
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Retaining individualities: the photodynamics of self-ordering porphyrin assemblies†
Wen-Dong
Quan
ab,
Anaïs
Pitto-Barry
 a,
Lewis A.
Baker
ab,
Eugen
Stulz
c,
Richard
Napier
d,
Rachel K.
O'Reilly
*a and
Vasilios G.
Stavros
*a
a,
Lewis A.
Baker
ab,
Eugen
Stulz
c,
Richard
Napier
d,
Rachel K.
O'Reilly
*a and
Vasilios G.
Stavros
*a
aDepartment of Chemistry, University of Warwick, Gibbet Hill Road, Coventry, UK. E-mail: v.stavros@warwick.ac.uk; rachel.oreilly@warwick.ac.uk
bMolecular Organisation and Assembly of Cells Doctoral Training Center (MOAC DTC), University of Warwick, Gibbet Hill Road, Coventry, UK
cSchool of Chemistry & Institute for Life Sciences, University of Southampton, Highfield, Southampton, UK
dSchool of Life Science, University of Warwick, Gibbet Hill Road, Coventry, UK
First published on 7th December 2015
Abstract
The retention of photochemical properties of individual chromophores is a key feature of biological light harvesting complexes. This is achieved despite extensive aggregation of the chromophores, which in synthetic chromophore assemblies often yields a change in spectral characteristics. As an alternative approach towards mimicking biological light harvesting complexes, we report the synthesis of porphyrin assemblies which retained the photochemical properties of the individual chromophore units despite their substantial aggregation. These new materials highlight a new bottom-up approach towards the design and understanding of more complex biomimetic and naturally occurring biological systems.
One of the most important processes for life on earth is photosynthesis, which is performed by plant and photosynthetic micro-organisms such as cyanobacteria. Nature uses light harvesting complexes (LHCs) to efficiently channel photoexcited energy on ultrafast time frames.1–5 This extraordinarily efficient process is enabled by the elegant and precise arrangement of chromophores,3,6–8 through sophisticated yet naturally-occurring self-assembly processes, and is an exemplar of evolution's phenomenal achievements. Natural LHCs have inspired numerous attempts to create synthetic mimics of such cyclic arrays of chromophores to enhance, for example, the efficiencies of photovoltaic cells.9–11 A template-directed synthetic method, elegantly demonstrated by the Anderson group, produced some of the closest mimics.12–14 These assembly processes and the requirement for covalent conjugation generally lead to altered spectral characteristics of the individual chromophores. Whilst this does extend the spectral coverage of single chromophoric systems, this is in contrast to biological LHCs, in which the UV-visible (UV-Vis) spectrum of the assembled system can usually be reconstructed by summing the UV-Vis spectra of the individual chromophores.8,15,16 Large aggregated and cross-linked systems are commonly utilised in biology for functions such as mechanical movements (actin filaments), photoprotection (melanin) and structural support (cross-linked cellulose and pectin), whereas the chromophores in LHCs are uniquely arranged through weak intermolecular interactions.4,7,8 Thus, the lack of covalent conjugations of these selected chromophores might be key to the functionalities of LHCs. Further, most of these synthetic assemblies require relatively high concentrations of the chromophores, and often challenging and complicated synthetic steps thus limiting their scalability and wider adoption.
In recent years, researchers have produced well-defined polymer-based self-assembled structures with relatively simple synthetic methods that are easily scaled up.17–21 In a handful of studies, various functionalised porphyrins, which are close mimics of some biological LHC chromophores, have been incorporated into these polymeric systems. These porphyrin–polymer conjugates were utilised in a range of applications such as photodynamic therapy (PDT),22–24 cell-imaging,25,26 initiators for complex polymers27 and simple proof-of-concept experiments for potential self-assembly methodologies.28,29 However, the only excited state dynamics studies performed involved long time scales (>nanoseconds), as the majority of the systems were oriented towards PDT applications.30,31 However, ultrafast dynamics of chromophores is a determinant of light energy harvesting efficiency.1–3,32–37 It is therefore crucial to understand the effects of such aggregation processes on the ultrafast photodynamics of individual chromophores to facilitate rational designs of LHC mimics based on non-covalent self-assembling polymers.
