Short peptide based self-assembled nanostructures: implications in drug delivery and tissue engineering
Jiban Jyoti
Panda
ab and
Virander Singh
Chauhan
*a
aInternational Centre for Genetic Engineering and Biotechnology, New Delhi 110067, India. E-mail: virander@icgeb.res.in
bInstitute of Nano Science and Technology, Mohali, 160062, Punjab, India
First published on 23rd April 2014
Abstract
Self-assembly of biomolecules facilitates the creation of a diverse range of hierarchical nanostructures from a wide range of polymeric and non-polymeric materials. Peptides and specifically short peptides are very attractive in this respect due to their unmatched biocompatibility, ease of synthesis, functionability as well as tunable bioactivity along with the availability of rich chemistry for fine-tuning the structure and function of peptides according to environmental conditions. Self-assembled peptide based nanostructures such as tubes, filaments, fibrils, hydrogels, vesicles, and monolayers have been studied by many research groups and found application as three-dimensional cell growing scaffolds, dental implants, neural tissue engineering scaffolds and as carriers for drugs, proteins and genes, and nucleotides. Nanostructures are also being developed from designed or modified amino acids to have enhanced cellular as well as in vivo stability. These modified nanostructures showed enhanced drug delivery properties both under in vivo and in vitro conditions.
Introduction
Self-assembly is defined as the autonomous organization of components into ordered patterns or structures.1 Living cells consist of many self-assembling systems that work in synchronization to achieve a defined goal. Self-assembly, as a process contributes significantly in biology such as maintaining cell integrity, performing important cellular functions as well as inducing abnormalities that cause diseases.1 Thus, understanding life necessitates a better understanding of self-assembly. Concepts of self-assembly have also been used in many disciplines for constructing useful materials. Molecular self-assembly is in fact a very practical way of making ensembles of nanostructures. The ubiquitous existence of self-assembly processes in living systems along with the prevalence of various non-covalent interactions (van der Waals, electrostatic, and hydrophobic metal–ligand, π–π stacking interactions, hydrogen and coordination bonds) in biology, have resulted in rapid development of self-assembling biomaterials as a promising research area.2–4 Self-assembly provides the flexibility of developing novel materials with tailored morphologies and desired functions through single-molecule design and engineering. This results in controlling the bulk properties of the resultant material by modulating individual monomeric building blocks. Thus, by modulating structural changes in constituent molecules it becomes possible to dictate the behavior of the end product.In recent past, many self-assembling nanomaterials have been generated from various organic polymers such as carbohydrates, nucleic acids, proteins etc. either to gain a better understanding of the phenomenon or to use them for applications ranging from molecular devices to delivery systems or scaffolds.1,2
For example, different polysaccharides due to their many merits have been investigated as templates for the synthesis of nanomaterials. Polysaccharides are of natural origin; they are biodegradable, non-toxic and safe. They have abundant natural resources and have a low cost of purification. Polysaccharides such as alginate, pectin, dextran, chitosan, hyaluronic acid etc. have been studied for constructing nanostructures. Polysaccharide nanostructures are prepared by methods such as covalent crosslinking, ionic crosslinking, polyelectrolyte complex formation, and by the self-assembly of polysaccharides modified with hydrophobic groups. However, cross-linking agents such as glutaraldehyde could be a limiting factor in the potential biomedical application of these nanostructures prepared by the cross-linking method. Polysaccharides like chitosan, dextran either in a native or modified form or in association with other polymers form nanoparticles that can encapsulate various drugs, genes, proteins, and peptides.5 Chitosan has been investigated for making a variety of nanocarriers,6,7 and chitosan nanocarriers have been shown to encapsulate drugs like retinol,8 utilized for DNA delivery.9 Also, calcium-alginate nanoparticles have been shown to encapsulate anti-tuberculosis drugs like isoniazid, pyrazinamide and rifampicin with high efficiency and loading resulting in increased bioavailability of these drugs.10
Furthermore, biomolecules like DNA, RNA and proteins are endowed with many desirable properties which make them suitable templates for constructing self-assembled nanostructures. DNA nanostructures with either two or three dimensions and with varied shapes like, icosahedral, tubular, tetrahedral, and Y-shaped geometries are being constructed and have been shown to deliver cargos like, drugs (anticancer, various antibiotics), oligo-nucleotides, dyes, inorganic nanoparticles, proteins etc. to different cells or to display groups that induce immunostimulation.11 The superiority of DNA nanoarchitechtures as compared to other self-assembling systems lies in the flexibility of fine tuning the geometry and size of DNA nanoarchitectures very accurately because of the well-known self-recognition properties of DNA and knowledge of the exact structure of the double helix on the atomic level. This allows the ability of controlling spatial distribution of cargoes and ligands on DNA nanostructures.
Multifunctional DNA nanostructures simultaneously decorated with targeting agents, drug payloads as well as imaging agents are also being investigated for theranostic applications.12 Hybrid nanoparticles like DNA–gold nanoparticles have additional features like magnetic properties, plasmonic effects or the ability of fluorescence quenching that could find application in imaging, detection and as transfection agents and gene regulation materials.13–15 DNA-block copolymer micelles (DBCs) have also been shown to have applications in biomaterial purification,16,17 templated synthesis18 and in nanoelectronics.19 For in vitro and in vivo experiments usually larger quantities of biomaterials are needed. However due to cost issues, it is important that while designing DNA nanostructures the number of strands per DNA nanostructure should be kept at the minimum and this is one of the major limitations of DNA nanostructures.
Like DNA, RNA molecules by virtue of their inter and intra molecular interactions can self-assemble to form nanostructures by both template or non-template mediated assembly. RNA self-assembly plays a significant role in nanofabrication by virtue of its ability to form 3D nanostructures, to produce reversible self-assembly and self-repair. RNA nanoparticles have shown application in pathogen detection, drug/gene delivery, and various other therapeutic applications.20 For instance, motor pRNA of the bacteriophage phi29 formed nanostructures of various shapes like twins, tetramers, rods, triangles, and 3D arrays of micron size via interaction of programmed helical regions and loops. These structures were resistant to a wide range of temperatures, salt concentrations, and pH and have shown potential to deliver oligonucleotides like ribozyme or siRNA to kill cancer cells both in vitro and in vivo or Hepatitis B virus.21,22
Like DNA, RNA and carbohydrates, proteins due to the existence of varied structures, shapes and chemical properties and availability of a large database can act as good templates for molecular self-assembly and many protein based nanostructures are being investigated.23–25
Though all these systems have their own demerits and limitations, their relative instability within living systems and other deleterious effects can hinder the potential applications of these systems for safe human use.
The idea of generating peptide based biomaterials essentially came from the observation that many soluble cellular proteins could self-assemble to form well-ordered tubular deposits in neurodegenerative diseases such as Alzheimer's, Parkinson's and in prion related diseases.26 Though, disastrous to the normal physiology,26 these protein assemblies formed the basis of developing novel nanobiomaterials.27 These observations have led to many efforts focusing on studying self-assembling peptides and several peptide based nanostructures have been described/developed.28–30
There are many advantages associated with using peptides as building blocks to make ensembles of nanostructures. These include the availability of detailed structural and functional information for many peptides and proteins, easy and rapid chemical synthesis, tunability to various environmental conditions (like pH, temperature, and ionic strength), and adoption of well defined helical or β-hairpin/sheet secondary structures that can energetically favor self-association and assembly.31 Peptides however generally are labile to enzymatic degradation, but if being developed as delivery agents, there are different strategies to circumvent this problem. For example, by introducing non-coded residues in the peptide design, the peptide based nanostructures can be made more resistant to enzymatic degradation.32,33
Nanostructures generated by peptide self-assembly
Peptides, based on their design, can assemble into different kinds of supramolecular architectures (Fig. 1) such as nanotubes34,35,36 and monolayers with a nanoscale order and as vesicular structure.28–30 Nanostructures have been developed from different types of peptides including cyclic peptides, amphiphilic peptides, peptides containing β-sheeted motifs, helical-structures or β-turns. | ||
| Fig. 1 Self-assembly of peptides into different types of nanostructures. | ||
Cyclic peptide based nanostructures
Ghadiri et al. for the first time developed peptide nanotubes based on cyclic polypeptides with an even number of alternating D- and L-amino acids. These peptides assembled by virtue of inter-molecular hydrogen bonding and formed self-assembled nanotubes with the internal diameter ranging from 7–8 Å (Fig. 2).37,38 Because of the alternating D and L-amino acid sequences, the peptide side chains in these assemblies lie on the outward direction creating a hollow tubular structure. The diameters of these nanotubes could be varied depending on the cyclic peptide ring size. Lanreotide growth hormone inhibitor, a natural cyclic peptide has also been shown to self-assemble to form ordered nanotubes by virtue of aromatic and hydrophobic interactions.39 These nanotubes showed potential for a wide range of applications as antimicrobial agents, biosensors, catalysts etc.40,41 | ||
| Fig. 2 Self-assembly of cyclic peptides with alternating D and L residues and their potential applications. | ||
Nanostructures generated from beta-sheeted or alpha-helical peptides
A large number of peptides have been shown to self-assemble by forming beta-sheeted secondary structures.42 Beta-sheeted peptides have been used to synthesize a variety of supramolecular architectures such as ribbons,43,44 nanotubes,36,45,46 monolayers with nanoscale order,47,48 and nanotubes with delocalized electronic states.49 An example of a pH responsive self-assembling beta sheeted peptide is P11-4 (QQRFEWEFEQQ). This peptide is pH sensitive due to the ionizable glutamate and arginine side chains. At concentrations below <10 mg ml−1 and at neutral pH, the peptide is soluble but adopts a hydrogel state at low pH (pI: 4.2). This occurs due the formation of anti-parallel β-sheet tapes, which then stack together to form fibrils. At its critical gelling concentration of 10 mg ml−1, the peptide also forms hydrogel like structures in culture medium or in the presence of 140 mM salt.43 Similarly, MAX1, a 20 residue long peptide, with two β-strands with alternating valine and lysine residues connected through a type II'β-turn, when kept under basic conditions, first folds into a β-hairpin like structure which then self-assembles into a hydrogel network.50Powers et al., also investigated the self-assembly of designed β-hairpin peptides and found that these peptides can self-assemble at the air/water interface to form various nanostructures.51 Other designed peptides like MAX-8, the modified version of MAX-1 with lysine at the 15th position being replaced by glutamic acid and MAX-3 with 7th, 12th and 16th valine residues of MAX-1 being replaced by threonine residues, have been shown to undergo triggered folding in response to pH and/or temperature changes, to adopt a β-hairpin conformation that undergoes self-assembly to form various nanostructures.52,53 Large polypeptides with alternating segments of polar and non-polar residues like the 16 residue peptides RADA-16 I and II have been shown to form β-sheeted structures that self-assembled into pH responsive hydrogels.54 RADA16 (RADARADARADARADA) is also marketed under the name of PuraMatrix™ (3DM, Inc., Cambridge, MA, USA). This 16 amino acid peptide readily self-assembles into a nanofiber network. Gel formation can also be triggered by an increase in ionic strength or by a change in pH. In another approach, linear peptides were assembled into nanotubes by utilization of highly directional metal–ligand interactions.55 The ability of these peptides to form polymeric β-sheets can be modulated by pH and salts in a manner that is dictated by the number and positioning of charged amino acid residues.
