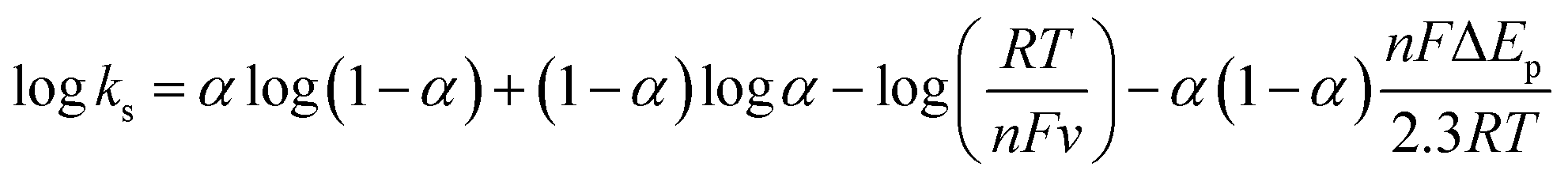DOI:
10.1039/C6RA00405A
(Paper)
RSC Adv., 2016,
6, 31807-31815
H2O/air plasma-functionalized carbon nanotubes decorated with MnO2 for glucose sensing
Received
6th January 2016
, Accepted 22nd March 2016
First published on 24th March 2016
Abstract
Multi-walled carbon nanotubes (MWCNTs) were functionalized using dielectric barrier discharge plasma in water vapor-saturated air at 70 °C. MnO2 was deposited on the MWCNTs by chronoamperometry, followed by glucose oxidase (GOx) immobilization, and the resulting GOx/MnO2/MWCNTs electrode was used for electrochemical detection of glucose. Structural, morphological, and elemental microanalysis was performed. Plasma-induced oxygen-based functional groups were confirmed on the MWCNT surfaces and improved their dispersion in aqueous solutions. The maximum amount of these groups was created at the optimum exposure time of 4 min. The GOx immobilized on the MnO2/MWCNTs hybrid showed a well-defined, reversible and surface-controlled redox wave around −0.45 V and a peak to peak separation of 0.04 V. The coefficient and rate constant for electron transfer of GOx were calculated as 0.41 and 1.08 s−1, respectively. The GOx/MnO2/MWCNT-modified electrode exhibited a linear behavior in the range of 0.1–3.2 mM glucose concentration with the competitive detection limit of 3.0 μM and a sensitivity of 24.2 μA mM−1 cm−2. This highly-stable glucose sensing electrode retained more than 76% of its initial faradic current value after 71 days. These results are relevant to the development of next-generation glucose sensors for diverse health- and food-related applications.
1. Introduction
Detection of the presence and precise measurements of glucose have important roles in numerous fields such as personalized medicine, clinical treatments, bioscience research, and industrial food processing. Although the electrochemical glucose sensors based on glucose oxidase (GOx) show high sensitivity and selectivity, their widespread applications are limited by several factors such as inefficient electron transfer and enzyme instability.1–3
Carbon nanotubes (CNTs) have been reported to enhance direct electron transfer between the active enzyme sites and electrode surfaces.4 Considering chemical stability, high aspect ratio, biocompatibility and good electron transfer rate of CNTs, they may be utilized as support for immobilization of GOx and can also provide a large electroactive surface area.5,6 In order to improve the analytical performance of CNTs-based enzymatic glucose sensors, metal (oxide) nanoparticles (NPs) such as Pd,3 Au,7 Pt5 and zinc oxide4 and bimetallics8 have been frequently used. Among different metal oxides NPs, manganese oxides due to their multiple oxidation states (i.e. MnO, Mn2O3, MnO2, Mn3O4), are promising for glucose biosensors. In particular, MnO2 which shows good electrochemical performance, high surface activity, strong adsorption ability and low toxicity can improve oxidation–reduction process in electrochemical analysis of glucose.9,10 Several methods for the synthesis of MnO2/CNT hybrids, including physical mixing,10 thermal decomposition,11 ball milling,12 electrochemical deposition10 and microwave-assisted reduction method,13 have been reported.
One of the limiting factors for preparing stable and effective metal/CNT hybrids is the poor dispersion ability of CNTs in aqueous solutions, which can be overcome by functionalization with hydrophilic functional groups such as oxygen-containing groups.6 Moreover, these acidic groups can create proper linkers for attachment of metal (oxide) NPs and also provide large capacity for loading different enzymes and improve the interaction between the enzymes and CNTs.14
One of the common methods for functionalization of CNTs is using wet chemical agents such as H2SO4 and HNO3. However, these highly-reactive oxidants may damage the nanotubes, e.g. by creating unwanted defects on their surfaces.15 Due to the reliability, energy efficiency and environmental friendliness, dielectric barrier discharge (DBD) plasmas represent a promising alternative. These plasmas dramatically reduce negative impact of ion bombardment common to many other plasma types and very effectively generate reactive functional groups.16 Precursor gases such as O2,6 H2S17 and NH3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 18 in DBD plasmas produce oxygen-containing, thiol and nitrogen-containing functional groups, respectively. DBD plasmas in water vapor-saturated air can be used to functionalize CNTs with oxygenated groups. In this way, the quantity of the functional groups may be controlled by the plasma parameters, such as the discharge power and exposure time.15
18 in DBD plasmas produce oxygen-containing, thiol and nitrogen-containing functional groups, respectively. DBD plasmas in water vapor-saturated air can be used to functionalize CNTs with oxygenated groups. In this way, the quantity of the functional groups may be controlled by the plasma parameters, such as the discharge power and exposure time.15
In this study, multi-walled carbon nanotubes (MWCNTs) were functionalized by DBD plasma in water vapor-saturated air, in the absence of any oxidizing acids, and the MnO2/MWCNT hybrids were synthesized using an electrochemical deposition method. A glucose biosensor was fabricated by immobilization of GOx on the surface of MnO2/MWCNT electrode and used in electrochemical detection of glucose.