In an effort to address the aforementioned synthesis and assembly challenges, as well as to obtain further insight into their ultrafast excited state dynamics, we designed a simple proof-of-concept porphyrin–polymer conjugate (Zn-dPP-pDMA, Fig. 1a) to exploit the natural solvophobicity-driven self-assembly of amphiphilic systems. This was based on well-optimised meso-functionalised porphyrin synthesis methodologies38–42 and reported polymerisation methods.43 The careful selection of synthetic techniques produced Zn-dPP-pDMA in gram scale quantities. The relatively large scale synthesis, together with a lowered concentration for solvophobicity-induced assembly, produced polymer assemblies in quantities sufficient for condensed phase ultrafast transient electronic absorption spectroscopy (TEAS). Together with static photochemical studies, we demonstrate that the photochemical properties of individual chromophores are retained in these extensively aggregated systems. These experiments fill a gap in our knowledge, serving as an intermediate case study system that bridges the gap between the photochemical studies of simple small bio-molecules and complex macro-biological and biomimetic systems.
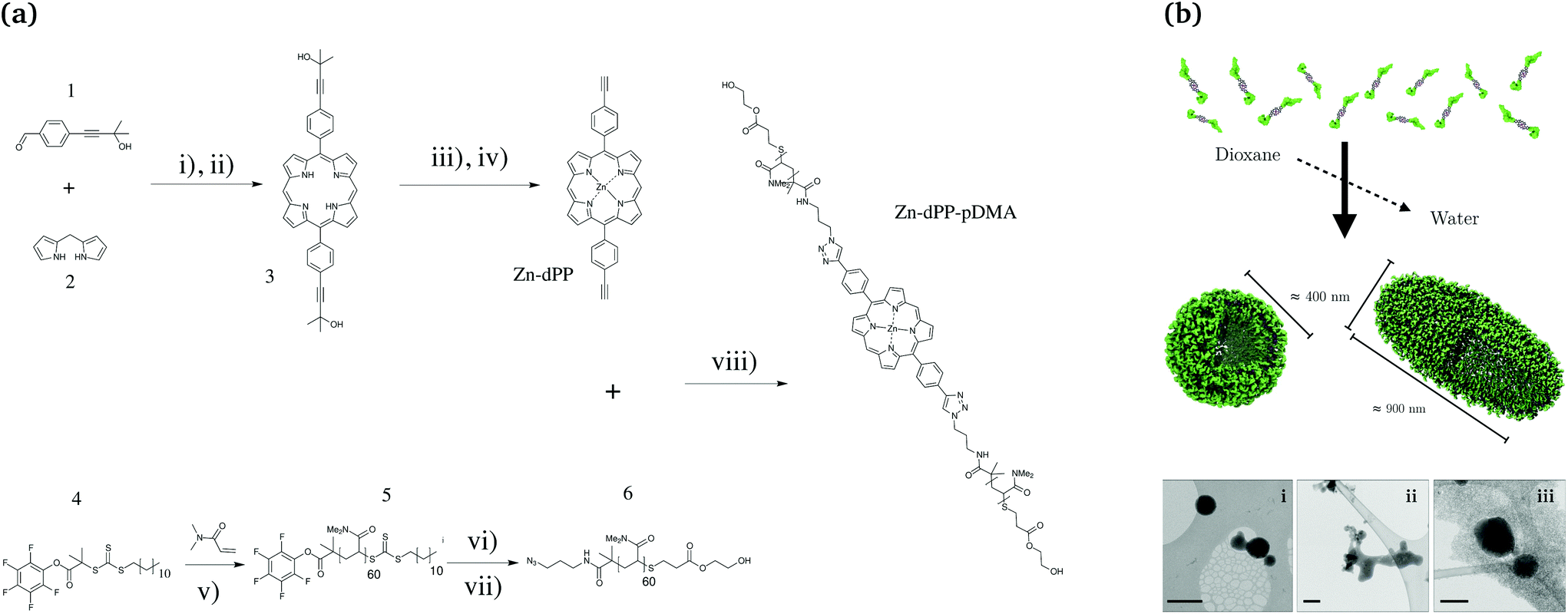 | ||
Fig. 1 (a) Scheme for the synthesis of poly(dimethylacrylamide) functionalised Zn-porphyrin (Zn-dPP-pDMA). (b) Cartoon representation of the assembly, and their visualisation under cryo-TEM (bottom, i–iii, scale bars = 500 nm). Conditions in (a): (i) BF3·Et2O, CH2Cl2, N2, RT, 45 min; (ii) DDQ, toluene, N2, reflux, 3 h; (iii) Zn(OAc)2·(H2O)2, CH2Cl2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :MeOH (8:1), N2, 35 °C, 20 min; (iv) NaOMe, toluene, N2, reflux, 18 h; (v) AIBN, 1,4-dioxane, 65 °C, 40 min (80% conversion); (vi) 3-azidopropan-amine, tetrahydrofuran (THF), N2, RT, 18 h; (vii) HEA, PBu3, N2, THF, RT, 24 h; (viii) Cu·P(OEt)3, dimethylformamide, N2, RT, 48 h. Detailed procedures are provided in the ESI.† :MeOH (8:1), N2, 35 °C, 20 min; (iv) NaOMe, toluene, N2, reflux, 18 h; (v) AIBN, 1,4-dioxane, 65 °C, 40 min (80% conversion); (vi) 3-azidopropan-amine, tetrahydrofuran (THF), N2, RT, 18 h; (vii) HEA, PBu3, N2, THF, RT, 24 h; (viii) Cu·P(OEt)3, dimethylformamide, N2, RT, 48 h. Detailed procedures are provided in the ESI.† | ||
