DOI:
10.1039/C4RA16605A
(Paper)
RSC Adv., 2015,
5, 15818-15830
Cu(OTf)2 catalyzed three component strategy for the synthesis of thienopyridine containing spirooxindoles and their cytotoxic evaluation†
Received
18th December 2014
, Accepted 28th January 2015
First published on 28th January 2015
Abstract
Synthesis of novel spirooxindoles via a three component reaction of thienopyridines, isatins and malononitriles under copper catalysis was accomplished. This one-pot, room temperature protocol allowed the synthesis of diversely substituted spirooxindoles in good to excellent yields. Cytotoxicity towards COLO320 cells revealed that compound 5v possessing a 2,6-difluorobenzyl group (IC50 of 49.1 μM) was found to be highly potent among the screened compounds. In addition, molecular docking of compound 5v into caspase-3 receptors exhibited the largest binding energy (−10.5 kcal mol−1) compared to other compounds. The formation of a DNA ladder for compound 5v also supports the experimental results.
Introduction
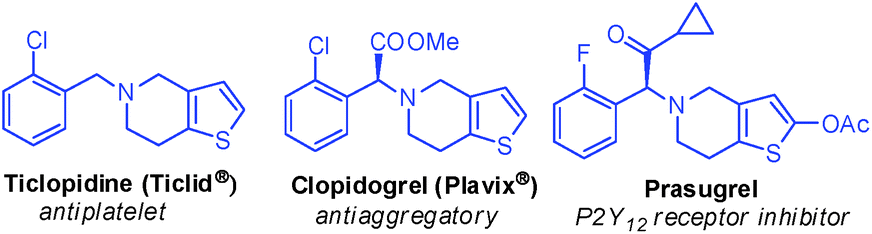
A growing number of naturally occurring alkaloids and pharmacological agents containing a spirooxindole core are continuously being discovered.1–6 Spirooxindole containing both indole and pyran rings has been shown to possess anticonvulsant,7 herbicidal,8 anticancer9 and antibacterial activities.10 In particular, sharing the indole 3-carbon atom in the formation of spirooxindole derivatives highly enhances the biological activity.11,12 On the other hand, thienopyridines represent a key structural unit in many synthetic pharmaceuticals (Fig. 1).13–16
 |
| | Fig. 1 Clinical drugs containing thienopyridine system. | |
Additionally, thiophene derivatives are excellent synthetic intermediates because of the unique electronic properties of sulphur as well as the steric constraints of a five-membered ring. They represent many bioactive agents and are considered as the bioisostere of benzene ring,17 since the distance between two neighboring carbon atoms in benzene is roughly equivalent to the diameter of sulphur atom in thiophene and the latter often displays pharmacological properties similar to that of benzene.18 Thus, one can anticipate that replacing the benzene ring19 of spiropyranones by thiophene would afford hybrid spirooxindoles (Fig. 2) of potential pharmacological interest.20 In addition, the model shown in Fig. 2 has four centers for the introduction of diversity into the target molecule. In view of the aforementioned knowledge based facts on different pharmacophores and in continuation of our research program on bio-active heterocycles,21–27 we envisioned a hybrid pharmacophore approach for the synthesis of potentially cytotoxic spirooxindoles. To this end, we prepared a series of novel spirooxindoles through the Cu(OTf)2 catalyzed three component reaction between isatin, malononitrile and thienopyridines. The cytotoxicity towards COLO320 cells of all the synthesized compounds was studied, supported by molecular docking and DNA fragmentation and all these results are disclosed in this article.
 |
| | Fig. 2 Design of targeted spirooxindoles via three component assemblage. | |
Results and discussion
Chemistry
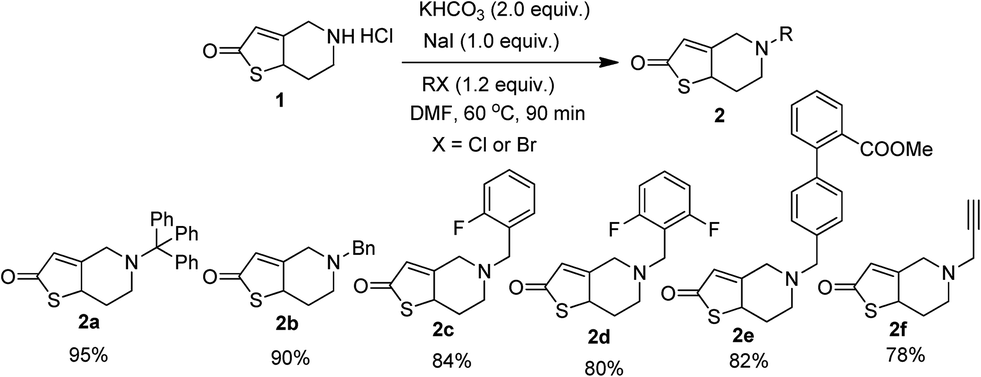
It is pertinent to note that, we have previously demonstrated the Cu(OTf)2 catalyzed three component condensation between isatin, malononitrile and kojic acid leading to novel spirooxindoles having cytotoxic potency.9 By taking the advantage of this methodology, the synthesis of targeted spirooxindole via three component reaction between thienopyridines, isatins and malononitriles was envisaged. Considering that thiene derivatives could potentially be introduced as substituents in the target molecule, we initially prepared six substituted thienes 2a–f in good to excellent yield by the modified literature procedure (Scheme 1).28 Since, we were interested in exploring the alteration of the N-substituent by a rigid benzyl function, substituent of various steric bulks 2a–e were synthesized. Additionally, substrate with a small hydrophobic group such as propargyl 2f was also prepared in order to compare the results with the benzyl substituents.
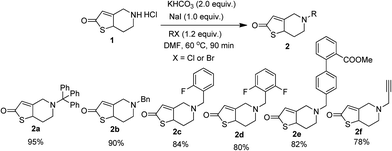 |
| | Scheme 1 Synthesis of N-alkyl thienopyridines 2a–2f. | |
Having obtained the requisite precursors in satisfactory yields, we then directed our attention towards the optimization of reaction conditions for the prototype reaction (Scheme 2 and Table 1). Couple of points regarding the optimization studies is worth noting. (i) Based on our previous experience with copper catalysis,9,24,29 we chose only copper catalysts for the screening studies. (ii) The solvent of choice was dichloromethane and dichloroethane, since MeOH, EtOH, MeCN could decrease the Lewis acidity of the catalyst and eventually the catalytic efficiency of the overall process. To begin our investigation, 1.0 mmol of thienopyridine 2a was allowed to react with isatin (1.0 mmol) and malononitrile (1.0 mmol) using 5 mol% of Cu(OTf)2 in 1,2-dichloroethane at 25 °C. To our delight, the reaction proceeded smoothly and afforded the product 5a in 85% yield after 2 h (entry 1). Effort to reduce the reaction time by refluxing the reaction for 30 min afforded the product in a yield reduced to 50%, which could be attributed to the partial decomposition of the thienopyridine 2a (entry 2). To assess the efficacy of Cu(OTf)2 in this three component reaction, a short screening with different copper catalysts were examined. To this end, Cu(OAc)2 led to the formation of product in 24% yield after 4 h at room temperature (entry 3). No significant observation could be observed, when CuCl2 was used as the catalyst in dichloromethane at refluxing condition (entry 4). Switching from the Cu(II)- to Cu(I)- sources such as copper(I) triflate·1/2benzene complex (entry 5), copper(I) chloride (entry 6), tetrakis(acetonitrile) copper(I) tetrafluoroborate (entry 7) or Cu(I)-thiopene-2-carboxylate (entry 8) completely failed to produce the product. Needless to say, in the absence of catalyst the reaction did not proceed at all and only the Knoevenagel product was obtained in quantitative yield. From the screening data, it is quite obvious that 5 mol% of Cu(OTf)2 in dichloroethane at room temperature for 2 h was effective for the desired transformation. Hence, the same reaction condition was employed for a wide range of substrates toward the preparation of structurally diverse spirooxindoles (Scheme 3 and Table 2). Under this condition, irrespective of the electronic nature of the peripheral substituents as well as variable steric bulks, all substrates underwent the reaction smoothly affording the products in good to excellent yield. The utility of our methodology was further manifested by its applicability to afford products with propargyl tether (5h, 5q, 5u, 5x, 5y and 5z) without any isomerization to the corresponding allenyl product.30–32 Conversely, subjecting the thienopyridine 1 devoid of N-substitution afforded no product at all and afforded only the isatylidene malononitrile along with trace amount of several unidentified by-products.
 |
| | Scheme 2 Optimization of reaction conditions for the model reaction. | |
Table 1 Optimization of Cu-based catalytic system for the three component assemblage
| Entrya |
[Cu] source |
Solvent |
Temp (°C) |
Time (h) |
Yieldb (%) |
| All reactions were performed using isatin (1.0 mmol), malononitrile (1.0 mmol), thienopyridine 2a (1.0 mmol), 5 mol% of [Cu] and 1 mL of solvent. Isolated yield after column chromatography and characterized by 1H NMR. No catalyst was employed. Isatylidene malononitrile was obtained quantitatively. |
| 1 |
Cu(OTf)2 |
(CH2)2Cl2 |
25 |
2 |
85 |
| 2 |
Cu(OTf)2 |
(CH2)2Cl2 |
Reflux |
0.5 |
50 |
| 3 |
Cu(OAc)2 |
(CH2)2Cl2 |
25 |
4 |
24 |
| 4 |
CuCl2 |
CH2Cl2 |
Reflux |
4 |
11 |
| 5 |
CuOTf·1/2benzene |
CH2Cl2 |
25 |
12 |
15 |
| 6 |
CuCl |
CH2Cl2 |
Reflux |
6 |
— |
| 7 |
Cu(MeCN)4BF4 |
CH2Cl2 |
25 |
12 |
— |
| 8 |
Cu(I)-thiophene-2-carboxylate |
(CH2)2Cl2 |
25 |
12 |
— |
| 9 |
—c |
(CH2)2Cl2 |
25 |
2 |
—d |
 |
| | Scheme 3 Cu(OTf)2 catalyzed three component assemblage. | |
Table 2 Synthesis of spirooxindole derivatives 5a–5z
| Entry |
R1 |
R2 |
Z |
Thienopyridinea |
Productb |
Yieldc (%) |
| Subjecting thienopyridine 1 afforded 50% of isatylidene malononitrile along with decomposed by-products. All products were characterized by IR 1H NMR 13C NMR and mass spectroscopy. Isolated yield after column chromatography. |
| 1 |
H |
H |
CN |
2a |
5a |
85 |
| 2 |
Cl |
H |
CN |
2a |
5b |
81 |
| 3 |
Cl |
H |
COOMe |
2a |
5c |
80 |
| 4 |
Br |
H |
COOMe |
2a |
5d |
78 |
| 5 |
H |
H |
COOEt |
2a |
5e |
84 |
| 6 |
H |
Ethyl |
CN |
2a |
5f |
85 |
| 7 |
H |
Allyl |
CN |
2a |
5g |
83 |
| 8 |
H |
Propargyl |
CN |
2a |
5h |
80 |
| 9 |
H |
Allyl |
COOEt |
2a |
5i |
79 |
| 10 |
H |
H |
CN |
2b |
5j |
88 |
| 11 |
H |
Ethyl |
CN |
2b |
5k |
89 |
| 12 |
H |
Benzyl |
CN |
2b |
5l |
90 |
| 13 |
Cl |
H |
CN |
2b |
5m |
90 |
| 14 |
H |
H |
COOMe |
2b |
5n |
95 |
| 15 |
H |
H |
CN |
2c |
5o |
84 |
| 16 |
H |
Benzyl |
CN |
2c |
5p |
91 |
| 17 |
H |
Propargyl |
CN |
2c |
5q |
79 |
| 18 |
Cl |
H |
COOMe |
2c |
5r |
89 |
| 19 |
H |
Methyl |
COOEt |
2c |
5s |
87 |
| 20 |
H |
H |
CN |
2d |
5t |
82 |
| 21 |
H |
Propargyl |
CN |
2d |
5u |
81 |
| 22 |
H |
H |
COOMe |
2d |
5v |
90 |
| 23 |
H |
H |
CN |
2e |
5w |
86 |
| 24 |
H |
H |
CN |
2f |
5x |
81 |
| 25 |
Me |
H |
CN |
2f |
5y |
84 |
| 26 |
NO2 |
H |
CN |
2f |
5z |
77 |
The structure of all products was confirmed by spectral data (FTIR, 1H NMR, 13C NMR and MS) and elemental analyses. As an illustrative example, the IR spectrum of compound 5t showed broad peaks at 3359 and 3181 cm−1, which revealed the presence of –NH2 and –NH functionalities, respectively. The sharp band at 2191 cm−1 confirmed the presence of cyano group. The stretching bands at 1708 cm−1 corresponds to the amide carbonyl group of oxindole moiety. The 1H NMR spectrum of compound 5t recorded in DMSO-d6 showed eighteen protons. The broad signals at δH 7.26 ppm and δH 10.67 ppm corresponds to –NH2 and –NH protons (D2O exchangeable), respectively. The two doublets at δH 2.08 ppm and δH 2.69 ppm with the geminal coupling constant of 1JH–H = 14.8 Hz correspond to the methylene protons of 2,6-difluorobenzyl group. A sharp singlet of two protons at δH 3.44 ppm corresponds to –NCH2– link and a multiplet of four protons at δH 2.64 ppm corresponds to –N(CH2)2– link. In 13C NMR spectrum, a low intense peak at δC 49.2 ppm indicates the presence of a spiro-carbon and a peak at δC 177.1 ppm corresponds to an amide carbonyl carbon. This observation was supported by DEPT-135 and 2D chemical shift correlation experiments (see ESI†). Mass spectrometry of compound 5t was acquired in the positive ionization FIA mode, which exhibited a peak at m/z = 477 [M + H]+ corresponds to the molecular mass of the compound. Finally, single crystal X-ray diffraction studies unambiguously confirmed the structure of the product 5t (Fig. 3).33
 |
| | Fig. 3 ORTEP diagram of 5t showing the atomic notation. | |
The proposed reaction mechanism is demonstrated in Scheme 4. The process represents a typical cascade reaction34–36 in which the isatin 3 first condenses with malononitrile 4 to afford isatylidene malononitrile derivative I. This step can be regarded as a fast Knoevenagel addition. The Lewis acidic Cu(OTf)2 interacts with the cyano group of I leading to the intermediate II. Nucleophilic attack of enolic form of 2 to the electron deficient C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C double bond of activated intermediate II leads to intermediate III, which is in equilibrium with IV. Thorpe–Ziegler type reaction of intermediate IV (nucleophilic attack of enolic hydroxyl to the activated cyano group) results in the cyclized oxonium intermediate V.37 Proton transposition of intermediate V leads to intermediate VI, which upon proto-demetallation affords spirooxindole 5.
