
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Artificial switchable catalysts
Victor
Blanco†
ab,
David A.
Leigh
*a and
Vanesa
Marcos†
a
aSchool of Chemistry, University of Manchester, Oxford Road, Manchester, M13 9PL, UK. E-mail: david.leigh@manchester.ac.uk
bDepartamento de Química Orgánica, Facultad de Ciencias, Universidad de Granada, Campus Fuentenueva S/N, 18071 Granada, Spain
First published on 12th May 2015
Abstract
Catalysis is key to the effective and efficient transformation of readily available building blocks into high value functional molecules and materials. For many years research in this field has largely focussed on the invention of new catalysts and the optimization of their performance to achieve high conversions and/or selectivities. However, inspired by Nature, chemists are beginning to turn their attention to the development of catalysts whose activity in different chemical processes can be switched by an external stimulus. Potential applications include using the states of multiple switchable catalysts to control sequences of transformations, producing different products from a pool of building blocks according to the order and type of stimuli applied. Here we outline the state-of-art in artificial switchable catalysis, classifying systems according to the trigger used to achieve control over the catalytic activity and stereochemical or other structural outcomes of the reaction.
 Victor Blanco | Victor Blanco was born in A Coruña (Spain) and obtained his BSc from the University of A Coruña. He obtained his PhD from the same University under the supervision of J. M. Quintela and C. Peinador focusing on the metal-directed self-assembly of N-monoalkyl-bipyridinium-based receptors and their inclusion complexes, catenanes and knots. He then joined David Leigh's group as a Marie Curie IEF Fellow and Post-Doctoral Research Associate working on transition metal molecular machines and rotaxane-based switchable catalysts. Since 2014 he holds a ‘Juan de la Cierva’ Post-Doctoral Fellowship at the University of Granada (Spain). |
 David A. Leigh | David Leigh was born in Birmingham (UK) and obtained his BSc and PhD from the University of Sheffield. After postdoctoral research at the NRC of Canada in Ottawa (1987–1989) he returned to the UK as a lecturer at UMIST. After periods at the University of Warwick (1998–2001) and the University of Edinburgh (2001–2012) he joined the University of Manchester in 2012 where he is currently the Sir Samuel Hall Chair of Chemistry. He was elected to the Fellowship of the Royal Society in 2009. His research interests include the development and applications of synthetic molecular machines. |
 Vanesa Marcos | Vanesa Marcos was born in Madrid (Spain) and obtained her BSc from the Universidad Autónoma de Madrid. She obtained her PhD from the same University under the Supervision of J. L. García Ruano and J. Alemán focusing on new diastereoselective syntheses using sulfinyl carbanions as chiral auxiliaries and on the development of new organocatalytic methodologies. She then joined N. Martín's group as a Post-Doctoral Researcher working on the organocatalytic synthesis of chiral fullerenes. In 2012 she joined David Leigh's group at the University of Manchester (UK) as a Research Associate working on the development of new molecular machines. |
1. Introduction
Different enzymatic syntheses within cells often occur in parallel. To ensure that the transformations proceed with the necessary spatial and temporal control, and without unwanted interference from other reaction pathways, enzyme activity is often modulated through feedback loops and a variety of trigger-induced effects.1,2 In contrast, the reactions promoted by man-made catalysts usually take place according to the initially chosen reaction conditions.3 Incorporating stimuli-responsive features into artificial catalysts can confer an additional ‘bio-like’ level of control over chemical transformations, potentially enabling such systems to perform tasks in synthesis that are difficult or impossible to accomplish in other ways. For example, switchable catalysts can be used to speed up or slow down the rate of a reaction according to the presence of a specific analyte or to change the stereochemical outcome of a reaction. In recent years their utility in performing more advanced tasks, such as turning ‘on’ and ‘off’ different catalysts to promote alternative reactions from a mixture of different building blocks, have started to be explored.In this review we discuss the state-of-the-art in the emerging field of artificial switchable catalysis, including systems whose activity and/or selectivity can be controlled in different chemical reactions, grouping them by the type of external triggering stimulus or other changes in the experimental conditions. We conclude by considering the future outlook for this area.
2. Light-driven switching
Photoresponsive processes, such as photosynthesis4 and vision,5 exemplify the effectiveness of light in initiating and regulating complex molecular and biochemical processes. The use of light to induce a change in catalyst state is particularly attractive as it is non-invasive, offers excellent temporal and spatial control, and can be precisely regulated with an appropriate light source. Artificial photoswitchable catalysts6 are generally based on photoactive units incorporated within the molecular architecture or through intermolecular interactions between a photochemical additive and the catalyst active site. In this review we classify these systems according to the principal type of effect used to achieve control: cooperative, steric or electronic effects, or the aggregation/dissociation of the catalyst.2.1 Cooperative effects
Cooperative effects in switchable catalysis are often based on a large geometrical change, typically occurring during an isomerization process, that induces a change in the distance or orientation of key sites in the catalyst. A seminal example of a cooperative template for catalysis was reported by Würthner and Rebek (Scheme 1),7 who used the isomerization of the azobenzene unit in E/Z-1 to change the relative positions of two carbazole-based adenine receptors to control the rate of the amide coupling reaction between aminoadenosine (2) and an adenosine-derived ester (3) (Scheme 1). In the extended E-1 form, the bound substrates (2, 3) are held too far apart to react. Upon UV isomerization to the folded form of the receptor, Z-1, bound 2 and 3 are held in close proximity and the amidation reaction is accelerated. By determining the binding constants of the substrates to the switchable catalyst and calculating the concentrations of the intermolecular complex, the authors showed that Z-1 is approximately 50 times more active than E-1. However, the system suffers from product inhibition: product 4 binds strongly to two imide groups of Z-1 whereas each of the reactants, 2 and 3, bind only to one imide group. Indeed, neither thermal nor photochemical reverse isomerization of the catalyst could be induced while 4 is bound to Z-1.8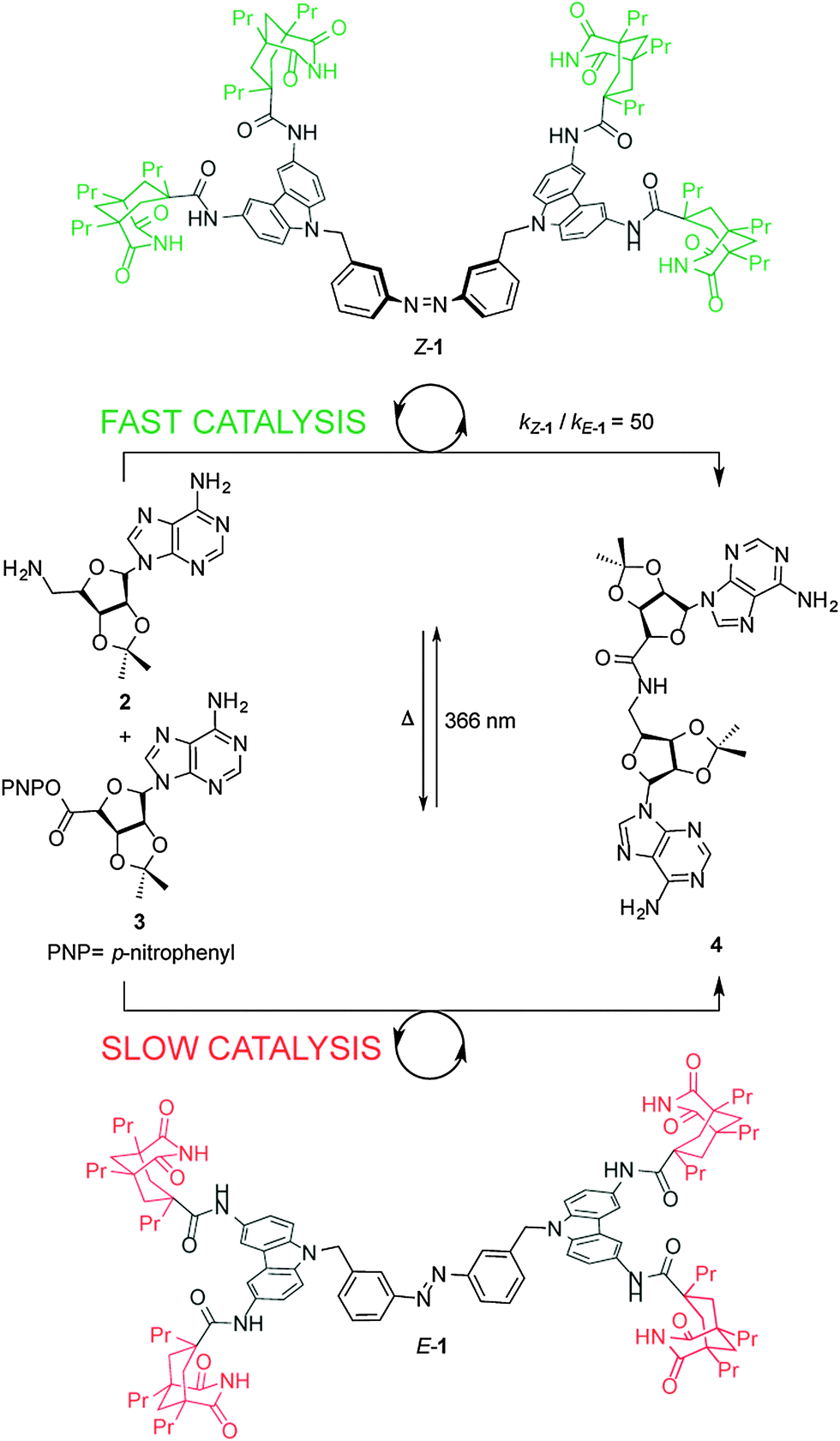 | ||
| Scheme 1 Controlling the rate of amidation using photoswitchable template E/Z-1.7 | ||
An example of a photoswitchable catalyst based on cooperative interactions was reported by Cacciapaglia and co-workers in 2003 (Scheme 2).9 The bis-barium(II) complex of azobis(benzo-18-crown-6) ether 5 was used to control the rate of basic ethanolysis of 4-carboxyacetanilide 6, exploiting the reversible E to Z isomerization of the azobenzene spacer. Efficient ethanolysis requires the participation of two metal binding sites, which is only feasible for Z-5, formed upon UV irradiation of the less active E-5 isomer (kZ-5/kE-5 up to 5). The authors demonstrated that the catalytic activity could be switched between ‘faster’ and ‘slower’ multiple times by alternating between exposure to UV or visible light, illustrating the reversibility of the photoisomerization reaction.
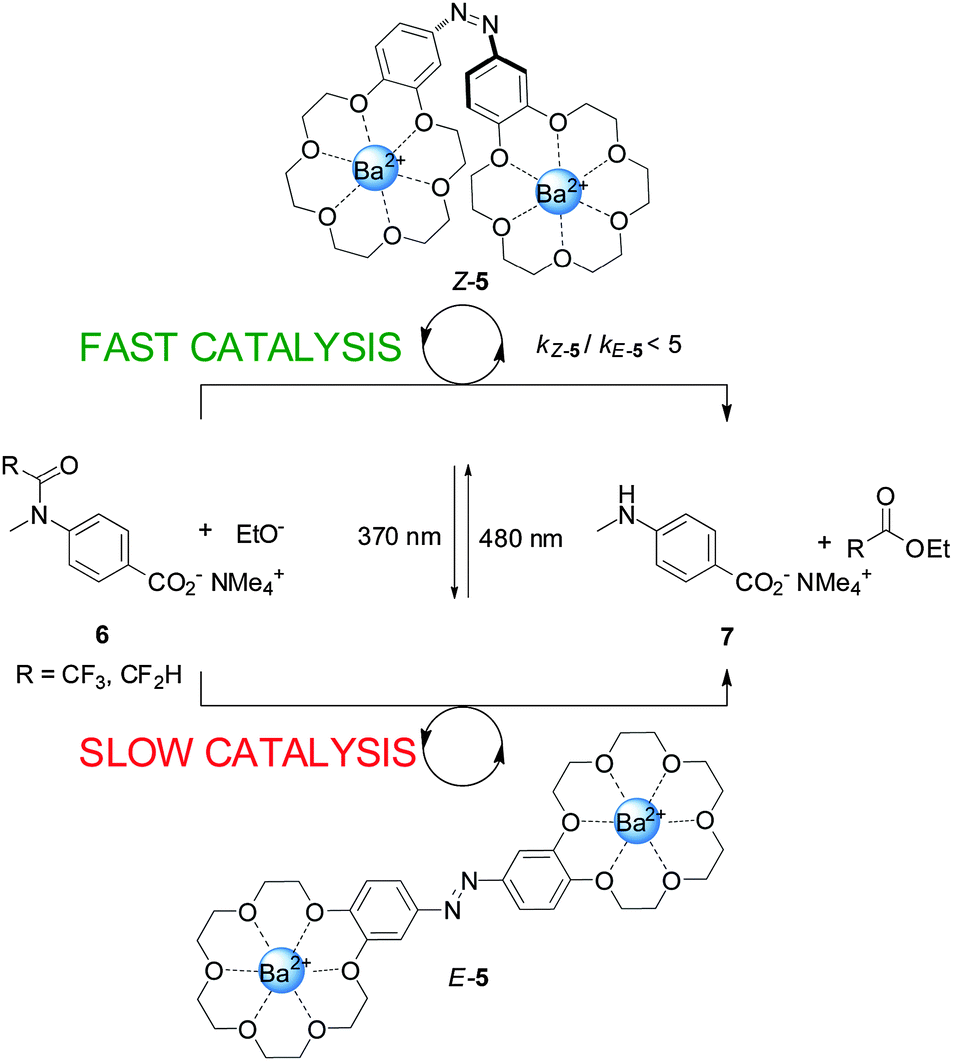 | ||
| Scheme 2 Controlling the rate of anilide ethanolysis using photoswitchable catalyst E/Z-5.9 | ||
Linking two triaryl alcohol groups through an azobenzene moiety resulted in cooperative bifunctional switchable catalyst E/Z-8, which can turn ‘on’ or ‘off’ the Morita–Baylis–Hillman reaction of 3-phenylpropanal (9) and 2-cyclopente-1-one (10) upon exposure to light (Scheme 3).10 In the E-8 isomer the hydroxyl groups are too far apart to engage in the intramolecular hydrogen bonding needed to increase the acidity of the trityl alcohol, affording 11 in 37% yield, only a small increase over the background reaction (27%). In contrast the Z-8 isomer, generated by UV irradiation of E-8, afforded 11 in 78% yield. The catalytic activity of Z-8 is significantly higher than other bis(trityl alcohol) derivatives similarly activated by intramolecular hydrogen bonding, suggesting that there are other cooperative enhancements in the catalysis mechanism due to having the two trityl alcohols in close proximity in Z-8.
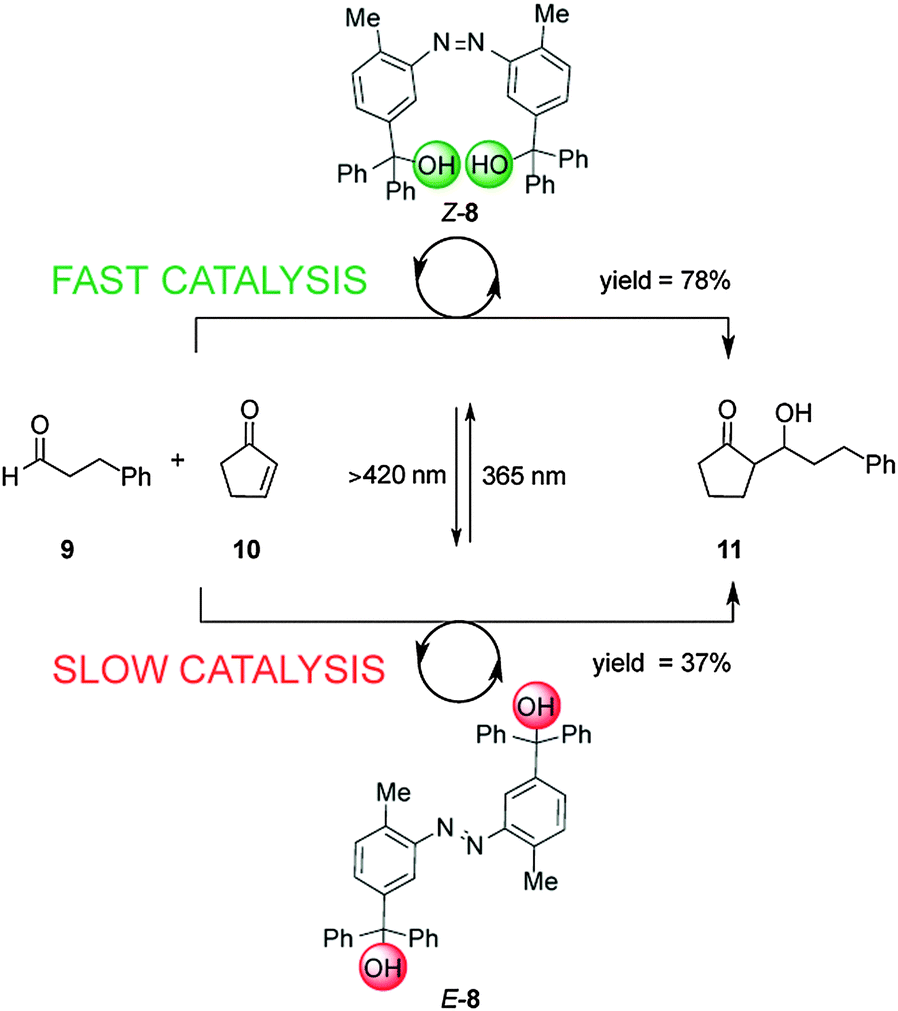 | ||
| Scheme 3 Controlling the rate of the Morita–Baylis–Hillman reaction using photoswitchable catalyst E/Z-8.10 | ||
Recently, a 3,3′-dicarboxylic acid enzyme mimic (E/Z-12) that acts as switchable catalyst with glycosidase activity has been described (Scheme 4).11 Isomers Z-12 and E-12 have significantly different pKa values. In E-12 both carboxylic acid groups ionize at roughly the same pH. In contrast, the deprotonation of Z-12 occurs in a stepwise fashion, with the carboxylic acid and carboxylate forms coexisting at pH values between 4.7 and 6.5. Z-12 showed a rate enhancement of six orders of magnitude for the hydrolysis of nitrophenyl glycoside 13 (giving 14) compared to E-12. The catalytic activity of E/Z-12 could be switched ‘on’ and ‘off’ multiple times over the course of the reaction by alternating exposure to UV or visible light.
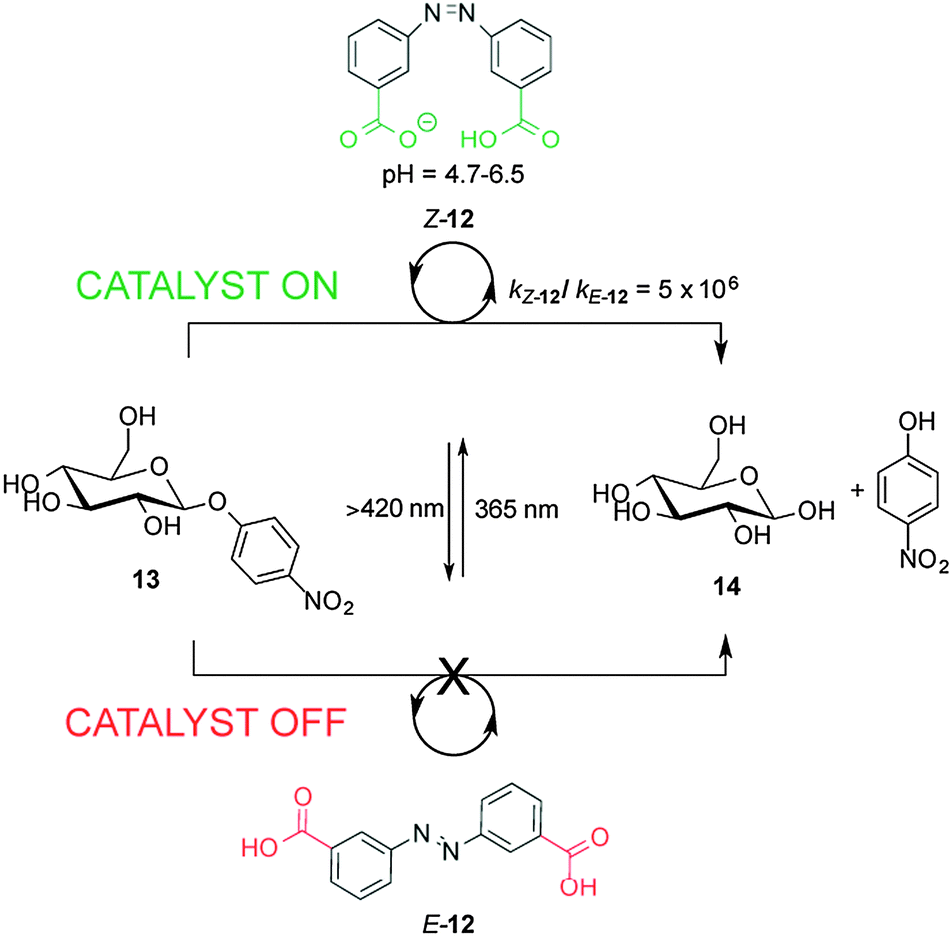 | ||
| Scheme 4 Controlling the rate of hydrolysis of nitrophenyl β-D-glucose 13 using photoswitchable catalyst E/Z-12.11 | ||
Cooperative interactions can also be used to change the stereochemical outcome of switchable catalysed reactions. Branda and co-workers successfully controlled the stereochemical outcome of the cyclopropanation of styrene (16) using a photoswitchable catalyst (Scheme 5).12 Ligand 15 consists of a dithienylethene-based chiral bis(oxazoline) which, in its open form (o-15), binds Cu(I) ions in a bidentate fashion. This results in a rigid chiral environment for the metal ion that subsequently catalyzes cyclopropanation of 16 with 17 to afford 18 as a trans–cis mixture with significant enantioselectivity (30–50% ee). In contrast, the closed form of the ligand (c-15; formed upon radiation at 313 nm), is only able to form monodentate complexes with Cu(I). The chiral environment is less well expressed in this complex and results in negligible enantioselectivity in the cyclopropanation reaction (5% ee). A limitation of this catalytic system is the low photocyclization efficiency in the presence of Cu(I), with only 23% of the closed form (c-15) present at the stationary state at 313 nm, meaning that switching the catalyst state only results in a small disruption in the stereoselectivity of the catalysed reaction.
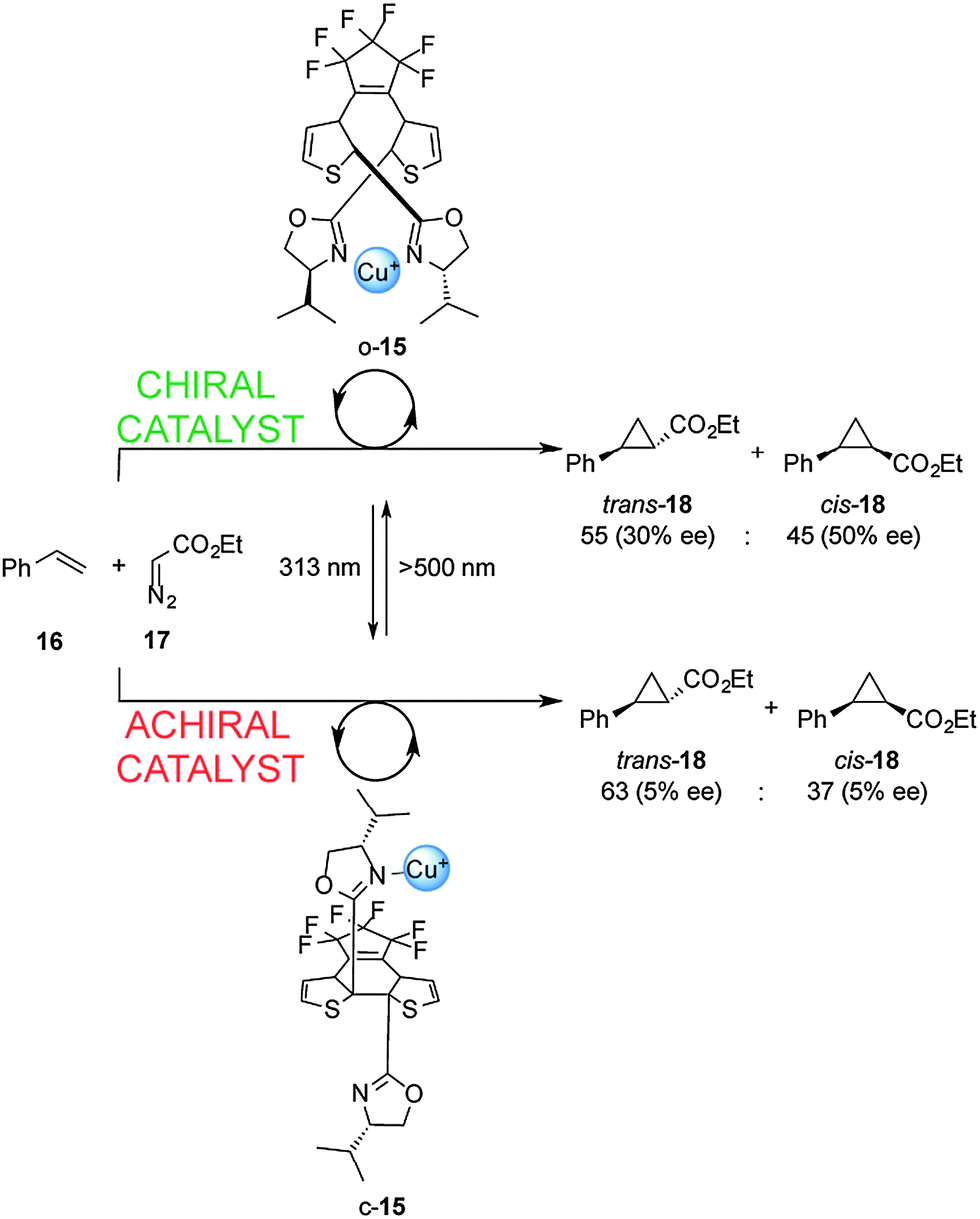 | ||
| Scheme 5 Controlling the stereoselectivity of the cyclopropanation of styrene using photoswitchable catalyst o/c-15.12 | ||
The first example of a cooperative photoswitchable catalyst able to control both catalytic activity and the stereoselectivity of the reaction was reported by Wang and Feringa (Scheme 6).13 A dimethylaminopyridine (DMAP) Brønsted base and a hydrogen bonding thiourea, which are known to cooperate in the catalysis of Michael additions, were attached to the rotor and stator of a light-driven unidirectional rotary motor (19). The resulting catalyst was able to alter both the rate and the stereochemical outcome of the Michael addition of aromatic thiol 21 to 2-cyclohexen-1-one (20). When the (P,P)-E-19 isomer is used as catalyst, the reaction is very slow (7% yield) and afforded the racemic Michael adduct (rac-22) (bottom, Scheme 6). Upon UV irradiation, E–Z isomerization takes place forming the (M,M)-Z-19 isomer, which shows a higher catalytic activity and moderate enantioselectivity (50% yield, 50% ee) in favour of the (S)-22 enantiomer (middle, Scheme 6). By thermally-activated helix inversion (P,P)-Z-19 is formed, which is also able to catalyse the reaction giving a higher yield and a similar degree of stereoselectivity (83% yield, 54% ee) but in favour of the (R)-22 enantiomer (top, Scheme 6). Subsequent photochemical and thermal isomerization can return the catalyst to its original state, (P,P)-E-19, via a fourth diastereoisomer (M,M)-E-19. This rotary motor organocatalyst allows for control over the rate and the selectivity in a reversible and sequential manner using light and temperature. The Feringa group later reported on a second generation molecular rotor where the phenyl spacers between the motor core and the catalytic units are absent.14 The resulting switchable catalyst is able to switch from a E-state with low catalytic activity and no stereocontrol in the Henry reaction of nitromethane with α,α,α-trifluoroketones, to two Z-states, in which the catalytic units are in close proximity, affording the corresponding products with opposite stereoselectivity in excellent yields and good enantioselectivity.14
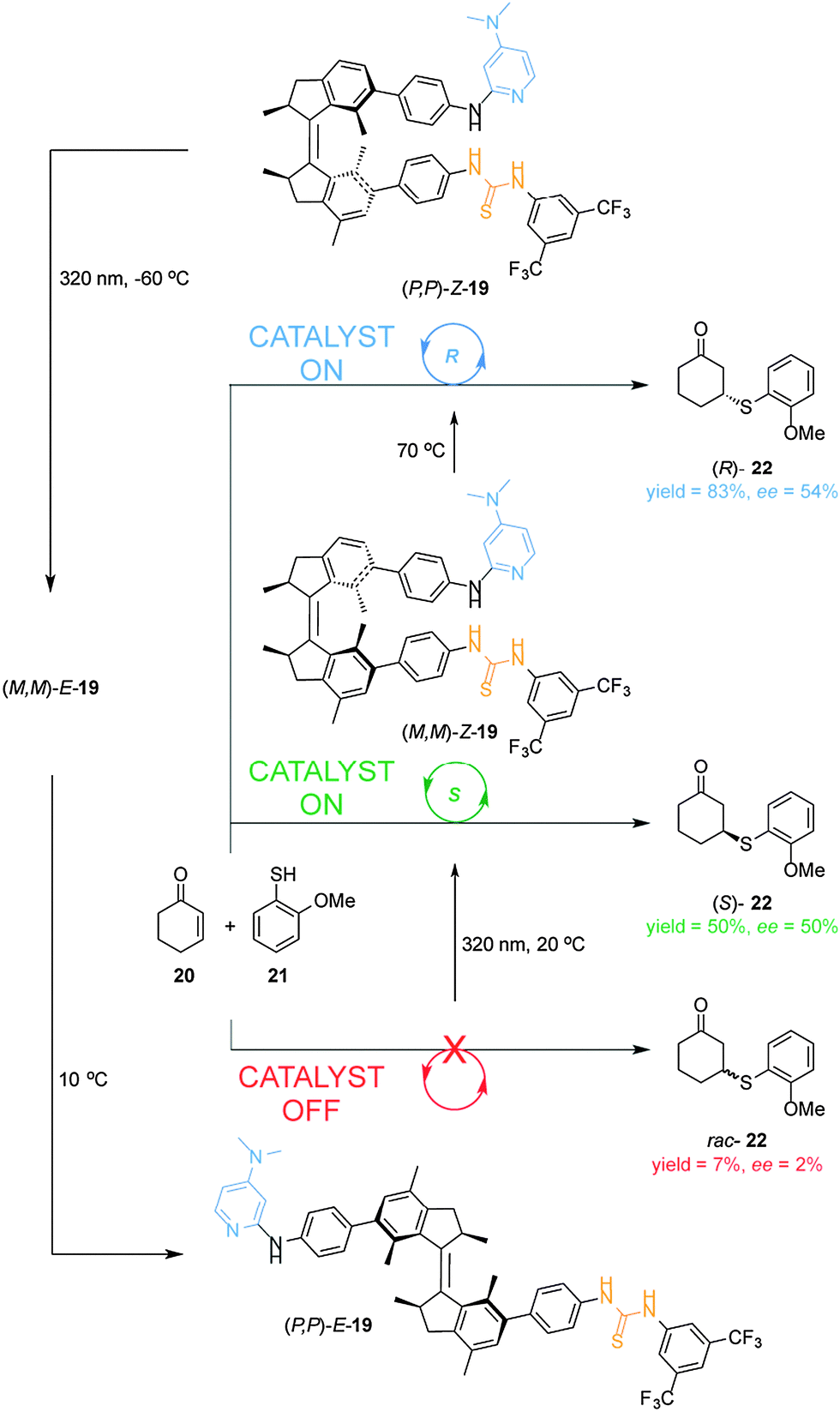 | ||
| Scheme 6 Controlling the rate and the stereoselectivity of a Michael addition using photoswitchable catalyst 19.13 | ||
The photoswitchable unit used in Feringa's rotary catalysts has also been exploited by Craig and colleagues in a switchable catalyst able to modulate the stereochemical outcome of asymmetric Heck arylations and Trost allylic alkylations.15 This catalyst consists of a macrocycle containing a chiral bis(phosphine) ligand linked to the photoswitchable unit. Modulation of the ligand geometry upon exposure to UV-light afforded isomers of the catalyst with different enantioselectivities for the Heck arylation and Trost allylic alkylation reactions.15 The Feringa group have also recently demonstrated dynamic control of chirality using phosphine ligands attached to their rotary molecular motor system.16
2.2 Steric effects
These catalyst systems feature a group that blocks access of the substrate to the active site in one state of the catalyst, normally through a large geometrical change during the isomerization process. Different approaches have been used to achieve this, such as host–guest binding, hydrogen bonding or simple steric bulk to shield the catalytic site. An early example using host–guest interactions to modulate catalytic activity was described by Ueno and co-workers (top, Scheme 7).17 Complexation of β-cyclodextrin (23) with 4-carboxyazobenzene (24) alters the rate of the hydrolysis of nitrophenol ester 25 in basic media. β-Cyclodextrin 23 acts as a host for the hydrophobic aryl substituent of 25, facilitating attack of one of the peripheral hydroxyl groups on the ester moiety. However, E-24 is a competitive inhibitor of the binding of 25 within 23, decreasing the rate of catalysis, whereas the Z isomer does not fit inside the cyclodextrin cavity. The system does not turnover and the catalyst and the azobenzene must exceed the substrate concentration by one and two orders of magnitude, respectively to compensate for unfavourable product inhibition.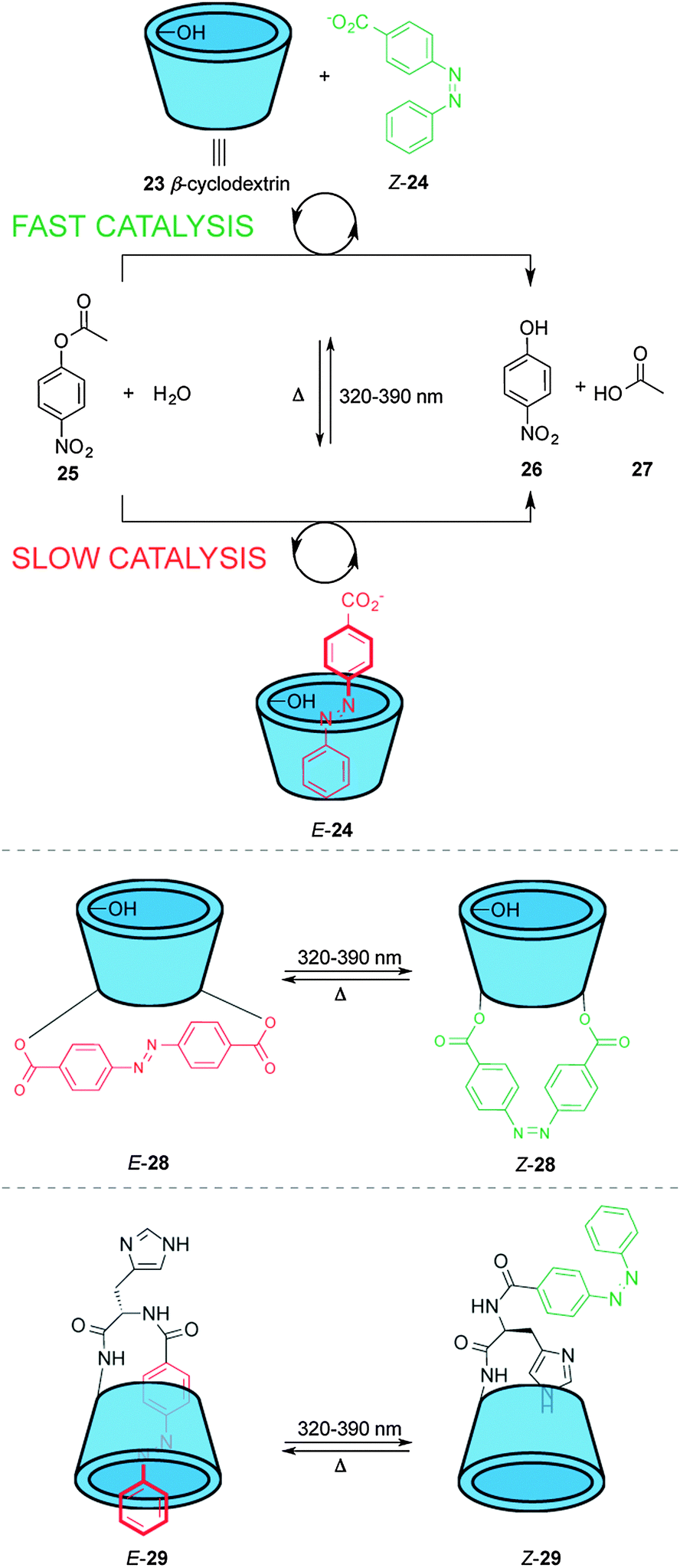 | ||
| Scheme 7 Controlling the rate of ester hydrolysis using photoswitchable β-cyclodextrin catalysts.17,18 | ||
The same group developed photoswitchable catalysts based on β-cyclodextrins by covalently linking azobenzene moieties to the lower rim of β-cyclodextrin (middle and bottom, Scheme 7). The extended linker in E-28 makes the internal cavity shallow, decreasing the binding of the ester substrate in the hydrophobic cavity (middle, Scheme 7).17 Isomerization to Z-28 creates a deeper cavity that offers stronger binding to the substrate resulting in a five-fold increase in the rate of catalysed ester hydrolysis. The group also investigated a switchable catalyst with an azobenzene unit attached to the β-cyclodextrin at a single point via a histidine residue (bottom, Scheme 7).18 In the ‘off’ state, E-29, the azobenzene unit occupies the cavity and the system displayed no catalytic activity. In Z-29 the azobenzene does not fit within the cavity and, therefore, the substrate can bind to the cyclodextrin turning ‘on’ the catalytic activity for the hydrolysis of various esters, including nitrophenyl acetate, Boc-D-alanine-p-nitrophenyl ester and Boc-L-alanine-p-nitrophenyl ester.
