
Towards modulating the colour hues of isoindigo-based electrochromic polymers through variation of thiophene-based donor groups†
Ming Hui
Chua
 a,
Sheng Heng Gerald
Toh
a,
Pin Jin
Ong
a,
Zhuang Mao
Png
a,
Qiang
Zhu
*a,
Shanxin
Xiong
b and
Jianwei
Xu
*ac
a,
Sheng Heng Gerald
Toh
a,
Pin Jin
Ong
a,
Zhuang Mao
Png
a,
Qiang
Zhu
*a,
Shanxin
Xiong
b and
Jianwei
Xu
*ac
aInstitute of Materials Research and Engineering, A*STAR (Agency for Science, Technology and Research), 2 Fusionopolis Way, Innovis, #08-03, 138634 Singapore. E-mail: jw-xu@imre.a-star.edu.sg; zhuq@imre.a-star.edu.sg
bCollege of Chemistry and Chemical Engineering, Xi'an University of Science and Technology, Xi'an 710054, China
cDepartment of Chemistry, National University of Singapore, 3 Science Drive 3, 117543 Singapore
First published on 11th January 2022
Abstract
Conjugated polymers containing isoindigo electron acceptor groups have gained attention for electrochromic (EC) applications in recent years. To obtain a deeper fundamental understanding of the EC properties of isoindigo-based conjugated polymers, a series of six isoindigo-based donor–acceptor alternating conjugated polymers containing thiophene, thieno[3,2-b]thiophene and 3,4-ethylenedioxythiophene (EDOT) donor units were synthesized, and their EC properties were studied. All six polymers revealed cyan green colours in the neutral states and bluish-grey colours in the oxidised states. Colourimetric studies (CIE 1976 L*a*b* colour space) showed that negative a* and b* values of the polymers in the neutral state converged towards the true black position of a*, b* = 0 upon electro-oxidation. An increase in transmissivity as evidenced by an increase in L* values was also observed. Notably, it was found that the hues of both neutral and oxidised colours could be tuned through variation of the numbers and types of donor groups. Furthermore, high colouration efficiency of up 626 cm2 C−1, which is by far the highest amongst all reported isoindigo-based EC polymers, can be achieved in the case of thieno[3,2-b]thiophene as a donor. The structure–property relationships in other EC switching properties such as optical contrasts, switching speeds and switching stabilities were also studied. These findings are believed to be useful towards the future development of better-performing EC polymers based on the isoindigo acceptor and the relevant colour tuning strategies.
1. Introduction
Ever since the first report of electrochromism in WO3 by Deb in 1969,1,2 there has been overwhelming research interest in the development of electrochromic (EC) materials owing to their promising applications in smart windows, optical displays anti-glare rear mirrors, and military camouflage coatings.3,4 The usefulness of such materials arises from the ability to modulate their electronic absorption spectroscopy and colours via applying an eternal electrical bias. For some EC materials, their photoluminescence properties may even be modulated, which is termed electroluminochromism.5,6 To date, different classes of EC materials have been reported and developed,7–9 including metal oxides,10–13 metal complexes,14–19 organic molecules20–24 and organic polymers.25–29 Compared to traditional inorganic EC materials, organic polymers offer the advantages of lower production cost and toxicity, better flexibility, lightweightness, and solution processability.For EC applications, conjugated polymers offer the additional advantage of colour tunability.30–32 This can be generally achieved via the use of different electron acceptor (A) and donor (D) groups, which can influence the frontier molecular orbital energies and optical bandgaps (Eg). In this regard, the conjugation of electron donor and acceptor groups in the polymer backbone creates a push–pull system that narrows the Eg and red-shifts the absorption wavelengths, and the extent of this in general greatly depends on the electron-withdrawing strength of the acceptor groups. A variety of acceptor groups, such as benzazoles, quinoxalines and diketopyrrolopyrroles, have been utilized and reported in EC donor–acceptor conjugated polymers.33–35 To date, there have been hundreds of EC conjugated polymers reported with colours spanning across the entire visible and near infrared (NIR) region.
Isoindigo is a structural isomer of the well-known indigo dye. Since its debut in 2009 as a promising building block for organic photovoltaics (PV) by Reynolds’ group,36,37 isoindigo has emerged as a popular choice of acceptor group in the construction of donor–acceptor type conjugated polymers for optoelectronic applications,38–41 namely organic field effect transistors (FETs) and PVs. Owing to its high electron-withdrawing nature, conjugated co-polymers with a low Eg value can be achieved.42 Isoindigo copolymers often exhibit intense broad absorption in the visible region with large onset absorption wavelengths extending into the NIR region,42 thus making them outstanding candidates for organic PVs with a high open circuit voltage.43–46 Depending on the alkyl side chains functionalized at the two N-positions, isoindigo copolymers also tend to experience substantial inter-chain π–π interactions which facilitate charge mobility in ambient-stable thin film FETs.47–50
Unlike organic FETs and PVs, isoindigo-based polymers have been less studied for EC applications in recent years. Earlier, in 2011, Reynolds’ group reported a homo-polymer of isoindigo as an EC material that switches from green to transmissive upon undergoing electrochemical oxidation.51 Several years later, Zhao et al. synthesized three isoindigo random co-polymers containing thiophene and propylenedioxythiophene (ProDOT) donors via the Stille polymerization, with different feed ratios of isoindigo and ProDOT precursors,52 in which the increasing ProDOT content was found to extend absorption such that black-to-transmissive EC switching can be achieved. Soon after, Xu and Lu et al. investigated the EC properties of isoindigo-bis(3,4-ethylene-dioxythiophene (EDOT)) polymers, prepared by electrochemical homo-polymerization of the D–A–D precursors, deposited over indium titanium oxide (ITO)-glass substrates.53 The polymers reveal green-to-blue EC switching with fast switching times as low as 0.5 s, and a high colouration efficiency of up to 362 cm2 C−1. Thereafter, Wang et al. synthesized a series of four isoindigo D–A co-polymers containing different 1,4-bis(diarylamino)arene donors which were found to exhibit multielectrochromic properties, switching from colourless to green or brown, then blue upon electrochemical oxidation.54 More recently, Zhao et al. successively developed two series of three-component isoindigo-based random co-polymers in which the indolo[3,2-b]carbazole and thiophene-containing polymers were found to exhibit cyan to grey colour switching,55 while the thieno[3,2-b]thiophene and cyclopentadithiophene-containing polymers exhibited a more diverse range of colours from neutral colour greenish-brown to violet and dark red, and changed to different shades of blue upon oxidation, with fast response times of 0.36 to 0.94 s and a high optical contrast of 56.9 to 80.8%.56 Table S2 (ESI†) summarises the key EC parameters of these reported isoindigo D–A polymers.
As isoindigo starts to gain attention as a promising building block for the construction of high performance EC materials, it is important to gain a deeper understanding of the structure–EC property relationships of isoindigo polymers to facilitate the design of better-performing materials. In particular, strategies towards tuning the switching-colours, hues and absorption range of isoindigo-based polymers will be of great importance, especially for the modulation of infrared transmittance in real-life applications such as smart windows for green buildings. While existing reports of isoindigo-based block copolymers provide insights into the potential of isoindigo acceptors in developing EC materials, they may not suffice in facilitating a deeper understanding through studying the EC switching properties of simple alternating polymers. Furthermore, the EC switching properties of these isoindigo-based polymers were studied in the solution electrolyte rather than in the ECD state.
Herein, we studied the EC properties of six simple isoindigo-based alternating copolymers: p(IID-T), p(IID-2T), p(IID-3T), p(IID-TT), p(IID-EDOT) and p(IID-2EDOT), which contains one, two and three thiophene donors, a thieno[3,2-b]thiophene donor, and one and two EDOT donors, respectively. The six organic-soluble polymers, whose chemical structures are shown in Scheme 1, were prepared by a straightforward Suzuki or Stille coupling polymerization between suitable acceptors and donor precursors. To our knowledge, the EC properties of these isoindigo-based polymers have not yet been extensively studied or reported. Even so for polymer p(IID-2EDOT) reported herein, structurally similar poly(bisEDOT-isoindigo)s reported by Xu and Lu et al.53 were prepared by electro-polymerization of monomer precursors on the ITO-glass substrates, which do not offer solution-processability, and subsequent EC switching performed in the solution electrolyte.
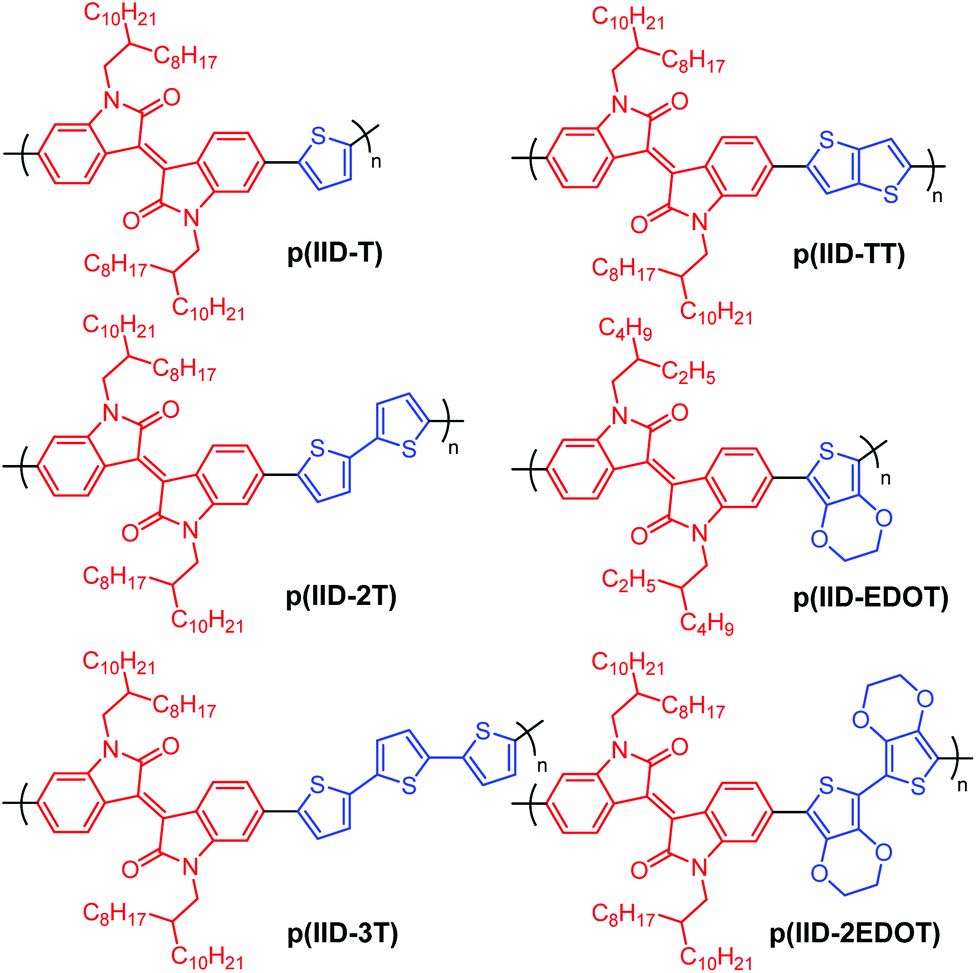 | ||
| Scheme 1 Chemical structures of isoindigo-based polymers synthesized for the study of EC properties. | ||
In this work, spectroelectrochemistry studies were performed on thin film EC devices (ECDs) fabricated from these six polymers, followed by the study of their kinetic EC switching performance to determine their optical contrasts, switching speeds and switching stabilities. Amongst the six polymers, p(IID-TT), bearing a thieno[3,2-b]thiophene donor, was found to exhibit the highest optical contrast (40.5%) and best switching stability (up to 1000 cycles) for the modulation of NIR wavelengths. More importantly, it was observed that by varying the type and number of thiophene-based donor groups used, EC switching colours can be tuned to afford different hues of cyan green neutral-state and bluish-grey oxidised-state colourations. This study is especially important towards the development of high-performance black electrochromes, the absorption of which covers the entire visible region indiscriminant of wavelengths, given the six isoindigo polymers were found to generally converge towards true black in the CIE 1976 L*a*b* colour space upon oxidation, with their bluish-grey oxidised state colourations.