C double bond of activated intermediate II leads to intermediate III, which is in equilibrium with IV. Thorpe–Ziegler type reaction of intermediate IV (nucleophilic attack of enolic hydroxyl to the activated cyano group) results in the cyclized oxonium intermediate V.37 Proton transposition of intermediate V leads to intermediate VI, which upon proto-demetallation affords spirooxindole 5.
 |
| | Scheme 4 Plausible mechanism for the Cu(OTf)2 catalyzed three component reaction. | |
Cytotoxic evaluation
The MTT [3-(4,5-dimethylthiazo-2-yl)-2,5-diphenyl-tetrazolium bromide] cell proliferation assay was used to evaluate the apoptosis-inducing activities of the spirooxindoles 5a–5z against COLO320 cell lines.38 The cytotoxic results were compared with reference drug cyclophosphamide, which showed 90% inhibition at a concentration 150 μg mL−1.39 As per the experimental results, all compounds showed maximum cytotoxicity against COLO320 cell line at 100 to 25 μg mL−1 concentration. All concentrations used in the experiment could decrease the cell viability significantly (P < 0.05) in a concentration-dependent manner. The IC50 values of all the tested compounds were summarized in Table 3. Attempts to understand the SAR of the spirooxindoles started with the substituent at the thienopyridine moiety (R), which revealed that compound 5v containing 2,6-difluorobenzyl group was highly potent with an IC50 of 49.1 μM. Compounds 5l and 5u possessing benzyl and 2,6-difluorobenzyl groups respectively were found to be almost active as 5v with an IC50 of 49.3 μM. The biphenyl analogue 5w (IC50 = 49.8 μM) had activity similar to that of 5l and 5u, suggesting that the additional phenyl group at those sites is tolerated. The benzyl analogue 5m, trityl analogue 5g and 2-fluorobenzyl analogue 5o, exhibited good inhibitory potency with IC50 of 50.3, 50.7 and 50.8 μM, respectively. The other substituted analogues 5a, 5c, 5e, 5f, 5i, 5j, 5p, 5q, 5s, 5t and 5x to 5z exhibited reduced but significant activities ranging from 51.2 to 57.3 μM. Compounds 5b, 5d, 5h, 5k, 5n and 5r emerged as the least active compounds among the tested series, exhibiting IC50 values of >100 μM. Among the substituents placed on the periphery of the oxindole moiety (R1 and R2), unsubstitution at C5 position (5a, IC50 = 51.2 μM) renders the molecule more active than those with chloro substitution (5b, IC50 = >100 μM). Replacement of C5–Cl (5c, IC50 = 53.4 μM) by a C5–Br (5d, IC50 = >100 μM) did reduced the potency to a great extent, which may be attributed to the differences in size and electronegativity. Interestingly, the replacement of a methyl group (5y, IC50 = 54.3 μM) and nitro group (5z, IC50 = 55.4 μM) on the C5 position did not exhibited a significant change despite the differences in polarity and steric bulk. Of the N-substituents on the oxindole, a decrease in inhibitory potency with ethyl (5f, IC50 = 53.9 μM) and increased potency with allyl (5g, IC50 = 50.7 μM) was observed respectively than the unsubstituted analogue (5a, IC50 = 51.2 μM). Among the same series, a drastically decreased cytotoxicity was observed for the N-propargyl group (5h, IC50 = >100 μM). However, an opposite trend was observed in the other series (5t and 5v), where the location of a propargyl group (5v, IC50 = 49.3 μM) demonstrated increased inhibitory potency than its unsubstituted counterpart (5t, IC50 = 53.1 μM). Compound with the N-benzyl group (5l, IC50 = 49.3 μM) emerged as the second most active among the screened compounds. Assessment of the SAR of polar substituents (Z) like CN, COOMe and COOEt located on the pyran scaffold was not easy as evidenced by their inconsistent IC50 values. For example, an enhanced cytotoxicity was observed for compound possessing COOMe group (5c, IC50 = 53.4 μM) than those containing CN group (5b, IC50 > 100 μM). However, a reverse trend was observed when the cytotoxicity was compared between compound 5m and 5n. Here, the compound with CN group (5m, IC50 = 50.3 μM) exhibited increased cytotoxicity against compound with COOMe group (5n, IC50 > 100 μM).
Table 3 IC50 value of compounds 5a–5z
| Entry |
Product |
IC50a (μM) |
| The reference, cyclophosphamide showed 90% inhibition (156 μg mL−1). |
| 1 |
5a |
51.2 |
| 2 |
5b |
>100 |
| 3 |
5c |
53.4 |
| 4 |
5d |
>100 |
| 5 |
5e |
55.2 |
| 6 |
5f |
53.9 |
| 7 |
5g |
50.7 |
| 8 |
5h |
>100 |
| 9 |
5i |
54.1 |
| 10 |
5j |
55.3 |
| 11 |
5k |
>100 |
| 12 |
5l |
49.3 |
| 13 |
5m |
50.3 |
| 14 |
5n |
>100 |
| 15 |
5o |
50.8 |
| 16 |
5p |
55.8 |
| 17 |
5q |
53.4 |
| 18 |
5r |
>100 |
| 19 |
5s |
54.4 |
| 20 |
5t |
53.1 |
| 21 |
5u |
49.3 |
| 22 |
5v |
49.1 |
| 23 |
5w |
49.8 |
| 24 |
5x |
57.3 |
| 25 |
5y |
54.3 |
| 26 |
5z |
55.4 |
Molecular docking
All the synthesized compounds 5a–5z were docked into the caspase-3 receptor from PDB entry 3DEJ. Ligand 2D structures were drawn using ChemDraw Ultra 7.0 (ChemOffice 2002). Chem3D Ultra 7.0 was used to convert 2D structure into 3D and the energy minimized using semi-empirical AM1 method. Minimize energy to minimum RMS gradient of 0.100 was set in each iteration. All structures were saved as .pdb file format for input to AutoDock Tools (ADT) version 1.5.4.40 All structures were then saved in PDBQT file format for input into AUTODOCK version 1.5.4.41 The co crystallized ligand42 in the caspase-3 structure was removed. For the protein structure, all hydrogen atoms were added, lower occupancy residue structures were deleted, and any incomplete side chains were replaced using the ADT version 1.5.4. Further ADT was used to remove crystal water, added Gagteiger charges to each atom, and merged the non-polar hydrogen atoms to the protein structure. The distance between donor and acceptor atoms that form a hydrogen bond was defined as 1.9 Å with a tolerance of 0.5 Å, and the acceptor–hydrogen–donor angle was not less than 120°. The structures were then saved in PDBQT file format for input into AUTODOCK version 1.5.4. A grid box with dimension of 40 × 40 × 40 Å3 centred on −46.0202, 15.0190 and −22.3194 was created around the binding site of co-crystal on caspase-3 protein using AutoDockTools. The centre of the box was set at co-crystal and grid energy calculations were carried out. For the AUTODOCK docking calculation, default parameters were used and 50 docked conformations were generated for each compound. In order to verify the reproducibility of docking calculations, the bound ligand was extracted from the complexes and submitted for one-ligand run calculation. This reproduced top scoring conformations of 10 falling within root-mean-square deviation (rmsd) values of 0.812 to 1.134 Å from bound X-ray conformation for caspase-3, suggesting this method is valid enough to be used for docking studies of other compounds. The outputs were exported to VMD and Pymol for visual inspection of the binding modes and interactions of the compounds with amino acid residues in the active sites (Fig. 4).
 |
| | Fig. 4 Docking of bound and docked ligand in caspase-3, where the docked ligand was discriminated by showing five hydrogen bond interactions. | |
Docking of different ligands to protein was performed using AUTODOCK, same protocols used in as that of validation study. All docking were taken into 2.5 million energy evaluations were performed for each of the test molecules. Docked ligand conformations were analyzed in terms of energy, hydrogen bonding and hydrophobic interaction between ligand and receptor protein caspase-3. Here it should be noted that the typical hydrogen bonding distance between a heteroatom and a hydrogen atom is 1.8 Å and 2.5 Å. We considered only the 3D view of ligand–protein interaction, which in turn influences the activity change. Usually topological features of the molecules will be considered in the 3D QSAR methodology or other molecular modelling methods where the 3D protein structure was not considered or available. The AUTODOCK scoring function is the composition of final intermolecular energy, Van der Waals force, hydrogen bonding, desolvation energy, electrostatic energy and torsional free energy. However hydrogen bond and hydrophobic interactions can be visualized as a picture, so we mentioned only those two interactions. Detailed analyses of the ligand–receptor interactions were carried out and final coordinates of the ligand as well as receptor were saved as pdb files. For display of the receptor with the ligand binding site, VMD software was used. From the docking scores, the free energy of binding (FEB) of all compounds were calculated (Table 4).
