 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Diastereoselective allylation and crotylation of N-tert-butanesulfinyl imines with allylic alcohols†‡
Olga Soares do Rego
Barros§
,
Juan Alberto
Sirvent
,
Francisco
Foubelo
* and
Miguel
Yus
*
Departamento de Química Orgánica, Facultad de Ciencias and Instituto de Síntesis Orgánica (ISO), Universidad de Alicante, Apdo. 99, 03080 Alicante, Spain. E-mail: foubelo@ua.es; yus@ua.es
First published on 24th April 2014
Abstract
The palladium-catalyzed allylation of N-tert-butanesulfinyl imines with allylic alcohols in the presence of InI as reducing reagent takes place with high diastereoselectivity in reasonable yields. The reaction with crotyl alcohol is totally regioselective, leading to the anti-diastereomer as the main reaction product.
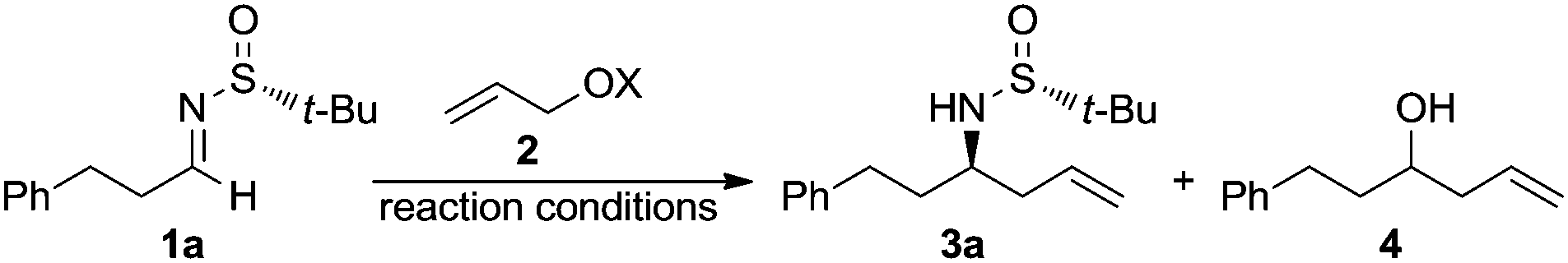
The stereoselective allylation of imines using organometallic compounds is of great interest in synthetic organic chemistry because the resulting homoallylic amines are valuable building blocks.1 For instance, the double bond of the allylic moiety can participate in a number of further synthetically useful transformations: hydroboration, hydration, epoxidation, hydroformylation, ozonolysis, olefin metathesis, etc.2 Among the stereoselective methodologies, the catalytic enantioselective allylations3 rely on the use of chiral Lewis acids or chiral Lewis bases. In addition, double activation could be also achieved by using chiral bifunctional catalysts4 by the simultaneous activation of both electrophilic and nucleophilic reaction partners through a cooperative action of different functionalities of the ligand. However, and in spite of the rapid evolution of catalytic methods in recent years, the use of stoichiometric reagents (including substrates and/or allylic organometallic partners) is still the favourite choice of organic chemists in the synthesis of key intermediates of natural products. In these diastereoselective allylations,5 the stereochemical information can be transferred by substrate control, including chiral auxiliaries, or through the use of chiral reagents (reagent control). Sometimes a double induction could also be involved in the process, a match/mismatch effect being possible. Performing the stereoselective allylation of imine derivatives with allylic halides in the presence of reducing metals under Barbier-type reaction conditions (metalation in the presence of the electrophile) is also of interest, because this strategy circumvents the need of isolating allyl metal species (the real nucleophiles), which usually are sensitive, and in many cases also toxic. The most commonly used metals in these transformations are chromium,6 indium7 and zinc.8 Particularly noteworthy is the use of allylic alcohols as allylating reagents of carbonyl compounds and imines under palladium catalysis in the presence of a reducing reagent.9 More recently, the use of the Kulinkovich reagent in the coupling of allylic alcohols with aldimines provides an alternative approach to regio-and stereodefined homoallylic amines.10 On the other hand, in the growing field of the synthetic applications of chiral N-tert-butanesulfinyl derivatives,11 we reported the diastereoselective allylation12 of N-tert-butanesulfinyl aldimines13 and ketimines14 with in situ generated allylindium reagents, and the first one-pot α-aminoallylation of aldehydes with chiral tert-butanesulfinamide, allylic bromides, and indium, which provides homoallylic amines with high chemo- and stereoselectivities.15 The chiral auxiliary is easily accessible in both enantiomeric forms in large-scale processes16 and can be easily removed under acidic conditions. Continuing our studies on the diastereoselective allylation of chiral sulfinyl imines, we report herein the use for the first time of allylic alcohols as allylating reagents of these chiral imines.