Though β-sheeted peptide motifs are being widely scrutinized as templates for forming self-assembled nanoscale materials,50,56 the design of α-helical nanoscale materials has seen only limited advancement. As models of fibrillar structures based on α-helical coiled coil motifs are prevalent in the body such as in the cytoskeleton and extracellular matrix,57 α-helical synthetic peptides have been attractive targets for de novo design of fibrillar nanostructures. For instance, filamentous nanostructures were generated from coiled-coil peptide motifs 25–50 residues long. These peptides were based on heptad repeats (abcdefg)n, with positions a and d being occupied by hydrophobic residues and polar residues anywhere else in the sequence. Hydrophobic residues formed an inter-helical hydrophobic core, providing a stabilizing interface between the helices. Charged residues at positions “e” and “g” participate in electrostatic interactions contributing to coiled-coil stability. These peptides form 2–5 helices that wrap around each other in a superhelical fashion to form nanoscale fibers.58–60 Woolfson and co-workers designed peptides (29–36 residue long) based on helical coiled-coils,61,62 that self-assembled into nanofibers. Their peptides were also based on a heptad sequence repeat, abcdefg, with hydrophobic residues isoleucine and leucine at the ‘a’ and ‘d’ sites, respectively. The hydrophobic residues promoted helix oligomerisation through hydrophobic collapse. To promote staggered assembly of peptides and fibril formation, lysines were incorporated at the ends of the peptides with central glutamates to allow ionic interactions resulting in a fibrous assembly.63,64 Hartgerink and co-workers have also used heptad repeats producing helical coiled-coils that form nanofibres in a concentration-dependent manner. They used pH and ionic strength as triggers for self-assembly with incorporation of isoleucine and leucine residues at positions ‘a’ and ‘d’ of the heptad, and glutamate at positions ‘e’ and ‘g’ providing an acidic region. Hence, at low pH ionic repulsion was eliminated and carboxylic acid side chains hydrogen bonded with each other to form assembly.64
Hydrogels are three dimensional (3D) networks with the capacity to imbibe and retain water. They have been shown to possess a wide range of biomedical applications.65,66 Hydrogels have also been developed from self-assembly of helical polypeptides based on ‘aba’ triblock motifs that are composed of a central random coil block flanked by two coiled-coil forming sequences. The self-assembly occurred as a balance between the oligomerization of the helical ends and swelling of the central water-soluble random coil segment.67–69 Interestingly, it was observed that hydrogel formation and their physical properties could be fine-tuned by changing the structure and length of the coiled-coil building blocks. Consequently, stimuli responsiveness (to temperature and/or pH, salt) could be introduced in the self-assembled hydrogels by modulating the amino acid sequence of the coiled-coil domains. Denaturation of coiled-coil domains by guanidine hydrochloride (GdnHCl) resulted in disassembly of the hydrogels and removal of GdnHCl by dialysis caused coiled-coil refolding and hydrogel reassembly.68
Various research groups have designed leucine zippers (a condition where the interacting surface between the helices contained leucine) for constructing nanostructures with different properties.70,71 O'Shea et al. were the first to propose a peptide design in which one strand with acidic residues and the other with basic residues yielded a parallel heterodimer to form a model called ‘Peptide Velcro’.70 In this design, a single asparagine residue in the sequence forms a hydrogen bond with a corresponding aspargine residue in the other subunit contributing to the directional specificity of the helix orientation and oligomerization state. Thus, it appears that simple sequences can be designed to have a very high preference to pair with each other to form useful structures. These ‘Peptide Velcro’s can be utilized to bring two molecules together, which may be useful in sensing applications.
Self-assembly of amphiphilic peptides
Zhang and co-workers demonstrated that 7–8 residue long surfactant-like peptides, characterized by well-defined hydrophilic and hydrophobic residues that could self-assemble in water to form well-ordered nanotubes and nanovesicles.72 The hydrophilic head groups of these peptides were composed of aspartic acids and their tails were composed of hydrophobic amino acids such as alanine, valine, or leucine. Like other surfactants, self-association by hydrophobic interactions was the major governing force for the self-assembly of these peptides.72Amphiphilic peptides generally consist of a hydrophilic peptide headgroup and a hydrophobic alkyl tail where the tail participates in aligning the head group to form various secondary, super-secondary and tertiary conformations.73–75 These peptides self-assembled to form a variety of morphological structures with nanodimensions such as micelles, vesicles or tubules.36,44,54,72 Tsonchev et al. demonstrated that the self-assembly of amphiphilic peptides was driven by both hydrophobic as well as electrostatic interactions.76
Bolaamphiphiles are also a class of amphiphilic molecules consisting of two hydrophilic groups flanked by a hydrocarbon chain.77 Their peptide segment undergo β-sheet hydrogen bonding to form various supramolecular nanostructures such as fibers, rods, tubes, ribbons, spheres etc.78–81 Bolaamphiphilic nanotubes have been shown to have several interesting applications such as use in viral assays,82,83 protein sensing84etc.
All of the above described strategies of making assembled structures have utilized large polypeptide sequences with the use of either purely L-amino acids, D-amino acids or both. However, the associated expense and complexity of synthesis of large linear peptides, cyclic and dendritic structures have strongly limited the applicability of such peptides for practical purposes. Moreover, their proteolytic instability has also been a major concern for their application under biological conditions or in in vivo situations.
Self-assembly of short peptides into different nanostructures
Lately, it was demonstrated that very small peptides could also self-assemble into various nanomorphologies, thereby minimising the difficulty and cost of their synthesis and simultaneously enhancing their stability (Fig. 3). These short peptide fragments were mostly discovered in a quest to determine the minimal required sequence for amyloid formation. An example for this is, NFGAIL (hIAPP22–27), a hexapeptide fragment of the islet amyloid polypeptide (IAPP) that forms well-ordered amyloid fibrils similar to those formed by the full length polypeptide.85 FGAIL (hIAPP23–27) a pentapeptide fragment of the IAPP polypeptide, also formed a fibrillar structure.85 Similarly, Westermark et al., discovered AILSS to be the strongly amyloidogenic region of IAPP and Reches et al. discovered NFGSVQ to be the minimally active amyloidogenic peptide fragments of the Aortic Medial Amyloid.86,87 | ||
| Fig. 3 Sequences of short peptides (≤5 amino acids long) that can self-assemble into nanostructures. | ||
KLVFF, a pentapeptide fragment of the amyloid β-peptide Aβ-42, also self-assembled in phosphate buffered saline (PBS) to form a hydrogel.88 It was hypothesized that shielding of the electrostatic charges of the peptide by salt ions favored β-sheet formation that self-associated to form a nanofibrillar gel network. Later, DFNKF, a pentapeptide fragment of human calcitonin, was also found to form well-ordered amyloid fibrils similar to those formed by the full length polypeptide.89 In a quest to find out the minimal requisite peptide sequences of the protein TAR–DNA binding protein (TDP 43) that are involved in aggregation and plaque formation in amyotrophic lateral sclerosis and frontotemporal lobar degeneration, Akash et al. discovered 10–13 residue peptides (like EDLIIKGISV, MNFGAFSINPAMM) that formed well ordered fibrillar structures similar to those formed by a full length protein.90 All these studies revealed that almost all of the amyloid forming peptides have minimally active shorter peptide fragments which carry the capacity to form amyloid fibrils similar to those formed by large native polypeptides. These studies also suggested that aromatic residues in general play a critical role in amyloid fibril formation.91
Hauser and co-workers created a unique class of natural tri- to heptapeptides made of simple, non-aromatic amino acids that self-assembled in water to form hydrogels.92 The amphiphilic peptide motif consisted of a tail of aliphatic nonpolar amino acids (N terminus) with decreasing hydrophobicity and a hydrophilic head group of acidic, neutral, or basic nonaromatic polar amino acids (C terminus). This assembly involved a conformational transition of the structurally unorganized monomers into metastable α-helical intermediates that terminated in cross-β structures. The peptides had a characteristic sequence motif consisting of an aliphatic amino acid tail of decreasing hydrophobicity capped by a polar head, which endowed them with amphiphilicity. A decrease in hydrophobicity from the N-terminus (acetylated to suppress charge effects) to the C-terminus strongly improved ease of self-assembly, stability and strength of nanostructures. It was observed that the length of the hydrophobic tail and the polarity of the head group were integral elements that supported facile hydrogel formation. Hexamers typically formed gels more readily than pentamers, tetramers, and trimers. Stronger gels were derived from head groups with acidic residues (D and E), followed by neutral (S and T) and basic (K) polar, non-aromatic amino acids.
Hauser and co-workers also carried out morphological evaluation of diverse nanostructures formed by varying the amino acid sequence and concentration of a class of small self-assembling peptides. They modified these peptides by replacing the aliphatic amino acid at the C-terminus with different aromatic amino acids. The best gelling hexamer LIVAGD and the smallest trimer peptide IVD, were modified by replacing the aspartic acid residue at the C-terminus with an aromatic amino acid (F, W and Y) residue and tracked for the effect of the introduced aromatic residues on the self-assembly and morphology of resulting nanostructures. Where, aliphatic peptides formed long, helical fibers that entangled into hydrogel meshes, the modified peptides contrastingly formed short, straight fibers with a flat morphology. No helical fibers were observed for the modified peptides. Such a study dealing with the assembly of small peptides derived from simple aliphatic amino acids is significant and may be relevant and helpful in understanding chemical evolution leading to the origin of life on Earth. These aliphatic peptide based self-assembled nanostructures were proposed to have a variety of potential applications in bioengineering and nanotechnology.93
Johansson and his group showed that even tetrapeptides are capable of forming amyloid fibrils. They hypothesized that hydrophobic interactions alone are not sufficient enough for the self-assembly of these peptides94 and there are other factors which favour peptide self-assembly into fibers.