Table 4 FEB value of compounds 5a–5z
| Entry |
Product |
FEBa (kcal mol−1) |
| Free energy of binding. |
| 1 |
5a |
−9.5 |
| 2 |
5b |
−9.8 |
| 3 |
5c |
−9.0 |
| 4 |
5d |
−9.6 |
| 5 |
5e |
−9.1 |
| 6 |
5f |
−9.5 |
| 7 |
5g |
−9.2 |
| 8 |
5h |
−9.7 |
| 9 |
5i |
−8.6 |
| 10 |
5j |
−9.0 |
| 11 |
5k |
−8.9 |
| 12 |
5l |
−9.4 |
| 13 |
5m |
−9.1 |
| 14 |
5n |
−8.4 |
| 15 |
5o |
−9.1 |
| 16 |
5p |
−8.6 |
| 17 |
5q |
−9.0 |
| 18 |
5r |
−8.5 |
| 19 |
5s |
−8.8 |
| 20 |
5t |
−9.1 |
| 21 |
5u |
−9.8 |
| 22 |
5v |
−10.5 |
| 23 |
5w |
−9.7 |
| 24 |
5x |
−8.2 |
| 25 |
5y |
−8.6 |
| 26 |
5z |
−8.3 |
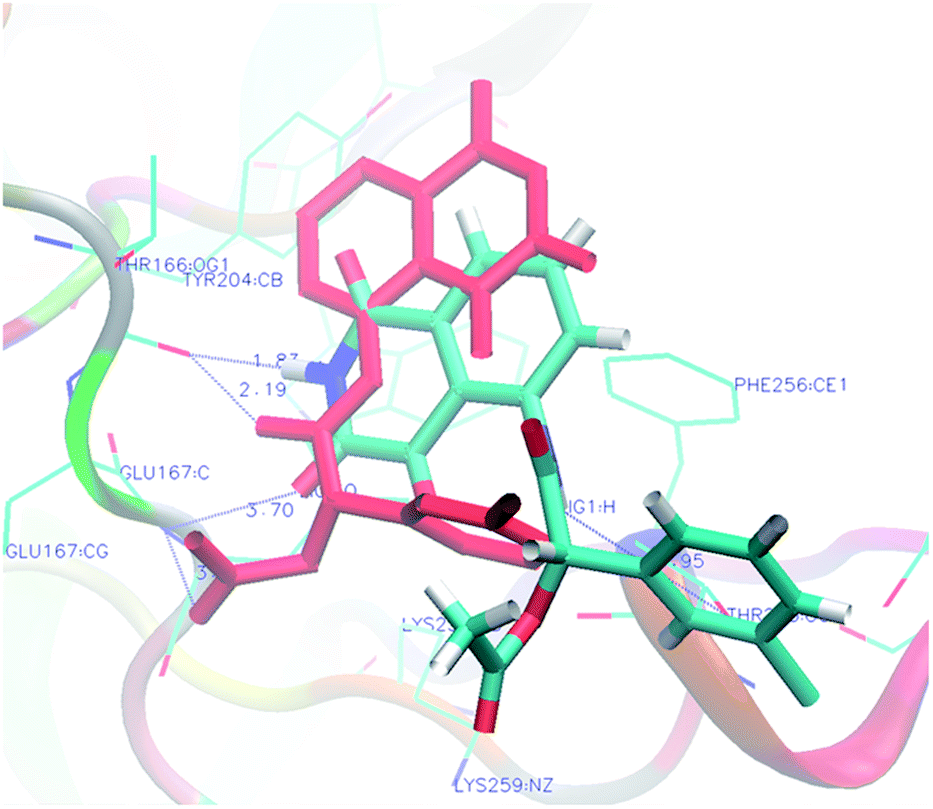
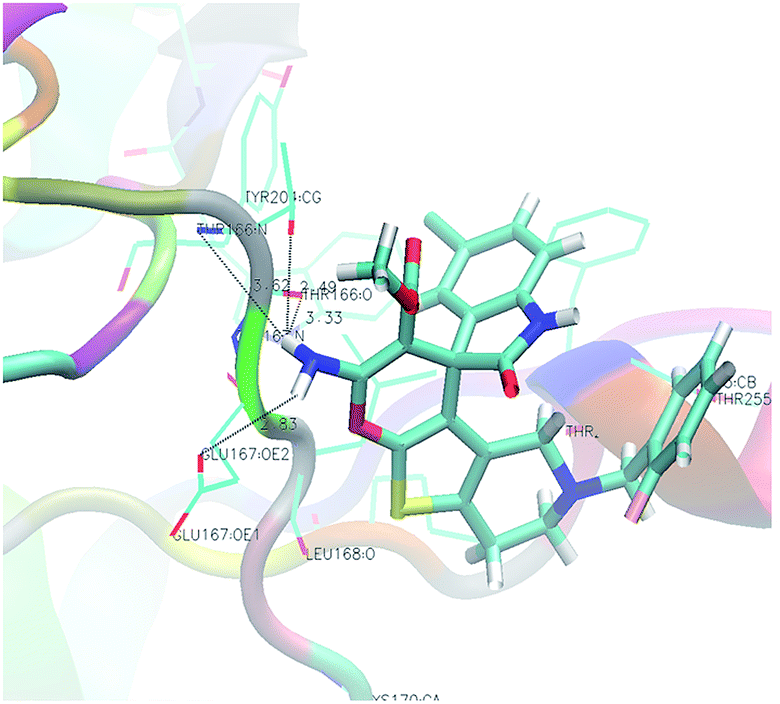
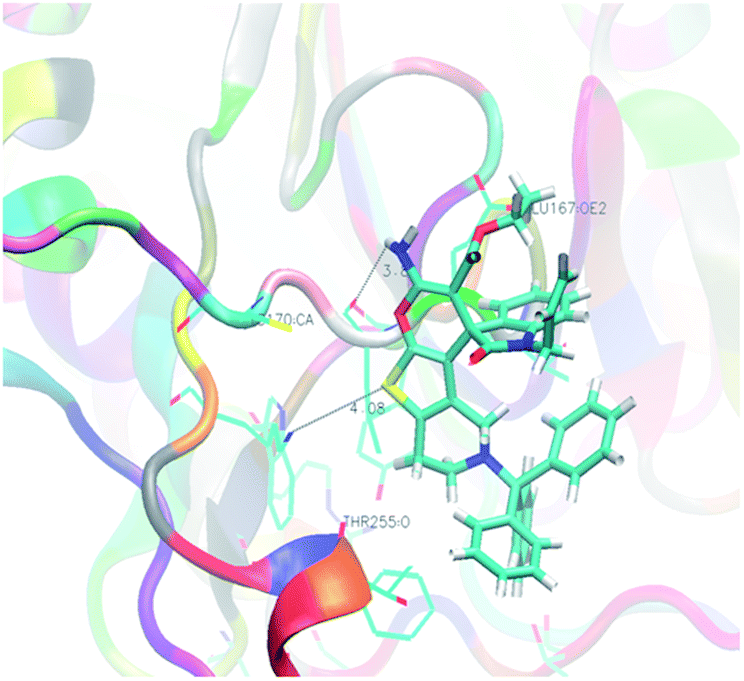
Analysis of the results revealed that all compounds exhibited docking scores between −8.2 and −10.5 kcal mol−1. Compound 5v emerged as the most active with a calculated binding energy of −10.5 kcal mol−1. The least binding energy was exhibited by compound 5x with a binding energy of −8.2 kcal mol−1. The intermediary active compound 5i showed −8.6 kcal mol−1 of binding energy. The binding interactions of these compounds were shown respectively in Fig. 5–7.
 |
| | Fig. 5 Docking of the most active compound 5v (FEB = −10.5 kcal mol−1) in caspase-3 (dotted lines showing hydrogen bond interactions). | |
 |
| | Fig. 6 Docking of the least active compound 5x (FEB = −8.2 kcal mol−1) in caspase-3 (dotted lines showing hydrogen bond interactions). | |
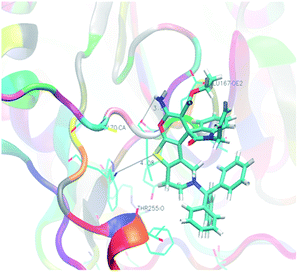 |
| | Fig. 7 Docking of the intermediary active compound 5i (FEB = −8.6 kcal mol−1) in caspase-3 (dotted lines showing hydrogen bond interactions). | |
All the reported molecules of caspase-3 inhibitors are allosteric. Crystal structure of caspase-3 reveals that the typical two-lobe receptor architecture connected by a 3 to 4 amino acid residue, the so called hinge region. The N-terminal lobe is comprised primarily of anti-parallel β-sheets and the C-terminal lobe is comprised mainly of α-helices. The unique features of caspase-3 receptor are a β-hairpin insert located at the N-terminal to helix α-C and most importantly the presence of a leucine residue at position 168 within the hinge region. Molecules which have trityl group (5a–5i) showed good to very good binding score (−9.8 to −9.1 kcal mol−1). This is because of the bulky nature of trityl group as well as its surface active nature which binds on the surface of caspase-3 receptor. Except molecule 5i (−8.6 kcal mol−1), all other molecules in the trityl series exhibited good binding score. The trityl group push the amino group towards the hinge region and the favourable interactions with the Leu 168, Lys 259 and Glu 167 was established by 5b in caspase-3. Instead of trityl group, compounds with benzyl group (5j–5n) showed moderate caspase-3 binding affinity (−9.0 to −8.4 kcal mol−1). This can be ascribed by the fact that benzyl group being less bulky and also shows less surface active nature than the trityl group. The molecule 5n shows poor inhibitory activity is mainly due to the presence of –COOMe group, which was sterically hindered by one of the C-terminal α-helices. However, it is to be noted that 5n forms necessary hydrogen bond interaction with Leu 168 of hinge region. In the next set of molecules, the benzyl ring was replaced with 2-fluorobenzyl group (5o–5s). The binding score (−9.1 to −8.5 kcal mol−1) of these molecules was not improved by the adding up of fluorine atom at the ortho-position. This group of molecules does not forms the essential hydrogen bond interactions, instead forms a strong hydrophobic as well as π-stacking interaction with Tyr204 residue. Further to enhance the inhibitory activity towards caspase-3, additional fluorine atom was introduced, which resulted in the series from 5t–5v. As expected, in this series, compound 5v showed enhanced binding energy of −10.5 kcal mol−1. This improved binding energy is mainly attributed to the increased electronic nature offered by the fluorine atoms as well as the formation of essential hydrogen bonding interactions with Leu 168, Lys 259 and Glu 167. The molecule 5w, which encompasses biphenyl ring showed pronounced binding energy of −9.7 kcal mol−1. This observation can be attributed to the biphenyl ring pushing the amino group of pyran ring towards the hinge region and forms the essential hydrogen bonding interactions with Leu 168 and biphenyl ring forms π-stacking interaction with Tyr204 residue. The final set of molecule (5x–5z) possessing propargyl group in the thienopyridine ring exhibited the binding energy of −8.6 to −8.2 kcal mol−1. The reduced binding energy is mainly due to the lack of surface interaction and less interactions with hinge region.
DNA fragmentation
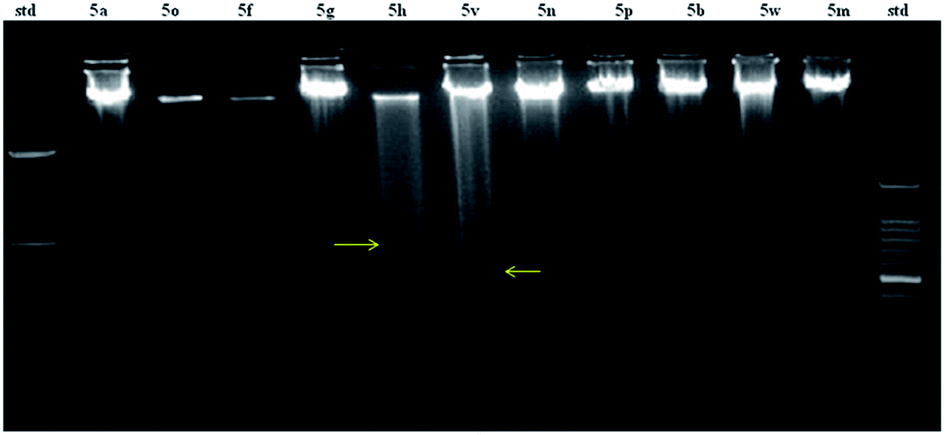
DNA fragmentation, which is a typical hallmark of the apoptotic cell death, has long been used to distinguish apoptosis from necrosis, and it is among the most reliable method for the detection of apoptotic cells. Since compound 5v emerged as the most active with an IC50 of 49.1 μM and FEB of −10.5 kcal mol−1, DNA fragmentation was analysed for the same in COLO320 cell lines. In order to compare the fragmentation pattern of 5v, compounds 5a, 5o, 5p, 5g, 5h, 5v, 5n, 5p, 5b, 5w and 5m were randomly selected and analyzed for their DNA fragmentation. Fig. 8 clearly shows the formation of DNA ladder (trailing) in compounds 5h and 5v which indicates that the DNA breakage or fragmentation has occurred. This can only happen when cell apoptosis takes place and therefore, the cytotoxic effect of compounds 5h and 5v is selectively mediated through the induction of apoptosis.
 |
| | Fig. 8 Agarose gel electrophoresis of DNA extracted from COLO320 cell. Cells were treated with 25 μg mL−1 of compounds and analysed after 24 h. (i) std on the extreme left lane represents 1 kb DNA ladder size marker. (ii) std on the extreme right lane represents 100 bp ladder. (iii) Trailing observed in compounds 5h and 5v. | |
Conclusions
For the first time, a series of hitherto unknown spirooxindoles bearing thienopyridine ring system was synthesized by Cu(OTf)2 catalyzed three component reaction. The chemistry is simple and efficient to prepare a wide range of spirooxindole derivatives in a single-step operation at good to excellent yields. All the synthesized compounds were evaluated for their cytotoxic potency towards COLO320 cells. Out of the 26 compounds screened, 20 compounds exhibited cytotoxicity with the IC50 values ranging between 49.1 and 57.3 μM. Our overall results from biological assay, molecular docking and DNA fragmentation analysis demonstrated that 5v is the most active compound interms of its low IC50 value (49.1 μM), largest free energy of binding (−10.5 kcal mol−1) and DNA trailing. Detailed mechanistic studies, lead optimization of the spirooxindole hybrids and evaluation of the most active compound 5v in in vivo model is underway in our laboratory and the results will be disclosed in due course.