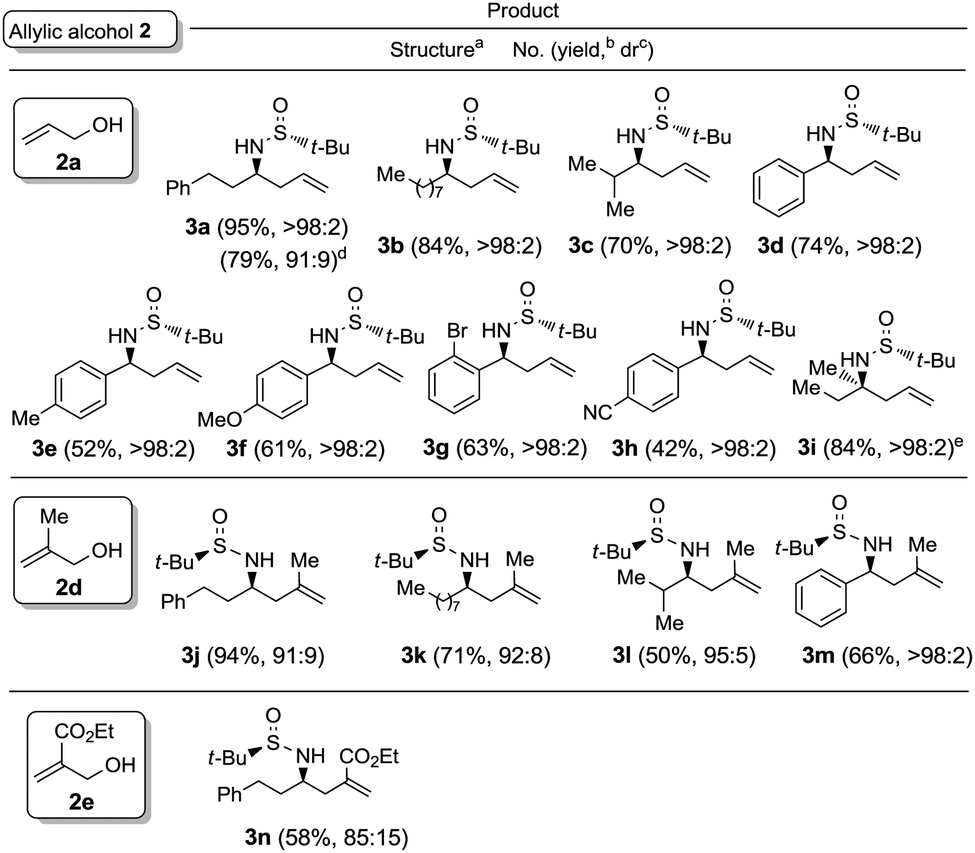
The chiral imine 1a derived from (R)-tert-butanesulfinamide and 3-phenylpropanal was taken as the imine model for the optimization of the reaction conditions in the palladium catalyzed allylation with allylic alcohols and derivatives. Although many assays were undertaken, only the most significant ones are compiled in Table 1. Thus, the reaction of allyl alcohol (2a) with imine 1a in the presence of InCl3 (1 equiv.), In (2 equiv.) and a substoichiometric amount of Pd(PPh3)4 (5 mol%), in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of THF and water,17 led to the expected reaction product 3a (84%) along with the homoallylic alcohol 4 resulting from the allylation of the aldehyde after hydrolysis of the starting imine (Table 1, entry 1). However, the allylation with allyl acetate (2b) under the same reaction conditions gave alcohol 4 as the major reaction product and only a small amount of the desired homoallylamine derivative 3a (Table 1, entry 2). It seems that allyl acetate (2b) is less reactive than allyl alcohol (2a) and hydrolysis occurred faster than allylation of the imine. On the other hand, the allylation with allyl acetate (2b) or diallyl carbonate (2c) proceeded very slowly when it was performed exclusively in THF at room temperature, and after 14 hours, 92 and 75%, respectively, of the starting material 1a remained unreacted (Table 1, entries 3 and 4). Total conversion was observed when the allylation with allyl acetate (2b) was performed in THF at 60 °C, producing in this case 3a and 4 in 95:5 ratio (Table 1, entry 5). Fortunately, we were please to find that switching from the combination In/InCl3 to InI18 as reducing reagent, the palladium catalyzed allylation of 1a with allyl alcohol (2a) occurred with total conversion and selectivity at room temperature (Table 1, entry 6), meanwhile with allyl acetate (2b) an almost 4:1 mixture of the desired amine derivative 3a and the homoallyl alcohol 4 was obtained (Table 1, entry 7). We studied next the scope of the allylation of N-tert-butylsulfinyl imines 1 with different allylic alcohols 2 by applying the optimized conditions depicted in Table 1, entry 6. The expected homoallylamine derivatives 3 were obtained in good yields for aliphatic aldimines and moderate-to-good yields in the case of aromatic aldimines using allyl (2a) and methallyl alcohol (2d) as allylating reagents (Table 2). It was also possible to carry out the allylation of the ketimine derived from butanone with allyl alcohol (2a) but under more demanding reaction conditions, 60 °C instead of room temperature in this case (Table 2, compound 3i). Regarding aromatic imines, electron-rich derivatives seemed to perform better than the electron-poor ones (Table 2, compare compounds 3f and 3h). Interestingly, this methodology is compatible with both the use of functionalized allylic alcohols, such as ethyl 2-(hydroxymethyl)acrylate (2e) (Table 2, compound 3n), and the presence of an active sp2 C–Br bond in palladium catalysis (Table 2, compound 3g). On Table 2, yield and diastereomeric ratio for the allylation of chiral imine 1a with allyl bromide in the presence of indium metal are also given in order to compare both methodologies. It could be said that palladium-catalyzed allylation of chiral imines 1 with allylic alcohols is superior to that using allylic halides taking into account stereoselectivities. Anyway, in both cases, they show the same sense of the stereochemical induction which is controlled by the configuration of the sulfur atom of the sulfinyl unit.
