 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceOn synergy between ultrahigh throughput screening and machine learning in biocatalyst engineering
Maximilian
Gantz†
 a,
Simon V.
Mathis†
b,
Friederike E. H.
Nintzel†
a,
Pietro
Lio
b and
Florian
Hollfelder
*a
a,
Simon V.
Mathis†
b,
Friederike E. H.
Nintzel†
a,
Pietro
Lio
b and
Florian
Hollfelder
*a
aDepartment of Biochemistry, University of Cambridge, 80 Tennis Court Road, Cambridge, CB2 1GA, UK
bDepartment of Computer Science, University of Cambridge, 15 JJ Thomson Avenue, Cambridge CB3 0FD, UK
First published on 23rd April 2024
Abstract
Protein design and directed evolution have separately contributed enormously to protein engineering. Without being mutually exclusive, the former relies on computation from first principles, while the latter is a combinatorial approach based on chance. Advances in ultrahigh throughput (uHT) screening, next generation sequencing and machine learning may create alternative routes to engineered proteins, where functional information linked to specific sequences is interpreted and extrapolated in silico. In particular, the miniaturisation of functional tests in water-in-oil emulsion droplets with picoliter volumes and their rapid generation and analysis (>1 kHz) allows screening of >107-membered libraries in a day. Subsequently, decoding the selected clones by short or long-read sequencing methods leads to large sequence-function datasets that may allow extrapolation from experimental directed evolution to further improved mutants beyond the observed hits. In this work, we explore experimental strategies for how to draw up ‘fitness landscapes’ in sequence space with uHT droplet microfluidics, review the current state of AI/ML in enzyme engineering and discuss how uHT datasets may be combined with AI/ML to make meaningful predictions and accelerate biocatalyst engineering.
1. Introduction
Protein engineering has made remarkable progress over the last decades, based on advances in recombinant DNA technology and site-directed mutagenesis,1 directed evolution,2 mechanistic and structural analysis3 and computational design.4 Nevertheless, the ambition of the original protein engineering, formulated as ‘designing tailor-made enzymes for every reaction”,5 has not been fulfilled so far, still warranting Jeremy Knowles’ warning about the premature use of the term ‘engineering’ in 1987.6 Two potentially crucial contributions have emerged more recently: on the one hand, development of new assay formats that make it possible to gain quantitative insight not only into one enzyme mutant at a time, but at a large number of them. When this comes at a low cost, by taking advantage of miniaturization – e.g. through microwells in microfluidic chambers7 or in in vitro compartments in microfluidics8 – much larger fractions of amino acid sequence space can be explored and functionally evaluated. On the other hand, machine learning offers increasingly capable algorithms suitable for interpreting such large datasets. Will it be possible to decipher complex combinatorial scenarios contained in these data and access mechanistic scenarios that would defy hypothesis-driven approaches, to enable extrapolations to inform biocatalyst engineering? Are these two approaches a natural match? Where do they synergise and how might they be combined to obtain insights on enzyme function and evolution that thus far have remained elusive?2. Microdroplets as in vitro compartments generated and screened in microfluidic devices – three examples for screening workflows
Screening diversity is central to protein engineering efforts and scale is crucial to identify rare hits in the vastness of sequence space. Water-in-oil emulsions promise several orders of magnitude higher throughput compared to traditional microtiter plate screening approaches by massively reducing the volume of an experiment without use of plasticware (>107-fold volume reduction compared to the regular 96-well plate format with a ∼200 μl volume) (Fig. 1). A large body of work in soft matter physics has equipped us with the ability to generate water-in-oil emulsion droplets in microfluidic devices at >kHz rates that allow analysis of >107 variants per day.9 Analytical interfaces exist to measure reaction progress at comparable scales, e.g. via detection by fluorescence10 or absorbance11,12 (or even label-free based on mass changes, albeit at lower throughput).13 When single emulsions are converted into double emulsions, commercial flow cytometers can be used for detection of fluorophores.14 Further, lab-on-a-chip devices miniaturise more complex liquid handling operations and coupled assays enlarge the range of reactions that can be assayed.11,15–17 Indeed, functional assays for all E. C. classes are already available.8 The above-mentioned scale and cost benefits of droplet experiments match recent advances in next generation sequencing technologies: thus large sequence-function datasets can be generated at much lower cost and with shorter lead times than conventional experimental formats (Fig. 1). Although sequence space is notoriously vast (for a 100 amino acid protein there are 1.3 × 10130 possible combinations), data in which sequence and function are correlated may contribute to rough descriptions of ‘fitness landscapes’ that track and inform the navigation across more or less interesting sections of sequence space.18,19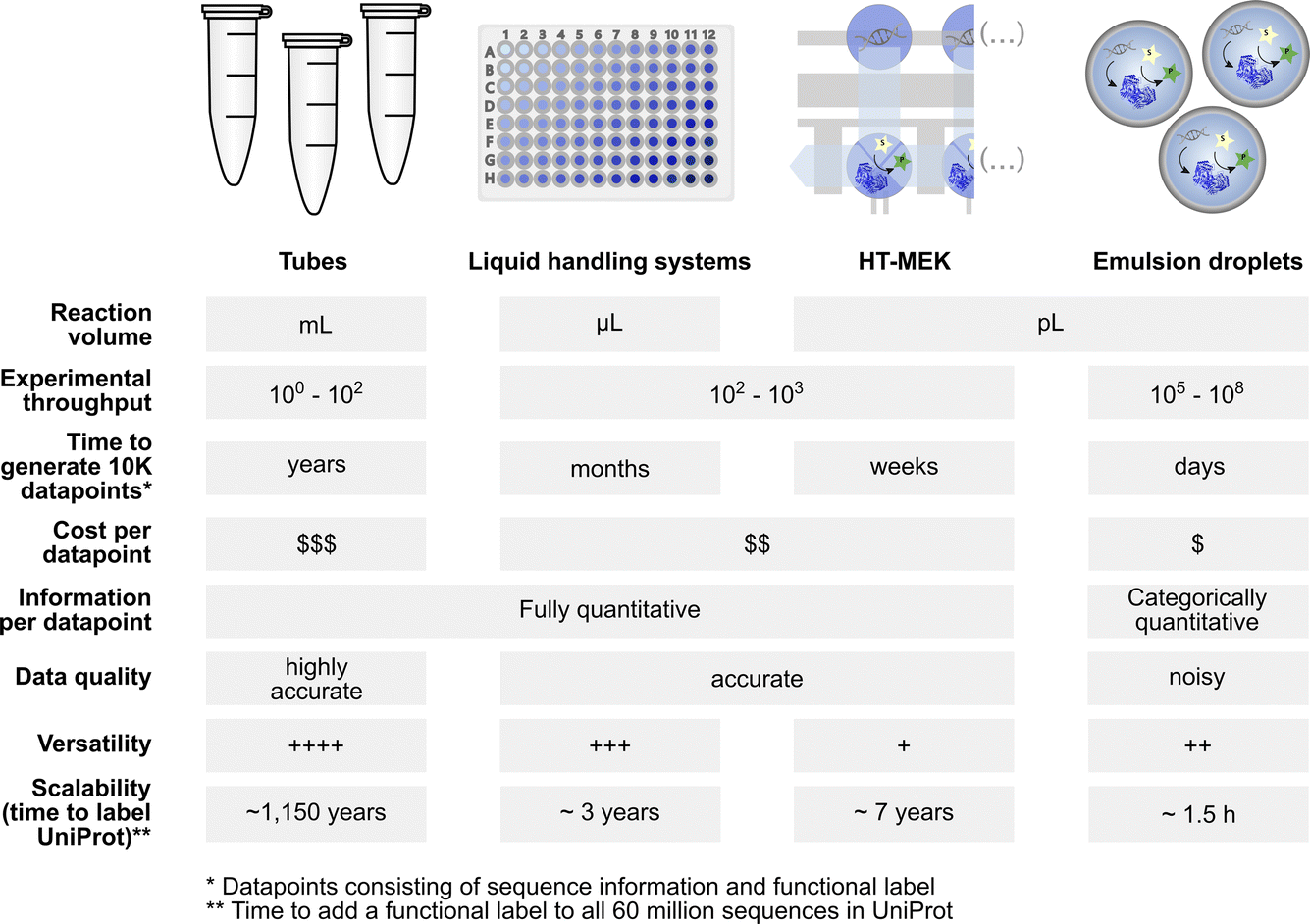 | ||
Fig. 1 Profiles of low to ultrahigh throughput experimentation systems (tubes, multiwell liquid handling systems, HT-MEK![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 7 and emulsion droplets8) that may be used as data generation tools for machine learning with their specific benefits and limitations. 7 and emulsion droplets8) that may be used as data generation tools for machine learning with their specific benefits and limitations. | ||
Three examples show this technology in action, taking library screening experiments in droplets (Fig. 2) all the way to ‘maps’ of sequence space (Fig. 3). Each example illustrates a distinct workflow (Fig. 2) in which large scale screening allows to infer information on fitness landscapes, generating substantial datasets that may be useful for AI/ML interpretation:
(A) Evolution of an amine dehydrogenase (AmDH), a valuable biocatalyst for the synthesis of chiral amines. Zurek et al.20 screened libraries of AmDH variants generated by error-prone PCR mutagenesis. Libraries were transformed into E. coli for expression. Single cells were encapsulated into droplets with substrates for a coupled assay using the dye WST-1 as a turnover sensor (Fig. 2A). Positive variants were selected based on an absorbance measurement (>105 droplets per hour, but faster systems are now available12,21) and DNA was recovered and sequenced using UMI-linked Oxford Nanopore sequencing (UMIC-seq) to achieve high-quality.