2. Experimental
2.1. Materials and instrumentation
All chemical reagents and solvents used in material synthesis and device fabrication were purchased from commercial suppliers (Sigma Aldrich (Merck), TCI and Alfa Aesar) and used without further purification, unless otherwise stated. ITO coated glass substrates (15 Ω sq−1; 30 mm × 35 mm × 1.1 mm) were obtained from Xin Yan Technology Ltd. They were successively washed with deionised water and acetone and ultra-sonicated in deionised water and acetone, followed by ozone surface treatment (100 °C for 10 minutes) prior to use.1H nuclear magnetic resonance (NMR) spectra of compounds were recorded in deuterated solvent (purchased from Cambridge Isotopes Laboratories) with tetramethylsilane (TMS) as an internal standard, recorded at 25 °C with a Jeol 500 MHz NMR spectrometer. The chemical shifts are expressed with a positive sign, in parts per million (ppm), relative to residual solvent signals as a reference. High resolution mass spectrometry (HRMS) of the synthesized intermediates was performed using an Agilent 7200 GC-QTOF mass spectrometry system. Gel permeation chromatography (GPC) analysis of polymer samples was recorded in THF solutions using an Agilent 1260 Infinity II GPC/SEC system with a refractive index detector, using polystyrene as a standard. The Fourier transform infrared (FTIR) spectra of the polymers were acquired on a PerkinElmer Spectrum 2000 FTIR spectrometer in KBr pellets. The ultraviolet-visible (UV-Vis) absorption spectra of the polymers, as well as spectroelectrochemistry and EC kinetic switching studies of the ECDs were obtained on a Shimadzu UV3600 UV-Vis-NIR spectrophotometer, while electrochemical studies were performed using a Metrohm Autolab PGSTAT128N potentiostat/galvanostat. Colourimetric analysis of the ECDs was conducted using a Hunterlab Ultrascan® Pro colorimeter. Scanning electron microscopy (SEM) of thin films was performed using a Jeol JSM-6700F SEM. The thickness of the spin-coated thin films was measured using a KLA-TENCOR P-10 surface profiler.
2.2. Polymer synthesis
Stepwise synthesis of the six polymers, p(IID-T), p(IID-2T), p(IID-3T), p(IID-TT), p(IID-EDOT) and p(IID-2EDOT), follows the synthetic route with slight modifications of the reported procedures as shown in Scheme 2.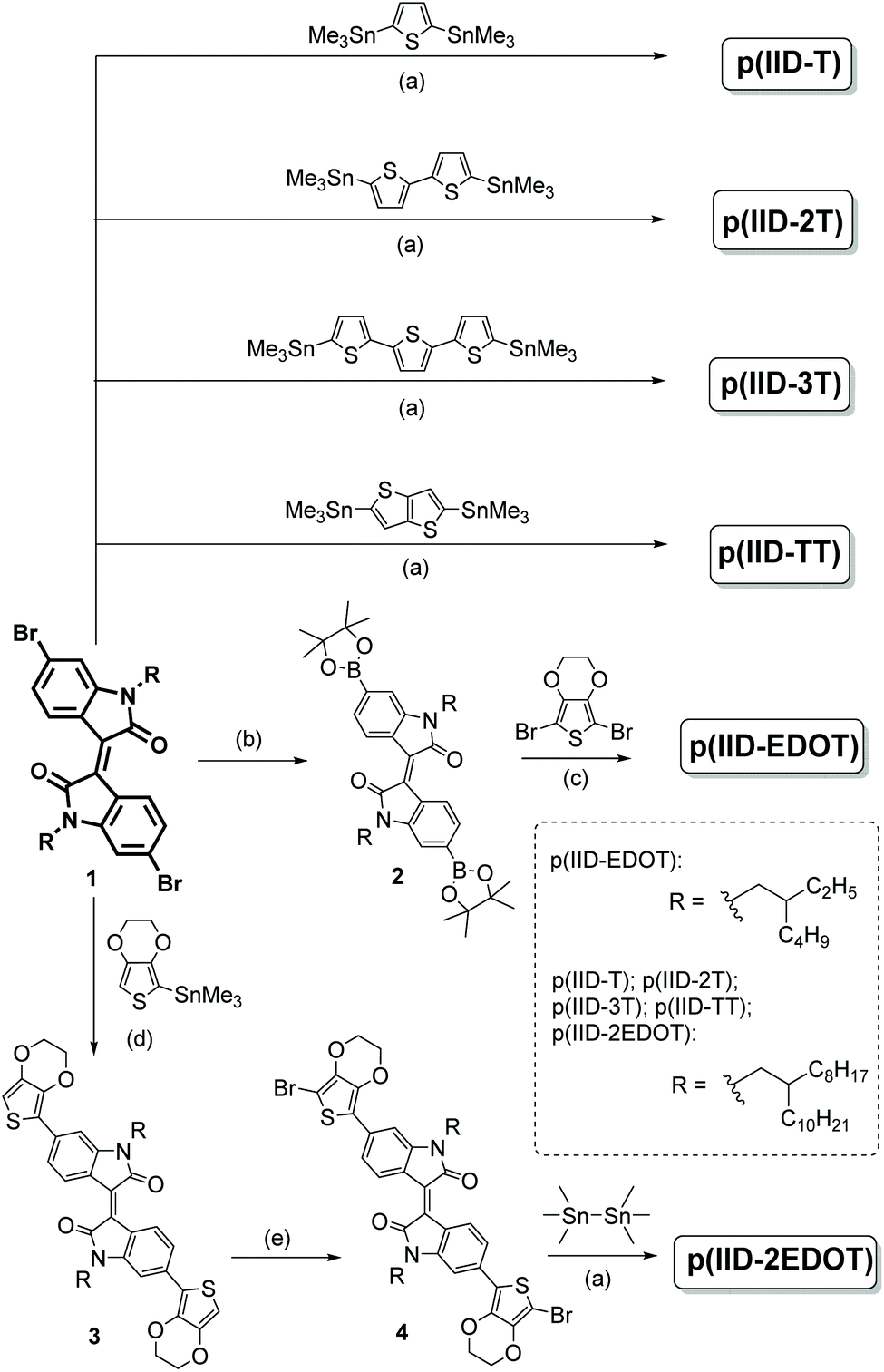 | ||
| Scheme 2 Synthesis of isoindigo-polymers p(IID-T), p(IID-2T), p(IID-3T), p(IID-TT), p(IID-EDOT) and p(IID-2EDOT). Reagents and conditions: (a) Pd(PPh3)4, toluene, 100 °C for 24 h; (b) B2(Pin)2, KOAc, Pd(dppf)Cl2·DCM, 1,4-dioxane, 80 °C for 18 h; (c) Pd(PPh3)4, K2CO3 in H2O, toluene, 85 °C for 72 h (d) Pd(PPh3)4, toluene, 90 °C for 16 h; (e) NBS, THF, 0 °C to r.t. for 24 h. | ||
The 6,6′-dibromoisoindigo precursor was first prepared on a gram scale via a condensation reaction performed in acetic acid under reflux conditions between 6-bromoisatin and 6-bromooxindole. Thereafter, 6,6′-dibromoisoindigo underwent N-alkylation by branched alkyl bromide in the presence of K2CO3 base at 100 °C in DMF. Here, branched alkyl chains, 2-ethylhexyl and 2-octyldodecyl, were chosen to enhance the polymer solubility. The as-prepared N,N′-dialkyl-6,6′-dibromoisoindigo precursor was then used to prepare polymers p(IID-T), p(IID-2T), p(IID-3T) and p(IID-TT) through a direct palladium-catalysed Stille polymerization with commercially available bis(trimethylstannyl) precursors thiophene, bithiophene, terthiophene and thienothiophene, respectively. Pre-functionalized EDOT donors were not commercially available and their preparations were non-trivial due to poor chemical stability. Unfortunately, attempts to prepare 2,5-bis(trimethylstannyl) EDOT from 2,5-dibromo(EDOT) via n-butyllithium lithiation followed by the addition of trimethyltin chloride, were unsuccessful, as mono-stannylated EDOT, 2-trimethylstannyl EDOT, was recovered instead. Therefore, the N,N′-dialkyl-6,6′-dibromoisoindigo precursor underwent a further Miyaura borylation step with bis(pinacolato)diboron catalyzed by Pd(dppf)Cl2·DCM to afford N,N′-dialkyl-6,6′-bis(pinacolboryl)isoindigo, which then underwent palladium-catalysed Suzuki polymerization with 2,5-dibromo(EDOT) to afford polymer p(IID-EDOT). For the preparation of p(IID-2EDOT), N,N′-dialkyl-6,6′-dibromoisoindigo first underwent Stille cross-coupling with 2-trimethylstannyl EDOT to afford the 6,6′-bis(EDOT)isoindigo intermediate, which was then brominated with NBS, and finally, underwent a palladium-catalysed Stille polymerization with hexylmethylditin to give polymer p(IID-2EDOT). All intermediates synthesized in the preparation of polymers were characterized by 1H and 13C NMR and mass spectrometry and the data were found to be in agreement with the reported literature (ESI†). All the six polymers synthesized were purified via Soxhlet extraction. The purified polymers were then characterized by 1H NMR, GPC, FTIR spectroscopy, UV-Vis solution absorption spectroscopy and cyclic voltammetry.