:1 mixture of THF and water,17 led to the expected reaction product 3a (84%) along with the homoallylic alcohol 4 resulting from the allylation of the aldehyde after hydrolysis of the starting imine (Table 1, entry 1). However, the allylation with allyl acetate (2b) under the same reaction conditions gave alcohol 4 as the major reaction product and only a small amount of the desired homoallylamine derivative 3a (Table 1, entry 2). It seems that allyl acetate (2b) is less reactive than allyl alcohol (2a) and hydrolysis occurred faster than allylation of the imine. On the other hand, the allylation with allyl acetate (2b) or diallyl carbonate (2c) proceeded very slowly when it was performed exclusively in THF at room temperature, and after 14 hours, 92 and 75%, respectively, of the starting material 1a remained unreacted (Table 1, entries 3 and 4). Total conversion was observed when the allylation with allyl acetate (2b) was performed in THF at 60 °C, producing in this case 3a and 4 in 95:5 ratio (Table 1, entry 5). Fortunately, we were please to find that switching from the combination In/InCl3 to InI18 as reducing reagent, the palladium catalyzed allylation of 1a with allyl alcohol (2a) occurred with total conversion and selectivity at room temperature (Table 1, entry 6), meanwhile with allyl acetate (2b) an almost 4:1 mixture of the desired amine derivative 3a and the homoallyl alcohol 4 was obtained (Table 1, entry 7). We studied next the scope of the allylation of N-tert-butylsulfinyl imines 1 with different allylic alcohols 2 by applying the optimized conditions depicted in Table 1, entry 6. The expected homoallylamine derivatives 3 were obtained in good yields for aliphatic aldimines and moderate-to-good yields in the case of aromatic aldimines using allyl (2a) and methallyl alcohol (2d) as allylating reagents (Table 2). It was also possible to carry out the allylation of the ketimine derived from butanone with allyl alcohol (2a) but under more demanding reaction conditions, 60 °C instead of room temperature in this case (Table 2, compound 3i). Regarding aromatic imines, electron-rich derivatives seemed to perform better than the electron-poor ones (Table 2, compare compounds 3f and 3h). Interestingly, this methodology is compatible with both the use of functionalized allylic alcohols, such as ethyl 2-(hydroxymethyl)acrylate (2e) (Table 2, compound 3n), and the presence of an active sp2 C–Br bond in palladium catalysis (Table 2, compound 3g). On Table 2, yield and diastereomeric ratio for the allylation of chiral imine 1a with allyl bromide in the presence of indium metal are also given in order to compare both methodologies. It could be said that palladium-catalyzed allylation of chiral imines 1 with allylic alcohols is superior to that using allylic halides taking into account stereoselectivities. Anyway, in both cases, they show the same sense of the stereochemical induction which is controlled by the configuration of the sulfur atom of the sulfinyl unit.