(B) Mutational scanning of a protein kinase involved in signaling networks. The human protein kinase MKK1 is an example of a broad class of phosphate transfer enzymes involved in signalling networks. In order to explore how these evolve, MKK1 (which targets ERK2) was randomised with a focus on six residues in its docking domain (D-domain), which mediates interaction with the downstream kinase ERK, activating its kinase activity. Each MKK1 variant was tested for its ability to bind and phosphorylate ERK2 in a coupled assay (Fig. 2B) exploring a scenario of neutral roaming in sequence space (i.e. a non-adaptive evolution experiment). The library was expressed (using a commercial in vitro transcription/translation system) in a polydisperse emulsion containing monoclonal magnetic beads. This cell free approach alleviates issues that frustrated previous in vivo kinase screens such as cellular background and functional redundancy, while it simultaneously benefits from the robust expression of kinases in an in vitro transcription/translation system. Selections were carried out in polydisperse emulsions (not even necessitating the use of microfluidics) and the gene as well as the substrate (giving a GFP readout when a kinase target sequence was protected by successful kinase action against proteolysis) were immobilized on a bead, so that flow cytometric sorting (FACS) could be used to identify active clones. Using next-generation sequencing (NGS) of the D-domain to calculate enrichment scores, functional combinations of D-domain variants were mapped out.
(C) Identifying promiscuous phosphotriesterases in metagenomic libraries. A metagenomic library with 1.25 million genomic inserts of mixed environmental origins (soil, degraded plant material and cow rumen) was screened using a fluorescent assay reporting on phosphotriesterase activity (Fig. 2C). The brightest 0.001% of droplets were sorted, sequenced using Sanger sequencing and characterised to reveal novel, uncharacterised “bridgeheads” in sequence space which is now functionally annotated in areas where homology-based classification would not have predicted phosphotriesterase activity.
 | ||
| Fig. 2 Ultra-high throughput screening workflows coupled to sequence data generation. (A) Single cells can be encapsulated in droplets with substrates and lysis agent. The amine dehydrogenase reaction is coupled to WST-1 reduction forming a colorimetric readout. Active variants are sorted using AADS (absorbance-activated droplet sorting) and the output is sequenced using high-quality nanopore sequencing (UMIC-seq). (B) Monoclonal beads carrying GFP linked via a chymotrypsin cleavage sequence are encapsulated and the kinase cascade is expressed via IVTT. A phosphorylated linker sequence is resistant to chymotrypsin cleavage. Active variants are sorted by FACS of the beads with multiple gates and the D-domain sequence and enrichment is read out via next generation sequencing (NGS). (C) Single E. coli cells expressing metagenomic library members are encapsulated into droplets along with a fluorogenic phosphotriester substrate. Phosphotriesterases hydrolyse the phosphotriester releasing fluorescent fluoresceine. Droplets containing active variants are sorted at >1 kHz using FADS (fluorescence-activated droplet sorting) and hits are revealed by Sanger sequencing of the selected clones. | ||
3. What kind of data do we generate in large scale droplet experiments?
Our objective is to reveal fitness landscapes to visualize the exploration of sequence space and ultimately steer ‘walks’ towards zones in which new or improved activities are more likely. Droplet experiments inform the maps by providing sequence information on individual variants correlated to a specific functional label (i.e. a qualitative or quantitative assessment of activity). While the variant sequence can be easily read out at the end of a screening workflow, functional labels can often only be obtained indirectly and require creative experimental design. Examples for such functional labels are variant identification after passing a set threshold for sorting in directed evolution or quantitative enrichment scores by deep mutational scanning,22 which has recently been integrated with high throughput screening and machine learning treatment.58 More generally, these labels can be categorized as binary data (e.g. selection is either successful or unsuccessful)23,24versus binned quantitative data (with granular enrichment scores).25 (These approaches are referred to as ‘categorically quantitative’ in Fig. 1.)The type of label that can be obtained from a large-scale droplet experiment is highly dependent on the chosen library size and design, the microfluidic workflow, and the choice of the sequencing strategy. NGS offers high enough sequencing depth to generate binned quantitative data (granular enrichment scores) for sequence-function mapping: reporting how often a variant occurs in the input vs. the output library. The technology used for sequencing determines the information content further. Short reads with only up to 600 bp read length (with 2 × 300 paired end sequencing) adequately describe mutational patterns in small proteins26 or functionally defined regions of proteins.25 However, long read sequencing technologies are necessary to reveal long-range epistatic effects in larger proteins. Corresponding datasets can be obtained with PacBio or Oxford Nanopore instruments. Oxford Nanopore sequencing is cheap (<1.1¢ per sequence)20 and can be carried out in any lab at low cost, while the capital expenditure for a PacBio (250000$ for PacBio vs. <1000$ for a MinION device) makes this impractical. The two technologies differ in their read quality, with PacBio giving high quality reads at single nucleotide resolution. Oxford Nanopore devices suffer from high error rates and are unable to pin-point single nucleotide mutations, but a workaround – consisting of UMI (unique molecular identifier) labelling followed by clonal amplification and consensus generation from multiple sequences (that are tagged by the same UMI)20 – exists to produce high quality sequences of even single amino acid mutants. While short-read NGS can be used to generate binned quantitative data with granular enrichment scores, long-read sequencing technologies operate at lower scale (90 Gb for PacBio, 50–110 Gb for Oxford Nanopore compared to up to 3000 Gb with Illumina sequencing) and are currently limited to the generation of binary data on variant identification per round of selection in directed evolution. (but see ref. 58 for new long read approach employing Oxford Nanopore devices).
Each of the three studies reviewed here (Fig. 3) uses different experimental designs, so the sequencing strategies are correspondingly different, but all three arrive at representations of hits in sequence space that can be interpreted as fitness landscapes:
(A) AmDH screening (Fig. 3A). In AmDH evolution long-read Nanopore sequencing (in a commercial MinION flow cell; Oxford Nanopore) was used to sequence 3000 hits with an activity higher than the threshold chosen for screening. A crucial accuracy improvement is achieved by tagging variants with unique molecular identifiers (UMIs): these are then amplified clonally, multiple nanopore sequences are generated and finally evaluated by deriving a consensus from many reads per amplified variant. In this way the sequencing accuracy was dramatically increased to >99.99%. The improved accuracy for cost efficient long-read nanopore sequencing is crucial for confidently resolving multiple mutations per variant and thus mapping evolutionary trajectories. The resulting dataset gives a fitness landscape shown in Fig. 3A that illustrates the evolution of a functional protein through three generations of ultrahigh throughput screening in directed evolution, in which the 3000 best hits of 250000 variants were sorted and sequenced. The apparent clustering reveals intra-gene cooperativity of mutations (epistasis), for which accurate long read sequencing was necessary and provided experimental evidence for sign epistasis. Information from multiple rounds of directed evolution constitutes a dataset conditioned by the combinability of mutations. The analysis of evolutionary trajectories in this way helps to extract features for further labelling and reconstructing or extrapolating functional evolution. Such features will be identified by their acquisition and conservation through rounds of evolution and may include residues with a catalytic function (located near the active site), but also enhancing solubility (conferred by residues the outside of the protein), stability (e.g. residues allowing improved packing or better hydrophobic interactions in the core of a globular protein), introduction of conformational flexibility or disorder (e.g. in order to facilitate recognition of new substrates or remove steric clashes) and finally patterns of the aforementioned epistatic interactions (i.e. long range interactions between often distant residues).
(B) Kinase screening (Fig. 3B). The narrow focus on the well-known docking domain (D-domain) of kinases made it possible to use the short reads provided by Illumina sequencing to draw up a fitness landscape. A starting library of 500000 mutants was generated from randomising six residues in the MKK1 docking domain (synthesised on beads by spit-and-mix assembly, with high quality and equal representation of nucleotides27). Library members were sorted into three bins according to activity. 2.9 × 104 MKK1 variants are functional, providing a rich dataset to explore cooperativity between the different randomised positions. Enrichment analyses identified patterns of interdependence between the randomized positions, highlighting the role of cooperative hydrophobic effects and charge balance. Taken together, the patterns are displayed in a fitness landscape in which transitions from one sequence motif to another are generally possible. Many well-connected variants capable of substrate binding and phosphorylation suggest high evolvability. The extensive well-labeled sequence dataset (Fig. 3B) carries information about implicit positive epistasis and may be further interpretable by ML in the future.
