
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Revisiting Li3V2(PO4)3 as an anode – an outstanding negative electrode for high power energy storage devices†
Xiaofei
Zhang‡
,
Ruben-Simon
Kühnel‡
,
Matthias
Schroeder
and
Andrea
Balducci
*
University of Muenster, MEET Battery Research Center & Institute of Physical Chemistry, Corrensstr. 28/30, 48149 Münster, Germany. E-mail: andrea.balducci@uni-muenster.de
First published on 3rd September 2014
Abstract
Monoclinic Li3V2(PO4)3 (LVP) has long been considered primarily as a cathode material for lithium-ion batteries (LIBs). However, due to its amphoteric nature, LVP can also host additional lithium ions. Nonetheless, its use as an anode material for LIBs has hardly been investigated. In this work, we synthesize a nanostructured Li3V2(PO4)3 material with an ionic liquid-derived carbon coating and test it as an anode material for LIBs. The nanostructured LVP shows excellent rate capability and delivers an exceptionally high capacity of about 100 mA h g−1 at 100 C. Fast lithiation/delithiation of the material is enabled by its nanorod-like structure, which allows rapid Li+ diffusion, and its high electronic conductivity due to an effective carbon coating. Furthermore, when cycled at 50 C, the capacity retention is 91% after 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cycles, and ex situ XRD shows a good preservation of the LVP structure. Due to its excellent high rate capacity and longterm stability, nanostructured LVP is a very promising candidate for the use as a negative electrode in lithium-ion capacitors (LICs). We show that a LIC containing LVP as a negative electrode and activated carbon as a positive one displays an energy density of 33 W h kg−1 at a power density of 16 kW kg−1; stable for 100000 cycles.
000 cycles, and ex situ XRD shows a good preservation of the LVP structure. Due to its excellent high rate capacity and longterm stability, nanostructured LVP is a very promising candidate for the use as a negative electrode in lithium-ion capacitors (LICs). We show that a LIC containing LVP as a negative electrode and activated carbon as a positive one displays an energy density of 33 W h kg−1 at a power density of 16 kW kg−1; stable for 100000 cycles.
1. Introduction
Electrochemical energy storage devices like lithium-ion batteries (LIBs) and supercapacitors (SCs) are in growing demand fuelled by new applications in areas such as electric mobility and renewable energies.1,2 In these latter applications not only high energy, but also high power performance might be required. SCs are the devices of choice for high power applications, but due to their storage mechanism their energy is considerably lower than that of LIBs.3,4 Therefore, in the last few years much effort has been made for the realization of advanced high power LIBs as well as on the development of innovative high power devices, e.g. lithium-ion capacitors (LICs), which are in most of the cases hybrids of LIBs and SCs.5–11High power LIBs presently available mainly contain anodes based on nanostructured lithium titanate (Li4Ti5O12, LTO). Nanostructured LTO can be lithiated/delithiated at high current rates, thus guaranteeing high power performance. However, the rather low capacity of this material and the high lithiation/delithiation potential of ca. 1.5 V vs. Li/Li+ limit the practical energy density of these high power LIBs.12 Graphite is used as an anode in conventional LIBs as well as for the negative electrode of the commercially available LICs. Graphite displays higher specific capacity than LTO and its use allows the realization of devices with high operative voltage (about 4 V). However, the rate capability of graphite, especially for the lithiation process, is rather limited due to long diffusion pathways and the hindrance towards Li+ intercalation caused by the staging mechanism, limiting the power performance of the devices containing this anode.13
Taking these points into account, the development of new anode materials with high ionic and electronic conductivities and with a relatively low average lithiation/delithiation potential (as close as possible to 0.0 V vs. Li/Li+) appears therefore of great importance for the realization of innovative high power devices.
Lithium vanadium phosphate (Li3V2(PO4)3; LVP) has attracted much attention as a cathode material owing to its high theoretical capacity of 197 mA h g−1 when charged up to 4.8 V vs. Li/Li+, high average potential, good stability and low costs.14 The structure of monoclinic LVP (space group: P21/n) is a three-dimensional network consisting of VO6 octahedra and PO4 tetrahedra linked together via common oxygen atoms to form a (V–O–P–O)n bonding arrangement, which houses Li+ ions in relatively large interstitial sites.15–17 As a consequence, Li+ ions can be reversibly extracted and re-inserted into the LVP structure with good ionic mobility, without causing too many structural changes of the LVP lattice. However, LVP displays a relatively low intrinsic electronic conductivity, which limits the performance of this material, particularly at high rates. Nonetheless, several studies showed that nanostructured and carbon-coated LVP particles might display high electronic conductivity and that these nanomaterials can be regarded as interesting materials also for high power applications.14,18
A very interesting feature of LVP is its amphoteric nature. LVP can also host additional Li+ ions and can therefore also be used as an anode material. LVP-based anodes can be used down to 0.0 V vs. Li/Li+, and they display two distinct potential regions of lithiation/delithiation: a two-phase region at high potentials (ca. 2.0–1.6 V vs. Li/Li+) and a single-phase region below ca. 1.6 V. So far, only a rather limited number of studies considered the use of LVP as a negative electrode material for energy storage devices.19–25 Nevertheless, considering the operative potential as well as the high ionic conductivity of LVP, anodes containing this amphoteric material could be of interest for the realization of innovative high power devices.
In a recent study, we reported about the ionic liquid-assisted synthesis of carbon coated LVP nanoparticles.18 We showed that the use of ionic liquids as a template for the realization of nanostructured LVP might lead to the realization of nanomaterials with high ionic and electronic conductivities, which are very promising in view of the realization of advanced high power devices.
In this work, we investigate the use of nanostructured LVP as an anode material for high power devices. In the first part of the manuscript, the diffusion processes as well as the structural changes occurring during the lithium insertion–extraction process on the LVP structure are investigated. In the second part, the rate performance and the cycling stability of LVP-based anodes are assessed. Finally, the use of LVP-based anodes for LICs is considered. The results of this study show that LVP-based negative electrodes might show excellent rate performance and cycle life and, therefore, are a promising candidate for the realization of advanced high power devices.
2. Experimental
Materials synthesis
Nanostructured Li3V2(PO4)3 (LVP) with a carbon coating based on the ionic liquid N-butyl-N-methylpyrrolidinium bis(trifluoromethanesulfonyl)imide was synthesized following our procedure described in ref. 18.Materials characterization
The crystalline structure of the LVP powder was characterized by X-ray diffraction (XRD) using Cu Kα radiation on a Bruker D8 Advance (Germany) for 4 s at each 0.02° step width from 15 to 60°. To analyze structural changes during cycling, ex situ XRD measurements were carried out for electrodes (see below) that were cycled for 10000 cycles between 3.0 and 0.0 V vs. Li/Li+ at a rate of 50 C. These electrodes were recovered from cycled cells stopped at 3.0 V. The cells were disassembled in an argon-filled glove box and carefully washed with DMC in order to remove electrolyte residues. For comparison, the XRD pattern of a pristine electrode was also recorded. For the XRD measurements of the pristine and cycled electrodes, the step time was set to 6 s, the step width to 0.01° and the 2θ range to 15–60°.