|
|
||
|---|---|---|
| Entry | Reaction conditions | Productsb |
|
1a:3ac:4 |
||
| a Reactions were carried out with 0.2 mmol of 1a. b Ratio was determined from 1H NMR spectrum of the crude reaction mixture. c Major diastereoisomer is shown. | ||
| 1 | 2a (X = H, 1.0 mmol), In (0.4 mmol), InCl3 (0.20 mmol), Pd(PPh3)4 (0.01 mmol), THF/H2O (0.3 mL/0.3 mL), 23 °C (14 h) | 0:84:16 |
| 2 | 2b (X = Ac, 1.0 mmol), In (0.4 mmol), InCl3 (0.20 mmol), Pd(PPh3)4 (0.01 mmol), THF/H2O (0.3 mL/0.3 mL), 23 °C (14 h) | 0:12:88 |
| 3 | 2b (X = Ac, 0.8 mmol), In (0.4 mmol), InCl3 (0.20 mmol), Pd(PPh3)4 (0.04 mmol), THF (1.0 mL), 23 °C (14 h) | 92:8:0 |
| 4 |
2c (X = CO2CH2CH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH2, 0.8 mmol), In (0.4 mmol), InCl3 (0.20 mmol), Pd(PPh3)4, (0.04 mmol), THF (1.0 mL), 23 °C (14 h) CH2, 0.8 mmol), In (0.4 mmol), InCl3 (0.20 mmol), Pd(PPh3)4, (0.04 mmol), THF (1.0 mL), 23 °C (14 h) |
75:15:10 |
| 5 | 2b (X = Ac, 0.8 mmol), In (0.4 mmol), InCl3 (0.20 mmol), Pd(PPh3)4 (0.04 mmol), THF (1.0 mL), 60 °C (14 h) | 0:95:5 |
| 6 | 2a (X = H, 0.8 mmol), InI (0.4 mmol), Pd(PPh3)4 (0.01 mmol), THF (1.0 mL), 23 °C (14 h) | 0:100:0 |
| 7 | 2b (X = Ac, 0.8 mmol), InI (0.4 mmol), Pd(PPh3)4 (0.01 mmol), THF (1.0 mL), 23 °C (14 h) | 0:79:21 |
|
|
|---|
| a Major diastereoisomer is shown. b Yields were determined for isolated compounds after column chromatography. c Diastereomeric ratios were determined by lH NMR analysis of the crude reaction mixture. d For comparison, yield and dr reported for compound 3a obtained by an indium mediated allylation with allyl bromide in THF at 60 °C.13 e The reaction was performed at 60 °C. |
|
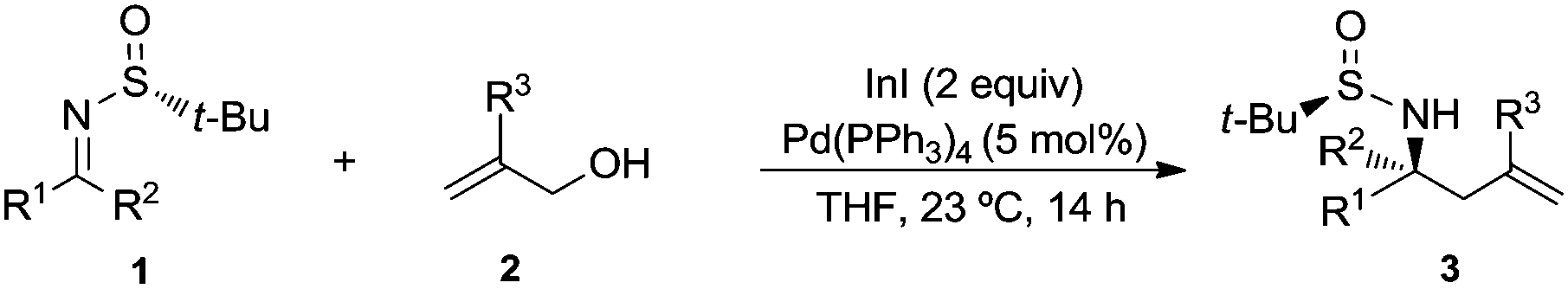
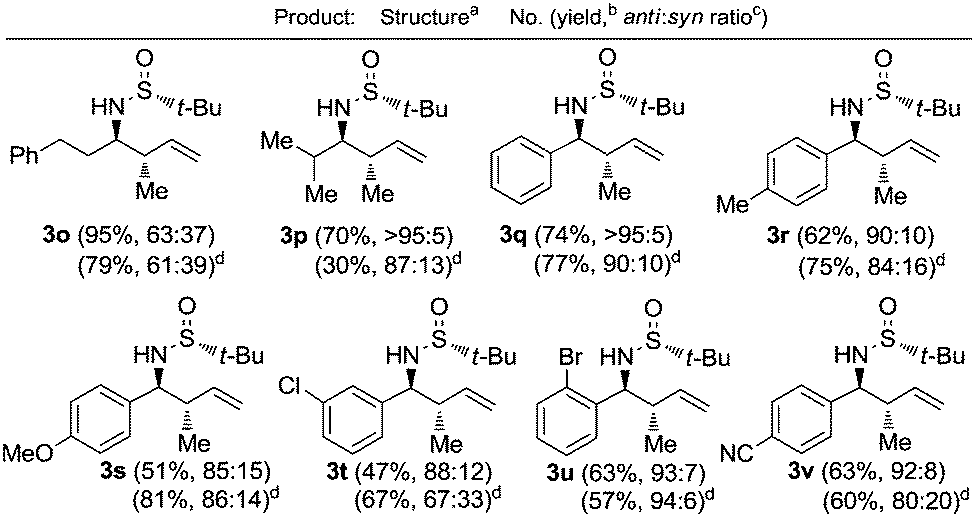
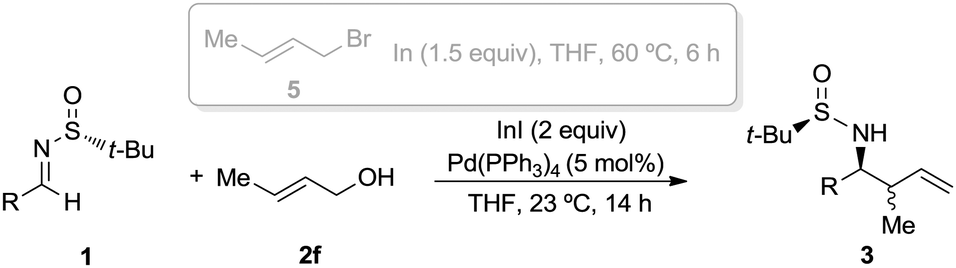
The crotylation of chiral imines 1 was also investigated. The reaction of our imine model compound 1a with crotyl alcohol (2f) under the standard conditions produced compound 3o in a regioselective process with almost total face selectivity and low anti:syn diastereoselectivity (63:37), meanwhile, the indium mediated crotylation of 1a