
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Highly efficient stabilisation of meta-ethynylpyridine polymers with amide side chains in water by coordination of rare-earth metals†
Hiroki
Makida
,
Hajime
Abe
* and
Masahiko
Inouye
*
Graduate School of Pharmaceutical Sciences, University of Toyama, Toyama 930-0194, Japan. E-mail: abeh@pha.u-toyama.ac.jp
First published on 4th December 2014
Abstract
An amphiphilic meta-ethynylpyridine polymer with chiral amide side chains was developed. The polymer was prepared by sequential Sonogashira reactions, and the product was soluble in polar and apolar solvents. The additive effects of metal salts on the polymer were examined in water and aqueous EtOH on the basis of UV-vis and CD spectra. The enhancement of the positive Cotton effect and hypochromism around 360 nm occurred by the addition of various metal salts, indicating the coordination of the cations to the amide side chains of the polymer to stabilise the helical structure. Among them, rare-earth metal salts, especially Sc(OTf)3 showed more efficient additive effects probably because of its strong coordination ability even in water. Positive cooperativity was observed for the coordination of Sc(OTf)3 to the polymer in aqueous EtOH.
Introduction
Chiral helical structures are seen in many kinds of biomacromolecules such as proteins, nucleic acids, and polysaccharides. To mimic such chiral helical strucures, a variety of artificial helical polymers and oligomers, also called as foldamers, have been developed.1 Since Moore and coworkers reported the sophisticated example of meta-phenylene ethynylenes in 1997, solvophobic interactions have been widely used as a driving force for the construction of helical structures of artificial foldamers.2–4We have developed meta-ethynylpyridine polymers and oligomers, in which a number of pyridine rings are linked with acetylene bonds at their 2 and 6 positions.5,6 These polymers and oligomers prefer to form transoid conformations under apolar conditions to suppress the repulsion caused by dipole moments at pyridine rings. On the other hand, spontaneous formation of a higher-order helical structure has been achieved under aqueous conditions by using amphiphilic meta-ethynylpyridine polymers with oligoethylene glycol side chains.6 Indeed, amphiphilic (R)- and (S)-1 with chiral centres could form circular dichroism (CD)-active biased helices (Fig. 1). Those helix formations were driven by solvophobic interaction, and the CD activity was found to be enhanced by the addition of a large excess amount of metal salts such as NaClO4 and Ca(ClO4)2.
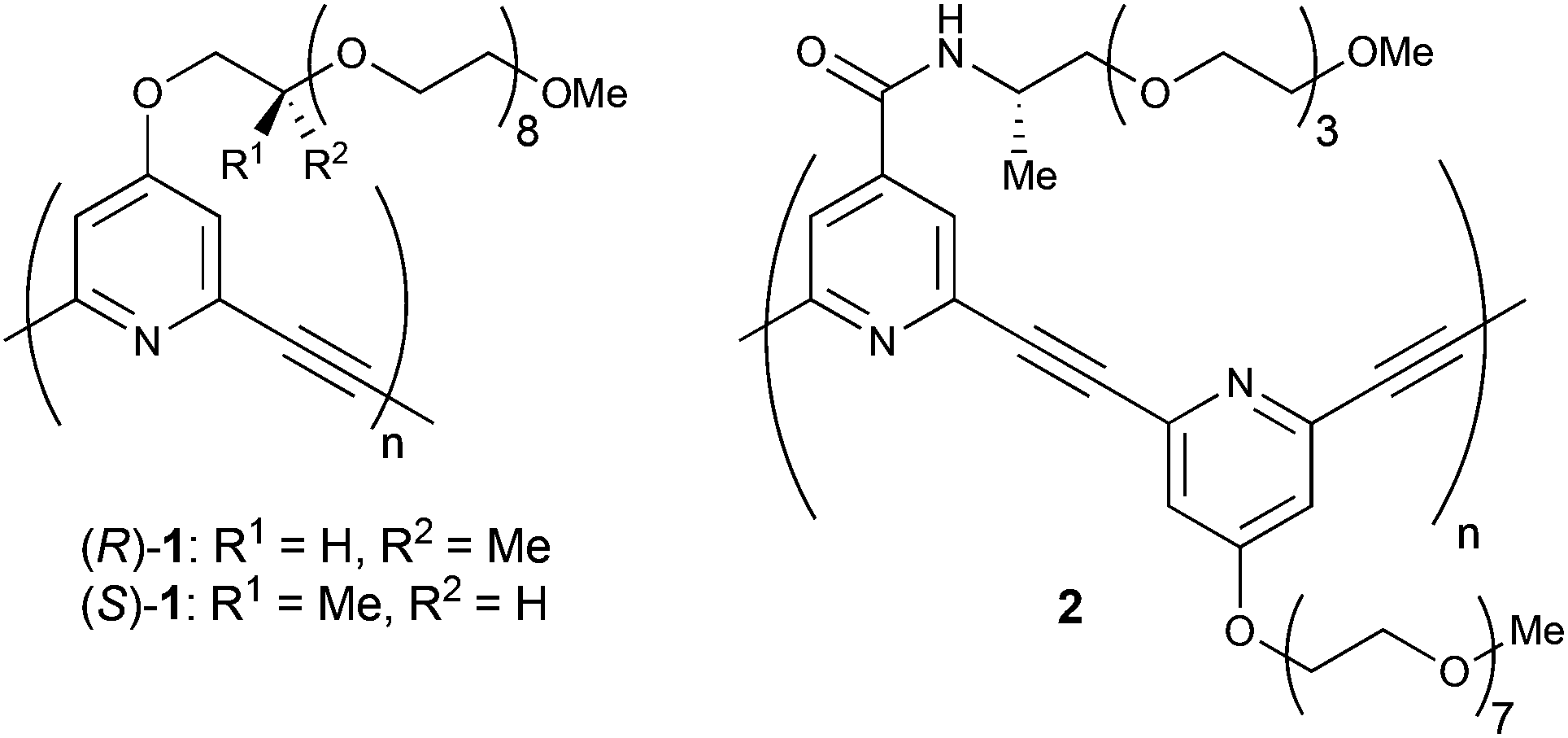 | ||
| Fig. 1 Chemical structures of previously reported (R)- and (S)-1, and newly designed chiral polymer 2. | ||
Herein, we report a new type of amphiphilic meta-ethynylpyridine polymer 2, poly(4-(((S)-1-methyl-3,6,9,12-tetraoxatridecyl)aminocarbonyl)-2,6-pyridylene ethynylene 4-(1,4,7,10,13,16,19,22-octaoxatricosanyl)-2,6-pyridylene ethynylene), in which chiral amide side chains were introduced as a coordination site for metal cations (Fig. 1). The polymer formed chiral helical structures not only in polar solvents such as H2O, MeOH, THF, and MeCN, but also in CH2Cl2. The chiral helical structure of the polymer can be stabilised by the addition of various kinds of metal salts, and in particular rare-earth metal salts were so efficient that only an equimolar amount of salts was needed even in H2O. The data of IR and 1H NMR analyses indicated that the coordination of the rare-earth cations to the amide carbonyl groups of 2 led to the efficient stabilisation of the helix.