Working with smaller peptides, Maji et al. demonstrated that tripeptides like Boc-AUV-OMe, Boc-AUI-OMe and Boc-AGV-OMe, where U is α-amino isobutyric acid, Boc is tert-butoxycarbonyl and Me is methyl, self-associated to form super-molecular β-sheet structures which further assembled into amyloid-like fibrils.95 In another piece of work Banerjee and co-workers demonstrated pH-responsive nanostructural transition of a tripeptide, TUA, from nanotubes to nanovesicles.96
Self-assembly of dipeptides and their modified versions into distinct nanostructures
Moving ahead in this direction researchers tried to explore the self-assembly of peptides as small as dipeptides (Fig. 4). The first report for dipeptide self-assembly came from Gazit's group who investigated the nature of self-assembly of the dipeptide, FF, a core motif of the amyloid β (Aβ) polypeptide segment. This dipeptide self-assembled into highly ordered nanotubes/microtubes,46,97–99 nanowires100 and nanoforests.101 Interestingly, FF nanotubes were found to be thermally stable, a unique property desired to be present in any biologically inspired material.102 Gazit et al. further used FF to construct arrays of nanotubes that can act as high surface-area electrodes for storing energy and making microfluidic chips.97 A newer type of assembly method based on vapor deposition was used for high scale production of FF nanotubes. This method was not only novel but also provided the handle to fine-tune the length and density of nanotubes by controlling monomer supply from the gas phase. This further demonstrated that the mode and nature of assembly of a given structure can be fine-tuned based on the method and conditions provided.97 FF based self-assembled nanotubes and nanowires have been investigated further for other mechanical applications such as construction of nanodevices, nanobiosensors and low resistance conducting nanowires.34,103 Ihee and his group demonstrated a fascinating morphological transformation between diphenylalanine based nanowires and nanotubes. Nanotubes were obtained by dissolving the peptide in water by sonication followed by heating, whereas, nanowires were obtained in water at high ionic strength. These two morphologies are inter-convertible.104 | ||
| Fig. 4 Formation and potential biomedical applications of dipeptide based nanostructures. | ||
Görbitz et al. showed that hydrophobic dipeptides like LL, LF, FL, and IL self-assembled into nanotubes by forming head to-tail (NH3+⋯–OOC) hydrogen bonds.105–109 Water-filled nanotubes of the dipeptide WG also demonstrated the ability of negative thermal expansion.110,111 These workers also showed that dipeptides VA, LS and FF can form nanoporous structures.105,108,109,112,113 Ripmeester's work further demonstrated that dipeptide-based nanoporous materials could adsorb inert gases, such as xenon.114–116
Gazit and co-workers further demonstrated that addition of a thiol group in FF changed its assembly from tubular to spherical structures.117 They further demonstrated the assembly of other aromatic homodipeptides into nano-spheres, nano-plates, nano-fibrils and hydrogels.118 These peptide nanostructures can be used as a casting mold for the fabrication of metallic nano-wires and coaxial nano-cables,46,119 and can have biomedical applications in biosensing, tissue engineering, molecular imaging etc.120–124
Crystalline dipeptides like AV, VA, IV and VI have been shown by Sozzani and his co-workers to self-assemble into nanoporous materials with the capacity to adsorb, separate, and store various gases such as methane, carbon dioxide, and hydrogen.125 Ventura and his co-workers reported that a dipeptide, IF, at 1.5 wt% and at pH 5.8 self-associated into a transparent, thermo-reversible gel composed of a network of nanofibrous structures in water.126
Modified dipeptides have also been explored as templates for making biologically functional self-assembled nano or microstructures owing to their enhanced proteolytic stability.127,128 Two modified dipeptides [β-AA; δ-Ava-F] containing an N-terminally positioned ω-amino acid residue [β-alanine β-A/δ-amino valeric acid (δAva)] self-associated to form nanotubes in the solid state as well as in an aqueous solution. TEM images of these two dipeptides revealed the formation of uniform and well ordered hollow nanotubular structures with varying dimensions.129 Interestingly, the nanotubular structures formed by these peptides in the solid state and in solution differed significantly demonstrating their differential assembly behavior and packing arrangements in these two states. These nanotubes were found to be stable over a wide range of pH values and temperature.129 The above studies indicated that water molecules by virtue of their intermolecular hydrogen bonding capacity, always play a pivotal role in the formation and stabilization of the nanotubular assemblies.129,130 In another study three water-soluble short peptides each having a common motif, a hybrid of β,α-amino acid residues (β-A-Xaa, Xaa = V/I/F), were found to self-assemble to form hollow nanotubes. These nanotubes could tolerate heat up to 80 °C, a wide range of pH (2–10), and were resistant to proteolytic degradation. These dipeptide-based robust crystalline nanotubes have been used as suitable templates for fabricating dipeptide-stabilized gold nanoparticles on their outer surfaces.131
Dipeptide-based nanoporous materials obtained from two water-soluble synthetic dipeptides namely β-A-Phg (Phg: phenylglycine) and Phg-β-A have shown the capacity to adsorb N2 gas. Interestingly, these nanoporous materials obtained from dipeptides were degradable by the soil bacterial consortium suggesting their ecofriendly nature.132 They were not only very different from the existing type of nanoporous materials generated from zeolites, metal organic frameworks (MOFs) and others, but also had the advantage of biodegradability and eco-friendliness generally lacking in other organic and polymeric systems.132
Fmoc-FF formed hydrogels with a nanofibrillar morphology in an aqueous solution with physical properties superior to those of hydrogels formed by longer polypeptides,128 whereas, an uncharged peptide analogue, Ac-FF-NH2, self-assembled into tubular structures.133 Gazit et al. further explored other amine and carboxyl modified diphenylalanine peptide analogues and revealed that these dipeptides formed ordered tubular structures at the nanometric scale.133 Ulijn and co-workers demonstrated that Fmoc dipeptides, derived from a combination of four different amino acids, namely glycine, alanine, leucine and phenylalanine, formed hydrogels whose structural and physical properties varied depending on the nature of the amino acids present in the peptide building blocks.134 Peptides containing aromatic moieties like Fmoc,128,135–137 beta or D-amino acid residues,138,139 and pyrene140,141 have been proven to be good templates for making nanofibrillar hydrogel networks by the virtue of π–π stacking and hydrophobic interactions. The dipeptide amphiphile Fmoc-LG has been shown to self-assemble into thin surface supported hydrogel gel films and gap-spanning hydrogel membranes whose thicknesses can be closely controlled from tens of nanometers to millimeters. The films and membranes were stable once formed and could be reversibly dried and collapsed, then reswollen to regain the gel structure.142
Dipeptides formed by the substitution of phenyl groups with napthyl goups such as di-D-1-Nal and di-D-2-Nal also formed ordered fibrillar nanostructures. Di-D-1-Nal (Nal: napthalene) formed fibers with 10 nm diameter. Di-D-2-Nal (Nal: naphthalene) peptides assembled into wider tubular structures with a diameter of about 50 nm and were more bundled than the fibrils formed by the di-D-1-Nal peptide. Interestingly, these naphthylalanine-based peptide structures showed single or low-number of walls as compared to multi-walled structures of the diphenylalanine tubes.118 Self-assembly of diphenylalanine peptides with nitro and phenyl groups has also been investigated. Di-para-nitro-Phe at a concentration of 5 mg ml−1 formed spherical nanostructures with different diameters embedded in fibrillar nanostructures. The di-4-phenyl-Phe homo-dipeptide self-assembled into square plates with various dimensions. These square plates appeared to be very thin and symmetrical with clear borders.118 It was also observed that the aromatic dipeptide, diphenylglycine, self-assembled into well ordered closed-caged nanospheres.143
Similar to FF, the dipeptide, (D)-F-(D)-F made up of two D-amino acids, self-assembled in water to form nanotubular structures with diameters ranging from 2 nm to 100 mm. Interestingly, both vesicles and nanotubes were found upon dilution of the solution with an appropriate volume of water suggesting that the concentrations of peptide can play a key role in determining the formation of nanotubes alone or formation of a mixture of nanotubes and nanovesicles.144 Junbai Li and his co-workers also showed nanotube to nanovesicle conversion of dipeptides. They demonstrated that a cationic dipeptide [FF-(NH2)·HCl] could form self-assembled nanotubes at physiological pH, which in turn spontaneously converted into spherical vesicle-like structures after dilution.145,146
Yet in another interesting example, the peptide Acp-YE (Acp, ε-amino caproic acid) demonstrated a concentration dependent nanovesicle to nanotube transformation. At a concentration of 6.9 mg ml−1, Acp-YE formed vesicles and nanotubular structures were obtained at a peptide concentration of 2.3 mg ml−1, whereas an intermediate concentration of 3.4 mg ml−1 of the peptide led to the formation of an array of fused vesicular structures that fused to form nanotubular structures upon dilution. Thus peptide concentration played a significant role in modulating the peptide assembly from nanovesicles to nanotubes or fused vesicular structures.147
Our own work has been focussed on the development and biomedical applications of dipeptides containing an unnatural amino acid α,β-dehydrophenylalanine (ΔPhe; ΔF) in the peptide backbone. We found that the dipeptide FΔF, similar to FF assembled into distinct tubular structures with a mean diameter of 27–30 nm in water (Fig. 5), which were stable over a broad range of pH conditions and also in the presence of proteases.32 FTIR and CD studies demonstrated that the dipeptides adopted a beta-turn like structure in the tubular state.32 High stability of the self-assembled tubes over a broad range of pH conditions and to a highly nonspecific proteolytic enzyme, proteinase K degradation, makes these tubes interesting candidates for future applications in drug delivery.
 | ||
| Fig. 5 TEM images of ΔF dipeptide nanostructures (nanotubes and nanovesicles). | ||
Two amphiphilic ΔF containing dipeptides, EΔF and KΔF, self-assembled into anionic and cationic vesicular structures respectively.148 EΔF assembled into pleomorphic spherical structures ranging from 50–200 nm with a mean diameter of ∼110 nm, whereas, vesicles formed by KΔF appeared mostly spherical with a mean diameter of ∼370 nm and a relatively narrow size range (250–450 nm). They were also stable to proteinase K. Light scattering studies of these particles demonstrated that EΔF formed particles with a mean size of approximately 370 ± 30 nm and KΔF formed vesicles with a mean hydrodynamic diameter of 400 ± 160 nm.