Polymers p(IID-T), p(IID-2T), p(IID-TT), p(IID-EDOT) and p(IID-2EDOT) were synthesized and obtained by Soxhlet extraction with chloroform in good yields of 70.4–99.0%. On the other hand, only a 14.0% yield of p(IID-3T) was extracted from chloroform during Soxhlet extraction, which is understandable given that most of the polymer formed will be much less soluble due to strong inter-chain π–π interactions and lower proportions of alkyl chains to aromatic units. In other words, what was extracted by chloroform during Soxhlet extraction was the more soluble shorter chain polymers of p(IID-3T), as verified by GPC analysis. These are nonetheless useful for us to study their EC properties and establish any possible structure–property relationship with the other five polymers synthesized. Amongst the other five polymers, the two containing EDOT donors p(IID-EDOT) and p(IID-2EDOT) appear to have relatively lower molecular weights as verified by GPC analysis probably due to the adoption polymerization approaches which are different from those of the other four polymers. Typically, Stille and Suzuki polymerization to form D–A type conjugated polymers involves di-brominated acceptor groups and di-stannylated or di-borylated donor groups, which is the case for the synthesis of p(IID-T), p(IID-2T), p(IID-3T) and p(IID-TT). However, due to the non-availability and non-trivial preparation of di-stannylated EDOT donor precursors, different synthetic approaches were adopted. Polymer p(IID-EDOT) was synthesized via a Suzuki polymerization between the relatively more stable dibromo-EDOT donor and di-boronic ester of the isoindigo acceptor, while p(IID-2EDOT) was prepared by using hexylmethylditin as a co-monomer to stannylate in situ and polymerize the dibrominated-bis(EDOT)isoindigo monomer. All six polymers obtained are highly soluble in organic solvents such as chloroform and chlorobenzene, making them solution-processable for the fabrication of EC thin film devices.
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3 v/v) eluent was performed to isolate the product as a deep red solid (100 mg, 87% yield). 1H NMR (500 MHz, CDCl3, δ): 0.88–0.94 (m, 12H), 1.32–1.44 (m + s, 40H), 1.90 (t, J = 5.8 Hz, 2H), 3.64–3.75 (m, 4H), 7.16 (s, 2H), 7.47 (dd, J = 8.0 Hz, 0.8 Hz, 2H), 9.12 (d, J = 8.0 Hz, 2H). 13C NMR (100 MHz, CDCl3, δ): 10.67, 14.11, 23.03, 24.07, 24.91, 28.55, 30.51, 37.41, 44.08, 83.49, 84.04, 113.32, 113.64, 124.19, 128.67, 128.85, 134.31, 144.41, 168.07. HRMS (ESI, m/z): [M + H]+ calculated for C44H65B2N2O6, 739.5023; measured, 739.5026.
:1 v/v to 0:1 v/v) mixture as an eluent to afford a dark purple solid product (227 mg, 81% yield). 1H NMR (500 MHz, CDCl3, δ): 0.84–0.88 (m, 12 H), 1.23–1.34 (m, 64 H), 1.92 (m, J = 2H), 3.69 (d, J = 7.3 Hz, 4H), 4.27 (m, 4H), 4.34 (m, 4H), 6.37 (s, 2H), 7.24 (d, J = 1.5 Hz, 2H), 7.33 (dd, J = 8.5 Hz, 2.7 Hz, 2H), 9.12 (d, J = 8.5 Hz, 2H). 13C NMR (100 MHz, CDCl3, δ): 14.10, 22.65, 22.67, 26.67, 29.30, 29.34, 29.59, 29.62, 29.65, 30.04, 31.81, 31.89, 36.50, 44.37, 64.35, 64.89, 99.21, 105.25, 117.79, 118.97, 120.17, 129.74, 131.69, 136.61, 139.54, 142.37, 145.33, 168.78. HRMS (ESI, m/z): [M]+ calculated for C68H98N2O6S2, 1103.6939; measured, 1103.6935.
:1 v/v as an eluent to afford a dark purple solid product (240 mg, 93% yield). 1H NMR (500 MHz, CD2Cl2, δ): 0.84–0.87 (m, 12 H), 1.23–1.33 (m, 64 H), 1.88 (m, 2H), 3.63 (d, J = 7.3 Hz, 4H), 4.34 (s, 8H), 7.14 (d, J = 1.6 Hz, 2H), 7.19 (dd, J = 8.5 Hz, 1.7 Hz, 2H), 9.08 (d, J = 8.5 Hz, 2H). 13C NMR (100 MHz, CD2Cl2, δ): 14.29, 23.08, 23.10, 27.04, 29.75, 29.77, 30.01, 30.05, 30.10, 30.46, 32.20, 32.30, 32.33, 36.85, 45.59, 65.30, 65.36, 87.85, 105.37, 118.19, 118.64, 120.69, 130.23, 131.85, 136.10, 139.49, 141.28, 145.85, 168.86. HRMS (ESI, m/z): [M]+ calculated for C68H96N2O6S2Br2, 1259.5149; measured, 1259.5141.
:3 v/v) eluent was performed to isolate the product as a deep red solid (100 mg, 87% yield). 1H NMR (500 MHz, CDCl3, δ): 0.88–0.94 (m, 12H), 1.32–1.44 (m + s, 40H), 1.90 (t, J = 5.8 Hz, 2H), 3.64–3.75 (m, 4H), 7.16 (s, 2H), 7.47 (dd, J = 8.0 Hz, 0.8 Hz, 2H), 9.12 (d, J = 8.0 Hz, 2H). 13C NMR (100 MHz, CDCl3, δ): 10.67, 14.11, 23.03, 24.07, 24.91, 28.55, 30.51, 37.41, 44.08, 83.49, 84.04, 113.32, 113.64, 124.19, 128.67, 128.85, 134.31, 144.41, 168.07. HRMS (ESI, m/z): [M + H]+ calculated for C44H65B2N2O6, 739.5023; measured, 739.5026.
:1 v/v to 0:1 v/v) mixture as an eluent to afford a dark purple solid product (227 mg, 81% yield). 1H NMR (500 MHz, CDCl3, δ): 0.84–0.88 (m, 12 H), 1.23–1.34 (m, 64 H), 1.92 (m, J = 2H), 3.69 (d, J = 7.3 Hz, 4H), 4.27 (m, 4H), 4.34 (m, 4H), 6.37 (s, 2H), 7.24 (d, J = 1.5 Hz, 2H), 7.33 (dd, J = 8.5 Hz, 2.7 Hz, 2H), 9.12 (d, J = 8.5 Hz, 2H). 13C NMR (100 MHz, CDCl3, δ): 14.10, 22.65, 22.67, 26.67, 29.30, 29.34, 29.59, 29.62, 29.65, 30.04, 31.81, 31.89, 36.50, 44.37, 64.35, 64.89, 99.21, 105.25, 117.79, 118.97, 120.17, 129.74, 131.69, 136.61, 139.54, 142.37, 145.33, 168.78. HRMS (ESI, m/z): [M]+ calculated for C68H98N2O6S2, 1103.6939; measured, 1103.6935.
:1 v/v as an eluent to afford a dark purple solid product (240 mg, 93% yield). 1H NMR (500 MHz, CD2Cl2, δ): 0.84–0.87 (m, 12 H), 1.23–1.33 (m, 64 H), 1.88 (m, 2H), 3.63 (d, J = 7.3 Hz, 4H), 4.34 (s, 8H), 7.14 (d, J = 1.6 Hz, 2H), 7.19 (dd, J = 8.5 Hz, 1.7 Hz, 2H), 9.08 (d, J = 8.5 Hz, 2H). 13C NMR (100 MHz, CD2Cl2, δ): 14.29, 23.08, 23.10, 27.04, 29.75, 29.77, 30.01, 30.05, 30.10, 30.46, 32.20, 32.30, 32.33, 36.85, 45.59, 65.30, 65.36, 87.85, 105.37, 118.19, 118.64, 120.69, 130.23, 131.85, 136.10, 139.49, 141.28, 145.85, 168.86. HRMS (ESI, m/z): [M]+ calculated for C68H96N2O6S2Br2, 1259.5149; measured, 1259.5141.
General procedure for Stille polymerization. To a flame-dried Schlenk flask connected to the Schlenk line, the dibromide and bis(trimethyltin) monomers were dissolved in anhydrous toluene under an inert gas atmosphere in 1
:1 molar equivalence ratio at a monomer concentration of 10 M. The Pd(PPh3)4 catalyst (0.05 equivalence) was added and the reaction mixture thoroughly degassed by the freeze–pump–thaw method. The reaction mixture was allowed to stir at 100 °C for 24 h, and thereafter upon cooling down to room temperature, the reaction mixture was added dropwise into a large excess of methanol to precipitate out the crude product. The crude product was collected via simple filtration, dried and purified using a Soxhlet extractor setup first by washing with hot methanol, acetone and hexane, and then extracted with chloroform. The solvent was removed using a rotary evaporator and the polymer product was washed with methanol, collected via simple filtration, and finally dried using a vacuum oven.