2. Experimental
2.1 MWCNT functionalization
MWCNTs were purchased from Shenzhen Nanotech Port Co. Ltd. The MWCNTs were heated up to 1000 °C in a He atmosphere to eliminate the possible functional groups formed during the synthesis and/or purification steps. Fig. 1a shows the functionalization process carried out using DBD plasma. 40.0 sccm air was saturated with water vapor in a bubbler at 70 °C and passed over MWCNT bed placed in 2.0 mm annular space between a 13.0 mm outer diameter stainless (SS) rod and a 15.0 mm inner diameter quartz tube, on which an SS foil was wrapped. A high voltage of 9.0 kV with 2.5 kHz frequency was applied between the rod and the foil, using PINTEK FG-32 function generator and TREK 10/40A amplifier, to generate the DBD plasma. The more details of this set-up have been reported elsewhere.19 In order to determine the optimum exposure time, the MWCNTs were exposed to plasma at constant power of 30.6 W for different times of 1, 2, 4 and 6 min. The plasma functionalized sample is denoted as F-MWCNTs.
 |
| | Fig. 1 The schematic of process steps for the preparation of GOx/MnO2/MWCNTs/GCE for glucose biosensor; (a) plasma functionalization, (b) electrochemical deposition of MnO2 and (c) immobilization of GOx. | |
2.2 Chronoamperometric deposition of MnO2 on F-MWCNTs
All electrochemical experiments were carried out using PalmSens potentiostat/galvanostat. A three-electrode system including a working bare/modified glassy carbon electrode (GCE), a saturated Ag/AgCl reference electrode, and a Pt wire counter electrode was used at ambient conditions. Prior to use, the GCE was polished with alumina slurry and sonicated in a mixture of deionized water and ethanol. To obtain F-MWCNTs/GCE, the F-MWCNTs were dispersed in 0.5 wt% Nafion aqueous solution and sonicated for 15 min. 10.0 μl of the F-MWCNT suspension (2.0 mg ml−1) was cast onto the surface of the GCE and dried subsequently. The F-MWCNTs/GCE was then immersed in 0.005 M of KMnO4 aqueous solution and the electrodeposition of MnO2 was carried out using chronoamperometry at a constant potential of −1.0 V for different times of 5, 10, 15 and 20 s (Fig. 1b).
The amount of MnO2 deposited on the nanotubes was controlled to obtain a porous nanostructure with a high surface area and charging current. 15 s was found to be an optimum deposition time. The MnO2/MWCNTs/GCE was then washed with deionized water. Afterwards, 5.0 μL of GOx solution (10.0 mg ml−1) was immobilized onto the surface of the MnO2/MWCNTs/GCE and dried at 4 °C (Fig. 1c) for 24 h. All the electrochemical tests were carried out in a phosphate buffer solution (PBS, 0.1 M, pH 7.0). The constructed electrode was stored in PBS at 4 °C when it was not in use. A glucose concentration range of 0.01–6.4 mM in the PBS at −0.55 V vs. Ag/AgCl, was selected for the amperometric detection of glucose. Selectivity of the proposed biosensor to glucose was investigated in presence of ascorbic acid (AA) and uric acid (UA).
2.3 Characterization
Morphology, structure and functional groups of MWCNT samples were studied using field emission scanning electron microscopy (FE-SEM, JEOL JSM-6700F, operating at 5–15 kV), X-ray diffraction (XRD, using Cu Kα radiation), FTIR (Fourier transform infrared spectroscopy, using KBr tablet, Bruker Vector 22 spectrometer), and X-ray photoelectron spectroscopy (XPS, Al Kα, CAE: 150.0 eV). In order to study the dispersion ability behavior of the MWCNT samples, 0.5 mg of the annealed sample and F-MWCNTs were dispersed in 5.0 ml water in an ultrasonic bath (Bandelin) for 10 min.
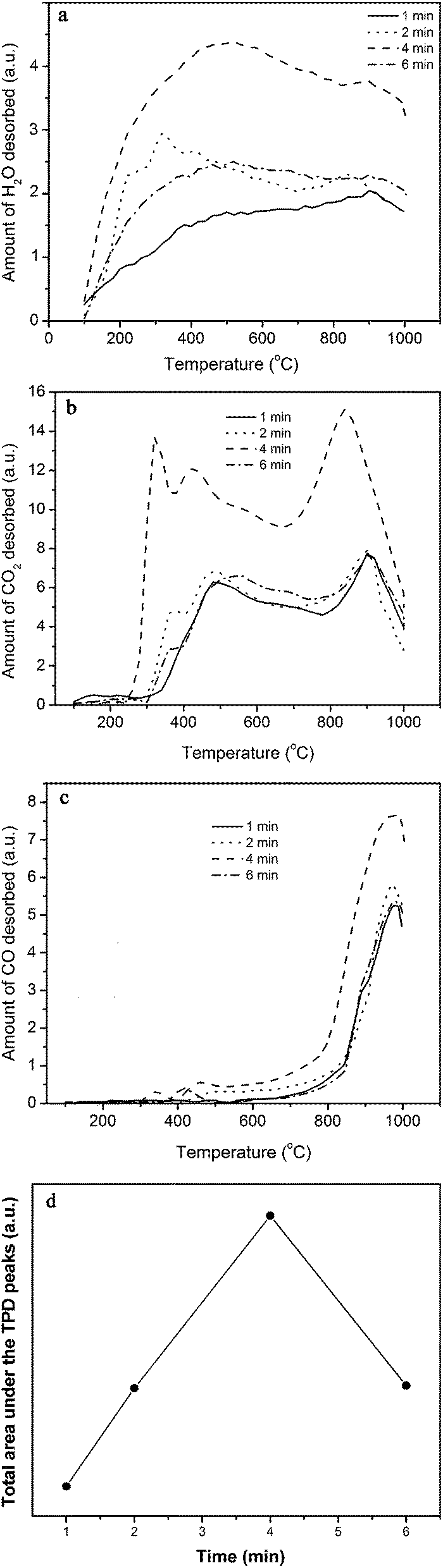
To determine the amount of functional groups generated on the surface of F-MWCNTs, temperature programmed desorption (TPD) technique was employed. In this technique, different F-MWCNT samples were heated with a rate of 10 °C min−1 from 100 up to 1000 °C under a flow of 20 sccm He and the gases evolved were analyzed using an FTIR equipped with a gas cell and/or a thermal conductivity detector (TCD). The FTIR was used in transmission mode with a resolution of 5 cm−1 in the range of 4000 to 400 cm−1. The wave numbers in the range of 2235–2030, 2400–2280 and 3650–3550 cm−1 were attributed to CO, CO2 and H2O, respectively.19