The synthetic scheme for the preparation of Zn-dPP-pDMA is shown in Fig. 1a. All the synthetic techniques employed were based on readily optimised procedures,38,39,41,43 and resulted in respectable to quantitative yields (see ESI,† for further details). The azide-functionalisation and Z-group removal44 of the starting pDMA (5) was performed with an improved one-pot two-step aminolysis method.45 The conjugation of 6 and Zn-5,15-bis(4-ethynylphenyl)-porphyrin (Zn-dPP) via copper-catalysed azide alkyne cycloaddition was completed at room temperature within 48 h. The excess pDMA was easily removed by preparative size-exclusion chromatography (prep-SEC) in dioxane as the conjugated product is strongly coloured; dioxane was then removed effectively by lyophilisation. The resulting Zn-dPP-pDMA was assembled at 3 mg mL−1 (230 μM) by solvent switch from dioxane with slow addition of 18.2 MΩ cm water (see ESI†). Although we expected the formation of small micelles, cryogenic transmission electron microscopy (cryo-TEM) revealed surprisingly large vesicular polymersomes with spherical (Fig. 1b, i) and ellipsoid morphologies (Fig. 1b, iii). The irregular structures observed (Fig. 1b, i and ii) suggested that the assemblies were dynamic and undergoing both fusion and fission processes, similar to other reported polymer-based vesicles.46 Static/dynamic light scattering (SLS/DLS) characterisations at room temperature (RT, 20 °C) identified aggregates with Rg (radius of gyration) ≈ 470 nm and Rh (hydrodynamic radius) ≈ 190 nm (Rg/Rh = 2.4), indicating that the majority of assemblies are ellipsoidal or undergoing the fusion/fission processes at RT.47 To verify that the large assemblies were indeed formed by the Zn-dPP-pDMA, we performed a series of characterisation experiments on samples filtered through membranes of different pore sizes. These studies showed that not only were large amounts of material remaining in the filter, the assemblies also underwent reorganisation, leading to significant change in size upon filtration (see ESI†). Thus, all photochemical experiments of these assemblies were performed with fresh, unfiltered samples, as shown in Fig. 1b.