Results and discussion
Synthesis of amide-introduced chiral polymer 2
Chiral polymer 2 was synthesised as shown in Scheme 1. The nucleophilic replacement of the nitro group of 2,6-dibromo-4-nitropyridine (3) with sodium monomethyl heptaethylene glycolate gave ether 4, which was converted to diiodide 5 by a copper-catalysed halogen exchange reaction.7 The building block 9 for the chiral side chain of 2 was prepared by assembling N-Boc-L-alaninol (6) and 10-iodo-2,5,8-trioxadecane (7).8 Condensation of 6 and the iodinated ether 7 gave Boc-protected amphiphilic chiral amine 8, and then the Boc group was removed to yield chiral amine 9. Hydrolysis of methyl 2,6-dibromoisonicotinate (10)9 with LiOH gave 2,6-dibromoisonicotinic acid (11), which was condensed with 9 to form chiral amide 12. Two bromine atoms of 12 were converted to ethynyl groups to give 14via TMS-protected intermediate 13 by the Sonogashira reaction with trimethylsilylacetylene (TMSA) followed by protiodesilylation. Trimeric block 15 was obtained by the Sonogashira reaction using 14 and an excess amount of diiodide 5. Finally, co-polymerisation of 14 and 15 furnished the target polymer 2 by applying the Sonogashira reaction under aqueous conditions.10 The crude product was purified by centrifugation, and the product was subjected to preparative gel permeation chromatography (GPC) using CHCl3 as the eluent to collect a fraction of relatively longer size. It was found that 2 was soluble in a wide range of solvents, such as CH2Cl2, CHCl3, THF, MeCN, MeOH, EtOH, and H2O. The number-average molecular weight (Mn) and dispersity (Mw/Mn) of each of the fractions were determined by analytical GPC using a DMF solution of LiBr (0.1 M) as the eluent. In the following studies, fractions from four lots (Mn = 1.2 × 104, 1.9 × 104, 5.3 × 104 and 5.6 × 104 g mol−1, see Table S1 in ESI†) were applied as a substrate. Among these fractions, no meaningful size-dependence could be noticed.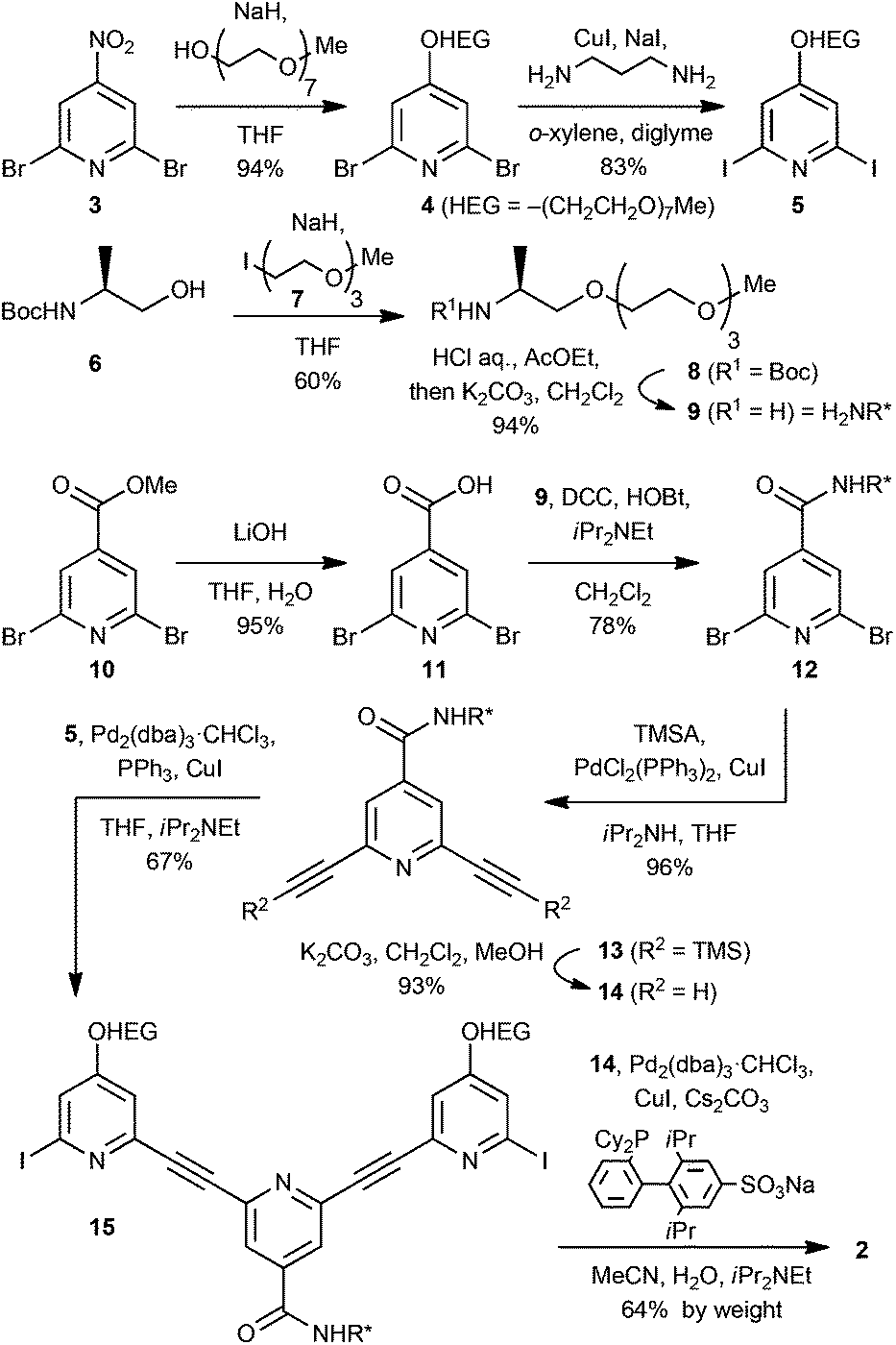 | ||
| Scheme 1 Preparation of chiral polymer 2. DCC = N,N′-dicyclohexylcarbodiimide, HOBt = 1-hydroxybenzotriazole, TMSA = trimethylsilylacetylene, dba = dibenzylideneacetone. | ||
Solvent effects on the chiral polymer 2
In advance, the concentration dependence of the polymer 2 was studied in MeCN and in H2O by UV-vis measurements. The results were summarised and displayed in Fig. S1 and S2 in ESI,† and the intermolecular interaction of 2 was considered to be negligible because the absorbance of 2 in MeCN (unit conc = 7.8 × 10−6 M to 3.6 × 10−3 M) and that in H2O (unit conc = 7.8 × 10−6 M to 4.0 × 10−3 M) were almost in proportion to the unit concentrations of 2.As mentioned above, the polymer 2 was expected to spontaneously form a chiral helical structure in H2O by hydrophobic interaction stabilising π-stacking as well as the precedent examples of helical foldamers reported by us6 and other groups.3,4 Here, solvent effects on the polymer 2 were investigated on the basis of UV-vis and CD spectra to collect information about the higher-order structures without metal salts (Fig. 2). In CH2Cl2, THF, MeCN, and MeOH, the first absorption bands were observed around 330 nm, while the remarkably red-shifted one was observed at λmax = 340 nm in H2O (Fig. 2a). In our previous study on amphiphilic ethynylpyridine polymers 1, a similar type of red shift of the absorption band was seen in H2O attributed to hydrophobic π-stacking interactions.6 Therefore, the red shift for 2 would also be caused by intramolecular π-stacking interaction among pyridine rings at an interval of one pitch in the helical structure.
