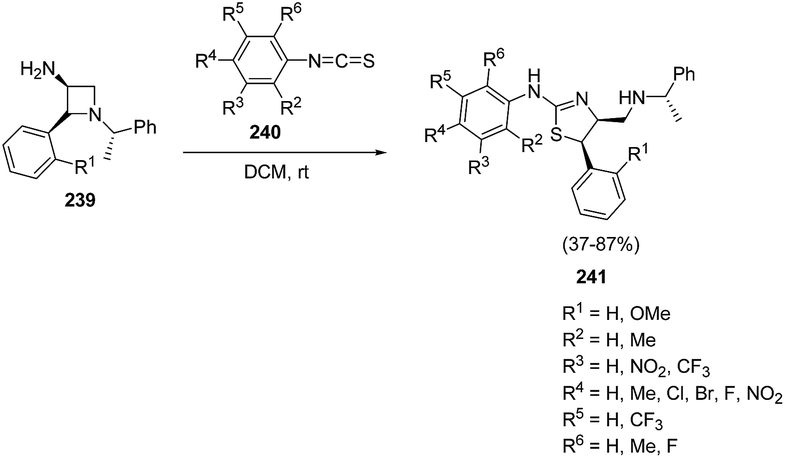 | ||
| Scheme 55 | ||
Mechanistically, the reaction was thought to proceed via the formation of thiourea as an intermediate which underwent intramolecular cyclization to afford corresponding dihydrothiazoles 241 as depicted in Scheme 56.63
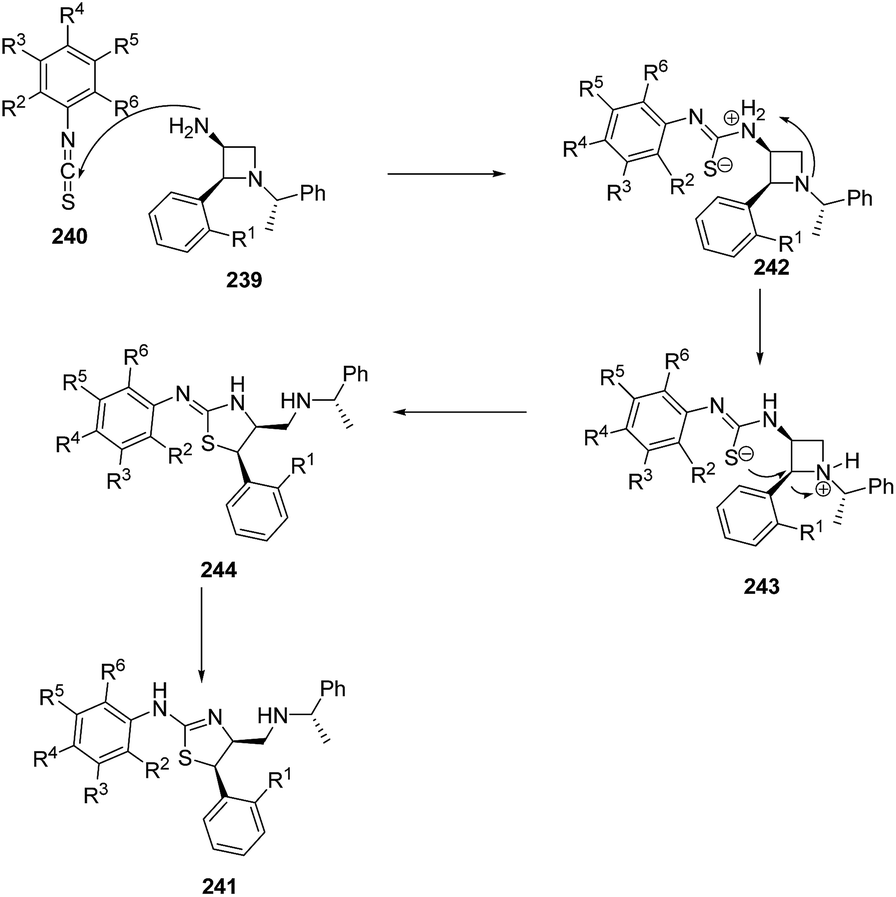 | ||
| Scheme 56 | ||
When 3-amino-4-aryl-azetidines 239 was treated with isocyanates 245 in dichloromethane at ambient temperature overnight, ureas 246 were the exclusive products without the isolation of corresponding dihydrooxazole 247 due to less nucleophilicity of oxygen than that of sulfur. The refluxing of 246 in dichloroethane in the presence of TFA, afforded the corresponding dihydrooxazole 247 as depicted in Scheme 57.63
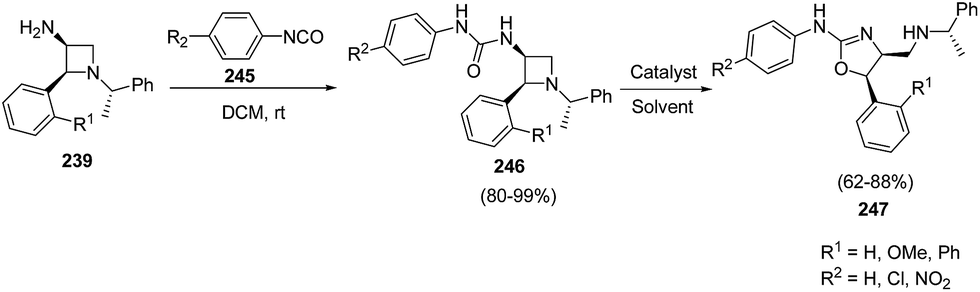 | ||
| Scheme 57 | ||
The pyrrole ring is one of the basic heteroaromatic motifs in numerous biologically active alkaloids, pharmaceutical products, or even in conducting polymers.64 2,4-Disubstituted pyrroles are useful intermediates for the synthesis of highly substituted pharmacologically relevant scaffolds.65 There are a plethora of methods such as the classical Hantzsch, Knorr, and Paal–Knorr reactions, and metal-based or 1,3-dipolar cycloadditions strategies66 for the synthesis of pyrroles. Zard et al.67 have developed a protocol for the synthesis of functionalized pyrroles having protected aminomethyl group at C-4 position via ring opening of Boc-protected azetidine. The treatment of Boc protected azetidine 248 with a range of primary amines or ammonia in presence of p-TsOH in hot dioxane furnished the corresponding pyrroles 249 as depicted in Scheme 58.