3. Results and discussion
3.1 Characterization
3.1.1 FTIR spectroscopy of F-MWCNT functional groups.
Fig. 2 shows FTIR spectra of the annealed MWCNTs and F-MWCNTs. The peak around 1560 cm−1 (Fig. 2a) is assigned to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C stretching bond.20 After functionalization the intensity of this peak has increased (Fig. 2b), as a result of the attachment of oxygen-containing groups and a decrease of the structure symmetry of carbon. The peaks at 1705 and 3450 cm−1 (Fig. 2b) which are attributed to acidic carbonyl and hydroxyl groups, respectively,20 indicate the formation of carboxyl groups on the surface of CNTs. The peak at 1270 cm−1 is assigned to the acidic C–O bond.21
C stretching bond.20 After functionalization the intensity of this peak has increased (Fig. 2b), as a result of the attachment of oxygen-containing groups and a decrease of the structure symmetry of carbon. The peaks at 1705 and 3450 cm−1 (Fig. 2b) which are attributed to acidic carbonyl and hydroxyl groups, respectively,20 indicate the formation of carboxyl groups on the surface of CNTs. The peak at 1270 cm−1 is assigned to the acidic C–O bond.21
 |
| | Fig. 2 FTIR spectra of (a) annealed MWCNTs and (b) F-MWCNTs. Plasma conditions: power = 30.6 W and exposure time = 4 min. | |
3.1.2 Colloidal dispersion stability.
The aqueous suspensions of annealed and F-MWCNT samples are shown in Fig. 3. The annealed MWCNTs without functionalization were dispersed in water, but precipitated immediately after sonication was stopped (Fig. 3a). The small amounts of oxygen-based groups remained after annealing caused the nanotubes to be soaked. However, generation of the oxygenated groups during water vapor-plasma functionalization makes the MWCNTs negatively charged (zeta potential = −35 mV)22 and leads to the formation of uniform suspension (Fig. 3b) which remained stable even after 6 h (Fig. 3c). These functional groups are polar and can induce hydrogen bonding with the water molecules.
 |
| | Fig. 3 Dispersion of (a) annealed and (b) F-MWCNT samples in H2O immediately after 1 min sonication, (c) stability of F-MWCNTs after 6 h. Plasma conditions: power = 30.6 W, exposure time = 4 min. The concentration of nanotubes is 0.1 mg ml−1. | |
3.1.3 TPD analyses of F-MWCNT functional groups: effects of plasma exposure time.
TPD profiles of water vapor-plasma functionalized samples are shown in Fig. 4. The evolution of H2O (Fig. 4a), which starts from 100 °C and passes through a maximum at around 400 °C, originates from decomposition of carboxyl and phenol groups below and above 400 °C, respectively. Two adjacent carboxyl groups may react during a condensation reaction and form carboxylic anhydride and H2O molecule.23
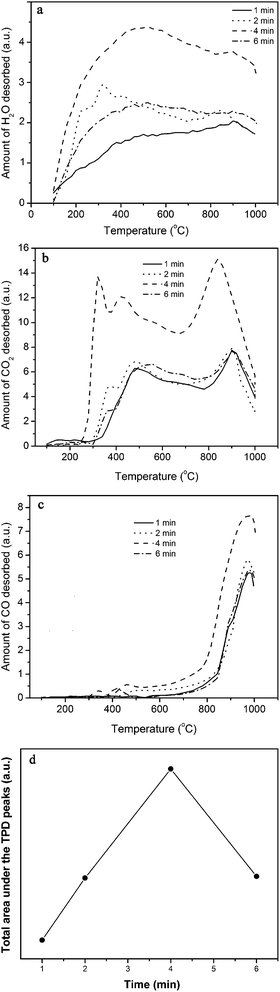 |
| | Fig. 4 (a) H2O, (b) CO2 and (c) CO gas evolution during TPD of the water vapor-plasma functionalized MWCNTs at different exposure times of 1, 2, 4 and 6 min. (d) The effect of the plasma exposure time on the amount of functional groups. Plasma conditions: power = 30.6 W and exposure time = 4 min. | |
The CO2 release (Fig. 4b) initiates at about 250 °C and displays three major peaks around 300, 450 and 850 °C, as a result of decomposition of carboxyl, anhydride and lactone functional groups, respectively.24 The CO evolution begins at around 400 °C and presents a sharp peak at 950 °C. The CO is formed from decomposition of anhydride at a temperature range of 400–600 °C, phenol at 600–700 °C and carbonyl/quinone/ether at 700–1000 °C.24
Similar to the proposed mechanisms for decomposition of carboxyl, carbonyl, ether, lactone and anhydride groups,19 some possible mechanisms for decomposition of other functional groups to H2O, CO2 and CO may be suggested as follows:
| | | 2 phenols → CO + H2O + Ca | (1) |
| | | 2 lactols → 2CO2 + CO + H2O + Ca | (2) |
where C
a denotes an active site on the nanotube surface. The oxygen-based functional groups may rearrange at around 300 °C as follows:
25| | | Carboxyl + phenol → lactone + H2O | (4) |
| | | 2 phenols → ether + H2O | (5) |
| | | Carboxyl + carbonyl → lactol | (6) |
The CO2 content of the evolved gases is higher than their CO content. One possible reason is that the CO molecules upon diffusion through pores of the CNTs may react with the surface oxygen-based groups and are desorbed mainly as CO2.23 In this reaction, the nanotubes with high surface area may act as catalyst. Total amounts of the evolved gases during the TPD of F-MWCNTs, estimated by integration of the TPD profiles measured by TCD (not shown here) are presented in Fig. 4d. The amount of evolved gases is the highest at the exposure time of 4 min. Thus, the sample functionalized for 4 min was selected to synthesize the MnO2/MWCNT hybrid.