In situ XRD measurements of LVP upon galvanostatic lithiation and delithiation were performed using a self-designed in situ cell. The cell body is made of stainless steel covered internally by a Mylar foil for electrical insulation. For the electrode preparation, 65 wt% LVP, 25 wt% conducting agent (Super C65, TIMCAL) and 10 wt% binder (polyvinylidene fluoride, PVDF) were mixed in N-methyl-2-pyrrolidone and stirred overnight. The obtained slurry was cast on a beryllium (Be) window, having a thickness of 250 μm (Brush Wellman), which served at the same time as a current collector and a “window” for the X-ray beam. The coated Be window was subsequently dried at 80 °C for 30 min and at 40 °C under vacuum overnight. Metallic lithium foil served as counter and reference electrodes. Two sheets of a Whatman glass fiber filter served as a separator and were drenched with 500 μL of electrolyte (1 M LiPF6 in EC:DMC (1:1 by weight)). The assembled cell was allowed to rest for 2 h. Subsequently, the cell was galvanostatically cycled at a rate of C/10 using a VSP potentiostat/galvanostat (Bio-Logic Science Instruments). XRD measurements were performed with the 2θ range set to 15–47°. A complete scan was recorded every 30 minutes, including a rest period at the beginning of every scan. After discharging to a lower cut-off potential of 0.0 V vs. Li/Li+, the cell was charged to an upper cut-off potential of 3.0 V.
The Raman spectrum of LVP was collected with a SENTERRA Raman microscope (Bruker Optics) as reported in ref. 18. The morphology and chemical composition of the carbon-coated LVP sample were characterized with a scanning electron microscope (SEM, AURIGA, Carl Zeiss, equipped with an energy-dispersive X-ray analyser (EDX)). The amount of carbon in the final product was evaluated by CHN analysis.
Electrochemical measurements
LVP electrodes were prepared by mixing 70 wt% LVP, 20 wt% conducting agent (Super C65, TIMCAL) and 10 wt% binder (polyvinylidene fluoride, PVDF) in N-methyl-2-pyrrolidone followed by stirring overnight. The obtained slurry was cast on dendritic copper foil (Schlenk, Germany) with a laboratory scale doctor blade set to a thickness of 150 μm. The electrode sheets were dried at 80 °C for 12 h. Disc electrodes with a diameter of 12 mm were cut out of the sheets and further dried at 120 °C under vacuum for 24 h. The mass loading of the electrodes was ca. 1–1.5 mg cm−2. All electrochemical measurements except for the in situ XRD measurements were carried out in 3-electrode Swagelok cells. The cells were assembled in an argon-filled glove box with oxygen and water levels below 1 ppm. The LVP electrodes were used as working electrodes and metallic lithium (Rockwood Lithium) was used as counter and reference electrodes. Whatman GF/D glass fiber filters drenched with 200 μL of electrolyte (1 M LiPF6 in EC:DMC (1:1 by weight)) were used as a separator.
Constant current cycling tests were performed on a MACCOR Battery tester 4300 in the potential range of 3.0 to 0.0 V vs. Li/Li+. The current rate of 1 C corresponds to a specific current of 266 mA g−1. The tests were carried out in climatic chambers set to 20 °C. For the rate test, five cycles were carried out at each current density. In Fig. 4, the potential profiles of each fifth cycle are shown. The discharge capacity of each fifth cycle was also used to calculate the capacity retention. Prior to the rate test, the cell was activated for five cycles at 0.1 C.
Cyclic voltammetry (CV) was performed on a VMP3 at scan rates of 0.05 to 0.6 mV s−1 in a potential range of 3.0 to 0.0 V vs. Li/Li+. The galvanostatic intermittent titration technique (GITT) was used to obtain diffusion coefficients of LVP over the whole potential range from 3.0 to 0.0 V vs. Li/Li+ during both lithiation and delithiation. The measurements were carried out with a MACCOR Battery tester 4300 after three charge–discharge cycles for activation carried out at a rate of 0.1 C with a VMP3. The electrodes were charged/discharged at 20 °C with a current density of 0.1 C for a time τ of 600 s followed by a relaxation period of 2 h at open-circuit potential (OCP). This charge–discharge step was continued until the desired cut-off potential of 0.0 or 3.0 V vs. Li/Li+ was reached. Lithium diffusion coefficients D were then calculated from the GITT measurements with the following equation:26
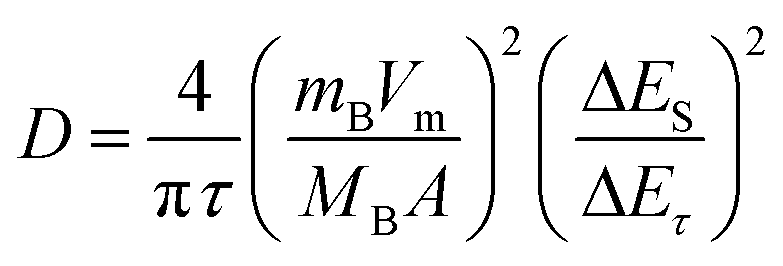 | (1) |
The LVP electrodes for the LIC experiments were prelithiated via a metallic Li electrode as described in the literature.27,28 The activated carbon (AC) positive electrodes were prepared similar to the LVP electrodes. The composition was 90 wt% AC (DLC Super 30, Norit, USA, specific BET surface area: 1400 m2 g−1), 5 wt% conducting agent (Super C65, TIMCAL) and 5 wt% sodium carboxymethylcellulose as a binder (CMC, Walocel CRT 2000 PA, Dow Wolff Cellulosics, Germany, dissolved in water). The balancing of the LIC full cells was based on the capacity achieved at a rate of 100 C for the LVP anodes and the corresponding current for the AC electrodes (around 25 mA in a potential window of 3.0 to 4.1 V vs. Li/Li+) and added up to a mass ratio of LVP:AC of 1:2.22. Similar to the half cell tests, Whatman glass fiber filters were used as a separator and 1 M LiPF6 in EC:DMC (1:1 by weight) was used as the electrolyte. All LICs were cycled using a VMP3 multichannel potentiostat/galvanostat (Bio-Logic Science Instruments) between 0.0 and 4.0 V. The used rates were 25 C, 50 C and 100 C. For better comparison, the balancing of all three rates was kept the same and the corresponding currents can be found in Table 1. The stated energy and power density were calculated from the constant current cycling results following a procedure reported before and are based on the active masses of both electrodes.27 Depending on the rate, 30000, 60000 and 100000 cycles were performed, respectively. The reported energy and power densities of the LIB and SC are taken from the literature.27 All potentials reported in this work refer to the Li/Li+ couple.