General procedure for Suzuki polymerization. To a flame dried Schlenk flask connected to the Schlenk line, the dibromide and bis(trimethyltin) monomers (0.5 mmol each) were dissolved in anhydrous toluene (28 mL) under an inert gas atmosphere. Aqueous K2CO3 solution (8.5 mL, 1 M) and Pd(PPh3)4 (0.01 mmol) were added and the mixture was thoroughly degassed via the freeze–pump–thaw method. The reaction mixture was allowed to stir at 85 °C for 72 h. Upon cooling to room temperature, the mixture was partitioned between water and chloroform. The combined organic layer was concentrated using a rotary evaporator, and added into a large excess of methanol to precipitate out the crude product. The crude product was collected via simple filtration, dried and purified using a Soxhlet extractor setup first by washing with hot acetone and hexane, and then extracted with chloroform. The solvent was removed using a rotary evaporator and the polymer product was washed with methanol, collected via simple filtration, and finally dried using a vacuum oven.
p(IID-T) was synthesized via Stille polymerization between N,N′-bis(2-octyldodecyl)-6,6′-dibromoisoindigo and 2,5-bis(trimethylstannyl)thiophene to afford a shiny purple solid (98.0% yield). 1H NMR (500 MHz, CDCl3, δ): 0.85 (m, 12H), 1.11–1.59 (m, broad, 64H), 1.89 (broad, 2H), 3.74 (broad, 4H), 6.67 (broad, 2H), 7.05 (broad, 2H), 7.47 (broad, 2H), 9.04 (broad, 2H). GPC using PS in THF as a standard (Mn = 34652, Mw = 58090, PDI = 1.676). FTIR (KBr): v (cm−1) = 2918 (s), 2849 (s), 1690 (m), 1607 (s), 1457 (m), 1112 (m), 790 (m).
p(IID-2T) was synthesized via Stille polymerization between N,N′-bis(2-octyldodecyl)-6,6′-dibromoisoindigo and 5,5′-bis(trimethylstannyl)-2,2′-bithiophene to afford a shiny purple solid (93.0% yield). 1H NMR (500 MHz, CDCl3, δ): 0.86 (broad, 12H), 1.25 (broad, 64 H), 3.73 (broad, 4H), 6.06–6.66 (m, broad, 8H), 8.62 (m, broad, 2H). GPC using PS in THF as a standard (Mn = 18835, Mw = 74570, PDI = 3.959). FTIR (KBr): v (cm−1) = 2921 (s), 2851 (s), 1687 (s), 1607 (s), 1453 (m), 1358 (m), 1111 (m), 786 (m).
p(IID-3T) was synthesized via Stille polymerization between N,N′-bis(2-octyldodecyl)-6,6′-dibromoisoindigo and 5,5′′-bis(trimethylstannyl)-2,2′:5′,2′′-terthiophene to afford a shiny purple solid (14.0% yield). 1H NMR (500 MHz, CDCl3, δ): 0.86 (s, broad, 12H), 1.25 (broad, 64 H), 1.84 (broad, 2H), 3.67 (broad, 4H), 6.89 (m, broad, 6H), 7.56 (m, broad, 4H), 9.01 (m, broad, 2H). GPC using PS in THF as a standard (Mn = 5098, Mw = 20700, PDI = 4.06). FTIR (KBr): v (cm−1) = 2921 (s), 2850 (s), 1687 (m), 1607 (s), 1456 (m), 1111 (m), 783 (m).
p(IID-TT) was synthesized via Stille polymerization between N,N′-bis(2-octyldodecyl)-6,6′-dibromoisoindigo and 2,5-bis(trimethylstannyl)thieno[3,2-b]thiophene to afford a shiny purple solid (99.0% yield). 1H NMR (500 MHz, CDCl3, δ): 0.84–0.88 (m, broad, 12H), 1.25 (broad, 64H), 4.02 (broad, 4H), 6.42 (broad, 4H), 7.47 (broad, 2H), 8.70 (broad, 2H). GPC using PS in THF as a standard (Mn = 74865, Mw = 225342, PDI = 3.01). FTIR (KBr): v (cm−1) = 2921 (s), 2852 (m), 1687 (m), 1607 (s), 1459 (m), 1112 (s), 801 (m).
p(IID-EDOT) was synthesized via Suzuki polymerization between 2,5-dibromo-3,4-ethylenedioxythiophene and N,N′-bis(2-ethylhexyl)-6,6′-bis(pinacolboryl)isoindigo to afford a shiny purple solid (70.4% yield). 1H NMR (500 MHz, CDCl3, δ): 0.93–0.99 (broad, 12H), 1.37 (broad, 16H), 1.84 (broad, 2H), 3.70 (broad, 4H), 4.44 (broad, 4H), 7.05–7.17 (m, broad, 2H), 7.47 (m, broad, 2H), 9.03–9.09 (m, broad, 2H). GPC using PS in THF as a standard (Mn = 8510, Mw = 31235, PDI = 3.67). FTIR (KBr): v (cm−1) = 2919 (m), 2854 (m), 1687 (s), 1604 (s), 1431 (s), 1351 (s), 1105 (s), 872 (m).
p(IID-2EDOT) was synthesized via Stille polymerization between hexylmethylditin and N,N′-bis(2-octyldodecyl)-6,6′-bis(5-bromo-3,4-ethylenedioxythiophen-2-yl)isoindigo to afford a shiny purple solid (90.0% yield). 1H NMR (500 MHz, CDCl3, δ): 0.86 (m, broad, 12H), 1.26 (m, broad, 64 H), 1.92 (broad, 2H), 3.70 (broad, 4H), 4.35–4.43 (m, 8H), 7.34 (m, broad, 2H), 7.47 (m, broad, 2H), 9.12 (broad, 2H). GPC using PS in THF as a standard (Mn = 11088, Mw = 21703, PDI = 1.957). FTIR (KBr): v (cm−1) = 2921 (s), 2851 (s), 1687 (m), 1607 (s), 1450 (s), 1358 (s), 1114 (s), 1082 (s).
2.3. EC device (ECD) fabrication
ITO/glass substrates were cleaned with water and acetone (washing and ultrasonication for 15 minutes) and then pre-treated with ozone (100 °C for 10 minutes) prior to usage. The polymers were dissolved in a chloroform/chlorobenzene (1:1 v/v) mixture at a concentration of ∼10 mg mL−1, and then filtered over a 0.45 μm PVDF filter. They were then deposited onto the ITO glass substrates by spin-coating ∼150 μL of polymer solution at 500 rpm for 30 s using a Laurell WS-400-LITE spin-coater. The edges of the spin-coated substrates were briefly cleaned and then were allowed to rest on a hotplate (∼80–85 °C) for a minute. Thereafter, the excessive polymer edges were removed by swabbing using a cotton bud damped with chloroform, to obtain an active area of 2 × 2 cm2. On another clean substrate, 250 μL of the gel electrolyte was pipetted out on a demarcated 2 × 2 cm2 area blocked using Parafilm, and left to dry under ambient conditions for 15 minutes. The gel electrolyte was prepared by dissolving 2.8 g of poly(methyl methacrylate) (Mw = 120000 g mol−1) in 28 mL of anhydrous acetonitrile with 0.512 g of lithium perchlorate (LiClO4) and 6.65 ml of propylene carbonate. The ECD was then assembled by sandwiching the polymer-coated substrate with the gel electrolyte-deposited substrate with approximately 0.3–0.5 cm of uncontacted edges on the left and right sides for electrical contact. Finally, the sandwiched device was firmly secured with sticky Scotch tape on the top and bottom edges.
3. Results and discussion
3.1. Optical and electrochemical properties
The solution and thin film absorption spectra of the six isoindigo polymers synthesized, p(IID-T), p(IID-2T), p(IID-3T), p(IID-TT), p(IID-EDOT) and p(IID-2EDOT), are shown in Fig. S1 (ESI†) and their absorption maximum wavelength (λabs) and onset wavelength (λonset), and calculated optical Eg (Eg = 1240/λonset) values in both solution and thin film states are summarized in Table 1. Generally, all six polymers exhibit a broad absorption band across the visible region and towards the far-red region. Each of them displays an intense lower energy absorption band with λabs between 600 and 800 nm, attributed to intramolecular charge transfer (ICT), and a less intense higher energy absorption band with λabs between 350 and 500 nm corresponding to the π–π transition. The positions of solution and thin film λabs were fairly consistent across all six polymers with only a slight blue-shift of the absorption λmax observed for p(IID-2T), p(IID-TT) and p(IID-EDOT), which could be attributed to the H-aggregation of polymer chains. It is also noted that the absorption spectra in thin films generally appear more resolved than that in solution, which could be attributed to the restriction of intramolecular motion. The λonset values of p(IID-T) and p(IID-EDOT) were slightly higher in the thin film than in solution, whereas the λonset values of p(IID-2T), p(IID-3T) and p(IID-TT) were slightly lower. Nonetheless, the calculated optical Eg values of the six polymers were fairly consistent between the solution and thin film states.| UV-Vis-NIR absorption spectroscopy | Cyclic voltammetry | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| λ Solnabs (nm) | λ Filmabs (nm) | ε (cm−1 M−1) | λ SolnOnset (nm) | E Opt, Solng (eV)b | λ FilmOnset (nm) |
E
Opt, filmgb (eV) |
E
Oxdc (eV) |
E HOMO (eV) | E LUMO (EV) |
E
elecgd (eV) |
|
| a Molar extinction coefficient. b Optical bandgap = 1240/λonset. c First oxidation potential. d Electrochemical bandgap = ELUMO − EHOMO. | |||||||||||
| p(IID-T) | 690 | 640, 690 | 29725 |
752 | 1.65 | 764 | 1.62 | 1.25 | −5.46 | −3.83 | 1.63 |
| p(IID-2T) | 645, 701 | 630, 692 | 47738 |
762 | 1.63 | 754 | 1.64 | 1.05 | −5.34 | −3.86 | 1.48 |
| p(IID-3T) | 630 | 625, 685 | 23373 |
764 | 1.62 | 756 | 1.64 | 1.02 | −5.38 | −3.91 | 1.47 |
| p(IID-TT) | 720 | 648, 713 | 39009 |
780 | 1.59 | 775 | 1.60 | 1.23 | −5.28 | −3.83 | 1.45 |
| p(IID-EDOT) | 700 | 675 | 53536 |
810 | 1.53 | 833 | 1.49 | 0.61 | −5.27 | −3.93 | 1.34 |
| p(IID-2EDOT) | 741 | 740 | 43163 |
937 | 1.32 | 935 | 1.32 | 0.60 | −5.06 | −4.05 | 1.01 |
As the number of thiophene donors increases from one to three, there is only a slight increase in solution λabs and λonset of p(IID-T), p(IID-2T), p(IID-3T), with only modest changes in optical Eg values. The relative intensities of lower-energy ICT peaks with respect to higher-energy π–π transition peaks decrease as the number of thiophene donors increases. Replacing the thiophene donor with thieno[3,2-b]thiophene led to a larger magnitude red-shift of λabs and λonset for p(IID-TT), corresponding to a smaller optical Eg. This is attributed to thieno[3,2-b]thiophene possessing a more extended π-conjugation than thiophene, as well as conferring greater structural co-planarity to the polymer. With the functionalization of the electron-rich ethylenedioxy group, EDOT is known to be a much stronger electron donor compared to thiophene. Polymers p(IID-EDOT) and p(IID-2EDOT) therefore have more red-shifted λabs and λonset values, and much smaller optical Eg values than p(IID-T) and p(IID-2T). While the red-shift in λmax from p(IID-T) to p(IID-EDOT) is modest in both solution and thin film states, probably due to the former having significantly larger Mn than the latter. p(IID-2EDOT) possesses the longest λmax and λonset values of 740 and 935 nm, respectively, and the narrowest optical Eg value of 1.32 eV amongst the six polymers. Similarly, the relative intensities of the ICT band with respect to the π–π transition band is larger for p(IID-EDOT) than p(IID-2EDOT) due to an increase in the number of EDOT donors, and the ICT absorption band of p(IID-2EDOT) is broader than that of p(IID-EDOT). The optical values derived herein for the six polymers are generally consistent with the reported values of polymers with similar structures, as summarized in Table S1, ESI.†