3.1.4 XPS analysis of F-MWCNTs and MnO2/MWCNTs.
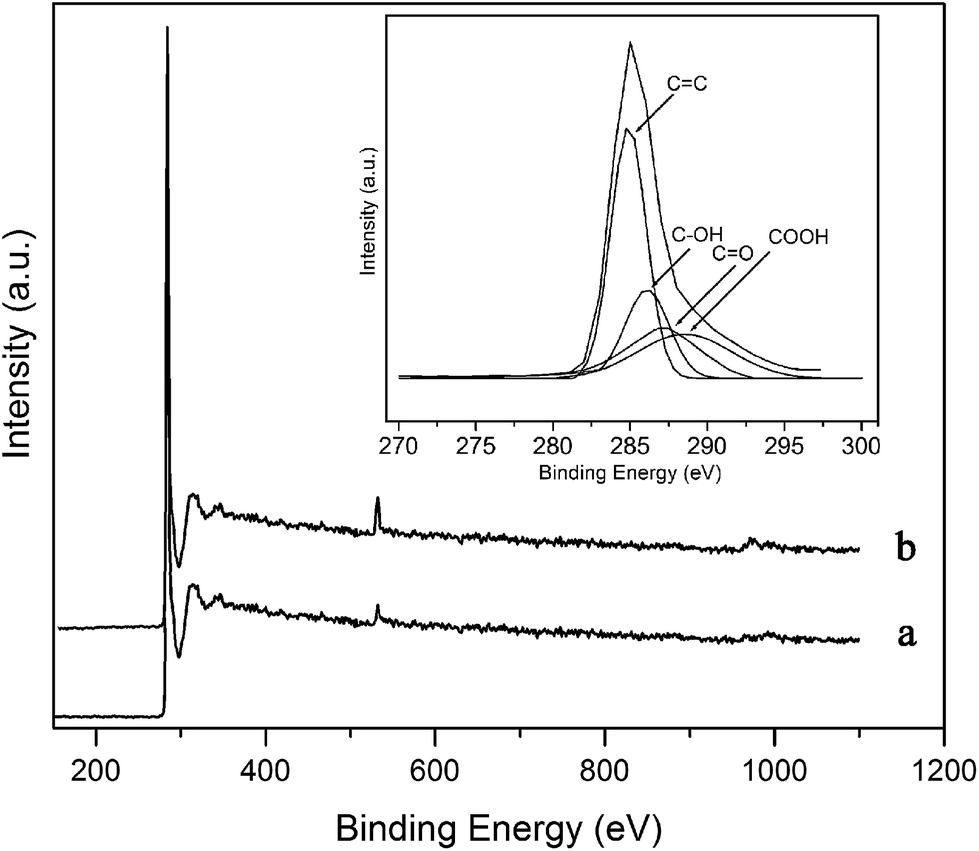
The XPS spectra of the annealed and F-MWCNT samples are presented in Fig. 5. The peak at 533 eV is utilized for calculating the percentage of oxygen atoms.26 This percentage was found to change from 1.4% for the annealed sample to 5.7% for the functionalized one.
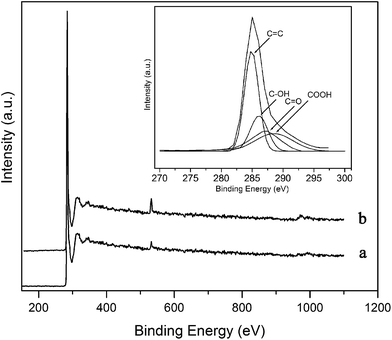 |
| | Fig. 5 XPS spectra of (a) annealed and (b) F-MWCNT samples. Inset: XPS C 1s spectra of F-MWCNTs has deconvoluted to CC, C–OH, CO and COOH peaks at 284.9, 286.3, 287.2 and 288.4 eV. Plasma conditions: power = 30.6 W and exposure time = 4 min. | |
To analyze the chemical bonds of the functional groups, the C 1s spectra is deconvoluted into four component peaks as shown in Fig. 5 inset. The main peak at 284.9 eV is attributed to sp2-carbon atoms of the nanotubes. Two other components centered at 286.3 and 287.2 eV are associated with C–OH and CO bonds, respectively. The peak at 288.4 eV can be assigned to COOH groups.26
Some of the radicals which can react with the opening ends and defect sites of the nanotubes and generate COOH, CO and C–O groups on the nanotube surface may be formed as follows:27
In the XPS spectra of the F-MWCNTs, no nitrogen-containing groups have been observed. Since the dissociation energy of an oxygen molecule is nearly a half of that for a nitrogen molecule and also because the bonding energy of C–O is higher compared to C–N,19 the oxygenated species are more likely to react with carbonaceous atoms. This finding is in agreement with the FTIR results.
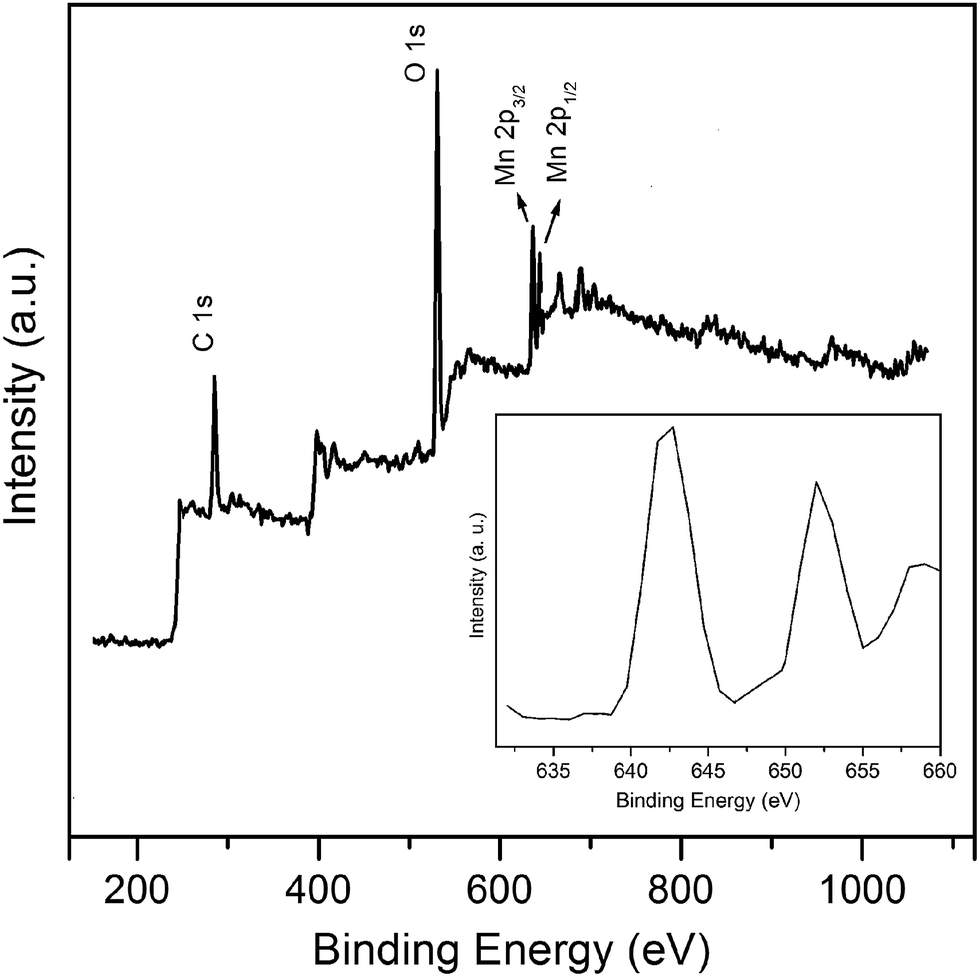
Fig. 6 presents the XPS spectra of the MnO2/MWCNT hybrid. The oxygen atoms percentage has increased to 15.4%. The peaks at 642.5 eV and 653.3 eV are assigned to Mn 2p3/2 and Mn 2p1/2, respectively.28 The percentage of manganese atoms in the MnO2/MWCNT sample was found to be approximately 4.8%.