(C) Triesterase screening (Fig. 3C). Screening of a metagenomic library (in binary mode for overcoming a phosphotriesterase activity threshold) yielded 8 hits, the majority of which had not been recognized as phosphotriesterases before. These new enzymes will constitute bridgeheads in sequence space for further annotation, being selected for function rather than found by sequence homology. New functional motifs were recognized, e.g. an α/β hydrolase fold, in which a catalytic triad (with a cystein nucleophile) served as a multiple turnover catalyst, despite its similarity to the target of phosphotriester as a toxin, an active site catalytic triad (containing serine) that is suicide-inhibited by the triester. Newly identified enzymes from this approach will be useful as a binary activity label for ML-based functional annotation to further annotate sequences in large metagenomic databases such as MGnify.28
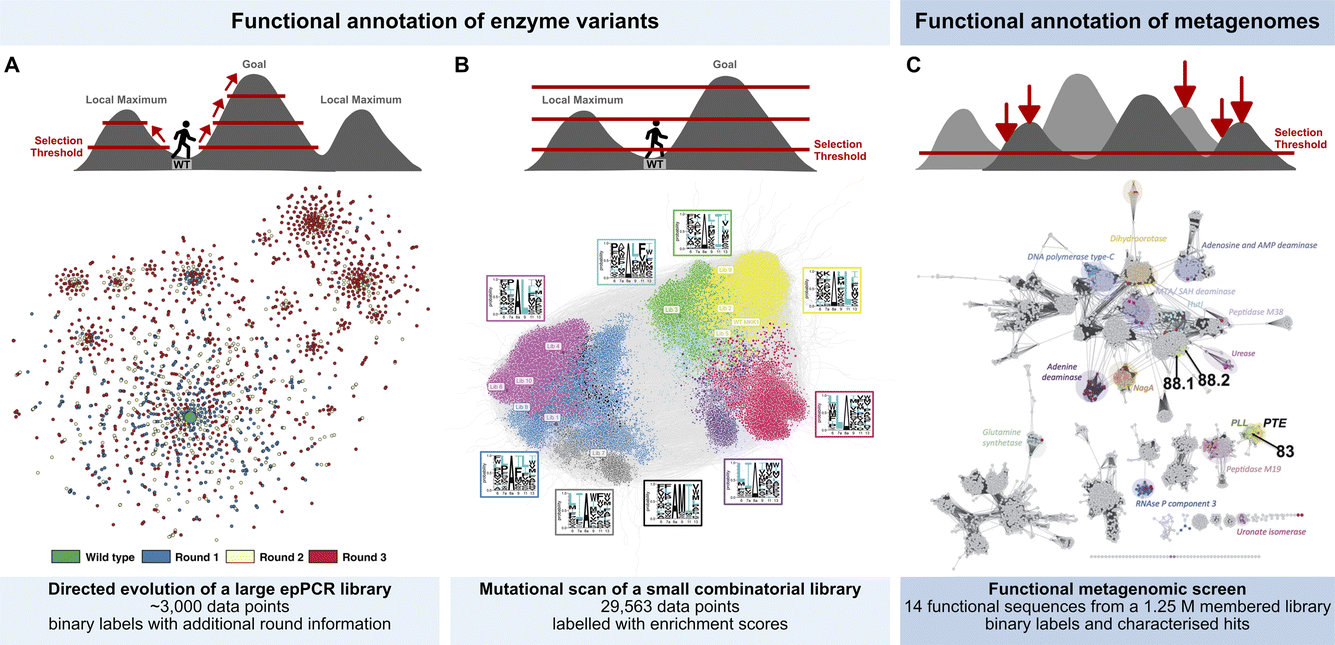 | ||
| Fig. 3 Functional annotation of sequence space. (A) Exploring productive trajectories on the fitness landscape of an amine dehydrogenase in three rounds of directed evolution.20 (B) Scanning the fitness landscape of a short kinase docking domain (D-domain) with increasing thresholds for comprehensive epistasis mapping.25 (C) Identifying islands of sulfatase and phosphotriesterase function in an unexplored landscape through functional metagenomics.23 | ||
The three campaigns provide examples for sequence space explorations, in which the experimental design and selection criterion shapes both, the area of sequence space that is explored and the functional readout that ultimately completes a fitness landscape by adding a third, functional dimension to sequence space (as represented by two notional dimensions).
(A) The case of kinase MKK1 is producing a dataset (Fig. 3B) focused on the small fraction of sequence space represented by docking domain mutagenesis and functionally annotated with granular enrichment scores that map a smooth fitness landscape with many overlapping functional motifs.
(B) The data on AmDH (Fig. 3A)20 covers mutations across the entire protein (being derived from an epPCR library) and thus samples a larger area of sequence space. The dataset can be interpreted as an exploration of sequence space in all directions, as long as the selection criterion of increasing AmDH activity is fulfilled (measured by a binary assay). The resulting fitness landscape is more complex and shaped by long-range epistatic effects that define founder mutations, with considerable ‘ruggedness’ of the fitness landscape (resulting in some mutational paths closed off due to sign epistasis), but also with evidence for positive epistasis across the protein structure (where the combined effect of two mutations can be larger than the sum of their individual contributions). Ruggedness in the fitness landscape with fewer paths for evolution suggests that transitions are more difficult and the evolvability potentially reduced, due to the intrinsic response of this protein to mutations.
(C) Finally the sequence context in which new phosphotriesterases are found is much broader (Fig. 3C),23 starting from a diverse metagenomic library (rather than a randomised single protein) and identifying peaks only in a binary screen. Additional surrounding sequences can be derived from sequence repositories, but as their function is inferred rather than tested, no inference about the shape of a fitness landscape can be made: it is simply annotated.
Interpreting large sequence collections rather than individual single mutants (e.g. a ‘winner’ of a selection or screening experiment) may offer additional insight. It is tempting to hope that the data can be used to reliably extrapolate from experimentally characterized variants and predict new ones with higher fitness. Cooperative epistatic effects define an evolutionary trajectory and may be inferred from information on groups of mutants (either as long-range intra-gene effects in ‘founder mutants’ of AmDH20 or as short range effects focused on the MKK1 kinase D-domain25) and its analysis may allow predictions.29 Even for metagenomic explorations,23,30 functionally annotated data can be the basis of prediction.