Cyclic voltammetry (CV) of the six polymers was performed with a three-electrode cell configuration (polymer-coated glassy carbon working electrode, platinum wire counter electrode and silver wire as a pseudo reference electrode) in a 0.1 M solution of LiClO4 in anhydrous acetonitrile and the measurements were calibrated against the ferrocene reference (Fc/Fc+). The cyclic voltammograms, measured at a scan rate of 50 mV s−1, are shown in Fig. S2 (ESI†) and the derived first oxidation potentials (EOxd), HOMO and LUMO energies, and electrochemical Eg are summarized in Table 1. As the isoindigo acceptor group is the same for the six polymers, the LUMO energy levels vary in the small range of −3.83 to −4.05 eV, which is rather close to and consistent with the reported values of −3.70 to −3.95 eV for structurally analogous isoindigo polymers (Table S1, ESI†). Likewise, the HOMO energy levels of the six polymers, which range from −5.46 to −5.06 eV, and correspond to the strength of the electron-donor groups with one thiophene being the weakest (hence lowest HOMO) and two EDOT the strongest (hence highest HOMO). Correspondingly, most of these values are also close to and consistent with the reported HOMO energies of structurally similar polymers (Table S1, ESI†). Similarly, the oxidation potentials of the polymers also correspond to their electron-donor strengths. The EOxd values decrease with an increase in the number of thiophene moieties from p(IID-T) to p(IID-3T), and polymers with EDOT moieties have significantly lower EOxd values than polymers with thiophene donors. Meanwhile, the EOxd value of p(IID-TT) appears comparable with that of p(IID-T). Consequently, the electrochemical Egs of the six polymers share a similar trend to that observed of the optical Eg derived from absorption onset wavelengths, with the Eg value of polymer p(IID-T) being the biggest and that of polymer p(IID-2EDOT) being the narrowest. With the exception of polymer p(IID-T), the electrochemical Egs of the remaining five polymers are generally smaller than their respective optical Eg values.
3.2. Film thickness and surface morphology
Using a surface profiler, the thicknesses of spin-coated thin films of polymers p(IID-T), p(IID-2T), p(IID-TT), p(IID-EDOT) and p(IID-2EDOT) were determined to be approximately 65, 161, 82, 156, 131 and 90 nm, respectively. Meanwhile, the surface morphologies of the spin-coated thin films for use in the fabrication of ECDs were also studied via SEM (Fig. S3 and S4, ESI†). Thin films of all six polymers in general appear to be rather homogeneous based on the images obtained on a 1 μm scale (Fig. S3†) A relatively large concentration of spherical nanoparticles can be seen homogeneously distributed over the surface of the polymer p(IID-T) film which may affect charge migration and hence the EC switching performance of the polymer. On the other hand, random spherical particles can be seen occasionally appearing in the films of polymers p(IID-2T) and p(IID-3T). On focusing-in further, SEM images of a 100 nm scale (Fig. S4†) reveal densely packed structures with regular cracks that are homogeneously distributed over the surfaces. The compactness of the film might have contributed to the less efficient inter-chain and interlayer charge transfer during electrochemical redox processes which may be the cause of long EC switching response times generally across all six polymers.57,583.3. Spectroelectrochemistry
The changes in the absorption spectra of the six polymers in response to electrochemical doping were studied with their respective thin film ECDs. The absorption spectra from 330 to 1600 nm were recorded using a UV-Vis-NIR absorption spectrophotometer in reference to a blank ECD containing only a gel electrolyte, and an external electrical potential was applied via simple wire connection from the ECDs to an external potentiostat. All six isoindigo polymers exhibit a strong ICT absorption band in the visible to far-red region in the neutral state between 500 and 800 nm, with that of p(IID-2EDOT) extending into the NIR region at ∼950 nm. Polymers containing thiophene and thieno[3,2-b]thiophene donors, p(IID-T), p(IID-2T), p(IID-3T) and p(IID-TT), display two distinct absorption peaks in this region at 640 and 690 nm, 630 and 692 nm, 625 and 685 nm, and 648 and 713 nm, respectively, in addition to the lower intensity π–π transition absorption peaks below 500 nm (at 475, 415, 435 and 380 nm, respectively). On the other hand, the two polymers, p(IID-EDOT) and p(IID-2EDOT), which contain EDOT donors display intense ICT absorption peaks at 675 and 740 nm, respectively, in addition to lower intensity π–π* transition absorption peaks at 470 and 419 nm, respectively, in the neutral state.The application of a positive electrical bias, which causes electrochemical oxidative doping, led to changes in the absorption spectra of all six polymers, as shown in Fig. 1. The application of a negative electrical bias, however, did not lead to any significant changes in their absorption spectra (Fig. S5, ESI†). This reflects the cathodically colouring nature of all six isoindigo polymers, which is consistent with other reported examples of isoindigo EC polymers. In general, the electrochemical oxidation of all six polymers led to the gradual collapse of both the intense ICT absorption bands between 500 and 950 nm, and the less intense π–π transition absorption bands between 350 and 500 nm. Concurrently, this led to the emergence of new absorption bands in the NIR region due to the formation of cationic polarons and bipolarons. The first set of newly emerged NIR absorption peaks for polymers p(IID-T), p(IID-2T), p(IID-3T), p(IID-TT), p(IID-EDOT) and p(IID-2EDOT) are located at 975, 900, 850, 920, 953 and 955 nm, respectively. It is also evident from Fig. 1 that another set of longer-wavelength NIR absorption bands emerge as well, with peak positions being located beyond 1600 nm, the absorbance intensities of which correspond to the increasing number of thiophene and EDOT donors across p(IID-T) to p(IID-3T) and p(IID-EDOT) to p(IID-2EDOT), respectively. Compared to the neutral absorption bands, these cationic absorption bands appear to be of lower intensity but much more broadened in nature (Table 1). Within the recorded absorption region of 330 to 1600 nm, the isosbestic points of polymers p(IID-T), p(IID-2T), p(IID-3T), p(IID-TT) and p(IID-EDOT) were located at 750, 735, 710, 760 and 800, respectively, whereas the isosbestic points of p(IID-2EDOT) were located at 473, 560 and 875 nm.
 | ||
| Fig. 1 Spectroelectrochemistry of isoindigo polymers (a) p(IID-T), (b) p(IID-2T), (c) p(IID-3T), (d) p(IID-TT), (e) p(IID-EDOT) and (f) p(IID-2EDOT) in fabricated ECDs, undergoing electrochemical oxidative doping. | ||
The onset of changes to the absorption spectra began at +1.2, +1.0 and +0.8 V applied potentials for polymers p(IID-T), p(IID-2T) and p(IID-3T), respectively, reflecting an increasing ease of oxidation with an increase in the number of thiophene donors. Likewise, the newly emerged NIR absorption peaks at 975, 900 and 850 nm reached maximum intensities at +2.1, +1.9 and +1.7 V for the three polymers, respectively, before starting to collapse when the applied potential was further increased. A trend in the blue-shift of the absorption maxima of the oxidised-state NIR absorption peak with an increasing number of thiophene donors is also evident herein. Changes to the absorption spectra of p(IID-TT) started and saturated at applied potentials of +1.0 and +1.9 V, respectively, suggesting an ease of oxidation somewhat similar to that of p(IID-2T). Meanwhile, changes to the absorption spectra of polymers p(IID-EDOT) and p(IID-2EDOT) started as soon as upon application of +0.2 V potential, before the newly emerged NIR absorption bands reached maximum intensities at +2.0 and +1.6 V, respectively, and thereafter started to collapse as the applied potential continued to increase. Compared to p(IID-T) and p(IID-2T), the use of electron-richer EDOT donors led to an increase in HOMO levels and narrowing of Eg values, causing polymers p(IID-EDOT) and p(IID-2EDOT) to be more easily oxidised. The effect of an increasing number of EDOT donors on increasing the ease of oxidation also appears to be more prominent herein as compared to that of thiophene donors as discussed above. The ease of oxidation observed herein can also be reflected by Eoxd of the respective polymers as reflected in Table 1.