We further extended these studies with remaining dipeptides with C-terminal ΔF but with varying N-terminal residues, where the N-terminal residue was any one of the 20 naturally occurring amino acids. Out of these, the ones with aromatic N-terminal amino acid formed nanotubes, whereas those with charged N-terminal residues formed vesicles. The dipeptides with hydrophobic N-terminal residues also formed vesicles (unpublished work) (Fig. 5). It was further observed that ΔF dipeptides with hydrophobic groups at their N-termini formed visible assemblies whereas those with hydrophilic N-termini formed assembled structures invisible to the naked eye. Such an observation is interesting and points towards flexibility in peptide design which can be exploited to modulate the overall nanostructure morphology. We also found some dipeptides like MΔF, IΔF and LΔF that self-assembled in a mixture of methanol and water (50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :50, v/v) to form various nanostructures can also load hydrophobic drugs. In a 50:50 mixture of methanol and water, LΔF formed large visible aggregates with a hydrodynamic diameter in the micrometer range, whereas IΔF formed nanostructures with a mean diameter of approximately 600 nm. The mean hydrodynamic diameter of MΔF nanoparticles was found to be approximately 160 nm. TEM showed that in a 50:50 mixture of methanol and water, all three dipeptides exhibited different assembly behaviour and formed nanostructures with varied morphologies. IΔF formed micelle-like structures with an average diameter of 20 nm. LΔF, with an equal molecular mass but differing in the position of a side chain methyl group of leucine, assembled into fibrillar structures, with diameters in the nanometer range. MΔF formed very regular vesicular structures with a mean diameter of ∼40 nm.149
:50, v/v) to form various nanostructures can also load hydrophobic drugs. In a 50:50 mixture of methanol and water, LΔF formed large visible aggregates with a hydrodynamic diameter in the micrometer range, whereas IΔF formed nanostructures with a mean diameter of approximately 600 nm. The mean hydrodynamic diameter of MΔF nanoparticles was found to be approximately 160 nm. TEM showed that in a 50:50 mixture of methanol and water, all three dipeptides exhibited different assembly behaviour and formed nanostructures with varied morphologies. IΔF formed micelle-like structures with an average diameter of 20 nm. LΔF, with an equal molecular mass but differing in the position of a side chain methyl group of leucine, assembled into fibrillar structures, with diameters in the nanometer range. MΔF formed very regular vesicular structures with a mean diameter of ∼40 nm.149
Another interesting study showed that the dipeptide FΔF, at a higher concentration and under appropriate assembling conditions could form stable hydrogels having dimensions in the nanometer range. The dipeptide gel was colorless and translucent in appearance. The gel was elastic in nature with higher storage modulus (G′ ∼ 209 kPa) than loss modulus (G′′ ∼ 19.7 kPa) and also had higher mechanical strength (evident from stable G′ and G′′ values at changing frequency). Electron micrographs further showed that the gel matrix was composed of a highly dense network of fibers, which provided it with high mechanical strength and solvent retention properties. The gel also showed responsiveness towards various environmental conditions such as salt concentration, temperature and pH, a desirable feature of hydrogels used in drug delivery.33
 | ||
| Fig. 6 Tumour targeted delivery by folic acid derivatized ΔF dipeptide nanoparticles (DNPS). | ||
Self-assembly behavior of single amino acids or modified single amino acids
Single amino acids with various chemical modifications have also been investigated for their potential to make self-assembled structures. Ryan et al. studied self-assembly of modified single amino acids and showed that Fmoc-F formed self-assembled hydrogels. They demonstrated that side chain functionalization of Fmoc-F can have a significant effect on its self-assembly and hydrogelation behaviour.150 Fluorinated derivatives of Fmoc-F, such as penta-fluorophenylalanine (5-Fl-Phe) and tri-fluorophenylalanine (3-Fl-Phe), when dissolved in water demonstrated spontaneous assembly into fibrils which later on formed a hydrogel network. Ryan et al. further investigated the effect of end group functionalization on the self-assembly and hydrogelation pattern of Fmoc-F derivatives by converting the C-terminal carboxylic acid moieties of Fmoc-5-Fl-F-OH and Fmoc-3-Fl-F-OH into amide and methyl ester groups. Their results depicted that though, C-terminal amide derivatives showed faster assembly than the parent carboxylic acids, the resultant hydrogels were weak and unstable to shear stress because of the lower water solubility of the amide functionality. On the other hand C-terminal esters owing to their high hydrophobicity, self-assembled into only a fibrous structure. From these results it became clear that the monomer/solvent interactions are in general very complex and they influence the self-assembly and hydrogelation pattern to a great extent. Overall, variation of either the fluorinated aromatic side chain or N-terminal functionalization influenced the hydrogelation pattern of these molecules. This also signified that fluorous and π–π interactions as the primary determinants for molecular recognition and self-assembly. A better understanding of these interactions would facilitate the development of optimal amino acid based low molecular weight (lmw) hydrogelators.150 Fmoc protected tyrosine (Fmoc-Y) also self-assembled into hydrogels by controlled enzyme-triggered dephosphorylation of Fmoc-phosphotyrosine.151 Such enzymatic hydrogelation of small molecules has also been investigated for various applications.152,153 In a very recent and interesting study, Gazit and co-workers demonstrated self-assembly of just a single amino acid phenylalanine into ordered fibrils at a pathological concentration found in the mental disorder phenylketonuria. The fibrils had an amyloid-like morphology and exhibited a well-ordered electron diffraction pattern. These assemblies exhibited cytotoxicity towards PC12 cells that was neutralized by the antibodies generated against the fibrils. These fibrils were shown to be present in the hippocampus mice model and in parietal cortex brain tissue from individuals with phenylketonuria.154Apart from experimental investigations a lot of work has also been done on the theoretical modelling and computer simulations of self-assembling systems. Schatz, Ratner, and group used bead and packing models to study the self-assembly of peptide amphiphiles (PAs).155–157 In their study of cone-shaped amphiphiles, Tsonchev et al. found that electrostatic interactions between charged residues of peptides induced a void volume in the hydrophobic tail region which guided the cluster to a cylindrical rather than a spherical shape.157 McCullagh and co-workers using molecular modelling demonstrated that fiber formation was dependent on the choice of peptide residues.156 Lee et al. studied the relaxation of self-assembled structures of 144 PA molecules, consisting of a hydrophobic alkyl chain attached to the N-terminus of the sequence SLSLAAAEIKVAV, into cylindrical nanofibers using atomistic molecular dynamics simulations in explicit water with physiological ion concentration. Self-assembly of the molecules is initiated in a cylindrical configuration based on prior experimental and theoretical investigations, and the resulting cylindrical configuration was found to be stable during 40 nanosecond simulations. It was observed that water and sodium ions can penetrate into the peptidic part of the fiber but not between the alkyl chains. The electrostatic interactions between the PAs and the sodium counterions and the van der Waals interactions between the PAs were found to be the most important interactions stabilizing the nanofiber architechture.158 Lee et al., also used a coarse-grained molecular dynamic simulation based on the MARTINI force-field to study the self-assembly process of 140 IKVAV epitope bearing PAs for 16 μs and found that PAs first formed spherical micelles (during first 0–0.05 μs), which then formed a three-dimensional network with neighboring micelles via van der Waals interactions, then the tails of different micelles merge and formed fibers.159 Velichko et al. developed a simplified coarse-grained model to study the influence of hydrogen-bond formation on the self-assembly of PAs and found that fibrous assemblies were formed by both hydrophobic interactions and the network of hydrogen bonds.160 It has been suggested that the formation of β-sheets parallel to the axis of the fiber is the driving force for the formation of cylindrical fibers rather than spherical micelles.160–163
In order to understand the Aβ fibrilization mechanism as well as structural properties of FF nanostructures, simulation studies were carried out using both all atom164,165 and coarse-grained peptide models.166,167 Tamamis et al. studied the association of FF peptides using an implicit water model, and found transient formation of ring-like structures which are reminiscent of the nanotubular structure.165 However, these studies used only a few peptide molecules (96 FF chains) or short simulation time and only resulted in disordered aggregates. Energy minimization168 simulations and molecular dynamics164 simulation studies of FF showed the formation of cylindrical structures which are reminiscent of tubular structures.169 Guo et al.170 used a coarse-grained peptide model to study FF nanostructures and their concentration dependence. They suggested that the FF peptide behaved like a surfactant that first formed vesicles and then these vesicles fused to form nanotubes. Their molecular dynamic trajectories showed the formation of ordered spherical, vesicular or tubular nanostructures and a formation of either vesicles or tubes was concentration dependent. At low concentrations fusion of vesicles or the fusion of vesicles with a bilayer occurred, whereas at high concentrations, first a bilayer was formed, that bent and closed to form tubes.170
Recently, Jeon et al. explored the initial stages of FF assembly by carrying out molecular dynamic simulations on zwitter ionic and capped FF nanotubes. They showed that electrostatic interactions between peptides led to the formation of ordered dimmers and trimers, whereas hydrophobic interactions between side chains were involved in deciding the structures of larger oligomers.171 Also FF peptides with charged termini because of electrostatic steering first formed dimer or trimer ladders that further facilitated hydrophobic association of side chains and formed more ordered and compact structures as compared to those of uncharged FF peptides that associated by hydrophobic interactions.171 Simulations of the crystal structure of FF suggested that the strongest interactions occurred between side chains and the charged termini formed salt bridges.171 All these studies along with experimental investigations shed light on the potential phenomena behind peptide assembly and significantly improved understanding of these systems.
Potential applications of peptide based self-assembled nanostructures in drug-delivery
Different types of biocompatible, inorganic nanomaterials have been developed for the drug delivery purpose.172 However, many of them contain potentially toxic elements173–176 and have been proven to be less promising for human use. For instance, positively charged lipid-based nanoparticles are known to trigger strong immune responses. Liposomes present technological limitations such as poor reproducibility and stability, and low drug entrapment efficiency and poor control of drug leaching. Polymer nanosystems may be potentially useful alternatives, but their surface functionalization for improving drug-targeting is usually complicated and rather ineffective. Besides, most nanostructures based on naturally occurring polymers have problems of eliciting unwanted immune response and also present lot to lot variability which makes it difficult to predict their behavior in living systems.Designed peptide based nanoparticles, due to their biocompatibility, ease of synthesis and functionability, in principle can be excellent candidates for drug and gene delivery. Encapsulation by self-assembled peptide nanostructures holds particular promise in the delivery of biological molecules, including DNA, water insoluble drugs and tagged molecules for imaging.
Drug delivery using nanostructures generated from long peptides
Tanaka et al. developed TV-XIIa, an 11-residual peptaibol derived carrier peptide for the delivery of antisense oligonucleotides. TV-XIIa was derivatised with a 10-mer of lysine at the C-terminus to make the designed carrier peptide, Ac-U-N-I-I-U-P-L-L-U-P-I-K-K-K-K-K-K-K-K-K-K-OH (U: α-aminoisobutyric acid), which electrostatically interacted with oligodeoxynucleotides (ODNs) and formed a complex with ODNs, capable of crossing membranes of NIH3T3 cells to accumulate in the cytoplasm and the nucleus.177A novel class of core–shell like self-assembled nanoparticles developed from an amphiphilic peptide, cholesterol-G3R6YGRKKRRQRRR, abbreviated as CG3R6TAT, where TAT is the transcriptional activator protein of the human immunodeficiency virus type-1, by Yang et al. demonstrated powerful antimicrobial activities against a variety of microbes (bacteria, yeasts and fungi).178 The nanoparticles possessed a broad spectrum of antimicrobial activities and were active against a variety of Gram-positive as well as drug-resistant Gram-positive bacteria, fungi and yeast with low minimal inhibitory concentration (MIC) values and very low haemolysis. More interestingly, the peptide nanoparticles could cross the blood brain barrier (BBB) in a Staphylococcus aureus-induced meningitis rabbit model and minimised bacterial growth in brain without causing any significant toxicity to the major organs. These properties indicate that nanoparticles can be developed as efficient antimicrobial agents in treating brain infections. Yang et al. further demonstrated that the efficacy of these peptide nanoparticles could be extended to other infectious diseases such as methicillin-resistant Staphylococcus aureus associated infections, Candida albicans-caused brain infections, and Stachybotrys chartarum infections. Taken together, these nanoparticles were found to be promising antimicrobial agents that can be used to treat brain infections and other infectious diseases.178 The cyclic peptide nanotubes developed by Ghadiri et al., also served as nanocontainers for effective drug delivery179 and also as anti-microbial agents.180,181
Peptide-Based-Nanoparticle Devices (PBNDs), which are described as short amphipathic peptides with a capacity to form stable nanoparticles with proteins and/or nucleic acids have also been explored for their potential as drug and gene delivery vehicles. Divita et al. developed a PBND named MPG, a 27-residue-long primary amphipathic peptide (acetyl-GALFLGFLGAAGSTMGAWSQPKKKRKVcysteamide), containing three distinct domains: an N-terminal hydrophobic motif (GALFLGFLGAAGSTMGA) derived from the fusion sequence of the HIV-1 gp 41 (glycoprotein 41) for interaction with the lipid moiety of the cell membrane, a hydrophilic domain (KKKRKV) derived from the nuclear localization sequence of simian virus 40 large T-antigen for promoting interactions with nucleic acids and intracellular trafficking of the cargo, and a linker domain (WSQP), which improves the flexibility and integrity of the hydrophobic and the hydrophilic domains. This PBND showed siRNA condensing properties by virtue of a basic peptide domain and membrane destabilizing properties due to the presence of a hydrophobic peptide sequence, formed stable nanoparticles with siRNA and entered the cell independent of the endosomal pathway to efficiently deliver siRNA into a variety of cell lines182,183 Other PBND-family peptides named CADY and PEP also acted as gene carriers that entered a variety of challenging cells independent of the endosomal pathway for efficient delivery.184
Nanocarriers generated from chondroitin sulfate A (CSA) coated and PEGylated poly-L-lysine-based dendrimers have also been developed for controlled and sustained delivery of an antimalarial drug chloroquine phosphate (CQ). Entrapment in the peptidic carrier led to a significant reduction in the cytotoxicity as well as hemolytic activity of the drug. Also a significant reduction in levels of ring and trophozoite stages of Plasmodium falciparum were found after being treated with drug loaded CSA coated dendrimers as compared to the free drug.185
Yang and co-workers developed self-assembled micelles generated from synthetic oligopeptide amphiphiles (i.e. A12H5K10 and A12H5K15) that acted as efficient gene delivery vehicles.186 In another study they developed nanostructures from an oligopeptide amphiphile, Ac-(AF)6-H5-K15 (FA32), and evaluated them as carriers for co-delivery of the anticancer drug doxorubicin (Dox) with a luciferase reporter and p53 genes. Co-delivery of the drug and genes using FA32 micelles demonstrated a synergistic cytotoxic effect between the p53 gene and Dox with an increase in the p53 mRNA expression level as well as end point cytotoxicity towards HepG2 cells.187
Drug delivery using nanostructures generated from short peptides
Peptide-based vectors owing to their easy functionability and better design strategies can be developed as efficient gene delivery vehicles. Zhang and his group developed a series of surfactant peptides like LLLLLLKK, which are composed of a hydrophobic tail attached to a polar head group at the C- or N-terminus. This peptide self-assembled into nanovesicles and nanotubes that acted as DNA delivery vehicles.188,189 In DNA solution, the positively charged peptides self-assembled into a tube that encapsulated the negatively charged DNA. This ‘minivan’ was then able to deliver DNA to growing cells, at least in some cases. These systems can further be developed as smarter materials by tagging the minivan surface with a cell specific marker.190 Using β-sheeted peptide-based nanoribbons as scaffolds, filament-shaped artificial viruses for gene and drug delivery have been developed.191Stimuli-responsive peptide nanostructures are particularly attractive as drug delivery vehicles since these can have stimuli triggered drug release at target sites. For example, stimuli responsive peptide nanovesicles can be loaded with drugs and other important biomolecules to release them in response to environmental systems like pH, temperature and others.147 Tripeptide derivatives conjugated with olsalazine, an anti-inflammatory prodrug, self-assembled in water to form prodrug-containing supramolecular hydrogels.192 It was also shown that the controlled release of an anti-inflammatory agent 5-aminosalicylic acid could be achieved from the gel by the disruption of the supramolecular hydrogel caused by the reduction of the azo group. This method can be generalised for developing new nanobiomaterials for site specific drug delivery.192 Banerjee et al.'s group developed nanovesicles from dipeptides containing glutamic acid residues at the C terminus. Though these vesicles were stable over a wide range of pH, they showed responsiveness towards the presence of calcium ions. These vesicles encapsulated the anticancer drug Dox, fluorescent dyes, various biologically active molecules and were capable of releasing them in response to calcium ions. They have also been shown to act as delivery agents for biologically active molecules, such as cyclic adenosine monophosphate within the cells, while preserving their biological activity.193 Another fascinating example of peptide nanostructure for drug delivery was the one generated from the oligopeptide Acp-YE (Acp, ε-amino caproic acid), which showed nanotube to vesicle transition and was responsive to calcium ions in the solution as well as to pH change.147 These nanovesicles could entrap Dox and release it in a triggered manner in the presence of calcium ions.