Experimental section
All commercially available solvents and reagents were used without further purification. Melting points were determined in capillary tubes and are uncorrected. Infrared (IR) spectra were recorded on a Perkin-Elmer FTIR spectrophotometer as KBr pellets. 1H and 13C NMR spectra were obtained in CDCl3, DMSO-d6 on a Bruker spectrometer at 400 and 100 MHz, respectively. Proton chemical shifts (δ) are relative to tetramethylsilane (TMS, δ = 0.00) as internal standard and expressed in parts per million. Spin multiplicities are given as s (singlet), d (doublet), t (triplet) and m (multiplet). Coupling constants (J) are given in hertz. Mass spectra were recorded on a PE-SCIEX API 3000 mass spectrometer. Elemental analyses were recorded using a Thermo Finnigan FLASH EA 1112CHN analyzer. All the compounds gave C, H and N analysis within ±0.5% of the theoretical values. Analytical TLC was performed on precoated plastic sheets of silica gel G/UV-254 of 0.2 mm thickness (Macherey-Nagel, Germany) using analytical grade solvents and visualized with iodine spray (10% (w/w) I2 in silica gel) or UV light (λ = 254 and 365 nm). Cytotoxicity was determined by measuring the absorbance at 540 nm in an ELISA reader and were statistically analyzed by Duncan multiple range test at P = 0.05 with the help of SPSS 11.5 version software package. COLO320 adenocarcinoma colorectal cancer cell line was obtained from National Institute of Cell Sciences, Pune. Cytotoxicity was determined by measuring the absorbance at 570 nm in an ELISA reader and were statistically analyzed by Duncan multiple range test at P = 0.05 with the help of SPSS 11.5 version software package. All computations for molecular docking studies were carried out on a Dell Desktop D510 personal computer (2.4 GHz Pentium 4 processor, Intel, Santa Clara, CA) running Red Hat Enterprise Linux Client release 5.5. The time required for each simulation run was on the order of Real = 1 h 13 m 54.67 s, CPU = 1 h 08 m 54.57 s, System = 11.75 s on the Pentium machine.
Typical experimental procedure for the N-alkylation of thiolactone
To a solution of 5,6,7,7a-tetrahydro-4H-thieno[3,2-c]-2-pyridone hydrochloride 1 (5.2 mmol) in 10 mL of DMF were added potassium bicarbonate (10.4 mmol) and sodium iodide (5.2 mmol). This reaction mixture was stirred for 30 minutes at room temperature and added alkyl halide (5.4 mmol) and heated to 60 °C for 90 minutes. After completion of the reaction as indicated by TLC, the reaction mixture was quenched with ice-cold water (60 mL) and extracted with ethyl acetate (3 × 20 mL). The combined organic layers were dried over anhydrous sodium sulphate, filtered and concentrated under reduced pressure. The resulting crude product was further purified by column chromatography on silica gel (100–200 mesh) using petroleum ether–EtOAc to afford analytically pure compound 2, which was stored at −2 °C under nitrogen atmosphere for further use.
5-Trityl-4,6,7,7a-tetrahydrothieno[3,2-c]pyridin-2-one (2a). Off-white solid; Mp 97–99 °C; 1H NMR (400 MHz, CDCl3): δH 1.63 (m, 1H), 2.04–2.14 (m, 1H), 2.15–2.18 (m, 1H), 2.34–2.39 (m, 1H), 3.30 (dd, J = 12.2 & 2.9 Hz, 1H), 3.93–3.97 (m, 1H), 4.09 (dd, J = 12.0 & 2.4 Hz, 1H), 6.07 (s, 1H), 7.12–7.19 (m, 4H), 7.26–7.29 (m, 4H), 7.45 (m, 7H); 13C NMR (100 MHz, CDCl3): δC 35.3, 47.4, 47.7, 50.8, 51.7, 125.6, 126.4, 127.8, 129.2, 129.3, 129.4, 170.0, 199.4; MS(ESI): m/z = 398 [M + H]+.
5-Benzyl-4,6,7,7a-tetrahydrothieno[3,2-c]pyridin-2-one (2b). Off-white solid; Mp 72–74 °C; 1H NMR (400 MHz, CDCl3): δH 1.83 (m, 1H), 2.35–2.41 (m, 2H), 2.89 (d, J = 12.0 Hz, 1H), 3.04–3.07 (m, 1H), 3.66 (s, 2H), 3.81 (d, J = 12.0 Hz, 1H), 4.13 (dd, J = 12.0 & 5.5 Hz, 1H), 6.02 (s, 1H), 7.27–7.34 (m, 5H); 13C NMR (100 MHz, CDCl3): δC 34.1, 51.4, 51.9, 54.7, 126.5, 127.6, 128.6 (2C), 129.1 (2C), 137.4, 168.4, 198.9; MS (ESI): m/z = 246 [M + H]+.
5-[(2-Fluorophenyl)methyl]-4,6,7,7a-tetrahydrothieno[3,2-c]pyridin-2-one (2c). Off-white solid; Mp 81–83 °C; 1H NMR (400 MHz, CDCl3): δH 1.82 (m, 1H), 2.28–2.34 (m, 1H), 2.45–2.48 (m, 1H), 2.94 (d, J = 12.0 Hz, 1H), 3.13 (m, 1H), 3.84 (s, 2H), 3.89 (d, J = 12.0 Hz, 1H), 4.08 (dd, J = 12.1 & 5.6 Hz, 1H), 6.05 (s, 1H), 7.21–7.33 (m, 2H), 7.48–7.55 (m, 2H); 13C NMR (100 MHz, CDCl3): δC 35.0, 47.4, 51.8, 53.7, 63.4, 115.4, 123.8, 125.6, 126.4, 127.8, 129.4, 161.4, 163.9, 170.0, 198.4; MS(ESI): m/z = 264 [M + H]+.
5-[(2,6-Difluorophenyl)methyl]-4,6,7,7a-tetrahydrothieno[3,2-c]pyridin-2-one (2d). White solid; Mp 90–92 °C; 1H NMR (400 MHz, CDCl3): δH 1.81 (m, 1H), 2.25–2.34 (m, 1H), 2.42–2.48 (m, 1H), 2.97 (d, J = 12.0 Hz, 1H), 3.09 (m, 1H), 3.84 (s, 2H), 3.86 (d, J = 12.0 Hz, 1H), 4.06 (dd, J = 12.1 & 5.6 Hz, 1H), 6.07 (s, 1H), 6.83–6.94 (m, 2H), 7.6129–7.33 (m, 1H); 13C NMR (100 MHz, CDCl3): δC 35.0, 47.2, 47.6, 50.8, 61.9, 109.8, 110.3 (2C), 122.8, 128.7, 160.2, 161.3, 163.8, 197.3; MS(ESI): m/z = 282 [M + H]+.
Methyl-2-[4-[(2-oxo-4,6,7,7a-tetrahydrothieno[3,2-c]pyridin-5-yl)methyl]phenyl]benzoate (2e). Pale yellow solid; Mp 112–114 °C; 1H NMR (400 MHz, CDCl3): δH 1.75 (m, 1H), 2.07–2.10 (m, 1H), 2.13–2.19 (m, 1H), 2.37–2.41 (m, 1H), 2.97 (d, J = 12.0 Hz, 1H), 3.35 (m, 1H), 3.90 (d, J = 12.0 Hz, 1H), 3.80–3.85 (m, 1H), 3.92 (s, 3H), 4.08 (dd, J = 12.0 & 2.4 Hz, 1H), 6.08 (s, 1H), 7.19 (d, J = 7.9 Hz, 2H), 7.26 (d, J = 7.9 Hz, 2H), 7.50 (m, 1H), 7.65 (m, 2H), 8.44 (d, J = 7.7 Hz, 1H); 13C NMR (100 MHz, CDCl3): δC 35.3, 47.0, 50.8, 51.7, 62.3, 62.5, 120.9, 123.0, 126.8, 127.8 (2C), 128.2, 129.3 (2C), 130.4, 133.3, 135.1, 137.9, 138.1, 163.2, 167.2, 198.3; MS(ESI): m/z = 380 [M + H]+.
5-(Prop-2-ynyl)-5,6,7,7a-tetrahydrothieno[3,2-c]pyridine-2(4H)-one (2f). Brown solid; Mp 59–61 °C; 1H NMR (400 MHz, CDCl3): δH 1.95 (m, 1H), 2.06–2.10 (m, 1H), 2.14–2.18 (m, 1H), 2.38–2.40 (m, 1H), 2.65 (m, 1H), 2.97 (d, J = 12.0 Hz, 1H), 3.07 (d, J = 12.0 Hz, 1H), 3.90 (s, 2H), 4.28 (m, 1H), 6.35 (s, 1H); 13C NMR (100 MHz, CDCl3): δC 34.3, 46.3, 47.0, 50.8, 61.7, 72.8, 79.2, 123.0, 163.2, 196.3; MS(ESI): m/z = 194 [M + H]+.
Typical experimental procedure for the synthesis of spirooxindoles
To an equimolar solution of isatin 3 (1.0 mmol), malononitrile 4 (1.0 mmol) in dichloroethane (1 mL) was added Cu(OTf)2 (0.018 mmol) and the reaction mixture was stirred at room temperature for 10 min. To this reaction mixture was added N-alkyl thienopyridinone 2 (1.0 mmol) and stirred at room temperature for 4 h. After completion of the reaction as indicated by TLC, the reaction mixture was quenched with water (10 mL) and extracted with dichloromethane (3 × 10 mL). The combined layers were dried over anhydrous sodium sulphate, filtered and concentrated under reduced pressure. The resulting crude material was purified by column chromatography over silica gel (100–200 mesh) using cyclohexane–EtOAc to afford the pure product of spirooxindole 5.
5′-Amino-2-oxo-12′-(triphenylmethyl)-1,2-dihydro-6′-oxa-8′-thia-12′-azaspiro[indole-3,3′-tricyclo[7.4.0.02,7]tridecane]-1′(9′),2′(7′),4′-triene-4′-carbonitrile (5a). Off-white solid; Mp 130–132 °C; IR (KBr): 3350, 3176, 2190, 1708, 1650, 1579, 1459, 1262, 1042, 761 cm−1. 1H NMR (400 MHz, DMSO-d6) δH 2.09–2.33 (m, 2H), 2.51 (m, 2H), 2.76 (m, 2H), 6.43 (d, J = 7.7 Hz, 1H), 6.86 (d, J = 7.4 Hz, 1H), 6.97 (d, J = 7.2 Hz, 1H), 7.03 (t, J = 7.2 Hz, 1H), 7.15 (m, 9H), 7.20 (m, 6H), 7.25 (bs, 2H, D2O exchangeable), 10.24 (bs, 1H, D2O exchangeable); 13C NMR (100 MHz, DMSO-d6) δC 25.3, 45.0, 45.7, 55.6, 56.1, 76.4, 109.3, 111.4, 118.1, 121.4, 122.6, 124.6, 126.0, 127.6, 128.5, 129.0, 129.2, 131.6, 141.3, 148.9, 161.3, 177.0; MS(ESI): m/z = 593 [M + H]+, anal. calcd for C37H28N4O2S: C, 74.98; H, 4.76; N, 9.45%, found C, 74.95; H, 4.81; N, 9.40%.
5′-Amino-5-chloro-2-oxo-12′-(triphenylmethyl)-1,2-dihydro-6′-oxa-8′-thia-12′-azaspiro[indole-3,3′-tricyclo[7.4.0.02,7]tridecane]-1′(9′),2′(7′),4′-triene-4′-carbonitrile (5b). Off-white solid; Mp 141–143 °C; IR (KBr): 3344, 31![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 666, 2191, 1707, 1651, 1569, 1459, 1252, 1045, 767 cm−1. 1H NMR (400 MHz, DMSO-d6) δH 2.12–2.37 (m, 2H), 2.52 (m, 2H), 2.80 (m, 2H), 6.42 (d, J = 7.8 Hz, 1H), 6.84 (d, J = 7.7 Hz, 1H), 6.94 (d, J = 1.7 Hz, 1H), 7.17 (m, 9H), 7.25 (m, 6H), 7.34 (bs, 2H, D2O exchangeable), 10.25 (bs, 1H, D2O exchangeable); 13C NMR (100 MHz, DMSO-d6) δC 25.4, 45.0, 45.6, 55.7, 56.3, 76.5, 109.3, 111.5, 118.2, 121.3, 122.5, 124.7, 126.1, 127.7, 128.3, 129.1, 129.3, 131.7, 141.0, 148.9, 161.0, 177.4; MS(ESI): m/z = 627 [35M + H]+, 629 [37M + H]+; anal. calcd for C37H27ClN4O2S: C, 70.86; H, 4.34; N, 8.93%, found C, 70.91; H, 4.37; N, 8.95%.