The azobenzene units were later tethered to Au nanoparticles (NPs) and used in conjunction with a Zn(II) coordinated β-cyclodextrin dimer as a heterogeneous photoswitchable catalyst to modulate the catalytic activity in ester hydrolysis.19
Following these strategies, in 2009 Harada and co-workers reported control over the polymerization of δ-valerolactone by photoisomerization of a E-cinnamoyl-α-cyclodextrin to Z-cinnamoyl-α-cyclodextrin, which turned ‘off’ the catalytic activity through the inhibition of the binding and inclusion of the δ-valerolactone monomer into the cyclodextrin cavity.20
Rebek et al. have described a light-responsive cavitand-piperidinium complex that acts a switchable catalyst for the Knoevenagel condensation of aromatic aldehydes with malonitrile (33) (Scheme 8).21 Cavitand 30 is armed with an isopropyl-substituted azobenzene unit that modulates the rate of the reaction. In the E state the cavitand binds to the piperidinium cation (31) and accelerates the rate of the reaction, producing up to 3.5-fold increase in rate compared to the reaction carried out with piperidinium acetate alone. Photoisomerization of the catalyst generates Z-30 for which the azobenzene unit can bind within the cavity, slightly inhibiting catalysis (E-30 accelerates the reaction ∼1.5× faster than Z-30). NOESY analysis indicates that the ammonium group of the piperidinium cation in the complex with E-30 protrudes from the open end of the cavitand. As the substrates are not guests in the complex, a wide range of aldehydes is tolerated by the mechanism of catalysis.
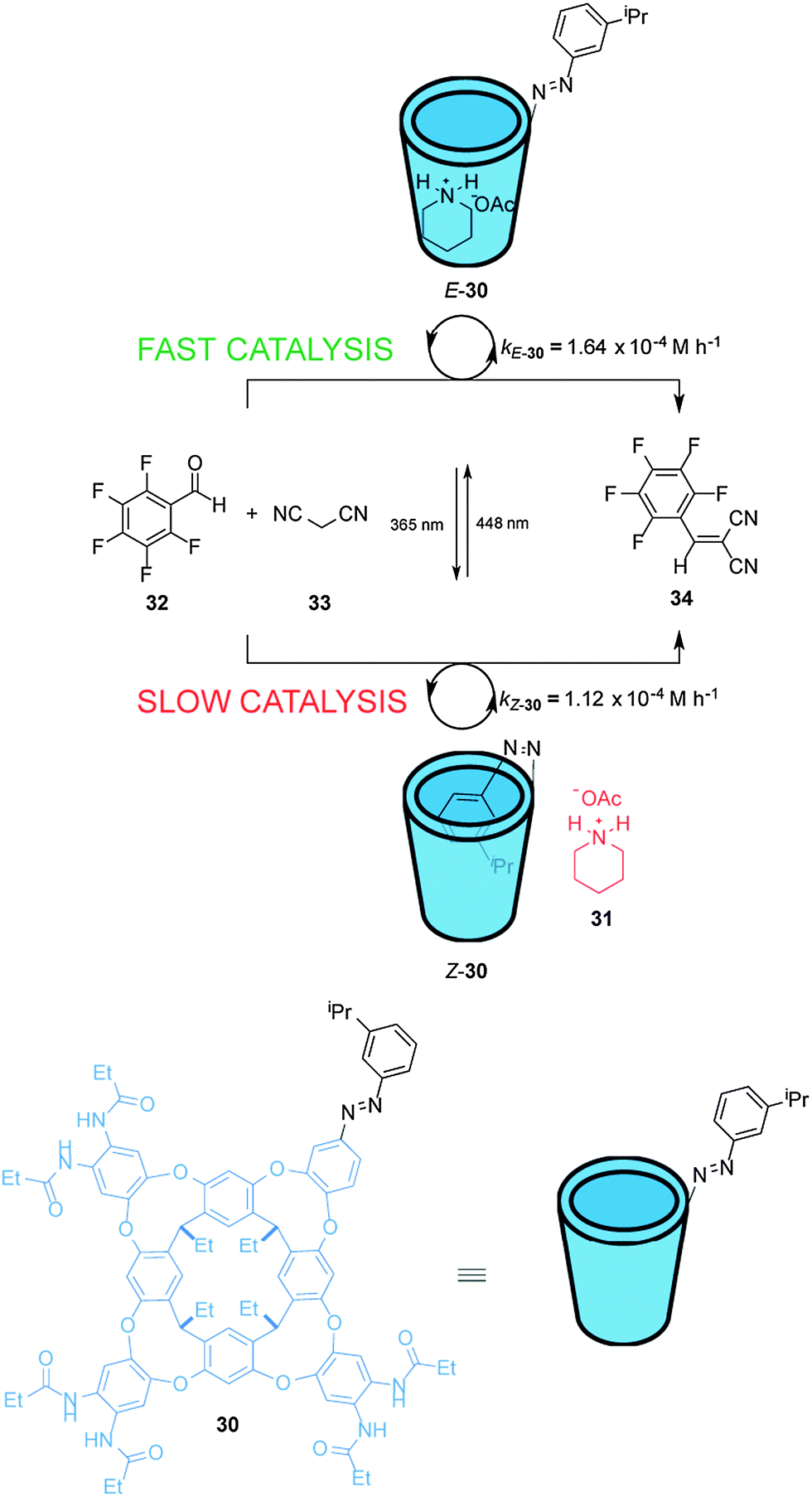 | ||
| Scheme 8 Controlling the rate of a Knoevenagel condensation using photoswitchable catalyst E/Z-30.21 | ||
Inoue and colleagues have exploited the reversible photoisomerization of stilbene derivatives in a switchable catalyst for the chemical fixation of carbon dioxide (Scheme 9).22 Control over catalysis of the condensation of carbon dioxide with 1,2-epoxypropane (37) was achieved by reversible coordination of 3,5-di-t-butyl-2-stilbazole (36) to aluminium porphyrin 35. Due to unfavourable steric interactions between the porphyrin ring system and the bulky tert-butyl groups in the E-2-stilbazole (E-36), the pyridine group is unable to effectively coordinate to the Al(III) centre in 35, resulting in a low yield of 38 (2% conversion over 18 h). Irradiation with UV light afforded the Z-2-stilbazole (Z-36), which bound to the aluminium porphyrin 35 enhancing the rate of the reaction (23% conversion over 18 h). Switching between UV and visible light multiple times allowed continuous modulation of the rate of the catalysed condensation reaction.
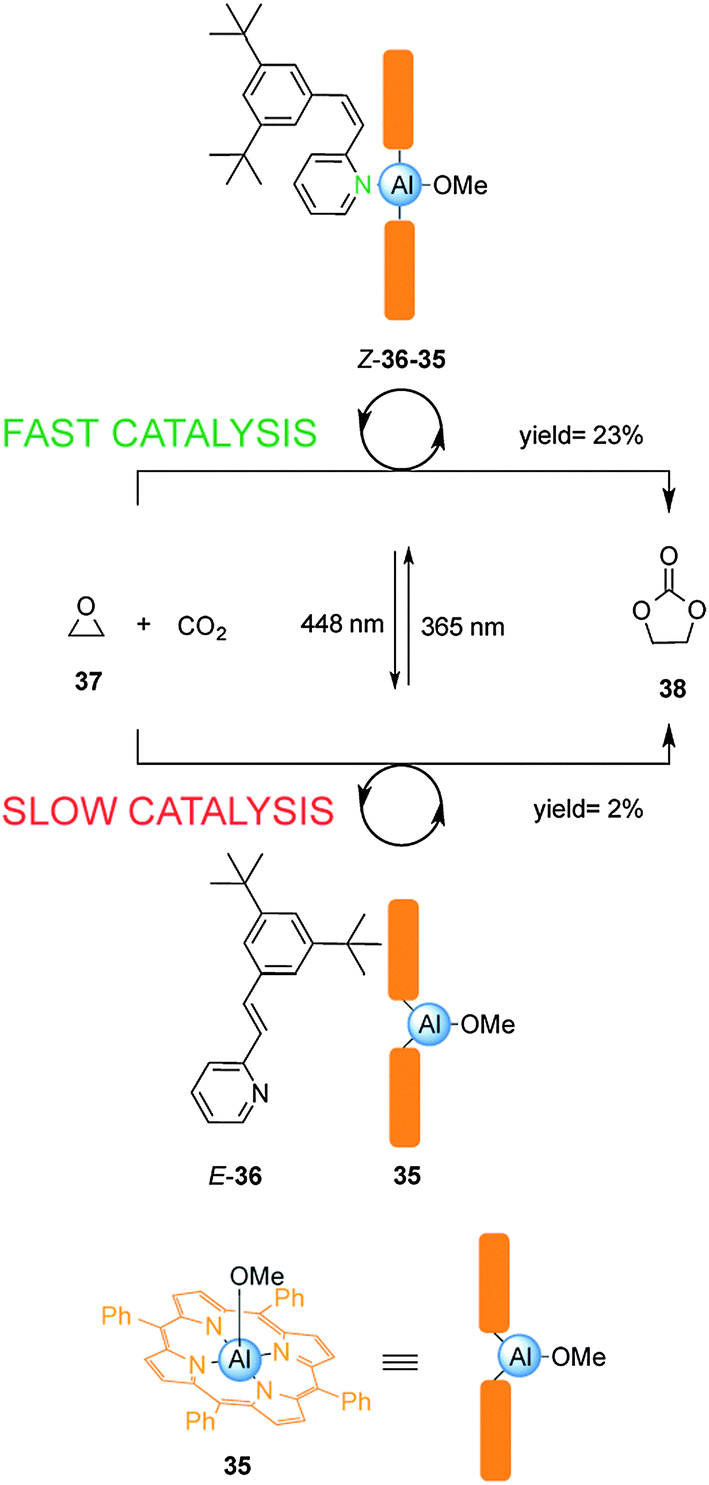 | ||
| Scheme 9 Controlling the rate of chemical fixation of carbon dioxide using reversible coordination of stilbene derivative 36 to aluminum porphyrin 35.22 | ||
Hecht and co-workers described a series of photoswitchable base catalysts (39a–c) able to control the rate of aza-Henry reactions (Scheme 10).23 Catalysts 39a–c comprise conformationally restricted tertiary piperidine bases as the catalytic unit and 3,5-disubstituted azobenzenes as a switchable steric shield. The E-39a–c catalysts have an inaccessible basic site and exhibit low basicity in acid–base titration experiments. UV irradiation affords the Z-39a–c isomers, in which the basic piperidine nitrogen atom is exposed, enhancing the basicity by almost one order of magnitude. Switchable catalyst 39c was able to effectively control the rate of the aza-Henry reaction of nitroethane (40) to p-nitroanisaldehyde (41) (kZ-39c/kE-39c = 35.5). Immobilization of the catalyst on silica gel particles enabled the system to be used as a heterogeneous switchable catalyst.24
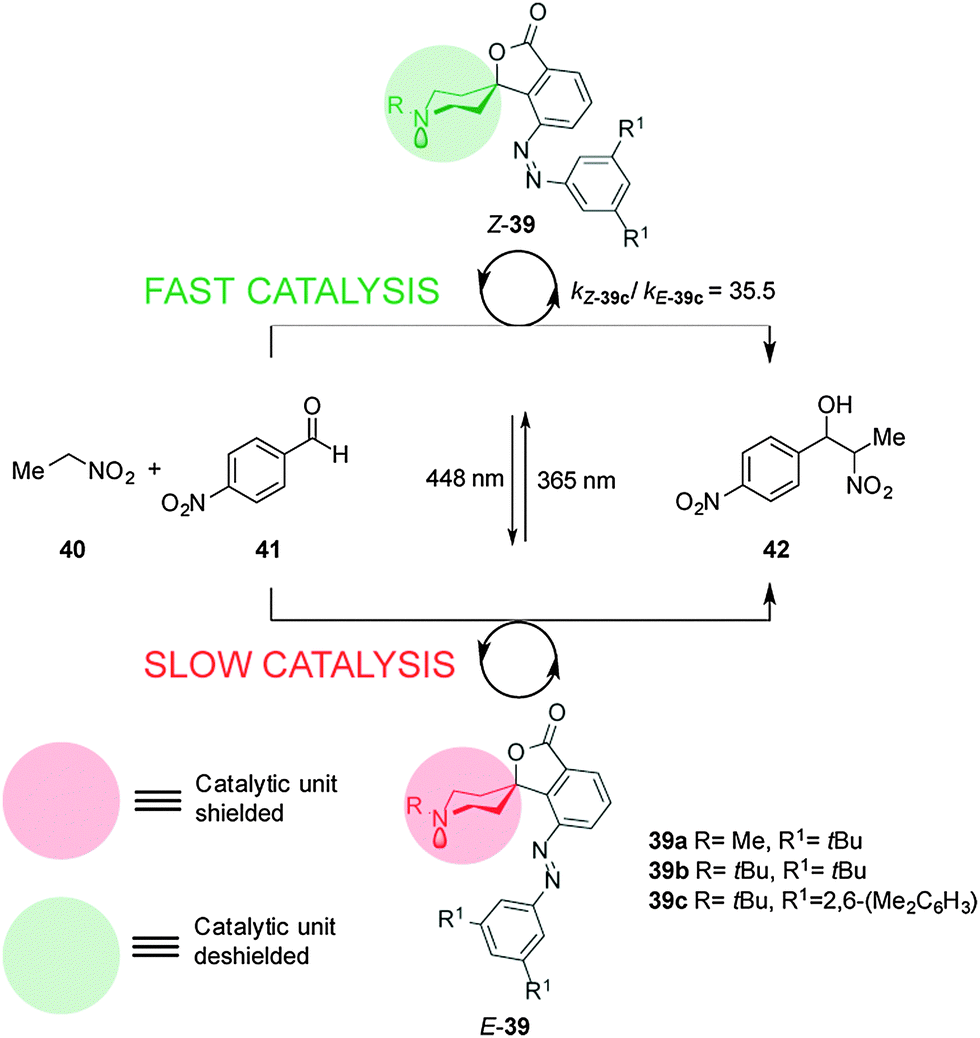 | ||
| Scheme 10 Controlling the rate of the aza-Henry reaction using switchable catalyst E/Z-39.23 | ||
Pericás and co-workers developed a switchable thiourea organocatalyst to control Michael addition reactions through hydrogen-bond shielding of the catalytic unit (Scheme 11).25 Catalyst 43 consists of a hydrogen bond donor (the thiourea unit) and a blocking moiety (a nitro group) located either side of an azobenzene unit. The thermodynamically stable E-43 diastereomer effectively catalyses the Michael addition of acetylacetone (44) to m-bromonitrostyrene (45) (full conversion after 19 h). UV irradiation forms Z-43, in which the nitro group binds to the thiourea, leading to significantly slower catalysis (23% conversion after 20 h).
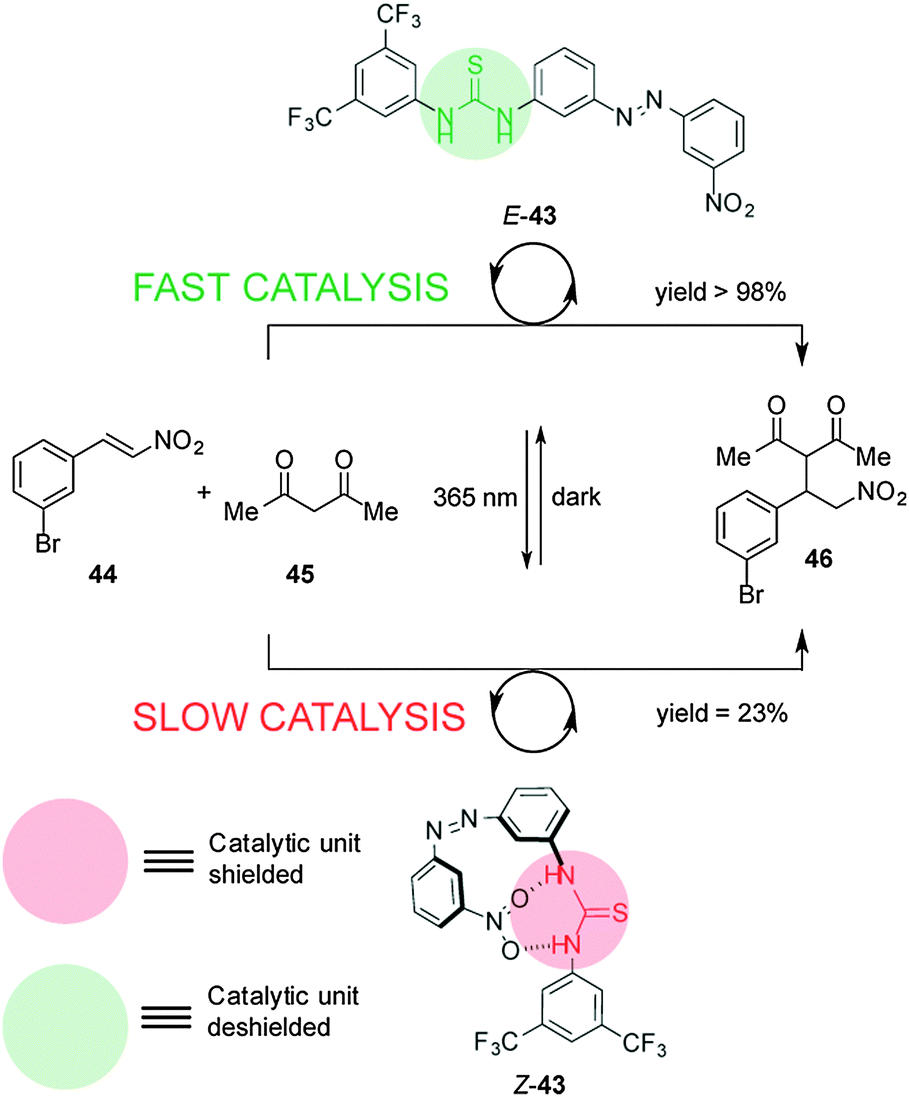 | ||
| Scheme 11 Controlling the rate of Michael addition using switchable catalyst E/Z-43.25 | ||
2.3 Electronic effects
Using photoinduced changes in electronic properties to alter the rate, chemo- and/or regioselectivity of catalysts has been less explored compared to steric and cooperative effects. The most popular concept is to use the breaking or forming of conjugation between the active site and an electronically activating group. The first example of photo-modulating catalytic activity via electronic changes was reported by Branda, using a photoactive diarylethene unit that undergoes ring-closing and ring opening reactions when exposed to UV and visible light, respectively (Scheme 12).26 The researchers developed a mimic of the bioactive form of vitamin B6, pyridoxal 5′-phosphate (PLP), in which the catalytic activity is dependent upon electronic connectivity between the functional groups. In the ring-open form of the catalyst (o-47), the pyridinium and aldehyde units are electronically isolated from each other, switching ‘off’ the catalytic activity for the racemization of L-alanine (less than 3% over 140 hours). However, UV irradiation to form the ring-closed isomer (c-47) results in a fully conjugated structure, turning ‘on’ catalysis and resulting in 30% deuterium exchange after 140 h and near-complete exchange (95%) after an additional 90 h. The in situ switching of the catalyst between its inactive and active forms was successfully demonstrated through alternating exposure to UV and visible light.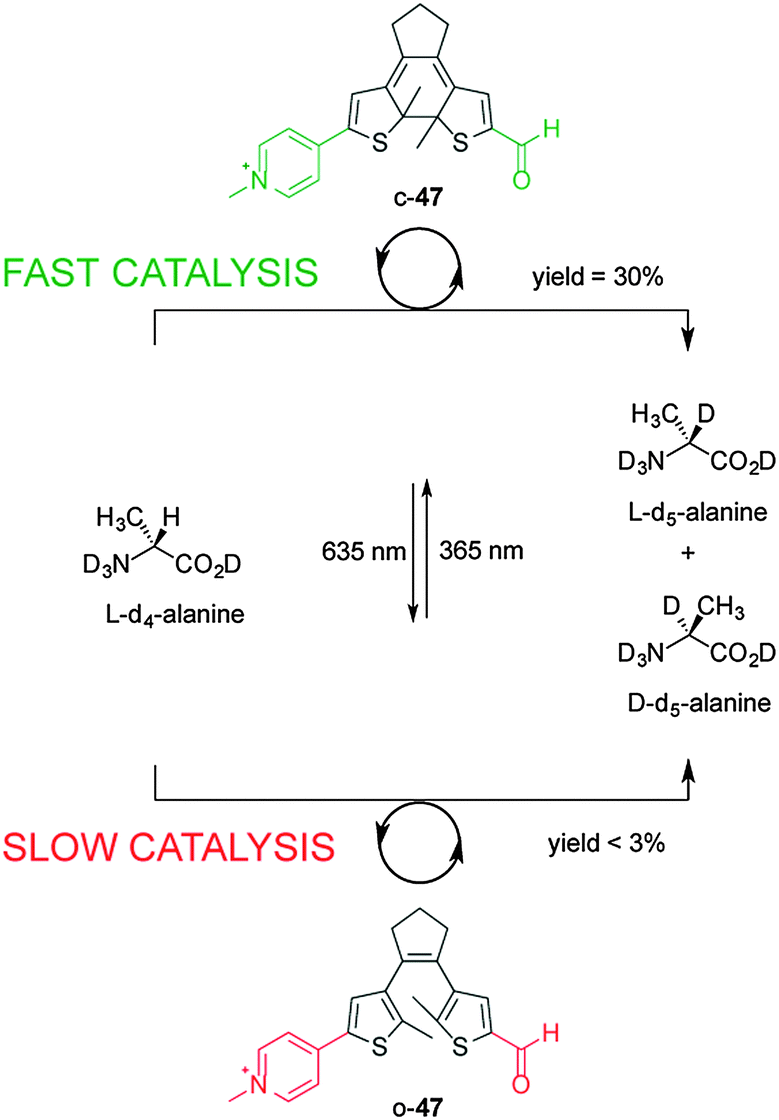 | ||
| Scheme 12 Controlling the rate of the racemization of L-alanine using switchable catalyst o/c-47.26 | ||
Bielawski and Neilson described control of the rate of transesterification and amidation reactions promoted by an electronically-modulated N-heterocyclic carbene (NHC) organocatalyst (Scheme 13).27 Switchable catalyst 48 is based on a NHC unit incorporated into a dithienylethene scaffold, where the length of the conjugated π-system modulates the electronic density at the carbenoid center of the imidazolium salt. In the presence of visible light and base the open form of the NHC catalyst, o-48, catalyzes transesterification and amidation reactions. Upon UV irradiation to the closed form, c-48, the rate of both transesterification and amidation reactions is significantly decreased (ko-48/kc-48 = 12.5 and 100, respectively). The difference in catalytic activity was rationalized on the basis of NMR experiments using an isotopic label at the C2 ‘carbene’ carbon that showed that the ring-open form (o-48) exists as an imidazolium species while the ring-closed species c-48 forms a less active alcohol adduct. The rate of the amidation reaction was successfully switched several times between the fast and slow states by alternating exposure to UV and visible light over the course of the reaction.
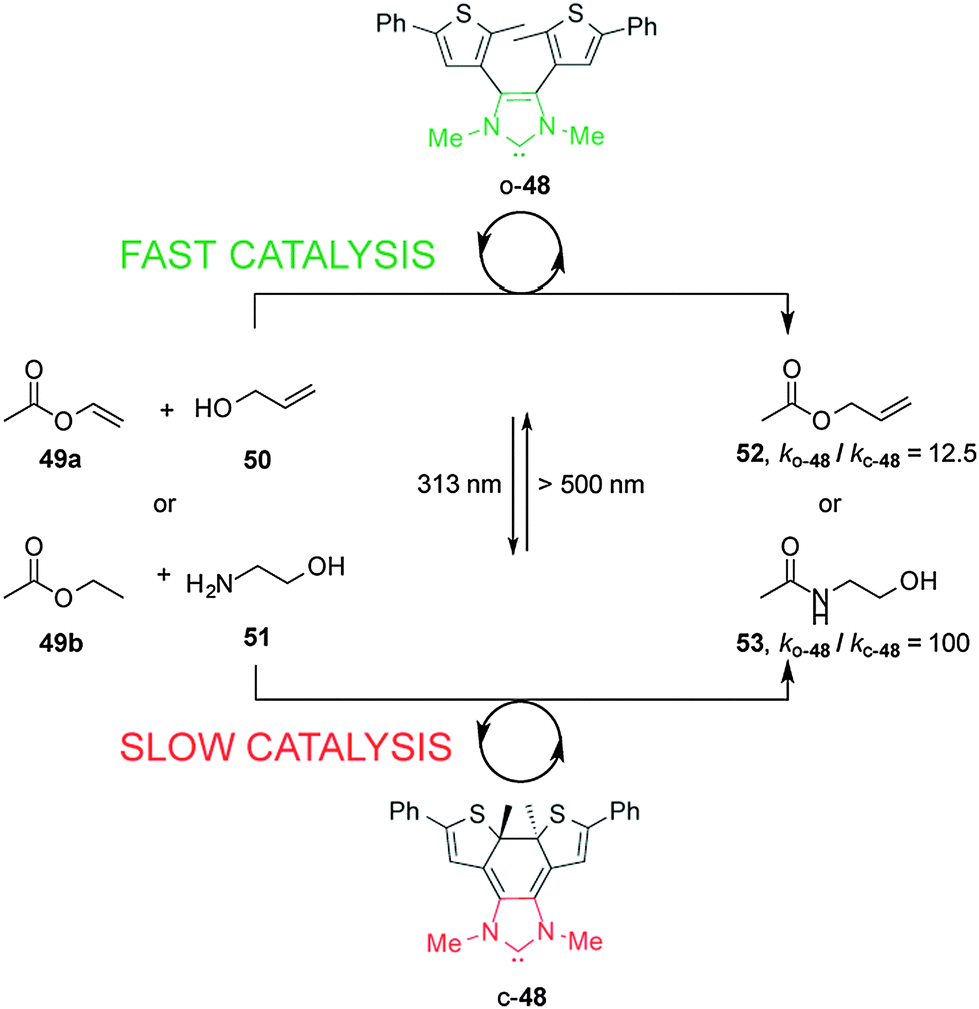 | ||
| Scheme 13 Controlling the rate of transesterification and amidation reactions using switchable catalyst o/c-48.27 | ||
The same group expanded the scope of the NHC switchable organocatalyst 48 to include the ring-opening polymerization of cyclic esters, such as δ-valerolactone and ε-caprolactone, achieving good control over the catalytic activity.28
The ability to photomodulate the electronic density of the NHC was further extended to control the rate of the rhodium(I)-catalyzed hydroboration of styrene (Scheme 14).29 Under visible light, open Rh(I) complex o-54 efficiently promoted the reaction. Exposure to UV light to form the close form, c-54, attenuated the rate of catalysis by almost one order of magnitude, an effect attributed to the decrease in donor strength of the NHC ligand upon photocyclization. The rate of the hydroboration of styrene with pinacolborane was switched several times over the course of a single reaction via alternating UV and visible irradiation.