Despite their extensive aggregation, the spectral features evidenced in the UV-Vis spectrum of Zn-dPP are largely retained in the assembled system (Fig. 2a). However, differences are apparent, which warrant discussion. Firstly, the Soret-band (≈ 414–420 nm, S2 ← S0) and Q-band (≈ 500–625 nm, S1 ← S0) are red-shifted by ca. 5 nm. Secondly, a broadening of the Soret band is evident. Lastly, there is an increase in Q-band relative to Soret-band intensities. These changes closely resemble that of a recently reported Mg(II)bisporphyrin system, in which the Mg⋯Mg non-bonding distance was determined to be ca. 6.5–7.5 Å.48 These observations, taken together with the near identical fluorescence spectra of all the present systems (see ESI†) and the absence of excitonic features, as seen in reported dimers and ordered aggregates,49,50 described by Kasha's exciton theory,51 leads us to propose that while the chromophores are held at close proximity to each other, extensive stacking is prevented, with the chromophores of the aggregates being weakly coupled. This is very likely the result of the repulsive interactions between the polymer chains.19,20
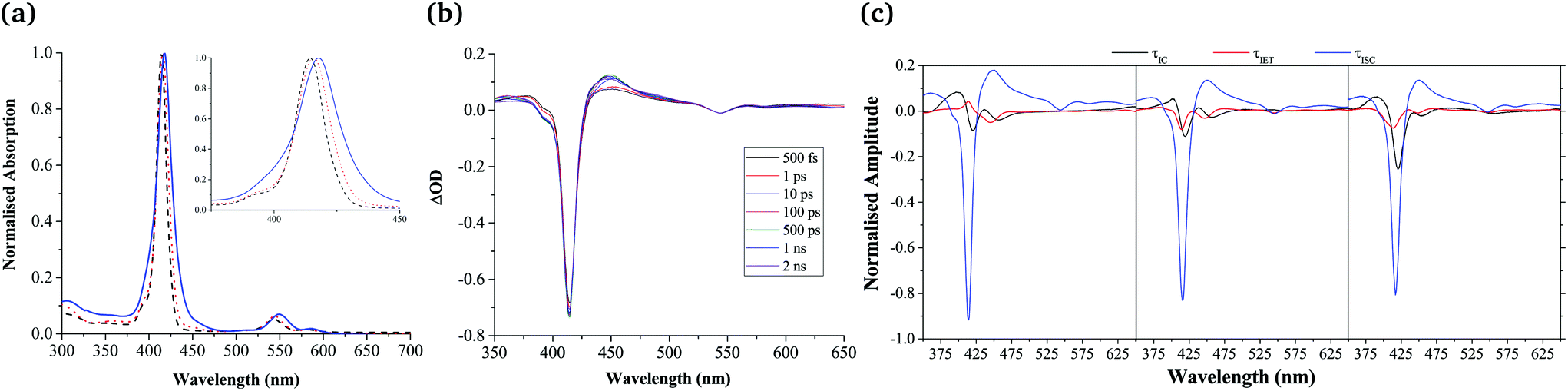 | ||
| Fig. 2 (a) Normalised UV-Vis spectra of Zn-dPP (black, dashed line), Zn-dPP-pDMA unimers in dioxane (red, dotted line) and Zn-dPP-pDMA assembled in water (blue, solid line). Inset shows zoomed-in Soret-band of each system. (b), TAS of Zn-dPP dissolved in dioxane photoexcited at 400 nm (2–5 mJ cm−2, 0.5 mm sample pathlength); ΔOD = change in optical density. (c) DAS of Zn-dPP (left), Zn-dPP-pDMA unimers (middle) fully solvated in dioxane and Zn-dPP-pDMA assembled in water (right). Amplitudes in (c) are normalised such that the sum of amplitudes at 416 nm equals to minus one. | ||
After having established the general chromophore arrangement, we determined the excited state dynamics of the three systems (Zn-dPP, Zn-dPP-pDMA unimers solvated in dioxane; and Zn-dPP-pDMA assembled in 18.2 MΩ cm water), using TEAS following photoexcitation to the S2 state with 400 nm radiation. We first examined Zn-dPP, in which the transient absorption spectra (TAS) show two dominant features: the large ground state bleach (GSB) in the Soret region (ca. 416 nm) and the excited state absorption shoulders (ESA, ca. 450 nm, Fig. 2b). Global fitting the TAS52–54 reveals two ultrafast processes (Table 1): internal conversion (IC) of S2 → S1 (τIC ≈ 1 ps) and intermolecular vibrational energy transfer (IET) between the Zn-dPP S1 excited state and the dioxane solvent bath (τIET ≈ 21.8 ps). These time constants and corresponding processes are comparable to the previously studied model Zn-tetraphenyl-porphyrin (Zn-tPP) reported by Zewail and co-workers (see ESI†).55 We note that in addition to these two extracted time-constants, there is a time-constant that extends beyond the temporal window of our measurements (2 ns) which, in accord with previous studies,55 we attribute to intersystem crossing (ISC) of S1 → Tn (τISC) (see Table 1, footnote a). The shapes of the decay associated spectra (DAS, Fig. 2c) are highly informative in guiding the interpretation of the TAS. In particular, any negative components correspond to an exponential rise in that population whilst any positive components correspond to an exponential decay in population. The flow of excited state populations can be visualised in the DAS when a positive and negative features appear concomitantly. This can be interpreted as either a change in electronic state (IC or ISC), or relaxation within a single electronically excited state (IET).52,56