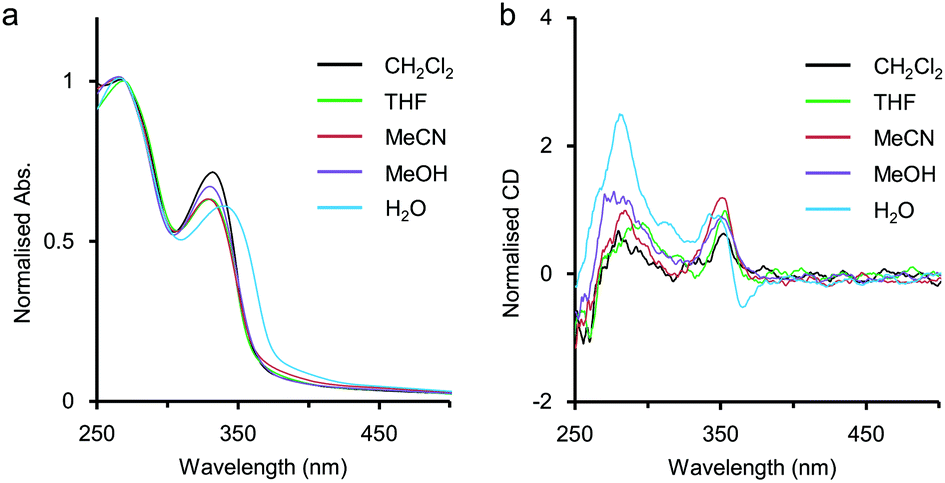 | ||
| Fig. 2 Solvent effects on (a) UV-vis and (b) CD spectra of 2. Conditions: 2 (Mn = 1.9 × 104 g mol−1, Mw = 2.7 × 104 g mol−1), 25 °C, path length = 1 mm. UV spectra were normalised at 270 nm. CD spectra were normalised as that the absorbance at 270 nm is 1.0. | ||
The polymer 2 showed a positive Cotton effect around 350 nm in CH2Cl2, THF, MeCN, and MeOH (Fig. 2b). In our previous study on chiral ethynylpyridine polymers and oligomers such as 1,6a no meaningful CD activity had been observed in less polar solvents such as CH2Cl2 and CHCl3. This was because the solvophobic intramolecular interaction was too weak to form a helix in less polar environments. On the other hand, 2 showed a weak but clearly perceivable CD activity around 350 nm even in CH2Cl2, indicating that some kind of intramolecular interaction other than the solvophobic one would work to stabilise the helical structure. The major structural difference of 2 against 1 is the presence of amide groups in the side chains. The hydrogen bonds between the amide groups in the side chains might stabilise the CD-active chiral helical structure. Recently, Sanda reported poly(meta-phenylene ethynylene)s containing hydrophobic chiral amino acid side chains,11 and Yashima reported poly(meta-phenylene ethynylene)s bearing hydrophilic L- or D-alanine-derived oligo(ethylene glycol) side chains.12 These polymers were reported to form chiral helical structures in less polar solvents such as CH2Cl2 and CHCl3, driven by the intramolecular hydrogen-bonding network between the amide groups of the side chains. In H2O, the shape of the CD spectrum of 2 was rather different from that of CD spectra in other solvents (Fig. 2b). Actually, a bisignate CD signal was observed at a range of about 330 nm to 380 nm, so that the polymer 2 would form a different kind of higher-order structure in H2O.
Additive effects of main-group metal cations for the polymer 2
Metal salts such as NaClO4 and Ca(ClO4)2 were found to stabilise the chiral helical structures of our previous (S)-1 and (R)-1 accompanied by displaying strong CD bands in H2O.5a,b,d,f,g,6a This stabilisation of the helical structure of 1 might be caused by complexation of metal cations at ethylene glycol side chains and/or at the nitrogen atoms of pyridine rings inside the helix. Herein the additive effect of main-group metal salts was also studied for aqueous solutions of the polymer 2 by UV-vis and CD measurements, and the data were compared to those in the cases of 1. As shown in Fig. 3a, the addition of a large excess amount of metal salts induced hypochromic effects on the first and second absorption bands around 340 nm and 265 nm, respectively. It has been reported that when foldamers form helical structures by hydrophobic interaction, hypochromism is often observed in the absorption spectra. The hypochromic changes shown in Fig. 3a would indicate that the addition of metal salts improved the hydrophobic π-stacking interaction to stabilise the chiral helical structure of 2. The coordination manner of the metal cations would be speculated to the following three possible complexation patterns among the functional groups in 2: (i) for ethylene glycol side chains; (ii) for amide carbonyl groups in the side chains; and (iii) for nitrogen of the pyridine ring inside the helix. We will discuss these possibilities on the basis of IR and 1H NMR studies later.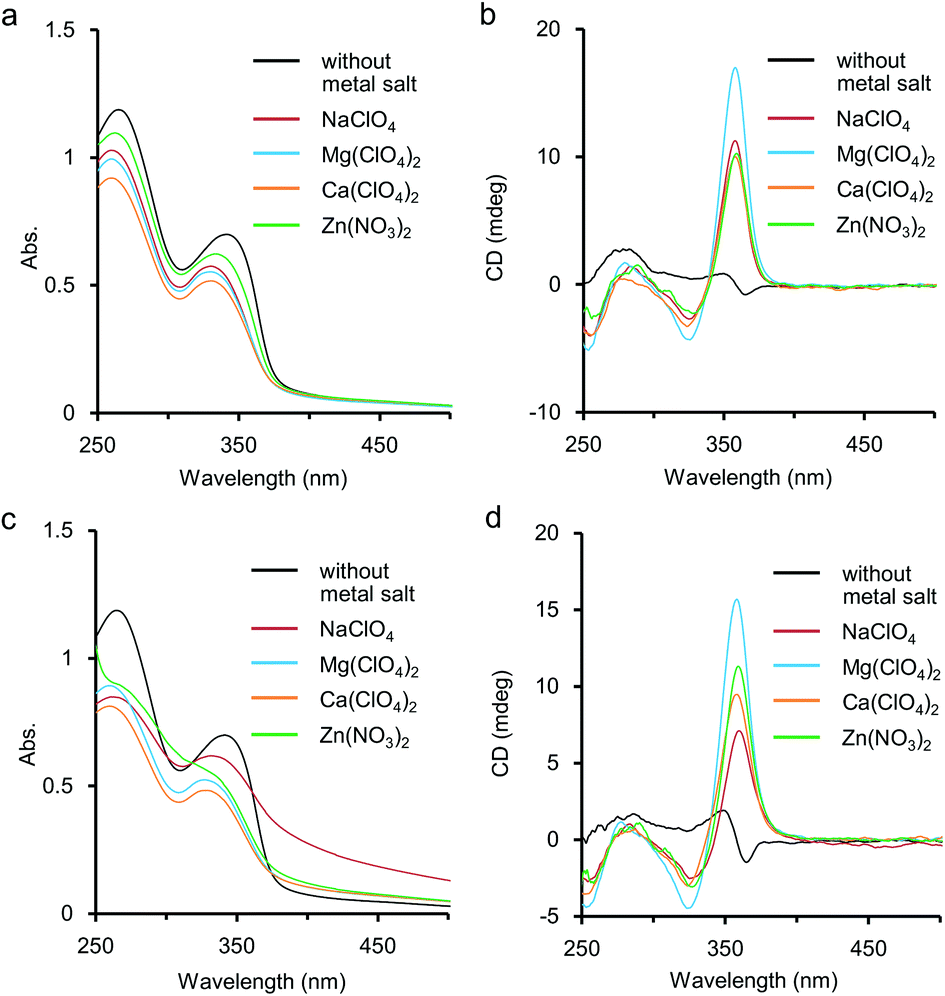 | ||
| Fig. 3 Additive effects of metal cations on aqueous solutions of 2. (a) UV-vis spectra at 25 °C. (b) CD spectra at 25 °C. (c) UV-vis spectra at 85 °C. (d) CD spectra at 85 °C. Conditions: [2] = 1.0 × 10−3 M (Mn = 1.2 × 104 g mol−1, Mw = 1.4 × 104 g mol−1, unit conc), [metal salt] = 1.0 M, H2O, path length = 1 mm. | ||