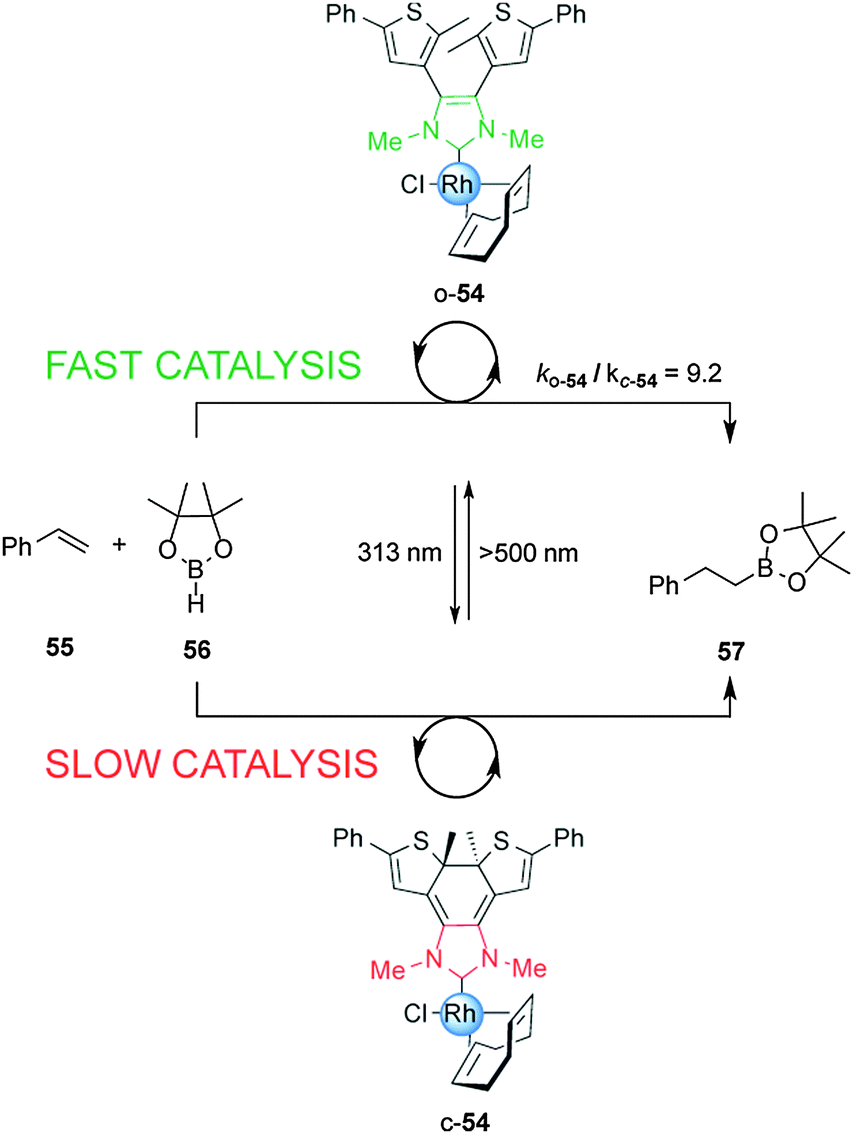 | ||
| Scheme 14 Controlling the rate of a hydroboration reaction using switchable rhodium catalyst o/c-54.29 | ||
Willner and co-workers reported on a surface-confined photoisomerizable monolayer coupled to negatively charged Pt nanoparticles for photoswitchable electrocatalysis and chemiluminescence (Scheme 15).30 The catalyst (58) is based on a photoswitchable nitrospiropyran linked to an indium tin oxide (ITO) electrode to form a monolayer that in closed form (c-58) lacks affinity for the negatively charged Pt nanoparticles and shows no catalytic activity towards H2O2 reduction. Upon UV irradiation the nitrospiropyran ring opens and picks up a proton to form a positively charged species (o-58), which attracts the Pt nanoparticles to the ITO electrode facilitating electrocatalytic H2O2 reduction. The reaction was monitored both electrochemically and via luminol addition which undergoes chemoluminescence upon UV irradiation in the presence of H2O2. The catalytic activity was successively switched ‘on’ and ‘off’ through alternating exposure to different wavelengths of light for up to ten cycles without detectable degradation of the heterogeneous catalyst.
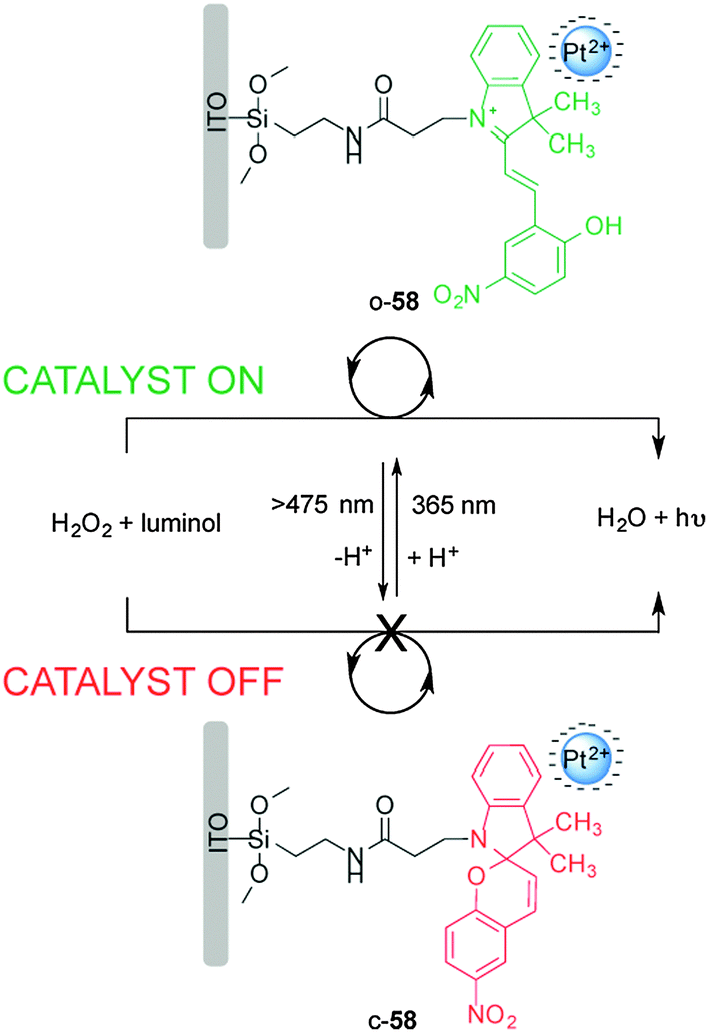 | ||
| Scheme 15 Heterogeneous photoswitchable catalyst o/c-58 for electrocatalysis and chemiluminescence.30 | ||
2.4 Aggregation/dissociation
The dynamic self-assembly of molecules into supramolecular ensembles is the basis of many functional systems in biology and has inspired the construction of synthetic systems with dynamic and responsive properties. Grzybowski's group reported the photoswitchable catalyzed hydrosilylation of p-anisaldehyde (60) and diphenylsilane (61), controlled by reversible aggregation/dispersion of catalytic Au nanoparticles (59) (Scheme 16).31 Heterogeneous catalyst 59 consists of Au nanoparticles decorated with a mixture of alkyl amines and azobenzene-terminated alkyl thiol moieties. Under visible light, the Au nanoparticles (E-59) remain dispersed and exposed to a large solvent surface area, efficiently promoting the hydrosilylation reaction (kE-59 = 9 × 10−4 mM−1 min−1). Irradiation with UV light isomerizes the E-azobenzene units of the thiol chains to the Z isomer, developing electric dipoles that cause aggregation of the Au nanoparticles (Z-59), decreasing their catalytic activity (kZ-59 = 1 × 10−5 mM−1 min−1; kE-59/kZ-59 = 90).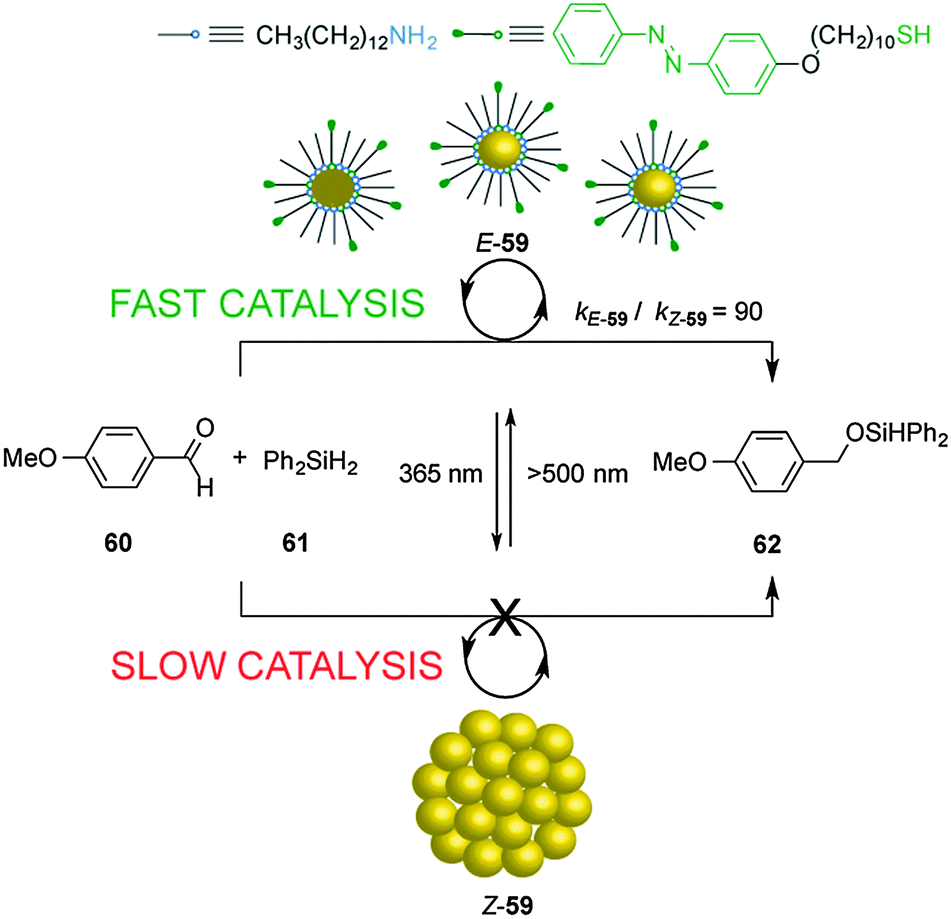 | ||
| Scheme 16 Heterogeneous photoswitchable catalyst E/Z-59 for the hydrosilylation of p-anisaldehyde (60).31 | ||
Shibasaki's group have developed a photoswitchable nucleophilic catalyst 63, that controls catalytic activity through switching between aggregated/dissociated states of a dipeptide derivative (Scheme 17).32 The switchable catalyst has an azobenzene unit linked through the peptide to a nucleophilic 4-aminopyridyl group. The aggregated/dissociated state varies according to the E/Z geometry of the azobenzene. The E-63 isomer has limited solubility due to extensive aggregation, almost switching ‘off’ the catalysed-rearrangement of 2-acyloxybenzofuran (64) (14% conversion after 100 min). In contrast Z-63 catalyzes the reaction effectively (80% conversion after 100 min). The switchable catalyst could also be used to promote the O-tert-butoxycarbonylation of 1-naphthol.
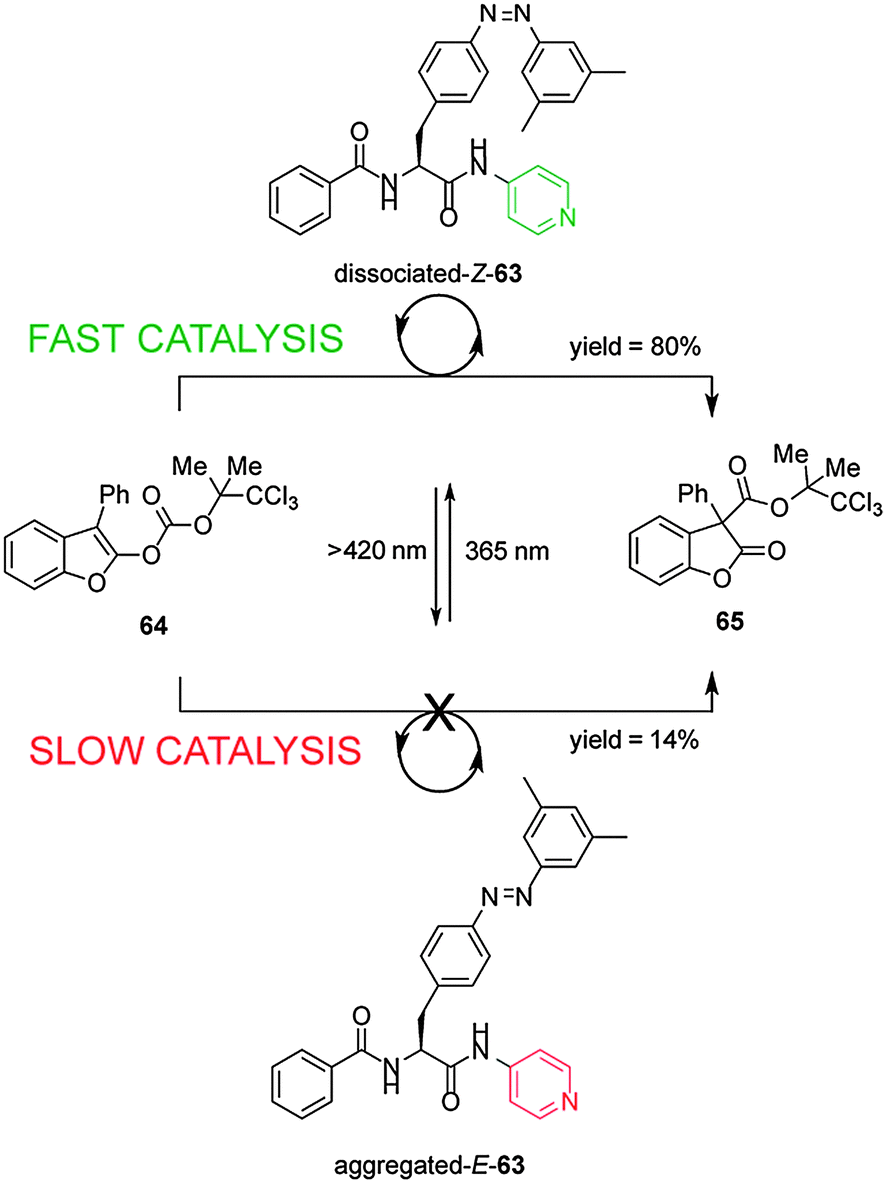 | ||
| Scheme 17 Controlling the rate of a rearrangement using photoswitchable catalyst E/Z-63.32 | ||
3. pH-driven switching
Chemically-driven processes driven by changes in pH are common in both nature2 and artificial molecular machines. However, in the context of controlling catalysis few examples have been reported thus far. An early use of a change in pH to control catalytic activity was reported by Schanz,33 who exploited the change in the electron-donating ability of N-heterocyclic carbene (NHC) ligands upon protonation to moderate the activity of Ru(II)-based olefin metathesis catalysts (Scheme 18).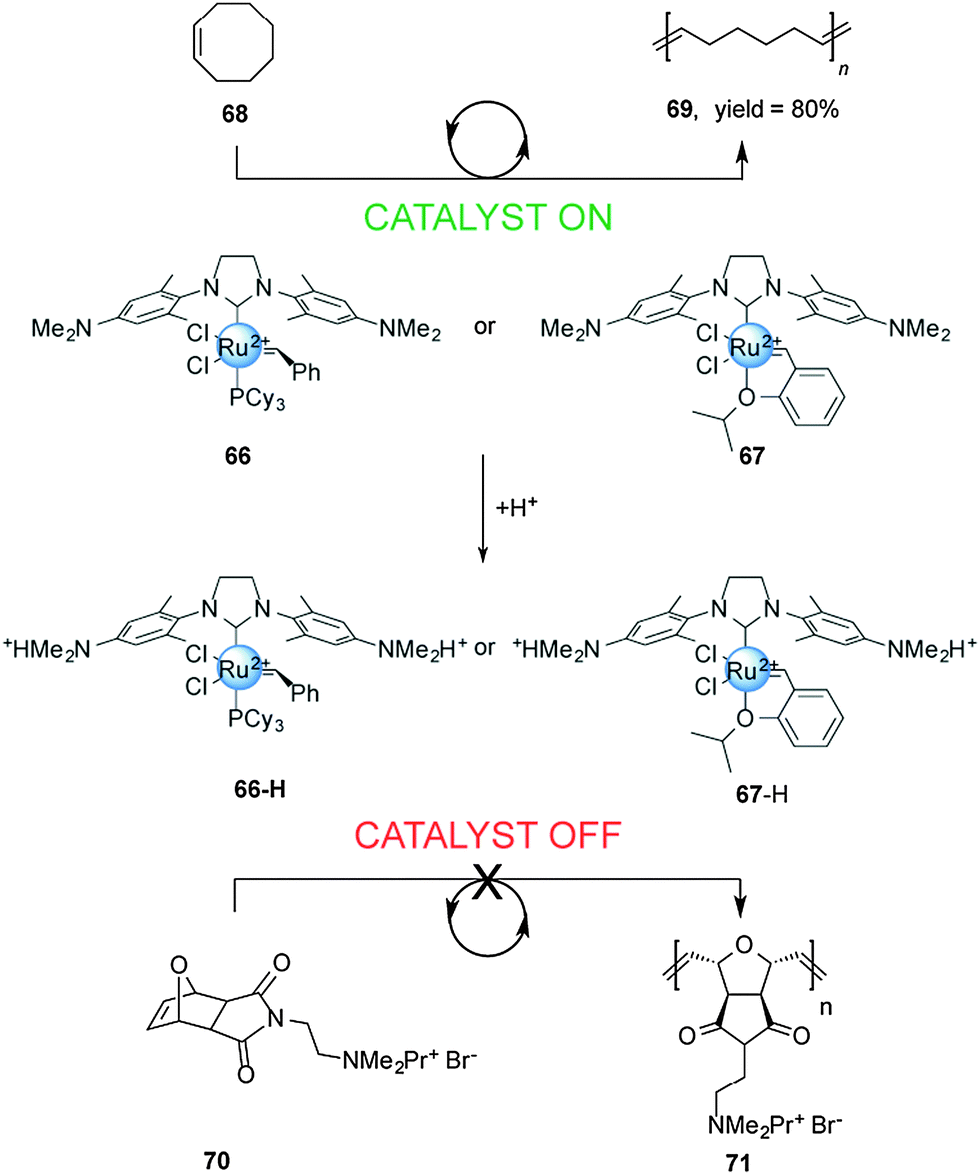 | ||
| Scheme 18 Controlling the rate of ROMP using pH-switchable catalysts 66 and 67.33 | ||
The first generation Grubbs-type 66 and Hoveyda–Grubbs-type 67 catalysts bear a pH-responsive H2ITap ligand (H2Tap = 1,3-bis(2′,6′-dimethylaminophenyl)-4,5-dihydroimidazol-2-ylidene). Control over the catalytic activity of these two pH-switchable ligands was investigated in ring-opening metathesis polymerization (ROMP) reactions. Under neutral conditions the NMe2 groups in the catalyst backbones are uncharged and the system showed excellent catalytic activity in the ROMP of cyclooctene (>80% conversion in 7 min with 66 and in 28 min with 67). Upon protonation with HCl, both catalysts become dicationic and the ROMP of cationic exo-7-oxanorbornene becomes negligible. The researchers also investigated the performance of these pH-responsive catalysts in the ring closing metathesis (RCM) of diallyl malonic acid. The non-protonated forms of 66 and 67 showed good conversion (45% and 59% after 60 min, respectively). In acid media, the RCM proceeded at slower rates and did not afford complete conversion.
In an extension of this work,34 the group demonstrated that gradual addition of p-toluenesulfonic acid (TsOH) reduced the rate of the ROMP reaction with the exo-7-norbornene derivative. DFT calculations of the atomic charge at the metal center of catalyst 66 and its mono- and dicationic forms confirmed that the propagation rates should be significantly affected upon protonation.
Plenio and co-workers subsequently described related Grubbs–Hoveyda complexes related to 66 and 67 that showed significant changes in the double-bond geometry (E/Z ratio = 0.78 to 1.04) and microstructure of the product in the ROMP of norbornene upon protonation of the catalyst.35
Recently, synthetic rotaxane catalysts36 in which the catalytic activity or the outcome of the reaction can be controlled by changes in pH have been reported.37 Rotaxane 72 consists of a dibenzo-24-crown-8 macrocycle and a thread bearing a dibenzylamine/ammonium unit, that acts as catalyst, and triazolium rings as alternative binding sites for the macrocycle (Scheme 19).38 The organocatalytic group can be concealed or revealed, and its activity turned ‘on’ and ‘off’, by the well-defined acid/base-promoted translocation of the macrocycle between the binding sites on the thread. In neutral conditions, the triazolium ring is the preferred binding site for the macrocycle in 72 and so the dibenzylamine residue is available for catalysis (Scheme 19, top). Upon protonation (72-H), the macrocycle preferentially binds the ammonium unit, switching ‘off’ the catalytic activity (Scheme 19, bottom). Switchable rotaxane catalyst 72 proved to very effectively control the rate of Michael addition of an aliphatic thiol (74) to trans-cinnamaldehyde (73), affording product 75 only when the reaction was catalyzed with the non-protonated rotaxane (72). The catalysis could be turned ‘on’ in situ through deprotonation of 72-H with similar effectiveness (Scheme 19).
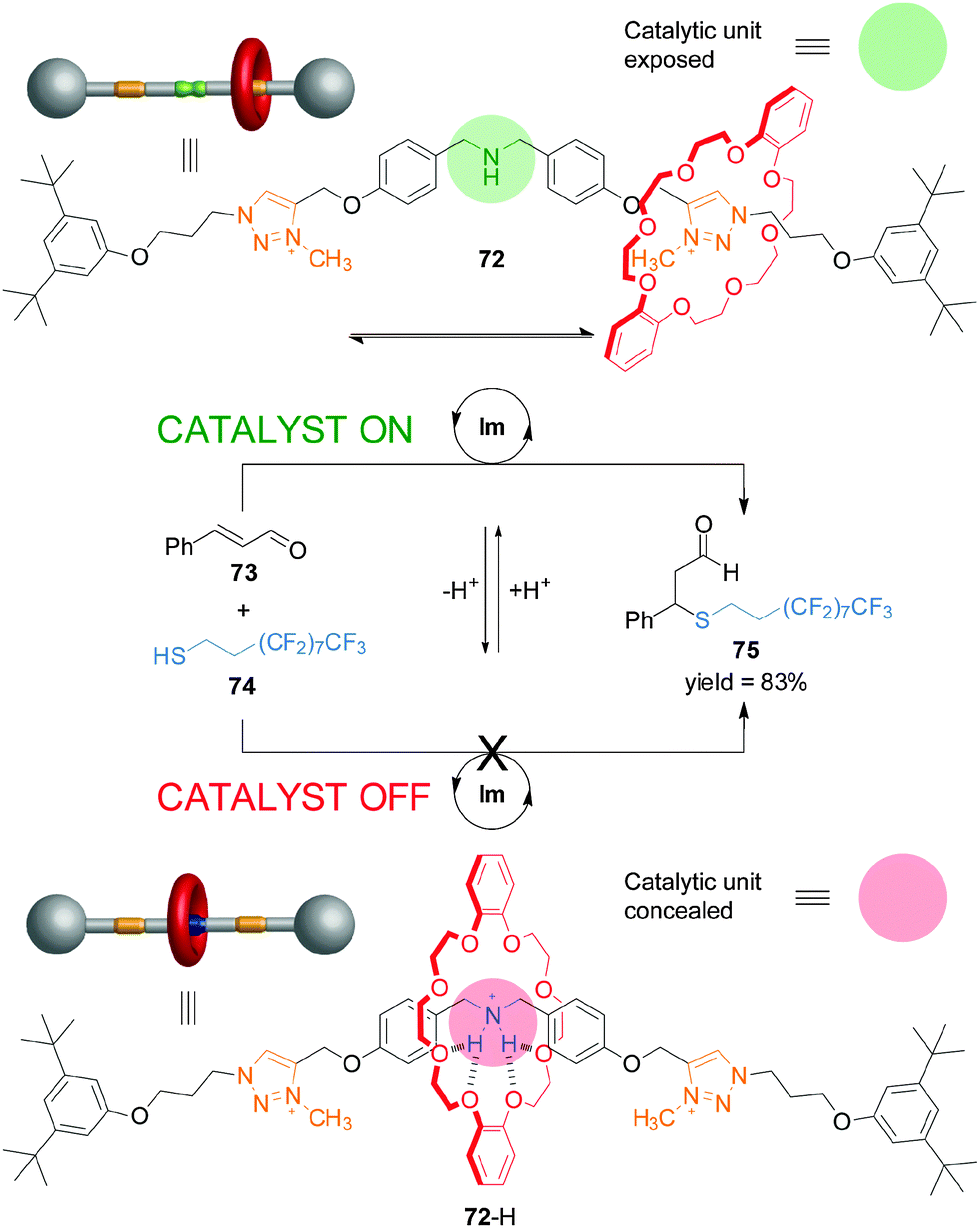 | ||
| Scheme 19 Controlling the rate of Michael addition using a pH-switchable rotaxane catalyst (72).38 | ||
Rotaxane 72 catalyzes the Michael addition reaction shown in Scheme 19 through iminium ion activation. However, secondary amines can also catalyse reactions through other activation pathways. One goal of switchable catalysts is to be able to use them to react selectively with particular building blocks within a pool of potential reactants. By switching ‘on’ and ‘off’ various types of catalyst in different orders by applying different stimulus it may then prove possible to carry out different sequences of reactions, generating different products from a common pool of building blocks. To accomplish this it is necessary to establish the scope and limitations of the switchable catalysts involved in the processes. Accordingly rotaxane 72 was investigated for its ability to catalyze a variety of reaction types through different activation modes (Fig. 1).39
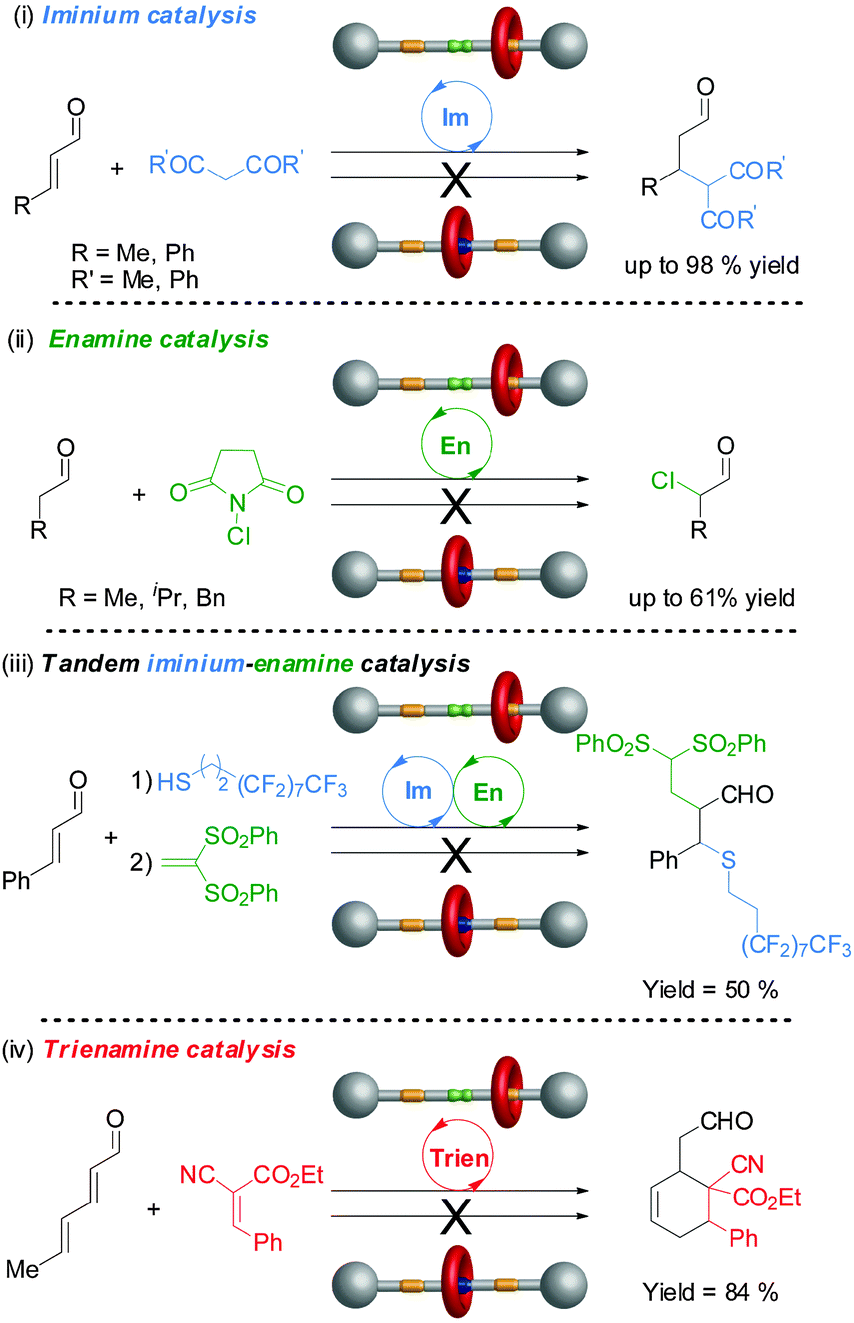 | ||
| Fig. 1 The scope of switchable rotaxane organocatalyst 72 through different activation mechanisms.39 | ||
The non-protonated rotaxane 72 (‘on-state’) very effectively catalyzed the β-functionalization of carbonyl compounds with C or S-nucleophiles through iminium activation (often 95–98% conversion), although the protonated form 72-H (‘off-state’) also promotes the most facile of these transformations to a lesser degree (up to 14% conversion) (Fig. 1(i)). Rotaxane 72 also promoted nucleophilic addition or substitution reactions via enamine catalysis, although with less effectiveness (40–61% conversions). However, switching is more effective with enamine catalysis, with the ‘off-state’ of the catalyst showing no detectable catalytic activity in these reactions (Fig. 1(ii)). The rotaxane catalyst is even able to promote tandem iminium-enamine reaction sequences with high efficiency (Fig. 1(iii)) and the Diels–Alder reaction of a dienal through a trienamine activation pathway (Fig. 1(iv)).
This type of switchable catalysis was extended to an asymmetric organocatalytic rotaxane (76) that features a simple acyclic chiral secondary amine housed within a rotaxane framework (Scheme 20).40 Switchable chiral rotaxane 76 was able to control the catalyzed Michael addition of 1,3-diphenylpropan-1,3-dione (78) to aliphatic α,β-unsaturated aldehydes (77a–c). The acyclic chiral secondary amine promotes the reaction with stereochemical control comparable to—or better than—commercial cyclic amine organocatalysts, albeit at the expense of slower rates of conversion. In situ switching between the catalyst states by alternating addition of acid and base afforded excellent control over both the rate and stereoselectivity of the reactions.