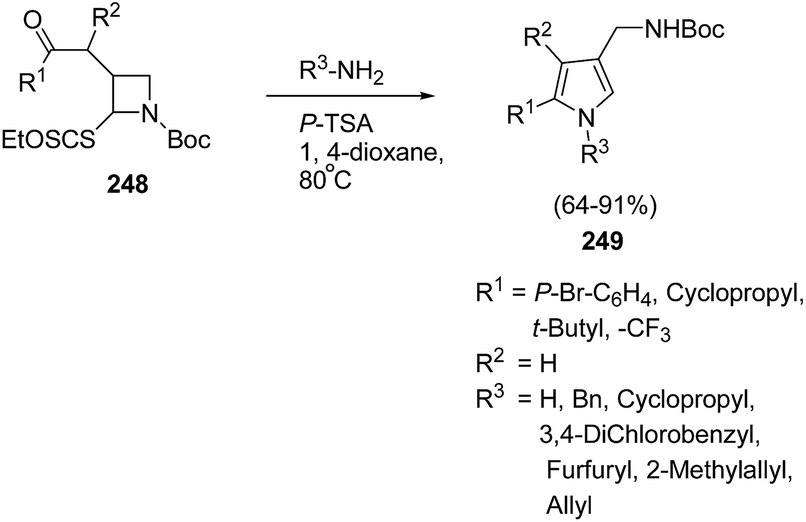 | ||
| Scheme 58 | ||
Mechanistically, the aminolysis of azetidine 248 resulted in the formation of thiol 250, which underwent ring-opening to generate thioaldehyde 252. The intramolecular condensation of amine with both the reactive thioaldehyde and the ketone afforded the desired pyrroles 249 (Scheme 59).67
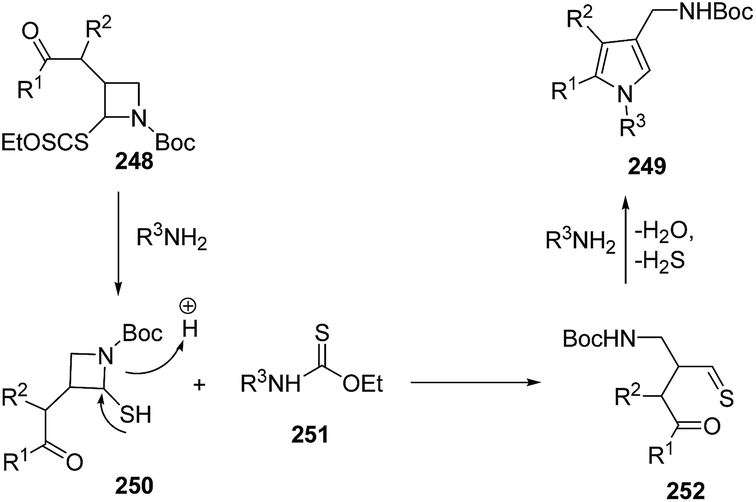 | ||
| Scheme 59 | ||
The synthesis of α-trifluoromethylated peptides, is still a challenging task68 due to steric hindrance and low nucleophilicity of the nitrogen atom. Stringent reaction conditions are required for the introduction of these amino acids into peptides. Brigaud and his co-workers have developed a convenient method for the syntheses of new enantiomerically pure nonproteogenic quaternary amino acid, α-Tfm-homoserines. The methodology involved the acidic hydrolysis of azetidine-2-carbonitriles viz. 72 and 73 resulting in concomitant hydrolysis of nitrile along with the removal of the phenylethanol (Scheme 60).21
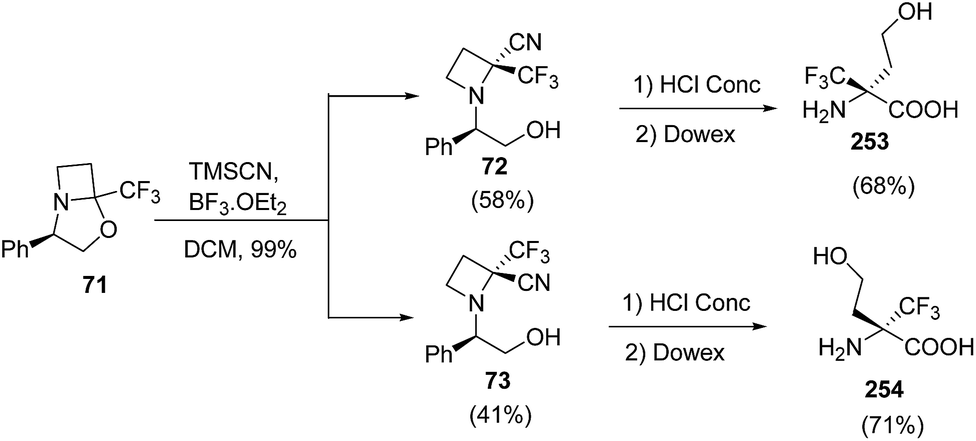 | ||
| Scheme 60 | ||
Azumaya et al.69 have recently explored the acid promoted aza-Claisen rearrangement of N-aryl-2-vinylazetidines into corresponding tetrahydrobenzazocines, which being unstable gets isomerized to vinyltetrahydroquinolines. The synthetic protocol involved the treatment of N-aryl-2-vinylazetidines 255 with different acids (H2SO4, TfOH and CF3COOH) in presence of different solvents (DCE, DCM and toluene) resulting in a mixture of tetrahydrobenzazocines 256 and 3-vinyl-1,2,3,4-tetrahydroquinolines 257 (Scheme 61).69
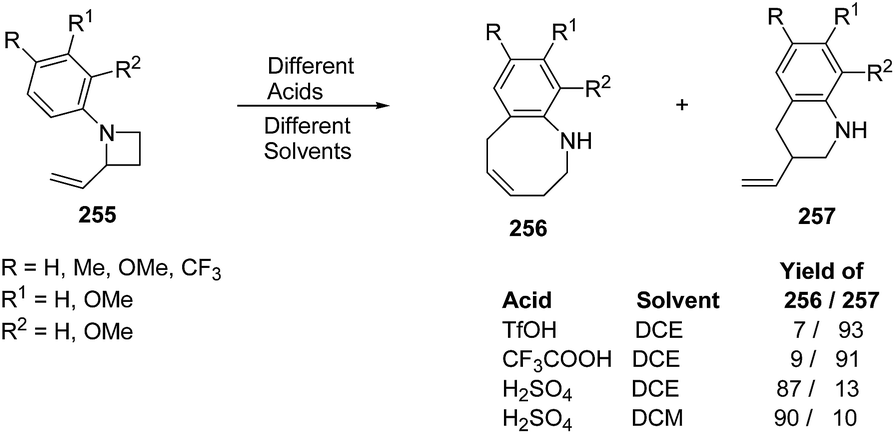 | ||
| Scheme 61 | ||