 |
| | Fig. 6 XPS spectra of MnO2/MWCNTs. Inset: the magnification of Mn 2p region, the peaks at 642.5 and 653.3 eV are assigned to Mn 2p3/2 and Mn 2p1/2. | |
3.1.5 Morphology of the F-MWCNTs and MnO2/MWCNTs.
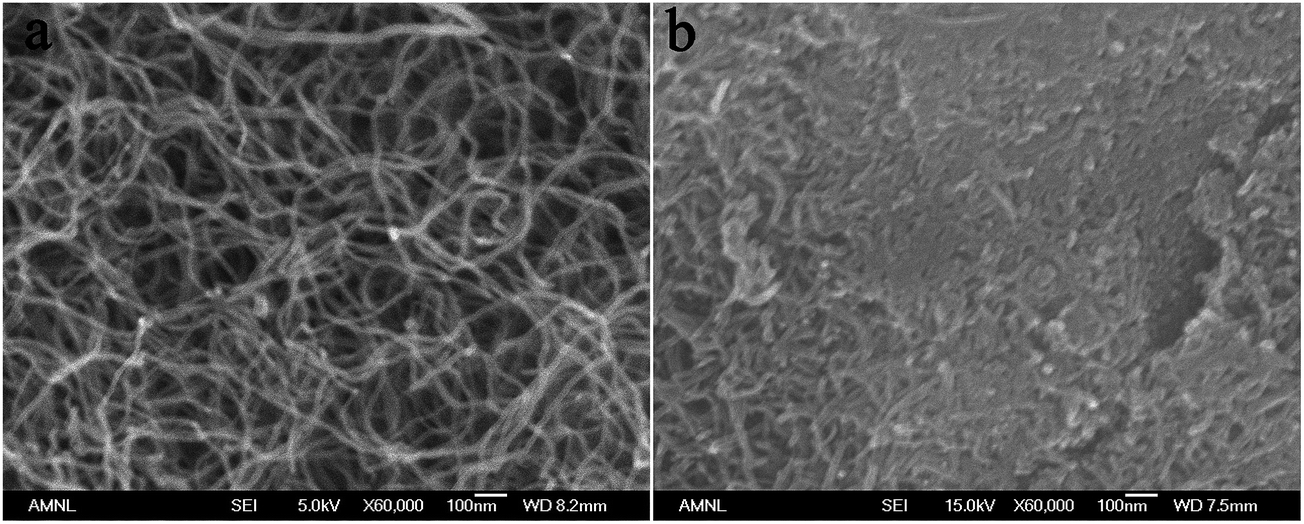
Fig. 7 displays the FE-SEM micrographs of F-MWCNTs and MnO2/MWCNTs. The nanotubes (Fig. 7a) show a twisted and tangled morphology with an average nanotube diameter of approximately 20 nm. The MnO2 which is thermodynamically the most stable structure of the manganese oxide species in aqueous solution has been deposited relatively uniform on the surface of MWCNTs (Fig. 7b) during the electrochemical reduction of MnO4− ions, as follows:29| | | MnO4− + 4H+ + 3e− → MnO2 + 2H2O | (14) |
 |
| | Fig. 7 SEM micrographs of (a) F-MWCNTs and (b) MnO2/MWCNTs. | |
The presence of carbonyl, carboxyl and/or hydroxyl groups on the surface of nanotubes which acts as a substrate, makes it more vulnerable to oxidation and facilitates the electron transfer from nanotubes to MnO4− ions and the deposition of thin layer of MnO2.13
3.1.6 XRD of the F-MWCNTs and MnO2/MWCNTs.
The XRD patterns of F-MWCNTs and MnO2/MWCNTs are presented in Fig. 8. The XRD pattern of F-MWCNTs (Fig. 8a) exhibits a sharp peak at 2θ of 26.35° which is assigned to (002) plane of graphitic carbon. The peaks at 28.9°, 43.4°, 56.9° and 59.1° (Fig. 8b) may be attributed to (110), (111), (211) and (220) planes of crystalline MnO2-NPs, respectively.28
 |
| | Fig. 8 XRD patterns of (a) F-MWCNTs and (b) MnO2/MWCNTs. The peaks at 28.9°, 43.4°, 56.9° and 59.1° are assigned to (110), (111), (211) and (220) planes of MnO2, respectively. | |
3.2 Performance of the glucose sensor
3.2.1 Enzyme immobilization.
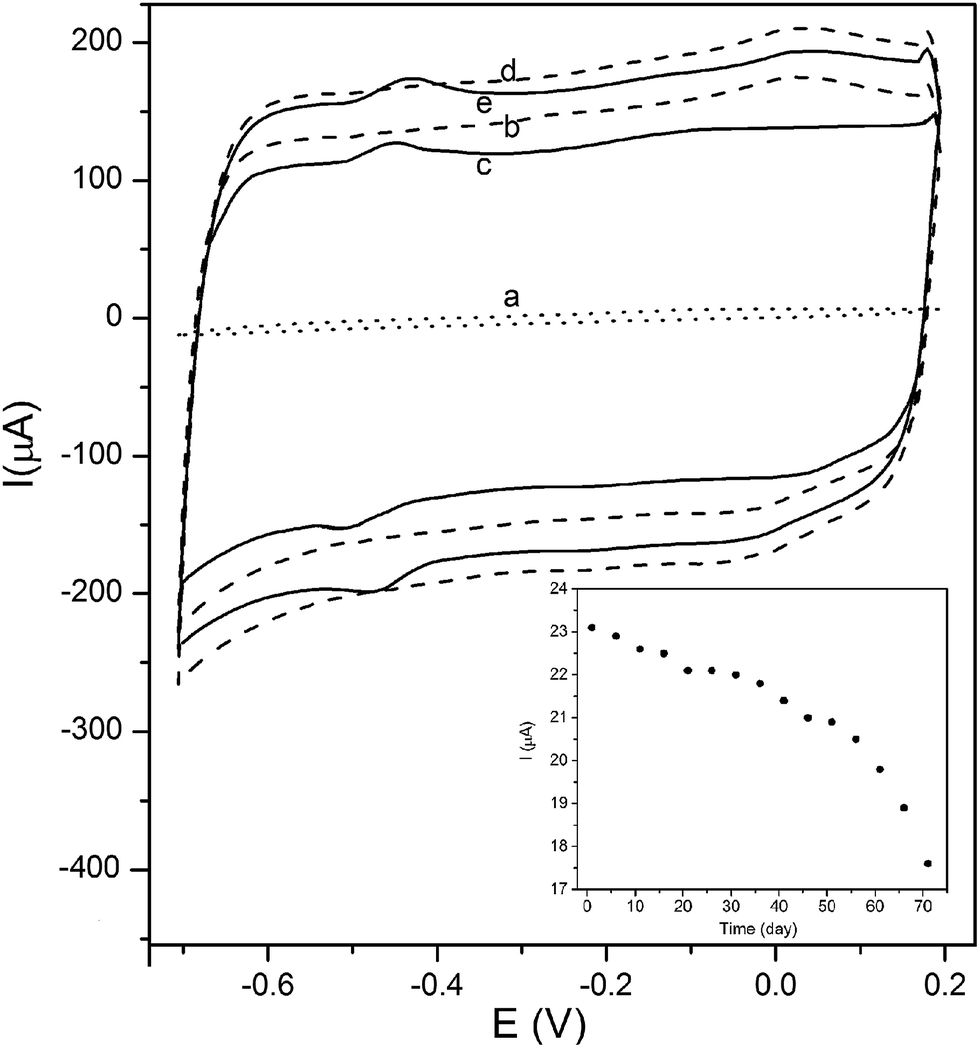
Fig. 9a shows the results of the cyclic voltammogram of the bare GCE. Addition of F-MWCNTs on surface of the bare electrode leads to a significant increase in the charging current (Fig. 9b). The GOx/F-MWCNTs/GCE nanocomposite (Fig. 9c) features a nearly symmetric redox wave around −0.49 V which is attributed to the bioactivity of GOx.30
 |