Remarkably, almost identical features are observed in the TAS of the functionalised systems (Zn-dPP-pDMA unimers in dioxane and assembled in water, see ESI†). Furthermore, these are fitted with almost identical time constants (Table 1) and the DAS (Fig. 2c) revealed no discernible differences in their features. Of these, τIC showed insignificant variations. The slightly faster τIET observed in the assembled system (15.2 ps cf. 20.3 ps in dioxane) suggests that the vibrational frequency match between the Franck–Condon active modes of the photoexcited Zn-tPP and the instantaneous normal modes of its surrounding molecules might be different between each system, as inferred in previous studies.55,57 However, the differences are within the 95% confidence interval of each other (Table 1 and ESI†), which makes this supposition tentative. The final ISC process demonstrated no discernible difference within the window of our experiments (Table 1 and Fig. 2c).
| System studied | τ IC | τ IET | τ ISC |
|---|---|---|---|
| a Due to the very large signal intensities attained at time zero (likely multicomponent in nature and attributed to linear and non-linear solvent-, glass-, and solute-only responses), which extend to ∼150 fs, this signal was excluded from the global fits. | |||
| Zn-dPP dioxane | 1.0 ± 0.3 ps | 21.8 ± 8 ps | ≫2 ns |
| Zn-dPP-pDMA unimers in dioxane | 1.0 ± 0.3 ps | 20.3 ± 8 ps | ≫2 ns |
| Zn-dPP-pDMA assembled in water | 0.8 ± 0.3 ps | 15.2 ± 6 ps | ≫2 ns |
In conclusion, we have presented a study on a basic system of solvated and aggregated porphyrin molecules assembled via solvophobicity. The photodynamic studies presented demonstrate that the individual Zn-dPP molecules retained their overall photochemical properties following the addition of a large polymer chain (pDMA), even following assemblies into macromolecular vesicles. The fact that the addition of such a large polymer has very little effect on the photochemical properties of the porphyrin adds credence to the ‘bottom-up’ approach towards understanding the photochemistry and photophysics of complex biological systems.58–61 Coupled with the relatively high yielding synthetic steps and simple assembly method, these types of polymer–chromophore conjugates could be opportune building blocks for more complex biomimetic systems. We propose that this proof of concept study should facilitate future modular designs of photo-active biomimetic arrays which do not rely on the complex covalent conjugation of multiple chromophores, thereby allowing full exploitation of individual pigment characteristics.
W.D.Q. thanks Dr A. M. Sanchez, Mr Ian Hands-Portman and Miss L. J. MacDougall (UoW) for their help and discussions on TEM and SEM instruments. The research leading to these results has received funding from the ERC under the EU 7th Framework Programme/ERC grant no. SCPs 615142; the EPSRC equipment grant EP/J007153; EPSRC studentship grant EP/F500378/1; and the RSURF scheme.