For CD, an intense positive CD band appeared around 360 nm after the addition of the metal salts, and the shapes of the CD spectra were typical for ethynylpyridine polymers and oligomers of chiral helical conformation (Fig. 3b).5,6 The thermal resistance about the chiral helical complexes of 2 and metal cations (Fig. 3c and 3d) was studied. When an aqueous solution containing 2 (1.0 × 10−3 M, unit conc) and Mg(ClO4)2 or Ca(ClO4)2 (1.0 M) was heated to 85 °C, the CD band around 360 nm was weakened only to some extent, and hypochromism was observed in the UV-vis spectrum. These changes suggested that the folded chiral helical structures of 2 were largely preserved even by heating up to subsequently high temperature. Without metal salts, enhancement of the CD band for 2 occurred by heating, accompanied by a small hypochromism (see also Fig. S3 in ESI†). For Zn(NO3)2 at 85 °C, the first CD band was slightly enhanced than at 25 °C, and the fine structures of absorption bands were almost lost. At that temperature the presence of NaClO4 (1.0 M) brought about remarkable tailing to over 700 nm as shown in Fig. 3c. This tailing would be due to scattering by aggregate of 2 mediated by the Na(I) cation. Indeed, by heating at 85 °C, clouding and precipitation subsequently occurred over time for the solution of 2 and NaClO4. Aggregation by heating has been reported not only for poly- and oligoethylene glycol13 but also for their derivatives,14e.g. oligoethylene glycol-introduced shape-persistent macrocycles.
Additive effects of rare-earth metal salts for the polymer 2
As mentioned above, although the addition of metal salts to 2 induced strong CD enhancement, a large excess amount of metal salts were needed to observe the phenomena. We then studied the additive effect of rare-earth metal salts, expecting that the multivalent coordination ability of the metal cations would stabilise the helical structure efficiently. Those salts have been known for their Lewis acidity compatible with aqueous environments and utilised as a potent catalyst for organic syntheses in aqueous media.15 To study the efficiency of rare-earth metal additives on the formation of a higher-order structure of 2, UV-vis and CD measurements of solutions of 2 (1.0 × 10−3 M, unit concentration) were carried out in the presence of an equimolar amount of rare-earth, main group, and transition metal salts (1.0 × 10−3 M) (Fig. 4). Under these experimental conditions, time-dependence was seen, so that the spectra were uniformly measured after 2 h from the preparation of the sample solutions (see below for the time-dependence issue). As shown in Fig. 4b, the addition of an equimolar amount of NaClO4 brought little CD change, and for Mg(OTf)2, Mg(ClO4)2, Cu(OTf)2, and Ni(OTf)2, only small changes of CD were observed. In the case that Sc(OTf)3, Y(OTf)3, Ce(OTf)3, Yb(OTf)3, or EuCl3 was present, hypochromism occurred for the absorption around 350 nm, compared to that in the absence of metal salts (Fig. 4a). It is noteworthy that the addition of rare-earth metal salts induced stronger positive CD bands around 360 nm than those induced by the addition of main group and transition metal salts (Fig. 4b). The use of a large excess amount of the rare-metal salts over the unit concentration of 2 was not necessary here, probably because of the strong Lewis acidity of the rare-earth metal salts even in H2O. Judging from the CD spectra in Fig. 4b, the extent of the additive effect to enhance CD varied with the kind of salt. The order of the efficiency of the additives was Sc(OTf)3 ≫ Yb(OTf)3 > Y(OTf)3 > Ce(OTf)3 > EuCl3. Recently, Imamoto and coworkers investigated the relative Lewis acidity of triflates of Sc(III), Y(III), lanthanide(III) (except Pm) by tandem mass spectrometry.16 From their reports, the order of Lewis acidity of the rare-earth metals is Sc(III) ≫ Yb(III) > Y(III) > Eu(III) ≫ Ce(III). For triflate additives, this order by Imamoto's group agrees well with the order of the CD enhancement efficiency shown in Fig. 4b. Therefore, the Lewis acidity or coordination ability was actually important to stabilise the chiral helical structure of 2. The position of EuCl3 did not match in the two orders because the chloride anions would have an effect on the coordination. The additive effects of Sc(NO3)3 and TfOH were also studied as control experiments. The addition of an equimolar amount of Sc(NO3)3 enhanced the CD of 2 similarly to Sc(OTf)3 (Fig. S4 in ESI†), showing the effectiveness of the Sc(III) cation. On the other hand, the addition of an equimolar amount of TfOH showed little change in the CD spectrum (Fig. S5 in ESI†), while an excess amount of TfOH (1.0 × 10−1 M, 100 equiv. to unit conc of 2) brought strong CD enhancement in the cases shown in Fig. 3. Thus, the strong Brønsted acidity could affect the polymer, however not so much as in the cases of the coordination with lanthanide salts.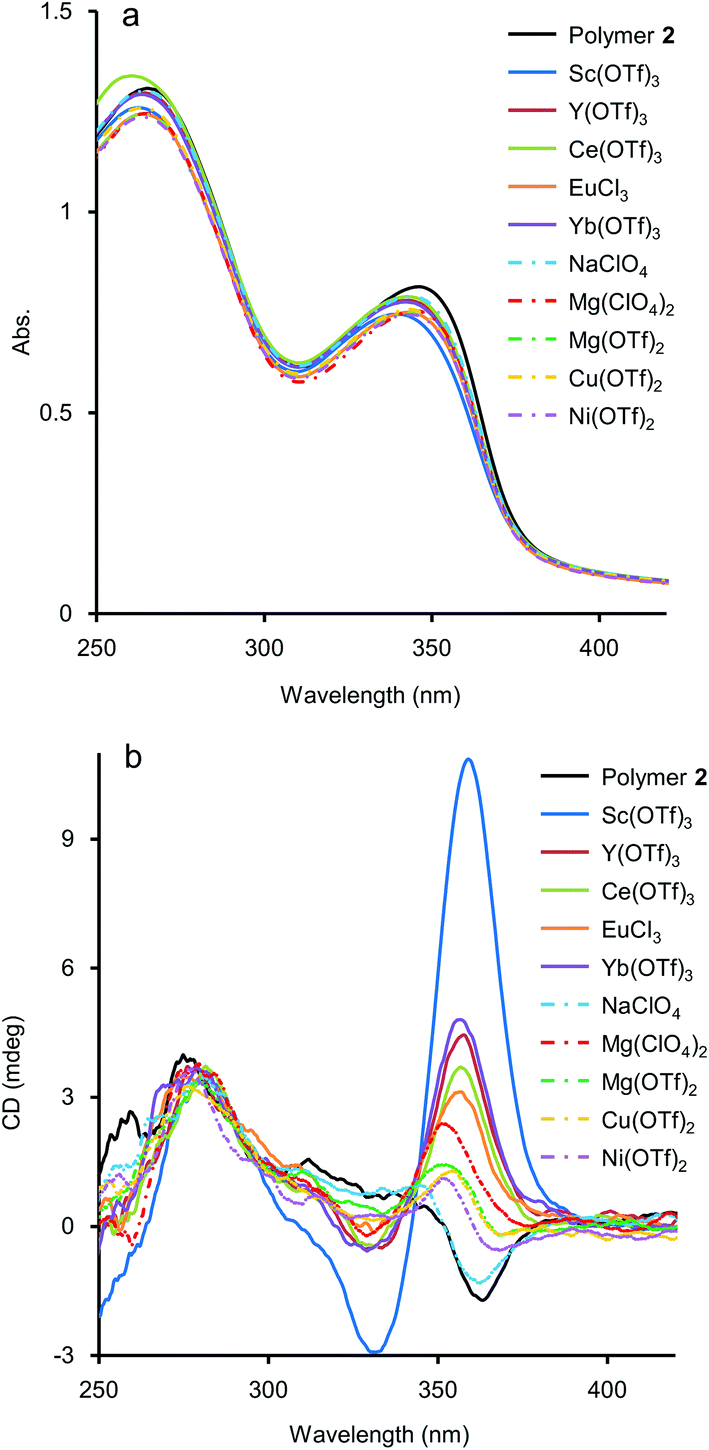 | ||