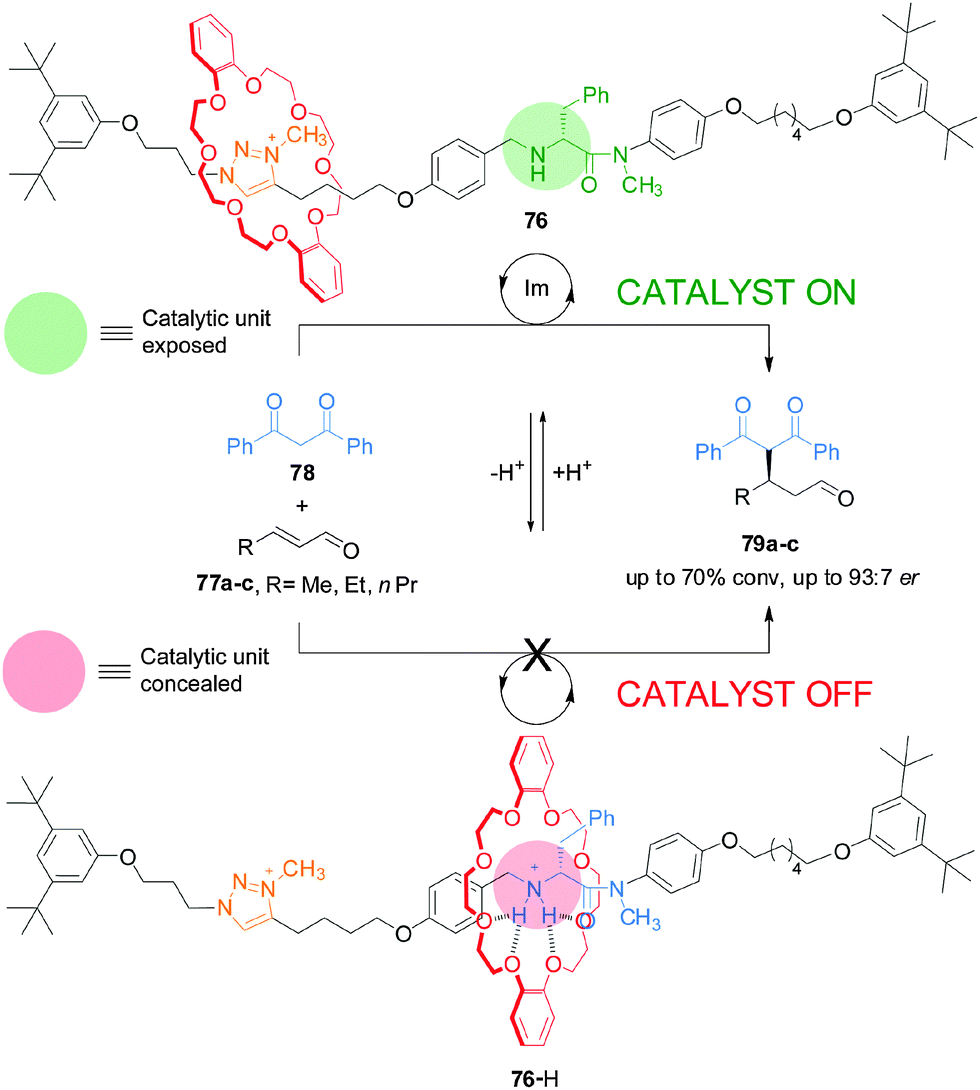 | ||
| Scheme 20 Controlling the rate and stereoselectivity of Michael addition using a switchable rotaxane asymmetric organocatalyst 76.40 | ||
Recently, a switchable rotaxane system was developed, 80, featuring two different organocatalytic sites: a squaramide moiety and a dibenzylamine group (Scheme 21).41 When the rotaxane is protonated (80-H), the macrocycle preferentially interacts with the ammonium unit revealing the squaramide unit, which can promote the Michael addition of 1,3-diphenylpropan-1,3-dione (78) to trans-β-nitrostyrene (81) through hydrogen bond catalysis (75% conversion after 18 h). In basic media the macrocycle preferentially resides over the squaramide, revealing the secondary amine which promotes the Michael addition of 1,3-diphenylpropan-1,3-dione (78) to crotonaldehyde (77a) via iminium ion catalysis (40% after 40 h). In this way the catalyst state controls which building blocks react together—and what product is formed—from a mixture of reactants.
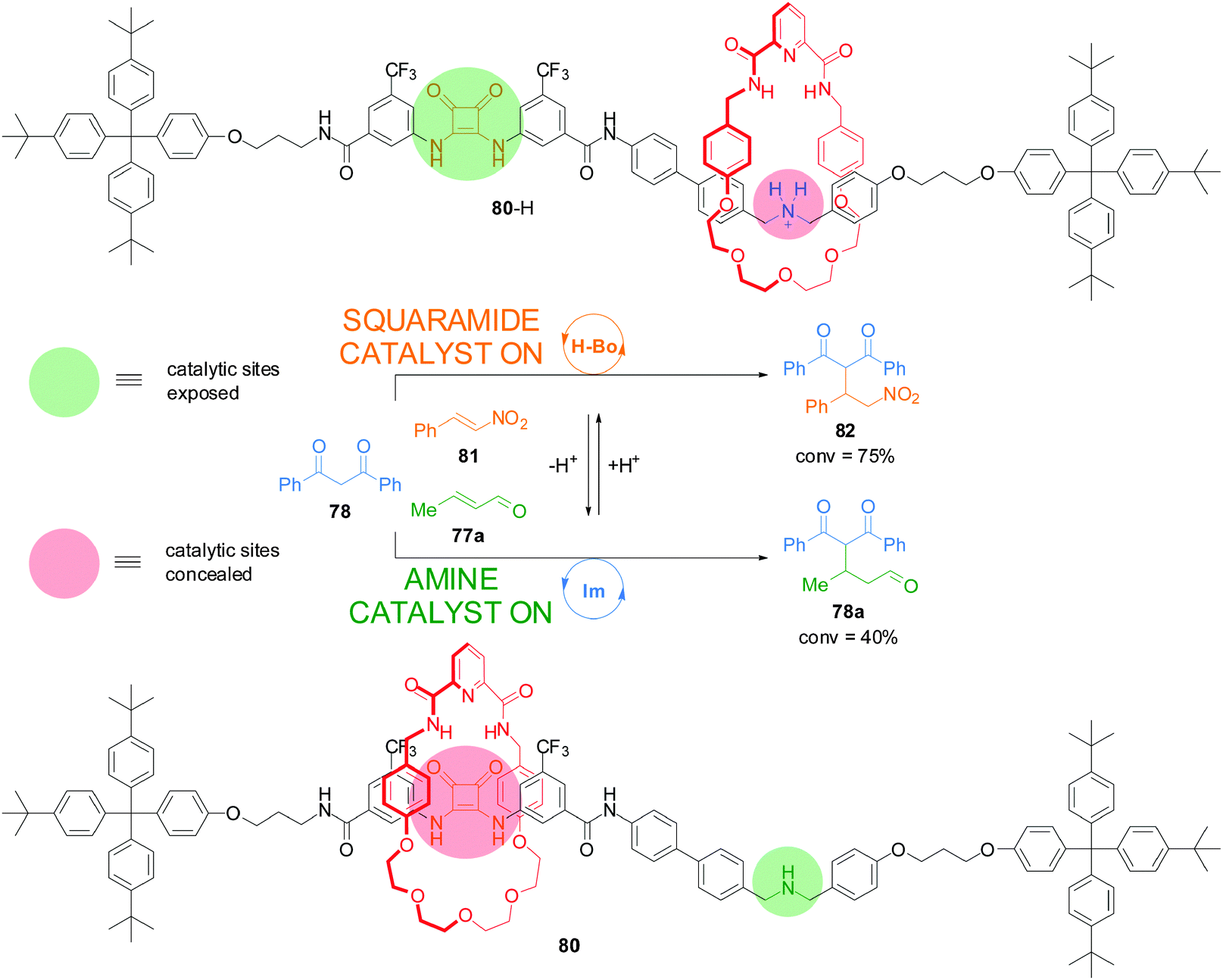 | ||
| Scheme 21 Controlling the reaction outcome from a mixture of building blocks using a switchable rotaxane featuring two different organocatalytic sites, 80.41 | ||
4. Coordination-driven switching
One of the first uses of metal-ion-coordination to enhance reactivity was reported by Shinkai and co-workers in 1983 (Scheme 22). They described a flavin derivatised with a crown ether (83) that reacted stoichiometrically with an NADH model (84) at an enhanced rate in the presence of alkali metal ions (up to 1.8-fold rate enhancement) or ammonium groups (2.4-fold acceleration).42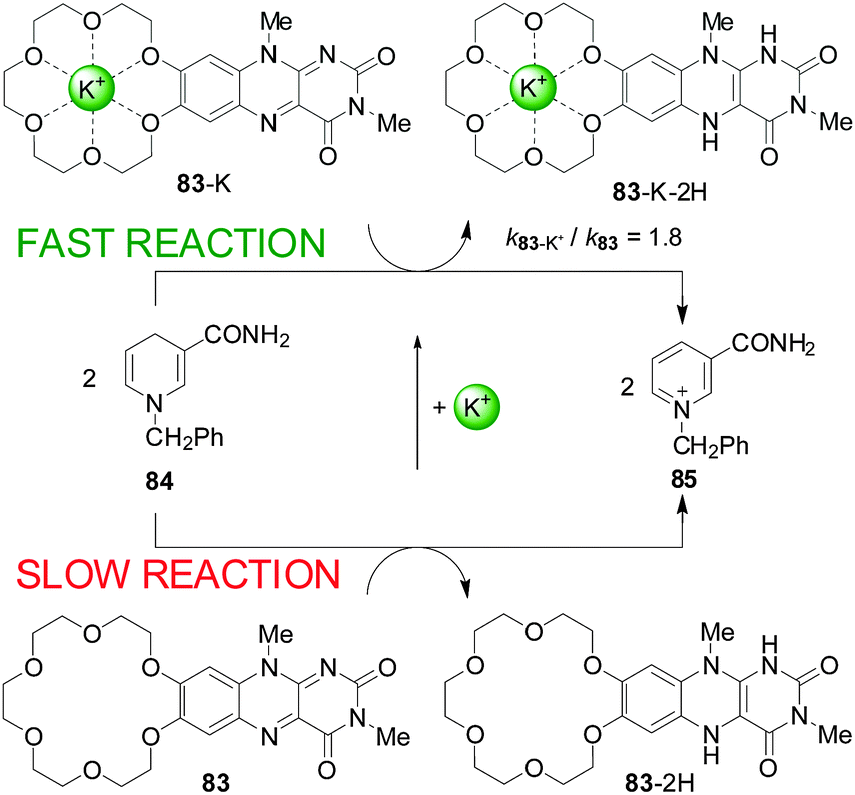 | ||
| Scheme 22 Acceleration of the redox reaction between 83 and 84 by coordination of an alkali metal ion to the crown ether.42 | ||
Many examples followed in which the activity of a catalytic centre could be influenced by tuning coordination binding events.43 Two main approaches have been developed for this purpose, the addition/removal of external ligands that can complex to a metal ion present in the catalyst (Section 4.1), and the addition/removal or metal ions that can coordinate to binding motifs in the structure of the catalytic system (Section 4.2).
4.1 Ligand-based switching
Mirkin and co-workers have developed a series of heterometallic macrocycles incorporating metal-based catalytic units whose activity can be regulated through a reversible coordination-induced conformational change in the molecule (e.g.Schemes 23–29).43 The switching mechanism is based on what the authors term the ‘weak link approach’.44 It involves metal complexation in side-arms additional to the catalytic site, through hemilabile ligands that typically bind to the auxiliary metal centres in a bidentate fashion in one state of the catalyst and in a monodentate mode in the other state. The metal centre is usually Rh(I), Cu(I) or Pt(II). In bidentate mode the molecule adopts a compact or ‘closed’ conformation. Small monodentate ligands (e.g. Cl− and/or CO) that bind to the metal centre more strongly than the ‘weak link’ (e.g. a thioether) of the bidentate ligand can displace it from the metal ion, inducing a conformational change in the molecule, which now adopts a more open conformation. The process can be reversed as removal of the small monodentate ligands allows the weak binding motif of the hemilabile ligand to re-coordinate to the metal centre, restoring the closed conformation.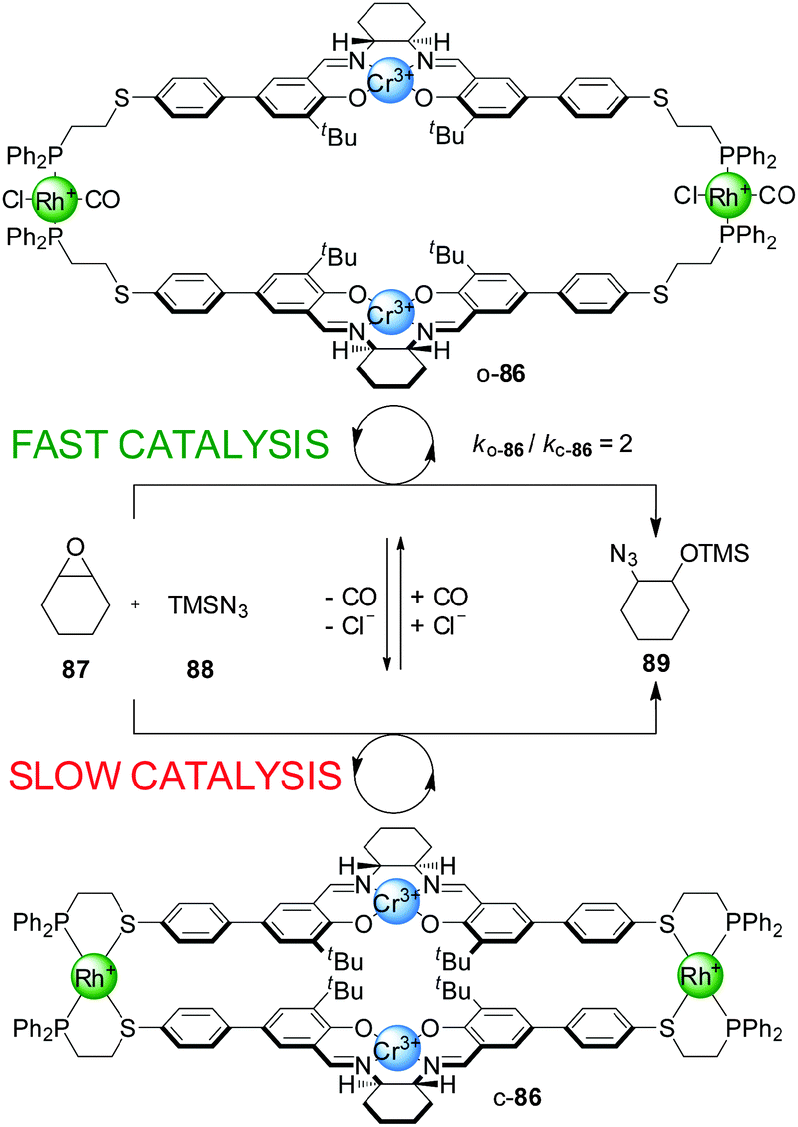 | ||
| Scheme 23 Control of the rate of an epoxide-opening reaction with catalyst o/c-86 through a change in catalyst conformation induced by coordination of small ligands to the side-arm metal complexes.45 | ||
 | ||
| Scheme 24 Coupling of coordination-controlled acetylation of 4-pyridylcarbinol (91) to a pH-sensitive fluorescence probe (94).46 | ||
 | ||
| Scheme 25 Regioselective control over the rate of acetylation of pyridine carbinol isomers using porphyrin-based switchable catalyst o/c-95.49 | ||
 | ||
| Scheme 26 Activation/deactivation by coordination-triggered switching between active ‘open’ and inactive ‘closed’ conformations of Zn(II) catalyst o/c-98.50 | ||
 | ||
| Scheme 27 Coordination-driven switching of the activity of tweezer catalyst o/c-102.51 | ||
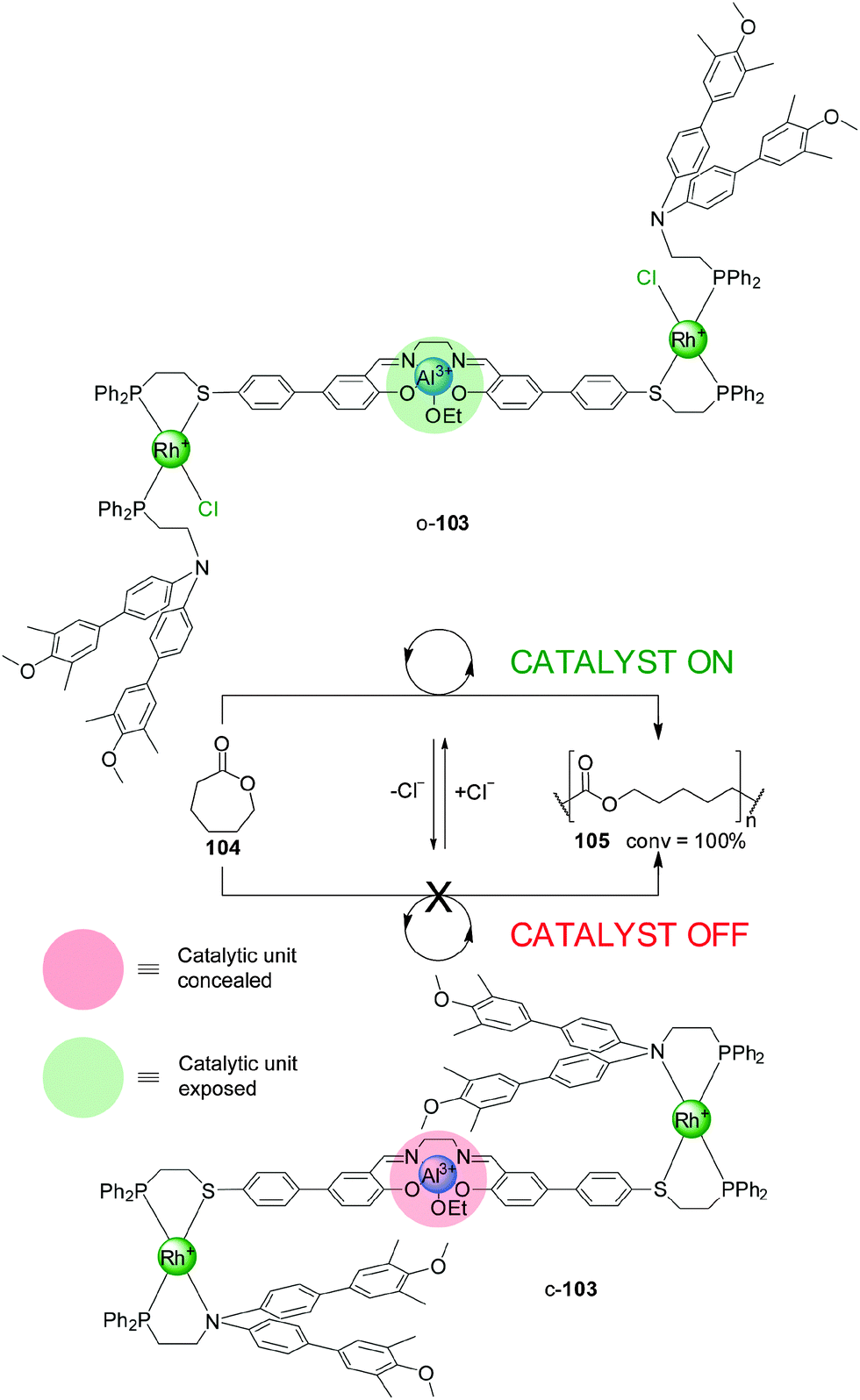 | ||
| Scheme 28 Control of the activity of Al(III)–salen catalyst o/c-103 through chloride binding.52 | ||
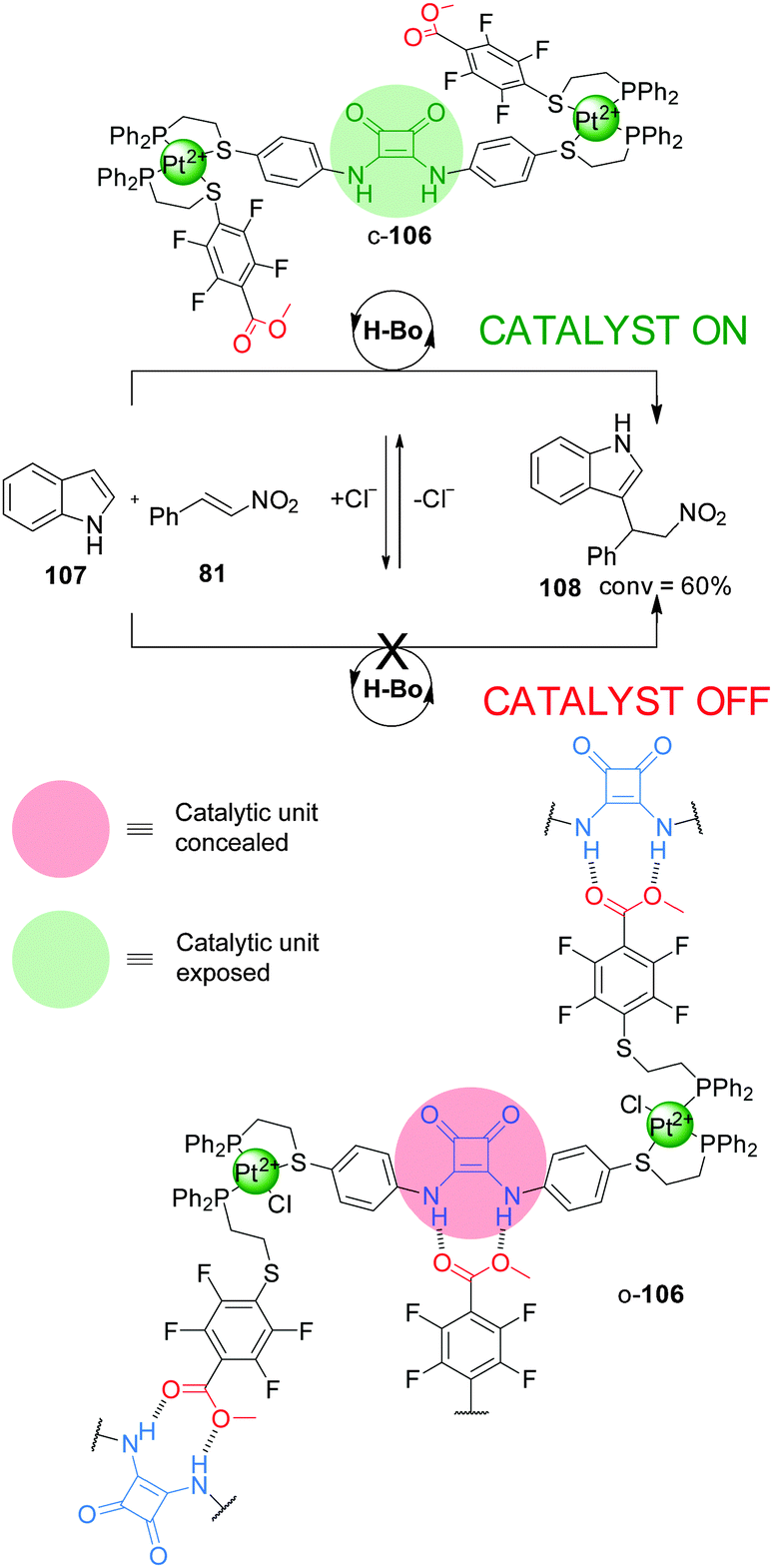 | ||
| Scheme 29 Switching ‘on’ and ‘off’ squaramide-based hydrogen-bond organocatalyst 106 by chloride-mediated aggregation.53 | ||
The rationale behind the application of this concept in switchable catalysis is that the control of the distance between the catalytic sites could influence their activity. The first such system developed consisted of a tetrametallic macrocycle with two Cr(III)–salen complexes as the catalytic units and two Rh(I) centres with P,S-bidentate ligands as the side-arm complexes (Scheme 23).45 When Cl− and CO were added to the reaction media, they bound to the Rh(I) ions replacing the thioether ligand and the system adopted the ‘open’ conformation (o-86). Removal of the CO under reduced pressure or through a N2 purge switched the catalyst back to the closed conformation (c-86), as both external ligands are required to displace Rh–S bonds. The catalytic activity of the two conformations was tested in the ring opening of cyclohexene oxide (87) with TMSN3 (88), which is catalyzed by Cr(III)–salen catalysts through a bimetallic intermediate. The ‘open’ conformation of the catalyst showed a modest two-fold enhancement of the reaction rate compared to the ‘closed’ conformation.
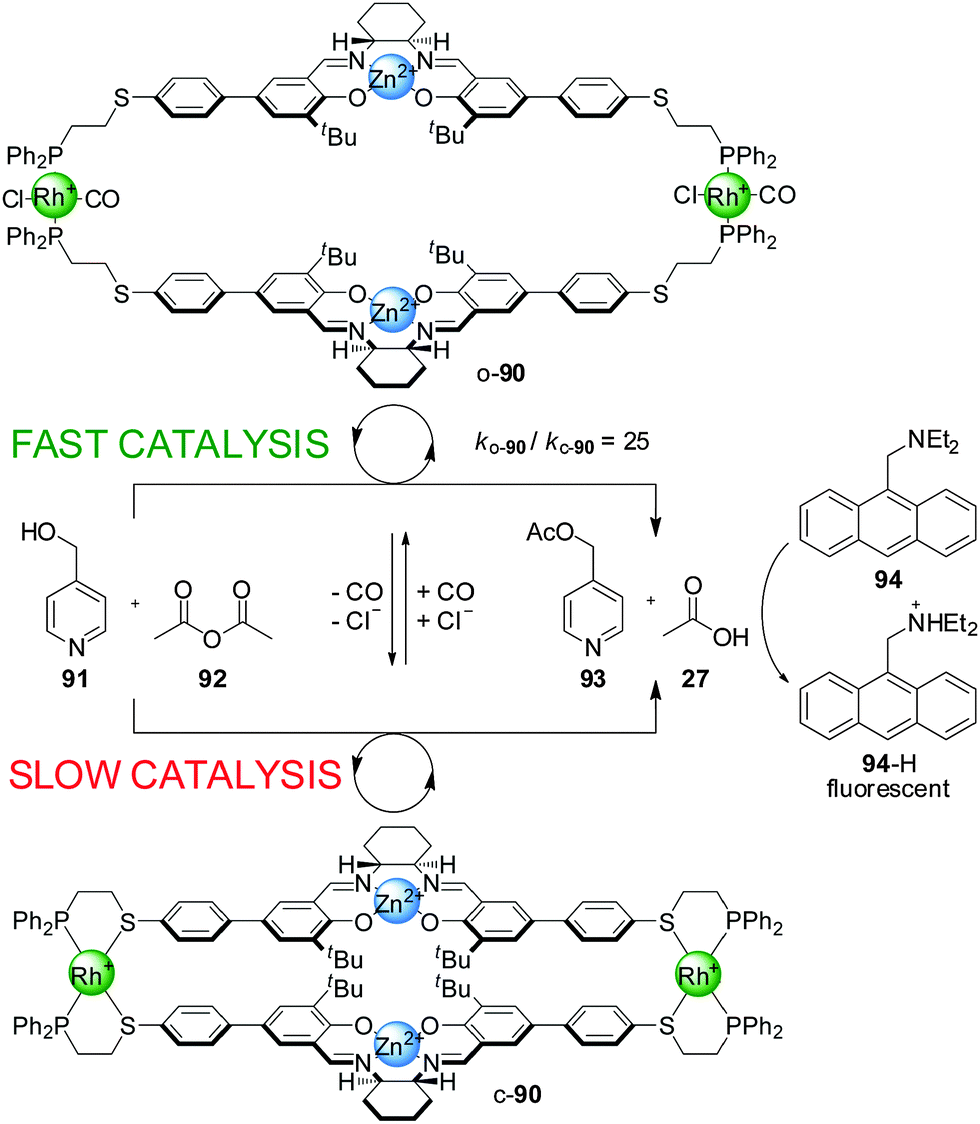
When a Zn(II)–salen complex was used instead of Cr(III) in switchable catalyst 90, the Cl− and CO-induced open–closed switching was more effective at controlling reactivity (Scheme 24).46 Acetylation of 4-pyridylcarbinol (91) with acetic anhydride (92) proceeded around 25× faster with the ‘on’ (‘open’) state than with the ‘off’ (‘closed’) state of the catalyst. The authors postulated that the closed state of the catalyst promoted the reaction through Lewis-acid-activation at a single site, while in the open state both metal centres participate in the catalysis, activating both the acetic anhydride and pyridylcarbinol substrates. The reaction was coupled to diethylaminomethylene anthracene (94), a pH-sensitive fluorophore, resulting in a fluorimetric sensor able to detect concentrations of Cl− as low as 800 nM (Scheme 24).
It was found that the switching of catalyst 90 was also possible using acetate anions instead of chloride, which could be used to establish autocatalytic rate acceleration. Under the reaction conditions the ‘off’ state of 90 catalyzed the acetylation of pyridine carbinol at a very slow rate. Upon addition of 10 mol% of tert-butylammonium acetate, a percentage of the inactive catalyst switched to its active form, which started to catalyze the reaction generating as the products 4-acetoxymethylpyridine (93) and acetic acid (27). The acetic acid is deprotonated by a base present (diethylaminomethylene anthracene, 94) and coordinates to additional Rh(I) centres, generating additional amounts of the active form of the catalyst, progressively accelerating the reaction until saturation at around 80% conversion. The autocatalytic sequence effectively amplifies detection of acetate ion.47
Cu(I) can also be used instead of Rh(I) in the switching mechanism. Bidentate ligands such as phenanthroline and 4,4′-bipyridine, and monodentate species such as tert-butylisonitrile, were found to promote the switching from the closed to the open conformation by coordination to the copper(I) centre.48 However, with Cu(I) the switching is irreversible and the original off state cannot be restored. The corresponding Cr(III)–salen-based catalyst with the Cu(I) side-arm complex had a 12-fold rate enhancement in the ‘on’ state compared to the ‘off’ state for the reaction between 4-pyridylcarbinol (91) and acetic anhydride (92).
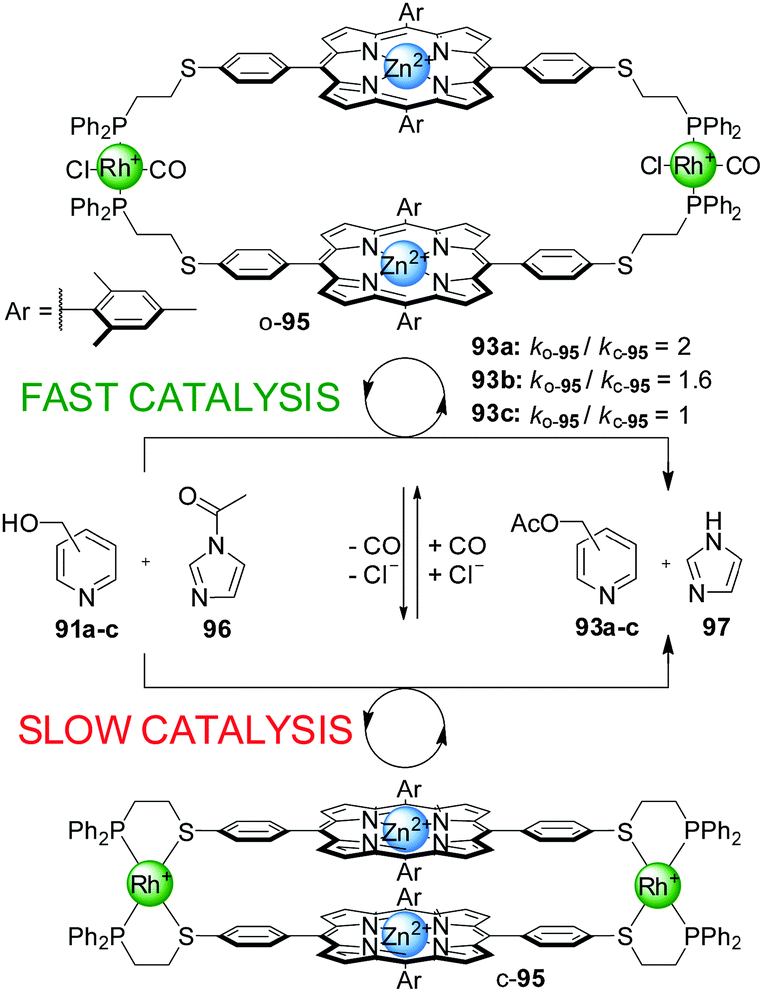
Switchable catalysts of this type based on catalytic units other that Cr(III)–salen complexes have also been explored. A system featuring Zn(II)–porphyrin catalytic centres (95) was tested in the acetylation of different isomers of pyridine carbinol (91a–c) with acetylimidazol (96), but did not afford better results than previous systems (only a 2-fold acceleration of the reaction, Scheme 25).49 However, the system displayed interesting regioselectivity. The open conformation of the switchable catalyst (o-95) showed enhancement of the catalytic activity with the para- and meta-isomers of pyridylcarbinol (91a and 91b, respectively), but not with ortho-pyridylcarbinol (91c). Therefore, discrimination of 2-pyridylcarbinol over the 3- and 4-isomers was achieved.