Peptide based self-assembled hydrogels in drug delivery
Hydrogels are an important class of biological materials with a wide range of applications in drug delivery and tissue engineering. Peptide hydrogels/nanogels are particularly important biomaterials as they do not use harmful chemicals (e.g., toxic cross-linkers etc.) to initiate gelation, they are non-toxic, non-immunogenic, biodegradable and degrade into natural amino acids. It has been shown that coiled-coil polypeptides based on, aba tri-block polymers self-associated to form hydrogels with potential application in sustained protein delivery.69Hydrogels prepared from MAX1 and MAX8 encapsulated and released model biomacromolecules in a controllable fashion. Fluorescence recovery after photobleaching (FRAP) measurements and bulk release studies of a series of FITC-labeled dextrans within gel networks of differing peptide weight percents, demonstrated that probe diffusion inside the hydrogel network depended on the probe size, the peptide sequence, and the mesh size of the gel. Further, the electrostatic interactions between a given macromolecule and the gel network also influenced their release pattern. This study suggested that the release of macromolecules with varied characteristics such as size and charges from these self-assembling β-hairpin peptide hydrogels could be controlled and fine-tuned by simply modifying the amino acid sequences and the peptide weight percent of the gel.194
Hydrogels generated from oligopeptides, GAlL and GFlL, demonstrated thermo and pH responsive behaviour. At their minimum gelling concentration, GAlL and GFlL gels were shown to entrap 8.62 × 10−3 (M) and 3.79 × 10−3 (M) of Dox respectively. They also showed controlled drug release behaviour with almost 85% and 90% of the drug molecules getting released from the gel matrix after 45 h respectively.195
Self-assembled hydrogels generated from Fmoc-diphenylalanine encapsulated various enzyme bioreceptors (e.g., glucose oxidase or horseradish peroxidase) and fluorescent reporters [e.g., CdTe and CdSe quantum dots (QDs)]. Enzyme loaded hydrogels were smartly used for biosensing purpose and for the detection of analytes such as glucose and toxic phenolic compounds by using a photoluminescence quenching of the hybridized QDs. These results suggest that the peptide hydrogels can act as intelligent optical biosensing platforms by virtue of their simple fabrication method (by self-assembly), efficient analyte diffusion, and high encapsulation efficiencies for fluorescent reporters and bioreceptors.196
Dipeptide hydrogels derived from beta-amino acids by Banerjee and co-workers have been shown to encapsulate vitamins like B2 and B12 and sustain their release for 3 days at physiological pH (7.46) and temperature (37 °C). This depicts the potential of these gel-based biomaterials for sustained release of drugs and other important biomolecules.197
Drug delivery using ΔF dipeptide nanostructures
Since the last decade, our group has been working on the design and synthesis of ΔF dipeptide based nanostructures for drug delivery applications. Dipeptide nanovesicles formed by amphipathic dipeptides KΔF and EΔF could encapsulate bioactive molecules such as vitamin B12, amodiaquin (antimalarial), ampicillin (antibiotic), mitoxantrone (anticancer), polypeptide insulin, synthetic anti-microbial peptides etc. The ability of these vesicles to entrap proteins of various sizes such as recombinant malaria vaccine candidates namely, merozoite surface protein-119 (Pf MSP-119; 11.2 kDa), merozoite surface protein-3N (Pf MSP-3N; 25 kDa), and Plasmodium falciparum Histidine Rich Protein-2 (Pf HRP-II; 32.9 kDa), in addition to chicken egg lysozyme (16.2 kDa), bovine serum albumin (BSA; 66.4 kDa), and anti-mouse goat IgG (150 kDa) were also investigated. Results showed that all proteins interacted with the nanovesicles to varying degrees. TEM imaging carried out to determine the position of proteins on the dipeptide vesicles showed that protein Pf HRP-II was encapsulated in both the vesicles, whereas Pf MSP-119 localized preferentially on the surface of the vesicles. However, TEM studies also suggested that some other proteins like BSA, lysozyme, Pf MSP-3N, and IgG destabilized the nanovesicles. The dipeptide vesicles were taken up by mammalian cells and were not cytotoxic.148Other ΔF dipeptides like RΔF, LΔF formed vesicular and micellar nanostructures. RΔF also formed self-assembled nanostructures with a mean hydrodynamic diameter of approximately 250 ± 50 nm. TEM showed that the dipeptide formed vesicular nanostructures with mean size ranging between 30 and 50 nm. LΔF with a hydrophobic N-terminus, assembled into micellar structures with a hydrodynamic diameter of ∼250 nm.198,199 Nanostructures formed by the FΔF, RΔF, LΔF and EΔF entrapped drugs like riboflavin, niacin, amodiaquin, mitoxantrone, ampicillin with varying entrapment efficiency. They also showed non-cytotoxicity towards a variety of mammalian cells. RΔF nanostructures could be labeled with the radioisotope Technetium-99 (Tc99) with stannous tartarate as the reducing agent. Biodistribution studies carried out using Tc-labeled RΔF nanostructures, showed accumulation in the kidney and bladder along with some radioactivity in the heart and blood vessels 1 hour post-injection. Interestingly, the biodistribution profile revealed that RΔF nanostructures can evade uptake by reticulo endothelial system (RES) organs such as the liver (no significant accumulation) to remain in blood circulation for a long time.198
Delivery of hydrophobic drugs has always been a challenge. Thus in order to improve the loading of hydrophobic drugs, we tried to develop novel self-assembled dipeptide nanostructures in an aqueous/organic mixture. We chose curcumin as the model drug and tried to load it in ΔF dipeptide nanoparticles. Among all the dehydrodipeptides tested, MΔF demonstrated the maximum curcumin loading efficiency and could release the drug in a sustained manner. Loading of curcumin in MΔF nanoparticles, increased its solubility and improved cellular availability. Curcumin–MΔF nanoparticles showed an enhanced cytotoxic effect in different cancerous cell lines such as human cervical cancer cell line (HeLa), human breast cancer cell line (MCF-7) and human hepatocarcinoma cell line (HuH-7), as compared to native curcumin. These nanostructures also enhanced curcumin's in vivo efficacy towards inhibiting tumor growth in Balb/c mice bearing a B16F10 melanoma tumor.149 Such dipeptide nanoparticles are also expected to improve the delivery of other potent hydrophobic drug molecules with poor cellular uptake, bioavailability, and efficacy.
Dipeptides EΔF, KΔF, RΔF and DΔF, with charged amino acids at N-termini, were synthesized and investigated for their assembly behavior. Out of the four, RΔF formed vesicular nanoparticles that could be easily derivatized with folic acid. Folic acid derivatized nanoparticles showed enhanced cellular-uptake in various cancer cells like human breast adenocarcinoma (MDA-MB-231)and HeLa that over-expressed folic acid receptors. Folic acid derivatized RΔF nanoparticles also exhibited high Dox encapsulation efficiency and Dox loaded nanoparticles demonstrated enhanced cytotoxicity towards cancer cells over-expressing folic acid receptors (Fig. 6). Biodistribution and tumor distribution studies carried out using Tc99 labeled RΔF and folic acid–RΔF nanoparticles in Ehrlich ascitic tumor bearing Balbc mice, exhibited enhanced tumor targeting and accumulation as compared to underivatized nanoparticles. In comparison with underivatized nanoparticles or native drug, Dox loaded folic acid–RΔF nanoparticles showed enhanced tumor regression in breast tumor bearing nude mice as well as ascitic tumor bearing Balbc mice.199
Recently, we have also shown the potential of FΔF nanotubes for intravitreal delivery of pazopanib, a multi-targeted tyrosine kinase inhibitor with efficacy for treating various cancers as well as ocular disease like choroidal neovascularisation in wet age related macular degeneration. The drug could be loaded in the nanotubes by both pre and post-loading methods. The pre-loading method was found to be more efficient in loading the drug in nanotubes (25% w/w pazopanib loading and ∼55% loading efficiency) compared to the post-loading (8% w/w pazopanib loading and ∼17% loading efficiency). Plain and peptide loaded nanotubes were non-cytotoxic to retinal pigment epithelial cells. The tubes sustained in vitro release of pazopanib for 35 days. They also demonstrated a sustainable eye delivery for a period of 15 days following intravitreal injection using a 33 gauge needle.200
We also investigated gene delivery potential of nanoparticles synthesized from cationic ΔF dipeptides RΔF and KΔF and found that the cationic dipeptides condensed plasmid DNA into discrete vesicular nanostructures with RΔF forming more regular and ordered vesicular nanostructures.201 Dipeptide nanoparticles were non-cytotoxic and showed enhanced cellular uptake. They protected DNAs condensed inside them from enzymatic degradation and could successfully deliver these DNAs to different types of cancer cells such as HeLa and HuH 7 (Fig. 7). GFP encoding plasmid DNA loaded dipeptide NPs showed positive gene transfection as well as gene expression in HuH 7 cells. By virtue of their simple dipeptide origin, ease of synthesis, high enzymatic stability as well biocompatibility, these nanostructures can be foreseen to be developed as vehicles for effective gene therapy.
 | ||
| Fig. 7 Cationic dipeptide–DNA nanoparticles for cellular delivery. | ||
Thus, our work showed that the ΔF dipeptide nanostructures can act as potential candidates for drug delivery applications. The most important point here is that these dipeptide nanostructures are biocompatible with no adverse side effects both in vitro and in vivo. Due to the presence of ΔF residue in the peptide design, these dipeptide nanostructures are expected to show enhanced stability to enzymatic degradation202,203 and thus would have high in vivo half-life.