| Fig. 4 Additive effects of an equimolar amount of metal salts in aqueous solutions on (a) UV-vis and (b) CD spectra. Conditions: [2] = 1.0 × 10−3 M (Mn = 5.6 × 104 g mol−1, Mw = 9.8 × 104 g mol−1, unit conc), [metal salts] = 1.0 × 10−3 M, H2O, 25 °C, path length = 1 mm. These spectra were uniformly measured after 2 h from the preparation of the sample solutions (see text). | ||
Time-dependence was observed in these experiments using an equimolar amount of metal salts. After the addition of the salts at 25 °C, the positive CD band in 2 around 360 nm grew up gradually and became static within a few days as shown in Fig. S6 in ESI.† In the cases using a large excess amount of additive salts (Fig. 3), time-dependence was negligible and the CD spectra became static within one hour (Fig. S7 in ESI†). Instead of waiting for several hours, the equilibrium state could be reached by annealing the mixture of 2 and the metal salts at 50 °C for 10 min. A CD titration experiment was performed for 2 with Sc(OTf)3 as a titrant with this annealing process to obtain a titration curve (Fig. 5), which could be fitted well with a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 binding isotherm, and the apparent binding constant17 was calculated as K′a = 3.4 × 103 M−1.
:1 binding isotherm, and the apparent binding constant17 was calculated as K′a = 3.4 × 103 M−1.
 | ||
| Fig. 5 (a) Changes of the CD spectrum of 2 during the titration with Sc(OTf)3 in H2O. (b) Titration curve of the changes of the CD at 360 nm (the range of [Sc(OTf)3] = 0 to 3.0 × 10−3 M is shown). Fitted solid line is drawn according to 1:1 binding isotherm. Conditions: [2] = 1.0 × 10−3 M (Mn = 5.3 × 104 g mol−1, Mw = 1.0 × 105 g mol−1, unit conc), [Sc(OTf)3] = 0 to 5.0 × 10−2 M, H2O, 25 °C, path length = 1 mm. | ||
Interestingly, in aqueous EtOH, a positive cooperative effect was observed for the coordination of Sc(III) to 2. UV-vis and CD measurements were carried out for an aqueous EtOH solution of 2 (1.0 × 10−3 M, unit conc, EtOH–H2O = 9:1) in the absence or presence of an equimolar amount of rare-earth metal salts (1.0 × 10−3 M). Unlike in H2O alone, the time-dependence was not so troublesome in aqueous EtOH that an equilibrium state could be reached within 2 h. Rapid molecular motion of 2 in aqueous EtOH might cause the shortening of the equilibrium period. The results are shown in Fig. S8 in ESI,† and the addition of rare-earth metal salts enhanced the positive CD band around 360 nm efficiently. The order of the efficiency of the additives was Sc(OTf)3 ≫ Yb(OTf)3 > Y(OTf)3 ≫ EuCl3 ≈ Ce(OTf)3, matching with that observed in Fig. 4b except EuCl3.
When titration experiments were carried out by using Sc(OTf)3 in an aqueous EtOH solution, the CD intensity at 360 nm was monitored (Fig. 6a). From the titration curve based on the change, we first attempted curve-fitting analyses based on the assumptions of 1:1 and 1:2 models. However, theoretical curves from such models did not fit sufficiently to the experimental titration curve (Fig. 6b). Because the polymer 2 possesses many binding sites in its side chain to coordinate with Sc(III), it is reasonable to assume that the initial complexation between 2 and Sc(III) may assist the further additional complexation by the conformational change of the ethynylpyridine main chain backbone. This interpretation means that the complexation may occur cooperatively among Sc(III) and the binding sites in the side chains of 2. Based on this hypothesis, we tried to study the binding manner by using the Hill equation (eqn (1)).18
| log(y/(1 − y)) = nlog[Sc(OTf)3] + nlogK′a | (1) |
 | ||
| Fig. 6 (a) Changes of CD spectrum of 2 during the titration with Sc(OTf)3 in aqueous EtOH. (b) Titration curve of the changes of the CD at 360 nm. (c) Hill plot of the data obtained from the titration curve. Fitted solid line is drawn according to the Hill equation (eqn (1)). Conditions: [2] = 1.0 × 10−3 M (Mn = 5.3 × 104 g mol−1, Mw = 1.0 × 105 g mol−1, unit conc), [Sc(OTf)3] = 0 to 5.0 × 10−2 M, EtOH–H2O = 9:1, 25 °C, path length = 1 mm. | ||
The apparent association binding constant K′a and the Hill coefficient n were estimated to be 1.7 × 104 M−1 and 1.3, respectively, from the least-square fitting shown in Fig. 6c.17 The estimated n value greater than 1 indicated that the binding was positively cooperative. That is, when one Sc(III) cation binds with 2, the next Sc(III) cation binds with 2 more strongly. Thus, the positive allosterism operated in the process of the complexation of Sc(III) on 2 in aqueous EtOH.