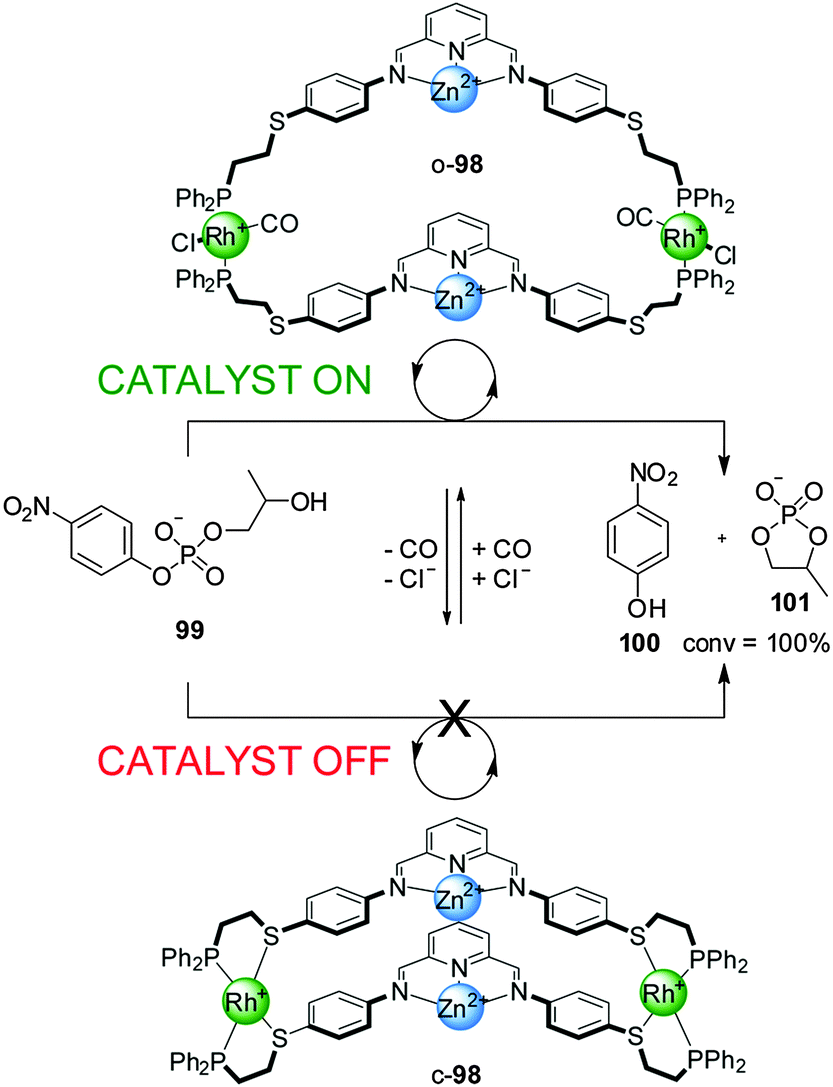
An unwelcome characteristic of most of these systems is that discrimination between the active and inactive states is limited by significant catalytic activity displayed by the ‘off’ closed state. This limitation was overcome in a system featuring Zn(II)–pyridine–bisimine catalytic motifs (98) with switching again triggered by the addition/removal of Cl− and CO (Scheme 26).50 In the hydrolysis of 2-(hydroxypropyl)-p-nitrophenyl phosphate (99) in a water/methanol 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3 mixture the closed state of the catalyst (c-98) was completely inactive while the open state (o-98) led to full conversion within 40 min. The absence of activity in the ‘off’ state was attributed to the short Zn–Zn distance that precluded the bimetallic intramolecular reaction, and to the presence of an acetate ion bridging the two Zn(II) centres hindering access of the substrates. In situ switching between both forms of the catalyst was also demonstrated.
:3 mixture the closed state of the catalyst (c-98) was completely inactive while the open state (o-98) led to full conversion within 40 min. The absence of activity in the ‘off’ state was attributed to the short Zn–Zn distance that precluded the bimetallic intramolecular reaction, and to the presence of an acetate ion bridging the two Zn(II) centres hindering access of the substrates. In situ switching between both forms of the catalyst was also demonstrated.
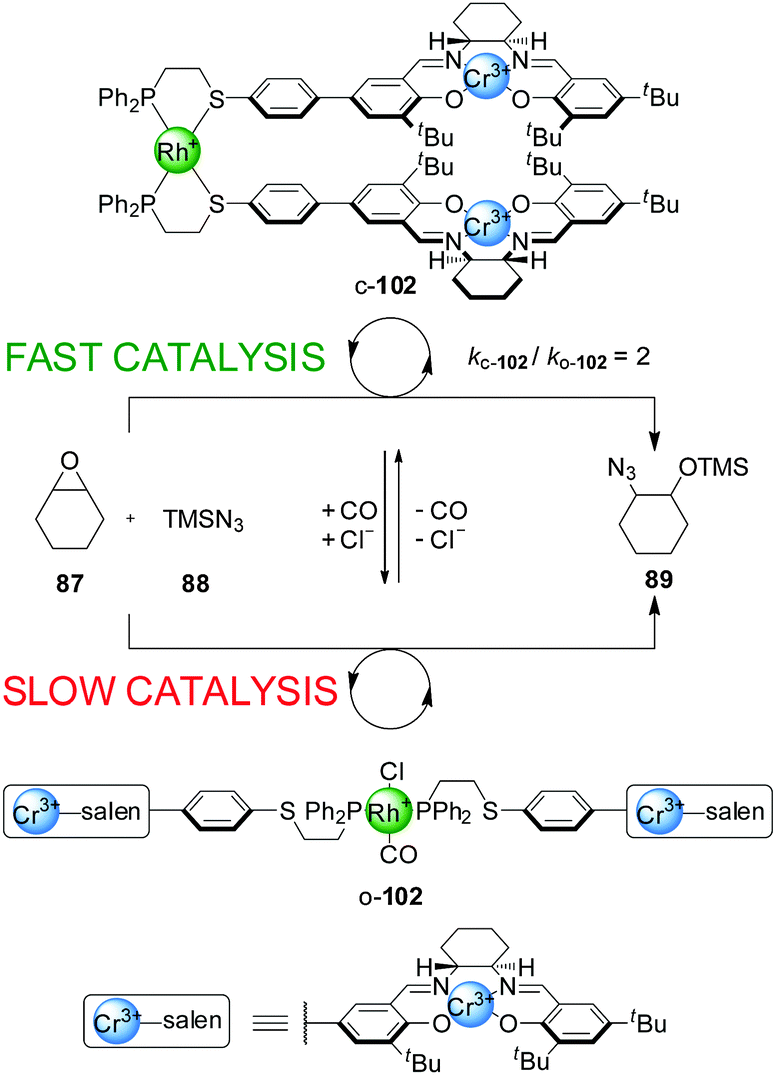
The same type of switching mechanism has been applied with acyclic catalytic assemblies, including a ‘tweezer’ catalyst, 102, incorporating two units of the Cr(III)–salen catalytic moiety and a Rh(I) centre coordinated to two P,S-hemilabile ligands (Scheme 27).51 Again, addition or removal of Cl− and CO ligands triggers switching between an ‘open’ and a ‘closed’ form. However, the catalytic activity of the system in the ring-opening reaction of cyclohexene oxide (87) with trimethylsilylazide (88) is reversed compared to the macrocyclic systems shown in Scheme 23. Here, the closed state (c-102) is the active form of the catalyst, as the Cr(III) catalytic centres are preorganized favouring the bimetallic catalysis. The open form (o-102) shows lower catalytic activity as a result of the more flexible arrangement of the salen sites, in part due to the phosphine ligands occupying a trans-coordination relationship across the Rh(I) centre. The system could be switched in situ by CO saturation/desaturation, alternating between reaction rates of ∼0.013 mM min−1 (less active, open form) and ∼0.026 mM min−1 (more active, closed form). The ‘on’ state afforded the product (89) with 80% ee.
An evolution of the switching mechanism was used in the design and operation of a triple-layer switchable catalyst, 103 (Scheme 28).52 This system consists of a Al(III)–salen motif—a catalyst for the living polymerization of lactones—linked to two P,S-bidentate ligands coordinated to Rh(I) square-planar complexes. The Rh(I) centres are also coordinated to P,N-bidentate ligands whose N atoms were derivatised with bulky blocking groups. Chloride ions or acetonitrile displace the nitrogen donor of the bidentate ligands. As a result the system can be switched from a closed conformation (c-103), in which the bulky groups block the catalytic unit (‘off’ state), to a ‘semi-open’ conformation (o-103), in which the catalytic site is exposed and can participate in catalysis (‘on’ state). Removal of the chloride ions with NaBArF switches the system back to the closed state. The system was tested in the Al(III)–salen-catalyzed ring-opening living polymerization of ε-caprolactone (104) (Scheme 28). The ‘on’ state of the catalyst gave full conversion of the monomer, while the ‘off’ state was almost completely inactive under the same reaction conditions (7% conversion to the polymer after 100 h). The active catalyst was in situ switched ‘off’ after 10 h, and the ‘on’ state restored with acetonitrile causing the reaction to resume.
The ‘weak link approach’ is not restricted to metal catalysis. A triple-layer design (106), which incorporates a squaramide organocatalytic group functionalised with P,S-bidentate ligands, has been used to control the rate of the Friedel–Crafts reaction between indole (107) and trans-β-nitrostyrene (81) (Scheme 29).53 The ligands are coordinated to a Pt(II) centre together with another hemilabile P,S-functionalised methyl tetrafluorobenzoate ligand. The system can be switched between the closed and semi-open forms by addition or removal of Cl−, which is able to displace the S atom of the weaker chelating S-tetrafluorobenzoate ligand. In the flexible semi-open form (o-106) the squaramide unit participates in intermolecular hydrogen bonding with a carboxylate group from another molecule of catalyst, resulting in aggregation and blocking of the active site (‘off’ state). In the rigid closed form (c-106) the intermolecular hydrogen bonding interactions are disfavoured due to steric hindrance and geometric constraints, so the squaramide unit can perform its catalytic function (‘on’ state). Catalyst c-106 afforded the Michael adduct 108 in ∼60% conversion after 24 h, while o-106 led to a less than 5% conversion, comparable to the background reaction (Scheme 29). Addition of two equivalents of tetrabutylammonium chloride switched the active form of the catalyst to its inactive form, stopping the reaction, which was resumed after restoring the system to the active form by addition of AgBArF.
A second system investigated consists of a tweezer structure with two urea groups (the hydrogen bond organocatalytic groups) linked by P,S-hemilabile ligands to a Pt(II) metal centre.54 The same mechanism used in 106 allows switching between the open and closed forms. The closed form is the active species of the catalyst for the Diels–Alder reaction between cyclopentadiene and methyl vinyl ketone. The open form is a poorer catalyst due to urea–urea intermolecular hydrogen bonding that blocks the active site.
Slugovc and co-workers have described a different type of catalyst that exhibits ligand-coordination-induced switching (Scheme 30).55 They prepared a Ru(II) olefin metathesis catalyst 109 bearing a benzylidene chelate, an N-heterocyclic carbene and two chlorides as ligands, which could catalyzed the ring-opening metathesis polymerisation of norbornene derivative 110 in >90% yield in 4 h. The system could be deactivated by replacing one of the chloride ions for pyridine followed by counterion exchange to PF6−. This ‘off’ state of the catalyst afforded only 10% conversion to polymer after 19 h under the same reaction conditions. Conditions to switch the system back to the original active state were not discussed.
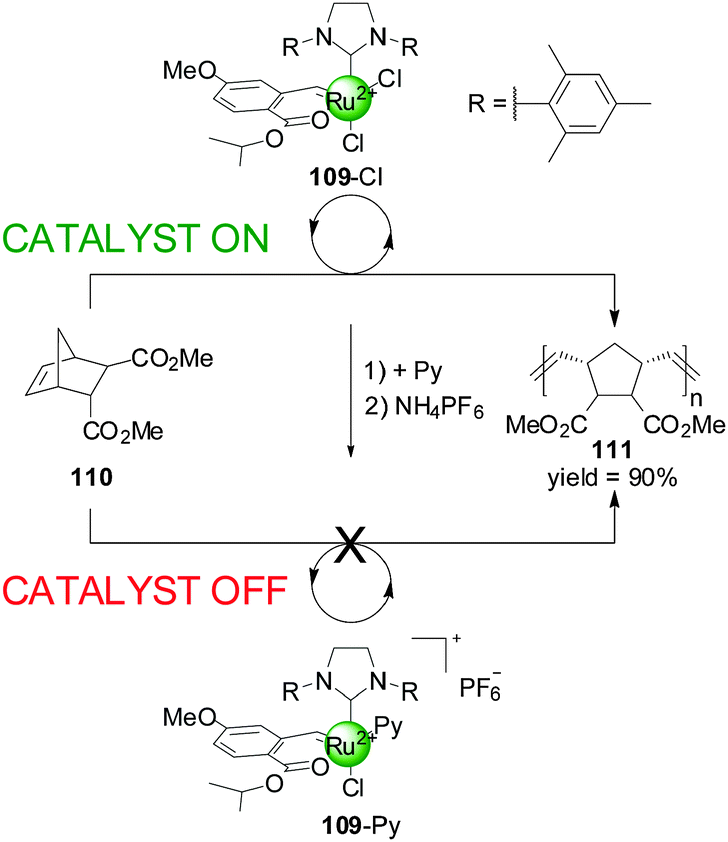 | ||
| Scheme 30 Deactivation of Ru(II) metathesis catalyst 109 promoted by Cl−–pyridine ligand exchange.55 | ||
4.2 Switching promoted by the addition/removal of metal cations
Following Shinkai's early example (Scheme 22), other systems were developed in which the binding of metal ions to crown-ether-like ligands modulated catalytic activity. Dervan and colleagues employed compound 112, which contains a Fe(II)-EDTA-type complex, to cleave DNA (Fig. 2a).56 In the absence of metal ions the system is not catalytically active, but in the presence of Ba2+ or Sr2+ the glycol chain binds to the ions adopting a conformation that bends the molecule and enables hydrogen bonding between the amide groups and the DNA bases, facilitating sequence-controlled cleavage of the strands. More recently, Kubo reported a bis(thiourea) dibenzo-diaza-30-crown-10 derivative (113, Fig. 2b)57 that catalysed the cleavage of 2-hydroxypropyl-p-nitrophenyl phosphate (99) in the presence of a stoichiometric amount of K+ 400 times faster than in the absence of the metal ion. The authors attributed this rate enhancement to the conformation adopted by the system upon coordination allowing the thioureas to bind the phosphate group, increasing the electrophility of the phosphorous centre.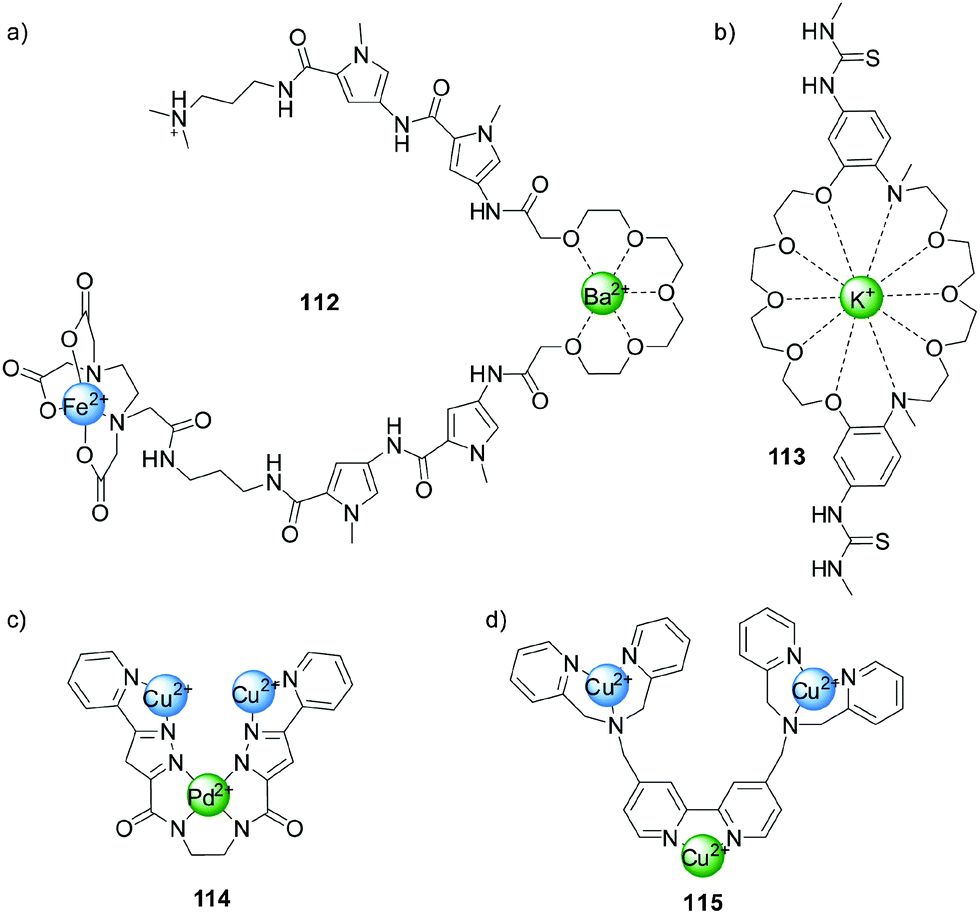 | ||
| Fig. 2 Catalysts activated by conformational changes induced by coordination of metal ions.56–61 | ||
Krämer has developed a series of systems in which the coordination of one metal ion (e.g. Pd(II) in compound 114, Fig. 2c)58 switches ‘on’ catalytic activity of another metal centre (Cu(II) in 114). Complex 114 displayed activity in the hydrolysis of nitrophenyl phosphate 99 when bound to Pd(II), but was inactive when coordinated to Pt(II).59
Catalysts featuring multiple metal ions have been prepared that display increased activity in phosphate diester cleavage reactions due to a cooperative effect between the metal centres.60 An example (115), shown in Fig. 2d, comprises a bidentate 2,2′-bipyridine ligand and two tridentate 2,2′-dipicolylamine units which bind more strongly to metal centres such as Cu(II) and Zn(II), allowing selective complexation at the different sites. When only the dipicolylamine groups are coordinated to metal centres the system displays poor phosphodiester cleavage activity. However, when the bipyridine unit is also bound catalysis was enhanced due to the coordination-induced change from the cisoid to the transoid conformation of the bidentate ligand, bringing together the Zn(II) or Cu(II) catalytic centres. A 19-fold increase of activity was achieved in the case of the Cu(II) system 115.61
These systems were switched ‘on’ by the external stimulus, but switching back to the ‘off’ state was not investigated. More recently, reversible switchable catalysts that can be controlled by coordination of metal ions to crown ethers present in their structure have been reported. Miller and co-workers developed an Iridium(III) complex with a pincer ligand bearing an aza-15-crown-5 moiety (116, Scheme 31).62 The activity of this complex for the activation of dihydrogen can be triggered by coordination of Li+ or Na+ to the crown ether. In the absence of alkali metal cations, the Ir(III) centre is hexa-coordinate with two oxygen atoms of the crown ether forming part of the coordination sphere of the metal (116-H). In this form, coordination of the substrate (the authors actually used deuterium in their experiments) was hampered by the crown ether (catalyst ‘off’) and dihydrogen activation, measured as H/D exchange at the metal centre, was slow (kobs = 1.2 × 10−6 s−1 in CH2Cl2). In the presence of Li+ or Na+ cations, however, the crown ether oxygen atoms were no longer coordinated to the Ir(III) centre, allowing the formation of a penta-coordinated Ir(III) complex with a dideuterium σ-complex (116-H-M, catalyst ‘on’). Thus, the addition of substoichiometric amounts of NaBArF (0.3 equiv.) and LiBArF (0.4 equiv.) resulted in a 20-fold and 250-fold rate enhancement of H/D exchange, respectively. The rate of the reaction could be modulated by in situ switching of the catalyst. Addition of chloride to the active form of the catalyst caused precipitation of the Na+ ions, switching the system ‘off’.
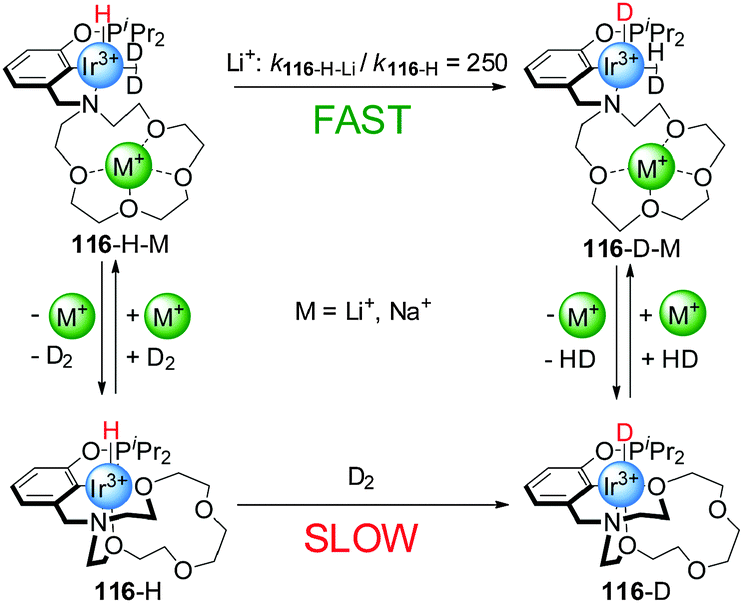 | ||
| Scheme 31 D2 activation by Ir(III) catalyst 116 triggered by reversible coordination of an alkali metal cation to a crown ether ligand.62 | ||
Fan and co-workers reported chiral aza-18-crown-6-phosphoramidite Rh(I) catalyst 117, whose activity in the asymmetric hydrogenation of dehydroamino acid esters (118) could be switched ‘on’ or ‘off’ by the reversible coordination of alkali metal cations to pendant crown ether groups (Scheme 32).63 In the absence of Na+ ions the O atoms of the crown ethers coordinate to the rhodium(I) centre and the system exhibits no catalytic activity (less than 1% conversion, catalyst ‘off’). Addition of two equiv. NaBArF results in the crown ethers disassociating from the Rh(I) centre (117-2Na). Complex 117-2Na catalyzes the hydrogenation with full conversion and excellent enantiomeric excess (92–98% ee) (catalyst ‘on’). Removal of Na+ with cryptand[2.2.2] restored the system to the inactive state. The switching cycle could be repeated without significant loss of activity.
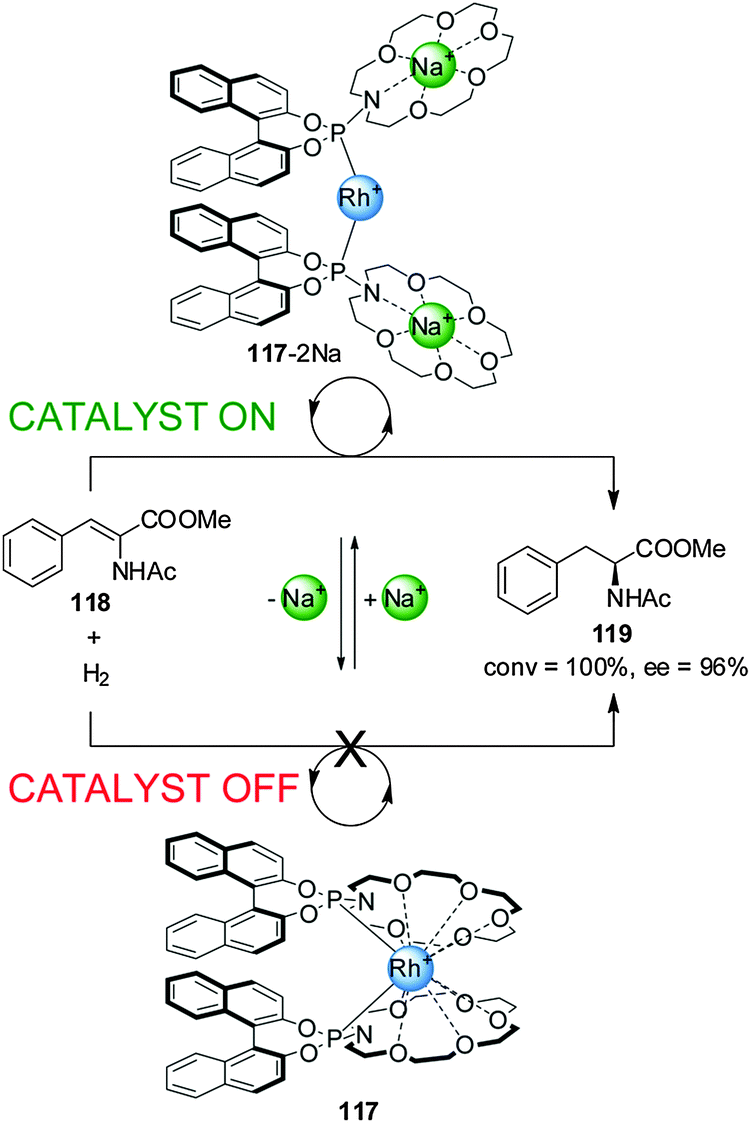 | ||
| Scheme 32 ‘On–off’ switching of Rh(I) catalyst 117 driven by addition or removal of Na+ ions.63 | ||
In 2012 Schmittel and co-workers reported a switchable catalyst system triggered by the addition or removal of Cu(I) ions (Scheme 33).64 The system consisted of three elements: a switch containing a Zn(II)–porphyrin unit and a 4-(2-pyridyl)pyrimidine moiety separated by an angular rigid oligophenylalkynyl spacer (120), a phenanthroline (phen) derivative (122) and piperidine (121). The latter is the active organocatalyst, able to promote the Knoevenagel reaction between 4-nitrobenzaldehyde (41) and diethyl malonate (123). In the absence of Cu(I), the pyrimidine unit coordinated to the Zn(II) porphyrin leaving the piperidine available for catalysis (120, catalyst ‘on’). When Cu(I) is added it forms a complex with the phenanthroline and pyridyl–pyrimidine group (120-Cu), and the Zn(II) porphyrin binds to the piperidine, preventing it acting as a catalyst (catalyst ‘off’). Removal of the Cu(I) ions with cyclam restored the system to the ‘on’ state. The reversibility of the system was demonstrated by three cycles of in situ switching.
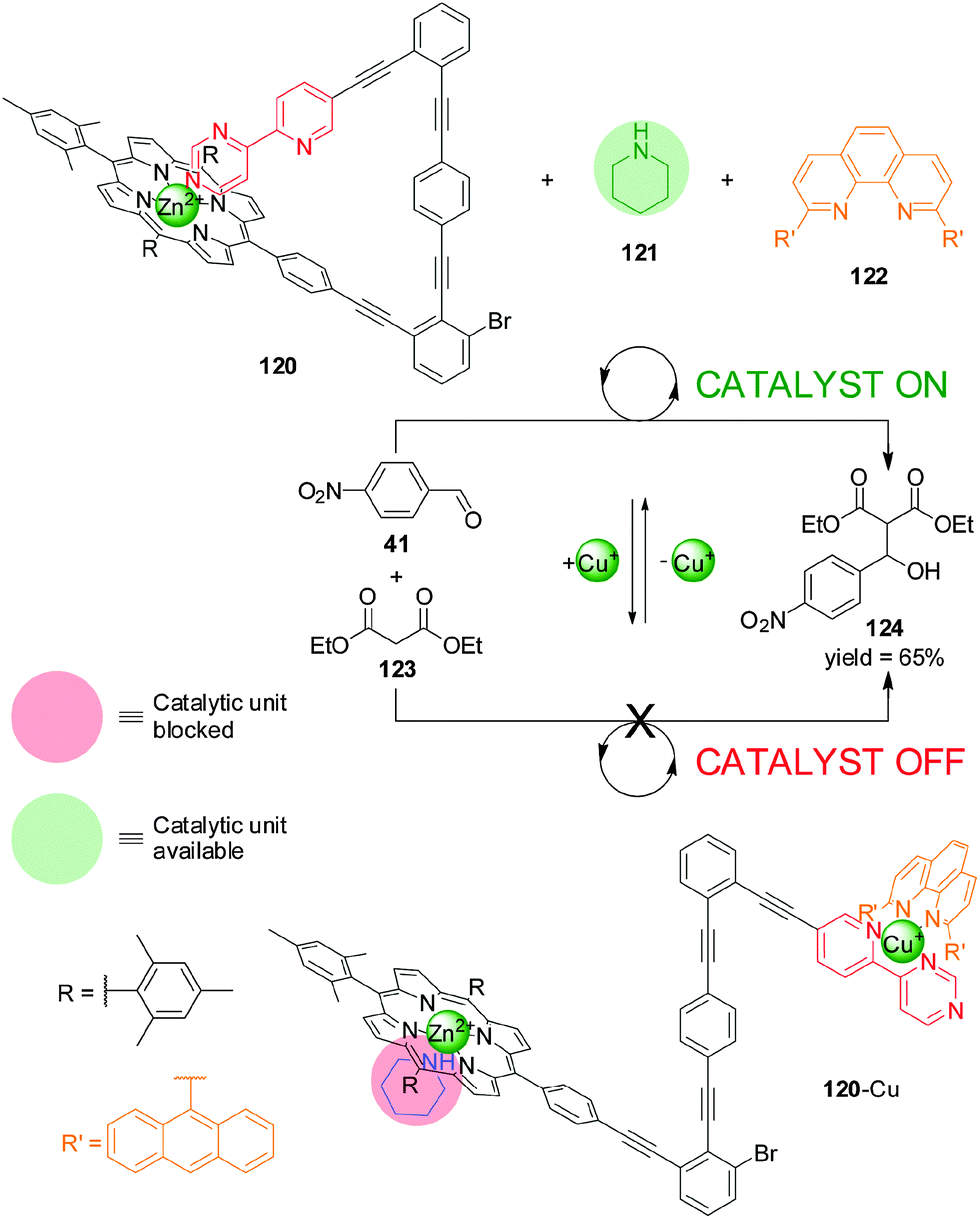 | ||
| Scheme 33 Switchable catalysis of a Knoevenagel reaction based on the capture/release of a piperidine catalyst (121) by Zn(II)–porphyrin 120 induced by the reversible coordination of Cu(I).64 | ||
A related catalyst (125), in which all the elements required for operation are included within a single molecule, has been used to promote the cis–trans photoisomerization of E-4-styrylpyrimidine (126) (Scheme 34).65 The Zn(II)–porphyrin moiety catalyzes the reaction in the presence of Cu(I) due to the metal ion forming a complex with the pyridyl–pyrimidine and the phen unit (‘on’ state; 125-Cu). Addition of cyclam extracts the Cu(I) and the pyrimidine ring coordinates to Zn(II), preventing the porphyrin from binding to the substrate (‘off’ state; 125). Significant differences in catalytic activity between the active and inactive states were demonstrated by in situ switching.