| Rate | I LVP [A g−1] | I full cell [A g−1] | I [mA cm−2] |
|---|---|---|---|
| 1 C | 0.266 | 0.0826 | 0.224 |
| 25 C | 6.65 | 2.07 | 5.59 |
| 50 C | 13.3 | 4.13 | 11.2 |
| 100 C | 26.6 | 8.26 | 22.36 |
3. Results and discussion
Fig. 1a shows an X-ray diffraction (XRD) pattern of the carbon-coated LVP nanostructure investigated in this study. The pattern clearly indicates the formation of a highly crystalline phase and the intense diffraction reflections are in excellent accordance with monoclinic Li3V2(PO4)3 (JCPDS card no. 96962). No carbon phase is detected in the LVP composite, indicating that the carbon generated from the ionic liquid-assisted synthesis is amorphous and its presence does not influence the crystal structure of LVP. According to the results of the elemental analysis, the carbon content of the final product is only 2.4 wt%. The scanning electron microscopy (SEM) images (Fig. 1c and d) show the general morphology of LVP. As is visible, the LVP particles display a nanorod like structure; they are on average 80–100 nm thick and about 1 μm long. In our previous work we showed that the use of an ionic liquid-assisted synthesis allows the realization of LVP nanoparticles with high electronic conductivity. The LVP nanoparticles shown in Fig. 1 display an electronic conductivity of 5.5 × 10−3 S cm−1. This value is more than four orders of magnitude higher than that of uncoated LVP and is also significantly higher than that of LVP nanoparticles obtained with conventional carbon precursors such as sucrose.18 The high electronic conductivity of the material is partially attributed to a relatively uniform carbon coating as evidenced by elemental mapping (Fig. S1†) and by the absence of strong signals corresponding to LVP (PO43− stretching vibrations)29 in the Raman spectrum of the material (Fig. 1b).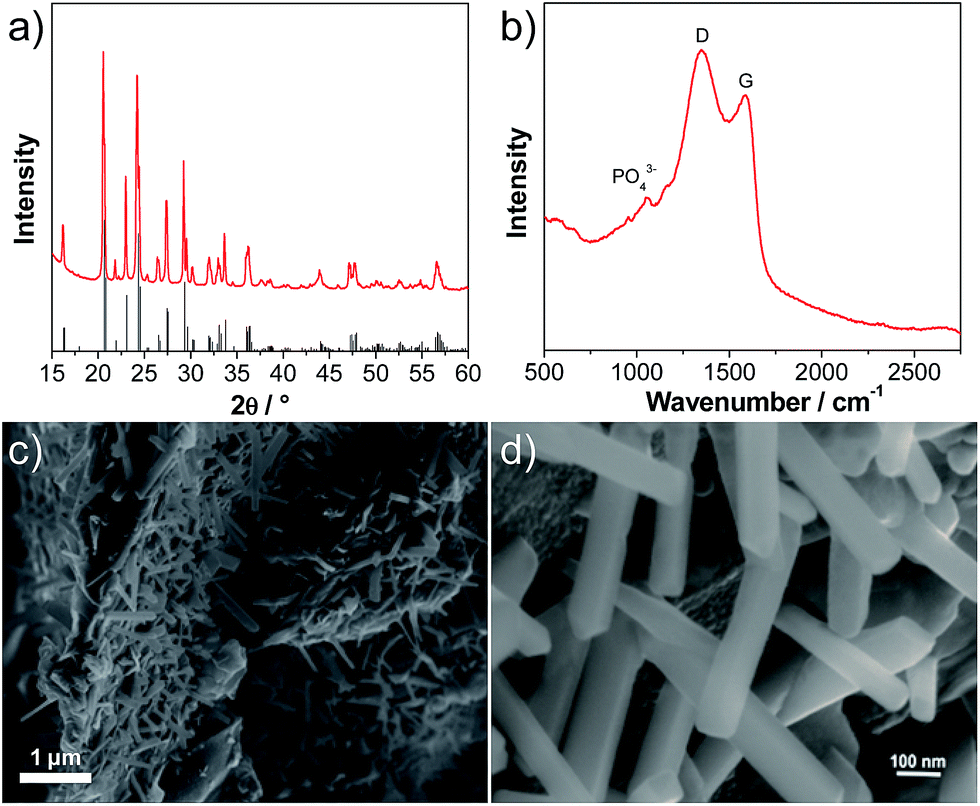 | ||
| Fig. 1 (a) Powder XRD pattern (the reference for monoclinic Li3V2(PO4)3 (JCPDS card no. 96962) is shown in the bottom), (b) Raman spectrum and (c and d) SEM images of the nanostructured Li3V2(PO4)3 (LVP). | ||
Fig. 2 shows a cyclic voltammogram of a LVP-based anode in the potential range from 3.0 to 0.0 V vs. Li/Li+ as obtained using a scan rate of 0.05 mV s−1. As shown in the figure, two distinct potential regions can be distinguished. From ca. 2.0 to 1.6 V four oxidation and four reduction peaks can be seen in the voltammogram. The existence of these peaks in the CV indicates that Li+ insertion/de-insertion takes place in a sequence of phase transitions in this potential region. It was suggested that approximately 0.5 Li+ is inserted at every step, corresponding to the following composition changes: Li3V2(PO4)3 → Li3.5V2(PO4)3 → Li4V2(PO4)3 → Li4.5V2(PO4)3 → Li5V2(PO4)3.19 At potentials below 1.6 V, reversible insertion–extraction of additional lithium takes place in a solid solution. It was suggested that another two Li+ can be reversibly inserted in this potential region, resulting in the formation of Li7V2(PO4)3.19 A wide irreversible current peak in the potential range from 0.9 to 0.6 V is visible in the voltammogram, which may be attributed to the decomposition of the electrolyte to form a solid electrolyte interphase (SEI) film, resulting in irreversible capacity. With an increasing scan rate (see the inset of Fig. 2), highly symmetrical and clearly splitting anodic/cathodic peaks can still be exhibited.