3.4. Colourimetric studies
Tuning EC polymers’ switching colours can be achieved through rational structural design. The effects of different donor groups on the colourimetric properties of the six polymers are therefore studied herein. Colourimetric analysis of the polymers in the neutral and oxidised states was performed using a colorimeter to determine their colour coordinates in the CIE 1976 L*a*b* colour space. For this colour scale, L* defines the lightness of the colour on a scale of 100 (darkest black) to 0 (brightest white), a* reflects the extent of redness (positive) to greenness (negative), and b* represents the extent of yellowness (positive) to blueness (negative) of the colour. The colour changes and the L*, a* and b* values for each polymer in the neutral and oxidised states are shown in Fig. 2a, and the 2-dimensional colour plot of the opponent-colour scales (a* vs. b*) is shown in Fig. 2b. All values are reported under outdoor daylight illumination (65/10°). The six polymers generally reveal cyan-green to bluish-grey colour changes upon undergoing electro-oxidation during EC switching (Fig. 2a). It can be clearly seen that the polymers reveal different hues of cyan and green in the neutral state and likewise, different hues of grey in the oxidised state.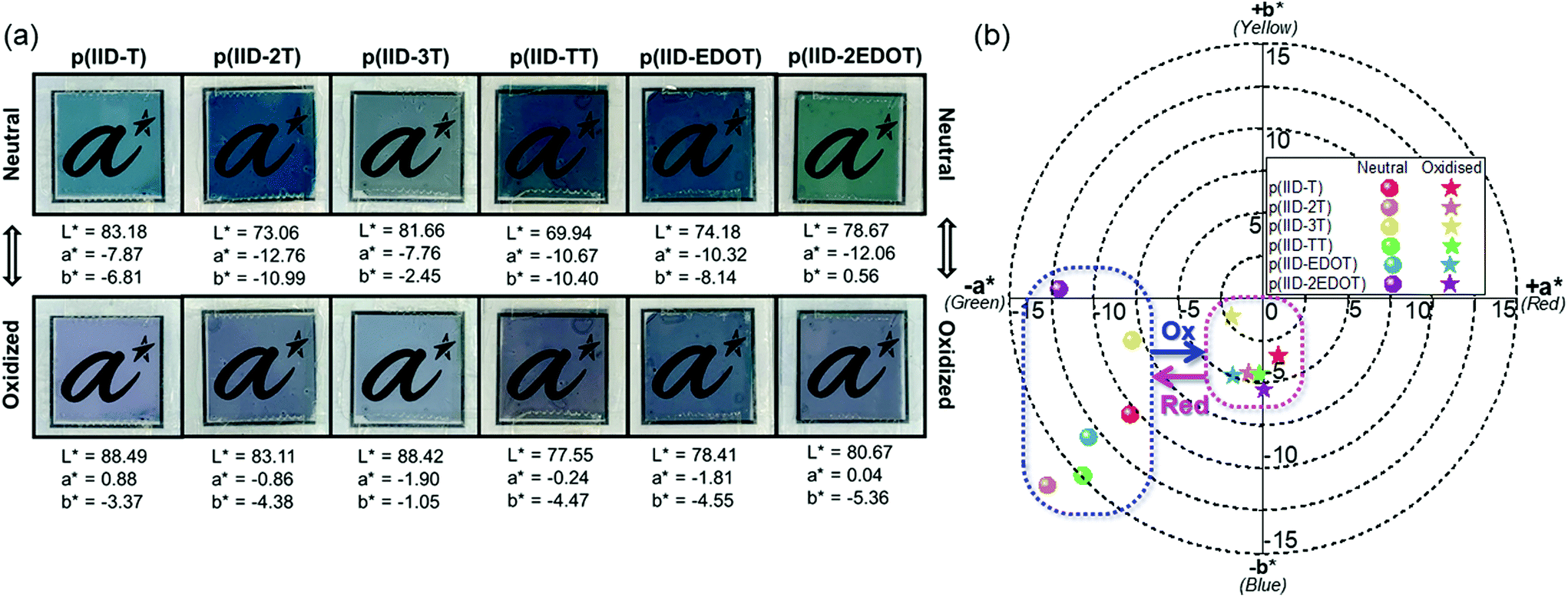 | ||
| Fig. 2 (a) Photos of the fabricated ECD polymers p(IID-T), p(IID-2T), p(IID-3T), p(IID-TT), p(IID-EDOT) and p(IID-2EDOT) in the neutral and oxidised states (at +2.0, +2.0, +1.9, +1.9, +1.9 and +1.6 V, respectively), with their respective L*, a* and b* colour coordinates provided. (b) 2D colour chart of a* vs. b* with plots of the six polymers in the neutral and oxidised states. | ||
The darkness/brightness of the colours of EC thin films can be influenced by film thickness and intrinsic molar absorptivity (ε) of the polymers. Herein, the L* values of the six polymers in the neutral state appear to correspond to the film thickness measured (section 3.2), as the thicker EC thin films of p(IID-2T) and p(IID-TT) have lower L* values while the thinner EC thin films of p(IID-T) and p(IID-3T) have relatively higher L* values. The lower L* values of the latter two films were also likely attributed to their lower ε values (Table 1) compared to the other four polymers as well. An overall increase in L* values was observed across all six polymers when they underwent electro-oxidation, as evidenced by an increase in transmissivity and brightness of the thin film colour. The increase in L* values (ΔL*), however, is rather modest with the smallest at +2.00 registered for polymer p(IID-2EDOT) and the largest at +10.05 registered for polymer p(IID-2T). Furthermore, polymers with electron-richer EDOT donors were found to exhibit a smaller increase in L* values compared to those with thieno[3,2-b]thiophene and thiophene donors. The increase in L* values can be rationalized by comparing the absorption spectra of the polymers in their respective neutral and oxidised states (Fig. 1), where the absorption intensities of the former state in the visible region are clearly higher than that of the latter state.
The opponent-colour scale (a* vs. b*) plots of the six polymers shows their respective colours and hues in both the neutral and oxidised state. All six polymers have negative a* values in the neutral state, indicating the general dominance of the green component in their neutral colours. Amongst them, p(IID-2T) has the largest green component with an a* value of −12.76, while p(IID-3T) has the least green component with an a* value of −7.76. All polymers except p(IID-2EDOT) have negative b* values in the neutral state, thus also revealing the presence of the blue component in their neutral colours and effectively placing them in the blue-green (also known as cyan) region of the two-dimensional (2D) colour plot. All six polymers exhibit intense absorption in the yellow to red region (Fig. 1) due to the strong ICT absorption band, thus giving rise to complementary neutral colours of blue and green. The extent of the blueness of their neutral colour may then be influenced by the positions and relative intensities of the lower wavelength π–π* transition absorption bands, which happen to absorb in the violet to blue region. Clearly, there is a trend of increasing intensities of the π–π* absorption band relative to that of the ICT absorption band, with an increase in the number of thiophene and EDOT donors, going from polymer p(IID-T) to p(IID-3T), and from p(IID-EDOT) to p(IID-2EDOT), respectively. As such, both polymers p(IID-3T) and p(IID-2EDOT), having a comparatively higher absorption in the blue region, exhibit the least negative and positive b* values, respectively. Meanwhile, the positions of π–π* absorption bands for polymer p(IID-TT) and p(IID-2T) are rather blue-shifted compared to the other four polymers, effectively absorbing in the violet rather than blue region. As a result, both polymers exhibit relatively low absorption in the blue and green regions, which explains their most negative a* and b* values in the neutral state. Green being one of three primary colours in the RGB colour scheme is necessary to achieve a full colour palette for ECDs according to “colour-mixing theory” principles,59–61 and the findings herein may be helpful in the design of neutral green polymeric electrochromes using the increasingly popular isoindigo group.
Meanwhile, compared to their neutral colours, the colour plots of the six polymers in the oxidised states converged towards the “true black” position at a* = 0 and b* = 0 (Fig. 2b). This is indicative of the different hues of grey colouration revealed by the polymers in the oxidised state. Among the six polymers, the oxidised colour of polymer p(IID-3T) has the least blue component with b* = −1.05, while that of p(IID-2EDOT) has the most blue component with b* = −5.36. Interestingly, the oxidised colour of polymer p(IID-T) has some red component with a positive a* value of +0.88, as compared to that of the other polymers with either close to zero or negative a* values. This is explainable from its absorption spectrum of the fully oxidised polymer, which shows two broad absorption bands of comparable intensities, with one at 975 nm and the other at 550 nm, resulting in lesser absorbance in the red region relative to the green region. It is worth noting that the other fully oxidised polymers reveal a small absorption band in the blue-green region as well, but their relative absorption intensities are much lower compared to the absorption of other parts of the visible region, and thus they did not reveal the red component in their oxidised colours. Given the importance of developing “true” black electrochromes in smart windows for the filtering of visible light indiscriminant of wavelengths,62–65 the bluish-grey oxidised colours revealed herein by the six isoindigo polymers potentially charted a way for isoindigo as an useful building block to develop “true” black electrochromes.
The colour changes of the six polymers were rather close to and consistent with several other reported isoindigo polymers containing thiophene-based donors such as ProDOT, EDOT and cyclopentadithiophene (Table S2, ESI†). Similarly, both reported electropolymerized poly(bisEDOT-isoindigo)s53 and our solution-processed p(IID-2EDOT) exhibit a green-to-blue colour change.
Overall, it has been demonstrated herein that the choice of simple thiophene-based donor groups can affect the hues of both neutral and oxidised colours of isoindigo-based EC polymers, with the effect on the former being more drastic than the latter. The type and number of thiophene-based donors used can determine the λmax, relative intensities and bandwidths of the absorption bands in both the neutral and oxidised states, thus giving rise to the subtle differences in colouration hues observed herein. The results herein can be useful to facilitate colour tuning in future designs of isoindigo-based EC polymers. The results also showed the potential of isoindigo-based polymers in the development of two important types of EC materials – green and black electrochromes. The tuning of the colours, hues and even brightness of the polymers in both neutral and oxidised states may therefore be further achieved through the rational selection of the type and even number of thiophene-based donor groups.