References
- L. Valkunas, J. Chmeliov, G. Trinkunas, C. D. P. Duffy, R. van Grondelle and A. V. Ruban, J. Phys. Chem. B, 2011, 115, 9252–9260 CrossRef CAS PubMed.
- J. Martiskainen, R. Kananavičius, J. Linnanto, H. Lehtivuori, M. Keränen, V. Aumanen, N. Tkachenko and J. Korppi-Tommola, Photosynth. Res., 2011, 107, 195–207 CrossRef CAS PubMed.
- N. Nelson and W. Junge, Annu. Rev. Biochem., 2015, 84, 659–683 CrossRef CAS PubMed.
- R. L. Leverenz, M. Sutter, A. Wilson, S. Gupta, A. Thurotte, C. Bourcier de Carbon, C. J. Petzold, C. Ralston, F. Perreau, D. Kirilovsky and C. A. Kerfeld, Science, 2015, 348, 1463–1466 CrossRef CAS PubMed.
- M. M. Enriquez, P. Akhtar, C. Zhang, G. Garab, P. H. Lambrev and H.-S. Tan, J. Chem. Phys., 2015, 142, 212432 CrossRef PubMed.
- A. V. Ruban, M. P. Johnson and C. D. P. Duffy, Energy Environ. Sci., 2011, 4, 1643–1650 CAS.
- L.-X. Shi, M. Hall, C. Funk and W. P. Schröder, Biochim. Biophys. Acta, Bioenerg., 2012, 1817, 13–25 CrossRef CAS PubMed.
- Y. Umena, K. Kawakami, J.-R. Shen and N. Kamiya, Nature, 2011, 473, 55–60 CrossRef CAS PubMed.
- R. E. Blankenship, D. M. Tiede, J. Barber, G. W. Brudvig, G. Fleming, M. Ghirardi, M. R. Gunner, W. Junge, D. M. Kramer, A. Melis, T. A. Moore, C. C. Moser, D. G. Nocera, A. J. Nozik, D. R. Ort, W. W. Parson, R. C. Prince and R. T. Sayre, Science, 2011, 332, 805–809 CrossRef CAS PubMed.
- N. Aratani, D. Kim and A. Osuka, Acc. Chem. Res., 2009, 42, 1922–1934 CrossRef CAS PubMed.
- Y. Yamamoto, G. Zhang, W. Jin, T. Fukushima, N. Ishii, A. Saeki, S. Seki, S. Tagawa, T. Minari, K. Tsukagoshi and T. Aida, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 21051–21056 CrossRef CAS PubMed.
- M. C. O'Sullivan, J. K. Sprafke, D. V. Kondratuk, C. Rinfray, T. D. W. Claridge, A. Saywell, M. O. Blunt, J. N. O'Shea, P. H. Beton, M. Malfois and H. L. Anderson, Nature, 2011, 469, 72–75 CrossRef PubMed.
- S. Liu, D. V. Kondratuk, S. A. L. Rousseaux, G. Gil-Ramírez, M. C. O'Sullivan, J. Cremers, T. D. W. Claridge and H. L. Anderson, Angew. Chem., Int. Ed., 2015, 54, 5355–5359 CrossRef CAS PubMed.
- D. V. Kondratuk, L. M. Perdigão, A. M. Esmail, J. N. O'Shea, P. H. Beton and H. L. Anderson, Nat. Chem., 2015, 7, 317–322 CrossRef CAS PubMed.
- F. Gan, S. Zhang, N. C. Rockwell, S. S. Martin, J. C. Lagarias and D. A. Bryant, Science, 2014, 345, 1312–1317 CrossRef CAS PubMed.
- M. Kato, J. Z. Zhang, N. Paul and E. Reisner, Chem. Soc. Rev., 2014, 43, 6485–6497 RSC.
- Y. Mai and A. Eisenberg, Chem. Soc. Rev., 2012, 41, 5969–5985 RSC.
- H.-A. Klok and S. Lecommandoux, Adv. Mater., 2001, 13, 1217–1229 CrossRef CAS.