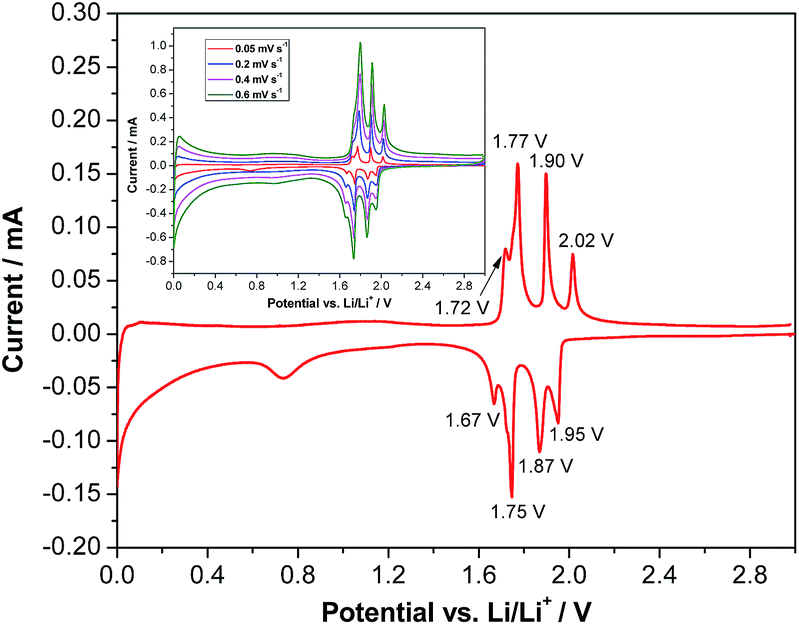 | ||
| Fig. 2 Cyclic voltammogram of LVP anodes recorded at a scan rate of 0.05 mV s−1 in the potential range of 3.0–0.0 V vs. Li/Li+. Inset: CV at different scan rates. | ||
As mentioned in the introduction, while a large number of studies have been dedicated to the investigation of LVP-based cathodes, only a very limited number of studies have been dedicated to LVP-based anodes. Particularly, only few studies investigated in detail the evolution of the lithium diffusion coefficient over the potential range in LVP-based anodes.20,24Fig. 3 shows the evolution of the diffusion coefficient over the potential from 3.0 to 0.0 V vs. Li/Li+ of the investigated LVP anodes, as obtained via GITT measurements. In order to obtain reliable results from GITT experiments, several requirements have to be fulfilled.13,26 It is important to note that in our investigation these requirements were only partially fulfilled. Therefore, as already reported in the literature, the results of such a type of investigation can be considered as an indication of the variation of the lithium diffusion coefficient over the potential only. Nevertheless, it is also important to remark that these results can be still analyzed qualitatively and, from them, reasonable conclusions concerning the general trend of the lithium insertion process can be made. Fig. 3 reports the values obtained for the investigated LVP-anodes. As shown, two regions can be distinguished: the two-phase region at high potentials and the single-phase region at low potentials. The two-phase processes are accompanied by four minima in the D vs. potential plots, corresponding to the current maxima visible in the CV (Fig. 2). In this region, the diffusion coefficient shows a wide variation from 8.2 × 10−11 to 1.9 × 10−14 cm2 s−1 during lithiation (Fig. 3a) and from 3.3 × 10−11 to 1.1 × 10−14 cm2 s−1 during delithiation (Fig. 3b). Similar minima in the chemical diffusion coefficient are also commonly observed for other materials which show phase transition when strong attractive interactions between the Li+ ions and the host matrix are present or some order–disorder transitions during lithiation/delithiation take place.30,31 A very different behavior was found for the low potential region (1.6–0.0 V vs. Li/Li+). Here, only small variations in the diffusion coefficient were found, indicating a continuous energy distribution for the Li+ insertion/deinsertion process. The diffusion coefficient of LVP tends to decrease during lithiation in this region, reaching a value of 1.2 × 10−11 cm2 s−1 at the cut-off potential of 0.0 V vs. Li/Li+. Delithiation of fully lithiated LVP is then easier with an initial D value of 1.5 × 10−10 cm2 s−1. However, the diffusion coefficient tends to decrease again with increasing potential, reaching a minimum of 4.3 × 10−12 cm2 s−1 before the beginning of the two-phase region. Diffusion coefficients were also calculated from CV measurements for the two-phase region (Fig. S2 and Table S1†). Values in the order of 10−11 to 10−10 cm2 s−1, similar to those obtained from the GITT measurements in the single-phase region, were found by the CV method.20
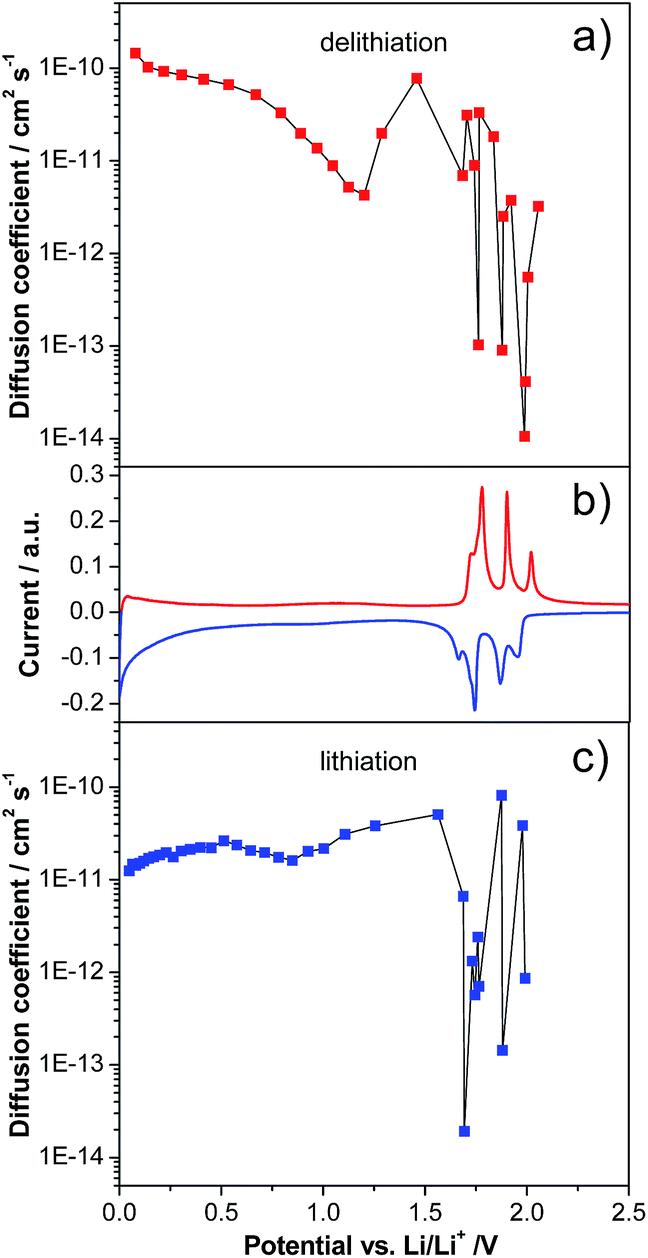 | ||
| Fig. 3 Li+ diffusion coefficients of LVP anodes derived from GITT measurements during (a) delithiation and (c) lithiation in the potential range of 3.0–0.0 V vs. Li/Li+. (b) Example of a cyclic voltammogram of LVP recorded in the same potential range. | ||
It is important to note that the lower potential region of LVP-based anodes appears especially interesting for high power applications. As recently shown for soft carbon-based anodes, the presence of a continuous distribution of diffusion coefficients, which indicates a less hindered lithiation/delithiation process compared to that in the two-phase region, can be advantageous during tests at high current densities.13 Taking this point into account, and considering the high electronic conductivity of the investigated LVP nanostructures, it is reasonable to expect a good power performance for such an anode material.