666, 2191, 1707, 1651, 1569, 1459, 1252, 1045, 767 cm−1. 1H NMR (400 MHz, DMSO-d6) δH 2.12–2.37 (m, 2H), 2.52 (m, 2H), 2.80 (m, 2H), 6.42 (d, J = 7.8 Hz, 1H), 6.84 (d, J = 7.7 Hz, 1H), 6.94 (d, J = 1.7 Hz, 1H), 7.17 (m, 9H), 7.25 (m, 6H), 7.34 (bs, 2H, D2O exchangeable), 10.25 (bs, 1H, D2O exchangeable); 13C NMR (100 MHz, DMSO-d6) δC 25.4, 45.0, 45.6, 55.7, 56.3, 76.5, 109.3, 111.5, 118.2, 121.3, 122.5, 124.7, 126.1, 127.7, 128.3, 129.1, 129.3, 131.7, 141.0, 148.9, 161.0, 177.4; MS(ESI): m/z = 627 [35M + H]+, 629 [37M + H]+; anal. calcd for C37H27ClN4O2S: C, 70.86; H, 4.34; N, 8.93%, found C, 70.91; H, 4.37; N, 8.95%.
Methyl-5′-amino-5-chloro-2-oxo-12′-(triphenylmethyl)-1,2-dihydro-6′-oxa-8′-thia-12′-azaspiro[indole-3,3′-tricyclo[7.4.0.02,7]tridecane]-1′(9′),2′(7′),4′-triene-4′-carboxylate (5c). Off-white solid; Mp 120–122 °C; IR (KBr): 3353, 3169, 1728, 1654, 1569, 1459, 1252, 1052, 767 cm−1. 1H NMR (400 MHz, DMSO-d6) δH 1.84 (m, 1H), 2.51–2.66 (m, 4H), 2.88 (m, 1H), 3.21 (s, 3H), 6.22 (d, J = 8.2 Hz, 1H), 6.84 (d, J = 7.6 Hz, 1H), 6.92 (d, J = 1.6 Hz, 1H), 7.22 (m, 15H), 7.89 (bs, D2O exchangeable, 2H), 10.26 (bs, D2O exchangeable, 1H); 13C NMR (100 MHz, DMSO-d6) δC 25.5, 44.9, 45.5, 50.2, 56.0, 74.4, 76.5, 109.3, 112.3, 120.9, 123.2, 125.5, 126.0, 127.6, 128.3, 129.3, 136.6, 141.1, 148.1, 161.1, 167.8, 178.6; MS(ESI): m/z = 658 [35M − H]−, 660 [37M − H]−; anal. calcd for C38H30ClN3O4S: C, 69.13; H, 4.58; N, 6.36%, found C, 68.98; H, 4.51; N, 6.40%.
Methyl-5′-amino-5-bromo-2-oxo-12′-(triphenylmethyl)-1,2-dihydro-6′-oxa-8′-thia-12′-azaspiro[indole-3,3′-tricyclo[7.4.0.02,7]tridecane]-1′(9′),2′(7′),4′-triene-4′-carboxylate (5d). Pale brown solid; Mp 155–157 °C; IR (KBr): 3434, 3085, 1720, 1686, 1505, 1472, 1259, 1011, 712 cm−1. 1H NMR (400 MHz, DMSO-d6) δH 1.83 (m, 1H), 2.61–2.88 (m, 5H), 3.21 (s, 3H), 6.19 (d, J = 8.2 Hz, 1H), 7.03 (m, 2H), 7.22 (m, 15H), 7.89 (bs, 2H, D2O exchangeable), 10.27 (bs, 1H, D2O exchangeable); 13C NMR (100 MHz, DMSO-d6) δC 25.5, 44.9, 45.5, 50.3, 56.1, 74.4, 76.5, 109.9, 112.3, 113.3, 120.9, 125.9, 127.7, 128.4, 129.3, 131.2, 136.9, 141.5, 148.1, 161.1, 167.8, 178.5; MS(ESI): m/z = 702 [79M − H]−, 704 [81M − H]−; anal. calcd for C38H30BrN3O4S: C, 64.77; H, 4.29; N, 5.96%, found C, 64.95; H, 4.31; N, 6.00%.
Ethyl-5′-amino-2-oxo-12′-(triphenylmethyl)-1,2-dihydro-6′-oxa-8′-thia-12′-azaspiro[indole-3,3′-tricyclo[7.4.0.02,7]tridecane]-1′(9′),2′(7′),4′-triene-4′-carboxylate (5e). Off white solid; Mp 187–189 °C; IR (KBr): 3444, 3089, 1729, 1680, 1507, 1462, 1255, 1019, 712 cm−1. 1H NMR (400 MHz, DMSO-d6) δH 0.58 (t, J = 7.0 Hz, 3H), 1.83 (m, 1H), 2.61–2.88 (m, 5H), 3.53 (m, 1H), 3.99 (m, 1H), 6.40 (d, J = 7.7 Hz, 1H), 6.86 (d, J = 7.7 Hz, 1H), 6.97 (d, J = 7.2 Hz, 1H), 7.03 (t, J = 7.2 Hz, 1H), 7.15 (m, 9H), 7.20 (m, 6H), 7.35 (bs, 2H, D2O exchangeable), 10.20 (bs, 1H, D2O exchangeable); 13C NMR (100 MHz, DMSO-d6) δC 13.7, 25.5, 46.2, 46.9, 51.0, 62.3, 74.4, 77.5, 109.9, 112.3, 113.3, 115.4, 120.9, 125.9, 127.7, 128.4, 129.3, 131.2, 136.9, 141.5, 148.1, 161.1, 167.8, 178.5; MS(ESI): m/z = 640 [M + H]+; anal. calcd for C39H33N3O4S: C, 73.22; H, 5.20; N, 6.57%, found C, 73.02; H, 5.16; N, 6.52%.
5′-Amino-1-ethyl-2-oxo-12′-(triphenylmethyl)-1,2-dihydro-6′-oxa-8′-thia-12′-azaspiro[indole-3,3′-tricyclo[7.4.0.02,7]tridecane]-1′(9′),2′(7′),4′-triene-4′-carbonitrile (5f). Off-white solid; Mp 171–173 °C; IR (KBr): 3489, 3163, 2192, 1698, 1634, 1485, 1399, 1251, 1007, 711 cm−1. 1H NMR (400 MHz, CDCl3) δH 1.14 (t, J = 7.1 Hz, 3H), 1.90–2.02 (m, 2H), 2.60–2.79 (m, 2H), 2.85 (m, 2H), 3.71 (m, 2H), 4.78 (bs, D2O exchangeable, 2H), 6.62 (d, J = 7.8 Hz, 1H), 6.95–7.02 (m, 2H), 7.08–7.11 (m, 3H), 7.15–7.18 (m, 6H), 7.23 (m, 7H); 13C NMR (100 MHz, DMSO-d6) δC 12.5, 25.3, 34.3, 44.9, 45.7, 48.7, 55.6, 76.4, 108.4, 111.0, 117.7, 121.7, 123.2, 124.5, 126.0, 127.7, 128.5, 128.8, 129.5, 131.5, 141.4, 149.0, 160.9, 174.8; MS(ESI): m/z = 643 [M + Na]+, anal. calcd for C39H32N4O2S: C, 75.46; H, 5.20; N, 9.03%, found C, 75.51; H, 5.17; N, 8.94%.
5′-Amino-2-oxo-1-(prop-2-en-1-yl)-12′-(triphenylmethyl)-1,2-dihydro-6′-oxa-8′-thia-12′-azaspiro[indole-3,3′-tricyclo[7.4.0.02,7]tridecane]-1′(9′),2′(7′),4′-triene-4′-carbonitrile (5g). Off-white solid; Mp 111–113 °C; IR (KBr): 3433, 3057, 2195, 1724, 1642, 1612, 1582, 1467, 1249, 1034, 712 cm−1. 1H NMR (400 MHz, DMSO-d6) δH 1.90 (m, 2H), 2.62 (m, 2H), 2.89 (m, 2H), 3.77 (m, 2H), 4.10 (d, J = 17.0 Hz, 1H), 5.06 (d, J = 10.3 Hz, 1H), 5.56 (d, J = 17.3 Hz, 1H), 6.71 (d, J = 7.7 Hz, 1H), 6.94–7.01 (m, 2H), 7.16 (m, 6H), 7.21 (m, 7H), 7.23 (m, 3H), 7.32 (bs, D2O exchangeable, 2H); 13C NMR (100 MHz, DMSO-d6) δC 25.3, 41.5, 45.0, 45.6, 45.7, 48.9, 55.2, 76.4, 108.9, 111.1, 116.5, 117.9, 121.8, 123.4, 124.5, 126.0, 126.2, 127.5, 127.7, 128.5, 128.7, 128.8, 129.4, 130.9, 131.3, 141.6, 149.5, 161.2, 175.1; MS(ESI): m/z = 655 [M + Na]+, anal. calcd for C40H32N4O2S: C, 75.92; H, 5.10; N, 8.85%, found C, 75.89; H, 5.05; N, 8.89%.
5′-Amino-2-oxo-1-(prop-2-yn-1-yl)-12′-(triphenylmethyl)-1,2-dihydro-6′-oxa-8′-thia-12′-azaspiro[indole-3,3′-tricyclo[7.4.0.02,7]tridecane]-1′(9′),2′(7′),4′-triene-4′-carbonitrile (5h). Off-white solid; Mp 152–154 °C; IR (KBr): 3402, 3164, 2193, 1704, 1640, 1559, 1489, 1242, 1037, 758 cm−1. 1H NMR (400 MHz, CDCl3) δH 2.56–2.59 (m, 2H), 2.97 (m, 2H), 3.06 (m, 1H), 3.43 (m, 2H), 3.47 (d, J = 13.6 Hz, 1H), 4.55 (d, J = 13.6 Hz, 1H), 4.83 (bs, D2O exchangeable, 2H), 6.65 (d, J = 7.7 Hz, 1H), 6.84 (t, J = 7.7 Hz, 1H), 6.93 (d, J = 7.2 Hz, 1H), 7.03 (t, J = 7.2 Hz, 1H), 7.15 (m, 9H), 7.20 (m, 6H); 13C NMR (100 MHz, DMSO-d6) δC 25.3, 32.6, 45.0, 49.3, 59.6, 68.9, 71.2, 76.4, 109.0, 111.5, 118.7, 121.0, 122.2, 124.2, 126.3, 127.8, 128.1, 129.0, 129.4, 131.7, 141.3, 148.5, 161.8, 177.4; MS(ESI): m/z = 653 [M + Na]+, anal. calcd for C40H30N4O2S: C, 76.17; H, 4.79; N, 8.88%, found C, 76.45; H, 4.81; N, 9.00%.
Ethyl-5′-amino-2-oxo-1-(prop-2-en-1-yl)-12′-(triphenylmethyl)-1,2-dihydro-6′-oxa-8′-thia-12′-azaspiro[indole-3,3′-tricyclo[7.4.0.02,7]tridecane]-1′(9′),2′(7′),4′-triene-4′-carboxylate (5i). Off-white solid; Mp 177–179 °C; IR (KBr): 3353, 3076, 1738, 1660, 1569, 1454, 1272, 1052, 759 cm−1. 1H NMR (400 MHz, CDCl3) δH 0.58 (t, J = 7.0 Hz, 3H), 1.89 (m, 2H), 2.60 (d, J = 14.7 Hz, 1H), 2.87 (m, 2H), 3.04 (m, 2H), 3.54 (m, 1H), 4.00 (m, 1H), 4.25 (d, J = 14.7 Hz, 1H), 4.79 (bs, D2O exchangeable, 2H), 5.18 (d, J = 10.1 Hz, 1H), 5.28 (d, J = 17.0 Hz, 1H), 5.57 (m. 1H), 6.57 (d, J = 7.8 Hz, 1H), 6.85 (m, 2H), 7.10–7.14 (m, 4H), 7.19–7.22 (m, 6H), 7.31 (m, 6H); 13C NMR (100 MHz, CDCl3) δC 13.9, 26.3, 43.1, 45.4, 45.8, 49.6, 61.2, 78.9, 107.9, 113.0, 118.9, 121.9, 122.9, 123.8, 126.0, 127.8, 127.9, 129.2, 129.7, 132.4, 135.3, 142.9, 148.6, 161.4, 168.6, 177.4; MS(ESI): m/z = 680 [M + H]+, anal. calcd for C42H37N3O4S: C, 74.20; H, 5.49; N, 6.18%, found C, 74.15; H, 5.55; N, 6.30%.