using crotyl bromide (5) occurred with high face-addition selectivity and a similar anti:syn ratio of 61:39 (Table 3). Surprisingly, the palladium-catalyzed crotylation of α-substituted (Table 3, compound 3p) and aromatic imines occurred with high anti-diastereoselectivity in all cases (Table 3, compounds 3q–v). On the other hand, the reaction of these aldimines with crotyl bromide (5) proceeded also with better diastereoselectivities than for 1a. A mechanism has been proposed for this palladium catalyzed allylations. First a π-allylpalladium complex I is formed. Indium salts facilitate the process by converting the hydroxy a better leaving group after coordination.17 Then, the initially formed π-allylpalladium(II) complex I is reductively transmetalated with indium(I) salts to give allylindium(III) species II, which is the real allylating reagent.18 Face selectivity could be explained by considering six-membered cyclic transition states III (R2 = H) or IV (R = Me) where the indium is coordinated to both the nitrogen and the oxygen atom of the sulfinyl imine, taking place a Si-face attack for imines with R-configuration at the sulfur atom. Regarding the regiochemistry and the diastereoselectivity in the crotylation reactions, the indium atom located at the terminal position in the allyl indium intermediate with E-configuration, the most stable carbanion, reacts at the γ-position through a cyclic boat-like six-membered transition state IV to produce the anti-isomer (Scheme 1). This kind of boat-like transition state IV has been proposed in a similar process to be preferred over the chair-like one III (in this case the syn-isomer would be produced), due to the steric repulsion between the methyl group and the substituent of the aldimine (Scheme 1).19 Higher diastereoselectivities are obtained for the palladium-catalyzed allylations with alcohols when the processes are performed at room temperature, instead of 60 °C. On the other, and according with the proposed working model, the bulkier the substituent of the starting aldimine 1, the higher diastereoselectivity is obtained (Table 3).