- A. Blanazs, S. P. Armes and A. J. Ryan, Macromol. Rapid Commun., 2009, 30, 267–277 CrossRef CAS PubMed.
- M. Stefik, S. Guldin, S. Vignolini, U. Wiesner and U. Steiner, Chem. Soc. Rev., 2015, 44, 5076–5091 RSC.
- R. K. O'Reilly, C. J. Hawker and K. L. Wooley, Chem. Soc. Rev., 2006, 35, 1068–1083 RSC.
- F. Li and K. Na, Biomacromolecules, 2011, 12, 1724–1730 CrossRef CAS PubMed.
- L. Xu, L. Liu, F. Liu, H. Cai and W. Zhang, Polym. Chem., 2015, 6, 2945–2954 RSC.
- X.-H. Dai, H. Jin, M.-H. Cai, H. Wang, Z.-P. Zhou, J.-M. Pan, X.-H. Wang, Y.-S. Yan, D.-M. Liu and L. Sun, React. Funct. Polym., 2015, 89, 9–17 CrossRef CAS.
- E. Huynh, J. F. Lovell, B. L. Helfield, M. Jeon, C. Kim, D. E. Goertz, B. C. Wilson and G. Zheng, J. Am. Chem. Soc., 2012, 134, 16464–16467 CrossRef CAS PubMed.
- T. V. Duncan, P. P. Ghoroghchian, I. V. Rubtsov, D. A. Hammer and M. J. Therien, J. Am. Chem. Soc., 2008, 130, 9773–9784 CrossRef CAS PubMed.
- L. R. H. High, S. J. Holder and H. V. Penfold, Macromolecules, 2007, 40, 7157–7165 CrossRef CAS.
- D. A. Roberts, M. J. Crossley and S. Perrier, Polym. Chem., 2014, 5, 4016–4021 RSC.
- D. A. Roberts, T. W. Schmidt, M. J. Crossley and S. Perrier, Chem. – Eur. J., 2013, 19, 12759–12770 CrossRef CAS PubMed.
- D. E. J. G. J. Dolmans, D. Fukumura and R. K. Jain, Nat. Rev. Cancer, 2003, 3, 380–387 CrossRef CAS PubMed.
- H. I. Pass, J. Natl. Cancer Inst., 1993, 85, 443–456 CrossRef CAS PubMed.
- J. M. Anna, G. D. Scholes and R. van Grondelle, BioScience, 2014, 64, 14–25 CrossRef.
- R. Moca, S. R. Meech and I. A. Heisler, J. Phys. Chem. B, 2015, 119, 8623–8630 CrossRef CAS PubMed.
- D. I. G. Bennett, K. Amarnath and G. R. Fleming, J. Am. Chem. Soc., 2013, 135, 9164–9173 CrossRef CAS PubMed.
- H. Liu, H. Zhang, D. M. Niedzwiedzki, M. Prado, G. He, M. L. Gross and R. E. Blankenship, Science, 2013, 342, 1104–1107 CrossRef CAS PubMed.
- M. Ballottari, M. J. P. Alcocer, C. D'Andrea, D. Viola, T. K. Ahn, A. Petrozza, D. Polli, G. R. Fleming, G. Cerullo and R. Bassi, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, E2431–E2438 CrossRef CAS PubMed.
- G. S. Engel, T. R. Calhoun, E. L. Read, T.-K. Ahn, T. Mancal, Y.-C. Cheng, R. E. Blankenship and G. R. Fleming, Nature, 2007, 446, 782–786 CrossRef CAS PubMed.
- B. J. Littler, M. A. Miller, C.-H. Hung, R. W. Wagner, D. F. O'Shea, P. D. Boyle and J. S. Lindsey, J. Org. Chem., 1999, 64, 1391–1396 CrossRef CAS.
- J. K. Laha, S. Dhanalekshmi, M. Taniguchi, A. Ambroise and J. S. Lindsey, Org. Process Res. Dev., 2003, 7, 799–812 CrossRef CAS.