Many attempts were made to improve the yield of tetrahydrobenzazocines 256 and the best results were achieved via stirring at 0 °C with subsequent stirring for 3 h at room temperature in presence of H2SO4 (Scheme 62).69
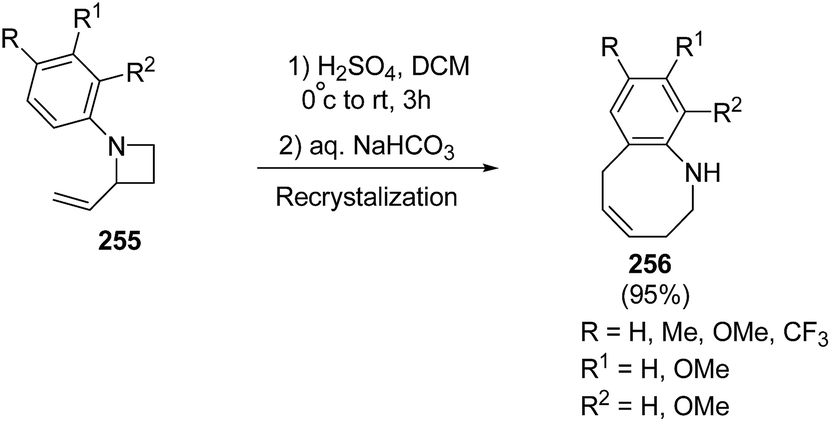 | ||
| Scheme 62 | ||
The stability of 256 was enhanced either via catalytic hydrogenation to afford hexahydrobenzazocine 258 or by protection of amino group through acylation to yield the acetylated benzazocine derivatives 259 (Scheme 63).69
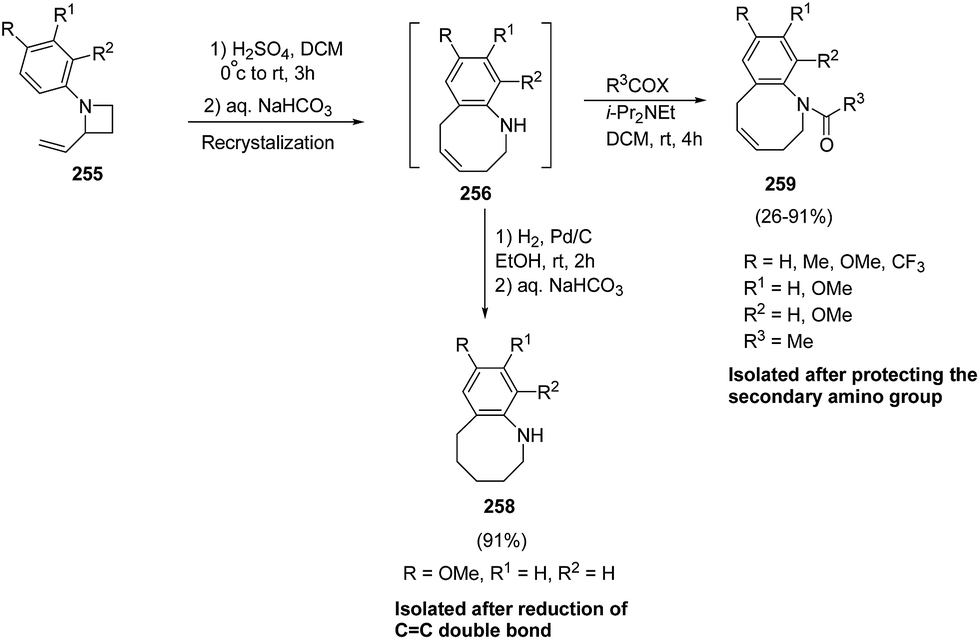 | ||
| Scheme 63 | ||
The treatment of 256 with silica gel gave 6,7-dimethoxy-3-vinyl-1,2,3,4-tetrahydroquinoline 257 in 81% yield. It was found that ring-contraction reaction was dramatically accelerated in the presence of formaldehyde. Thus, the treatment of 256 with formaldehyde in presence of triflic acid at room temperature yielded 6,7-dimethoxy-3-vinyl-1,2,3,4-tetrahydroquinoline 257 in 76% yield (Scheme 64).69
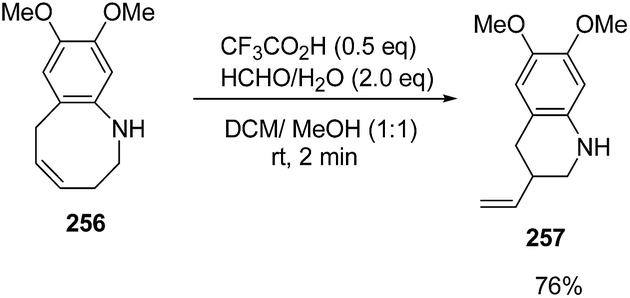 | ||
| Scheme 64 | ||
Couty and co-workers70 have utilized azetidinium triflate in a one-pot two-steps sequence to generate azetidinium ylides 261 with subsequent regioselective Hofmann elimination to yield acrylonitrile 262 which was utilized in DBU-catalyzed conjugate addition of amines yielding 4-amino-2-aminomethyl-butyronitriles 263–265. The synthetic methodology involved the treatment of azetidinium ion 260 with LiHMDS in THF at −78 °C with subsequent methylation resulting in the synthesis of acrylonitrile 261 which underwent ring opening to yield alkene 262. It has been found that 3 equiv. of base and 3 equiv. of methyl iodide resulted in good yields of alkene 262. DBU catalyzed conjugate addition of secondary amines to acrylonitrile 262 gave good yields of diamines 263–265, having fair levels of diastereoselectivity (Scheme 65).