|
|
|---|
|
a Major diastereoisomer is shown.
b Yields were determined for isolated compounds after column chromatograph.
c
anti:syn ratios were determined by 1H NMR analysis of the crude reaction mixture.
d For comparison, yield and dr reported for compounds 3 obtained by an indium mediated allylation with crotyl bromide in THF at 60 °C.
|
|
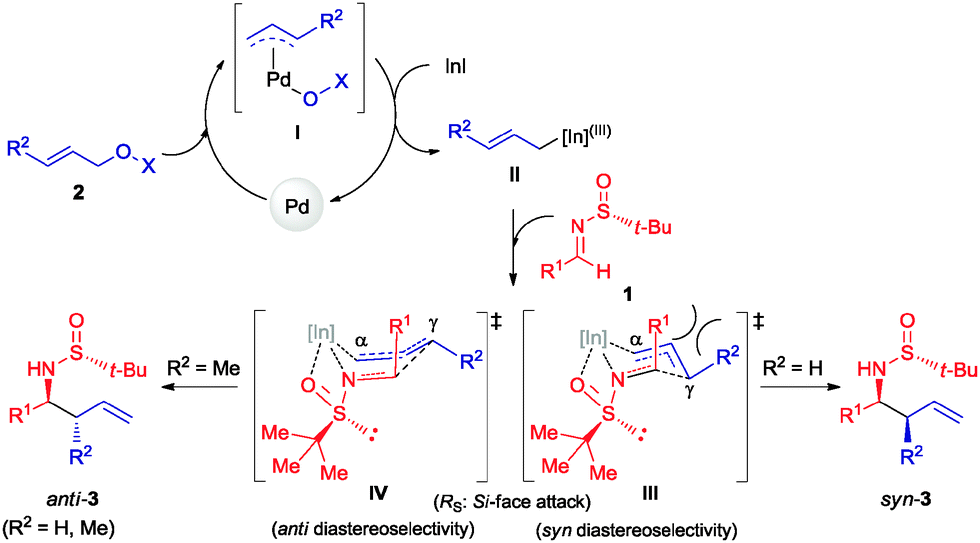 | ||
| Scheme 1 Proposed reaction mechanism. | ||
Finally, the tert-butanesulfinyl group was easily removed from compounds 3q after treatment with a 6 M HCl aqueous solution in THF to give the known homoallylic amine 620 with anti-relative configuration (Scheme 2).