- E. Stulz, S. M. Scott, Y.-F. Ng, A. D. Bond, S. J. Teat, S. L. Darling, N. Feeder and J. K. M. Sanders, Inorg. Chem., 2003, 42, 6564–6574 CrossRef CAS PubMed.
- P. D. Rao, S. Dhanalekshmi, B. J. Littler and J. S. Lindsey, J. Org. Chem., 2000, 65, 7323–7344 CrossRef CAS PubMed.
- M. O. Senge, Chem. Commun., 2011, 47, 1943–1960 RSC.
- T. R. Wilks, J. Bath, J. W. de Vries, J. E. Raymond, A. Herrmann, A. J. Turberfield and R. K. O'Reilly, ACS Nano, 2013, 7, 8561–8572 CrossRef CAS PubMed.
- H. Willcock and R. K. O'Reilly, Polym. Chem., 2010, 1, 149–157 RSC.
- K. E. B. Doncom, C. F. Hansell, P. Theato and R. K. O'Reilly, Polym. Chem., 2012, 3, 3007–3015 RSC.
- D. E. Discher and A. Eisenberg, Science, 2002, 297, 967–973 CrossRef CAS PubMed.
- J. P. Patterson, M. P. Robin, C. Chassenieux, O. Colombani and R. K. O'Reilly, Chem. Soc. Rev., 2014, 43, 2412–2425 RSC.
- S. A. Ikbal, A. Dhamija and S. P. Rath, Chem. Commun., 2015, 51, 14107–14110 RSC.
- I.-W. Hwang, M. Park, T. K. Ahn, Z. S. Yoon, D. M. Ko, D. Kim, F. Ito, Y. Ishibashi, S. R. Khan, Y. Nagasawa, H. Miyasaka, C. Ikeda, R. Takahashi, K. Ogawa, A. Satake and Y. Kobuke, Chem. – Eur. J., 2005, 11, 3753–3761 CrossRef CAS PubMed.
- S. Verma, A. Ghosh, A. Das and H. N. Ghosh, J. Phys. Chem. B, 2010, 114, 8327–8334 CrossRef CAS PubMed.
- M. Kasha, H. Rawls and M. Ashrafel-Bayoumi, Pure Appl. Chem., 1965, 11, 371–392 CrossRef CAS.
- A. S. Chatterley, C. W. West, V. G. Stavros and J. R. R. Verlet, Chem. Sci., 2014, 5, 3963–3975 RSC.
- L. A. Baker, M. D. Horbury, S. E. Greenough, P. M. Coulter, T. N. V. Karsili, G. M. Roberts, A. J. Orr-Ewing, M. N. R. Ashfold and V. G. Stavros, J. Phys. Chem. Lett., 2015, 6, 1363–1368 CrossRef CAS PubMed.
- J. R. Lakowicz, Principles of Fluorescence Spectroscopy, Springer Science+Business Media, 3rd edn, 2006 Search PubMed.
- H.-Z. Yu, J. S. Baskin and A. H. Zewail, J. Phys. Chem. A, 2002, 106, 9845–9854 CrossRef CAS.
- C. R. S. Mooney, D. A. Horke, A. S. Chatterley, A. Simperler, H. H. Fielding and J. R. R. Verlet, Chem. Sci., 2013, 4, 921–927 RSC.
- R. M. Stratt and M. Maroncelli, J. Phys. Chem., 1996, 100, 12981–12996 CrossRef CAS.
- G. M. Roberts and V. G. Stavros, Chem. Sci., 2014, 5, 1698–1722 RSC.
- J. R. R. Verlet, Chem. Soc. Rev., 2008, 37, 505–517 RSC.
- S. J. Harris, D. Murdock, Y. Zhang, T. A. A. Oliver, M. P. Grubb, A. J. Orr-Ewing, G. M. Greetham, I. P. Clark, M. Towrie, S. E. Bradforth and M. N. R. Ashfold, Phys. Chem. Chem. Phys., 2013, 15, 6567–6582 RSC.
- P.-Y. Cheng, J. S. Baskin and A. H. Zewail, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 10570–10576 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5cc09095d |
| This journal is © The Royal Society of Chemistry 2016 |