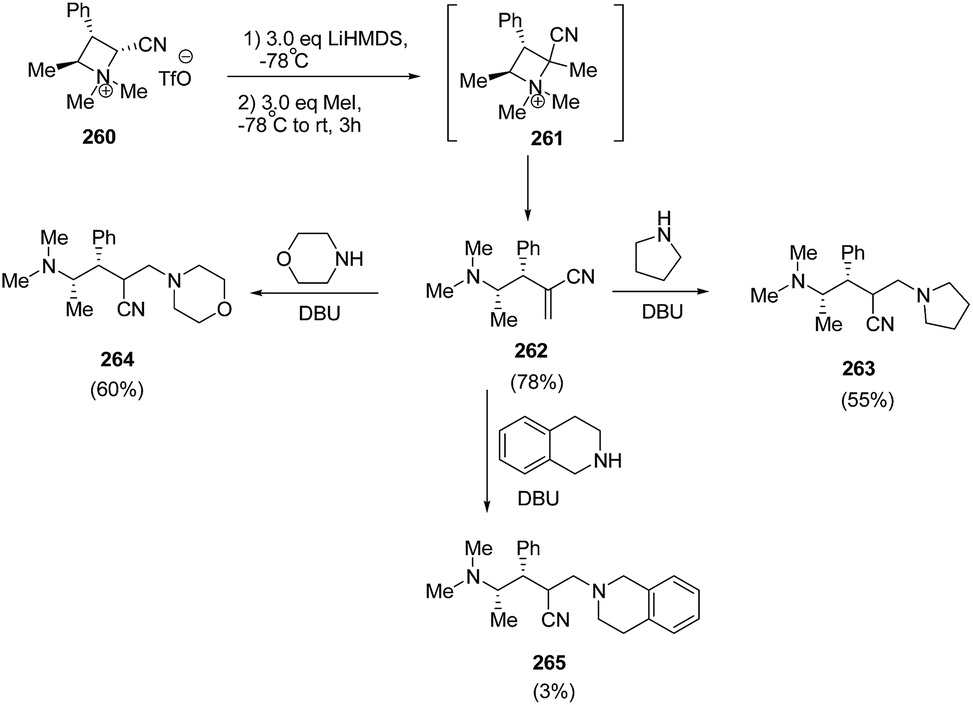 | ||
| Scheme 65 | ||
Further, the conjugate addition of azetidinium ylide derived from 260 on 266 yielded the C2 symmetrical cyclopropanes 267 in good yield as depicted in Scheme 66.70
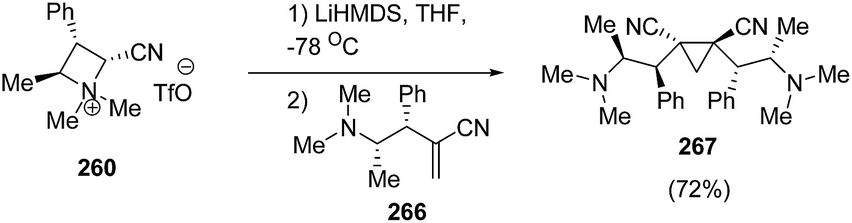 | ||
| Scheme 66 | ||
Bhattacharya and co-workers71 have developed a route for synthesis of enantiopure γ-amino ethers via copper(II)triflate-catalyzed ring-opening of chiral azetidines with alcohols. The synthetic methodology involved the ring-opening of enantiopure (S)-2-phenyl-N-tosylazetidine 268 with different alcohols in the presence of catalytic amount of Lewis acid and stoichiometric amount of quaternary ammonium salt at 0 °C to afford γ-amino ethers 269 (Scheme 67).
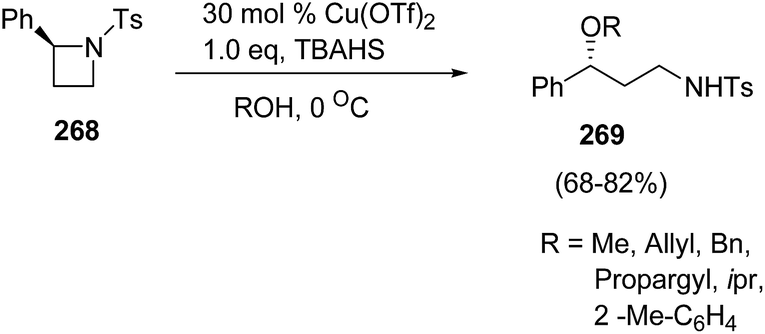 | ||
| Scheme 67 | ||
The methodology was further extended towards methanol promoted ring-opening of enantiopure cis- and trans-2,4-disubstituted azetidines 270–273 affording the corresponding 1,3-amino ethers 274–277. The ring-opening of enantiomerically pure trans-(2R,4S)-2-allyl-4-phenyl-N-tosyl-azetidine 272 afforded amino ether 276 as the major diastereomer having syn configuration, whereas cis-273 afforded amino ether 277 having anti configuration as the major product as depicted in Scheme 68.71
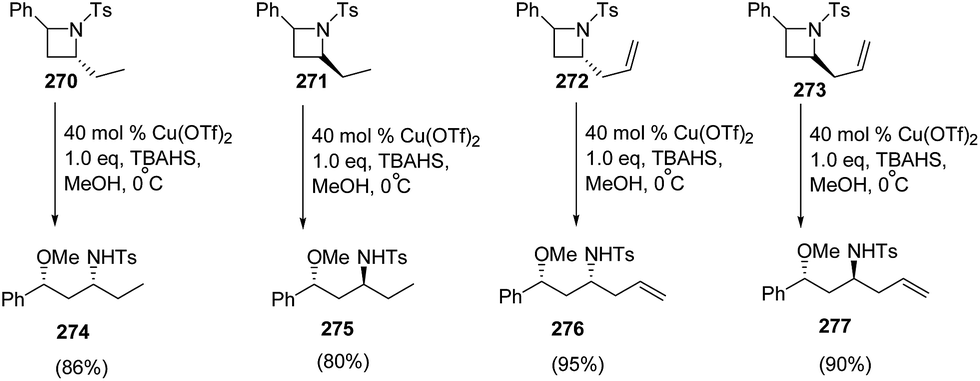 | ||
| Scheme 68 | ||
The ring-opening of 268 with 3-chloropropanol in the presence of 40 mol% of Cu(OTf)2 with stoichiometric amount of TBAHS at 0 °C in dichloromethane, afforded oxazocane 279, having two carbon higher homologue of morpholines (Scheme 69).71
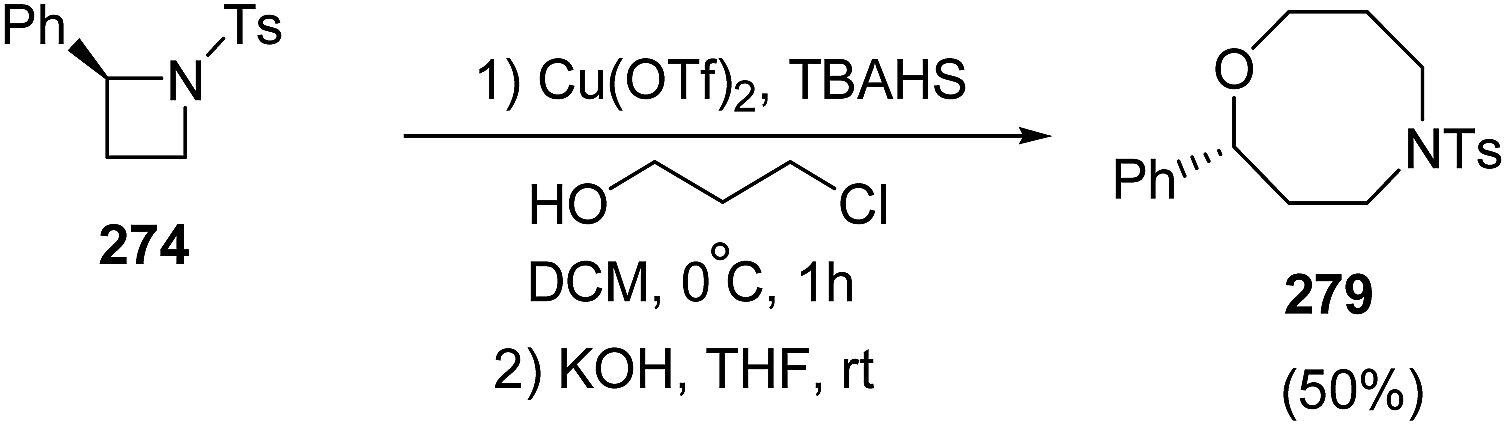 | ||
| Scheme 69 | ||
4. Conclusion
The inspiring work contained in the present review article attests the importance of azetidine in heterocyclic synthesis. Literary rationale has witnessed a recent upsurge of different synthetic methods including intramolecular ring closure, β-lactam-synthon approach as well as the use of metal/organocatalysis for their stereoselective synthesis. Nevertheless, this synthetically arduous four-membered ring with immense importance both in biological/medicinal chemistry and as a versatile synthon will continue to pose challenges to synthetic chemists for developing efficient strategies.Conflicts of interest