5′-Amino-12′-benzyl-2-oxo-1,2-dihydro-6′-oxa-8′-thia-12′-azaspiro[indole-3,3′-tricyclo[7.4.0.02,7]tridecane]-1′(9′),2′(7′),4′-triene-4′-carbonitrile (5j). Off-white solid; Mp 194–196 °C; IR (KBr): 3454, 3129, 2190, 1688, 1655, 1640, 1621, 1463, 1379, 1256, 1154, 760 cm−1. 1H NMR (400 MHz, CDCl3) δH 2.23 (d, J = 13.8 Hz, 1H), 2.71 (m, 4H), 2.83 (d, J = 13.8 Hz, 1H), 3.42 (m, 2H), 4.83 (bs, D2O exchangeable, 2H), 6.74 (d, J = 7.6 Hz, 1H), 7.01 (t, J = 7.6 Hz, 1H), 7.09–7.13 (m, 3H), 7.19 (t, J = 7.6 Hz, 1H), 7.27 (m, 3H), 7.49 (bs, D2O exchangeable, 1H); 13C NMR (100 MHz, CDCl3) δC 24.8, 49.6, 49.7, 49.8, 57.8, 61.9, 110.2, 111.0, 117.9, 122.5, 123.1, 125.1, 127.0, 128.2, 128.3, 129.0, 129.3, 131.8, 137.4, 140.8, 149.6, 161.1, 177.7; MS(ESI): m/z = 441 [M + H]+, anal. calcd for C25H20N4O2S: C, 68.16; H, 4.58; N, 12.72%, found C, 68.08; H, 4.55; N, 12.67%.
5′-Amino-12′-benzyl-1-ethyl-2-oxo-1,2-dihydro-6′-oxa-8′-thia-12′-azaspiro[indole-3,3′-tricyclo[7.4.0.02,7]tridecane]-1′(9′),2′(7′),4′-triene-4′-carbonitrile (5k). Off-white solid; Mp 181–183 °C; IR (KBr): 3459, 3134, 2193, 1698, 1650, 1642, 1611, 1468, 1369, 1246, 1156, 764 cm−1. 1H NMR (400 MHz, CDCl3) δH 1.18 (t, J = 7.2 Hz, 1H), 2.12 (d, J = 14.6 Hz, 1H), 2.62 (d, J = 14.6 Hz, 1H), 2.67 (m, 4H), 3.33 (td, J = 13.6 Hz, 2H), 3.49–3.73 (m, 2H), 4.88 (bs, 2H, D2O exchangeable), 6.69 (d, J = 7.8 Hz, 1H), 6.99 (t, J = 7.4 Hz, 1H), 7.07 (m, 3H), 7.23 (m, 4H); 13C NMR (100 MHz, CDCl3) δC 12.7, 24.9, 35.2, 49.3, 49.9, 50.4, 59.9, 62.4, 108.8, 111.1, 117.4, 123.2, 123.7, 125.4, 127.3, 128.3, 128.4 (2C), 129.1 (2C), 129.7, 131.6, 137.8, 141.9, 149.9, 160.9, 175.6; MS(ESI): m/z = 469 [M + H]+. Anal. calcd for C27H24N4O2S: C, 69.21; H, 5.16; N, 11.96%, found C, 69.11; H, 5.09; N, 11.84%.
5′-Amino-1,12′-dibenzyl-2-oxo-1,2-dihydro-6′-oxa-8′-thia-12′-azaspiro[indole-3,3′-tricyclo[7.4.0.02,7]tridecane]-1′(9′),2′(7′),4′-triene-4′-carbonitrile (5l). Off-white solid; Mp 169–171 °C; IR (KBr): 3412, 3063, 2195, 1715, 1650, 1621, 1466, 1394, 1246, 1154, 757 cm−1. 1H NMR (400 MHz, CDCl3) δH 1.96 (d, J = 14.6 Hz, 1H), 2.51 (d, J = 14.6 Hz, 1H), 2.64 (m, 4H), 3.19 (td, J = 14.7 Hz, 2H), 4.38 (d, J = 15.6 Hz, 1H), 4.84 (bs, 2H, D2O exchangeable), 4.91 (d, J = 15.6 Hz, 1H), 6.57 (d, J = 7.7 Hz, 1H), 6.96 (t, J = 7.4 Hz, 1H), 7.02 (m, 2H), 7.08 (d, J = 7.4 Hz, 1H), 7.10 (t, J = 8.3 Hz, 1H), 7.25 (m, 3H), 7.32 (m, 5H); 13C NMR (100 MHz, CDCl3) δC 24.9, 44.2, 49.3, 49.8, 50.2, 59.9, 62.2, 109.5, 110.6, 111.1, 117.4, 123.1, 123.7, 125.1, 126.9, 127.1, 127.5, 127.7, 127.8, 128.2, 128.3, 128.9, 129.0, 129.1, 129.5, 131.1, 135.3, 137.8, 141.9, 149.6, 160.7, 175.8; MS(ESI): m/z = 531 [M + H]+. Anal. calcd for C32H26N4O2S: C, 72.43; H, 4.94; N, 10.56%, found C, 72.21; H, 4.90; N, 10.44%.
5′-Amino-12′-benzyl-5-chloro-2-oxo-1,2-dihydro-6′-oxa-8′-thia-12′-azaspiro[indole-3,3′-tricyclo[7.4.0.02,7]tridecane]-1′(9′),2′(7′),4′-triene-4′-carbonitrile (5m). Pale brown solid; Mp 138–140 °C; IR (KBr): 3450, 3120, 2191, 1678, 1665, 1650, 1629, 1473, 1372, 1251, 1150, 769 cm−1. 1H NMR (400 MHz, CDCl3) δH 2.20 (d, J = 13.7 Hz, 1H), 2.69 (m, 4H), 2.88 (d, J = 13.7 Hz, 1H), 3.39 (m, 2H), 4.80 (bs, D2O exchangeable, 2H), 6.76 (d, J = 7.6 Hz, 1H), 7.04 (d, J = 1.6 Hz, 1H), 7.09–7.13 (m, 3H), 7.19 (m, 1H), 7.27 (m, 2H), 7.51 (bs, D2O exchangeable, 1H); 13C NMR (100 MHz, CDCl3) δC 24.5, 49.2, 49.5, 49.9, 58.8, 61.9, 110.2, 111.0, 117.9, 120.5, 123.3, 125.5, 127.1, 128.5 (2C), 128.7, 129.0 (2C), 129.4, 133.9, 137.4, 140.8, 149.6, 162.7, 178.7; MS(ESI): m/z = 475 [35M + H]+, 477 [37M + H]+; anal. calcd for C25H19ClN4O2S: C, 63.22; H, 4.03; N, 11.80%, found C, 63.08; H, 4.05; N, 11.67%.
Methyl-5′-amino-12′-benzyl-2-oxo-1,2-dihydro-6′-oxa-8′-thia-12′-azaspiro[indole-3,3′-tricyclo[7.4.0.02,7]tridecane]-1′(9′),2′(7′),4′-triene-4′-carbonitrile (5n). Off-white solid; Mp 141–143 °C; IR (KBr): 3338, 3070, 1738, 1675, 1660, 1649, 1479, 1382, 1250, 1156, 760 cm−1. 1H NMR (400 MHz, CDCl3) δH 2.18 (d, J = 13.5 Hz, 1H), 2.62 (m, 4H), 2.85 (d, J = 13.5 Hz, 1H), 3.21 (s, 3H), 3.49 (m, 2H), 4.78 (bs, D2O exchangeable, 2H), 6.73 (d, J = 7.9 Hz, 1H), 7.00 (t, J = 7.8 Hz, 1H), 7.09–7.13 (m, 3H), 7.19 (t, J = 7.6 Hz, 1H), 7.27 (m, 3H), 7.53 (bs, D2O exchangeable, 1H); 13C NMR (100 MHz, CDCl3) δC 24.5, 48.2, 49.5, 49.9, 52.8, 58.7, 79.3, 110.6, 111.3, 117.4, 120.2, 122.0, 123.6, 125.1, 127.5, 128.1, 128.3, 129.3, 129.7, 133.7, 137.5, 140.9, 149.5, 162.7, 168.2, 178.7; MS (ESI): m/z = 474 [M + H]+; anal. calcd for C26H23N3O4S: C, 65.94; H, 4.90; N, 8.87%, found C, 65.90; H, 4.89; N, 8.82%.
5′-Amino-12′-[(2-fluorophenyl)methyl]-2-oxo-1,2-dihydro-6′-oxa-8′-thia-12′-azaspiro[indole-3,3′-tricyclo[7.4.0.02,7]tridecane]-1′(9′),2′(7′),4′-triene-4′-carbonitrile (5o). Off-white solid; Mp 153–155 °C; IR (KBr): 3357, 3175, 2193, 1705, 1650, 1576, 1458, 1403, 1268, 1045, 761 cm−1. 1H NMR (400 MHz, CDCl3) δH 2.29 (d, J = 14.7 Hz, 1H), 2.69–2.75 (m, 4H), 2.87 (d, J = 14.7 Hz, 1H), 3.49 (s, 2H), 4.77 (bs, D2O exchangeable, 2H), 6.79 (t, J = 7.8 Hz, 1H), 6.95–7.04 (m, 2H), 7.07–7.13 (m, 2H), 7.18–7.22 (m, 3H), 7.39 (bs, D2O exchangeable, 1H); 13C NMR (100 MHz, CDCl3) δC 24.4, 48.8, 49.9, 53.9, 55.7, 56.0, 109.9, 111.2, 115.1, 115.3, 118.1, 121.7, 122.5, 123.5, 123.7, 124.1, 124.6, 128.1, 129.2, 129.3, 131.3, 131.4, 132.0, 141.3, 148.9, 159.4, 161.0, 161.8, 177.2; MS(ESI): m/z = 459 [M + H]+, anal. calcd for C25H19FN4O2S: C, 65.49; H, 4.18; N, 12.22%, found C, 65.42; H, 4.13; N, 12.29%.
5′-Amino-1-benzyl-12′-[(2-fluorophenyl)methyl]-2-oxo-1,2-dihydro-6′-oxa-8′-thia-12′-azaspiro[indole-3,3′-tricyclo[7.4.0.02,7]tridecane]-1′(9′),2′(7′),4′-triene-4′-carbonitrile (5p). Off-white solid; Mp 119–121 °C; IR (KBr): 3350, 3165, 2191, 1700, 1647, 1586, 1459, 1400, 1263, 1043, 769 cm−1. 1H NMR (400 MHz, CDCl3) δH 2.28 (d, J = 14.6 Hz, 1H), 2.63–2.70 (m, 4H), 2.88 (d, J = 14.6 Hz, 1H), 3.14 (td, J = 14.4 Hz, 2H), 3.39 (s, 2H), 4.79 (bs, D2O exchangeable, 2H), 6.77 (t, J = 7.8 Hz, 1H), 6.95–7.04 (m, 2H), 7.07–7.13 (m, 3H), 7.18–7.22 (m, 3H), 7.39 (m, 2H), 7.44 (m, 2H); 13C NMR (100 MHz, CDCl3) δC 24.4, 47.8, 49.4, 49.9, 55.3, 55.7, 59.8, 109.7, 111.5, 115.1, 115.3, 118.1, 121.4, 122.7, 123.7, 123.9, 124.1, 124.7, 126.5, 126.9 (2C), 128.1 (2C), 128.4, 129.4, 129.5, 131.4, 131.5, 132.2, 136.2, 141.4, 148.6, 159.5, 161.0, 161.9, 177.4; MS(ESI): m/z = 548 [M + H]+, anal. calcd for C32H25FN4O2S: C, 70.05; H, 4.59; N, 10.21%, found C, 70.03; H, 4.53; N, 10.29%.
5′-Amino-12′-[(2-fluorophenyl)methyl]-2-oxo-1-(prop-2-yn-1-yl)-1,2-dihydro-6′-oxa-8′-thia-12′-azaspiro[indole-3,3′-tricyclo[7.4.0.02,7]tridecane]-1′(9′),2′(7′),4′-triene-4′-carbonitrile (5q). Off-white solid; Mp 145–147 °C; IR (KBr): 3355, 3169, 2190, 1708, 1653, 1573, 1468, 1413, 1267, 1043, 760 cm−1; 1H NMR (400 MHz, CDCl3) δH 2.28 (d, J = 14.7 Hz, 1H), 2.65–2.77 (m, 4H), 2.81 (d, J = 14.7 Hz, 1H), 3.03 (t, J = 2.3 Hz, 1H), 3.49 (s, 2H), 4.55 (d, J = 2.2 Hz, 2H), 4.83 (bs, D2O exchangeable, 2H), 6.77 (t, J = 7.6 Hz, 1H), 6.90–7.04 (m, 3H), 7.09–7.18 (m, 2H), 7.18–7.22 (m, 2H); 13C NMR (100 MHz, CDCl3) δC 23.9, 32.8, 48.8, 51.9, 52.4, 53.0, 59.9, 62.2, 73.4, 109.9, 110.2, 115.2, 115.4, 117.9, 120.7, 122.8, 123.6, 123.8, 124.3, 124.7, 128.2, 129.3, 129.5, 131.3, 131.4, 132.6, 141.3, 148.8, 159.1, 161.1, 161.6, 177.9; MS(ESI): m/z = 497 [M + H]+, anal. calcd for C28H21FN4O2S: C, 67.73; H, 4.26; N, 11.28%, found C, 67.62; H, 4.23; N, 11.33%.