Similarly, the FΔF hydrogel also showed drug loading and release behavior (Fig. 8). The peptide gel could effectively entrap vitamins like ascorbic acid, riboflavin, and vitamin B12, antibiotics like ampicillin and chloramphenicol, antimalarial drugs such as amodiaquin, anticancer drugs like fludarabine and mitoxantrone and anti-tuberculosis drugs like L-cycloserine and isoniazid. The polypeptide insulin was also entrapped in the dipeptide gel. All the entrapped molecules in the hydrogel, exhibited sustained release behavior from the gel matrix with their diffusion coefficient (D) values ranging between D= 2.9 × 10−11 to 5.6 × 10−10. The release rates of drugs from the gel matrix (D values) were also found to be dependent on the nature of drugs like their molecular weight, ClogP values and net charge. The gel further demonstrated stimuli responsive behavior and released entrapped molecules in response to changes in environmental pH or salt-concentration.33 The ability of the dipeptide hydrogel to encapsulate a diverse kind of bioactive molecules within its matrix and their “depot-like” sustained release behavior, as well as increased proteolytic stability, makes the described dipeptide gel a promising candidate for potential drug delivery applications.
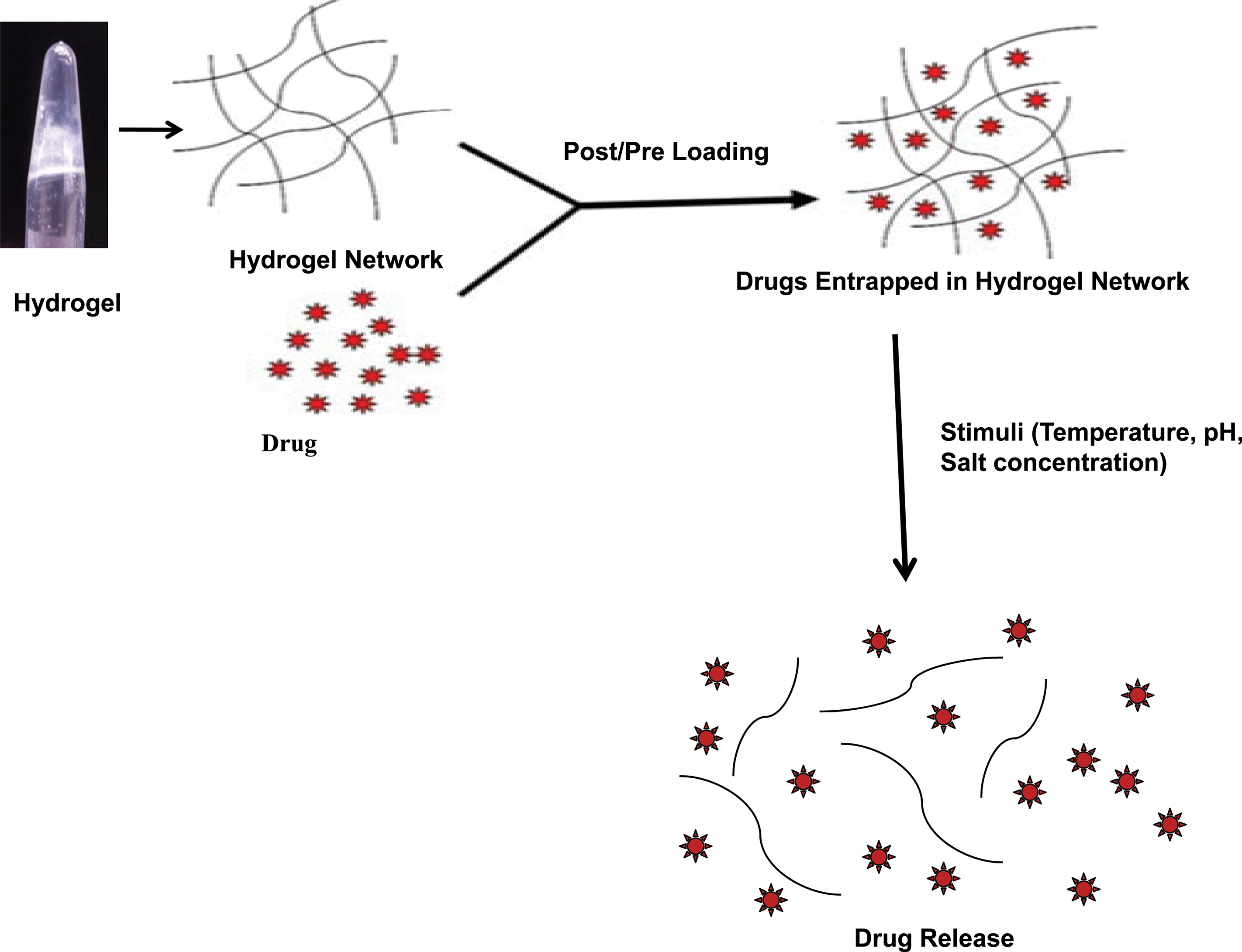 | ||
| Fig. 8 Schematic diagram showing drug entrapment and release in a peptide (FΔF) hydrogel. | ||
Potential application of self-assembling peptides in tissue engineering
Successful tissue regeneration requires that cells should be provided with an environment appropriate for regeneration, induction, and development of neo-tissue. The behavior pattern of cells changes when they are constrained to two dimensional (2D) culture in Petri dishes, after being isolated from their complex native tissue environment. Hence, it is necessary that cells should be grown in a three dimensional (3D) matrix to maintain their natural phenotypic shape and behavior patterns. Scaffolds required for the 3D growth of cells to generate specific organs should ideally be biodegradable with nontoxic degradation products easily eliminated by the host. One important aim in the design of synthetic tissue scaffolds is to mimic the structure and function of the extracellular matrix (ECM) for providing the cells a microenvironment similar to that encountered in vivo. The ECM organizes cells into tissue, affects overall cellular architecture, provides pathways for migratory cells, participates in signal transduction pathways and strengthens tissues. The architecture of the natural ECM components has been an inspiration for researchers to synthesize materials with a similar structure for biomedical applications such as hydrogels, for cell-based therapeutics204,205 and soft tissue engineering scaffolds.206The present day cell-based therapeutic hydrogels are either derived from natural biopolymers (such as collagen, hyaluronate, fibrin, alginate, agarose, and chitosan) or from synthetic polymers (such as poly(acrylic acid), poly(ethylene oxide), poly(vinyl alcohol), and polyphosphazene).206 The natural polymers suffer from limitations such as the presence of variable properties based on their source of origin or presence of viral contaminants, whereas, synthetic polymers usually lack functional sites for cellular interactions and may show adverse side-effects.
Self-assembling peptides generally show favorable properties concerning biocompatibility, immunogenicity and biodegradability, producing non-toxic waste products. In recent past several self-assembled peptide based hydrogels have been explored as scaffolds for 3D cell growth and tissue engineering purpose.207 Self-assembling peptide nanofiber scaffolds with amino terminal with sequences derived from collagen IV or laminin demonstrated enhanced adhesion and spreading of human aortic endothelial cells.208 In order to mimic the ECM structure and porosity, peptide nanofibers were further combined with epithelial growth factors. The modified nanofibers showed an improved rate of wound healing on a human skin equivalent tissue model.209 Such modified scaffolds have also showed enhanced growth properties in mouse neural stem cells210 and pre-osteoblasts.211 Zhang et al. developed a unique type of peptide based scaffolds which could be injected into the myocardium, creating an intra-myocardial microenvironment for mouse endothelial cells that promoted vascular cell recruitment.212
Hartgerink et al. developed various peptide amphiphiles which could form different types of nanofibers.213 Peptide amphiphile molecules were derivatized with biologically active peptides containing RGD for promoting cell adhesion and phosphorylated serine residues for promoting hydroxyapatite mineralization. Linear and cyclic RGDS214 as well as various other bioactive signals such as biotin215 were also presented on the N-terminal of peptide amphiphile nanofibers to promote better cell growth and differentiation. Peptide nanofibers were further assembled to from branched peptide amphiphiles, leading to greater accessibility of binding sites. These nanofibers were developed as bioactive scaffolds with improved epitope recognition for enhanced cell adhesion214,216 for tissue engineering applications,217,218 and magnetic resonance imaging.219 Sargeant et al. fabricated hybrid bone implants by self-assembling peptide amphiphile nanofibers within porous titanium.220 These implants demonstrated cell encapsulation, vascularization and mineralization of calcium phosphate with a calcium:phosphate ratio similar to that of hydroxyapatite.
Self-assembling nanofibers have also been shown to act as scaffolds for neural progenitor cells,221 dental stem cells222 as well as have been used for cell entrapment223 and stimulation of angiogenesis.224 RGD containing β-sheet forming peptide sequences with capacity to self-assemble into nanoribbons, have been shown to encapsulate hydrophobic molecules as well as cells through integrin receptors. Hydrogel scaffolds produced by the peptide Ac-(KLDL)3–CONH2 have been shown to support the growth and differentiation of chondrocytes along with stimulating the synthesis and accumulation of the extracellular matrix.225 Self-assembling peptides were also being developed as scaffolds for cartilage repair and promotion of nerve cell growth.226 A peptide amphiphile, C16-G3A4-IKVAV, shown to promote the re-growth of nerve cells in rats was made by including a neurite-promoting laminin epitope tag IKVAV.227
The β-hairpin hydrogel developed from the peptide MAX 1, promoted growth and proliferation of fibroblast cells.228 Another study demonstrated hydrogel formation from an N-terminally protected peptide sequence napthalene-FFGRGD.229 This was used for surface coating on poly(3-caprolactone) (PCL) films, and to promote cell attachment and growth.