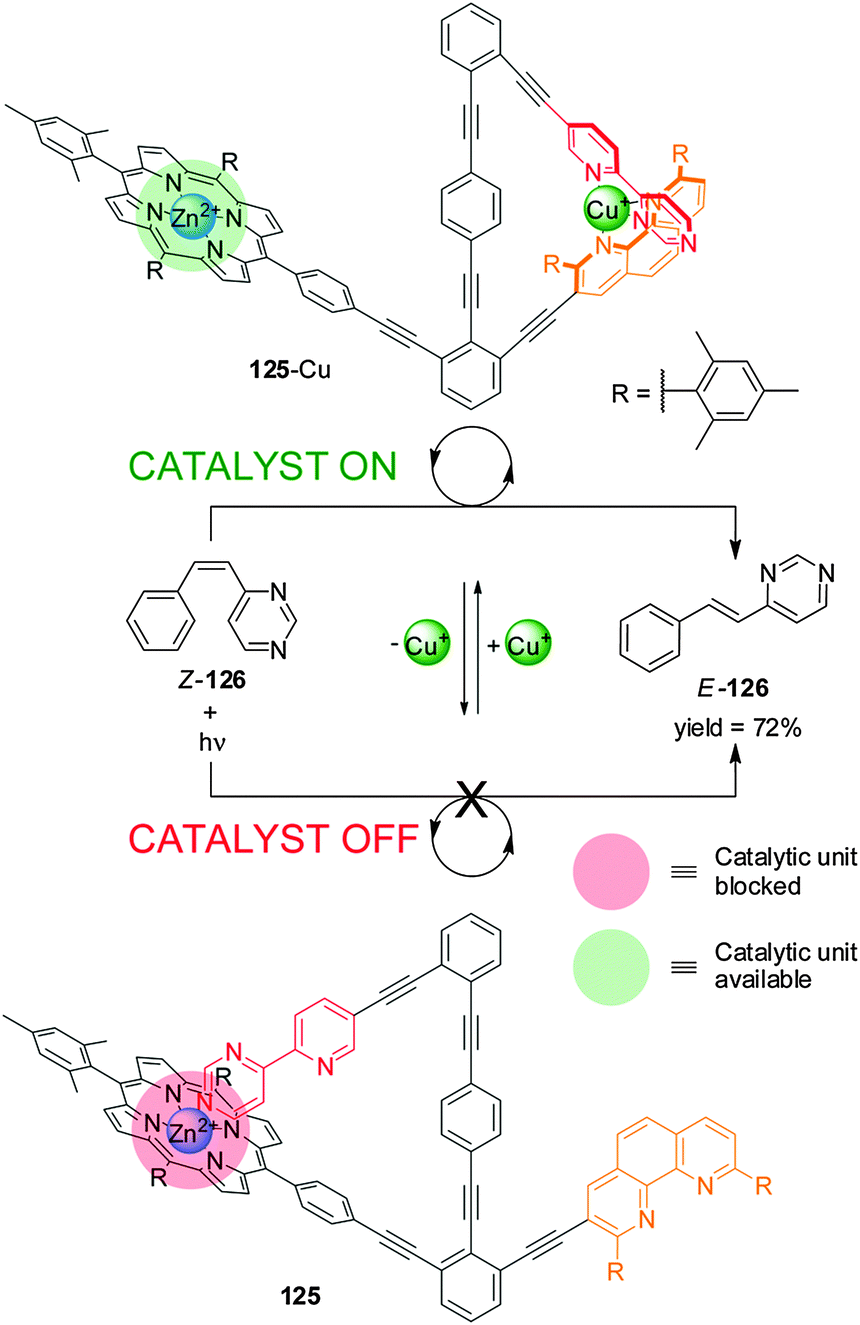 | ||
| Scheme 34 Control of the activity of catalyst 125 in a cis–trans isomerization process triggered by the addition/removal of Cu(I).65 | ||
A different metal ion provided the external stimulus in a design (127) comprising a phenanthroline and terpyridine (terpy) ligands coordinated to a Cu(I) centre (Scheme 35).66 Addition of Fe(II) resulted in the formation of a dimeric species (1272-Fe) in which two terpyridine ligands are coordinated to the Fe(II) centre, leaving accessible coordination sites on the Cu(I)–phen complex available to catalyse the cyclopropanation of Z-cyclooctene (68) with ethyldiazoacetate (17) (catalyst ‘on’; 1272-Fe). Extracting the Fe(II) with 4-N,N-dimethylaminoterpyridine restores the system to its inactive monomeric form (catalyst ‘off’; 127).
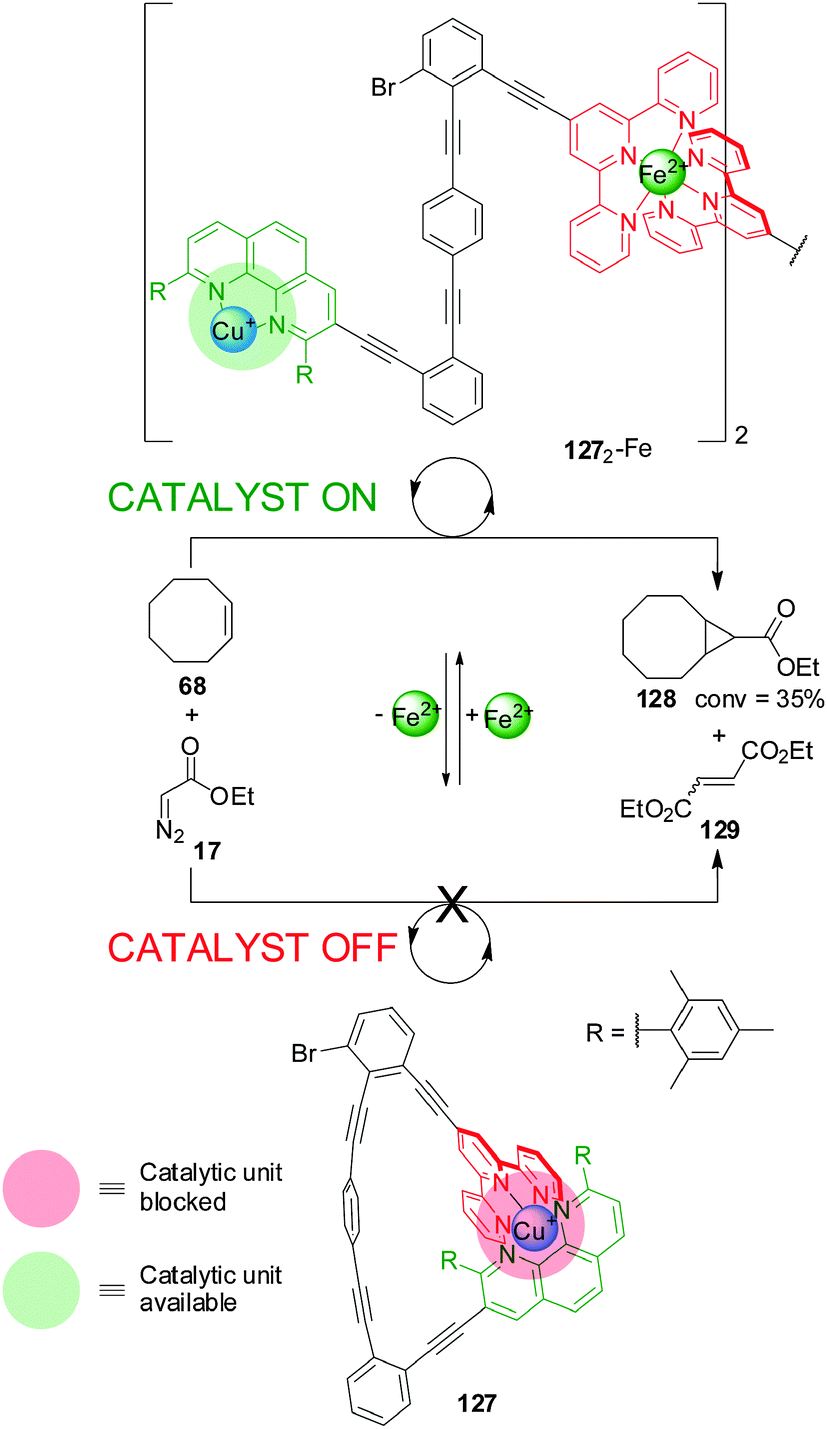 | ||
| Scheme 35 Control of the activity of Cu(I) catalyst 127 by switching between a monomeric and a dimeric form promoted by the reversible coordination of Fe(II).66 | ||
System 125 (Scheme 34) was used in combination with piperidine (Pip, 121) to develop a switchable catalyst (130-Cu-Pip), which is able to control not only the rate but also the chemoselectivity of catalysis (Scheme 36).67 The authors sequentially applied two different chemical stimuli (Cu(I) and phen derivative 131) to the system formed from 130 and piperidine. The system starts in an ‘off’ state in which the potential catalytic species, piperidine and Cu(I), are coordinated to binding sites preventing them from being catalytically active (130-Cu-Pip). Addition of an external phen ligand (131) displaces the pyridyl–pyrimidine arm from the Cu(I) complex, which then binds to the porphyrin, displacing the piperidine from the Zn(II) centre (130-Cu-131). The liberated piperidine can catalyze the Knoevenagel reaction in a similar fashion to Scheme 33 (piperidine catalysis ‘on’). In this state the Cu(I) is coordinatively saturated with bidentate ligands and cannot participate in catalysis (Cu catalysis ‘off’). However, if a second equivalent of Cu(I) is added, an intramolecular complex is formed and the catalytically active Cu(I)–phen complex (131-Cu) is released and able to promote the Cu-catalyzed azide–alkyne cycloaddition (CuAAC) ‘click’ reaction between benzyl azide (132) and 4-nitrophenylacetylene (133) (Cu catalyst ‘on’). As the pyridyl–pyrimidine ligand is no longer bound to the Zn porphyrin, its position is occupied by the piperidine, which is no longer available and therefore the Knoevenagel reaction does not take place (piperidine catalysis ‘off’). Addition of one equivalent of 131 turned ‘off’ both catalytic activities sending the system back to its original state. The sequential addition of the different chemical stimuli switches the different activities with a high degree of control; only one reaction type is observed in each state.
 | ||
| Scheme 36 Switching of catalytic system 130 between an ‘off’ state and two ‘on’ states with orthogonal catalytic processes through the sequential addition of phenanthroline derivative 131 and Cu(I).67 | ||
Aprahamian and co-workers recently reported a switch (135) that is able to liberate or extract a proton (the catalytically active species) to and from the reaction media through the reversible coordination of Zn(II) (Scheme 37).68 The system was used to catalyze imine bond hydrolysis. In the absence of Zn(II) a proton (shown in blue in Scheme 37) is covalently bound to the hydrazone group of the switch (135) and cannot participate in catalysis (catalyst ‘off’). Upon addition of Zn(II), the system (135-Zn) acts as a tridentate ligand that coordinates to the metal centre through the nitrogen atoms of the hydrazone, the quinoline and imidazol units releasing a proton. This was used to catalyse the hydrolysis of anthracen-9-yl-N-(4-nitrophenyl)-methanimine (136) into p-nitroaniline (137) and 9-anthraldehyde (138), which proceeded to completion (catalyst ‘on’). Removal of Zn(II) with CN− switched the system back to the ‘off’ state stopping the reaction. Upon addition of more Zn(II) the catalytic activity resumed and full conversion was achieved.
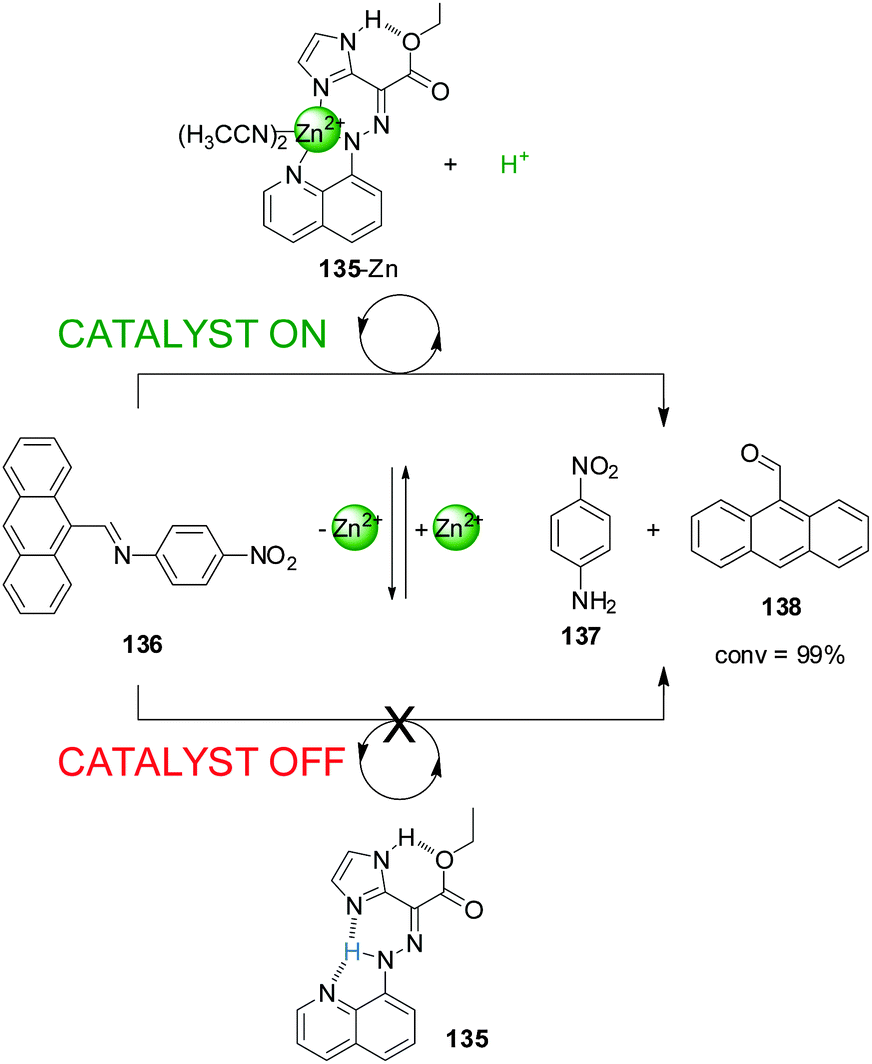 | ||
| Scheme 37 Controlling the hydrolysis of imine 136 by switchable H+ capture/release through coordination of 135 to Zn(II).68 | ||
5. Redox switching
One of the most explored redox mechanisms to influence the activity of a catalyst is the control of the oxidation state of redox-active ligands, often through incorporating ferrocene or cobaltocene groups into catalysts. In 1995 Wrighton and co-workers, developed a Rh(I) catalyst (139) with a 1,1′-bis(diphenylphosphino)cobaltocene (dppc) ligand in which the change of the cobalt oxidation state could be exploited to accelerate or slow down the rate of reactions catalyzed with this system (Scheme 38).69 Interestingly, the ‘on’ and ‘off’ states of the catalyst can correspond to either form of the catalyst depending on the particular reaction. Hydrogenation of cyclohexane proceeds at 3.5 mM min−1 in the presence of the reduced form of the catalyst (‘on’ state), while with the oxidized form (139ox) only a rate of 0.23 mM min−1 is observed. In contrast, the hydrosilylation of acetone was faster in the presence of 139ox than with 139red. In situ switching was achieved by oxidation or reduction of the cobalt complex with 1 equiv. of decamethylferrocenium or cobaltocene.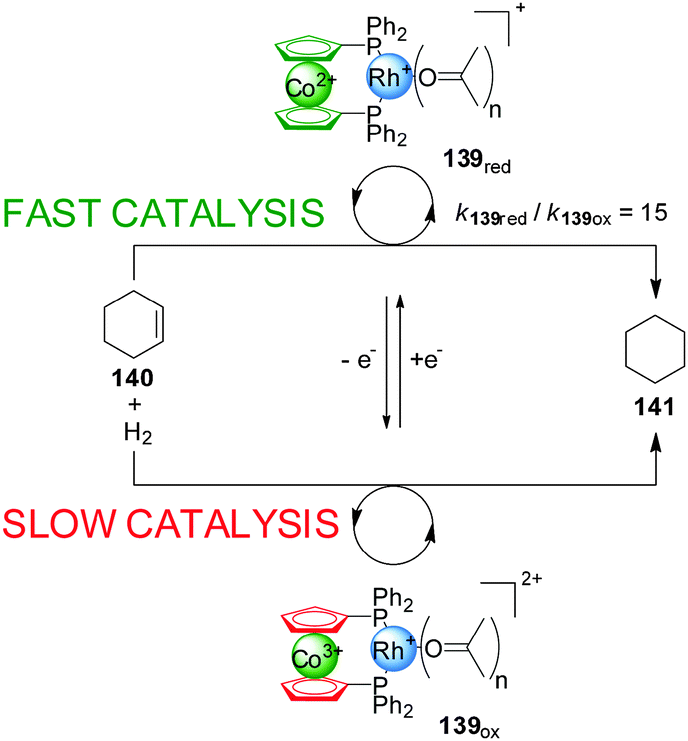 | ||
| Scheme 38 Co(II)/Co(III) redox control over the activity of a Rh(I) hydrogenation catalyst 139.69 | ||
Ferrocene ligands have been used to regulate the activity of metal catalysts in a similar manner. Mirkin and colleagues developed a Rh(I) catalyst with ferrocene–arene ligands that showed switchable activity in the isomerization of allyl groups to the corresponding vinyl derivatives.70 When the Fe(II) in the ferrocene groups was oxidized to Fe(III), the initial rate of the isomerization reaction was increased 2.2–3.3-fold depending on the substrate-to-catalyst ratio. The authors attributed this difference to the increased η6-arene ligand lability of the Fe(III) system. However, the enhanced catalytic rate was not maintained throughout the reaction.
Gibson and Long demonstrated that the activity of Ti(IV)–salen catalyst 142 could be modulated by oxidation/reduction of the redox-active ferrocene ligands (Scheme 39).71 The reduced ferrocene-containing initiator (142red, ‘on’ state) promoted the ring-opening polymerization of rac-lactide (rac-143) 30× faster than the oxidized ferrocenium-containing form (142ox, ‘off’ state) under the same reaction conditions, resulting in 95% conversion after 70 h. The catalytic activity could be controlled by in situ switching of the system: catalyst 142 was switched ‘off’ by oxidation with AgOTf, markedly slowing the rate of polymerization; subsequent reduction with decamethylferrocene restored the ‘on’ state.
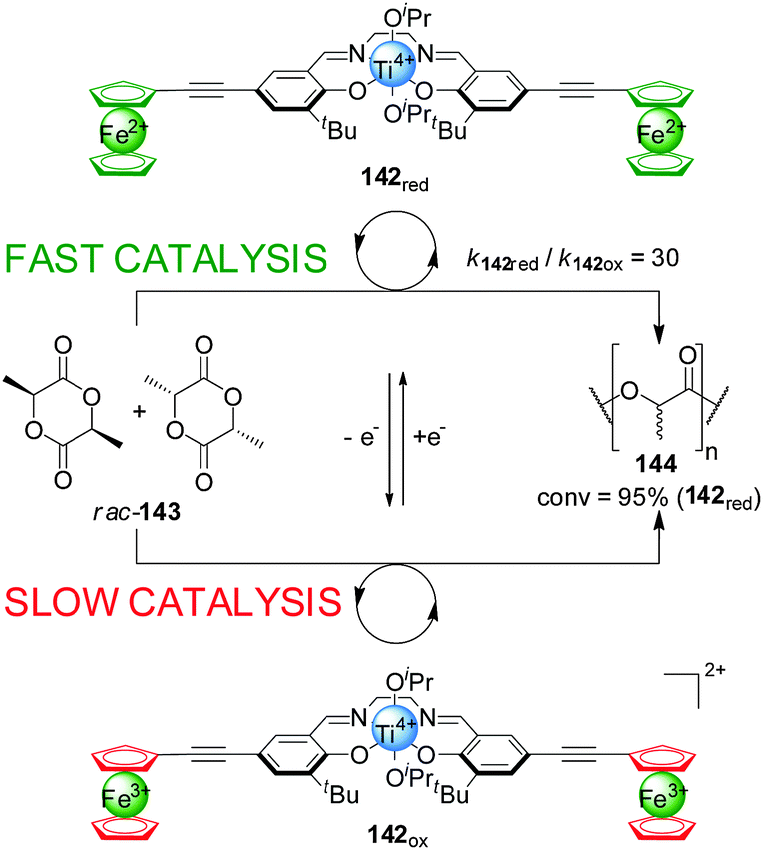 | ||
| Scheme 39 Influencing the rate of polymerization of rac-lactide (143) by oxidation/reduction of ferrocene groups pendant to a Ti(IV)–salen catalyst (142).71 | ||
Following a similar strategy, Diaconescu and co-workers developed an Ytrium(III) complex, 145, that displayed redox-controllable activity (Scheme 40).72 In a similar way to previous examples in this Section, the reduction or oxidation of the iron centre in a phosphimine ferrocene ligand was exploited to activate or deactivate the metal catalytic centre in the polymerization of L-lactide (L-143). The active state of the system, 145red, catalyzed the reaction with 74% conversion after 3 h, while the Fe(III)-oxidized form (145ox, ‘off’ state) did not give any conversion to the polymer. In situ switching allowed an excellent control over the polymerization rate with a high integrity in the ‘off’ state and full recovery of catalytic activity upon subsequent reduction. Replacing Y(III) with In(III) changes which of the redox states is active catalytically: the oxidized form of the catalyst gives 49% conversion of trimethylene carbonate into the corresponding polymer, while the reduced form afforded only 2% conversion under the same conditions.
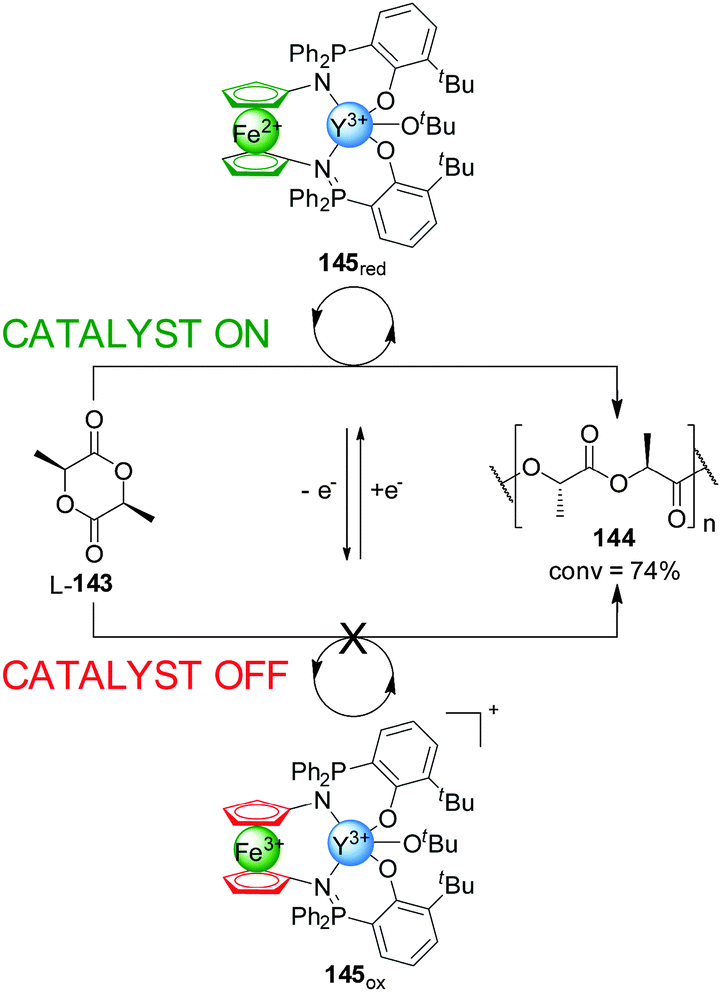 | ||
| Scheme 40 ‘On–off’ switching of catalyst 145 promoted by reversible ferrocene/ferrocenium redox reactions.72 | ||
Bielawski and colleagues extended this concept to ferrocene-containing monodentate ligands, in particular ferrocene NHC ligands for Ru(II) olefin metathesis catalysts (Scheme 41).73 Ru(II) complex 146 was tested in the ring-closing olefin metathesis of diethyl diallylmalonate (147). In its reduced form the system catalyzed the reaction with 80% conversion within 1 h. Oxidation of ferrocene to ferrocenium reduced the activity of the catalyst, affording 7% conversion in the same period. In situ switching was performed but, although switching ‘off’ catalysis was effective, subsequent reactivation of the catalyst restored only 13% of the catalytic activity, attributed to some ruthenium oxidation. To overcome this a more robust catalyst incorporating a 1′,2′,3′,4′,5′-pentamethylferrocene-substituted NHC ligand was investigated. This system gave less than 10% of the rate enhancement in its ‘on’ state as the previous catalyst, but 94% of this activity was recovered after in situ switching.
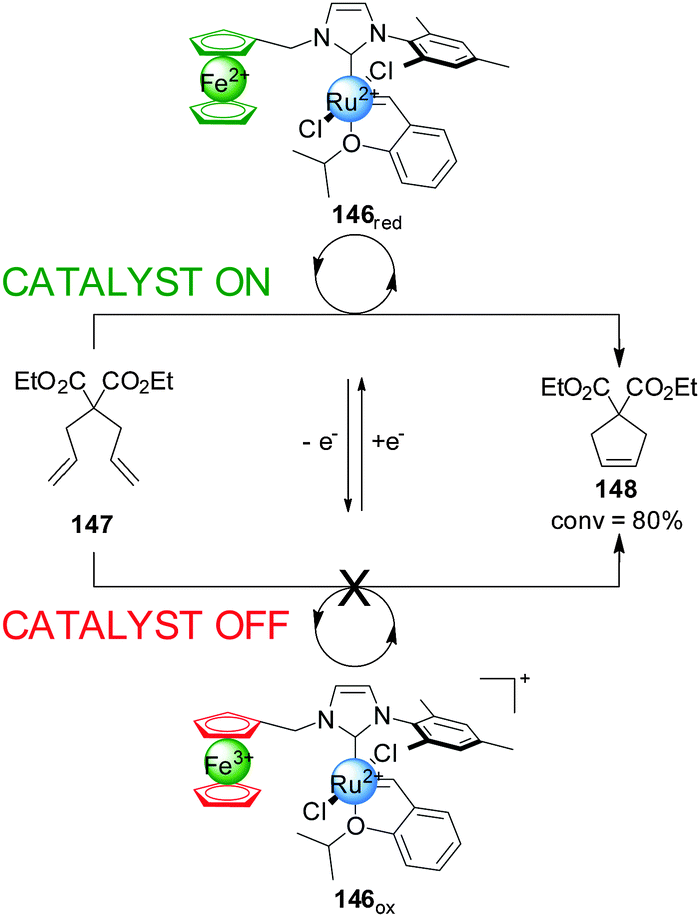 | ||
| Scheme 41 Control over the activity of metathesis catalyst 146 by change of the oxidation state of a ferrocene iron centre.73 | ||
The rationale behind the control of reactivity of the metal catalysts discussed in this Section is that the change in the oxidation state of the metallocene results in a change in the donating ability of the ligands, increasing or decreasing the electronic density at the metal centre. However, the redox chemistry of ferrocene has also been exploited to control catalyst activity in other ways. In 2005 Plenio and colleagues fucntionalized a Grubbs–Hoveyda Ru(II) olefin metathesis catalyst with ferrocene groups.74 The activity of this catalyst could be triggered by oxidation/reduction of the ferrocene, which caused precipitation/solubilisation of the catalyst in an apolar solvent. In the reduced state the catalyst (soluble in toluene) catalysed the ring-closing metathesis of N-tosyldiallylamide (catalyst ‘on’). Oxidation precipitated the complex, stopping the reaction (catalyst ‘off’). Reduction of the ferrocenium groups redissolved the complex restoring the catalytic activity. This behaviour allowed recycling of the catalyst by filtration of the insoluble oxidized form and reuse after reduction.
Hey-Hawkins and co-workers incorporated ferrocene-containing phosphine ligands into a dendritic structure (149) to take advantage of two switching mechanisms available for redox-active ferrocene ligands (Scheme 42).75 The dendrimer comprised twelve ferrocenylphosphane units coordinated to Ru(II) centres to form (η6-arene)RuCl2(L) complexes able to catalyze the isomerization of allyl alcohols to ketones. The reduced form of the catalyst (149red, ‘on’ state) efficiently catalyzed the isomerization of 1-octen-3-ol (150) (full conversion within 40 min). Interestingly, the observed activity with the dendrimer catalyst was higher than that of a monomeric ferrocene-containing Ru(II) complex at the same metal loading. When the dendritic catalyst was oxidized (149ox, ‘off’ state) its activity dropped by almost an order of magnitude. The lower activity of the oxidised form was attributed to a combination of electronic and solubility effects. Reduction of the oxidised form restored the system to the ‘on’ state, recovering the catalytic activity.
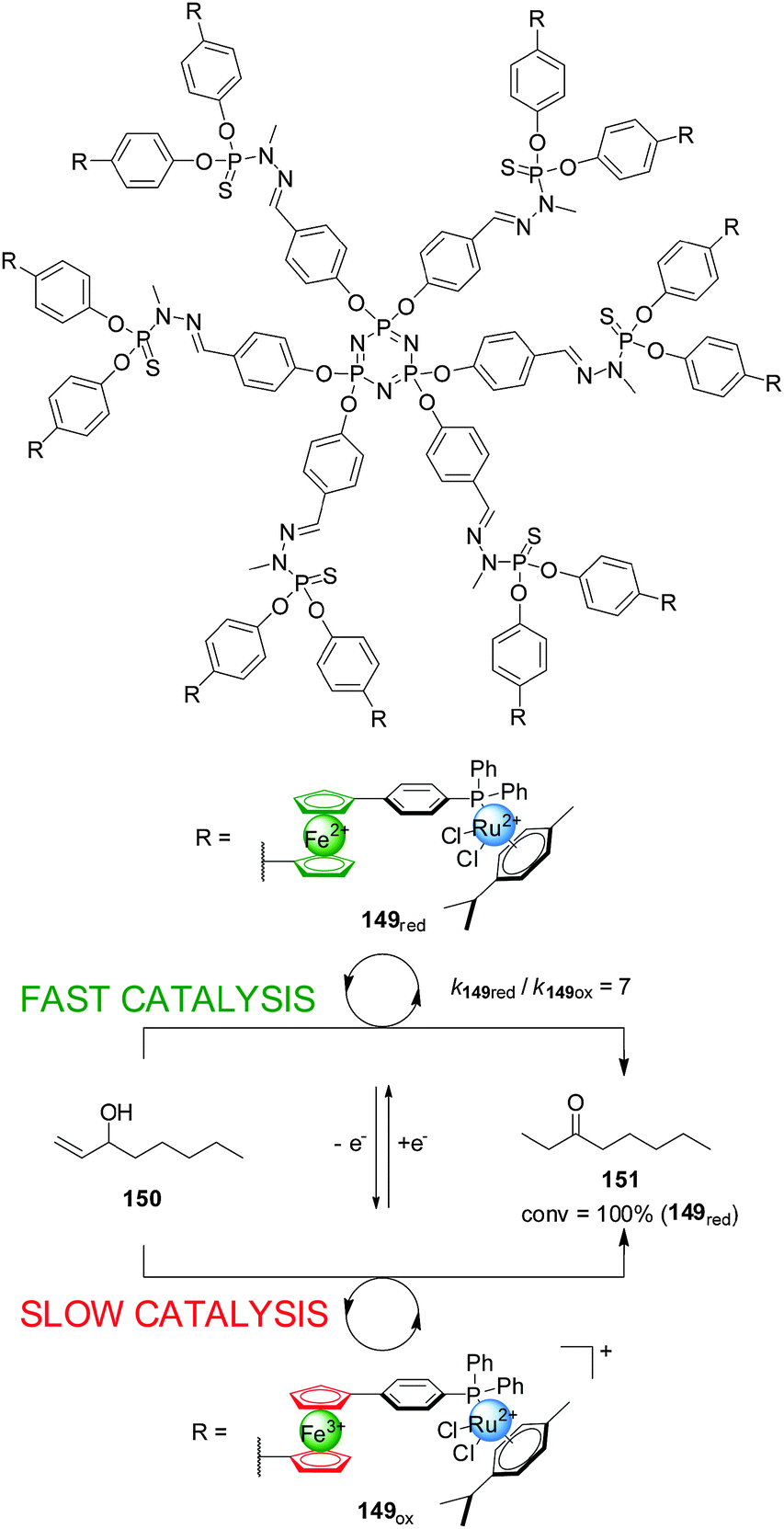 | ||
| Scheme 42 Redox-switching of the rate of isomerization of allyl alcohol 150 using dendritic Ru(II)-catalyst 149.75 | ||
Although the change in oxidation state of pendant metallocene ligands is the most explored mechanism for redox-switchable catalysts, control over the activity of a metal catalyst can also be achieved by a direct change in the oxidation state of the catalytic metal centre. In 2011, Matyjaszewski and co-workers described a remarkable Cu(I)/Cu(II) system for atom-transfer radical polymerization that can be controlled by electrochemical switching of the oxidation state of the copper centre (Scheme 43).76 The catalyst (or activator, ‘on’ state) Cu(I)BrMe6TREN (Me6TREN = tris[2-(dimethylamino)ethyl]amine) reacts with alkyl bromides to form Cu(II)Br2Me6TREN (a deactivator, ‘off’ state) and a radical species that undergoes monomer addition giving rise to a polymerization reaction. However, the different species are in equilibrium as the Cu(II)Br2Me6TREN can be reduced by radicals reforming Cu(I)BrMe6TREN and the alkyl bromide. In the absence of other factors, the reduction is the fastest process and the equilibrium favours the starting materials and so polymerization, which depends on the Cu(I)/Cu(II) ratio, is not observed. However, the equilibrium can be influenced electrochemically. By applying a potential of −0.69 V (vs. Ag/Ag+), Cu(II)Br2Me6TREN formed during the reaction is reduced to Cu(I)BrMe6TREN which can react again leading to a 80% conversion of methyl acrylate over 2 h. With a more positive potential (E = −0.40 V), the equilibrium is shifted towards the Cu(II) complex, which reduces the number of radicals, preventing further polymerization. The system could be switched in situ by changing the applied potentials. Varying the applied potential between E = −0.69 V and E = −0.40 V the polymerization reaction could be switched ‘on’ or ‘off’, respectively, as the Cu(I)/Cu(II) equilibrium is shifted towards either Cu(I) or Cu(II) species.