The cooperativity observed here could be rationalised as follows: since rare-earth metal cations have multivalent coordination ability, they can favourably coordinate with two or more side chains of the polymer 2. So the cations are able to be a bridge between the side chains. It could be meaningless if such a bridge was thrown between the side chains of the adjacent two pyridine units, however, the distance between the units was kept by the rigidity of acetylene bonds. Therefore, the coordination bridge would be thrown between the side chains of the pyridine units with a separation of one helix pitch, stabilising the helical conformation. After one bridge was built, a subsequent bridging would become favourable, because other pyridine units also get nearer one another to show the positive allosterism.
IR and 1H NMR studies for the complexation
The spectral change of IR absorption caused by the addition of Sc(OTf)3 was studied to explore which functional group of 2 works as a coordination site. The samples for IR measurements were prepared by drop-casting CHCl3 solutions of the polymer 2 with or without Sc(OTf)3 on an NaCl plate. In the absence of Sc(OTf)3, the polymer 2 showed IR absorption bands at 1654 cm−1 for the amide carbonyl group and at 1541 cm−1 for the pyridine rings (Fig. 7, blue). The presence of Sc(OTf)3 made the band for the amide carbonyl group shifted to 1635 cm−1, whereas the band for the pyridine rings was observed at 1541 cm−1 without a meaningful shift (Fig. 7, red). This finding suggests that the Sc(III) cation prefers to coordinate with the amide group in the polymer 2.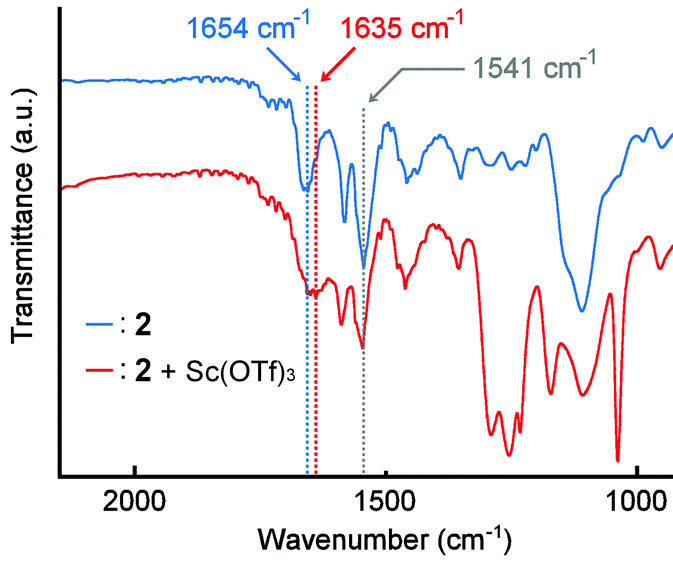 | ||
| Fig. 7 IR spectra of polymer 2 (blue) without Sc(OTf)3 and (red) with Sc(OTf)3 (0.5 eq. to unit conc of 2) casted on an NaCl plate. 2 (Mn = 5.3 × 104 g mol−1, Mw = 1.0 × 105 g mol−1), NaCl. | ||
The chemoselectivity of the coordination of Sc(III) on 2 was further studied by 1H NMR measurements using diacetylenic 14 (Fig. 8) as a model compound, showing a signal for the amide N–H proton at 7.35 ppm in CDCl3–DMSO-d6 = 300:1. In the presence of 0.5 × 10−3 and 1.0 × 10−3 M of Sc(OTf)3, the chemical shift of the signal was observed at 7.36 and 7.38 ppm, respectively, with a small downfield shift (Fig. 8). On the other hand, the chemical shifts for the C–H protons at ethynyl groups, the pyridine ring, and the ethylene glycol side chain were kept constant by the addition of Sc(OTf)3. These IR and 1H NMR experiments indicate that the Sc(III) cation tends to interact with the amide groups, probably carbonyl oxygen atoms, of the polymer 2. Therefore, the chiral centres nearby the amide groups biased helical sense of the complex to induce strong CDs.
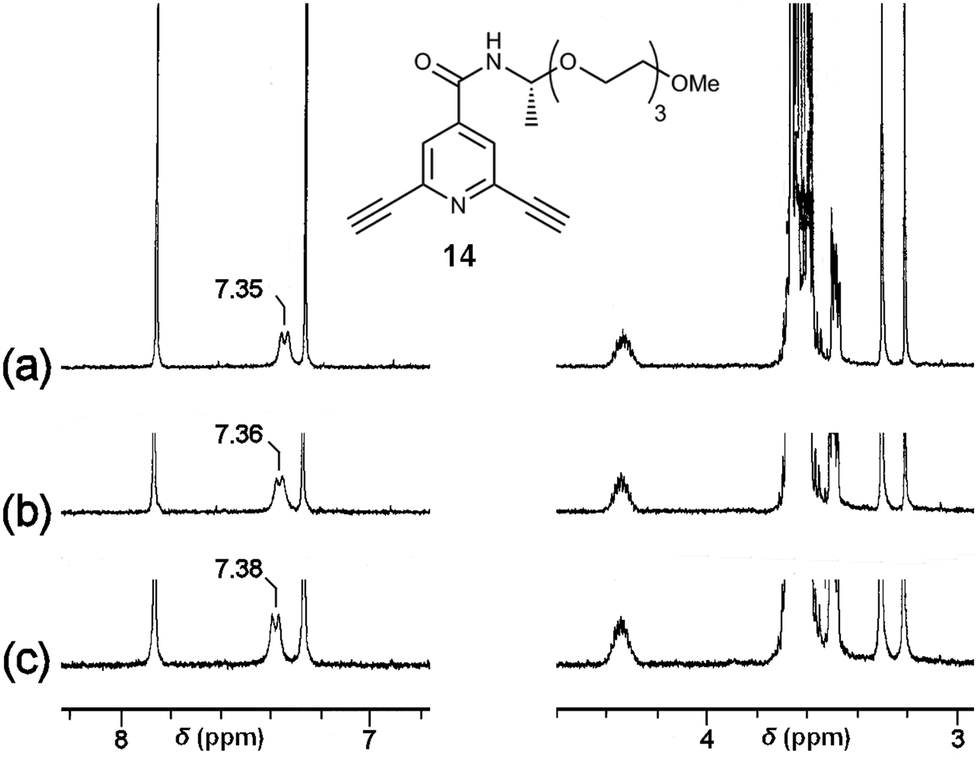 | ||
| Fig. 8 The additive effect of Sc(OTf)3 on 1H NMR (300 MHz) spectrum of diacetylene 14. The chemical shifts for the amide proton shifted downfield by the addition of Sc(OTf)3. (a) 14 (1.0 × 10−2 M); (b) 14 (1.0 × 10−2 M) + Sc(OTf)3 (5.0 × 10−4 M); (c) 14 (1.0 × 10−2 M) + Sc(OTf)3 (1.0 × 10−3 M). Conditions: 300 MHz, CDCl3–DMSO-d6 = 300:1, 23 °C. | ||
Conclusions
We have prepared a new type of meta-ethynylpyridine polymer 2 with amphiphilic amide side chains having chiral centres. The polymer 2 was soluble in a variety of solvents and showed a weak CD by itself. When the additive effects of metal salts on 2 were examined in water, several kinds of salts were found to enhance the positive CD band around 360 nm with hypochromism in UV-vis spectra. The additive effects of rare-earth metal salts, especially Sc(OTf)3 were noticeable, so that only an equimolar amount of Sc(OTf)3 was almost sufficient to bring about the maximum enhancement. These strong CD enhancements and UV-vis hypochromism would be due to the stabilisation of the helical structure of 2 by the coordination of Sc(III) to the amide carbonyl groups of the side chains in the polymer. The titration experiment and Hill analyses of an aqueous EtOH solution of 2 with Sc(OTf)3 revealed that the cation coordinates with 2 and enhances its CD in a cooperative manner.Experimental