Similarly self-assembled fibers generated from the RADA16 peptide were used in a wide range of biomaterial applications, including cartilage tissue repair,226 osteoblast proliferation and differentiation,211 bone regeneration230 and axon regeneration.231 Cardiomyocytes or non-differentiated stem cells loaded fibril-forming peptide gels when injected into damaged heart tissues led to the improvement of transplanted cell survival and wound healing after a myocardial infarction.212,232
Scaffolds derived from peptide-amphiphiles have been shown to promote the attachment of primary human bladder cells, demonstrating the potential biological application of PAs for augmenting the biocompatibility of polymeric materials conventionally used for tissue engineering.218
Another wonderful application of PA-based nanofibers includes the adhesion and migration of neural cells in vitro.227 Remarkably, these PA-based nanofibers exhibited extremely promising results in an animal model based on a spinal cord injury.233 Another amphiphilic peptide construct, containing a heparin-binding site, exhibited very exciting results in promoting angiogenesis.234 These types of peptides were further modified with biotin215 and a Gd3+ metal-chelating moiety suitable for detection by magnetic resonance imaging (MRI).219
Vescovi and group reported about short assembling peptides containing the bone marrow homing peptide 1 (BMHP 1) functional motif (PFSSTKT) (BMHP1-SAPs). These peptides assembled to form nano or microstructures of varied shape like tubular fibers, twisted ribbons, tubes and sheets. Interestingly, despite having a heterogeneous nanostructure morphology and varied scaffold stiffness, all the BMHP1-SAPs exhibited β-sheets and β-turns like the secondary structure. A few of these 10-mer peptides promoted adhesion, differentiation, and proliferation of human neural stem cells. These SAPs could be derivatized with the cell adhesion promoting motif like RGDGG, for promoting enhanced cell attachment. In vivo experiments carried out using these SAPs exhibited negligible side reactions towards host nervous tissue.210 Hydrogels prepared from the P11-4 peptide were also found to have tissue engineering applications such as enamel remineralization,235 injectable scaffolds236 and joint lubricants.237
Zhou et al. reported a peptide based bioactive hydrogel using molecular self-assembly of a mixture of two aromatic short peptide derivatives: Fmoc-FF (fluorenylmethoxycarbonyl diphenylalanine) and Fmoc-RGD (arginine–glycine–aspartate). This biomimetic nanofibrous hydrogel acted as a 3D-scaffold for anchorage-dependent cells.134
An array of FF nanofibers grown on gold microelectrodes led to the development combined cell culture and biosensing platforms. Peptide nanowires were modified with conductive polymers for enabling detection of dopamine at physiological concentrations as well as to grow different cell lines such as PC12 and HeLa.238
Our group has also reported 3D growth of mammalian cells [HeLa and fibroblast (L929)] on a chemically functionalized FΔF hydrogel. The peptide hydrogel was functionalized with the cell adhesion motif “RGD”containing pentapeptide to facilitate cell growth and proliferation. This functionalized hydrogel provided a wonderful support for 3D cell growth for more than two weeks with maintaining cell viability and spreading. This study provides an excellent example of a simple peptide based-hydrogel to attain increased cell growth promoting properties, with high enzymatic stability.239 This gel-based soft material acts as a convenient template for 3D cell growth with probable use in tissue engineering and cell biology.
All these results encourage the use of peptide-based biomaterials in regenerative medicine and pave the way for the development of novel self-assembling peptide sequences that may be useful for materials science and regenerative medicine applications. In addition to the above mentioned applications peptide based self-assembled structures can have many more applications such as materials for surface engineering, biosensing devices and many more to be discovered in near future.29
Conclusion
Self-assembly is a marvelous strategy to make an ensemble of nanostructures. Small peptide-based self-assembled nanostructures, due to their inherent biocompatibility, easy tunability, simple and cost-effective synthesis could offer a myriad of potential uses in biomedical applications. Peptides could spontaneously self-assemble into tubular or fibrillar or vesicular nanostructures. The nanostructures have been shown to entrap a wide range of bioactive molecules with a controlled release pattern. Peptide based hydrogels with a nanofibrillar morphology have been shown to support 3D growth of various cell types along with promoting cell differentiation in many cases. Thus small peptides have tremendous potential to be developed as intelligent biomedical scaffolds with many biomedical applications. Thus peptide self-assembly is emerging as a new area of research and has spurred intensive interest in the fabrication of nanoscale devices and demand for miniaturization in both academia and industries necessitate progress in this area.Acknowledgements
This work is supported from a Nanoinitiative grant from the Department of Science and Technology, India, a Nanotechnology grant from the Department of Biotechnology, India and core funding at the ICGEB, New Delhi, India. JJP also thanks UNESCO-L'oreal for women in science for the fellowship.References
- G. M. Whitesides and B. Grzybowski, Science, 2002, 295, 2418–2421 CrossRef CAS PubMed.
- S. Zhang, Nat. Biotechnol., 2003, 21, 1171–1178 CrossRef CAS PubMed.
- S. Stupp, Nano Lett., 2010, 10, 4783–4786 CrossRef CAS PubMed.
- G. M. Whitesides, J. P. Mathias and C. T. Seto, Science, 1991, 254, 1312–1319 CAS.
- Z. Liu, Y. Jiao, Y. Wang, C. Zhou and Z. Zhang, Adv. Drug Delivery Rev., 2008, 60, 1650–1662 CrossRef CAS PubMed.
- K. A. Janes, P. Calvo and M. J. Alonso, Adv. Drug Delivery Rev., 2001, 47, 83–97 CrossRef CAS.
- M. Prabaharan and J. F. Mano, Drug Delivery, 2005, 12, 41–57 CrossRef CAS.
- D. G. Kim, Y. I. Jeong, C. Choi, S. H. Roh, S. K. Kang, M. K. Jang and J. W. Nah, Int. J. Pharm., 2006, 319, 130–138 CrossRef CAS PubMed.
- Z. R. Cui and R. J. Mumper, J. Controlled Release, 2001, 75, 409–419 CrossRef CAS.
- A. Zahoor, S. Sharma and G. K. Khuller, Int. J. Antimicrob. Agents, 2005, 26, 298–303 CrossRef CAS PubMed.
- J. W. de Vries, F. Zhang and A. Herrmann, J. Controlled Release, 2013, 172, 467–483 CrossRef CAS PubMed.
- D. Kim, Y. Y. Jeong and S. Jon, ACS Nano, 2010, 4, 3689–3696 CrossRef CAS PubMed.
- J. I. Cutler, E. Auyeung and C. A. Mirkin, J. Am. Chem. Soc., 2012, 134, 1376–1391 CrossRef CAS PubMed.
- N. L. Rosi, Science, 2006, 312, 1027–1030 CrossRef CAS PubMed.
- J. J. Storhoff, R. Elghanian, R. C. Mucic, C. A. Mirkin and R. L. Letsinger, J. Am. Chem. Soc., 1998, 120, 1959–1964 CrossRef CAS.
- M. D. Costioli, I. Fisch, F. Garret-Flaudy, F. Hilbrig and R. Freitag, Biotechnol. Bioeng., 2003, 81, 535–545 CrossRef CAS PubMed.
- M. Safak, F. E. Alemdaroglu, Y. Li, E. Ergen and A. Herrmann, Adv. Mater., 2007, 19, 1499–1505 CrossRef CAS.
- F. E. Alemdaroglu, K. Ding, R. Berger and A. Herrmann, Angew. Chem., Int. Ed., 2006, 45, 4206–4210 CrossRef CAS PubMed.
- M. Kwak, J. Gao, D. K. Prusty, A. J. Musser, V. A. Markov, N. Tombros, M. C. A. Stuart, W. R. Browne, E. J. Boekema, G. ten Brinke, H. T. Jonkman, B. J. van Wees, M. A. Loi and A. Herrmann, Angew. Chem., Int. Ed., 2011, 50, 3206–3210 CrossRef CAS PubMed.
- P. Guo, J. Nanosci. Nanotechnol., 2005, 5, 1964–1982 CrossRef CAS PubMed.
- S. Guo, N. Tschammer, S. Mohammed and P. Guo, Hum. Gene Ther., 2005, 16, 1097–1109 CrossRef CAS PubMed.
- A. Khaled, S. Guo, F. Li and P. Guo, Nano Lett., 2005, 5, 1797–1808 CrossRef CAS PubMed.
- E. N. Marsh and W. F. DeGrado, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 5150–5154 CrossRef CAS PubMed.
- H. Gradišar and R. Jerala, J. Nanobiotechnol., 2014, 12, 1–9 CrossRef PubMed.
- C.-J. Tsai, J. Zheng, D. Zanuy, N. Haspel, H. Wolfson, C. Alema and R. Nussinov, Proteins: Struct., Funct., Bioinf., 2007, 68, 1–12 CrossRef CAS PubMed.
- E. Gazit, Drugs Future, 2004, 29, 613–619 CrossRef CAS.
- I. W. Hamley, Angew. Chem., Int. Ed., 2007, 46, 8128–8147 CrossRef CAS PubMed.
- E. Gazit, Chem. Soc. Rev., 2007, 36, 1263–1269 RSC.
- A. Lakshmanan, S. Zhang and C. A. Hauser, Trends Biotechnol., 2012, 30, 155–165 CrossRef CAS PubMed.
- K. Rajagopal and J. P. Schneider, Curr. Opin. Struct. Biol., 2004, 14, 480–486 CrossRef CAS PubMed.
- T. J. Deming, Adv. Drug Delivery Rev., 2002, 54, 1145–1155 CrossRef CAS.
- M. Gupta, A. Bagaria, A. Mishra, P. Mathur, A. Basu, S. Ramakumar and V. S. Chauhan, Adv. Mater., 2007, 19, 858 CrossRef CAS.
- J. J. Panda, A. Mishra, A. Basu and V. S. Chauhan, Biomacromolecules, 2008, 9, 2244–2250 CrossRef CAS PubMed.
- S. Scanlon and A. Aggeli, Nano Today, 2008, 3, 22–30 CrossRef CAS.
- D. Ranganathan, M. P. Samant and I. L. Karle, J. Am. Chem. Soc., 2001, 123, 5619–5624 CrossRef CAS PubMed.
- S. Santoso, W. Hwang, H. Hartman and S. Zhang, Nano Lett., 2002, 2, 687–691 CrossRef CAS.
- M. R. Ghadiri, J. R. Granja, R. A. Milligan, D. E. McRee and N. Khazanovich, Nature, 1993, 366, 324–327 CrossRef CAS PubMed.
- J. D. Hartgerink, J. R. Granja, R. A. Milligan and M. R. Ghadiri, J. Am. Chem. Soc., 1996, 118, 43–50 CrossRef CAS.
- C. Valery, M. Paternostre, B. Robert, T. Gulik-Krzywicki, T. Narayanan, J. C. Dedieu, G. Keller, M. L. Torres, R. Cherif-Cheikh, P. Calvo and F. Artzner, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 10258–10262 CrossRef CAS PubMed.
- S. Fernandez-Lopez, H. S. Kim, E. C. Choi, M. Delgado, J. R. Granja, A. Khasanov, K. Kraehenbuehl, G. Long, D. A. Weinberger, K. M. Wilcoxen and M. R. Ghadiri, Nature, 2001, 412, 452–455 CrossRef CAS PubMed.
- R. J. Brea, C. Reiriz and J. R. Granja, Chem. Soc. Rev., 2010, 39, 1448–1456 RSC.
- A. Aggeli, I. A. Nyrkova, M. Bell, R. Harding, L. Carrick, T. C. McLeish, A. N. Semenov and N. Boden, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 11857–11862 CrossRef CAS PubMed.
- A. Aggeli, M. Bell, L. M. Carrick, C. W. Fishwick, R. Harding, P. J. Mawer, S. E. Radford, A. E. Strong and N. Boden, J. Am. Chem. Soc., 2003, 125, 9619–9628 CrossRef CAS PubMed.
- D. M. Marini, W. Hwang, D. A. Lauffenburger, S. Zhang and R. D. Kamm, Nano Lett., 2002, 2, 295–299 CrossRef CAS.
- K. Lu, J. Jacob, P. Thiyagarajan, V. P. Conticello and D. G. Lynn, J. Am. Chem. Soc., 2003, 125, 6391–6393 CrossRef CAS PubMed.
- M. Reches and E. Gazit, Science, 2003, 300, 625–627 CrossRef CAS PubMed.
- S. Colfer, J. W. Kelly and E. T. Powers, Langmuir, 2003, 19, 1312–1318 CrossRef CAS.