Indeed, the LVP-based anodes exhibit outstanding rate capabilities. Fig. 4a shows the capacity retention obtained from rate tests of the anode in half-cell configuration. At 1 C, the LVP-anode displays a capacity of 239 mA h g−1, which is close to the theoretical capacity of this anode material (equal to 266 mA h g−1 for the reversible insertion–extraction of four Li+ ions). This value of capacity is not particularly impressive, as it is significantly lower than that of the state of the art anode material graphite. Nevertheless, as shown in the figure, the LVP anode displays outstanding capacity retention when increasing the applied current. When a current density corresponding to 10 C is applied, the LVP anode displays a discharge capacity of 181 mA h g−1, which corresponds to 76% of the capacity at a rate of 1 C. When the current density is increased to a value corresponding to 100 C, which is a value of current in the range of SC applications, the discharge capacity of LVP is 99 mA h g−1, corresponding to a capacity retention of 41%. At low current densities, especially for the delithiation process, the plateaus corresponding to the two-phase process of lithiation/delithiation are visible in the potential profiles (Fig. 4b). With increasing current densities, the potential plateaus become less pronounced, especially during lithiation. Furthermore, it is clearly visible that the capacity retention in the single-phase region is higher than that in the two-phase region. Hence, the average delithiation potential does not significantly increase with the applied current density, which should help to improve the energy retention of a device using LVP as a negative electrode.
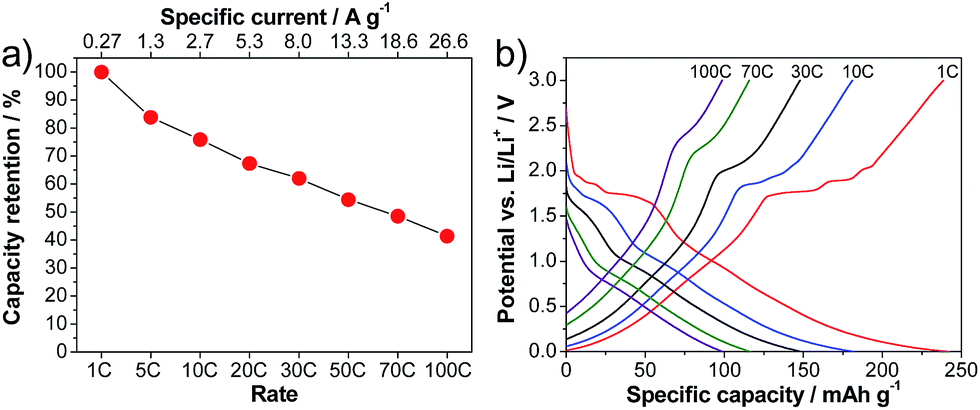 | ||
| Fig. 4 Rate performance of LVP anodes cycled in the potential range of 3.0–0.0 V vs. Li/Li+. (a) Delithiation capacity retention. (b) Corresponding potential profiles. | ||
The high rate performance shown by the LVP anodes cannot be achieved by conventional anode materials such as graphite and, to the best of our knowledge, is among the highest reported for non-conventional anode materials during tests at high current densities. The unique combination of morphology, high ionic and electronic conductivities of the investigated nanoparticles is the origin of the impressive performance at high C-rates displayed by this material. The presence of nanorod like structures shortens the diffusion paths and enlarges the contact area between the active material and the electrolyte, leading to fast Li+ ion diffusion. Furthermore, particularly below 1 V vs. Li/Li+, the lithium diffusion process is not highly hindered and does not limit the electrode performance when high current densities are applied.13 At the same time, as the considered nanostructures display high electronic conductivity (see above), the electronic conductivity of the material does not limit the performance at high current density. About the latter point it is important to note that the investigated LVP anodes only have a carbon content of 2.4 wt%, indicating that the carbon coating of the LVP nanoparticles, which has been made using an ionic liquid as a precursor, can be considered extremely effective.18 Considering these results, our LVP anode can certainly be considered as a very promising candidate for the realization of innovative high power devices. Importantly, as this anode can be cycled down to 0 V vs. Li/Li+, it is expected that devices containing this material will also display interesting values of energy.
Besides high charge and discharge capacities, a negative electrode material for high power applications should also display a high cycling stability, considering the high number of charge–discharge cycles high power LIBs or LICs are usually subjected to. The cycling stability of a material is strongly related to the mechanism of insertion and extraction of lithium into its crystalline structure. In order to have high cycling stability, this process should cause neither dramatic structural changes, nor huge volumetric expansion.
In order to investigate the structural variation of LVP anodes during lithiation and delithiation, we carried out an in situ XRD experiment. The voltage profile of the first lithiation step down to 0.0 V is presented in Fig. 5b. The corresponding XRD patterns are presented in Fig. 5a. As shown above for the CV measurements, the two-phase and the solid solution region can be clearly distinguished also in the voltage profiles. In the two phase region (3–1.6 V vs. Li/Li+), several changes in the XRD patterns clearly indicate phase transitions (see the inset of Fig. 5b). Upon lithiation of LVP in this potential region, the intensity of several reflections strongly decreases and new reflections appear. For example, the intensities of the reflections at 20.4, 22.8, 24.1, 27.2, and 29.1° decrease, and new reflections at 19.9, 23.1 and 26.8° appear, while the reflections at 29.4, 31.7, 33.0 and 33.5° disappear upon lithiation.
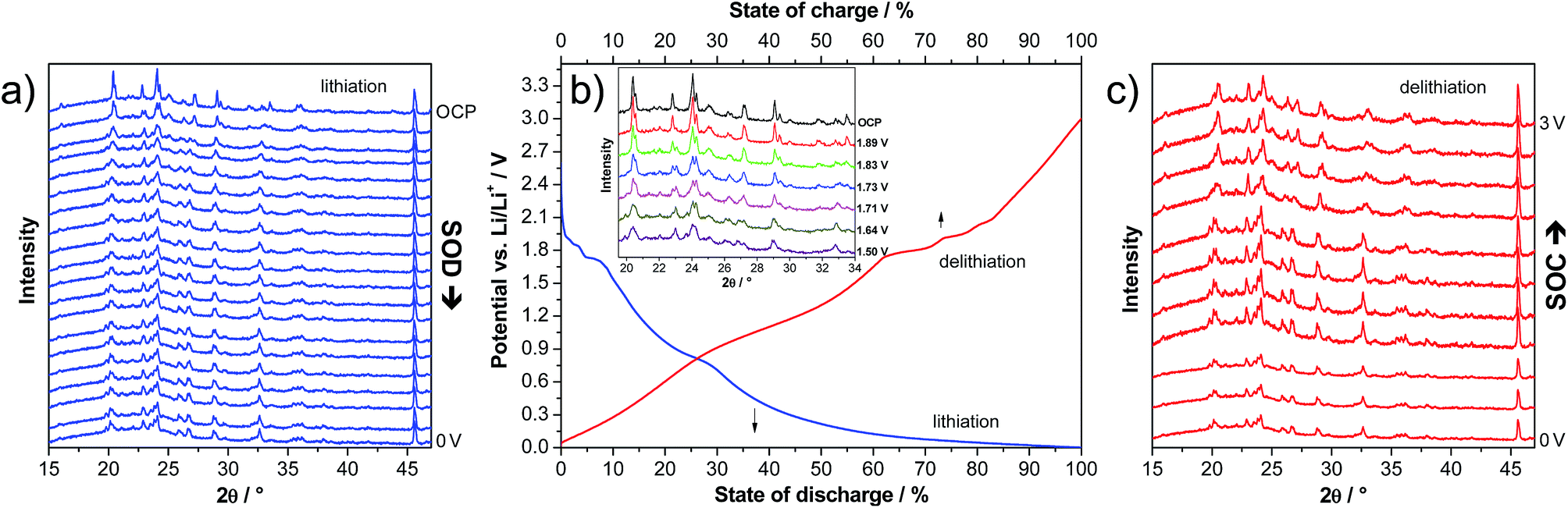 | ||
| Fig. 5 In situ XRD experiment of LVP anodes recorded during the first full cycle in the potential range of 3.0–0.0 V vs. Li/Li+. XRD patterns (a and c) and corresponding potential profiles (b). Inset: magnification of the XRD patterns during lithiation in the two-phase region. | ||
When the cell is further lithiated from 1.6 to 0.0 V vs. Li/Li+, the XRD pattern remains almost constant, in line with the sloppy potential profile in this potential region, which indicates solid-solution behaviour. There are only some reflections shifting to slightly lower 2θ angles, indicating a small expansion of the structure due to the ongoing lithiation process. For example, the reflection at 23.0° shifts to 22.9° upon lithiation of the material from 1.5 to 0.0 V.