| | Fig. 9 Cyclic voltammograms of (a) bare GCE, (b) F-MWCNTs/GCE, (c) GOx/F-MWCNTs/GCE, (d) MnO2/MWCNTs/GCE and (e) GOx/MnO2/MWCNTs/GCE in 0.1 M deoxygenated PBS at a scan rate of 50 mV s−1. Inset: the faradic current of anodic peak of GOx/MnO2/MWCNTs/GCE measured every 5 days. | |
As a result of the presence of MnO2-NPs on the surface of F-MWCNTs/GCE, the charging current increases (Fig. 9d), indicating an increase in the surface area of the electrode. The higher surface area of the electrode may lead to the higher loading of the enzyme. Compared to GOx/F-MWCNTs/GCE structure, the GOx/MnO2/MWCNTs/GCE composite (Fig. 9e) displays an enhanced GOx redox wave at −0.45 V which suggests that MnO2 increases redox activity of GOx. The peak to peak potential separation (ΔEp) for the GOx/MnO2/MWCNTs/GCE and the GOx/F-MWCNTs/GCE composites are 0.04 and 0.06 V, respectively.
The amount of charge transferred during the anodic reaction (the base-to-base area under the anodic peak) of GOx/MnO2/MWCNTs/GCE is about 1.4 times higher than that of GOx/F-MWCNTs/GCE. The presence of crystalline MnO2-NPs decreases formal potential and ΔEp and increases the charge transfer of redox wave of GOx. These enhancements can be attributed to the good performance of MnO2 in electron transfer and ion exchange.31
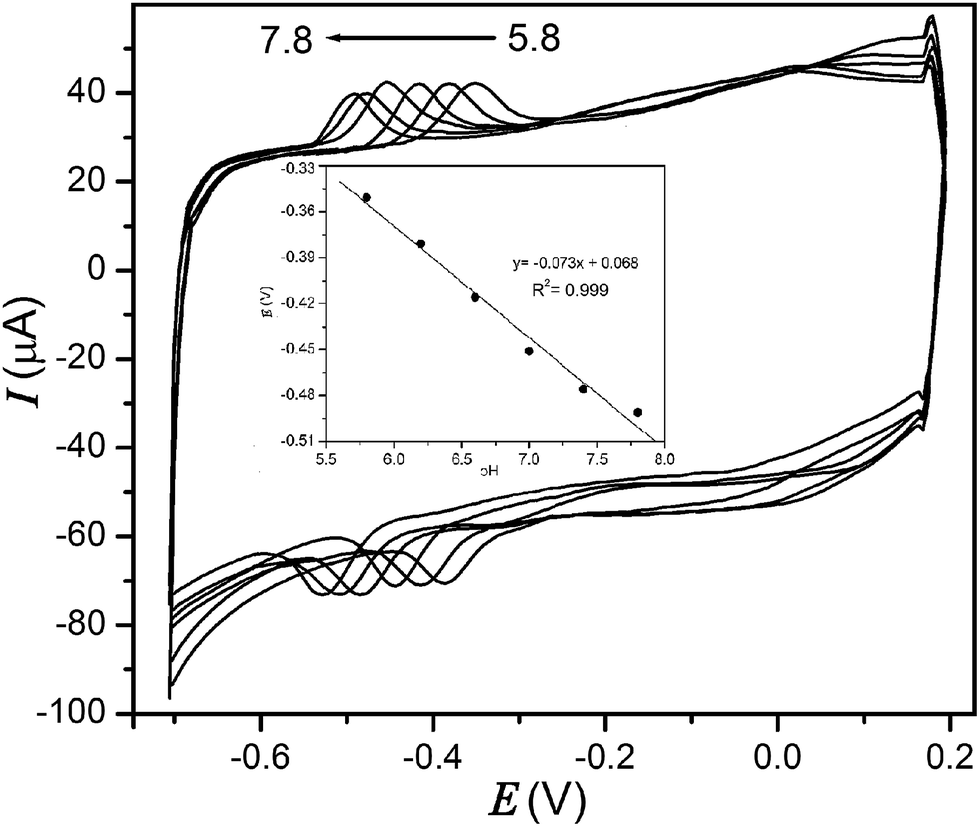
3.2.3 Effect of pH.
The influence of pH value on GOx redox wave of modified electrode in various buffer solutions (pH: 5.8–7.8) is presented in Fig. 11. The peak potentials shift negatively with an increase of the solution pH. The plot of Epversus pH (Fig. 11 inset) shows a linear relation (R2 = 0.999) over entire range of pH. The slope of this plot is −0.073 V per pH, which is close to the theoretical value of Nernstian equation for equal number of electron and proton transfer process.34 Therefore, the FAD/FADH2 redox couple of GOx/MnO2/MWCNTs/GCE comprises 2 electrons and 2 protons based on the following reaction:| | | GOx(FAD) + 2e− + 2H+ ⇆ GOx(FADH2) | (16) |
 |
| | Fig. 11 Cyclic voltammograms of GOx/MnO2/MWCNTs/GCE in various deoxygenated PBS (pH of 5.8, 6.2, 6.6, 7.0, 7.4 and 7.8) at a scan rate of 50 mV s−1. Inset: effect of pH on potential of electrode. | |