Preparation of chiral amide-introduced polymer 2 (typical procedure for synthesis of the polymer 2)
To a solution of 14 (11 mg, 30 μmol) and 15 (35 mg, 24 μmol) in MeCN (1.2 mL) and H2O (0.6 mL) was added a mixture of Pd2(dba)3·CHCl3 (3.1 mg, 3.0 μmol), sodium 2′-(dicyclohexylphosphino)-2,6-diisopropylbiphenyl-4-sulphonate19 (6.4 mg, 12 μmol), CuI (0.58 mg, 3.0 μmol), and Cs2CO3 (47 mg, 0.14 mmol) in MeCN (0.1 mL), H2O (0.1 mL), and iPr2NEt (0.2 mL). This reaction mixture was stirred for 3 days at 30 °C and treated with 3-aminopropyl-functionalised silica gel (9.0 mg). The resulting suspension was filtered, and the filtrate was concentrated by a rotary evaporator. The residue was treated with a Sephadex LH-20 column by using CHCl3 as the eluent. The resulting CHCl3 solution of crude 2 was diluted with CHCl3 up to 40 mL, washed with distilled H2O (20 mL × 2), and concentrated by a rotary evaporator. The resulting residue was dissolved in distilled H2O (12 mL), and the aqueous solution was washed with hexane (15 mL), Et2O (15 mL), and hexane–AcOEt = 3:1 (24 mL) successively. The combined organic layer was concentrated by a rotary evaporator to give a brown viscous oil. This viscous oil was again dissolved in CHCl3 (1.8 mL), and the solution was poured dropwise into ice-cold Et2O (18 mL) to give a turbid emulsion, from which 2 was obtained by centrifugation. The obtained product was a brown viscous oil and was subjected to preparative GPC (Shodex K-2002 and K-2002.5, eluent; CHCl3) to divide it into several fractions. The weight yield of 2 was 30 mg (64% by weight, the total amount of the fractions (1), (2), and (3) in Fig. S9 in ESI†). The fraction eluted at an earlier retention time (the fraction (2) in Fig. S9,†Mn = 5.3 × 104 g mol−1, 9.8 mg, 21% yield by weight) was used for the following analyses. 1H NMR (CDCl3, 300 MHz) δ 1.32 (br d, 3n H), 3.29 (br s, 3n H), 3.36 (br s, 3n H), 3.48–3.90 (br m, 40n H), 4.18–4.26 (br m, 2n H), 4.30–4.42 (br m, n H), 7.20 (br s, 2n H), 8.03 (br s, 2n H); IR (neat) ν 3346, 2918, 2872, 1662, 1654, 1579, 1541 cm−1. The molecular weights of the fractions of the polymer 2 were evaluated by analytical GPC using TOSOH TSKgel G2000HHR and TSKgel G3000HHR columns with a DMF solution of 0.1 M LiBr as the eluent. In this report, four fractions from four lots (Table S1 in ESI†) were applied as a substrate.
Notes and references
- For recent reviews for helical foldamers, see: (a) E. Yashima, K. Maeda, H. Iida, Y. Furusho and K. Nagai, Chem. Rev., 2009, 109, 6102–6211 CrossRef CAS PubMed; (b) E. Yashima and K. Maeda, in Foldamers, ed. S. Hecht and I. Huc, Wiley-VCH, Weinheim, 2007, pp. 331–366 Search PubMed; (c) D. J. Hill, M. J. Mio, R. B. Prince, T. S. Hughes and J. S. Moore, Chem. Rev., 2001, 101, 3893–4011 CrossRef CAS PubMed; (d) T. Sierra, in Chirality at the Nanoscale, ed. D. B. Amabilino, Wiley-VCH, Weinheim, 2009, pp. 115–189 Search PubMed; (e) G. Guichard and I. Huc, Chem. Commun., 2011, 47, 5933–5941 RSC; (f) H. Juwarker, J. Suk and K.-S. Jeong, Chem. Soc. Rev., 2009, 38, 3316–3325 RSC; (g) B.-B. Ni, Q. Yan, Y. Ma and D. Zhao, Coord. Chem. Rev., 2010, 254, 954–971 CrossRef CAS PubMed; (h) R. A. Smaldone and J. S. Moore, Chem. – Eur. J., 2008, 14, 2650–2657 CrossRef CAS PubMed.
- For examples of helical foldamers utilising solvophobic interaction developed by Moore and coworkers, see: (a) J. C. Nelson, J. G. Saven, J. S. Moore and P. G. Wolynes, Science, 1997, 277, 1793–1796 CrossRef CAS; (b) Y. Zhao and J. S. Moore, in Foldamers, ed. S. Hecht and I. Huc, Wiley-VCH, Weinheim, 2007, pp. 75–108 Search PubMed; (c) C. R. Ray and J. S. Moore, Adv. Polym. Sci., 2005, 177, 91–149 CrossRef CAS; (d) M. T. Stone, J. M. Heemstra and J. S. Moore, Acc. Chem. Res., 2006, 39, 11–20 CrossRef CAS PubMed.