It is important to note that upon delithiation to 3.0 V vs. Li/Li+, the XRD pattern approximately returns to the pristine state, suggesting reversible changes of the LVP structure during charge and discharge (Fig. 5c). There are only some small differences of the XRD pattern at 3.0 V compared to the one of the pristine electrode (Fig. 5a). For example, the ratios of the intensities of the two double reflections at ca. 20.5 and 24.1° are inversed. A similar trend in the positions of the major diffraction reflections is observed before and after the completion of the second cycle within the potential range of 3.0 to 0.0 V vs. Li/Li+. This finding supports the conclusion of a recent investigation that the structural integrity of the LVP anode is maintained in spite of the two-phase reaction mechanism.19 Often, Li+ ion insertion accompanies a pronounced volumetric change, which can lead to limited cycling stability and practical capacity. However, the in situ XRD investigation indicates that LVP undergoes only small volume changes, and hence good reversibility upon full charging–discharging of LVP-based anodes can be expected.
Fig. 6a shows the variation of the discharge capacity of the LVP anode during prolonged charge–discharge cycling carried out at current density corresponding to 50 C. As shown in the figure, during the initial cycles, the capacity of the LVP anode decreases from a value of ca. 220 mA h g−1 to a value of 150 mA h g−1. During these initial cycles, the coulombic efficiency of the charge–discharge process is lower than 90%. This low value is most likely related to decomposition processes of the electrolyte and SEI formation.32 After these initial cycles, the capacity of the LVP anode becomes extremely stable and after 10000 cycles the electrode is still able to deliver a capacity of about 135 mA h g−1, corresponding to a capacity retention of more than 90% compared to the 100th cycle. During all these cycles, the coulombic efficiency of the charge–discharge process was always close to 100%. The XRD pattern of the cycled electrode (Fig. 6b) indicates that some changes of the LVP structure occurred during this high number of cycles. As shown in the figure, all major reflections of partially delithiated LVP according to the in situ XRD experiment are also found for the electrode subjected to 10000 cycles of charge–discharge. However, the lower intensity and the broadening of the reflections indicate a reduction of the crystallinity of the material. Nevertheless, this reduction does not seem to have a pronounced effect on the electrochemical behavior of the LVP anode.
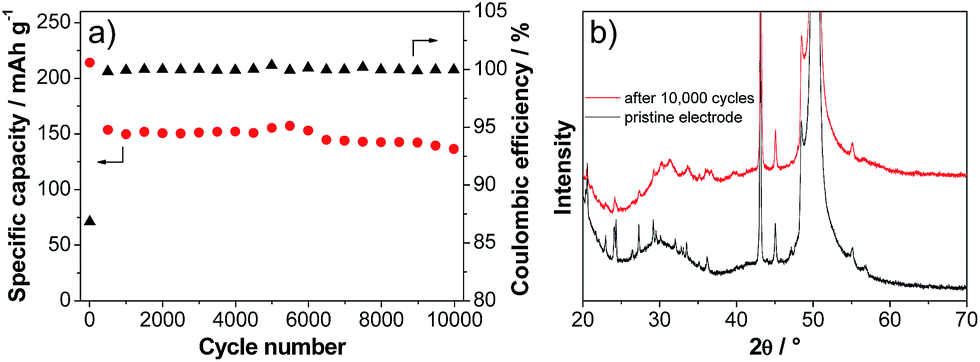 | ||
| Fig. 6 (a) Longterm cycling performance of LVP anodes cycled between 3.0 and 0.0 V vs. Li/Li+ at a current corresponding to 50 C. (b) XRD pattern of a pristine electrode and the electrode subjected to 10000 cycles at 50 C. | ||
The results reported above clearly indicate that our LVP anode not only displays very high capacity during tests carried out at a high C-rate but also an extraordinary cycling stability. Consequently, this anode material can be regarded as one of the most promising candidates for the realization of innovative high power devices.
With the aim to verify the performance of this material in a high power device, we realized a LIC containing LVP as a negative electrode and activated carbon (AC) as a positive electrode. In this kind of setup, prelithiation of the negative electrode is always necessary to introduce lithium into the system and to handle the irreversible capacity. Therefore, the lower initial efficiency of LVP does not represent an obstacle to its introduction in such devices. The LVP electrodes in the LIC full cells were prelithiated via the lithium reference electrode as described in the literature.27,28 Afterwards, the LIC full cells were cycled between 0.0 and 4.0 V. Fig. 7a shows the evolution of the energy density and energy efficiency over cycling of the LVP-based LIC at different current densities of 2.07, 4.13 and 8.26 A g−1 (corresponding to a C-rate of the LVP negative electrode of 25 C, 50 C and 100 C, respectively and based on both active material masses). The number of cycles considered in these tests was dependent on the investigated current density (the higher the current, the higher the cycle number). As shown, after a small fading of the energy density in the beginning, all investigated cells possess a very stable energy density over the following several ten thousands of cycles. It is important to note that the balancing of all cells is based on the half cell capacities at 100 C, which does not lead to a worsened performance of the 25 C and 50 C cells. As expected, due to the proportionality of the overpotential and voltage with the current, the energy density of the devices decreases with increasing applied current. Nevertheless, all LICs display outstanding energy density (referred to the weight of both active materials) as well as cycling stability. As shown in the figure, after 30000 cycles at 2.07 A g−1 the LVP-based LIC displays an energy density of 45 W h kg−1. The same type of device displays an energy density of 40 W h kg−1 after 60000 cycles at 4.13 A g−1. When the current is increased to 8.26 A g−1, the LVP-based LIC is able to deliver, after 100000 cycles, an energy density of more than 30 W h kg−1. The evolution of the energy efficiency of the three investigated cells is also given in the figure. Only a slight decrease of the energy efficiency can be observed at all three currents. Fig. 7b and c compare the voltage and potential profiles of the LIC cycled at 100 C in the beginning and at the end of cycling. As shown in the figure, all profiles represent each other very well and only minor changes can be detected. The biggest difference is a small upward shift of the end-of-discharge potentials of both electrodes (2.42 V vs. Li/Li+ to 2.66 V vs. Li/Li+). Very importantly, no further overpotentials (e.g. caused by calendar life or aging) evolve over cycling, which would certainly lead to a more severely decreased energy efficiency.