Methyl-5′-amino-5-chloro-12′-[(2-fluorophenyl)methyl]-2-oxo-1,2-dihydro-6′-oxa-8′-thia-12′-azaspiro[indole-3,3′-tricyclo[7.4.0.02,7]tridecane]-1′(9′),2′(7′),4′-triene-4′-carboxylate (5r). Off-white solid; Mp 131–133 °C; IR (KBr): 3402, 3205, 1735, 1670, 1570, 1438, 1413, 1288, 1043, 751 cm−1. 1H NMR (400 MHz, CDCl3) δH 2.28 (d, J = 14.7 Hz, 1H), 2.32–2.43 (m, 4H), 2.84 (d, J = 14.7 Hz, 1H), 3.20 (s, 3H), 3.52 (s, 2H), 4.85 (bs, D2O exchangeable, 2H), 6.80 (t, J = 7.8 Hz, 1H), 6.90 (d, J = 1.7 Hz, 1H), 7.07–7.13 (m, 2H), 7.15 (m, 1H), 7.21–7.25 (m, 2H), 7.44 (bs, D2O exchangeable, 1H); 13C NMR (100 MHz, CDCl3) δC 24.7, 48.8, 51.2, 52.3, 55.7, 59.7, 76.8, 109.8, 111.3, 115.1, 115.3, 118.1, 121.5, 122.4, 123.6, 123.8, 124.1, 124.7, 128.1, 129.2, 129.3, 131.4, 131.5, 132.0, 141.4, 148.5, 159.2, 161.0, 161.8, 178.2; MS(ESI): m/z = 524 [35M − H]−, 526 [37M − H]−; anal. calcd for C26H21ClFN3O4S: C, 59.37; H, 4.02; N, 7.99%, found C, 59.30; H, 4.03; N, 7.39%.
Ethyl-5′-amino-12′-[(2-fluorophenyl)methyl]-1-methyl-2-oxo-1,2-dihydro-6′-oxa-8′-thia-12′-azaspiro[indole-3,3′-tricyclo[7.4.0.02,7]tridecane]-1′(9′),2′(7′),4′-triene-4′-carboxylate (5s). Off-white solid; Mp 129–131 °C; IR (KBr): 3452, 3200, 1736, 1672, 1571, 1448, 1403, 1289, 1043, 751 cm−1. 1H NMR (400 MHz, CDCl3) δH 0.73 (t, J = 7.1 Hz, 3H), 2.26 (d, J = 14.5 Hz, 1H), 2.22–2.33 (m, 4H), 2.80 (d, J = 14.5 Hz, 1H), 3.40 (s, 3H), 3.45 (m, 1H), 3.52 (s, 2H), 3.98 (m, 1H), 4.85 (bs, D2O exchangeable, 2H), 6.89 (t, J = 7.6 Hz, 1H), 7.07–7.13 (m, 2H), 7.15–18 (m, 2H), 7.21–7.25 (m, 3H); 13C NMR (100 MHz, CDCl3) δC 14.0, 24.4, 35.2, 48.8, 51.2, 52.3, 52.5, 59.7, 76.8, 109.5, 111.2, 115.3, 115.6, 118.1, 121.4, 122.4, 123.4, 123.7, 124.0, 124.3, 128.0, 129.2, 129.4, 131.3, 131.4, 132.3, 141.7, 148.5, 159.6, 161.2, 162.0, 178.0; MS (ESI): m/z = 520 [M + H]+; anal. calcd for C28H26FN3O4S: C, 64.72; H, 5.04; N, 8.09%, found C, 64.70; H, 5.03; N, 8.12%.
5′-Amino-12′-[(2,6-difluorophenyl)methyl]-2-oxo-1,2-dihydro-6′-oxa-8′-thia-12′-azaspiro[indole-3,3′-tricyclo[7.4.0.02,7]tridecane]-1′(9′),2′(7′),4′-triene-4′-carbonitrile (5t). Off-white solid; Mp 125–127 °C; IR (KBr): 3359, 3181, 2191, 1736, 1708, 1653, 1586, 1469, 1400, 1258, 1036, 751 cm−1. 1H NMR (400 MHz, DMSO-d6) δH 2.08 & 2.69 (2 × d, J = 14.8 Hz, 2H), 2.64 (m, 4H), 3.44 (s, 2H), 6.85–6.89 (m, 2H), 6.95 (d, J = 7.2 Hz, 1H), 6.99 (t, J = 7.2 Hz, 2H), 7.17 (t, J = 7.6 Hz, 1H), 7.26 (bs, D2O exchangeable, 2H), 7.34–7.41 (m, 1H), 10.67 (bs, D2O exchangeable, 1H); 13C NMR (100 MHz, DMSO-d6) δC 24.6, 47.6, 48.1, 49.9, 55.7, 55.9, 110.1, 110.9, 111.2, 111.4, 111.7, 111.9, 112.1, 117.9, 121.6, 122.2, 124.4, 127.9, 129.2, 129.7, 129.8, 129.9, 131.9, 141.2, 148.9, 159.7, 159.8, 160.8, 162.1, 162.2, 177.1; MS(ESI): m/z = 477 [M + H]+, anal. calcd for C25H18F2N4O2S: C, 63.02; H, 3.81; N, 11.76%, found C, 63.09; H, 3.79; N, 11.79%.
5′-Amino-12′-[(2,6-difluorophenyl)methyl]-2-oxo-1-(prop-2-yn-1-yl)-1,2-dihydro-6′-oxa-8′-thia-12′-azaspiro[indole-3,3′-tricyclo[7.4.0.02,7]tridecane]-1′(9′),2′(7′),4′-triene-4′-carbonitrile (5u). Off-white solid; Mp 174–176 °C; IR (KBr): 3357, 3175, 2193, 1705, 1650, 1576, 1458, 1403, 1268, 1045, 761 cm−1. 1H NMR (400 MHz, CDCl3) δH 2.26–2.29 (m, 2H), 2.71 (m, 4H), 2.82 (d, J = 14.7 Hz, 1H), 3.49 (td, J = 12.0 Hz, 2H), 4.55 (s, 2H), 4.79 (bs, D2O exchangeable, 2H), 6.79 (t, J = 7.8 Hz, 2H), 7.04 (t, J = 7.4 Hz, 1H), 7.11 (m, 2H), 7.20 (m, 1H), 7.32 (t, J = 7.7 Hz, 1H); 13C NMR (100 MHz, CDCl3) δC 25.0, 29.9, 47.9, 49.2, 49.5, 58.6, 59.4, 73.0, 76.0, 109.8, 110.8, 111.1, 111.2, 111.3, 111.4, 112.6, 112.8, 112.9, 117.4, 123.2, 124.3, 125.3, 128.1, 129.3, 129.4, 129.5, 129.8, 131.1, 141.1, 149.9, 160.7, 160.8, 161.0, 163.1, 163.2, 175.3; MS(ESI): m/z = 515 [M + H]+, anal. calcd for C28H20F2N4O2S: C, 65.36; H, 3.92; N, 10.89%, found C, 65.41; H, 3.89; N, 10.79%.
Methyl-5′-amino-12′-[(2,6-difluorophenyl)methyl]-2-oxo-1,2-dihydro-6′-oxa-8′-thia-12′-azaspiro[indole-3,3′-tricyclo[7.4.0.02,7]tridecane]-1′(9′),2′(7′),4′-triene-4′-carboxylate (5v). Off-white solid; Mp 180–182 °C; IR (KBr): 3350, 3171, 1736, 1728, 1663, 1589, 1479, 1401, 1248, 1046, 750 cm−1. 1H NMR (400 MHz, DMSO-d6) δH 2.09 & 2.71 (td, J = 14.7 Hz, 2H), 2.60 (s, 4H), 3.23 (s, 3H), 3.50 (m, 2H), 6.89 (d, J = 7.8 Hz, 2H), 6.92 (d, J = 7.4 Hz, 1H), 6.98 (t, J = 7.4 Hz, 1H), 7.36 (bs, D2O exchangeable, 2H), 7.39–7.44 (m, 3H), 10.57 (bs, D2O exchangeable, 1H); 13C NMR (100 MHz, DMSO-d6) δC 24.6, 47.3, 48.0, 49.3, 50.2, 55.4, 75.9, 110.2, 110.7, 111.3, 111.5, 111.7, 111.8, 112.2, 117.8, 121.5, 122.4, 124.6, 127.6, 129.5, 129.3, 129.6, 129.9, 131.8, 141.4, 148.8, 159.6, 159.9, 160.8, 162.1, 162.3, 168.3, 178.0; MS(ESI): m/z = 510 [M + H]+, anal. calcd for C26H21F2N3O4S: C, 61.29; H, 4.15; N, 8.25%, found C, 61.19; H, 4.19; N, 8.22%.
Methyl-2-(4-{5′-amino-4′-cyano-2-oxo-1,2-dihydro-6′-oxa-8′-thia-12′-azaspiro[indole-3,3′-tricyclo[7.4.0.02,7]tridecane]-1′(9′),2′(7′),4′-trien-12′-ylmethyl}phenyl)benzoate (5w). Off-white solid; Mp 193–195 °C; IR (KBr): 3433, 3199, 2194, 1720, 1643, 1619, 1472, 1291, 125, 1158, 762 cm−1. 1H NMR (400 MHz, CDCl3) δH 1.59 (m, 1H), 2.46 (m, 1H), 2.56 (m, 1H), 2.86–2.93 (m, 2H), 2.99 (d, J = 12.2 Hz, 1H), 3.06–3.08 (m, 1H), 3.83 (d, J = 12.2 Hz, 1H), 3.95 (s, 3H), 4.86 (bs, 2H, D2O exchangeable), 6.66 (d, J = 7.6 Hz, 1H), 6.99 (t, J = 7.2 Hz, 1H), 7.06 (d, J = 7.2 Hz, 1H), 7.15–7.21 (m, 3H), 7.24 (m, 2H), 7.36 (d, J = 7.5 Hz, 1H), 7.42 (t, J = 7.4 Hz, 1H), 7.55 (t, J = 7.4 Hz, 1H), 7.88 (d, J = 7.5 Hz, 1H), 8.86 (bs, 1H, D2O exchangeable); 13C NMR (100 MHz, CDCl3) δC 24.9, 48.9, 49.8, 51.7, 53.2, 59.3, 62.2, 110.9, 111.6, 117.8, 122.8, 123.3, 125.3, 127.4, 128.6 (3C), 129.1 (2C), 129.4, 130.0, 130.2, 131.3, 131.9, 132.1, 137.0, 140.4, 141.2, 143.5, 149.4, 161.3, 169.3, 177.2; MS(ESI): m/z = 575 [M + H]+. anal. calcd for C33H26N4O4S: C, 68.97; H, 4.56; N, 9.75%, found C, 68.93; H, 4.50; N, 9.81%.
5′-Amino-2-oxo-12′-(prop-2-yn-1-yl)-1,2-dihydro-6′-oxa-8′-thia-12′-azaspiro[indole-3,3′-tricyclo[7.4.0.02,7]tridecane]-1′(9′),2′(7′),4′-triene-4′-carbonitrile (5x). Off-white solid; Mp 197–199 °C; IR (KBr): 3402, 3176, 2190, 1704, 1638, 1569, 1469, 1269, 1044, 761 cm−1. 1H NMR (400 MHz, DMSO-d6) δH 2.12–2.32 (m, 2H), 2.54 (m, 2H), 2.66 (t, J = 2.2 Hz, 1H), 2.76 (m, 2H), 3.55 (d, 2.2 Hz, 2H), 6.79 (d, J = 7.7 Hz, 1H), 6.90 (d, J = 7.7 Hz, 1H), 6.91 (d, J = 7.6 Hz, 1H), 7.06 (t, J = 7.2 Hz, 1H), 7.250 (bs, 2H, D2O exchangeable), 10.26 (bs, 1H, D2O exchangeable); 13C NMR (100 MHz, DMSO-d6) δC 24.3, 45.7, 49.3, 51.1, 52.6, 59.1, 71.2, 78.4, 114.9, 116.8, 117.6, 118.9, 124.7, 127.6, 127.8, 127.9, 131.6, 141.3, 148.9, 161.3, 177.0; MS(ESI): m/z = 389 [M + H]+, anal. calcd for C21H16N4O2S: C, 64.93; H, 4.15; N, 14.42%, found C, 64.95; H, 4.11; N, 14.22%.