3.2.4 Determination of glucose using GOx/MnO2/MWCNTs electrode.
Fig. 12 presents the amperometric response of the GOx/MnO2/MWCNTs/GC electrode upon successive addition of glucose in PBS by applying potential of −0.55 V. The GOx acts as biocatalyst for oxidation of glucose and formation of H2O2.35 The electrochemical oxidation of H2O2 and electron generation process which may be catalyzed by MnO2 (eqn (17) and (18)) shows a well-defined and step-rise current response.36,37| | | MnO2 + H2O2 → Mn(OH)2 + O2 | (17) |
| | | Mn(OH)2 + 2OH− → MnO2 + 2H2O + 2e− | (18) |
 |
| | Fig. 12 Amperometric response of GOx/MnO2/MWCNTs/GCE to glucose at a potential of −0.55 V in PBS. The concentrations of glucose after each addition were 0.01, 0.05, 0.1, 0.2, 0.4, 0.8, 1.6, 3.2 and 6.4 mM, respectively. Inset: the calibration curve for glucose concentrations. | |
The modified electrode shows a linear behavior (Fig. 12 inset) in the range of 0.1–3.2 mM with a sensitivity of 24.2 μA mM−1 cm−2 and response time of less than 10 s. The detection limit was estimated to be 3.0 μM (S/N = 3).
Table 1 compares the analytical features of the GOx/MnO2/MWCNTs/GCE with the other glucose biosensors, which have used manganese oxides as a part of the immobilization matrix. Although the linear range of GOx/MnO2/MWCNTs/GCE is not wide, it exhibits higher sensitivity as is compared to the other electrodes. It should be noticed that, the electrode fabrication method in this report is quite simpler and faster than that of other works.
Table 1 Comparison of the analytical parameters of GOx/MnO2/MWCNTs/GCE with some other glucose biosensors
| Electrode material |
Detection potential (V) |
Linear range (mM) |
Sensitivity |
Detection limit (μM) |
Ref. |
|
Carbon fiber electrode.
Carbon paste electrode.
Poly(diallyldimethylammonium).
Octahedral molecular sieves.
|
| GOx–mesoMnO2–gelatin/GCE |
0.6 |
0.0009–2.73 |
24.2 μA mM−1 cm−2 |
0.18 |
36
|
| MnO2–GOx–CFEa |
0.58 |
1.5–15 |
1 nA mM−1 |
800 |
38
|
| GOx–La0.66Sr0.33MnO3–CPEb |
0.7 |
— |
0.16 μA mM−1 |
— |
39
|
| GOx/PDDAc/manganese oxide–graphite electrode |
−0.5 |
0.02–2.8 |
1.4 μA mM−1 |
9.8 |
40
|
| GOx–manganese oxide OMSd–CPE |
0.3 |
0.1–3.5 |
0.44 μA mM−1 |
0.1 |
41
|
| GOx/MnO2/MWCNTs/GCE |
−0.55 |
0.1–3.2 |
2.2 μA mM−1 |
3.0 μM |
This work |
3.2.5 Selectivity of the modified electrode toward glucose.
The typical amperometric response of GOx/MnO2/MWCNTs/GCE to subsequent addition of glucose and AA and UA is shown in Fig. 13. A significant current increase is observed upon addition of 2.0 mM glucose, while no response to 0.1 mM AA and UA is detected. This reveals that presence of AA and UA does not interfere with glucose sensing and therefore the proposed biosensor is fairly selective to glucose.
 |
| | Fig. 13 Amperometric response of GOx/MnO2/MWCNTs/GCE to successive addition of 2.0 mM glucose and 0.1 mM AA and UA in PBS at a potential of −0.55 V. | |
3.2.6 Stability of the GOx/MnO2/MWCNTs/GCE.
The long-term stability of the fabricated electrode was quantified by the measurement of the anodic peak current of the modified electrode every 5 days (Fig. 9 inset). The initial faradic current of the modified electrode was 23.1 μA and decreased by 24% after 71 days, indicating a relatively high stability in redox activity of the fabricated biosensor. Moreover, the charging current of GOx/MnO2/MWCNT electrode remained unchanged, indicating its good mechanical adhesion to the surface of the electrode. This stability may be due to the effect of the DBD plasma treatment and the presence of MnO2-NPs35 which improve immobilization of GOx on the surface of the electrode. The reproducibility of GOx/MnO2/MWCNTs/GCE construction was estimated from the response to 1.0 mM glucose at three electrodes prepared under the same conditions. The results show that the electrode has satisfying reproducibility with relative standard derivation of 3.6%.
4. Conclusions
MWCNTs were effectively functionalized in DBD plasma in the presence of humid air, MnO2 nanoparticles were deposited by chronoamperometry, GOx was immobilized, and the resulting GOx/MnO2/MWCNT nano-hybrid structures were used for fabricating a highly-stable glucose biosensor. Maximum amount of oxygenated groups, such as carboxyl and carbonyl ones are formed on the F-MWCNTs during 4 min exposure time in the DBD plasma. These functional groups are hydrophilic and improve dispersion ability and stability of the nanotubes in water. Desorption of these groups at high temperatures under helium atmosphere evolves H2O, CO2 and CO gases. The bioactivity of GOx/MnO2/MWCNTs/GCE is surface controlled, quasi-reversible, and also clearly shows long-term stability. The presence of crystalline MnO2-NPs on the surface of the nanotubes enhances charge transfer efficiency and reversibility of the GOx mediated redox processes.
Acknowledgements
This work was partially supported by the Nanobiomedicine Center of Excellence, Nanoscience and Nanotechnology Research Center and Center of Excellence in Biothermodynamics of the University of Tehran. Microanalysis equipment use, fruitful discussions, and technical assistance kindly provided by the members of the Plasma Sources and Applications Centre (PSAC) of NIE, Nanyang Technological University (Singapore), in particular Professor S. Xu is gratefully acknowledged.
References
- C. Chen, Q. Xie, D. Yang, H. Xiao, Y. Fu, Y. Tan and S. Yao, RSC Adv., 2013, 3, 4473–4491 RSC.
- S. Masoomi-Godarzi, A. A. Khodadadi, M. Vesali-Naseh and Y. Mortazavi, J. Electrochem. Soc., 2014, 161, B19–B25 CrossRef CAS.
- M. Baghayeri, H. Veisi, H. Veisi, B. Maleki, H. Karimi-Maleh and H. Beitollahi, RSC Adv., 2014, 4, 49595–49604 RSC.