3.5. Electrochromic switching studies
EC switching performances of the six polymers were studied via chronoabsorptometry, where transmittances at particular wavelengths were monitored as a function of time, while stepped potentials are being applied to their ECDs. In accordance with the findings from the spectroelectrochemical studies performed, EC switching studies were performed based on the collapse (bleaching) of the neutral-state absorption band in the visible region, and the emergence (colouration) of the oxidised-state absorption band in the NIR region, upon undergoing electrochemical oxidation. For the former, the wavelengths of study were 690, 690, 625, 710, 675 and 740 nm for polymers p(IID-T), p(IID-2T), p(IID-3T), p(IID-TT), p(IID-EDOT) and p(IID-2EDOT), respectively, whereas for the latter, the wavelengths of study were 975, 900, 850, 920, 950 and 1040 nm, respectively. These wavelengths of study were chosen due to significant differences in the absorption intensities between the neutral and fully oxidised states. Herein, the optical contrast, colouration efficiency, response time and switching stability of the six polymers were determined and studied, and the results are summarised in Table 2.| Polymer | Visible – far red | NIR | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
|
λ
absa (nm) |
Δ%Tb (%) |
τ
bc (s) |
τ
cd (s) |
CEe (cm2 C−1) |
λ
absa (nm) |
Δ%Tb (%) |
τ
bc (s) |
τ
cd (s) |
CEe (cm2 C−1) | |
| a Absorption wavelength of study for EC switching. b Maximum optical contrast. c Bleaching time. d Colouration time. e Colouration efficiency. | ||||||||||
| p(IID-T) | 690 | 22.7 | 27.1 | 2.6 | 336 | 975 | 12.0 | 15.0 | 3.7 | 186 |
| p(IID-2T) | 690 | 33.4 | 18.0 | 3.4 | 412 | 900 | 39.3 | 30.5 | 2.5 | 370 |
| p(IID-3T) | 625 | 16.2 | 26.7 | 1.2 | 143 | 850 | 28.7 | 24.5 | 0.9 | 357 |
| p(IID-TT) | 710 | 36.0 | 15.1 | 3.3 | 73.5 | 920 | 55.7 | 31.7 | 3.6 | 626 |
| p(IID-EDOT) | 675 | 25.2 | 29.0 | 0.5 | 279 | 950 | 39.3 | 20.3 | 0.4 | 77 |
| p(IID-2EDOT) | 740 | 17.8 | 16.0 | 6.1 | 375 | 1040 | 38.7 | 8.7 | 3.7 | 433 |
Generally, the maximum Δ%T values of these six polymers may be influenced not only by their intrinsic absorption properties in both neutral and oxidised states, but also voltages applied and the film thickness. For instance, the Δ%T of p(IID-2EDOT) is lower in both 740 and 1040 nm compared to Δ%T of p(IID-2T) at 690 and 900 nm, which could be attributed to the former having a much thinner film than the latter as mentioned earlier (section 3.2). Similarly, the relatively large maximum Δ%T of polymer p(IID-TT) could also be partially attributed to its largest film thickness of 156 nm.
where Tox and Tred refer to the absolute transmittance (%) in the bleached and coloured states, respectively; q is the charge (in Coulombs, C) injected or ejected during the switching process, and A is the area of the polymer thin film on the ECDs. The six isoindigo polymers generally exhibit moderate to high CE based on the same redox-switching potentials and wavelengths for each polymer stated above. For the EC switching of visible wavelengths, polymer p(IID-2T) exhibits the highest CE of 412 cm2 C−1, followed by that of polymer p(IID-2EDOT) at 375 cm2 C−1. Polymer p(IID-TT) has the lowest CE of 73.5 cm2 C−1. However, for the EC switching of NIR wavelengths, polymer p(IID-TT) achieves the highest CE at 626 cm2 C−1, which is probably the highest CE reported for any isoindigo-based polymers thus far. This is followed by that of polymer p(IID-2EDOT) at 433 cm2 C−1, while polymer p(IID-EDOT) exhibits the lowest CE at 77 cm2 C−1.

 | ||
| Fig. 3 Square-wave potential step absorptiometry of ECDs containing polymers (a) p(IID-T), (b) p(IID-2T), (c) p(IID-3T), (d) p(IID-TT), (e) p(IID-EDOT) and (f) p(IID-2EDOT) undergoing five cycles of EC switching at different voltages at 30 s/10 s oxidation/reduction intervals. | ||
 | ||
| Fig. 4 Optical memory retention of the six polymers at their respective visible/far-red (a) and NIR (b) wavelengths-of-interest as reflected by the changes in normalized transmittance (T) after oxidation, under open-circuit conditions over 200 s. The vertical dotted lines in both graphs at time = 50 s and 250 s indicate the time points where the ECDs were fully charged (oxidised) and subjected to open-circuit conditions and after 200 s. | ||
 | ||
| Fig. 5 Changes in the Δ%Ts of the ECDs bearing the six polymers over the number of EC switching cycles for both visible/far-red (a) and NIR (b) wavelengths of study. | ||
To begin with, the Δ%T value of ECDs bearing polymer p(IID-T) experienced a steady decrease over the course of EC switching cycling, reaching less than 1% at 690 and 1.3% at 1190 nm by the 800th and 750th cycle, respectively. The decrease in Δ%T at 690 nm was gradual up to the 500th cycle followed by a sharper drop, whereas the decrease in Δ%T at 1190 nm was constant after a sharp decrease over the first 50 cycles. Both Tox and Tred of p(IID-T) at 690 nm were found to increase as the number of switching cycles increases, whereas at 975 nm, only Tox increased over the number of cycles while Tred remained relatively constant at ∼94% (Fig. S7†).
ECDs bearing polymer p(IID-2T) also exhibit relatively good EC switching stability at 900 nm. While its Δ%T at 690 nm gradually slides after the 100th cycle, recording an overall 11.2% drop in absolute Δ%T, the Δ%T at 900 nm experienced only a 6.4% decrease after 800 cycles as it stabilised around 33% after the 250th cycle. This translates to retention values of 51.2 and 83.6% of the initial Δ%Ts at 690 and 800 nm, respectively.
Meanwhile, ECDs bearing polymer p(IID-3T) showed a modest decrease in Δ%T of 4.3% at 625 nm but a more drastic decrease in Δ%T of 23.4% at 850 nm after 800 cycles. This translates to the polymer retaining 74.6% of its initial Δ%T at 625 nm but only 14.8% of its initial Δ%T at 850 nm. The decrease in Δ%T at 625 nm appeared to stabilise and taper off after 250 cycles whereas at 850 nm, the drop was more constant, especially after 200 cycles. The Tred value of p(IID-3T) at 850 nm was observed to slightly decrease over the cycling process while the Tox value increased to a larger extent, contributing to a decrease in Δ%T (Fig. S9†).
ECDs bearing polymer p(IID-TT) generally appears to exhibit the best EC switching stability in both visible/far-red and NIR wavelengths. The Δ%T at 710 nm remained relatively constant around 22% even after 800 cycles whereas the Δ%T at 920 nm dropped modestly by 7.4%, translating to a retention of 81.7% of its initial Δ%T at that wavelength.
On the other hand, the Δ%Ts of ECDs bearing polymers p(IID-EDOT) and p(IID-2EDOT) at 675 and 740 nm, respectively, deteriorated at a decreasing rate before becoming relatively constant after the 300th cycle. The decrease was more drastic for p(IID-2EDOT) compared to the p(IID-EDOT), with the former retaining only 32.7%, while the latter retains 57.9% of their initial Δ%T by the 800th cycle. Similar trends of decrease in Δ%Ts were observed for the two polymers at 950 and 1040 nm, respectively. Polymer p(IID-EDOT) suffered a 16.8% drop in absolute Δ%T whereas polymer p(IID-2EDOT) suffered an even larger 28.2% decrease by the 800th cycle, thus effectively retaining only 46.7 and 26.7% of their initial Δ%Ts, respectively. Interestingly, square-wave potential step absorptiometry showed a significant increase in Tred at 740 nm and an increase in Tred at 1040 nm for polymer p(IID-2EDOT) within the first 100 cycles with only a slight change to Tox (Fig. S12†), indicating signs of early polymer degradation. Meanwhile, polymer p(IID-EDOT) exhibits slight changes in Tred over the cycling process for both wavelengths of study, in addition to the moderate attenuation of Tox (Fig. S11†).
Nonetheless, it is also noted that some minor discrepancies in EC switching stabilities were observed between visible/far-red and NIR wavelengths. These may be attributed to several reasons, such as changes in the polymer structure over the course of EC switching, differences in thin-film morphology between ECDs, minor inconsistencies between different ECDs arising from the process of fabrication.
4. Conclusion
In summary, we synthesized six simple isoindigo-based D–A alternating copolymers and studied their EC properties. All six isoindigo polymers reveal neutral-state cyan-green colours of different hues, which change to different hues of bluish-grey upon undergoing electrochemical oxidation. The polymers generally exhibit higher Δ%T for EC switching in the NIR region compared to the visible/far-red region. Colourimetric analysis shows that all six polymers reveal negative a* values in the neutral state. The widely scattered 2D colour plots in the green region converge towards true black (a*, b* = 0) upon oxidation, with all polymers having negative b* values thereafter. The six polymers exhibit moderate to high CEs in both visible/far-red and NIR regions, and they show a much faster colouration time than the bleaching time. Subsequent EC switching stability studies were performed, revealing that p(IID-TT) exhibits the best cycling stability in the visible/far-red wavelength with almost no change in absolute Δ%T at 710 nm. Furthermore, this work demonstrated the ability to tune subtly the hues of both neutral and oxidized state colours of isoindigo-based alternating D–A copolymers. Given the rich green component in neutral colours of isoindigo polymers and the trend of convergence towards true black of their oxidised colours, the isoindigo acceptor is therefore a highly promising building block to develop future green and black electrochromes. Taking into account that the EC properties of isoindigo-based copolymers have not been extensively examined previously in the form of fabricated ECDs, the findings in this work therefore would further validate the usefulness and potential of isoindigo-based EC polymers for practical applications such as EC-based smart windows and smart eyewear.Conflicts of interest
The authors declare no conflicting interests, whether financial or personal, that may exert any form of influence on the results and conclusion of this work.Acknowledgements
The authors would like to acknowledge the A*STAR, SERC Thermoelectric Materials Program (grant numbers: 1527200019 and 1527200021) and the A*STAR 2020 Career Development Fund (grant number: C210112042) for the financial support of this work.References
- S. K. Deb, Appl. Opt., 1969, 8, 192–195 CrossRef PubMed.
- S. K. Deb, Philos. Mag., 1973, 27, 801–822 CrossRef CAS.
- M. H. Chua, T. Tang, K. H. Ong, W. T. Neo and J. W. Xu, in Electrochromic Smart Materials: Fabrication and Applications, The Royal Society of Chemistry, 2019, pp. 1–21, 10.1039/9781788016667-00001.
- J. Kim, M. Rémond, D. Kim, H. Jang and E. Kim, Adv. Mater. Technol., 2020, 5, 1900890 CrossRef CAS.
- M. H. Chua, Q. Zhu, K. W. Shah and J. Xu, Polymers, 2019, 11, 98 CrossRef PubMed.
- G. A. Corrente and A. Beneduci, Adv. Opt. Mater., 2020, 8, 2000887 CrossRef CAS.
- P. R. Somani and S. Radhakrishnan, Mater. Chem. Phys., 2003, 77, 117–133 CrossRef CAS.