4. The current state of AI for enzyme engineering
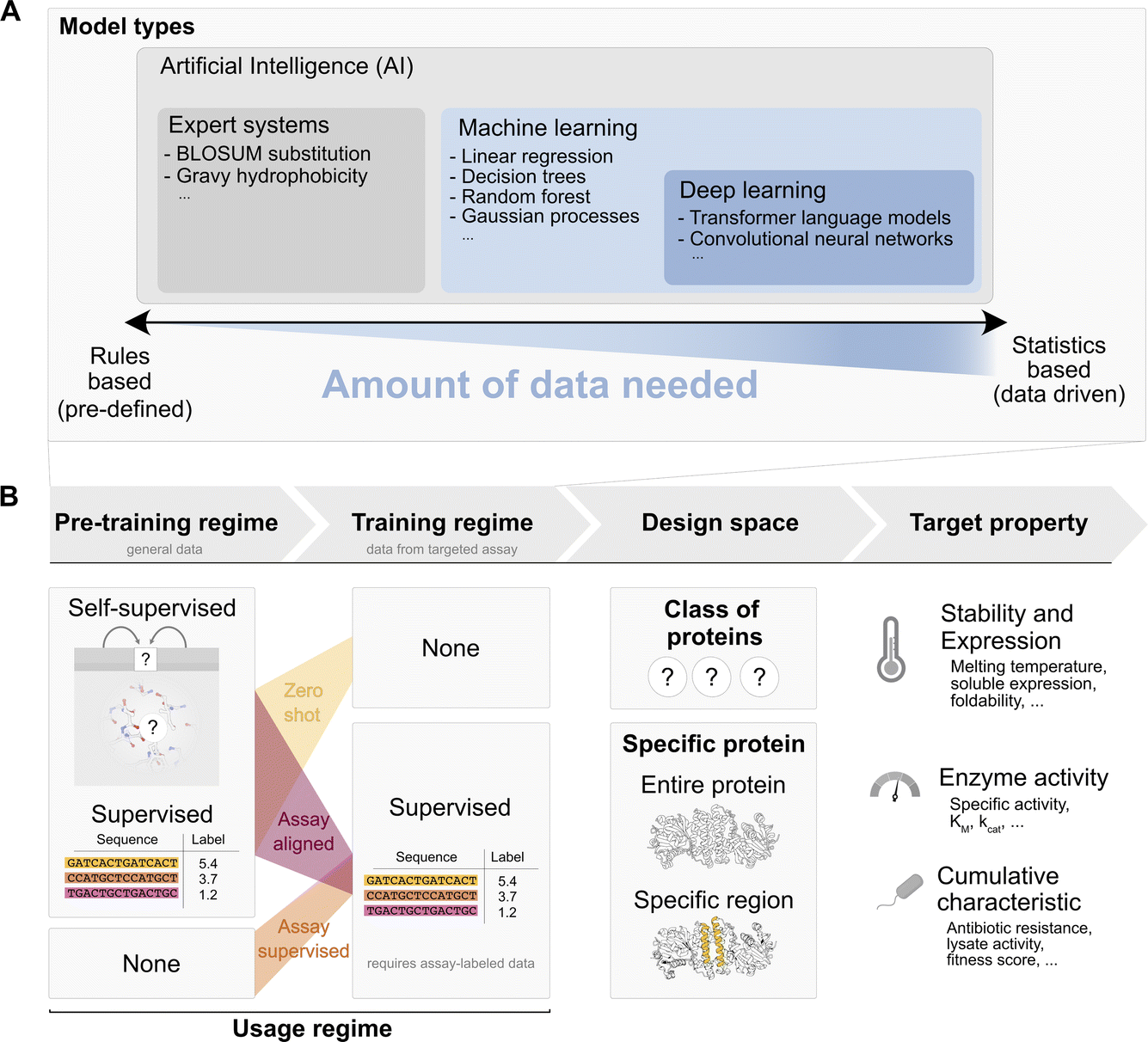
Given the demand for green, carbon neutral biocatalytic processes that require new or improved enzymes, it is tempting for protein engineers to dream of an algorithmic black box that automatically and reliably produces instructions for enhanced activity improvements, as easily as AlphaFold2 (ref. 31) comes up with a structural model. However, structure is easier to predict than activity, with the latter requiring sub-Angstrom precision in the active site and orchestrating a number of catalytic effects just in time to cross the transition state. Furthermore, the input dataset for AlphaFold2 is the well-established, rigorously quality-controlled, and systematically organized PDB which was built up in a community effort over years. In contrast, there is no such systematic framework for functional characterisation of large enzyme libraries yet. While substantial organizational effort has been put into EnzymeML,32 this data exchange format is based on STRENDA, ready to receive high quality data on a few enzymes (or mutants) rather than necessarily shallower ultrahigh throughput data on fitness in larger libraries.To discuss the interface between ultrahigh-throughput experiments and AI, we must understand the AI enzyme engineering landscape (Tables 1–3.40,44–56 AI models differ in the extent to which they rely on rules derived from prior knowledge or autonomously identify statistical patterns in data without user input. A useful distinction can be made between expert systems that make decisions based on rules drawn up by a human expert (e.g. gravy hydrophobicity33 or BLOSUM substitution34). In contrast, machine learning is an umbrella term for techniques that do not rely on such rules, but instead derive rules from data (the “learning” aspect in machine learning, e.g. linear regression, random forest, etc.). Deep learning is a subclass of machine learning and is loosely distinguished from general machine learning by its large count of learnable parameters: often of similar or larger order of magnitude to the available datapoints (or beyond). Many contemporary neural network approaches, such as transformers35 (the main component of modern language models36), AlphaFold2 (ref. 31) and convolutional networks,37 belong to this category. The amount of data available is a first criterion in the choice of a model, with deep learning approaches being more data hungry, while general machine learning techniques can live with fewer data inputs. The parameters of these models are then tuned in one or more ‘training’ steps.
| Enzyme | Data | Achievement | Ref. | |||||
|---|---|---|---|---|---|---|---|---|
| Reaction | Library screening | Library type | Data type | Total data points | Data points used for ML | Improvement top variant | % success (>wt) | |
| a Training data heavily biased towards single mutations. A more sophisticated structure guided model that is less biased on single mutation data is also presented and shows similar improvements in conversion but no specific activity is reported. b Possible to express 168/200 ordered double/triple mutants. c Training data: (9 single mutants + 7 higher order combinations of those 9 singles from a previous DE campaign); improvement for same pH as training data (claimed 121-fold improvement at different pH). d Two variant engineered (S) & (R) specific: 93% ee/79% ee for (S/R) respectively, starting from 76% ee (S). | ||||||||
| Imine reductase (IRED) | Robotic screening (plate) | 20 random singlesa | Conversion | 11303 |
20* | Specific activity: ∼wt conversion: 4.6-fold | n.a.b | 44 |
| Mixed (singles + 1 EPCR round)a | ∼5000 | Specific activity: 1.3-fold conversion: 8.3-fold | ||||||
| Mixed (singles 2 epCR rounds)a | ∼8000 | Specific activity: 1.3-fold conversion: 7.1-fold | ||||||
| Microfluidics | epPCR | Conversion | 17143 |
10860 |
k cat/KM: 16-fold | 70% | 58 | |
| k cat: 23-fold | ||||||||
| Glucose oxidase | Spectro photometer (plate) | Focused – from a previous campaign | Michaelis–Menten | 16c | 16c | k cat/KM: 12.1-foldc | One variant | 46 |
| k cat: 4.8-fold | ||||||||
| Halogenase | LC-MS (plate) | Focused (3 sites) | Conversion | 504 | 504 | Conversion: 16-fold | 100% | 47 |
| k cat/KM: 82-fold | ||||||||
| k cat: 93-fold | ||||||||
| Hydroxylase XylM | Biosensor (plate) | Focused (5 sites) | Sensor coupled to fluorescent protein | Round 1: 126 | Round 1: 126 | Yield: 15-fold | Sensor: 94% | 48 |
| Round 2: 126 + 50 | Round 2: 126 + 50 | |||||||
| Nitric oxide dioxygenase | Plate assay | Focused | Enantiomeric excess | Round 1: 124 | Round 1: 124 | Lysate activity: 3.2-fold; | n.d. 360 predictions | 49 |
| Round 2: 155/166 | Round 2: 155/166 | e.e.: 1.2-fold and reversedd | ||||||
| Luciferase | Bioluminescence (plate) | Focused (non-cons regions) | Bioluminescence | 164 | 164 | Specific activity: 7.8-fold | 72% (26/36) | 50 |
| Beta lactamase | Antibiotic resistance | Error-prone PCR | Antibiotic resistance | 96 and 24 | 96 and 24 | Enrichment up to ∼40-fold vs wildtype | 2.5% | 53 |
| Enzyme | AI model specification | Achievement | Ref. | |||||
|---|---|---|---|---|---|---|---|---|
| Reaction | Model type | Training regime | Usage regime | Design space | Target property | Improvement top variant | % Success (>wt) | |
| a Starting with a panel of models from scikit-learn, the top three model types were selected and used to identify the top 1000 sequences in each predicted library. b Presumably doubles/triples of the 20 input singles were considered. c Possible to express 168/200 ordered double/triple mutants. d Random forest on UniRep 1900 descriptors. Note: UniRep1900 is in principle a self-supervised trained language model, so it could be argued the training regime was supervised + self-supervised and the usage regime was assay aligned rather than assay supervised. e Training data: (9 single mutants + 7 higher order combinations of those 9 singles from a previous DE campaign); improvement for same pH as training data (claimed 121-fold improvement at different pH). f The selection of these 3 sites was based on (1) docking studies with the structure and (2) previously published literature results and (3) previous knowledge of the enzyme. This is not trivial to replicate for any enzyme. g This study used 2 models: a more shallow machine learning based one and a deep learning based one. Specific region: 5 determined via alanine scan and 50 variants were tested in each round. h Two variant engineered (S) & (R) specific: 93% ee/79% ee for (S/R) respectively, starting from 76% ee (S). i Two rounds of evolution performed, while most other studies listed here perform one round. | ||||||||
| Imine reductase (IRED) | Random forest | Supervisedd | Assay supervised | Specific regionb | Specific activity & conversion | specific activity: ∼wt conversion: 4.6-fold | n.a.c | 44 |
| Random forest | Specific protein | specific activity: 1.3-fold conversion: 8.3-fold | ||||||
| Random forest & structure-informed | Specific proteinb | specific activity: 1.3-fold conversion: 7.1-fold | ||||||
| Augumented ridge regression & decision tree with rational engineering | Supervised | Assay supervised | Entire protein | k cat and kcat/KM | k cat/KM: 16-fold | 70% | 58 | |
| k cat: 23-fold | ||||||||
| Glucose oxidase | Machine learning (partial least squares) | Supervised | Assay supervised | Specific region | Michaelis–Menten | k cat/KM: 12.1-folde | One variant | 46 |
| k cat: 4.8-fold | ||||||||
| Halogenase | Machine learning (Gaussian Process) | Supervised | Assay supervised | Specific regionf | Conversion | conversion: 16-fold | 100% | 47 |
| k cat/KM: 82-fold | ||||||||
| k cat: 93-fold | ||||||||
| Hydroxylase XylM | Machine learning & deep learningg | Supervised & self-supervised | Assay supervised & assay aligned | Specific regiong | Sensor/yield | Yield: 15-fold | Sensor: 94% | 48 |
| Nitric oxide dioxygenase | Machine learninga | Supervised | Assay supervisedi | Specific region | Lysate activity and stereoselectivity | lysate activity: 3.2-fold; | n.d. 360 predictions | 49 |
| e.e.: 1.2-fold and reversedh | ||||||||
| Luciferase | Machine learning (Gaussian process & self-play reinforcement learning) | Supervised | Assay supervised | Specific region | Bioluminescence | Specific activity: 7.8-fold | 72% (26/36) | 50 |
| Beta lactamase | Deep learning (LSTM language model) | Self-supervised | Assay aligned | Specific region | Enrichment under Amp selection | Enrichment up to ∼40-fold vs wildtype | 2.5% | 53 |
| Enzyme | AI model specification | Achievement | Ref. | |||||
|---|---|---|---|---|---|---|---|---|