5′-Amino-5-methyl-2-oxo-12′-(prop-2-yn-1-yl)-1,2-dihydro-6′-oxa-8′-thia-12′-azaspiro[indole-3,3′-tricyclo[7.4.0.02,7]tridecane]-1′(9′),2′(7′),4′-triene-4′-carbonitrile (5y). Off-white solid; Mp 187–189 °C; IR (KBr): 3403, 3166, 2195, 1714, 1658, 1579, 1479, 1279, 1049, 766 cm−1. 1H NMR (400 MHz, DMSO-d6) δH 2.14–2.32 (m, 2H), 2.39 (s, 3H), 2.56 (m, 2H), 2.68 (t, J = 2.2 Hz, 1H), 2.79 (m, 2H), 3.56 (d, 2.2 Hz, 2H), 6.81 (d, J = 7.7 Hz, 1H), 6.95 (d, J = 1.6 Hz, 1H), 7.16 (t, J = 7.2 Hz, 1H), 7.35 (bs, 2H, D2O exchangeable), 10.26 (bs, 1H, D2O exchangeable); 13C NMR (100 MHz, DMSO-d6) δC 22.3, 24.8, 45.4, 49.5, 51.1, 52.8, 59.4, 71.8, 78.0, 114.6, 116.7, 117.9, 124.9, 126.5, 127.8, 127.9, 128.0, 131.7, 141.5, 148.7, 164.4, 177.0; MS(ESI): m/z = 403 [M + H]+, anal. calcd for C22H18N4O2S: C, 65.65; H, 4.51; N, 13.92%, found C, 65.63; H, 4.41; N, 13.82%.
5′-Amino-5-nitro-2-oxo-12′-(prop-2-yn-1-yl)-1,2-dihydro-6′-oxa-8′-thia-12′-azaspiro[indole-3,3′-tricyclo[7.4.0.02,7]tridecane]-1′(9′),2′(7′),4′-triene-4′-carbonitrile (5z). Off-white solid; Mp 211–213 °C; IR (KBr): 3400, 3160, 2196, 1710, 1655, 1574, 1478, 1289, 1059, 766 cm−1. 1H NMR (400 MHz, DMSO-d6) δH 2.20–2.30 (m, 2H), 2.53 (m, 2H), 2.71 (t, J = 2.2 Hz, 1H), 2.81 (m, 2H), 3.58 (d, 2.2 Hz, 2H), 7.25 (bs, D2O exchangeable, 2H), 7.51 (d, J = 7.7 Hz, 1H), 7.99 (d, J = 1.6 Hz, 1H), 8.18 (m, 1H), 10.29 (bs, 1H, D2O exchangeable); 13C NMR (100 MHz, DMSO-d6) δC 24.6, 45.7, 49.4, 52.6, 52.9, 59.5, 71.6, 78.6, 109.7, 117.4, 118.3, 121.4, 124.6, 127.5, 127.7, 128.3, 131.6, 141.3, 148.7, 163.4, 177.0; MS(ESI): m/z = 434 [M + H]+, anal. calcd for C21H15N5O4S: C, 58.19; H, 3.49; N, 16.16%, found C, 58.23; H, 3.41; N, 16.12%.
Evaluation of cytotoxicity by MTT assay
COLO320 adenocarcinoma colorectal cancer cell line was maintained in complete tissue culture medium RPMI with 10% fetal bovine serum and 2 mM L-glutamine, along with antibiotics (about 100 IU per mL of penicillin, 100 μg per mL of streptomycin) with the pH adjusted to 7.2. Cells (5 × 105) were seeded in 96 well plates containing medium with different concentrations such as 500 μg mL−1, 250 μg mL−1 and 100 μg mL−1. The cells were cultivated at 37 °C, 5% CO2, 95% air and 100% relative humidity. After various durations of cultivation, the solution in the medium was removed. An aliquot of 100 μL of medium containing 1 mg per mL of 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl-tetrazolium bromide (MTT) was loaded to the plate. The cells were cultured for 4 h and then the solution in the medium was removed. An aliquot of 100 μL of DMSO was added to the plate, which was shaken until the crystals were dissolved. The cytotoxicity against cancer cells was determined by measuring the absorbance of the converted dye at 570 nm in an ELISA reader. Cytotoxicity of each sample was expressed as IC50 value. The IC50 value is the concentration of test sample that causes 50% inhibition of cell growth, averaged from three replicate experiments.
DNA fragmentation assay
For the DNA fragmentation assay, COLO320 colorectal adenocarcinoma cells (1 × 106 cells; control and treated with isolated compound) were collected by centrifugation at 2000 rpm for 10 min and washed twice with phosphate-buffered saline (PBS, Ambion USA). The cell pellet was suspended in 100 μL of cell lysis buffer (10 mM Tris–HCl buffer, pH 7.4 containing 10 mM EDTA and 0.5% triton X-100), kept at 4 °C for 15 min and the cell lysate was centrifuged at 16000 rpm for 20 min. The supernatants were incubated with proteinase K (0.4 mg mL−1; Sigma-Aldrich) at 60 °C for 60 min, then incubated with RNase A (0.4 mg mL−1; Sigma-Aldrich) at 37 °C for 60 min. The supernatants were mixed with 20 μL of 5 MNaCl solution and 120 μL of isopropyl alcohol overnight at −20 °C. The supernatants were then collected by centrifugation at 16000 rpm for 15 min. DNA samples were dissolved in TE buffer (10 mM Tris–HCl, pH 7.4 and 1 mM EDTA, pH 8.0), and separated by 2% agarose gel electrophoresis.
Acknowledgements
The authors acknowledge the Department of Chemistry, Indian Institute of Technology-Madras (IITM), Chennai, India for single crystal X-ray diffraction analysis. One of the authors, K.P. is grateful to the management of Orchid Chemicals and Pharmaceuticals Ltd.
References
- S. T. Hilton, T. C. T. Ho, G. Pljevaljcic and K. Jones, Org. Lett., 2000, 2, 2639 CrossRef CAS.
- S. P. Baran and R. M. Richter, J. Am. Chem. Soc., 2005, 127, 15394 CrossRef PubMed.
- M.-Y. Chang, C.-L. Pai and Y.-H. Kung, Tetrahedron Lett., 2005, 46, 8463 CrossRef CAS PubMed.
- J. Ma and S. M. Hecht, Chem. Commun., 2004, 1190 RSC.
- S. Edmondson, S. J. Danishefsky, L. Sepplorenzinol and N. Rosen, J. Am. Chem. Soc., 1999, 121, 2147 CrossRef CAS.
- R. S. Kumar, S. M. Rajesh, S. Perumal, D. Banerjee, P. Yogeeswari and D. Sriram, Eur. J. Med. Chem., 2010, 45, 411 CrossRef CAS PubMed.
- K. C. Joshi, R. Jain, K. Sharma, S. K. Bhattacharya and R. K. Goel, J. Indian Chem. Soc., 1988, 115, 202 Search PubMed.
- K. C. Joshi, R. Jain and S. Arora, J. Indian Chem. Soc., 1988, 65, 277 CAS.
- K. Parthasarathy, C. Praveen, C. Balachandran, P. S. Kumar, S. Ignacimuthu and P. T. Perumal, Bioorg. Med. Chem. Lett., 2013, 23, 2708–2713 CrossRef CAS PubMed.
- K. Higashiyama and H. Otomasu, Chem. Pharm. Bull., 1980, 28, 648 CrossRef CAS.
- L.-M. Wang, N. Jiao, J. Qiu, J.-J. Yu, J.-Q. Liu, F.-L. Guo and Y. Liu, Tetrahedron, 2010, 66, 339 CrossRef CAS PubMed.
- A. H. Abdel, E. M. Keshk, M. A. Hanna and S. M. El-Bady, Bioorg. Med. Chem., 2004, 12, 2483 CrossRef PubMed.
- J. Yang, W.-Y. Hua, F.-X. Wang, Z.-Y. Wang and X. Wang, Bioorg. Med. Chem., 2004, 12, 6547 CrossRef CAS PubMed.
- G. Chow and R. C. Ziegelstein, Am. J. Cardiovasc. Drugs, 2007, 7, 169 CrossRef CAS PubMed.
- N. E. Schwartz and G. W. Albers, Curr. Neurol. Neurosci. Rep., 2008, 8, 29 CrossRef CAS PubMed.
- P. Savi, J.-L. Zachayus, N. Delesque-Touchard, C. Labouret, C. Hervé, M.-F. Uzabiaga, J.-M. Pereillo, J.-M. Culouscou, F. Bono, P. Ferrara and J.-M. Herbert, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 11069 CrossRef CAS PubMed.
- For related discussion, see: S. Raju, V. R. Batchu, N. K. Swamy, R. V. Dev, B. R. Srikanth, J. M. Babu, K. Vyas, P. R. Kumar, K. Mukkanti, P. Annamalai and M. Pal, Tetrahedron, 2006, 62, 9554 CrossRef CAS PubMed.
- A. Burger, Prog. Drug Res., 1991, 37, 287 CAS.
- It is noteworthy to mention that the corresponding benzo-analog (4-aryl-4H-chromenes) induces apoptosis, see: W. Kemnitzer, J. Drewe, S. Jiang, H. Zhang, J. Zhao, C. Crogan-Grundy, L. Xu, S. Lamothe, H. Gourdeau, R. Denis, B. Tseng, S. Kasibhatla and S. X. Cai, J. Med. Chem., 2007, 50, 2858 CrossRef CAS PubMed.
- P. J. Sanfilippo, J. J. McNally, J. B. Press, L. J. Fitzpatrick, M. J. Urbanski, L. B. Katz, E. Giardino, R. Falotico, J. Salata, J. B. Moore Jr and W. Miller, J. Med. Chem., 1992, 35, 4425 CrossRef CAS.
- C. Praveen, P. DheenKumar, D. Muralidharan and P. T. Perumal, Bioorg. Med. Chem. Lett., 2010, 20, 7292 CrossRef CAS PubMed.
- C. Praveen, A. Ayyanar and P. T. Perumal, Bioorg. Med. Chem. Lett., 2011, 21, 4072 CrossRef CAS PubMed.
- C. Praveen, A. Ayyanar and P. T. Perumal, Bioorg. Med. Chem. Lett., 2011, 21, 4170 CrossRef CAS PubMed.
- C. Praveen, C. Iyyappan, K. Girija, K. S. Kumar and P. T. Perumal, J. Chem. Sci., 2012, 124, 451 CrossRef CAS.
- C. Praveen, A. Nandakumar, P. Dheenkumar, D. Muralidharan and P. T. Perumal, J. Chem. Sci., 2012, 124, 609 CrossRef CAS.
- C. Praveen, P. Dheenkumar and P. T. Perumal, J. Chem. Sci., 2013, 125, 71 CrossRef CAS PubMed.
- K. Karthikeyan, P. M. Sivakumar, M. Doble and P. T. Perumal, Eur. J. Med. Chem., 2010, 45, 3446 CrossRef CAS PubMed.
- A. Badorc, D. Frehel, J.-P. Maffrand and E. Vallee, US Pat. 4740510, 1988.
- C. Praveen, C. Iyyappan and P. T. Perumal, Tetrahedron Lett., 2010, 51, 4767 CrossRef CAS PubMed.
- X. Pan, R. Huang, J. Zhang, L. Ding, W. Li, Q. Zhang and F. Liu, Tetrahedron Lett., 2012, 53, 5364 CrossRef CAS PubMed.
- G. Abbiati, V. Canevari, S. Caimi and E. Rossi, Tetrahedron Lett., 2005, 46, 7117 CrossRef CAS PubMed.
- M. Noguchi, H. Okada, M. Watanabe, K. Okuda and O. Nakamura, Tetrahedron, 1996, 52, 6581 CrossRef CAS.
- ESI.†.
- A. Dömling, E. Herdtweck and I. Ugi, Acta Chem. Scand., 1998, 52, 107 CrossRef PubMed.
- C. O. Kappe, Tetrahedron, 1993, 49, 6937 CrossRef CAS.
- L. Weber, K. Illegen and M. Almstetter, Synlett, 1999, 366 CrossRef CAS PubMed.
- N. Martin, C. Seoane and J. L. Soto, Tetrahedron, 1988, 44, 5861 CrossRef CAS.
- T. Mosssman, J. Immunol. Methods, 1983, 65, 55 CrossRef.
- M. K. Raj, C. Balachandran, V. Duraipandiyan, P. Agastian, S. Ignacimuthu and A. Vijayakumar, Med. Chem. Res., 2013, 22, 3823 CrossRef.
- http://mgltools.scripps.edu/.
- http://vina.scripps.edu/.
- (1S)-1-(3-Chlorophenyl)-2-oxo-2-[(1,3,4-trioxo-1,2,3,4-tetrahydroisoquinolin-5-yl)amino]ethyl acetate was used as the co-crystal.
Footnote |
| † Electronic supplementary information (ESI) available: 1H and 13C NMR Spectrum of all compounds and NOESY spectrum of compounds 7d and 8d. CCDC 936526. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c4ra16605a |
|
| This journal is © The Royal Society of Chemistry 2015 |
Click here to see how this site uses Cookies. View our privacy policy here.