- W. T. Neo, M. H. Chua and J. W. Xu, in Electrochromic Smart Materials: Fabrication and Applications, The Royal Society of Chemistry, 2019, pp. 22–50, 10.1039/9781788016667-00022.
- R. J. Mortimer, Annu. Rev. Mater. Res., 2011, 41, 241–268 CrossRef CAS.
- R. Reisfeld, M. Zayat, H. Minti and A. Zastrow, Sol. Energy Mater. Sol. Cells, 1998, 54, 109–120 CrossRef CAS.
- T. Murayama, W. Kuwagata, K. Koike, Y. Harada, S. Sasa, M. Yano, S. Kobayashi and K. Inaba, In Electrochromic properties of single-crystalline tungsten trioxide films grown by molecular beam epitaxy, 2016 IEEE International Meeting for Future of Electron Devices, Kansai (IMFEDK), 23–24 June 2016, 2016, pp. 1–2 Search PubMed.
- W. Wu, M. Wang, J. Ma, Y. Cao and Y. Deng, Adv. Electron. Mater., 2018, 4, 1800185 CrossRef.
- W. C. Dautremont-Smith, Displays, 1982, 3, 3–22 CrossRef CAS.
- R. J. Mortimer and J. R. Reynolds, J. Mater. Chem., 2005, 15, 2226–2233 RSC.
- K. P. Rajan and V. D. Neff, J. Phys. Chem., 1982, 86, 4361–4368 CrossRef CAS.
- M. L'Her, Y. Cozien and J. Courtot-Coupez, J. Electroanal. Chem. Interfacial Electrochem., 1983, 157, 183–187 Search PubMed.
- H. Li and T. F. Guarr, J. Electroanal. Chem. Interfacial Electrochem., 1991, 297, 169–183 CrossRef CAS.
- R. Banasz and M. Wałęsa-Chorab, Coord. Chem. Rev., 2019, 389, 1–18 CrossRef CAS.
- Y. Zhuang, S. Guo, Y. Deng, S. Liu and Q. Zhao, Chem. – Asian J., 2019, 14, 3791–3802 CrossRef CAS PubMed.
- K. Madasamy, D. Velayutham, V. Suryanarayanan, M. Kathiresan and K.-C. Ho, J. Mater. Chem. C, 2019, 7, 4622–4637 RSC.
- Y. Shi, G. Wang, Q. Chen, J. Zheng and C. Xu, Sol. Energy Mater. Sol. Cells, 2020, 208, 110413 CrossRef CAS.
- Y.-M. Zhang, X. Wang, W. Zhang, W. Li, X. Fang, B. Yang, M. Li and S. X.-A. Zhang, Light: Sci. Appl., 2015, 4, e249–e249 CrossRef CAS.
- M. Stolar, Pure Appl. Chem., 2020, 92, 717–731 CrossRef CAS.
- Y. Jiang, Z. Xian, Y. Meng, G. Zhou, C. Cabanetos, J. Roncali, J.-M. Liu and J. Gao, Dyes Pigm., 2019, 162, 697–703 CrossRef CAS.
- W. T. Neo, Q. Ye, S.-J. Chua and J. Xu, J. Mater. Chem. C, 2016, 4, 7364–7376 RSC.
- G. Gunbas and L. Toppare, Chem. Commun., 2012, 48, 1083–1101 RSC.
- T. Abidin, Q. Zhang, K.-L. Wang and D.-J. Liaw, Polymer, 2014, 55, 5293–5304 CrossRef CAS.
- W. T. Neo, Q. Ye, M. H. Chua, Q. Zhu and J. Xu, Macromol. Rapid Commun., 2020, 41, 2000156 CrossRef CAS PubMed.
- H.-J. Yen and G.-S. Liou, Polym. Chem., 2018, 9, 3001–3018 RSC.
- P. M. Beaujuge and J. R. Reynolds, Chem. Rev., 2010, 110, 268–320 CrossRef CAS PubMed.
- C. M. Amb, A. L. Dyer and J. R. Reynolds, Chem. Mater., 2011, 23, 397–415 CrossRef CAS.
- W. T. Neo, L. M. Loo, J. Song, X. Wang, C. M. Cho, H. S. On Chan, Y. Zong and J. Xu, Polym. Chem., 2013, 4, 4663–4675 RSC.
- M. H. Chua, Q. Zhu, T. Tang, K. W. Shah and J. Xu, Sol. Energy Mater. Sol. Cells, 2019, 197, 32–75 CrossRef CAS.
- X. Lv, W. Li, M. Ouyang, Y. Zhang, D. S. Wright and C. Zhang, J. Mater. Chem. C, 2017, 5, 12–28 RSC.
- J. H. L. Ngai, X. Gao and Y. Li, in Electrochromic Smart Materials: Fabrication and Applications, The Royal Society of Chemistry, 2019, pp. 103–128, 10.1039/9781788016667-00103.
- R. Stalder, J. Mei and J. R. Reynolds, Macromolecules, 2010, 43, 8348–8352 CrossRef CAS.
- J. Mei, K. R. Graham, R. Stalder and J. R. Reynolds, Org. Lett., 2010, 12, 660–663 CrossRef CAS PubMed.
- R. Stalder, J. Mei, K. R. Graham, L. A. Estrada and J. R. Reynolds, Chem. Mater., 2014, 26, 664–678 CrossRef CAS.
- X. Wei, W. Zhang and G. Yu, Adv. Funct. Mater., 2021, 31, 2010979 CrossRef CAS.
- C. Lu, H.-C. Chen, W.-T. Chuang, Y.-H. Hsu, W.-C. Chen and P.-T. Chou, Chem. Mater., 2015, 27, 6837–6847 CrossRef CAS.
- P. Deng and Q. Zhang, Polym. Chem., 2014, 5, 3298–3305 RSC.
- T. Lei, J.-Y. Wang and J. Pei, Acc. Chem. Res., 2014, 47, 1117–1126 CrossRef CAS PubMed.
- G. Zhang, Y. Fu, Z. Xie and Q. Zhang, Macromolecules, 2011, 44, 1414–1420 CrossRef CAS.
- L. Zhu, M. Wang, B. Li, C. Jiang and Q. Li, J. Mater. Chem. A, 2016, 4, 16064–16072 RSC.
- L. Fang, Y. Zhou, Y.-X. Yao, Y. Diao, W.-Y. Lee, A. L. Appleton, R. Allen, J. Reinspach, S. C. B. Mannsfeld and Z. Bao, Chem. Mater., 2013, 25, 4874–4880 CrossRef CAS.
- E. Wang, Z. Ma, Z. Zhang, P. Henriksson, O. Inganäs, F. Zhang and M. R. Andersson, Chem. Commun., 2011, 47, 4908–4910 RSC.
- T. Lei, Y. Cao, Y. Fan, C.-J. Liu, S.-C. Yuan and J. Pei, J. Am. Chem. Soc., 2011, 133, 6099–6101 CrossRef CAS PubMed.
- T. Lei, J.-H. Dou and J. Pei, Adv. Mater., 2012, 24, 6457–6461 CrossRef CAS PubMed.
- T. Lei, Y. Cao, X. Zhou, Y. Peng, J. Bian and J. Pei, Chem. Mater., 2012, 24, 1762–1770 CrossRef CAS.
- Y. Wang, E. Zeglio, H. Liao, J. Xu, F. Liu, Z. Li, I. P. Maria, D. Mawad, A. Herland, I. McCulloch and W. Yue, Chem. Mater., 2019, 31, 9797–9806 CrossRef CAS.
- R. Stalder, J. Mei, J. Subbiah, C. Grand, L. A. Estrada, F. So and J. R. Reynolds, Macromolecules, 2011, 44, 6303–6310 CrossRef CAS.
- H. Xie, M. Wang, L. Kong, Y. Zhang, X. Ju and J. Zhao, RSC Adv., 2017, 7, 11840–11851 RSC.
- H. Gu, S. Ming, K. Lin, S. Chen, X. Liu, B. Lu and J. Xu, Electrochim. Acta, 2018, 260, 772–782 CrossRef CAS.
- Q. Lu, X. Zhang, W. Cai, Y. Wang, C. Yang, Y. Chen, W. Zhang, Z. Zhang, H. Niu and W. Wang, Sol. Energy Mater. Sol. Cells, 2019, 200, 109979 CrossRef CAS.
- Y. Zhang, S. Chen, Y. Zhang, H. Du and J. Zhao, Polymers, 2019, 11, 1626 CrossRef PubMed.
- X. Cheng, Y. Ma, X. Ju, W. Zhao, J. Zhao, Q. Li, Z. Sang, H. Du and Y. Zhang, Synth. Met., 2020, 270, 116589 CrossRef CAS.
- W. T. Neo, Q. Ye, Z. Shi, S.-J. Chua and J. Xu, J. Polym. Res., 2018, 25, 68 CrossRef.
- W. T. Neo, Z. Shi, C. M. Cho, S.-J. Chua and J. Xu, ChemPlusChem, 2015, 80, 1298–1305 CrossRef CAS PubMed.
- R. H. Bulloch, J. A. Kerszulis, A. L. Dyer and J. R. Reynolds, ACS Appl. Mater. Interfaces, 2015, 7, 1406–1412 CrossRef CAS PubMed.
- L. You, J. He and J. Mei, Polym. Chem., 2018, 9, 5262–5267 RSC.
- P. M. Beaujuge, S. V. Vasilyeva, D. Y. Liu, S. Ellinger, T. D. McCarley and J. R. Reynolds, Chem. Mater., 2012, 24, 255–268 CrossRef CAS.
- W. T. Neo, C. M. Cho, Z. Shi, S.-J. Chua and J. Xu, J. Mater. Chem. C, 2016, 4, 28–32 RSC.
- C.-Y. Hsu, J. Zhang, T. Sato, S. Moriyama and M. Higuchi, ACS Appl. Mater. Interfaces, 2015, 7, 18266–18272 CrossRef CAS PubMed.
- H.-S. Liu, B.-C. Pan, D.-C. Huang, Y.-R. Kung, C.-M. Leu and G.-S. Liou, NPG Asia Mater., 2017, 9, e388–e388 CrossRef CAS.
- M. D. Hossain, C. Chakraborty, U. Rana, S. Mondal, H.-J. Holdt and M. Higuchi, ACS Appl. Polym. Mater., 2020, 2, 4449–4454 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/d1py01531a |
| This journal is © The Royal Society of Chemistry 2022 |