There are no conflicts to declare.Acknowledgements
AA wishes to thank University Grants Commission, New Delhi, India for financial support under grant no. ROMRP-NRCB-CHEM-2015-16-87198.Notes and references
- (a) D. Antermite, L. Degennaro and R. Luisi, Org. Biomol. Chem., 2017, 15, 34 RSC; (b) J. Dolfen, N. De Kimpe and M. D’hooghe, Synlett, 2016, 27, 1486 CrossRef CAS; (c) F. Couty, B. Drouillat, G. Evano and O. David, Eur. J. Org. Chem., 2013, 2045 CrossRef CAS; (d) B. Alcaide, P. Almendros and C. Aragoncillo, Curr. Opin. Drug Discovery Dev., 2010, 13, 685 CAS; (e) A. Brandi, S. Cicchi and F. M. Cordero, Chem. Rev., 2008, 108, 3988 CrossRef CAS PubMed; (f) F. Couty and G. Evano, Synlett, 2009, 19, 3053 CrossRef; (g) G. S. Singh, M. D'Hooghe and N. De Kimpe, Azetidines, Azetines and Azetes: Monocyclic, in Comprehensive Heterocyclic Chemistry III, ed. A. R. Katritzky, Elsevier, 2008, vol. 2, ch. 2.01, pp. 1–110 Search PubMed; (h) G. B. Evans, R. H. Furneaux, B. Greatrex, A. S. Murkin, V. L. Schramm and P. C. Tyler, J. Med. Chem., 2008, 51, 948 CrossRef CAS PubMed; (i) T. M. Bott and F. G. West, Heterocycles, 2012, 84, 223 CrossRef CAS; (j) G. B. Evans, R. H. Furneaux, B. Greatrex, A. S. Murkin, V. L. Schramm and P. C. Tyler, J. Med. Chem., 2008, 51, 948 CrossRef CAS PubMed; (k) D. Honcharenko, J. Barman, O. P. Varghese and J. Chattopadhyaya, Biochemistry, 2007, 46, 5635 CrossRef CAS PubMed; (l) J. Slade, J. Bajwa, H. Liu, D. Parker, J. Vivelo, G. P. Chen, J. Calienni, E. Villhauer, K. Prasad, O. Repic and T. J. Blacklock, Org. Process Res. Dev., 2007, 11, 825 CrossRef CAS; (m) N. Kolocouris, G. Zoidis, G. B. Foscolos, G. Fytas, S. R. Prathalingham, J. M. Kelly, L. Naesensc and E. De Clercq, Bioorg. Med. Chem. Lett., 2007, 17, 4358 CrossRef CAS PubMed.
- F. Couty and G. Evano, Org. Prep. Proced. Int., 2006, 38, 427 CrossRef CAS.