| Reaction | Model type | Training regime | Usage regime | Design space | Target property | Improvement top variant | % Success (>wt) | |
| a No wild type comparison available, % active variants used, all ordered (not only soluble) enzymes considered; engineering for pH stability. b Based on endonuclease structure and sequence conservation data. c Direct AI prediction is a single mutant (A53M) leading to 3-fold reduced side product formation, which was then combined with other predictions (rationally/assuming additivity) to get their 17-fold reduced off product formation. d Self-supervised: masked AA prediction in microenvironment; supervised: model selected based on correlation of zero-shot fitness with DeltaTM of single mutants in FireProtDB. | ||||||||
| Malate dehydro-genease | Deep learning (protein GAN) | Self-supervised | Zero-shotd | Class of proteins | Specific activity | Wild-type like specific activity | 22a% | 45 |
| Methyltransferase | Deep learning (MutComputeX) | Self-supervised & supervised | Zero-shot | Specific protein | Product titer | Conversion: 1.6-foldc | n.d. | 51 |
| Beta lactamase | Deep learning (MutCompute) | Self-supervised | Zero-shot | Specific protein | BLA activity | Antibiotic resistance >wt, no quant measurement | 30% | 52 |
| TEV protease | Deep learning (protein MPNN) | Self-supervised | Zero-shot | Specific region | Fluorogenic substrate | k cat/KM: 26-fold (but mainly tied to solubility/thermostability) | 3 out of 144 designs | 54 |
| PETase | Deep learning (MutCompute) | Self-supervised | Zero-shot | Specific protein | PET hydrolysis activity | Specific activity: 29-fold | 80% | 40 |
| Endonuclease (Ago Proteins – KmAgo) | Deep learning (CPDiffusion) | Self-supervised (on family-focussed dataset) | Zero-shot | Specific proteinb | ssDNA cleavage assay | DNA cleavage activity: up to 8.6-fold | 75% | 55 |
| Lysozyme | Deep learning (ProGen language model) | Self-supervised | Zero-shot | Class of proteins | Michaelis–Menten kinetics | Wildtype-like activity | n.a. | 56 |
The training steps determine the data used and how it informs the model’s parameters. We distinguish between pre-training steps, which use general data such as the observed sequences on UniProt or general thermostability annotations from FireProtDB38, and assay specific training, which uses data from the targeted assay. A pre-training step may precede self-supervised or supervised learning: in the self-supervised mode only sequence or structure are available, while a functional label, e.g. an activity measurement, is absent. Instead of functional labels, “pseudo-labels” unrelated to function are created by masking parts of the sequence or structure and predicting the amino acids that should occupy the masked positions. This approach is called “self” supervised, because the labels are generated from the datapoint itself, through a masking process. This pre-training mode is used e.g. for protein language models39 and also for methods that take the structural environment into account.40,41 By integrating this information, the model learns to pick up on common sequence or structural motifs. Alternatively, when we have access to experimental mapping of sequence to function or a relevant proxy, a model may be pre-trained in a supervised way given the annotation. In contrast to general pre-training, assay-specific training, requires labels from the assay of interest and is therefore only possible in a supervised mode.
Pre-training steps and assay-specific training can be combined. Workflows may include pre-training steps (self-supervised or supervised) along with assay-specific training. The combinations of pre-training and assay specific training give rise to three broad usage regimes for a model to predict a target property (or generate a sequence with a desired target property value) that is probed by a specific assay run in the lab:
(i) Zero-shot: in this case a model is only pre-trained on general data and is used “as is” without supervised training on any assay labelled data to predict a target property. For example, a language model (such as ESM) might be trained through self-supervision (sequence masking) on all sequences observed in UniProt, and subsequently used in a “zero-shot” way by evaluating the probability that ESM assigns to a sequence containing a given mutation vs. the probability of the wildtype sequence. This assumes that the target property correlates with the self-supervision task that was used during training (e.g. thermostability, because ‘natural’ motifs in UniProt must be at least marginally thermostable to be observed in living organisms). As another example, we might pre-train a linear regression model “supervised” on cDNA display proteolysis data42 from general proteins, and then task the model to predict thermostability of our target protein “as is” (zero-shot). (ii) Assay aligned (also referred to as ‘transfer learning’ or ‘task-specific fine-tuning’ in the ML community): in the assay aligned regime, a model that was previously trained (=“pre-trained model”) on general data through self-supervision or supervision is “aligned” to the assay specific data through additional supervised training on a, commonly smaller, assay specific dataset. For instance, this may be achieved using the same model (e.g. ESM) and updating its parameters slightly based on the assay labelled sequence-to-function data (‘fine-tuning’). As another example, one may use another model which uses representations or outputs from the pre-trained model as some of its inputs and train it on the assay labelled data (‘feature extraction’). This process is illustrated for example in Hsu et al.43 where the output of ESM is used as input to a smaller linear regression. In essence, “assay aligned” usage takes an existing pre-trained model and trains it further with assay specific data. The loose idea is that this allows “motifs” and “patterns” that can efficiently be represented by the pre-trained model to be “re-mapped” to the assay data and thereby better extract which motifs might improve or decrease the targeted property. (iii) Assay supervised: in this case the given model is trained in a supervised way directly on assay data without pre-training on other data. Since the amount of available assay data is often very low, the types of models in this approach tend to be general machine learning models (not deep-learning models).
The functional coordinate defined by the assay determines the target property that is to be predicted, e.g. thermostability, solubility and expression, enzyme activity and cumulative characteristics (i.e. a mixed set of properties including general fitness, growth rate in the presence of antibiotic or lysate activity).
Finally design space restrictions can be incorporated, e.g. by explicitly restricting options based on expert knowledge, such as evolutionary or structural data at the following levels: (a) assignment to a specific class of proteins, e.g. an EC category or a particular fold; (b) sequences derived from a specific protein: starting from the WT sequence improvements in the target property are sought by mutating any position or combination of positions in the wildtype enzyme; (c) specific regions of a starting protein are considered preferentially – e.g. mutations in a subregion of the wildtype defined from an evolutionary conservation threshold from an MSA, expert knowledge of key positions or an enzyme structure.
5. Recent machine learning studies in enzyme engineering
The large datasets emerging from ultrahigh throughput screens will be prime candidates for machine learning analyses. Especially, unbiased datasets with large coverage of design space promise to hold solutions for problems that are difficult to access with traditional hypothesis driven research. However, the current studies on enzyme engineering that involve ML are using far smaller datasets. Nevertheless, even with smaller datasets remarkable progress has been made, highlighting what the use of ML has achieved and what one may expect if more data can be fed into the algorithms.Four groups of common workflow have been tested experimentally (see Tables 1–3) and can be characterized by their primary variations in usage regime and design space (Fig. 4B). We classify these as zero-shot approaches with focused (ZSF) or broad design space (ZSB) on the one hand, and, on the other hand, assay labeled regimes with focused (ALF) or broad design space (ALB). Assay labelled regimes with focused design space are usually informed by data from focused libraries targeting selected positions or regions in the protein only, in contrast to modes with a broader design space which, among others, include random mutagenesis (e.g. by error-prone PCR) across the entire protein.