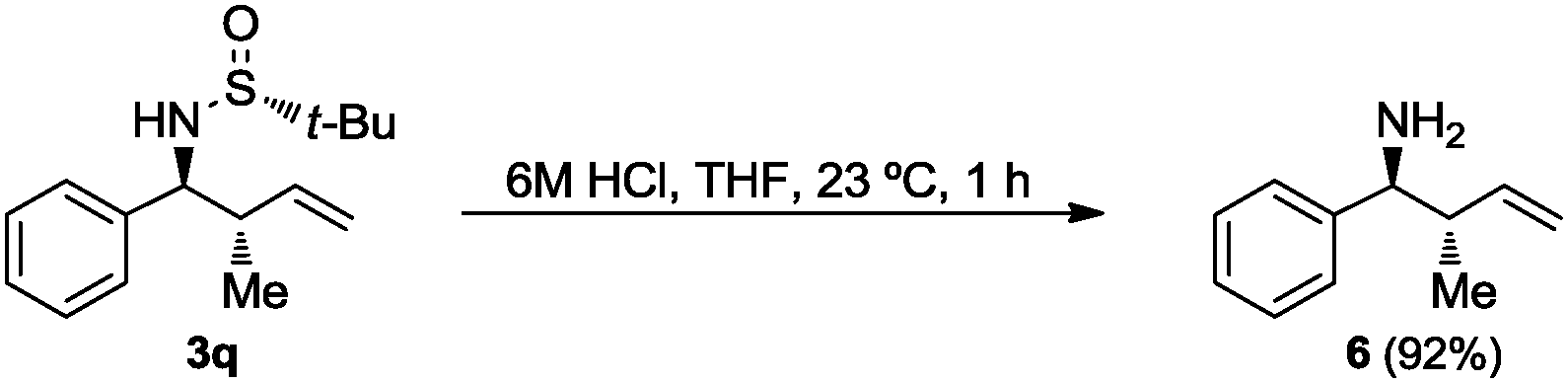 | ||
| Scheme 2 Desulfinylation of 3q. | ||
In summary, allylation of N-tert-butanesulfinyl imines was performed with high diastereoselectivity with allylic alcohols under palladium-catalysis. The reaction with crotyl alcohol occurred also in reasonable yields and high anti-diastereoselectivity in a regioselective manner. In addition, and comparing to allylic halides, allylic alcohols are preferable allylating reagents by taking into account environmental (less toxic), economic (less expensive) and availability (numerous allylic alcohols are commercially available) considerations.
We thank the Spanish Ministerio de Ciencia e Innovación (Grant No. CTQ2011-24165), the Generalitat Valenciana (Grant No. PROMETEO/2009/039 and FEDER) and the University of Alicante for financial support. OSRB thanks CNPq of Brazil for a fellowship.
Notes and references
- For reviews on stereoselective allylations, see: (a) S. E. Denmark and J. Fu, Chem. Rev., 2003, 103, 2763 CrossRef CAS PubMed; (b) P. Merino, T. Tejero, J. I. Delso and V. Mannucci, Curr. Org. Synth., 2005, 2, 479 CrossRef CAS; (c) H. Ding and G. K. Friestad, Synthesis, 2005, 2815 CAS; (d) G. K. Friestad and A. K. Mathies, Tetrahedron, 2007, 63, 2541 CrossRef CAS PubMed; (e) R. B. Kargbo and G. R. Cook, Curr. Org. Chem., 2007, 11, 1287 CrossRef CAS; (f) H. Yamamoto and M. Wadamoto, Chem. – Asian J., 2007, 2, 692 CrossRef CAS PubMed; (g) M. Kanai, R. Wada, T. Shibuguci and M. Shibasaki, Pure Appl. Chem., 2008, 80, 1055 CrossRef CAS; (h) S. Kobayashi, Y. Mori, J. S. Fossey and M. M. Salter, Chem. Rev., 2011, 111, 2626 CrossRef CAS PubMed.
- E. M. Carreira and L. Kvaerno, Classics in Stereoselective Synthesis, Wiley-VCH, Weinheim, 2009, pp. 153–185 Search PubMed.
- For a review, see: M. Yus, J. C. González-Gómez and F. Foubelo, Chem. Rev., 2011, 111, 7774 CrossRef CAS PubMed.
- D. H. Paull, C. J. Abraham, M. T. Scerba, E. Alden-Danforth and T. Lectka, Acc. Chem. Res., 2008, 41, 655 CrossRef CAS PubMed.
- For a review, see: M. Yus, J. C. González-Gómez and F. Foubelo, Chem. Rev., 2013, 113, 5595 CrossRef CAS PubMed.
- (a) Y. Okude, S. Hirano, T. Hiyama and H. Nozaki, J. Am. Chem. Soc., 1977, 99, 3179 CrossRef CAS; (b) A. Fürstner and N. Shi, J. Am. Chem. Soc., 1996, 118, 12349 CrossRef; (c) M. Bandini, P. G. Cozzi, P. Melchiorre and A. Umani-Ronchi, Angew. Chem., Int. Ed., 1999, 38, 3357 CrossRef CAS; (d) A. Berkessel, D. Menche, C. A. Sklorz, M. Schröder and I. Paterson, Angew. Chem., Int. Ed., 2003, 42, 1032 CrossRef CAS PubMed; (e) M. Inoue, T. Suzuki and M. Nakada, J. Am. Chem. Soc., 2003, 125, 1140 CrossRef CAS PubMed.