- (a) H. Brauner-Osborne, L. Bunch, N. Chopin, F. Couty, G. Evano, A. A. Jensen, M. Kusk, B. Nielsen and N. Rabasso, Org. Biomol. Chem., 2005, 3, 3926 RSC; (b) B. Drouillat, K. Wright, J. Marrot and F. Couty, Tetrahedron: Asymmetry, 2012, 23, 690 CrossRef CAS; (c) D. Honcharenko, C. Zhou and J. Chattopadhyaya, J. Org. Chem., 2008, 73, 2829 CrossRef CAS PubMed; (d) O. Plashkevych, S. Chatterjee, D. Honcharenko, W. Pathmasiri and J. Chattopadhyaya, J. Org. Chem., 2007, 72, 4716 CrossRef CAS PubMed; (e) D. Honcharenko, O. P. Varghese, O. Plashkevych, J. Barman and J. Chattopadhyaya, J. Org. Chem., 2006, 71, 299 CrossRef CAS PubMed.
- C. Agami, F. Couty and N. Rabasso, Tetrahedron Lett., 2002, 43, 4633 CrossRef CAS.
- (a) T. M. Bott, J. A. Vanecko and F. G. West, J. Org. Chem., 2009, 74, 2832 CrossRef CAS PubMed; (b) W. Van Brabandt, Y. Dejaegher, R. Van Landeghem and N. De Kimpe, Org. Lett., 2006, 8, 1101 CrossRef CAS PubMed; (c) F. Couty, F. Durrat, G. Evano and J. Marrot, Eur. J. Org. Chem., 2006, 4214 CrossRef CAS.
- (a) S. Purser, P. R. Moore, S. Swallow and V. Gouverneur, Chem. Soc. Rev., 2008, 37, 320 RSC; (b) W. K. Hagmann, J. Med. Chem., 2008, 51, 4359 CrossRef CAS PubMed.
- S. Kenis, M. D’hooghe, G. Verniest, T. A. D. Thi, C. P. The, T. V. Nguyen and N. De Kimpe, J. Org. Chem., 2012, 77, 5982 CrossRef CAS PubMed.
- A. Feula, S. S. Dhillon, R. Byravan, M. Sangha, R. Ebanks, M. A. H. Salih, N. Spencer, L. Male, I. Magyary, W.-P. Deng, F. Muller and J. S. Fossey, Org. Biomol. Chem., 2013, 11, 5083 CAS.
- F. Colpaert, S. Mangelinckx, S. De Brabandere and N. De Kimpe, J. Org. Chem., 2011, 76, 2204 CrossRef CAS PubMed.
- M. S. Dowling, D. P. Fernando, J. Hou, B. Liu and A. C. Smith, J. Org. Chem., 2016, 81, 3031 CrossRef CAS PubMed.
- A. Zukauskaite, S. Mangelinckx, V. Buinauskaite, A. Sackus and N. De Kimpe, Amino Acids, 2011, 41, 541 CrossRef CAS PubMed.
- L. Provins, B. Christophe, P. Danhaive, J. Dulieu, M. Gillard, L. Quere and K. Stebbins, Bioorg. Med. Chem. Lett., 2007, 17, 3077 CrossRef CAS PubMed.
- S. T. Murphy, H. L. Case, E. Ellsworth, S. Hagen, M. Huband, T. Joannides, C. Limberakis, K. R. Marotti, A. M. Ottolini, M. Rauckhorst, J. Starr, M. Stier, C. Taylor, T. Zhu, A. Blaser, W. A. Denny, G. L. Lu, J. B. Smaill and F. Rivault, Bioorg. Med. Chem. Lett., 2007, 17, 2150 CrossRef CAS PubMed.
- S. Stankovic, M. D’hooghe, K. A. Tehrani and N. De Kimpe, Tetrahedron Lett., 2012, 53, 107 CrossRef CAS.
- T. K. Pradhan, K. S. Krishnan, J.-L. Vasse and J. Szymoniak, Org. Lett., 2011, 13, 1793 CrossRef CAS PubMed.
- (a) H. O'Dowd, J. G. Lewis, J. Trias, R. Asano, J. Blais, S. Lopez, C. K. Park, C. Wu, W. Wang and M. F. Gordeev, Bioorg. Med. Chem. Lett., 2008, 18, 2645 CrossRef PubMed; (b) M. R. Faust, G. Hoefner, J. Pabel and K. T. Wanner, Eur. J. Med. Chem., 2010, 45, 2453 CrossRef CAS PubMed.
- S. Mangelinckx, B. D. Sterck, F. Colpaert, S. Catak, J. Jacobs, S. Rooryck, M. Waroquier, V. V. Speybroeck and N. De Kimpe, J. Org. Chem., 2012, 77, 3415 CrossRef CAS PubMed.
- S. T. M. Orr, S. Cabral, D. P. Fernando and T. Makowski, Tetrahedron Lett., 2011, 52, 3618 CrossRef CAS.
- T. J. Senter, M. C. O'Reilly, K. M. Chong, G. A. Sulikowski and C. W. Lindsley, Tetrahedron Lett., 2015, 56, 1276 CrossRef CAS PubMed.
- A. A. Kudale, P. Anaspure, F. Goswami and M. Voss, Tetrahedron Lett., 2014, 55, 7219 CrossRef CAS.
- N. Lensen, J. Marais and T. Brigaud, Org. Lett., 2015, 17, 342 CrossRef CAS PubMed.
- Y. Ishida, N. Iwahashi, N. Nishizono and K. Saigo, Tetrahedron Lett., 2009, 50, 1889 CrossRef CAS.
- G. Horne, F. X. Wilson, J. Tinsley, D. H. Williams and R. Storer, Drug Discov. Today, 2011, 16, 107 CrossRef CAS PubMed.
- P. R. Markad, D. P. Sonawane, S. Ghosh, B. A. Chopade, N. Kumbhar, T. Louat, J. Herman, M. Waer, P. Herdewijn and D. D. Dhavale, Bioorg. Med. Chem., 2014, 22, 5776 CrossRef CAS PubMed.
- P. P. Lawande, V. A. Sontakke, R. J. Nair, A. Khan, S. G. Sabharwal and V. S. Shinde, Tetrahedron, 2015, 71, 5085 CrossRef CAS.