5.1 Zero-shot regime without assay-labeled data (yellow sections in Tables 1–3)
(i) MutCompute. MutCompute is a deep learning approach (3D convolutional network) that was pre-trained in a self-supervised way based on structures in the Protein Database, by masking out amino acids in a given structure and predicting the identity of the masked amino acid based on the local context (a structural microenvironment defined by a 20 Å cube centered around the masked amino acid). MutCompute was successfully applied to the improvement of a plastic-degrading PETase by Lu et al.40 in zero-shot mode, coming up with 159 variants that were experimentally tested. Combinability studies of the best mutations from this panel yielded FAST-PETase, improved by more than an order of magnitude. Enhancements are larger at higher temperatures, suggesting that temperature adaptation is the main source of catalytic improvement. Additionally, MutCompute was successfully applied with a methyltransferase51 and a β-lactamase.52
(ii) ProteinMPNN (Fig. 4A). ProteinMPNN is another deep learning model (graph neural network) originally created for sequence-redesign given a backbone structure. It is pre-trained in a self-supervised mode by ‘deleting’ the side-chain and amino acid information in a given structure and then re-predicting the correct sequence – position by position (autoregressively) – based only on the backbone and Cβ coordinates, as well as the amino acid types that it already predicted.41 At usage time, a wildtype backbone structure, and optionally the amino acid types for a few fixed positions in the sequence, can be used as input and the remaining sequence is re-designed to fold into that target backbone. ProteinMPNN’s pre-training has been shown to correlate with solubility and thermostability41 (Fig. 4A). The rationale is that ProteinMPNN’s pre-training was based on general protein structures in which certain backbone fragments and motifs re-appear with slightly varied amino acids, such that for a given backbone fragment plausible (but diverse) amino acids are inferred at usage time. Since ProteinMPNN has been trained on structures in the PDB, which predominantly come from crystals and therefore need to be at least modestly stable and soluble, it is thought to predict stable and soluble solutions. Existing protein structures are biased towards these properties simply by virtue of being stable enough to be observed.
 | ||
| Fig. 4 Breakdown of key aspects of AI enzyme engineering efforts. We distinguish between pre-training (i.e. training on general data) and assay specific training (i.e. training on data from the targeted assay), which gives rise to three usage regimes: (i) zero-shot = pre-training only. (ii) Assay supervised = training only. (iii) Assay aligned = pre-training + training. Note that multiple pre-training steps are possible. Further details are explained in the text. | ||
A successful zero-shot application of ProteinMPNN for enzyme engineering is the work of Sumida et al.,54 who improved the solubility and stability of TEV protease. In order not to disturb the functionally relevant constituents of the protein, evolutionarily conserved and active site residues were exempted from randomization (Fig. 5A). 129/144 designs exhibited higher levels of soluble expression than the starting point and 64/144 designs showed some activity with a model substrate. The top three designs were further characterised on the model substrate and all showed higher catalytic efficiencies than the parent (up to 26-fold improvements) and the top hit (hyperTEV60) has 40 °C increase in melting temperature Tm. At 30 °C, hyperTEV60 retains 90% of its activity over 4 h, while the parent enzyme only retains 15% activity (Fig. 5B). These observations are consistent with the studies involving MutCompute,40 namely that biophysical robustness brings about an increased ability to form product. Observing an effect on reaction kinetics (with the actual native protease substrate) would provide more direct evidence for transition state stabilization (as opposed to improving the availability of a “competent state”, either by increased Tm or backbone rigidification).
 | ||
| Fig. 5 Machine learning informed engineering of a TEV protease54 and a halogenase.47 (A) Design strategy for TEV protease engineering. Based on structural and evolutionary constraints as input, the design space was defined by fixing the amino acid identities of the active site residues and conserved residues. ProteinMPNN was used to redesign the remaining residues and generate the designed sequences as output. (B) Stability assay. The best design hyperTEV60 shows improved benchtop stability compared to the native TEVd when incubated at 30 °C over time. (C) Identification of engineering sites for WelO5* halogenase. The target substrate soraphen A was docked into WelO5* and three positions were chosen for generating a full randomization library. (D) Activity assays for WelO5* variants. Hits from the combinatorial library (red) and from the ML predictions (green and blue) were tested in biotransformations with cell lysate. Results are displayed as fold increase compared to the parent GAP. The best hit in the combinatorial screen was SLP and the best hit in the ML predictions was VLA. | ||
The studies provide evidence that, when used in a focussed zero-shot way, models such as Mutcompute and ProteinMPNN can yield catalysts able to generate more reaction product. While biophysical characteristics are improved, the current data is less clear on improvements to the catalytic machinery. It is possible that the emphasis on stability in the pre-training data for self-supervision, which is from the general PDB and may not contain much signal on catalytic proficiency, is responsible for generating proteins mainly improved in structural integrity or solubility. If this is so, then initially unstable proteins should benefit most from these approaches and would make promising targets for ZSF machine learning approaches, although other excellent stability-enhancing algorithms already exist.57 However, such an approach will miss out on potentially destabilizing mutations that may nevertheless be crucial for catalytic activation. Mutations at sites in the protein that were often deliberately excluded in these models (first shell residues, conserved residues) will not be suggested. This conservative bias in the designs may decrease the chance to find designs with improved catalysis, and may be overcome by feeding data on directed evolution trajectories (e.g. from droplet screens) into the algorithms. Higher throughput data from catalytic selections (e.g. in microdroplets) may enhance the value of models currently used in ZSF packages. It remains to be seen whether learning input from comprehensive activity screens (Fig. 3) would give less conservative solutions, overcoming a possible learning bias from the preponderance of stable structures in the training data, and allow better extrapolation towards solutions for catalysis beyond the conditio sine qua non of stability.
5.2 Models incorporating assay-labelled data (red in Tables 1–3)
Studies using assay-labelled data are most likely to benefit from larger amounts of screening data, especially when their design space is large. Screening datasets – the larger the better (e.g. obtained in microdroplets) – will drive the success of this area. However, compared to the entirety of possible solutions the “coverage of the problem space” is still small. Focussing on a few sites for randomization may cover a large part of the relevant space, so even when less training data are available, solutions may be found. The number of datapoints used thus far varies between 7 (InnovSar46) and 5000.44 These numbers are small, even when making optimistic assumptions about the hit rates of functional proteins in sequence space. More high-quality data (covering the relevant space through a good amount of diverse datapoints that are individually reliable) might be needed, although no natural threshold for reliable predictions seems to exist (Tables 1–3).
6. Implications and conclusions
More data are always better, but library design, screening technology, labeling method and sequencing approach determine interpretability.‘Smart’ libraries limit the design space to a few randomized residues that can be oversampled, but rely on a reductionist model of protein function that might not reflect reality: mutations far away from the active site and unknown hotspots are often playing unanticipated roles and proteins are typically cooperative (highlighted by the relevance of intra-gene epistasis). ML approaches will play a key role in uncovering these complex higher order phenomena that are often overlooked in traditional experiments. Instead of deep and focused, broad and unbiased coverage of sequence space may be more valuable input data for such ML endeavors.
The experimental approach used for screening determines what type of label can be attached to library members evaluated in a screening experiment. Fully quantitative datasets require cumbersome plate screening or use of high-throughput microfluidic enzyme kinetics (HT-MEK): information on multiple parameters (e.g. activity, specificity, stability) provides excellent input for ML, but the numbers of library members that can be characterized in such detail is practically limited to a few thousands. Higher throughput may aid better predictions, because the increased coverage of sequence space will give ML interpretation and extrapolation a better grounding. Experimental binning of survivors in ultra-high throughput screenings is practically straightforward (e.g. when using FACS25) and provides a ranking based on ‘quantitative categories’. Experimental noise (e.g. overlap of separate bins) may compromise the data quality, but the high throughput and coverage in a microfluidic screen will mitigate this problem to some extent. Binary data, where survivors are merely measured against a threshold activity, avoids possibly experimentally elusive differences between bins and simply labels survivors based on occurrence. Binned and binary data can be obtained straightforwardly in ultrahigh throughput droplet screening, where multimillion membered libraries can be interrogated to come to grips with the combinatorial explosion of higher order interactions. The nature of the quantitative data plays a role: rankings based on lysate assays vs. expression-normalised assays, long-term conversion vs. initial rates, turnover of (undemanding) model substrates vs. (unreactive) natural substrates etc. will be different, so ML interpretations will be biased accordingly. Interpretations of these datasets need to deconvolute the combined effects of stability and activity that contribute differently to the range of quantitative descriptors outlined above. Finally, the experimental approach for sequencing determines the information content further: short reads neglect long-range interactions, but provide deeper information on limited complexity. One objective in this phase of research at the interface of ML and experiment will be to reflect on how these set-up considerations impact interpretations, even though more data must always be best.
Both approaches discussed here, ultrahigh throughput screening and machine learning, have thus far mainly been used as powerful discovery engines of new and improved proteins. To be more than discovery tools, the current challenge is to coordinate the ability of ultrahigh throughput screening to generate large datasets with ML’s potential to read and interpret complex messages, be it on catalysis, molecular recognition or protein evolution. To be useful in this respect, datasets need to be large, well-labelled, diverse and of good quality. Noisy data needs to be paired with robust ML algorithms, to avoid overfitting the noise inherent in the data. Open access protocols for both ML and uHT screening should be made available, to make data compatible and interpretations comparable.
Once the screening/ML interface becomes more established it will be interesting to probe whether alternative models applied to the same dataset lead to similar molecular conclusions: if current predictions already reliably yield robust and stable proteins (e.g. with higher Tm), will the molecular patterns that lead to higher catalytic efficiency also be revealed? The two properties are intertwined (e.g. stability enables catalytic improvement through epistatic interactions) and may be difficult to disaggregate. However, obtaining multiple datasets under different conditions – at varying temperatures or pH or with different substrates – would lead to sequence–function relationships familiar from traditional lower throughput research (e.g. pH-rate profiles, temperature denaturation curves, physical organic analysis of molecular recognition of substrates with varying reactivity or steric requirements), but apply them to many enzyme mutants in one go. If it becomes possible to isolate and understand the molecular responses to such variations, then ML will have made ultrahigh throughput screening a mechanistic tool, able to deal with the challenge of enormous complexity that thus far has made protein engineering more difficult than the original protein engineers envisaged.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors would like to thank Chaitanya Joshi and Charlie Harris for feedback. M. G. holds a Trinity College Benn W Levy studentship, S. V. M. received funding from the UKRI Centre for Doctoral Training in Application of Artificial Intelligence to the Study of Environmental Risks (EP/S022961/1). F. E. H. N. is supported by the European Union through the Marie-Curie network ‘Oligomed’ (956070). F. H. was an ERC Advanced Grant holder (695669).References
- M. Smith, In vitro mutagenesis, Annu. Rev. Genet., 1985, 19(1), 423–462 CrossRef CAS PubMed.