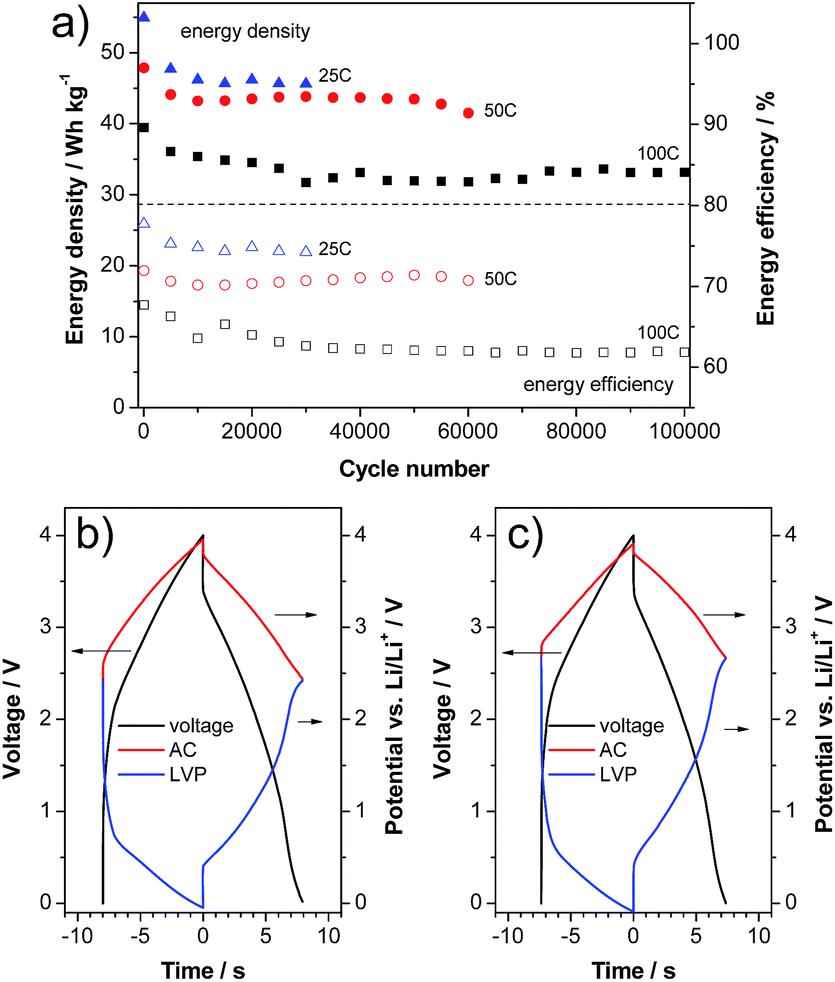 | ||
| Fig. 7 Constant current cycling of a LVP/AC based LIC: (a) energy density and efficiency vs. cycle number. Voltage and potential profiles of the (b) 5000th and (c) 100000th cycles at 100 C. | ||
The energy density and the cycling stability of the investigated LVP-based LIC are among the highest so far reported for such type of high power devices, and, to the best of our knowledge, are the highest for systems containing non-carbonaceous anodes. Fig. 8 compares in a Ragone like plot the energy and power densities of the investigated devices, in the beginning, in the middle and in the end of cycling, with those of an activated carbon based supercapacitor (SC) (0.0–2.8 V, 1 M Et4NBF4 in PC) and a graphite/LiCoO2 based LIB (3.0–4.2 V, LP30). It is important to outline that all three devices are lab-made, they have a comparable weight and they have been tested under similar conditions. The values of energy and power reported in the figure refer to the active materials only. Therefore, such a comparison has to be seen only as an indication about the characteristics of these devices. Nevertheless, this plot clearly shows that, using the LVP anode, it is possible to realize high power devices that are able to display very interesting values of energy and power that fill the gap between LIBs and SCs. The energy efficiency of the LIB and SC shown here are also given in the ESI (Table S2†). As shown above, these devices are able to display this promising performance over several ten thousands of cycles, as requested for high power devices.
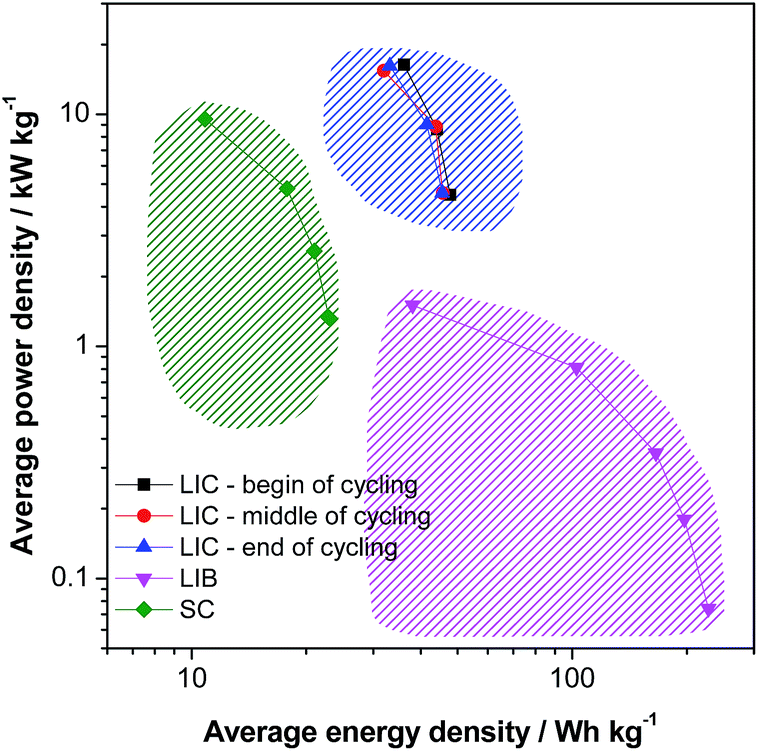 | ||
| Fig. 8 Ragone like plot (begin: 5000th cycle, middle: 15000th, 30000th & 60000th cycles, end: 30000th, 60000th and 100000th cycles for 25, 50 and 100 C, respectively). LIC: lithium-ion capacitor; LIB: lithium ion battery; SC: supercapacitor. | ||
4. Conclusion
LVP is not only a promising cathode material for LIBs, but it can also host additional Li+ ions due to its amphoteric nature. Lithium insertion into LVP in the anode potential region takes place with two different mechanisms. At high potentials, LVP undergoes a series of phase transitions. At lower potentials, LVP shows solid solution behavior. Lithium diffusion in this potential region is very fast, offering the opportunity to design a high power LVP material. Our nanorod-like carbon-coated LVP synthesized by an ionic liquid assisted method displays high electronic conductivity, hence, displaying outstanding rate capability as an anode material. At the very high current of 100 C, nanostructured LVP anodes display a capacity of about 100 mA h g−1. Furthermore, the LVP anode displays superior longterm cycling stability: 91% capacity retention after 10000 cycles at 50 C. The excellent high rate capacity and cycling stability of our LVP also make this material an attractive candidate for the use as a negative electrode material in lithium-ion capacitors. We demonstrate here an LVP/activated carbon hybrid device with a similar power performance and a much improved energy density compared to that of conventional supercapacitors.