- V. U. Pawar, S. T. Chavan, S. G. Sabharwal and V. S. Shinde, Bioorg. Med. Chem., 2010, 18, 7799 CrossRef CAS PubMed.
- B. V. S. Reddy, C. Kishore and A. S. Reddy, Tetrahedron Lett., 2014, 55, 49 CrossRef CAS.
- N. Araujo, S. F. Jenkinson, R. F. Martınez, A. F. G. Glawar, M. R. Wormald, T. D. Butters, S. Nakagawa, I. Adachi, A. Kato, A. Yoshihara, K. Akimitsu, K. Izumori and G. W. J. Fleet, Org. Lett., 2012, 14, 4174 CrossRef CAS PubMed.
- A. Weber, J. Med. Chem., 2004, 47, 4135 CrossRef CAS PubMed.
- (a) C. M. L. Delpiccolo, M. M. Amezaga and E. G. Mata, Recent Approaches Toward the Generation of Molecular Diversity Based on β-Lactam Structures, in Beta-Lactams-Novel Synthetic Pathway and Applications, ed. B. K. Banik, Springer International Publishing AG, 2017, pp. 129–162 Search PubMed; (b) B. Alcaide, P. Almendros and C. Aragoncillo, Synthesis of Five-Membered Heterocycles Through β-Lactam Ring-Expansion Reaction, in Beta-Lactams-Novel Synthetic Pathway and Applications, ed. B. K. Banik, Springer International Publishing AG, 2017, pp. 163–218 Search PubMed.
- (a) V. Mehra, P. Singh, K. Bisetty and V. Kumar, RSC Adv., 2014, 4, 41793 RSC; (b) V. Mehra and V. Kumar, Tetrahedron Lett., 2014, 55, 845 CrossRef CAS; (c) V. Mehra, P. Singh, N. Manhas and V. Kumar, Synlett, 2014, 25, 1124 CrossRef; (d) V. Mehra and V. Kumar, Tetrahedron, 2013, 69, 3857 CrossRef CAS; (e) A. Anand, V. Mehra and V. Kumar, Synlett, 2013, 24, 865 CrossRef CAS; (f) V. Mehra, P. Singh and V. Kumar, Tetrahedron, 2012, 68, 8395 CrossRef CAS; (g) A. Kamath and I. Ojima, Tetrahedron, 2012, 68, 10640 CrossRef CAS PubMed; (h) S. Dekeukeleire, M. D’hooghe, M. Vanwalleghem, W. V. Brabandt and N. De Kimpe, Tetrahedron, 2012, 68, 10827 CrossRef CASR. Raj, V. Mehra, P. Singh, V. Kumar, G. Bhargava, M. P. Mahajan, S. Handa and L. Slaughter, Eur. J. Org. Chem., 2011, 2697 CrossRef CAS.
- (a) B. Van Driessche, W. Van Brabandt, M. D'Hooghe, Y. Dejaegher and N. De Kimpe, Tetrahedron, 2006, 62, 6882 CrossRef CAS; (b) L. Kvaerno, M. Werder, H. Hauser and E. M. Carreira, Org. Lett., 2005, 7, 1145 CrossRef PubMed.
- V. Mehra, Neetu and V. Kumar, Tetrahedron Lett., 2013, 54, 4763 CrossRef CAS.
- S. Stankovic, H. Goossens, S. Catak, M. Tezcan, M. Waroquier, V. V. Speybroeck, M. D’hooghe and N. De Kimpe, J. Org. Chem., 2012, 77, 3181 CrossRef CAS PubMed.
- T. Soeta, Y. Miyamoto, S. Fujinami and Y. Ukaji, Tetrahedron, 2014, 70, 6623 CrossRef CAS.
- X. Wei, D. Liu, Q. An and W. Zhang, Org. Lett., 2015, 17, 5768 CrossRef CAS PubMed.
- P. R. Payne, P. Garcia, P. Eisenberger, J. C.-H. Yim and L. L. Schafer, Org. Lett., 2013, 15, 2182 CrossRef CAS PubMed.
- Ferrocenes: Ligands, Materials and Biomolecules, ed. P. Stepnicka, John Wiley &Sons, New York, 2008, p. 39 Search PubMed.
- (a) V. S. Sudhir, N. B. R. Baig and S. Chandrasekharan, Eur. J. Org. Chem., 2009, 5365 CrossRef CAS; (b) J. Tauchman and P. Stepnicka, Inorg. Chem. Comm., 2010, 13, 149 CrossRef CAS; (c) A. Garcia, B. Insuasty, M. A. Herranz, R. Martinez-Alvarez and N. Martin, Org. Lett., 2009, 11, 5398 CrossRef CAS PubMed.
- (a) Q. Li, C.-H. Ding, X.-L. Hou and L.-X. Dai, Org. Lett., 2010, 12, 1080 CrossRef CAS PubMed; (b) S. L. Marquard, D. C. Rosenfeld and J. F Hartwig, Angew. Chem., Int. Ed., 2010, 49, 793 CrossRef CAS PubMed.
- S. Gouthaman, P. Shanmugam and A. B. Mandal, Tetrahedron Lett., 2013, 54, 3007 CrossRef CAS.
- Y. Shang, K. Liao, X. He and J. Hu, Tetrahedron, 2013, 69, 10134 CrossRef CAS.
- A. Rai and L. D. S. Yadav, Tetrahedron Lett., 2013, 54, 3127 CrossRef CAS.
- S. Takizawa, F. A. Arteaga, Y. Yoshida, M. Suzuki and H. Sasai, Org. Lett., 2013, 15, 4142 CrossRef CAS PubMed.
- M. Amongero and T. S. Kaufman, Tetrahedron Lett., 2013, 54, 1924 CrossRef CAS.
- N. Kern, A.-S. Felten, J.-M. Weibel, P. Pale and A. Blanc, Org. Lett., 2014, 16, 6104 CrossRef CAS PubMed.
- H.-H. Zhang, Y.-C. Luo, H.-P. Wang, W. Chen and P.-F. Xu, Org. Lett., 2014, 16, 4896 CrossRef CAS PubMed.