- F. Hu, S. Chen, C. Wang, R. Yuan, Y. Chai, Y. Xiang and C. Wang, J. Mol. Catal. B: Enzym., 2011, 72, 298–304 CrossRef CAS.
- M. Yang, Y. Yang, Y. Liu, G. Shen and R. Yu, Biosens. Bioelectron., 2006, 21, 125–1131 Search PubMed.
- J. H. Kim, M. J. Song, C. J. Lee, J. H. Lee, J. H. Kim and N. K. Min, Carbon, 2013, 52, 398–407 CrossRef CAS.
- B. Haghighi, S. Bozorgzadeh and L. Gorton, Sens. Actuators, B, 2011, 155, 577–583 CrossRef CAS.
- P. Si, Y. Huang, T. Wang and J. Ma, RSC Adv., 2013, 3, 3487–3502 RSC.
- J. Qu, L. Shi, C. He, F. Gao, B. Li, Q. Zhou, H. Hu, G. Shao, X. Wang and J. Qiu, Carbon, 2014, 66, 485–492 CrossRef CAS.
- Y. Pan, Z. Mei, Z. Yang, W. Zhang, B. Pei and H. Yao, Chem. Eng. J., 2014, 242, 397–403 CrossRef CAS.
- Z. Fan, J. H. Chen, M. Y. Wang, K. Z. Cui, H. H. Zhou and W. Kuang, Diamond Relat. Mater., 2006, 15, 1478–1483 CrossRef CAS.
- Y. K. Zhou, B. L. He, F. B. Zhang and H. L. Li, J. Solid State Electrochem., 2004, 8, 482–487 CrossRef CAS.
- J. Yan, Z. Fan, T. Wei, W. Qian, M. Zhang and F. Wei, Carbon, 2010, 48, 3825–3833 CrossRef CAS.
- Y. S. Chen, J. H. Huang and C. C. Chuang, Carbon, 2009, 47, 3106–3112 CrossRef CAS.
- F. Pourfayaz, Y. Mortazavi, A. A. Khodadadi, S. H. Jafari, S. Boroun and M. Vesali-Naseh, Appl. Surf. Sci., 2014, 295, 66–70 CrossRef CAS.
- K. Ostrikov, E. C. Neyts and M. Meyyappan, Adv. Phys., 2013, 62, 113–224 CrossRef CAS.
- M. Vesali-Naseh, Y. Mortazavi, A. A. Khodadadi, P. Parsaeian and A. A. Moosavi-Movahedi, Sens. Actuators, B, 2013, 188, 488–495 CrossRef CAS.
- L. TuŠek, M. Nitschke, C. Werner, K. Stana-Kleinschek and V. Ribitsch, Colloids Surf., A, 2001, 195, 81–95 CrossRef.
- M. Vesali-Naseh, A. A. Khodadadi, Y. Mortazavi, F. Pourfayaz, O. Alizadeh and M. Maghrebi, Carbon, 2010, 48, 1369–1379 CrossRef.
- Y. H. Yan, J. Cui, M. B. Chan-Park, X. Wang and Q. Y. Wu, Nanotechnology, 2007, 18, 115712–115717 CrossRef.
-
D. Pavia, C. M. Lampman and G. S. Kriz, in Introduction to spectroscopy, Saunders, Washington, 3rd edn, 1979, vol. 2, pp. 13–101 Search PubMed.
- Y. Liu, L. Gao, J. Sun and Y. Wang, J. Ceram. Process. Res., 2010, 11, 120–122 Search PubMed.
- H. P. Boehm, Carbon, 2002, 40, 145–149 CrossRef CAS.
- J. L. Figueiredo and M. F. Pereira, Catal. Today, 2010, 150, 2–7 CrossRef CAS.
- G. D. Puente, J. J. Pis, J. A. Menhdez and P. Grange, J. Anal. Appl. Pyrolysis, 1997, 43, 125–138 CrossRef.
- R. Ionescu, E. H. Espinosa, E. Sotter, E. Llobet, X. Vilanova, X. Correig, A. Felten, C. Bittencourt, G. Van-Lier, J. C. Charlier and J. J. Pireaux, Sens. Actuators, B, 2006, 113, 36–46 CrossRef CAS.
-
R. A. Wolf, Atmospheric pressure plasma for surface modification, Wiley, New York, 2012 Search PubMed.
- C. Yu, G. Li, L. Wei, Q. Fan, Q. Shu and J. C. Yu, Catal. Today, 2014, 224, 154–162 CrossRef CAS.
- S. B. Ma, K. Y. Ahn, E. S. Lee, K. H. Oh and K. B. Kim, Carbon, 2007, 45, 375–382 CrossRef CAS.
- F. Li, J. Song, F. Li, X. Wang, Q. Zhang, D. Han, A. Ivaska and L. Niu, Biosens. Bioelectron., 2009, 25, 883–888 CrossRef CAS PubMed.
- D. P. Dubal, D. S. Dhawale, R. R. Salunkhe and C. D. Lokh, J. Electroanal. Chem., 2010, 647, 60–65 CrossRef CAS.
- M. Yousef-Elahi, A. A. Khodadadi and Y. Mortazavi, J. Electrochem. Soc., 2014, 161, B81–B87 CrossRef.
- E. Laviron, J. Electroanal. Chem., 1979, 101, 19–28 CrossRef CAS.
- D. Wen, Y. Liu, G. Yang and S. Dong, Electrochim. Acta, 2007, 5312–5317 CrossRef CAS.
- F. Gao, J. Yin, Z. Yao, M. Li and L. Wang, J. Electrochem. Soc., 2010, 157, F35–F39 CrossRef CAS.
- J. Yu, T. Zhao and B. Zeng, Electrochem. Commun., 2008, 10, 1318–1321 CrossRef CAS.
- B. Xu, M. L. Ye, Y. X. Yu and W. D. Zhang, Anal. Chim. Acta, 2010, 20–26 Search PubMed.
- S. B. Hocevar, B. Ogorevc, K. Schachl and K. Kalcher, Electroanalysis, 2004, 16, 1711–1716 CrossRef CAS.
- G. L. Luque, N. F. Ferreyra, A. G. Leyva and G. A. Rivas, Sens. Actuators, B, 2009, 142, 331–336 CrossRef CAS.
- J. J. Xu, J. J. Feng, X. Zhong and H. Y. Chen, Electroanalysis, 2008, 20, 507–512 CrossRef CAS.
- X. Cui, G. Liu and Y. Lin, J. Nanomed. Nanotechnol., 2005, 1, 130–135 CrossRef CAS PubMed.
|
| This journal is © The Royal Society of Chemistry 2016 |
Click here to see how this site uses Cookies. View our privacy policy here.