- F. H. Arnold, Innovation by evolution: bringing new chemistry to life (Nobel Lecture), Angew. Chem., Int. Ed., 2019, 58(41), 14420–14426 CrossRef CAS PubMed.
- A. R. Fersht, Structure and Mechanism in Protein Science, Freeman, New York, 1999 Search PubMed.
- X. Pan and T. Kortemme, Recent advances in de novo protein design: Principles, methods, and applications, J. Biol. Chem., 2021, 296, 100558 CrossRef CAS PubMed.
- K. M. Ulmer, Protein engineering, Science, 1983, 219(4585), 666–671 CrossRef CAS PubMed.
- J. R. Knowles, Tinkering with enzymes: what are we learning?, Science, 1987, 236(4806), 1252–1258 CrossRef CAS PubMed.
- C. J. Markin, D. A. Mokhtari, F. Sunden, M. J. Appel, E. Akiva, S. A. Longwell, C. Sabatti, D. Herschlag and P. M. Fordyce, Revealing enzyme functional architecture via high-throughput microfluidic enzyme kinetics, Science, 2021, 373(6553), 391 CrossRef PubMed.
- M. Gantz, S. Neun, E. J. Medcalf, L. D. van Vliet and F. Hollfelder, Ultrahigh-Throughput Enzyme Engineering and Discovery in In Vitro Compartments, Chem. Rev., 2023, 123(9), 5571–5611 CrossRef CAS PubMed.
- J. J. Agresti, E. Antipov, A. R. Abate, K. Ahn, A. C. Rowat, J.-C. Baret, M. Marquez, A. M. Klibanov, A. D. Griffiths and D. A. Weitz, Ultrahigh-throughput screening in drop-based microfluidics for directed evolution, Proc. Natl. Acad. Sci. U. S. A., 2010, 107(9), 4004–4009 CrossRef CAS PubMed.
- J.-C. Baret, O. J. Miller, V. Taly, M. Ryckelynck, A. El-Harrak, L. Frenz, C. Rick, M. L. Samuels, J. B. Hutchison and J. J. Agresti, et al., Fluorescence-activated droplet sorting (FADS): efficient microfluidic cell sorting based on enzymatic activity, Lab Chip, 2009, 9(13), 1850–1858 RSC.
- F. Gielen, R. Hours, S. Emond, M. Fischlechner, U. Schell and F. Hollfelder, Ultrahigh-throughput-directed enzyme evolution by absorbance-activated droplet sorting (AADS), Proc. Natl. Acad. Sci. U. S. A., 2016, 113(47), 7383–7389 CrossRef PubMed.
- E. J. Medcalf, M. Gantz, T. S. Kaminski and F. Hollfelder, Ultra-High-Throughput Absorbance-Activated Droplet Sorting for Enzyme Screening at Kilohertz Frequencies, Anal. Chem., 2023, 95(10), 4597–4604 CrossRef CAS PubMed.
- D. A. Holland-Moritz, M. K. Wismer, B. F. Mann, I. Farasat, P. Devine, E. D. Guetschow, I. Mangion, C. J. Welch, J. C. Moore and S. Sun, et al., Mass Activated Droplet Sorting (MADS) Enables High-Throughput Screening of Enzymatic Reactions at Nanoliter Scale, Angew. Chem., Int. Ed., 2020, 59(11), 4470–4477 CrossRef CAS PubMed.
- A. Zinchenko, S. R. Devenish, B. Kintses, P. Y. Colin, M. Fischlechner and F. Hollfelder, One in a million: flow cytometric sorting of single cell-lysate assays in monodisperse picolitre double emulsion droplets for directed evolution, Anal. Chem., 2014, 86(5), 2526–2533 CrossRef CAS PubMed.
- S. Ladeveze, P. J. Zurek, T. S. Kaminski, S. Emond and F. Hollfelder, Versatile Product Detection via Coupled Assays for Ultrahigh-Throughput Screening of Carbohydrate-Active Enzymes in Microfluidic Droplets, ACS Catal., 2023, 13(15), 10232–10243 CrossRef CAS PubMed.
- R. Scheele, Y. Weber, F. Nintzel, M. Herger, T. S. Kaminski and F. Hollfelder, Ultrahigh throughput evolution of tryptophan synthase in droplets via an aptamer-biosensor, ACS Catal., 2024, 18(8), 6259–6271 CrossRef PubMed.
- M. Penner, O. J. Klein, M. Gantz, S. Boss, P. Barker, P. Dupree and F. Hollfelder, Sub-single-turnover quantification of enzyme catalysis at ultrahigh throughput via a versatile NAD(P)H coupled assay in microdroplets, bioRxiv, 2023, preprint, DOI:10.1101/2023.11.22.568356.
- J. Maynard Smith, Natural selection and the concept of a protein space, Nature, 1970, 225(5232), 563–564 CrossRef PubMed.
- E. Svensson and R. Calsbeek, The Adaptive Landscape in Evolutionary Biology, OUP Oxford, 2012 Search PubMed.
- P. J. Zurek, P. Knyphausen, K. Neufeld, A. Pushpanath and F. Hollfelder, UMI-linked consensus sequencing enables phylogenetic analysis of directed evolution, Nat. Commun., 2020, 11(1), 6023 CrossRef CAS PubMed.
- E. S. Richter, A. Link, J. S. McGrath, R. W. Sparrow, M. Gantz, E. J. Medcalf, F. Hollfelder and T. Franke, Acoustic sorting of microfluidic droplets at kHz rates using optical absorbance, Lab Chip, 2023, 23(1), 195–202 RSC.
- P. A. Romero, T. M. Tran and A. R. Abate, Dissecting enzyme function with microfluidic-based deep mutational scanning, Proc. Natl. Acad. Sci. U. S. A., 2015, 112(23), 7159–7164 CrossRef CAS PubMed.
- P. Y. Colin, B. Kintses, F. Gielen, C. M. Miton, G. Fischer, M. F. Mohamed, M. Hyvonen, D. P. Morgavi, D. B. Janssen and F. Hollfelder, Ultrahigh-throughput discovery of promiscuous enzymes by picodroplet functional metagenomics, Nat. Commun., 2015, 6, 10008 CrossRef CAS PubMed.
- B. Kintses, C. Hein, M. F. Mohamed, M. Fischlechner, F. Courtois, C. Laine and F. Hollfelder, Picoliter cell lysate assays in microfluidic droplet compartments for directed enzyme evolution, Chem. Biol., 2012, 19(8), 1001–1009 CrossRef CAS PubMed.
- R. A. Scheele, L. H. Lindenburg, M. Petek, M. Schober, K. N. Dalby and F. Hollfelder, Droplet-based screening of phosphate transfer catalysis reveals how epistasis shapes MAP kinase interactions with substrates, Nat. Commun., 2022, 13(1), 844 CrossRef CAS PubMed.
- J. D. Schnettler, M. Wang, M. Gantz, A. A. Bunzel, C. Karas, F. Hollfelder and M. H. Hecht, Selection of a Promiscuous Minimalist cAMP Phosphodiesterase from a Library of De Novo Designed Proteins, Nat. Chem., 2024 DOI:10.1038/s41557-024-01490-4.
- L. Lindenburg, T. Huovinen, K. van de Wiel, M. Herger, M. R. Snaith and F. Hollfelder, Split & mix assembly of DNA libraries for ultrahigh throughput on-bead screening of functional proteins, Nucleic Acids Res., 2020, 48(11), e63 CrossRef CAS PubMed.
- A. L. Mitchell, A. Almeida, M. Beracochea, M. Boland, J. Burgin, G. Cochrane, M. R. Crusoe, V. Kale, S. C. Potter and L. J. Richardson, et al., MGnify: the microbiome analysis resource in 2020, Nucleic Acids Res., 2020, 48(D1), D570–D578 CAS.
- C. M. Miton and N. Tokuriki, How mutational epistasis impairs predictability in protein evolution and design, Protein Sci., 2016, 25(7), 1260–1272 CrossRef CAS PubMed.
- S. Neun, P. Brear, E. Campbell, T. Tryfona, K. El Omari, A. Wagner, P. Dupree, M. Hyvonen and F. Hollfelder, Functional metagenomic screening identifies an unexpected beta-glucuronidase, Nat. Chem. Biol., 2022, 18(10), 1096–1103 CrossRef CAS PubMed.