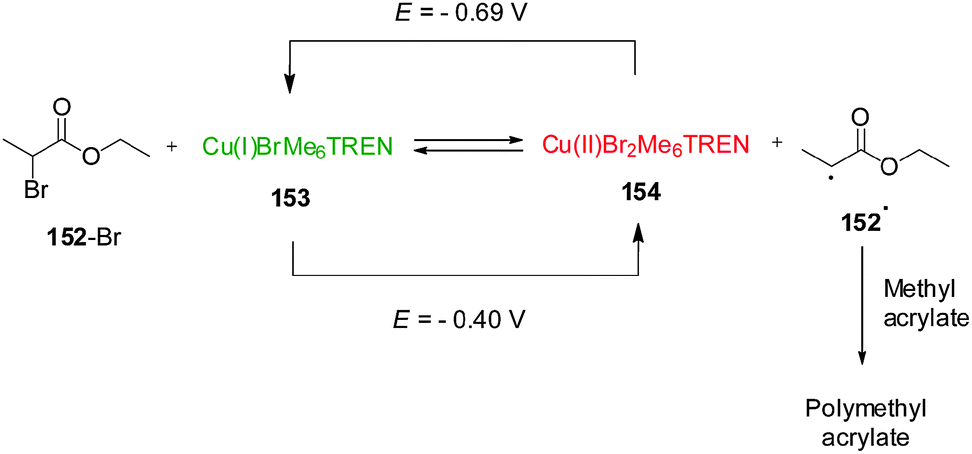 | ||
| Scheme 43 Electrochemical control over the activation of a polymerization reaction by reduction/oxidation of Cu(I)/Cu(II).76 | ||
Diaconescu and co-workers have reported a switchable polymerization catalyst 155 based on a Ce(III)–salen complex (Scheme 44).77 Oxidation or reduction of the cerium centre was used to activate or deactivate the catalyst. The Ce(III) complex was catalytically active in the ring-opening polymerization of L-lactide (L-143), resulting in 85% conversion over 2 h. In contrast, the BArF salt of the oxidized Ce(IV) complex showed no polymerization activity. The system could be switched in situ. After some polymer had formed, the Ce(III) complex was treated with ferrocenium BArF, forming the corresponding Ce(IV) complex and stopping further polymerization. The active state could be restored by reduction with cobaltocene, with no significant loss of catalytic activity.
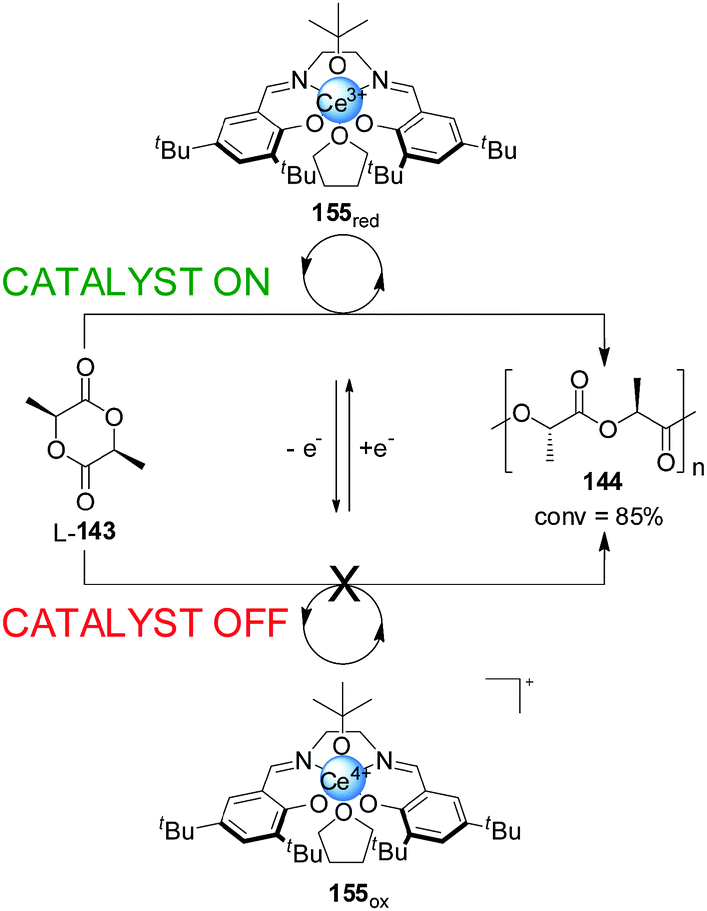 | ||
| Scheme 44 ‘On–off’ switching of lactide polymerization catalyst 155 triggered by reduction/oxidation of the cerium catalytic centre.77 | ||
Another switchable catalyst based on the oxidation/reduction of a cerium catalytic centre was reported by Okuda and colleagues.78 The system comprises two tetradentate OSSO ligands coordinated to cerium. Here again, the Ce(III) complex was active (catalyst ‘on’) in the polymerization of meso-lactide, affording 78% conversion after 7 h, while the Ce(IV) system showed much lower activity (catalyst ‘off’) giving only 10% conversion under the same conditions.
Byers and co-workers developed a readily accessible bis(imino)pyridine iron complex (156) which was found to catalyze the polymerization of (rac)-lactide depending on the oxidation state of the iron centre (Scheme 45).79 While the Fe(II) (156red, catalyst ‘on’) complex catalyzed the formation of the polylactide (144) effectively (95% conversion over 24 h), the Fe(III) complex (156ox) did not show any catalytic activity under the same reaction conditions (catalyst ‘off’). The lack of activity of 156ox was attributed to the electron-poor character of the complex. Switching of the catalytic activity could be performed in situ. After 40 min of polymerization in the presence of 156red, the Fe(II) centre was oxidized with ferrocenium hexafluorophosphate forming 156ox. The polymerization did not proceed further until cobaltocene was added to restore the active form of the catalyst.
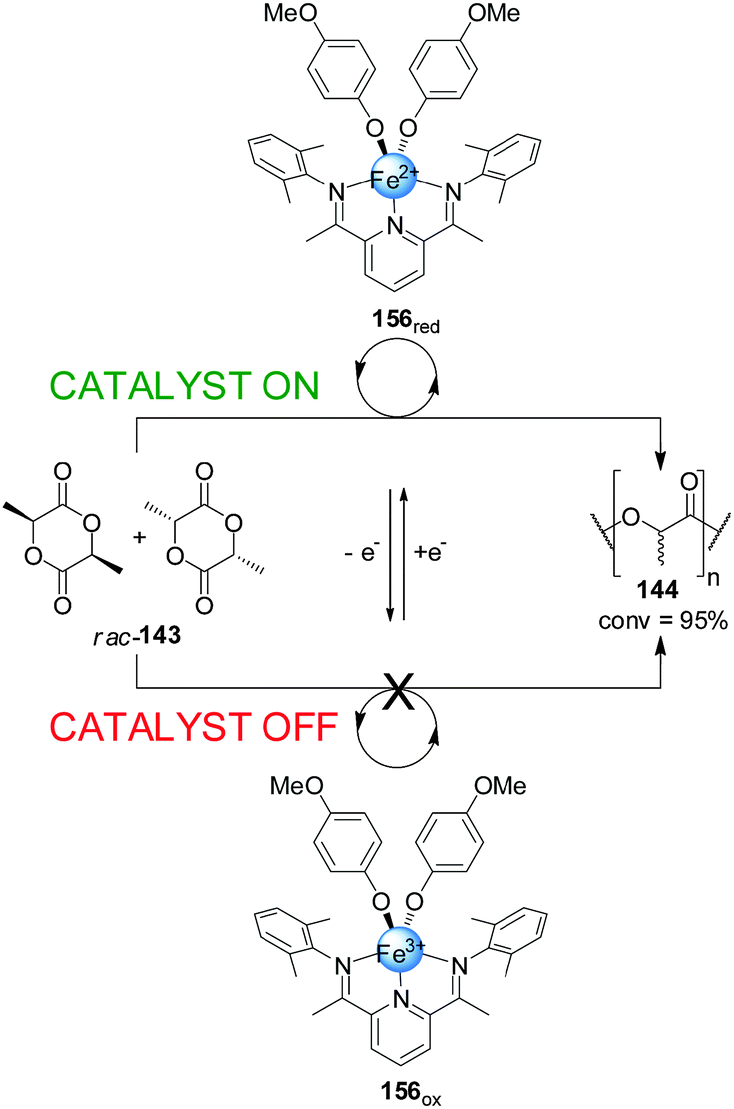 | ||
| Scheme 45 Iron-based redox-switchable catalyst 156 for the polymerization of rac-lactide.79 | ||
A similar system was reported by Lang and co-workers, in which a change in the oxidation state of an iron centre bound to 2,6-di(1H-pyrazol-3-yl)pyridine and chloride ligands was used to switch ‘on’ or ‘off’ the catalytic activity of the complex in the ring-opening polymerization of ε-caprolactone.80 While the Fe(III) complex can catalyse the polymerization in >90% yield in the presence of an tert-butanol, the reduced form of the catalyst containing an Fe(II) centre, proved to be inactive under the same reaction conditions. In situ switching of the catalyst could be achieved by addition of cobaltocene and ferrocenium hexafluorophosphate.
The change in oxidation state of a metal ion can also be used to control selectivity. In 2012, Canary and colleagues reported bisurea catalyst 157 incorporating a ligand motif derived from L-methionine (Scheme 46).81 A multidentate pocket is formed that is able to complex either Cu(I) or Cu(II). The different coordination geometries of the different oxidation states induces chirality of different handedness for the two structures (157red and 157ox). The inversion in helicity is transferred through to the spatial arrangements of the pendant urea groups and their selectivity as organocatalysts for the asymmetric Michael addition of diethyl malonate (123) to trans-β-nitrostyrene (81). With the copper centre in its oxidized state (Cu(II)), 157ox catalyzed the formation of (S)-158 in 55% yield (75% conversion) and 72% ee. In contrast complex 157red afforded (R)-158 in 40% yield and 70% ee. However, a definitive explanation for the helix inversion and its influence on the handedness of the products of catalysis remains elusive.
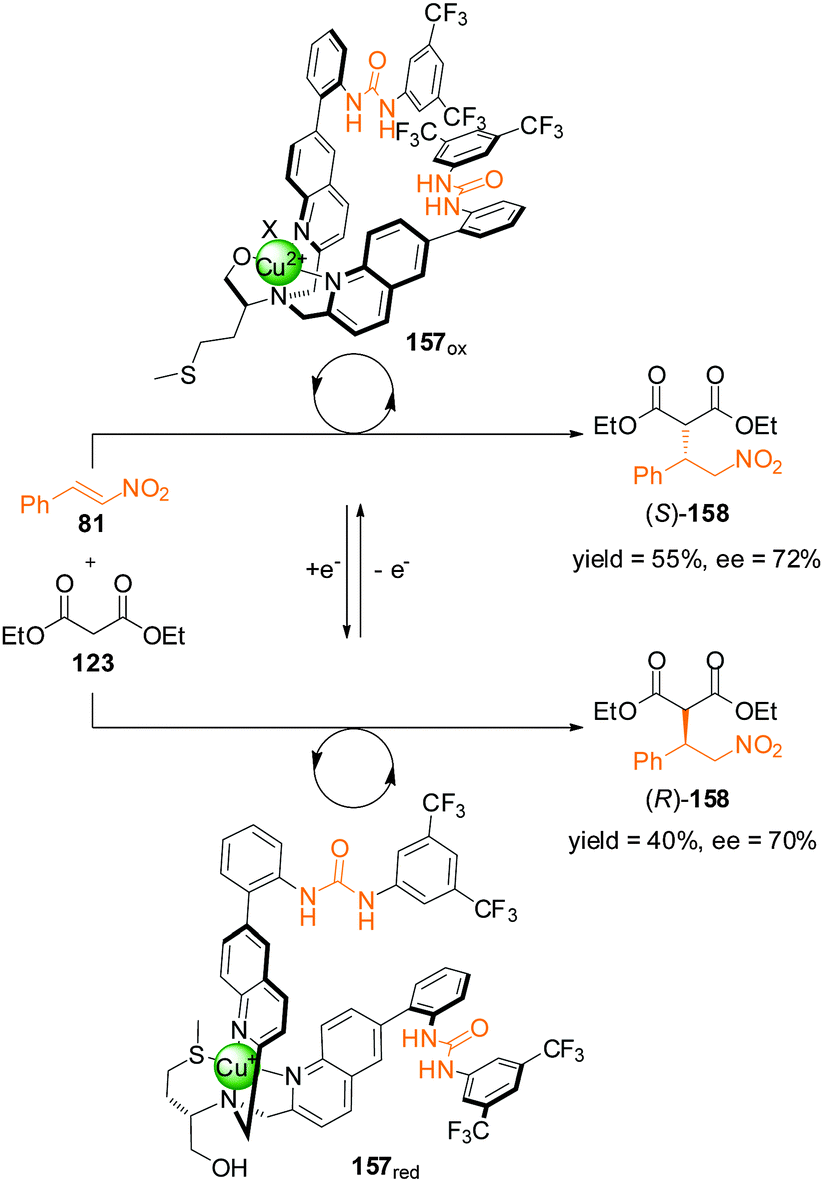 | ||
| Scheme 46 Control of the stereoselectivity of the Michael addition of diethyl malonate to β-nitrostyrene promoted by redox-switchable catalyst 157.81 | ||
Control over the particular substrate reacting from a mixture of building blocks has also been achieved using redox switching. Diaconescu and co-workers reported a series of Zr(IV) and Ti(IV) complexes bearing a ferrocene alkoxide ligand (Scheme 47).82 These systems catalyse the polymerization of L-lactide (L-143) and ε-caprolactone (104) with a monomer preference dependent on the oxidation state of the ferrocene unit. The reduced form of the catalysts are selective for the polymerization of L-lactide (L-143), with excellent conversions (82% with 159red) and only minor incorporation of the caprolactone (105) (<5% with 159red). In contrast, the ferrocenium-containing oxidized catalysts preferentially promoted ε-caprolactone polymerization, affording high conversions to 105 (the lowest being 48% with 159ox) and, in general, small amounts of incorporation of L-143 (<5% with 159ox). Each of the catalysts could be switched in situ when only one of the monomers (104 or L-143) was present, but only the Ti(IV) complex 159 proved able to switch control of the polymerization reactions when both substrates were present (Scheme 47). By switching the oxidation state of the iron centre, the one-pot copolymerization of both monomers was achieved. Starting with 159red, L-143 was polymerized with 58% conversion after 36 h with minor incorporation of 104. Then the selectivity of the catalyst was switched by oxidation, subsequently leading to polymerization of ε-caprolactone (18% conversion after 2 h) and affording a block copolymer poly-(143-minor-104)-(104-minor-143).
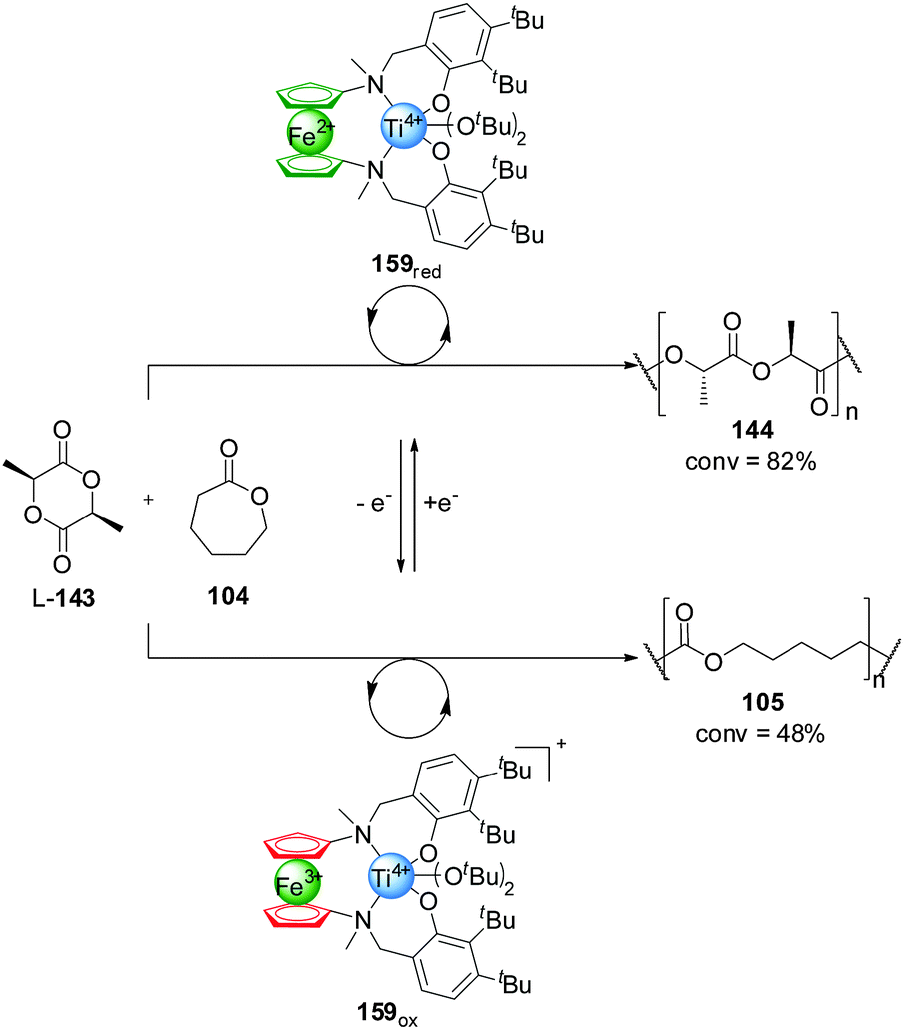 | ||
| Scheme 47 Switching of monomer selectivity by polymerization catalyst 159, triggered by oxidation/reduction of the pendant ferrocene group.82 | ||
Wu and co-workers developed a primary amine organocatalyst functionalized with a ferrocene unit whose activity and diastereo- and enantioselectivities in the aldol reaction between cyclohexenone and p-nitrobenzaldehyde depends on the oxidation state of the ferrocene. Using the reduced form of the catalyst, the reaction proceeded with low yields and poor enantio- and diastereoselectivity. However, with the oxidised Fe(III) ferrocenium moiety the catalyst afforded excellent yields (up to 95%), enantio-(up to 94% ee) and diastereoselectivities (up to 96:4 dr). The origin of the stereocontrol was attributed to the ability of ferrocenium to act as a Lewis acid, coordinating to the aldehyde and providing a well-expressed chiral environment for the substrate within the complex. The scope of the catalyst was extended to a range of different aldol reactions.83
Although most efforts on redox-triggered catalysts have focused on metal centres, reduction/oxidation of organic groups has also been used to modulate the activity of switchable catalysts:
Fujiwara and colleagues prepared an Al-functionalized mesoporous material (160) with a catalytically-active inner cavity able to promote the dimerization of α-methylstyrene (161) (Scheme 48).84 Access to the cavity could be blocked by oxidation of thiol groups placed at the pore openings (160ox; catalyst ‘off’). When the disulfide groups were reduced in situ (160red), the catalytic sites became accessible (catalyst ‘on’) and product 162 was formed in up to 97% yield. The material could be reused with no loss of catalytic activity.
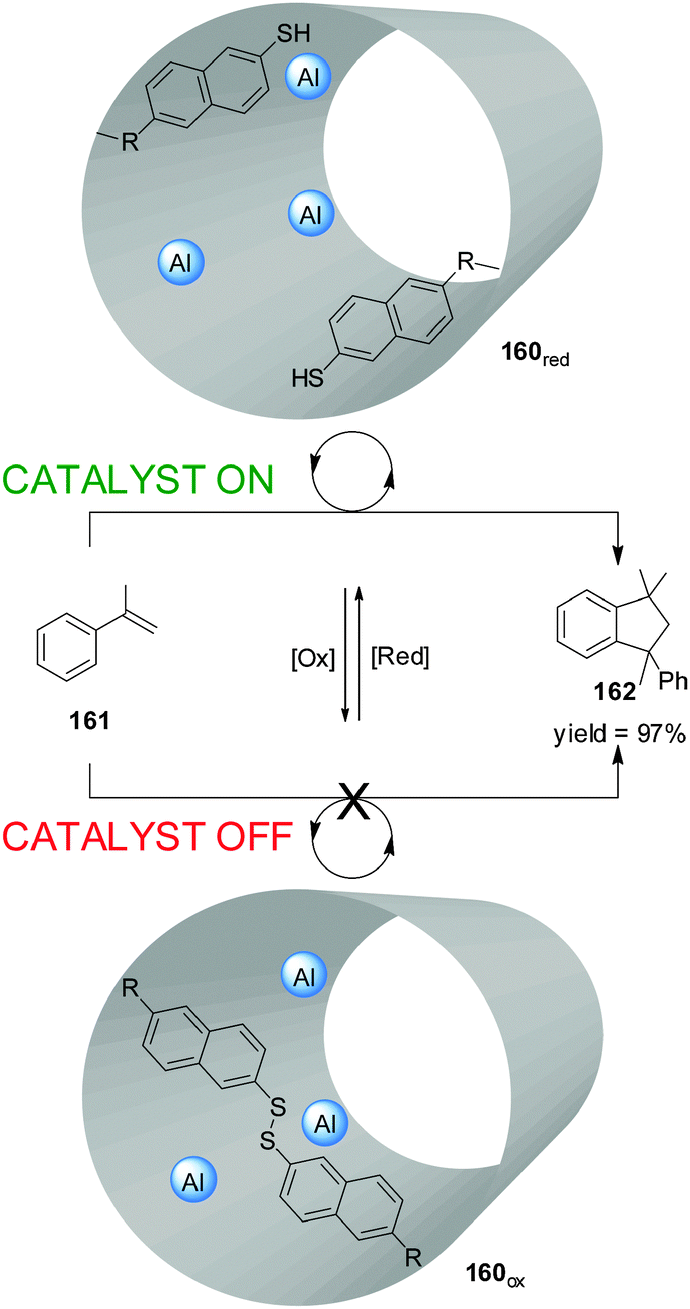 | ||
| Scheme 48 Control over the activity of heterogeneous catalyst 160 through the reversible redox cleavage or formation of disulfide bonds.84 | ||
In 2010, Bielawski et al. reported a Ni(II) complex comprising two NHC ligands bearing a redox-active naphthoquinone motif.85 With the ligand in the oxidised form (catalyst ‘on’) the complex efficiently catalyzed the Kumada reaction between phenylmagnesium chloride and p-bromoarenes, but showed lower activity in reduced form (catalyst ‘off’). The rationale for this behaviour is that after oxidative-addition of the bromoarene to the Ni(0) complex (formed by reduction of the Ni(II) catalyst with the Grignard reagent), that complex could be reduced with cobaltocene, affording an electron-rich complex that undergoes the transmetallation and reductive-elimination steps at significantly slower rates.
6. Switching driven by mechanical forces
Recently efforts have been made to employ mechanical force as a switching mechanism for catalysis.86 Mechanical forces are usually applied in solution through the use of ultrasound, which induces hydrodynamic force fields around collapsing cavitation bubbles. Due to the high strain rates, polymer chains uncoil and stretch under these conditions which can cause the weakest bonds in the chain to rupture. A limitation in using this stimulus to control catalytic activity is that only sufficiently long polymer chains can be affected in this way.Sijbesma envisaged that metal coordination bonds of complexes with polymer-functionalized ligands could be selectively dissociated with ultrasound leading to catalytically-active ligands or metal complexes.86 In 2008 they reported a polytetrahydrofuran (pTHF) polymer with an embedded bis-N-heterocyclic carbene–silver(I) complex (Ag-(163)2, Scheme 49, top) where the selective scission of the Ag(I)–NHC coordination bond could be achieved by sonication, exposing the active NHC–silver(I) complex 163 (catalyst ‘on’).87 The system, having two pTHF ligands of ∼5 kDa, was able to catalyze the transesterification of benzyl alcohol with vinyl acetate with 65% conversion over 60 min. Without applying ultrasound, the metal–ligand bond is not dissociated significantly and the metal catalyst is concealed (catalyst ‘off’) giving than 3% conversion under the same reaction conditions.88 To demonstrate the generality of the approach, the authors also used a Ru(II)–benzylidene complex with NHC ligands functionalized with pTHF chains (Ru-(163)2, Scheme 49, bottom).88 These complexes were used to promote the ring closing olefin metathesis (RCM) of diethyl diallylmalonate. Without sonication the complexes with pTHF groups of different molecular weight did not show any catalytic activity, while the reaction proceeded efficiently when the system was sonicated. In situ switching of the catalyst was achieved by turning the sonication ‘on’ and ‘off’, showing no increase in the degree of polymerization when ultrasound was not applied. However, the authors pointed that the activation of the catalyst is actually irreversible as the benzylidene group does not coordinate back to the Ru(II) centre to restore the initial species when sonication is ceased. Quick decomposition of the active catalysts was proposed to explain the lack of activity during the ‘off’ period.
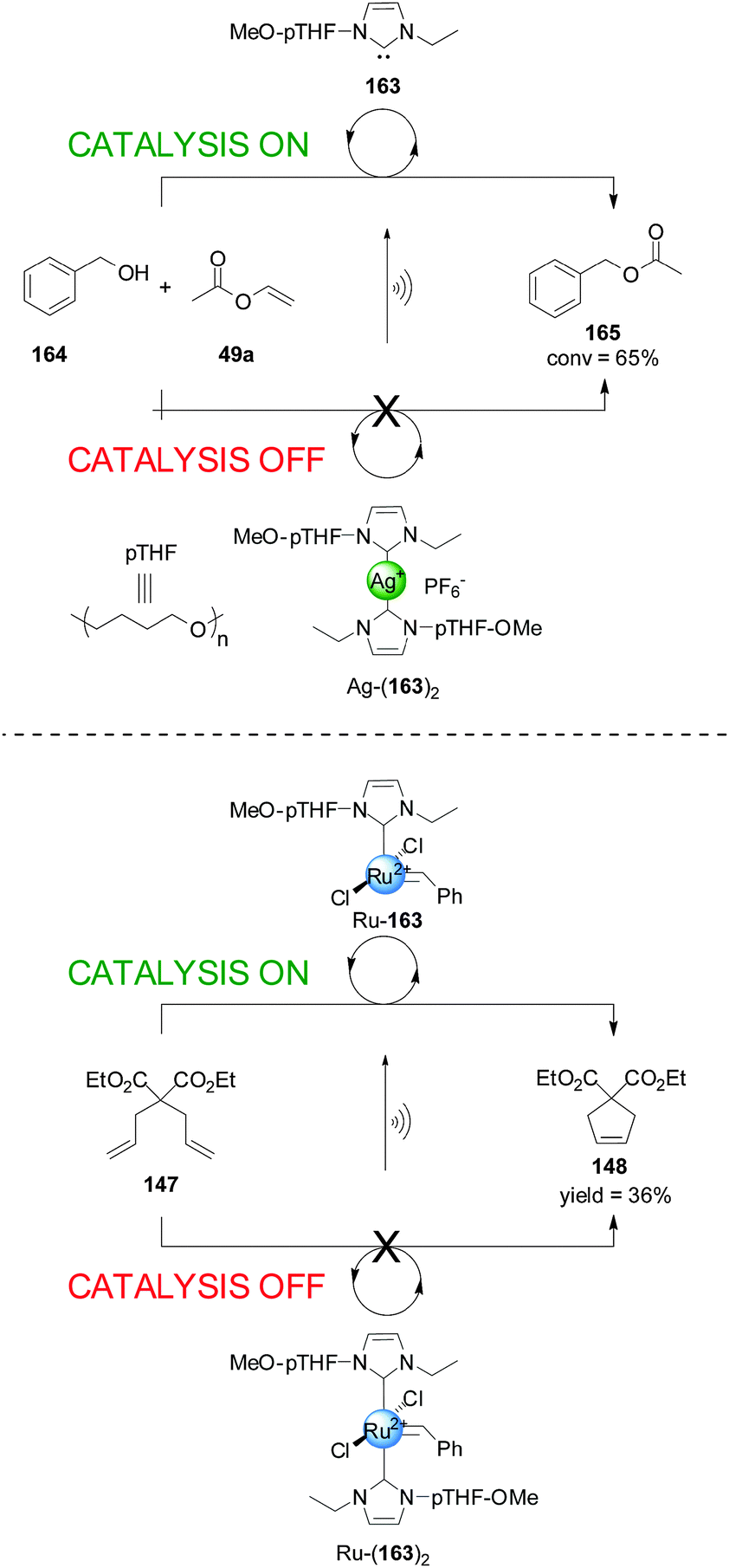 | ||
| Scheme 49 Polymer-functionalized catalysts activated by ultrasound-induced breaking of metal–ligand coordination bonds.87,88 | ||
In both these systems the mechanical activation pathway was consistent with the higher conversions obtained using higher molecular weight polymer chains on the ligands, and through the lack of catalytic activity of similar complexes with non-polymeric ligands. Further studies with polymers containing the NHC–silver(I) complex confirmed these results.89 Direct correlation between the conversion achieved and the concentration of the catalyst was explained by the catalytic species being deactivated by radicals thermally generated during sonication. Suppression of these species, e.g. by performing the reaction under a methane rather than argon atmosphere, increased the catalytic activity of both the Ag(I)- and Ru(II)-based catalysts.90 The activity of the NHC–Ru(II) containing catalyst in ring-opening olefin metathesis polymerization (ROMP) reactions was not as clear-cut as with RCM. Although the catalysed ROMP of cyclooctene stopped when sonication was ceased under some conditions, when the ROMP was performed under optimized reaction conditions the polymerization continued for several hours after sonication ceased due to the increased stability of the active species.91 Although useful for the application of these systems in self-healing materials,92 this may hinder its use in other area of switchable catalysis.
7. Switching driven by changes in reaction conditions
In Sections 1–6 of this Review we documented how control over the rate or selectivity of catalysis has been achieved with catalysts whose structure or reactivity is changed by a stimulus. Recently, significant changes in the chemo- or stereo-selectivity of catalyzed reactions brought about by altering reaction conditions have been reported. In this Section we describe some of the more remarkable examples of this phenomena and briefly highlight stereodivergent synthesis modulated by changes in experimental parameters that do not involve altering the chirality of the catalyst itself.93Jessop and co-workers found that a 1:1 mixture of a strong base, such as DBU (1,8-diazobicyclo[5.4.0]undadec-7-ene), and a primary alcohol, adds to CO2 to generate an ionic iminium carbonate, which upon purging with N2, reverts back to its non-ionic form.94 In 2014 Coulembier and colleagues exploited this process in developing a CO2-switchable ROP reaction (Scheme 50).95 The authors used DBU, TBD (triazabicyclodecene) and primary alcohol 164 (10:1:10) as the catalytic system, which is known to promote the ROP of cyclic esters such as 104. Under a N2 atmosphere (‘on’ state), the ROP polymerization of 104 to form 166 proceeded in excellent yield (96%), while simple exposure to CO2 formed DBUH+-165 and completely turned ‘off’ the catalytic activity. The authors demonstrated that the catalytic activity could be switched ‘on’ and ‘off’ multiple times by alternating exposure to N2 or CO2, providing the ability to regulate the molar mass of the resulting polymer.