- For recent examples of foldamers forming single helical structures by hydrophobic interaction, see: (a) M. T. Stone and J. S. Moore, Org. Lett., 2004, 6, 469–472 CrossRef CAS PubMed; (b) R. M. Meudtner and S. Hecht, Angew. Chem., Int. Ed., 2008, 47, 4926–4930 CrossRef CAS PubMed; (c) Y. Wang, F. Li, Y. Han, F. Wang and H. Jiang, Chem. – Eur. J., 2009, 15, 9424–9433 CrossRef CAS PubMed; (d) R. Pfukwa, P. H. J. Kouwer, A. E. Rowan and B. Klumperman, Angew. Chem., Int. Ed., 2013, 52, 11040–11044 CrossRef CAS PubMed; (e) Y. Hua, Y. Liu, C.-H. Chen and A. H. Flood, J. Am. Chem. Soc., 2013, 135, 14401–14412 CrossRef CAS PubMed.
- For recent examples on foldamers forming double helical structures by hydrophobic interactions, see: (a) S. J. Dawson, Á. Mészáros, L. Pethő, C. Colombo, M. Csékei, A. Kotschy and I. Huc, Eur. J. Org. Chem., 2014, 4265–4275 CrossRef CAS; (b) J. Shang, Q. Gan, S. J. Dawson, F. Rosu, H. Jiang, Y. Ferrand and I. Huc, Org. Lett., 2014, 16, 4992–4995 CrossRef CAS PubMed; (c) H. Goto, H. Katagiri, Y. Furusho and E. Yashima, J. Am. Chem. Soc., 2006, 128, 7176–7178 CrossRef CAS PubMed; (d) H. Goto, Y. Furusho and E. Yashima, J. Am. Chem. Soc., 2007, 129, 109–112 CrossRef CAS PubMed; (e) H. Goto, Y. Furusho, K. Miwa and E. Yashima, J. Am. Chem. Soc., 2009, 131, 4710–4719 CrossRef CAS PubMed; (f) T. Ben, Y. Furusho, H. Goto, K. Miwa and E. Yashima, Org. Biomol. Chem., 2009, 7, 2509–2512 RSC.
- (a) S. Takashima, H. Abe and M. Inouye, Tetrahedron: Asymmetry, 2013, 24, 527–531 CrossRef CAS PubMed; (b) F. Kayamori, H. Abe and M. Inouye, Eur. J. Org. Chem., 2013, 1677–1682 CrossRef CAS; (c) H. Abe, H. Makida and M. Inouye, Heterocycles, 2012, 86, 955–963 CrossRef CAS PubMed; (d) H. Abe, Y. Ohishi and M. Inouye, J. Org. Chem., 2012, 77, 5209–5214 CrossRef CAS PubMed; (e) H. Abe, H. Makida and M. Inouye, Tetrahedron, 2012, 68, 4353–4361 CrossRef CAS PubMed; (f) S. Takashima, H. Abe and M. Inouye, Chem. Commun., 2012, 48, 3330–3332 RSC; (g) S. Takashima, H. Abe and M. Inouye, Chem. Commun., 2011, 47, 7455–7457 RSC; (h) H. Abe, S. Takashima, T. Yamamoto and M. Inouye, Chem. Commun., 2009, 2121–2123 RSC; (i) H. Abe, D. Murayama, F. Kayamori and M. Inouye, Macromolecules, 2008, 41, 6903–6909 CrossRef CAS; (j) H. Abe, N. Masuda, M. Waki and M. Inouye, J. Am. Chem. Soc., 2005, 127, 16189–16196 CrossRef CAS PubMed; (k) M. Inouye, M. Waki and H. Abe, J. Am. Chem. Soc., 2004, 126, 2022–2027 CrossRef CAS PubMed.
- (a) H. Abe, K. Okada, H. Makida and M. Inouye, Org. Biomol. Chem., 2012, 10, 6930–6936 RSC; (b) M. Waki, H. Abe and M. Inouye, Angew. Chem., Int. Ed., 2007, 46, 3059–3061 CrossRef CAS PubMed; (c) M. Waki, H. Abe and M. Inouye, Chem. – Eur. J., 2006, 12, 7839–7847 CrossRef CAS PubMed.
- A. Klapars and S. L. Buchwald, J. Am. Chem. Soc., 2002, 124, 14844–14845 CrossRef CAS PubMed.
- P. H. J. Kouwer and T. M. Swager, J. Am. Chem. Soc., 2007, 129, 14042–14052 CrossRef CAS PubMed.
- C. M. Amb and S. C. Rasmussen, J. Org. Chem., 2006, 71, 4696–4699 CrossRef CAS PubMed.
- K. W. Anderson and S. L. Buchwald, Angew. Chem., Int. Ed., 2005, 44, 6173–6177 CrossRef CAS PubMed.
- H. Sogawa, M. Shiotsuki, H. Matsuoka and F. Sanda, Macromolecules, 2011, 44, 3338–3345 CrossRef CAS.
- M. Banno, T. Yamaguchi, K. Nagai, C. Kaiser, S. Hecht and E. Yashima, J. Am. Chem. Soc., 2012, 134, 8718–8728 CrossRef CAS PubMed.
- (a) G. D. Smith and D. Bedrov, J. Phys. Chem. B, 2003, 107, 3095–3057 CrossRef CAS; (b) M. Ataman, Colloid Polym. Sci., 1987, 265, 19–25 CrossRef CAS.
- (a) L. Li, Y. Che, D. E. Gross, H. Huang, J. S. Moore and L. Zang, ACS Macro Lett., 2012, 1, 1335–1338 CrossRef CAS; (b) H.-J. Kim, T. Kim and M. Lee, Acc. Chem. Res., 2011, 44, 72–82 CrossRef CAS PubMed; (c) S. Li, K. Liu, G. Kuang, T. Masuda and A. Zhang, Macromolecules, 2014, 47, 3288–3296 CrossRef CAS.
- (a) S. Kobayashi, Chem. Lett., 1991, 20, 2187–2190 CrossRef; (b) S. Kobayashi, Eur. J. Org. Chem., 1999, 15–27 CrossRef CAS; (c) S. Kobayashi, M. Sugiura, H. Kitagawa and W. W.-L. Lam, Chem. Rev., 2002, 102, 2227–2302 CrossRef CAS PubMed.
- Studies of Lewis acidity of rare-earth metal cations: (a) H. Tsuruta, T. Imamoto and K. Yamaguchi, Chem. Commun., 1999, 1703–1704 RSC; (b) H. Tsuruta, K. Yamaguchi and T. Imamoto, Tetrahedron, 2003, 59, 10419–10438 CrossRef CAS PubMed.
- Because the polymer 2 is a mixture of molecules of various lengths, the binding constant was obtained as an averaged value, apparent binding constant K′a. Similarly, the value of n was also obtained as an averaged value.
- K. A. Connors, Binding Constants, John Wiley & Sons, New York, 1987 Search PubMed.
- K. W. Anderson and S. L. Buchwald, Angew. Chem., Int. Ed., 2005, 44, 6173–6177 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Fig. S1–S9, Table S1, experimental details of the synthetic route to 2, and 1H and 13C NMR spectra for compounds 4, 5, 8, 12–15, and 2. See DOI: 10.1039/c4ob02129k |
| This journal is © The Royal Society of Chemistry 2015 |