- J. Jumper, R. Evans, A. Pritzel, T. Green, M. Figurnov, O. Ronneberger, K. Tunyasuvunakool, R. Bates, A. Žídek and A. Potapenko, Highly accurate protein structure prediction with AlphaFold, Nature, 2021, 596(7873), 583–589 CrossRef CAS PubMed.
- S. Lauterbach, H. Dienhart, J. Range, S. Malzacher, J. D. Sporing, D. Rother, M. F. Pinto, P. Martins, C. E. Lagerman and A. S. Bommarius, et al., EnzymeML: seamless data flow and modeling of enzymatic data, Nat. Methods, 2023, 20(3), 400–402 CAS.
- J. Kyte and R. F. Doolittle, A simple method for displaying the hydropathic character of a protein, J. Mol. Biol., 1982, 157(1), 105–132 CrossRef CAS PubMed.
- S. Henikoff and J. G. Henikoff, Amino acid substitution matrices from protein blocks, Proc. Natl. Acad. Sci. U. S. A., 1992, 89(22), 10915–10919 CrossRef CAS PubMed.
- A. Vaswani, N. Shazeer, N. Parmar, J. Uszkoreit, L. Jones, A. N. Gomez, Ł. Kaiser and I. Polosukhin, Attention is all you need, Advances in neural information processing systems 30, 2017 Search PubMed.
- T. Brown, B. Mann, N. Ryder, M. Subbiah, J. D. Kaplan, P. Dhariwal, A. Neelakantan, P. Shyam, G. Sastry and A. Askell, Language models are few-shot learners, Adv. Neural Inf. Process. Syst., 2020, 33, 1877–1901 Search PubMed.
- Y. LeCun and Y. Bengio, Convolutional networks for images, speech, and time series, In The Handbook of Brain Theory and Neural Networks, 1998, pp 255–258 Search PubMed.
- J. Stourac, J. Dubrava, M. Musil, J. Horackova, J. Damborsky, S. Mazurenko and D. Bednar, FireProtDB: database of manually curated protein stability data, Nucleic Acids Res., 2021, 49(D1), D319–D324 CrossRef CAS PubMed.
- Z. Lin, H. Akin, R. Rao, B. Hie, Z. Zhu, W. Lu, N. Smetanin, R. Verkuil, O. Kabeli and Y. Shmueli, Evolutionary-scale prediction of atomic-level protein structure with a language model, Science, 2023, 379(6637), 1123–1130 CrossRef CAS PubMed.
- H. Lu, D. J. Diaz, N. J. Czarnecki, C. Zhu, W. Kim, R. Shroff, D. J. Acosta, B. R. Alexander, H. O. Cole and Y. Zhang, et al., Machine learning-aided engineering of hydrolases for PET depolymerization, Nature, 2022, 604(7907), 662–667 CrossRef CAS PubMed.
- J. Dauparas, I. Anishchenko, N. Bennett, H. Bai, R. J. Ragotte, L. F. Milles, B. I. Wicky, A. Courbet, R. J. de Haas and N. Bethel, Robust deep learning–based protein sequence design using ProteinMPNN, Science, 2022, 378(6615), 49–56 CrossRef CAS PubMed.
- K. Tsuboyama, J. Dauparas, J. Chen, E. Laine, Y. Mohseni Behbahani, J. J. Weinstein, N. M. Mangan, S. Ovchinnikov and G. J. Rocklin, Mega-scale experimental analysis of protein folding stability in biology and design, Nature, 2023, 620(7973), 434–444 CrossRef CAS PubMed.
- C. Hsu, H. Nisonoff, C. Fannjiang and J. Listgarten, Learning protein fitness models from evolutionary and assay-labeled data, Nat. Biotechnol., 2022, 40(7), 1114–1122 CrossRef CAS PubMed.
- E. J. Ma, E. Siirola, C. Moore, A. Kummer, M. Stoeckli, M. Faller, C. Bouquet, F. Eggimann, M. Ligibel and D. Huynh, et al., Machine-Directed Evolution of an Imine Reductase for Activity and Stereoselectivity, ACS Catal., 2021, 11(20), 12433–12445 CrossRef CAS.
- D. Repecka, V. Jauniskis, L. Karpus, E. Rembeza, I. Rokaitis, J. Zrimec, S. Poviloniene, A. Laurynenas, S. Viknander and W. Abuajwa, et al., Expanding functional protein sequence spaces using generative adversarial networks, Nat. Mach. Intell., 2021, 3(4), 324–333 CrossRef.
- R. Ostafe, N. Fontaine, D. Frank, M. Ng Fuk Chong, R. Prodanovic, R. Pandjaitan, B. Offmann, F. Cadet and R. Fischer, One-shot optimization of multiple enzyme parameters: Tailoring glucose oxidase for pH and electron mediators, Biotechnol. Bioeng., 2020, 117(1), 17–29 CrossRef CAS PubMed.
- J. Buchler, S. H. Malca, D. Patsch, M. Voss, N. J. Turner, U. T. Bornscheuer, O. Allemann, C. Le Chapelain, A. Lumbroso and O. Loiseleur, et al., Algorithm-aided engineering of aliphatic halogenase WelO5* for the asymmetric late-stage functionalization of soraphens, Nat. Commun., 2022, 13(1), 371 CrossRef CAS PubMed.
- Y. Ogawa, Y. Saito, H. Yamaguchi, Y. Katsuyama and Y. Ohnishi, Engineering the Substrate Specificity of Toluene Degrading Enzyme XylM Using Biosensor XylS and Machine Learning, ACS Synth. Biol., 2023, 12(2), 572–582 CrossRef CAS PubMed.
- Z. Wu, S. J. Kan, R. D. Lewis, B. J. Wittmann and F. H. Arnold, Machine learning-assisted directed protein evolution with combinatorial libraries, Proc. Natl. Acad. Sci. U. S. A., 2019, 116(18), 8852–8858 CrossRef CAS PubMed.
- Y. Wang, H. Tang, L. Huang, L. Pan, L. Yang, H. Yang, F. Mu and M. Yang, Self-play reinforcement learning guides protein engineering, Nat. Mach. Intell., 2023, 5(8), 845–860 CrossRef.
- S. d’Oelsnitz, D. Diaz, D. Acosta, M. Schechter, M. Minus, J. Howard, J. Loy, H. Do, H. S. Alper and A. D. Ellington, Nat. Commun., 2024, 15, 2084 CrossRef PubMed.
- R. Shroff, A. W. Cole, D. J. Diaz, B. R. Morrow, I. Donnell, A. Annapareddy, J. Gollihar, A. D. Ellington and R. Thyer, Discovery of Novel Gain-of-Function Mutations Guided by Structure-Based Deep Learning, ACS Synth. Biol., 2020, 9(11), 2927–2935 CrossRef CAS PubMed.
- S. Biswas, G. Khimulya, E. C. Alley, K. M. Esvelt and G. M. Church, Low-N protein engineering with data-efficient deep learning, Nat. Methods, 2021, 18(4), 389–396 CrossRef CAS PubMed.
- K. H. Sumida, R. Núñez-Franco, I. Kalvet, S. J. Pellock, B. I. Wicky, L. F. Milles, J. Dauparas, J. Wang, Y. Kipnis, N. Jameson and A. Kang, Improving protein expression, stability, and function with ProteinMPNN, J. Am. Chem. Soc., 2024, 146(3), 2054–2061 CrossRef CAS PubMed.
- B. Zhou; L. Zheng; B. Wu; K. Yi; B. Zhong; P. Lio; L. HongConditional Protein Denoising Diffusion Generates Programmable Endonucleases, bioRxiv, 2023, DOI:10.1101/2023.08.10.552783.
- A. Madani, B. Krause, E. R. Greene, S. Subramanian, B. P. Mohr, J. M. Holton, J. L. Olmos, C. Xiong, Z. Z. Sun and R. Socher, Large language models generate functional protein sequences across diverse families, Nat. Biotechnol., 2023, 41(8), 1099–1106 CrossRef CAS PubMed.
- O. Khersonsky, R. Lipsh, Z. Avizemer, Y. Ashani, M. Goldsmith, H. Leader, O. Dym, S. Rogotner, D. L. Trudeau and J. Prilusky, et al., Automated Design of Efficient and Functionally Diverse Enzyme Repertoires, Mol. Cell, 2018, 72(1), 178–186 CrossRef CAS PubMed.
- M. Gantz, V. Mathis, F. E. H. Nintzel, P. J. Zurek, T. Knaus, E. Patel, D. Boros, F.-M. Weberling, M. R. A. Kenneth, O. J. Klein, E. J. Medcalf, J. Moss, M. Herger, T. S. Kaminski, F. G. Mutti, P. Lio, F. Hollfelder, Microdroplet screening rapidly profiles a biocatalyst to enable its AI-assisted engineering, bioRxiv, 2024, preprint, DOI:10.1101/2024.1104.1108.588565.
Footnote |
| † Equal contribution. |
| This journal is © The Royal Society of Chemistry 2024 |