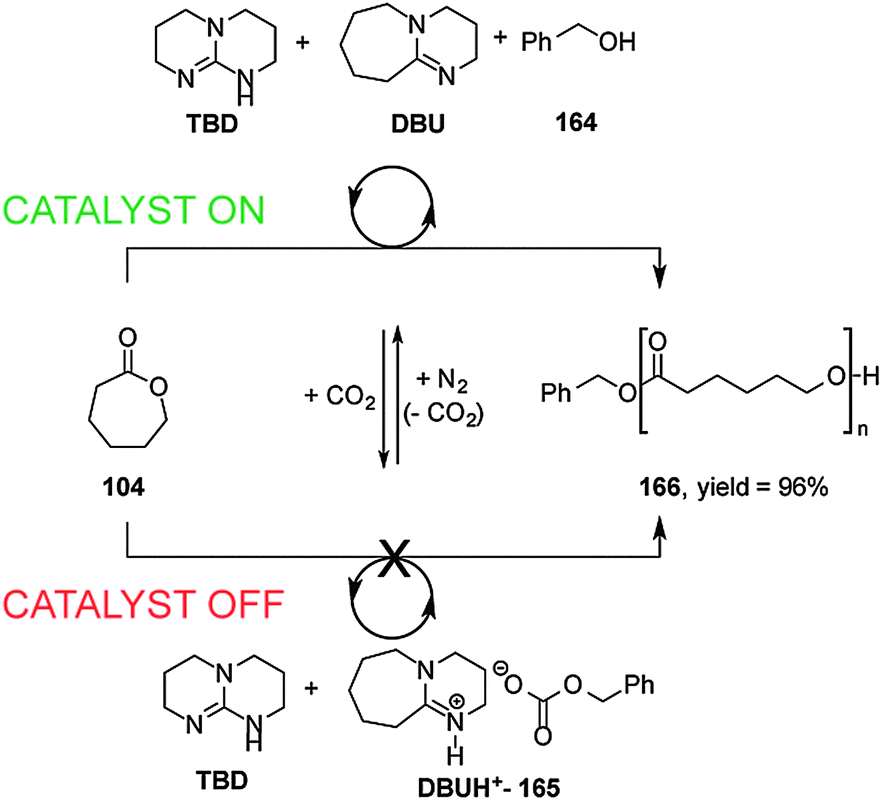 | ||
| Scheme 50 CO2-switchable catalytic system for the ROP of caprolactone (104).95 | ||
Williams and co-workers have recently developed a CO2-switchable metal catalyst, which is able to chemoselectively control both the ring opening polymerization of lactones and the ring-opening copolymerization of epoxides by using a N2 or CO2 atmosphere, respectively.96 By altering between these conditions—in a one pot procedure—the authors achieved the selective preparation of poly(carbonate-block-ester) copolymers.
In 2010, Sohtome and Nagasawa described an enantiodivergent organocatalyzed Mannich-type reaction using a conformationally flexible guanidine/bisthiourea catalyst (169) (Scheme 51).97 When the reaction between aromatic N-Boc-imines (167a–i) and dimethylmalonate (168) was carried out in nonpolar solvents, such as m-xylene, excellent yields and enantiocontrol (99% yield, 97% ee) in favour of the (S)-enantiomer were achieved. In contrast, when the reaction was performed using polar aprotic solvents, such as acetonitrile, the (R)-enantiomer was formed ((R)-170) in similar yield and enantioselectivity. Mechanistic studies suggested that the solvent-dependent stereodiscrimination exhibited by catalyst 169 is not controlled by aggregation effects or functional group masking by the solvent. Rather, it is likely a consequence of compensating differences in the enthalpies and entropies of activation. The S-stereoselectivity is controlled by differences in the entropies of activation, whereas the R-stereoselectivity arises from differences in the enthalpies of activation.
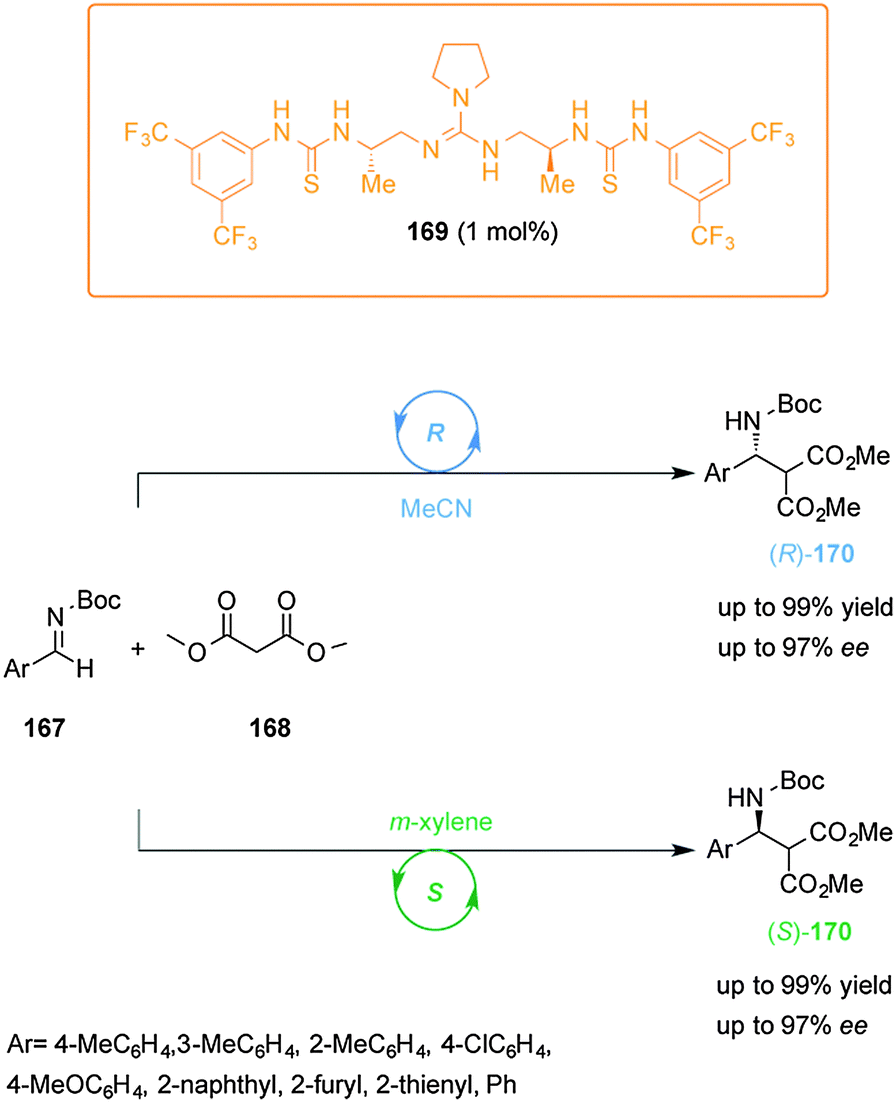 | ||
| Scheme 51 A solvent-dependant enantiodivergent Mannich-type reaction using a conformationally flexible guanidine/bisthiourea catalyst 169.97 | ||
In another impressive example, Melchiorre and co-workers described the diastereodivergent Michael addition of alkyl thiols (171) to α,β-unsaturated ketones (172) catalyzed by cinchona alkaloid derivative 173, in which the sense of the diastereoselectivity could be reversed by the choice of the solvent and the acid additive employed (Scheme 52).98 Using 2-fluorobenzoic acid in CHCl3, the syn-Michael products (syn-174) are obtained in high yield and enantioselectivity. However, using diphenyl phosphate (DPP) as the additive in acetone, led to the formation of the anti-Michael adducts (anti-174) in comparable yield and enantioselectivity. The switching in diastereoselectivity is likely a consequence of a change in the structure of the iminium intermediate caused by the counter-anion (which comes from the acid additive) and the solvent polarity. In situ switching in the diastereoselectivity from syn-174 to anti-174 was also demonstrated.
 | ||
| Scheme 52 Solvent- and acid-dependant diastereodivergent Michael addition of alkyl thiols to α,β-unsaturated ketones catalyzed by cinchona alkaloid derivative 173.98 | ||
Solvent-dependent stereoselectivity of other catalytic systems have also been reported.99 Changes in a variety of experimental parameters, including changes in temperature,100 the use of different metal ions with the same chiral ligand,101 change of the counter-anion102 or cation,103 the use of different acids as additives,104 or other kind of additives105 have all been successfully used to efficiently modulate the enantioselectivity of reactions.106
8. Conclusions and outlook
Switchable catalysts triggered by a range of stimuli, including light, pH, coordination events, redox processes, mechanical forces and changes in reaction conditions, are starting to prove their utility in the control of a range of different types of chemical transformations. Artificial switchable catalysts are already able to alter the rate, stereochemical outcome and chemoselectivity of different types of reaction. Although the field is still in its infancy it is growing rapidly, fuelled by developments in supramolecular chemistry, metal catalysis and organocatalysis. Most efforts are currently focussed on the invention of new types of switchable catalysts with different forms of stimuli-response units incorporated within their architectures in order to achieve control over the catalytic activity and regio-, chemo- and stereocontrol of various chemical reactions. A significant limitation in the majority (but by no means all) of the systems developed to date is the modest differences in the activities of the ‘on’ and ‘off’ states of the catalysts. In order to approach the level of control exhibited by enzymatic synthesis, artificial switchable catalysts need to be developed that accelerate reactions strongly in the ‘on’ state and not-at-all in the ‘off’ state.Two key challenges in particular remain unsolved: (i) the development of multi-responsive switchable catalysts that can control multiple catalytic processes by applying stimulus in a specific order; and (ii) the use of multiple (compatible) switchable catalysts with orthogonal responses, that can control which building blocks in a mixture react together, the order in which they react and the chemo- and regioselectivity of reactions and the outcome of one-pot tandem reaction sequences, without the need for further intervention.
This new area of catalysis research offers great potential for advanced functions and should ultimately be able to solve problems that are difficult or impossible to achieve using conventional methodologies. The next generation of switchable catalysts—featuring improved ‘on’/‘off’ ratios, greater substrate scope and the ability to catalyze multiple classes of reaction—should bring these goals tantalisingly within reach.
Acknowledgements
We thank the EPSRC for funding our research in this area. V. B. thanks Ministerio de Economía y Competitividad (Spain) for a ‘Juan de la Cierva’ postdoctoral contract.Notes and references
- T. Traut, Enzyme Activity: Allosteric Regulation, in eLS, John Wiley & Sons, 2001 Search PubMed.
- T. Traut, Allosteric Regulatory Enzymes, Springer, New York, 2008 Search PubMed.
- P. W. N. M. van Leeuwen, Homogeneous Catalysis. Understanding the Art, Springer, Dordrecht, 2004 Search PubMed; Catalysis for Fine Chemical Synthesis, ed. S. M. Roberts, John Wiley & Sons, 2007 Search PubMed.
- J. Barber, Chem. Soc. Rev., 2009, 38, 185–196 RSC.
- D. O'Brien, Science, 1982, 218, 961–966 Search PubMed.
- R. S. Stoll and S. Hecht, Angew. Chem., Int. Ed., 2010, 49, 5054–5075 CrossRef CAS PubMed; B. M. Neilson and C. W. Bielawski, ACS Catal., 2013, 3, 1874–1885 CrossRef; R. Göstl, A. Senf and S. Hecht, Chem. Soc. Rev., 2014, 43, 1982–1996 RSC; T. Imahori and S. Kurihara, Chem. Lett., 2014, 43, 1524–1531 CrossRef.
- F. Würthner and J. Rebek, Angew. Chem., Int. Ed. Engl., 1995, 34, 446–448 CrossRef PubMed.
- F. Würthner and J. Rebek, J. Chem. Soc., Perkin Trans. 2, 1995, 1727–1734 RSC.
- R. Cacciapaglia, S. Di Stefano and L. Mandolini, J. Am. Chem. Soc., 2003, 125, 2224–2227 CrossRef CAS PubMed.
- T. Imahori, R. Yamaguchi and S. Kurihara, Chem. – Eur. J., 2012, 18, 10802–10807 CrossRef CAS PubMed.
- M. Samanta, V. S. Rama Krishna and S. Bandyopadhyay, Chem. Commun., 2014, 50, 10577–10579 RSC.
- D. Sud, T. B. Norsten and N. R. Branda, Angew. Chem., Int. Ed., 2005, 44, 2019–2021 CrossRef CAS PubMed.
- J. Wang and B. L. Feringa, Science, 2011, 331, 1429–1432 CrossRef CAS PubMed.
- M. Vlatković, L. Bernardi, E. Otten and B. L. Feringa, Chem. Commun., 2014, 50, 7773–7775 RSC.
- Z. S. Kean, S. Akbulatov, Y. Tian, R. A. Widenhoefer, R. Boulatov and S. L. Craig, Angew. Chem., Int. Ed., 2014, 53, 14508–14511 CrossRef CAS PubMed.
- D. Zhao, T. M. Neubauer and B. L. Feringa, Nat. Commun., 2015, 6, 6652 CrossRef PubMed.
- A. Ueno, K. Takahashi and T. Osa, J. Chem. Soc., Chem. Commun., 1980, 837–838 RSC; A. Ueno, K. Takahashi and T. Osa, J. Chem. Soc., Chem. Commun., 1981, 94–96 RSC.
- W.-S. Lee and A. Ueno, Macromol. Rapid Commun., 2001, 22, 448–450 CrossRef CAS.
- L. Zhu, H. Yan, C. Y. Ang, K. T. Nguyen, M. Li and Y. Zhao, Chem. – Eur. J., 2012, 18, 13979–13983 CrossRef CAS PubMed.
- M. Osaki, Y. Takashima, H. Yamaguchi and A. Harada, Org. Biomol. Chem., 2009, 7, 1646–1651 CAS.
- O. B. Berryman, A. C. Sather, A. Lledó and J. Rebek, Angew. Chem., Int. Ed., 2011, 50, 9400–9403 CrossRef CAS PubMed.
- H. Sugimoto, T. Kimura and S. Inoue, J. Am. Chem. Soc., 1999, 121, 2325–2326 CrossRef CAS.
- M. V. Peters, R. S. Stoll, A. Kühn and S. Hecht, Angew. Chem., Int. Ed., 2008, 47, 5968–5972 CrossRef CAS PubMed; R. S. Stoll, M. V. Peters, A. Kühn, S. Heiles, R. Goddard, M. Bühl, C. M. Thiele and S. Hecht, J. Am. Chem. Soc., 2009, 131, 357–367 CrossRef PubMed.
- R. S. Stoll and S. Hecht, Org. Lett., 2009, 11, 4790–4793 CrossRef CAS PubMed.
- L. Osorio-Planes, C. Rodríguez-Escrich and M. A. Pericàs, Org. Lett., 2014, 16, 1704–1707 CrossRef CAS PubMed.
- D. Wilson and N. R. Branda, Angew. Chem., Int. Ed., 2012, 51, 5431–5434 CrossRef CAS PubMed.
- B. M. Neilson and C. W. Bielawski, J. Am. Chem. Soc., 2012, 134, 12693–12699 CrossRef CAS PubMed.
- B. M. Neilson and C. W. Bielawski, Chem. Commun., 2013, 49, 5453 RSC.
- B. M. Neilson and C. W. Bielawski, Organometallics, 2013, 32, 3121–3128 CrossRef CAS.
- T. Niazov, B. Shlyahovsky and I. Willner, J. Am. Chem. Soc., 2007, 129, 6374–6375 CrossRef CAS PubMed.
- Y. Wei, S. Han, J. Kim, S. Soh and B. A. Grzybowski, J. Am. Chem. Soc., 2010, 132, 11018–11020 CrossRef CAS PubMed.
- A. Nojiri, N. Kumagai and M. Shibasaki, Chem. Commun., 2013, 49, 4628–4630 RSC.
- S. L. Balof, S. J. P'Pool, N. J. Berger, E. J. Valente, A. M. Shiller and H.-J. Schanz, Dalton Trans., 2008, 5791–5799 RSC.
- S. L. Balof, B. Yu, A. B. Lowe, Y. Ling, Y. Zhang and H.-J. Schanz, Eur. J. Inorg. Chem., 2009, 1717–1722 CrossRef CAS PubMed.
- L. H. Peeck, S. Leuthäusser and H. Plenio, Organometallics, 2010, 29, 4339–4345 CrossRef CAS.
- For other rotaxanes incorporating catalytic units, see: P. Thordarson, E. J. A. Bijsterveld, A. E. Rowan and R. J. M. Nolte, Nature, 2003, 424, 915–918 CrossRef CAS PubMed; Y. Tachibana, N. Kihara and T. Takata, J. Am. Chem. Soc., 2004, 126, 3438–3439 CrossRef PubMed; G. Hattori, T. Hori, Y. Miyake and Y. Nishibayashi, J. Am. Chem. Soc., 2007, 129, 12930–12931 CrossRef PubMed; Y. Li, Y. Feng, Y.-M. He, F. Chen, J. Pan and Q.-H. Fan, Tetrahedron Lett., 2008, 49, 2878–2881 CrossRef PubMed; J. Berná, M. Alajarín and R.-A. Orenes, J. Am. Chem. Soc., 2010, 132, 10741–10747 CrossRef PubMed; N. Miyagawa, M. Watanabe, T. Matsuyama, Y. Koyama, T. Moriuchi, T. Hirao, Y. Furusho and T. Takata, Chem. Commun., 2010, 46, 1920–1922 RSC; Y. Suzaki, K. Shimada, E. Chihara, T. Saito, Y. Tsuchido and K. Osakada, Org. Lett., 2011, 13, 3774–3777 CrossRef PubMed; B. Lewandowski, G. De Bo, J. W. Ward, M. Papmeyer, S. Kuschel, M. J. Aldegunde, P. M. E. Gramlich, D. Heckmann, S. M. Goldup, D. M. D'Souza, A. E. Fernandes and D. A. Leigh, Science, 2013, 339, 189–193 CrossRef PubMed.
- U. Lüning, Angew. Chem., Int. Ed., 2012, 51, 8163–8165 CrossRef CAS PubMed; D. A. Leigh, V. Marcos and M. R. Wilson, ACS Catal., 2014, 4, 4490–4497 CrossRef.
- V. Blanco, A. Carlone, K. D. Hänni, D. A. Leigh and B. Lewandowski, Angew. Chem., Int. Ed., 2012, 51, 5166–5169 CrossRef CAS PubMed.
- V. Blanco, D. A. Leigh, U. Lewandowska, B. Lewandowski and V. Marcos, J. Am. Chem. Soc., 2014, 136, 15775–15780 CrossRef CAS PubMed.
- V. Blanco, D. A. Leigh, V. Marcos, J. A. Morales-Serna and A. L. Nussbaumer, J. Am. Chem. Soc., 2014, 136, 4905–4908 CrossRef CAS PubMed.
- J. Beswick, V. Blanco, G. De Bo, D. A. Leigh, U. Lewandowska, B. Lewandowski and K. Mishiro, Chem. Sci., 2015, 6, 140–143 RSC.
- S. Shinkai, Y. Ishikawa, H. Shinkai, T. Tsuno and O. Manabe, Tetrahedron Lett., 1983, 24, 1539–1542 CrossRef CAS; S. Shinkai, Y. Ishikawa, H. Shinkai, T. Tsuno, H. Makishima, K. Ueda and O. Manabe, J. Am. Chem. Soc., 1984, 106, 1801–1808 CrossRef; S. Shinkai, K. Kameoka, K. Ueda and O. Manabe, J. Am. Chem. Soc., 1987, 109, 923–924 CrossRef.
- M. J. Wiester, P. A. Ulmann and C. A. Mirkin, Angew. Chem., Int. Ed., 2011, 50, 114–137 CrossRef CAS PubMed.
- N. C. Gianneschi, M. S. Masar and C. A. Mirkin, Acc. Chem. Res., 2005, 38, 825–837 CrossRef CAS PubMed.
- N. C. Gianneschi, P. A. Bertin, S. T. Nguyen, C. A. Mirkin, L. N. Zakharov and A. L. Rheingold, J. Am. Chem. Soc., 2003, 125, 10508–10509 CrossRef CAS PubMed.
- N. C. Gianneschi, S. T. Nguyen and C. A. Mirkin, J. Am. Chem. Soc., 2005, 127, 1644–1645 CrossRef CAS PubMed.
- H. J. Yoon and C. A. Mirkin, J. Am. Chem. Soc., 2008, 130, 11590–11591 CrossRef CAS PubMed.
- M. S. Masar, N. C. Gianneschi, C. G. Oliveri, C. L. Stern, S. T. Nguyen and C. A. Mirkin, J. Am. Chem. Soc., 2007, 129, 10149–10158 CrossRef CAS PubMed.
- C. G. Oliveri, N. C. Gianneschi, S. T. Nguyen, C. A. Mirkin, C. L. Stern, Z. Wawrzak and M. Pink, J. Am. Chem. Soc., 2006, 128, 16286–16296 CrossRef CAS PubMed.
- H. J. Yoon, J. Heo and C. A. Mirkin, J. Am. Chem. Soc., 2007, 129, 14182–14183 CrossRef CAS PubMed.
- N. C. Gianneschi, S.-H. Cho, S. T. Nguyen and C. A. Mirkin, Angew. Chem., Int. Ed., 2004, 43, 5503–5507 CrossRef CAS PubMed.
- H. J. Yoon, J. Kuwabara, J. H. Kim and C. A. Mirkin, Science, 2010, 330, 66–69 CrossRef CAS PubMed.
- C. M. McGuirk, J. Mendez-Arroyo, A. M. Lifschitz and C. A. Mirkin, J. Am. Chem. Soc., 2014, 136, 16594–16601 CrossRef CAS PubMed.
- C. M. McGuirk, C. L. Stern and C. A. Mirkin, J. Am. Chem. Soc., 2014, 136, 4689–4696 CrossRef CAS PubMed.
- M. Zirngast, E. Pump, A. Leitgeb, J. H. Albering and C. Slugovc, Chem. Commun., 2011, 47, 2261–2263 RSC.
- J. H. Griffin and P. B. Dervan, J. Am. Chem. Soc., 1987, 109, 6840–6842 CrossRef CAS.
- T. Tozawa, S. Tokita and Y. Kubo, Tetrahedron Lett., 2002, 43, 3455–3457 CrossRef CAS.
- L. Kovbasyuk and R. Krämer, Chem. Rev., 2004, 104, 3161–3188 CrossRef CAS PubMed; I. O. Fritsky, R. Ott and R. Krämer, Angew. Chem., Int. Ed., 2000, 39, 3255–3258 CrossRef; I. O. Fritsky, R. Ott, H. Pritzkow and R. Krämer, Chem. – Eur. J., 2001, 7, 1221–1231 CrossRef; I. O. Fritsky, R. Ott, H. Pritzkow and R. Krämer, Inorg. Chim. Acta, 2003, 346, 111–118 CrossRef; K. P. Strotmeyer, I. O. Fritsky, R. Ott, H. Pritzkow and R. Krämer, Supramol. Chem., 2003, 15, 529–547 CrossRef PubMed.
- L. Kovbasyuk, H. Pritzkow, R. Krämer and I. O. Fritsky, Chem. Commun., 2004, 880–881 RSC.
- A. Scarso, G. Zaupa, F. B. Houillon, L. J. Prins and P. Scrimin, J. Org. Chem., 2007, 72, 376–385 CrossRef CAS PubMed.
- S. Takebayashi, M. Ikeda, M. Takeuchi and S. Shinkai, Chem. Commun., 2004, 420–421 RSC; S. Takebayashi, S. Shinkai, M. Ikeda and M. Takeuchi, Org. Biomol. Chem., 2008, 6, 493–499 Search PubMed.
- M. R. Kita and A. J. M. Miller, J. Am. Chem. Soc., 2014, 136, 14519–14529 CrossRef CAS PubMed.
- G.-H. Ouyang, Y.-M. He, Y. Li, J.-F. Xiang and Q.-H. Fan, Angew. Chem., Int. Ed., 2015, 54, 4334–4337 CrossRef CAS PubMed.
- M. Schmittel, S. De and S. Pramanik, Angew. Chem., Int. Ed., 2012, 51, 3832–3836 CrossRef CAS PubMed.
- M. Schmittel, S. Pramanik and S. De, Chem. Commun., 2012, 48, 11730–11732 RSC.
- S. De, S. Pramanik and M. Schmittel, Dalton Trans., 2014, 43, 10977–10982 RSC.
- S. De, S. Pramanik and M. Schmittel, Angew. Chem., Int. Ed., 2014, 53, 14255–14259 CrossRef CAS PubMed.
- J. T. Foy, D. Ray and I. Aprahamian, Chem. Sci., 2015, 6, 209–213 RSC.
- I. M. Lorkovic, R. R. Duff and M. S. Wrighton, J. Am. Chem. Soc., 1995, 117, 3617–3618 CrossRef CAS.
- C. S. Slone, C. A. Mirkin, G. P. A. Yap, I. A. Guzei and A. L. Rheingold, J. Am. Chem. Soc., 1997, 119, 10743–10753 CrossRef CAS.
- C. K. A. Gregson, V. C. Gibson, N. J. Long, E. L. Marshall, P. J. Oxford and A. J. P. White, J. Am. Chem. Soc., 2006, 128, 7410–7411 CrossRef CAS PubMed.
- E. M. Broderick, N. Guo, C. S. Vogel, C. Xu, J. Sutter, J. T. Miller, K. Meyer, P. Mehrkhodavandi and P. L. Diaconescu, J. Am. Chem. Soc., 2011, 133, 9278–9281 CrossRef CAS PubMed.
- K. Arumugam, C. D. Varnado, S. Sproules, V. M. Lynch and C. W. Bielawski, Chem. – Eur. J., 2013, 19, 10866–10875 CrossRef CAS PubMed.
- M. Süßner and H. Plenio, Angew. Chem., Int. Ed., 2005, 44, 6885–6888 CrossRef PubMed.
- P. Neumann, H. Dib, A.-M. Caminade and E. Hey-Hawkins, Angew. Chem., Int. Ed., 2015, 54, 311–314 CrossRef CAS PubMed.
- A. J. D. Magenau, N. C. Strandwitz, A. Gennaro and K. Matyjaszewski, Science, 2011, 332, 81–84 CrossRef CAS PubMed.
- E. M. Broderick, N. Guo, T. Wu, C. S. Vogel, C. Xu, J. Sutter, J. T. Miller, K. Meyer, T. Cantat and P. L. Diaconescu, Chem. Commun., 2011, 47, 9897–9899 RSC.
- A. Sauer, J.-C. Buffet, T. P. Spaniol, H. Nagae, K. Mashima and J. Okuda, ChemCatChem, 2013, 5, 1088–1091 CrossRef CAS PubMed.
- A. B. Biernesser, B. Li and J. A. Byers, J. Am. Chem. Soc., 2013, 135, 16553–16560 CrossRef CAS PubMed.
- Y.-Y. Fang, W.-J. Gong, X.-J. Shang, H.-X. Li, J. Gao and J.-P. Lang, Dalton Trans., 2014, 43, 8282–8289 RSC.
- S. Mortezaei, N. R. Catarineu and J. W. Canary, J. Am. Chem. Soc., 2012, 134, 8054–8057 CrossRef CAS PubMed.
- X. Wang, A. Thevenon, J. L. Brosmer, I. Yu, S. I. Khan, P. Mehrkhodavandi and P. L. Diaconescu, J. Am. Chem. Soc., 2014, 136, 11264–11267 CrossRef CAS PubMed.
- Q. Zhang, X. Cui, L. Zhang, S. Luo, H. Wang and Y. Wu, Angew. Chem., Int. Ed., 2015, 54, 5210–5213 CrossRef CAS PubMed.
- M. Fujiwara, S. Terashima, Y. Endo, K. Shiokawa and H. Ohue, Chem. Commun., 2006, 4635–4637 RSC.
- A. G. Tennyson, V. M. Lynch and C. W. Bielawski, J. Am. Chem. Soc., 2010, 132, 9420–9429 CrossRef CAS PubMed.
- R. Groote, R. T. M. Jakobs and R. P. Sijbesma, Polym. Chem., 2013, 4, 4846–4859 RSC.
- S. Karthikeyan, S. L. Potisek, A. Piermattei and R. P. Sijbesma, J. Am. Chem. Soc., 2008, 130, 14968–14969 CrossRef CAS PubMed.
- A. Piermattei, S. Karthikeyan and R. P. Sijbesma, Nat. Chem., 2009, 1, 133–137 CrossRef CAS PubMed.
- R. Groote, L. van Haandel and R. P. Sijbesma, J. Polym. Sci., Part A: Polym. Chem., 2012, 50, 4929–4935 CrossRef CAS PubMed.
- R. Groote, R. T. M. Jakobs and R. P. Sijbesma, ACS Macro Lett., 2012, 1, 1012–1015 CrossRef CAS.
- R. T. M. Jakobs and R. P. Sijbesma, Organometallics, 2012, 31, 2476–2481 CrossRef CAS.
- R. T. M. Jakobs, S. Ma and R. P. Sijbesma, ACS Macro Lett., 2013, 2, 613–616 CrossRef CAS.
- Y. H. Kim, Acc. Chem. Res., 2001, 34, 955–962 CrossRef CAS PubMed; M. Bartók, Chem. Rev., 2010, 110, 1663–1705 CrossRef PubMed; Z. Dai, J. Lee and W. Zhang, Molecules, 2012, 17, 1247–1277 CrossRef PubMed; N. Kumagai and M. Shibasaki, Catal. Sci. Technol., 2013, 3, 41–57 Search PubMed.
- P. G. Jessop, D. J. Heldebrant, X. Li, C. A. Eckert and C. L. Liotta, Nature, 2005, 436, 1102 CrossRef CAS PubMed.
- O. Coulembier, S. Moins, R. Todd and P. Dubois, Macromolecules, 2014, 47, 486–491 CrossRef CAS.
- C. Romain and C. K. Williams, Angew. Chem., Int. Ed., 2014, 53, 1607–1610 CrossRef CAS PubMed.
- Y. Sohtome, S. Tanaka, K. Takada, T. Yamaguchi and K. Nagasawa, Angew. Chem., Int. Ed., 2010, 49, 9254–9257 CrossRef CAS PubMed.
- X. Tian, C. Cassani, Y. Liu, A. Moran, A. Urakawa, P. Galzerano, E. Arceo and P. Melchiorre, J. Am. Chem. Soc., 2011, 133, 17934–17941 CrossRef CAS PubMed.
- S. Arseniyadis, A. Valleix, A. Wagner and C. Mioskowski, Angew. Chem., Int. Ed., 2004, 43, 3314–3317 CrossRef CAS PubMed; A. B. Northrup and D. W. C. MacMillan, Science, 2004, 305, 1752–1755 CrossRef PubMed; T. Yamamoto, T. Yamada, Y. Nagata and M. Suginome, J. Am. Chem. Soc., 2010, 132, 7899–7901 CrossRef PubMed; M. Messerer and H. Wennemers, Synlett, 2011, 499–502 Search PubMed; R. J. Chew, X.-R. Li, Y. Li, S. A. Pullarkat and P.-H. Leung, Chem. – Eur. J., 2015, 21, 4800–4804 CrossRef PubMed.
- G. Storch and O. Trapp, Angew. Chem., Int. Ed., 2015, 54, 3580–3586 CrossRef CAS PubMed.
- A. Nojiri, N. Kumagai and M. Shibasaki, J. Am. Chem. Soc., 2009, 131, 3779–3784 CrossRef CAS PubMed; G. Lu, T. Yoshino, H. Morimoto, S. Matsunaga and M. Shibasaki, Angew. Chem., Int. Ed., 2011, 50, 4382–4385 CrossRef PubMed.
- J. Lv, L. Zhang, Y. Zhou, Z. Nie, S. Luo and J.-P. Cheng, Angew. Chem., Int. Ed., 2011, 50, 6610–6614 CrossRef CAS PubMed.
- P. Mazón, R. Chinchilla, C. Nájera, G. Guillena, R. Kreiter, R. J. M. Klein Gebbink and G. van Koten, Tetrahedron: Asymmetry, 2002, 13, 2181–2185 CrossRef.
- J. Gao, S. Bai, Q. Gao, Y. Liu and Q. Yang, Chem. Commun., 2011, 47, 6716–6718 RSC; R. Doran, M. P. Carroll, R. Akula, B. F. Hogan, M. Martins, S. Fanning and P. J. Guiry, Chem. – Eur. J., 2014, 20, 15354–15359 CrossRef CAS PubMed; R. Doran and P. J. Guiry, J. Org. Chem., 2014, 79, 9112–9124 CrossRef PubMed.
- S.-H. Chen, B.-C. Hong, C.-F. Su and S. Sarshar, Tetrahedron Lett., 2005, 46, 8899–8903 CrossRef CAS PubMed; N. Abermil, G. Masson and J. Zhu, Org. Lett., 2009, 11, 4648–4651 CrossRef PubMed.
- M. Bartók, Chem. Rev., 2010, 110, 1663–1705 CrossRef CAS PubMed; J. Escorihuela, M. I. Burguete and S. V. Luis, Chem. Soc. Rev., 2013, 42, 5595–5617 RSC.
Footnote |
| † These authors contributed equally to the writing of this review. |
| This journal is © The Royal Society of Chemistry 2015 |