- T. O. Painter, J. R. Bunn, F. J. Schoenen, J. T. Douglas, V. W. Day and C. Santini, J. Org. Chem., 2013, 78, 3720 CrossRef CAS PubMed.
- X. Ma, J.-X. Wang, S. Li, K.-H. Wang and D. Huang, Tetrahedron, 2009, 65, 8683 CrossRef CAS.
- S. E. Denmark and G. L. Beutner, Angew. Chem., Int. Ed., 2008, 47, 1560 CrossRef CAS PubMed.
- (a) F. Lopez and J. L. Mascarennas, Chem.–Eur. J., 2011, 17, 418 CrossRef CAS PubMed; (b) C.-K. Pei and M. Shi, Chem.–Eur. J., 2012, 18, 6712 CrossRef CAS PubMed; (c) Q.-Y. Zhao, Z. Lian, Y. Wei and M. Shi, Chem. Commun., 2012, 48, 1724 RSC; (d) Y. Wei and M. Shi, Chem. Rev., 2013, 113, 6659 CrossRef CAS PubMed.
- L.-J. Yang, S. Li, S. Wang, J. Nie and J.-A. Ma, J. Org. Chem., 2014, 79, 3547 CrossRef CAS PubMed.
- (a) T. A. Brandt, S. Caron, D. B. Damon, J. Di Brino, A. Ghosh, D. A. Griffith, S. Kedia, J. A. Ragan, P. R. Rose, B. C. Vanderplas and L. Wei, Tetrahedron, 2009, 65, 3292 CrossRef CAS; (b) Y. Han, M. Han, D. Shin, C. Song and H.-G. Hahn, J. Med. Chem., 2012, 55, 8188 CrossRef CAS PubMed.
- (a) J. Xiao and S. W. Wright, Tetrahedron Lett., 2013, 54, 2502 CrossRef CAS; (b) P.-A. Nocquet, D. Hazelard and P. Compain, Eur. J. Org. Chem., 2011, 6619 CrossRef CAS.
- K. Mollet, M. D’hooghe, L. Broeckx, B. Danneels, T. Desmet and N. De Kimpe, Tetrahedron, 2013, 69, 2603 CrossRef CAS.
- L. Menguy, B. Drouillat and F. Couty, Tetrahedron Lett., 2015, 56, 6625 CrossRef CAS.
- P.-A. Nocquet, D. Hazelard and P. Compain, Tetrahedron, 2012, 68, 4117 CrossRef CAS.
- F. Couty, F. Durrat and G. Evano, in Targets in Heterocyclic Systems-Chemistry and Properties, ed. O. A. Attanasi and D. Spinelli, Italian Society of Chemistry, Rome, 2005, 9, p. 186 Search PubMed.
- (a) D. C. Blakemore, J.-Y. Chiva and I. Thistlethwaite, Synlett, 2011, 1101 CAS; (b) K. Mollet, S. Catak, M. Waroquier, V. Van Speybroeck, M. D’hooghe and N. De Kimpe, J. Org. Chem., 2011, 76, 8364 CrossRef CAS PubMed; (c) K. Mollet, L. Broeckx, M. D’hooghe and N. De Kimpe, Heterocycles, 2012, 84, 431 CrossRef CAS.
- T. R. Hoye, V. Dvornikovs and E. Sizova, Org. Lett., 2006, 8, 5191 CrossRef CAS PubMed.
- F. D. King, A. Aliev, S. Caddick and D. Tocher, Tetrahedron, 2015, 71, 3411 CrossRef CAS.
- L. Menguy, B. Drouillat, J. Marrot and F. Couty, Tetrahedron Lett., 2012, 53, 4697 CrossRef CAS.
- X. Zhou, X. Xu, Y. Li, Z. Zhang and Z.-B. Zheng, Tetrahedron Lett., 2016, 57, 1236 CrossRef CAS.
- (a) I. S. Young, P. D. Thornton and A. Thompson, Nat. Prod. Rep., 2010, 27, 1801 RSC; (b) H. Fan, J. Peng, M. T. Hamann and J.-F. Hu, Chem. Rev., 2008, 108, 264 CrossRef CAS PubMed.
- (a) S. Han, D. S. Siegel, K. C. Morrison, P. J. Hergenrother and M. Movassaghi, J. Org. Chem., 2013, 78, 11970 CrossRef CAS PubMed; (b) R. Raju, A. M. Piggott, L. X. Barrientos Diaz, Z. Khalil and R. J. Capon, Org. Lett., 2010, 12, 5158 CrossRef CAS PubMed.
- (a) R. Yan, X. Kang, X. Zhou, X. Li, X. Liu, L. Xiang, Y. Li and G. Huang, J. Org. Chem., 2014, 79, 465 CrossRef CAS PubMed; (b) M.-C. P. Yeh, M.-N. Lin, C.-H. Hsu and C. J. Liang, J. Org. Chem., 2013, 78, 12381 CrossRef CAS PubMed; (c) Y. Zhou, X. Yan, C. Chen and C. Xi, Organometallics, 2013, 32, 6182 CrossRef CAS; (d) Y. Shi and V. Gevorgyan, Org. Lett., 2013, 15, 5394 CrossRef CAS PubMed; (e) R. Chaudhuri and U. Uli Kazmaier, Organometallics, 2013, 32, 5546 CrossRef CAS.
- S. Han and S. Z. Zard, Org. Lett., 2014, 16, 1992 CrossRef CAS PubMed.
- (a) J. L. Acena, A. E. Sorochinsky and V. A. Soloshonok, Synthesis, 2012, 44, 1591 CrossRef CAS; (b) X.-L. Qiu and F.-L. Qing, Eur. J. Org. Chem., 2011, 3261 CrossRef CAS.
- T. Shimizu, S. Koya, R. Yamasaki, Y. Mutoh, I. Azumaya, K. Katagiri and S. Saito, J. Org. Chem., 2014, 79, 4367 CrossRef CAS PubMed.
- C. Lo, O. David and F. Couty, Tetrahedron Lett., 2014, 55, 535 CrossRef CAS.
- M. K. Ghorai, D. Shukla and A. Bhattacharyya, J. Org. Chem., 2012, 77, 3740 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2017 |