Acknowledgements
The authors wish to thank the University of Muenster and the Ministry of Innovation, Science and Research of North Rhine-Westphalia (MIWF) within the project “Superkondensatoren und Lithium-Ionen-Hybrid-Superkondensatoren auf der Basis ionischer Flüssigkeiten” and the Bundesministerium für Bildung und Forschung (BMBF) within the project IES (contract number 03EK3010) for the financial support.Notes and references
- N. S. Choi, Z. Chen, S. A. Freunberger, X. Ji, Y.-K. Sun, K. Amine, G. Yushin, L. F. Nazar, J. Cho and P. G. Bruce, Angew. Chem., Int. Ed., 2012, 51, 9994 CrossRef CAS PubMed.
- B. Scrosati and J. Garche, J. Power Sources, 2010, 195, 2419 CrossRef CAS PubMed.
- P. Simon and Y. Gogotsi, Nat. Mater., 2008, 7, 845 CrossRef CAS PubMed.
- P. Simon, Y. Gogotsi and B. Dunn, Science, 2014, 343, 1210 CrossRef CAS PubMed.
- D. Cericola and R. Kötz, Electrochim. Acta, 2012, 72, 1 CrossRef CAS PubMed.
- V. Khomenko, E. Raymundo-Piñero and F. Béguin, J. Power Sources, 2008, 177, 643 CrossRef CAS PubMed.
- K. Naoi and P. Simon, Electrochem. Soc. Interface, 2008, 17, 34 CAS.
- W. J. Cao and J. P. Zheng, J. Power Sources, 2012, 213, 180 CrossRef CAS PubMed.
- S. R. Sivakkumar and A. G. Pandolfo, Electrochim. Acta, 2012, 65, 280 CrossRef CAS PubMed.
- Y. Wang, P. He and H. Zhou, Energy Environ. Sci., 2011, 4, 4994 CAS.
- L. Li and A. Manthiram, J. Mater. Chem. A, 2013, 1, 5121 CAS.
- L. Shen, X. Zhang, E. Uchaker, C. Yuan and G. Cao, Adv. Energy Mater., 2012, 2, 691 CrossRef CAS.
- M. Schroeder, S. Menne, J. Ségalini, D. Saurel, M. Casas-Cabanas, S. Passerini, M. Winter and A. Balducci, J. Power Sources, 2014, 266, 250 CrossRef CAS PubMed.
- H. Liu, P. Gao, J. Fang and G. Yang, Chem. Commun., 2011, 47, 9110 RSC.
- H. Huang, S.-C. Yin, T. Kerr, N. Taylor and L. F. Nazar, Adv. Mater., 2002, 14, 1525 CrossRef CAS.
- S.-C. Yin, H. Grondey, P. Strobel, M. Anne and L. F. Nazar, J. Am. Chem. Soc., 2003, 125, 10402 CrossRef CAS PubMed.
- S.-C. Yin, H. Grondey, P. Strobel, H. Huang and L. F. Nazar, J. Am. Chem. Soc., 2003, 125, 326 CrossRef CAS PubMed.
- X. Zhang, N. Böckenfeld, F. Berkemeier and A. Balducci, ChemSusChem, 2014, 7, 1710 CrossRef CAS PubMed.
- X. H. Rui, N. Yesibolati and C. H. Chen, J. Power Sources, 2011, 196, 2279 CrossRef CAS PubMed.
- X. H. Rui, N. Yesibolati, S. R. Li, C. C. Yuan and C. H. Chen, Solid State Ionics, 2011, 187, 58 CrossRef CAS PubMed.
- W.-F. Mao, H.-Q. Tang, Z.-Y. Tang, J. Yan and Q. Xu, ECS Electrochem. Lett., 2013, 2, A69 CrossRef CAS PubMed.
- E. Kobayashi, A. Kitajou, S. Okada and J.-I. Yamaki, J. Power Sources, 2013, 244, 312 CrossRef CAS PubMed.
- N. Böckenfeld and A. Balducci, J. Power Sources, 2013, 235, 265 CrossRef PubMed.
- N. Böckenfeld and A. Balducci, J. Appl. Electrochem., 2014, 44, 467 CrossRef.
- A. Shahul Hameed, M. V. Reddy, B. V. R. Chowdari and J. J. Vittal, Electrochim. Acta, 2014, 128, 184 CrossRef PubMed.
- W. Weppner and R. A. Huggins, J. Electrochem. Soc., 1977, 124, 1569 CrossRef CAS PubMed.
- M. Schroeder, M. Winter, S. Passerini and A. Balducci, J. Electrochem. Soc., 2012, 159, A1240 CrossRef CAS PubMed.
- M. Schroeder, M. Winter, S. Passerini and A. Balducci, J. Power Sources, 2013, 238, 388 CrossRef CAS PubMed.
- N. Membreño, P. Xiao, K.-S. Park, J. B. Goodenough, G. Henkelman and K. J. Stevenson, J. Phys. Chem. C, 2013, 117, 11994 Search PubMed.
- P. P. Prosini, M. Lisi, D. Zane and M. Pasquali, Solid State Ionics, 2002, 148, 45 CrossRef CAS.
- K. M. Shaju, G. V. Subba Rao and B. V. R. Chowdari, Electrochim. Acta, 2003, 48, 2691 CrossRef CAS.
- X. W. Lou, J. S. Chen, P. Chen and L. A. Archer, Chem. Mater., 2009, 21, 2868 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Elemental mapping of the LVP material, calculation of apparent lithium diffusion coefficients from CV measurements and information about the power, energy and energy efficiency of the devices presented in the Ragone like plot. See DOI: 10.1039/c4ta03845b |
| ‡ Xiaofei Zhang and Ruben-Simon Kühnel contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2014 |