- For a review, see: Z.-L. Shen, S.-Y. Wang, Y.-K. Chok, Y.-H. Xu and T.-P. Loh, Chem. Rev., 2013, 113, 271 CrossRef CAS PubMed.
- For recent papers on this topic, see: (a) A. Shen, Z.-T. He, H.-J. Yu, Y. Fukui, P. Tian and G.-Q. Lin, Synthesis, 2013, 1649 CAS; (b) D. Chen and M.-H. Xu, Chem. Commun., 2013, 49, 1327 RSC.
- For a review, see: Y. Tamaru, Eur. J. Org. Chem., 2005, 2647 CrossRef CAS.
- (a) I. L. Lysenko, H. G. Lee and J. K. Cha, Org. Lett., 2009, 11, 3132 CrossRef CAS PubMed; (b) M. Takahashi, M. McLaughlin and G. C. Micalizio, Angew. Chem., Int. Ed., 2009, 48, 3648 CrossRef CAS PubMed; (c) M. A. Tarselli and G. C. Micalizio, Org. Lett., 2009, 11, 4596 CrossRef CAS PubMed; (d) M. Z. Chen, M. McLaughlin, M. Takahashi, M. A. Tarselli, D. Yang, S. Umemura and G. C. Micalizio, J. Org. Chem., 2010, 75, 8048 CrossRef CAS PubMed.
- For recent reviews, see: (a) G.-Q. Lin, M.-H. Xu, Y.-W. Zhong and X.-W. Sun, Acc. Chem. Res., 2008, 41, 831 CrossRef CAS PubMed; (b) F. Ferreira, C. Botuha, F. Chemla and A. Pérez-Luna, Chem. Soc. Rev., 2009, 38, 1162 RSC; (c) M. A. T. Robak, M. A. Herbage and J. A. Ellman, Chem. Rev., 2010, 110, 3600 CrossRef CAS PubMed.
- For a review, see: F. Foubelo and M. Yus, Eur. J. Org. Chem., 2014, 485 CrossRef CAS.
- F. Foubelo and M. Yus, Tetrahedron: Asymmetry, 2004, 15, 3823 CrossRef CAS PubMed.
- J. A. Sirvent, F. Foubelo and M. Yus, Chem. Commun., 2012, 48, 2543 RSC.
- (a) J. C. González-Gómez, M. Medjahdi, F. Foubelo and M. Yus, J. Org. Chem., 2010, 75, 6308 CrossRef PubMed; (b) J. C. González-Gómez, F. Foubelo and M. Yus, Org. Synth., 2012, 89, 88 CrossRef.
- (a) D. J. Weix and J. A. Ellman, Org. Lett., 2003, 5, 1317 CrossRef CAS PubMed; (b) D. J. Weix and J. A. Ellman, Org. Synth., 2005, 82, 157 CrossRef CAS.
- T. Jang, G. Keum, S. B. Kang, B. Y. Chung and Y. Kim, Synthesis, 2003, 775 CAS.
- S. Araki, T. Kamei, T. Hirashita, H. Yamamura and M. Kawai, Org. Lett., 2000, 2, 847 CrossRef CAS PubMed.
- M. Liu, X.-W. Sun, M.-H. Xu and G.-Q. Lin, Chem. – Eur. J., 2009, 15, 10217 CrossRef CAS PubMed.
- P. V. Ramachandran, T. E. Burghardt and L. Bland-Berry, J. Org. Chem., 2005, 70, 7911 CrossRef CAS PubMed.
Footnotes |
| † This paper is dedicated to Professor Richard J. K. Taylor on occasion of his 65 birthday. |
| ‡ Electronic supplementary information (ESI) available: Experimental procedures, full characterization, and copies of 1H and 13C NMR spectra of compounds 3 and 6. See DOI: 10.1039/c4cc02317j |
| § Permanent address: Instituto de Química, Universidade Federal de Goiás, Campus Samambaia, CEP 74001-970, Goiána, Goiás, Brazil. |
| This journal is © The Royal Society of Chemistry 2014 |