- G. Xu, W. Wang, J. T. Groves and M. H. Hecht, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 3652–3657 CrossRef CAS PubMed.
- N. Ashkenasy, W. S. Horne and M. R. Ghadiri, Small, 2006, 2, 99–102 CrossRef CAS PubMed.
- J. P. Schneider, D. J. Pochan, B. Ozbas, K. Rajagopal, L. Pakstis and J. Kretsinger, J. Am. Chem. Soc., 2002, 124, 15030–15037 CrossRef CAS PubMed.
- E. T. Powers, S. I. Yang, C. M. Lieber and J. W. Kelly, Angew. Chem., Int. Ed. Engl., 2002, 41, 127–130 CrossRef CAS.
- L. Haines-Butterick, K. Rajagopal, M. Branco, D. Salick, R. Rughani, M. Pilarz, M. S. Lamm, D. J. Pochan and J. P. Schneider, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 7791–7796 CrossRef CAS PubMed.
- D. J. Pochan, J. P. Schneider, J. Kretsinger, B. Ozbas, K. Rajagopal and L. Haines, J. Am. Chem. Soc., 2003, 125, 11802–11803 CrossRef CAS PubMed.
- T. C. Holmes, S. de Lacalle, X. Su, G. Liu, A. Rich and S. Zhang, Proc. Natl. Acad. Sci. U. S. A., 2000, 97, 6728–6733 CrossRef CAS.
- M. V. Tsurkan and M. Y. Ogawa, Chem. Commun., 2004, 2092–2093 RSC.
- A. Aggeli, M. Bell, N. Boden, J. N. Keen, P. F. Knowles, T. C. McLeish, M. Pitkeathly and S. E. Radford, Nature, 1997, 386, 259–262 CrossRef CAS PubMed.
- K. Beck and B. Brodsky, J. Struct. Biol., 1998, 122, 17–29 CrossRef CAS PubMed.
- R. Fairman and K. S. Akerfeldt, Curr. Opin. Struct. Biol., 2005, 15, 453–463 CrossRef CAS PubMed.
- S. A. Potekhin, T. N. Melnik, V. Popov, N. F. Lanina, A. A. Vazina, P. Rigler, A. S. Verdini, G. Corradin and A. V. Kajava, Chem. Biol., 2001, 8, 1025–1032 CrossRef CAS.
- D. E. Wagner, C. L. Phillips, W. M. Ali, G. E. Nybakken, E. D. Crawford, A. D. Schwab, W. F. Smith and R. Fairman, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 12656–12661 CrossRef CAS PubMed.
- E. Moutevelis and D. N. Woolfson, J. Mol. Biol., 2009, 385, 726–732 CrossRef CAS PubMed.
- D. Papapostolou, A. M. Smith, E. D. Atkins, S. J. Oliver, M. G. Ryadnov, L. C. Serpell and D. N. Woolfson, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 10853–10858 CrossRef CAS PubMed.
- C. Gribbon, K. J. Channon, W. Zhang, E. F. Banwell, E. H. Bromley, J. B. Chaudhuri, R. O. Oreffo and D. N. Woolfson, Biochemistry, 2008, 47, 10365–10371 CrossRef CAS PubMed.
- H. Dong, S. E. Paramonov and J. D. Hartgerink, J. Am. Chem. Soc., 2008, 130, 13691–13695 CrossRef CAS PubMed.
- A. S. Hoffman, Adv. Drug Delivery Rev., 2002, 54, 3–12 CrossRef CAS.
- P. Gupta, K. Vermani and S. Garg, Drug Discovery Today, 2002, 7, 569–579 CrossRef CAS.
- W. A. Petka, J. L. Harden, K. P. McGrath, D. Wirtz and D. A. Tirrell, Science, 1998, 281, 389–392 CrossRef CAS.
- C. Xu, V. Breedveld and J. Kopecek, Biomacromolecules, 2005, 6, 1739–1749 CrossRef CAS PubMed.
- C. Xu and J. Kopecek, Pharm. Res., 2008, 25, 674–682 CrossRef CAS PubMed.
- E. K. O'Shea, K. J. Lumb and P. S. Kim, Curr. Biol., 1993, 3, 658–667 CrossRef CAS.
- M. G. Ryadnov, B. Ceyhan, C. M. Niemeyer and D. N. Woolfson, J. Am. Chem. Soc., 2003, 125, 9388–9394 CrossRef CAS PubMed.
- S. Vauthey, S. Santoso, H. Gong, N. Watson and S. Zhang, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 5355–5360 CrossRef CAS PubMed.
- T. Gore, Y. Dori, Y. Talmon, M. Tirrell and H. Bianco-Peled, Langmuir, 2001, 17, 5352–5360 CrossRef CAS.
- R. S. Tu and M. Tirrell, Adv. Drug Delivery Rev., 2004, 56, 1537–1563 CrossRef CAS PubMed.
- J. Schneider, P. Berndt, K. Haverstick, S. Kumar, S. Chiruvolu and M. Tirrell, J. Am. Chem. Soc., 1998, 120, 3508–3509 CrossRef CAS.
- S. Tsonchev, K. L. Niece, G. C. Schatz, M. A. Ratner and S. I. Stupp, J. Phys. Chem. B, 2008, 112, 441–447 CrossRef CAS PubMed.
- R. M. Fuoss and D. Edelson, J. Am. Chem. Soc., 1951, 73, 269–273 CrossRef CAS.
- R. C. Claussen, B. M. Rabatic and S. I. Stupp, J. Am. Chem. Soc., 2003, 125, 12680–12681 CrossRef CAS PubMed.
- M. Kogiso, Y. Okada, T. Hanada, K. Yase and T. Shimizu, Biochim. Biophys. Acta, 2000, 1475, 346–352 CrossRef CAS.
- F. Qiu, Y. Chen, C. Tang, Q. Zhou, C. Wang, Y. K. Shi and X. Zhao, Macromol. Biosci., 2008, 8, 1053–1059 CrossRef CAS PubMed.
- M. Z. Menzenski and I. A. Banerjee, New J. Chem., 2007, 31, 1674–1680 RSC.
- R. I. MacCuspie, I. A. Banerjee, C. Pejoux, S. Gummalla, H. S. Mostowski, P. R. Krause and H. Matsui, Soft Matter, 2008, 4, 833–839 RSC.
- R. de la Rica, E. Mendoza, L. M. Lechuga and H. Matsui, Angew. Chem., Int. Ed. Engl., 2008, 47, 9752–9755 CrossRef CAS PubMed.
- N. Kameta, M. Masuda, G. Mizuno, N. Morii and T. Shimizu, Small, 2008, 4, 561–565 CrossRef CAS PubMed.
- K. Tenidis, M. Waldner, J. Bernhagen, W. Fischle, M. Bergmann, M. Weber, M. L. Merkle, W. Voelter, H. Brunner and A. Kapurniotu, J. Mol. Biol., 2000, 295, 1055–1071 CrossRef CAS PubMed.
- P. Westermark, U. Engstrom, K. H. Johnson, G. T. Westermark and C. Betsholtz, Proc. Natl. Acad. Sci. U. S. A., 1990, 87, 5036–5040 CrossRef CAS.
- M. Reches and E. Gazit, Amyloid, 2004, 11, 81–89 CrossRef CAS.
- M. J. Krysmann, V. Castelletto, A. Kelarakis, I. W. Hamley, R. A. Hule and D. J. Pochan, Biochemistry, 2008, 47, 4597–4605 CrossRef CAS PubMed.
- M. Reches, Y. Porat and E. Gazit, J. Biol. Chem., 2002, 277, 35475–35480 CrossRef CAS PubMed.
- A. Saini and V. S. Chauhan, ChemBioChem, 2001, 12, 2495–2501 CrossRef PubMed.
- M. Kamihira, A. Naito, S. Tuzi, A. Y. Nosaka and H. Saito, Protein Sci., 2000, 9, 867–877 CrossRef CAS PubMed.
- C. A. Hauser, R. Deng, A. Mishra, Y. Loo, U. Khoe, F. Zhuang, D. W. Cheong, A. Accardo, M. B. Sullivan, C. Riekel, J. Y. Ying and U. A. Hauser, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 1361–1366 CrossRef CAS PubMed.
- A. Lakshmanan and C. A. Hauser, Int. J. Mol. Sci., 2011, 12, 5736–5746 CrossRef CAS PubMed.
- L. Tjernberg, W. Hosia, N. Bark, J. Thyberg and J. Johansson, J. Biol. Chem., 2002, 277, 43243–43246 CrossRef CAS PubMed.
- S. K. Maji, D. Haldar, M. G. B. Drew, A. Banerjee, A. K. Das and A. Banerjee, Tetrahedron, 2004, 60, 3251–3259 CrossRef CAS PubMed.
- P. P. Bose, A. K. Das, R. P. Hegde, N. Shamala and A. Banerjee, Chem. Mater., 2007, 19, 6150–6157 CrossRef CAS.
- L. Adler-Abramovich, D. Aronov, P. Beker, M. Yevnin, S. Stempler, L. Buzhansky, G. Rosenman and E. Gazit, Nat. Nanotechnol., 2009, 4, 849–854 CrossRef CAS PubMed.
- M. Wang, L. Du, X. Wu, S. Xiong and P. K. Chu, ACS Nano, 2011, 5, 4448–4454 CrossRef CAS PubMed.
- P. Zhu, X. Yan, Y. Su, Y. Yang and J. Li, Chemistry, 2010, 16, 3176–3183 CrossRef CAS PubMed.
- J. Ryu and C. B. Park, Adv. Mater., 2008, 20, 3754–3758 CrossRef CAS.
- M. Reches and E. Gazit, Nat. Nanotechnol., 2006, 1, 195–200 CrossRef CAS PubMed.
- L. Adler-Abramovich, M. Reches, V. L. Sedman, S. Allen, S. J. Tendler and E. Gazit, Langmuir, 2006, 22, 1313–1320 CrossRef CAS PubMed.
- N. Hendler, N. Sidelman, M. Reches, E. Gazit, Y. Rosenberg and S. Richter, Adv. Mater., 2007, 19, 1485–1488 CrossRef CAS.
- J. Kim, T. H. Han, Y. I. Kim, J. S. Park, J. Choi, D. G. Churchill, S. O. Kim and H. Ihee, Adv. Mater., 2010, 22, 583–587 CrossRef CAS PubMed.
- C. H. Görbitz, New J. Chem., 2003, 27, 1789–1793 RSC.
- C. H. Görbitz, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2004, 60, o626–o628 Search PubMed.
- C. H. Gorbitz, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2006, 62, o328–330 Search PubMed.
- C. H. Gorbitz, Chemistry, 2001, 7, 5153–5159 CrossRef CAS.
- C. H. Gorbitz, Acta Crystallogr., Sect. B: Struct. Sci., 2002, 58, 849–854 Search PubMed.
- H. Birkedal, D. Schwarzenbach and P. Pattison, Angew. Chem., Int. Ed. Engl., 2002, 41, 754–756 CrossRef CAS.
- Y. Pan, H. Birkedal, P. Pattison, D. Brown and G. Chapuis, J. Phys. Chem. B, 2004, 108, 6458–6466 CrossRef CAS PubMed.
- C. H. Görbitz, M. Nilsen, K. Szeto and L. W. Tangen, Chem. Commun., 2005, 4288–4290 RSC.
- C. H. Gorbitz, Chemistry, 2007, 13, 1022–1031 CrossRef PubMed.
- D. V. Soldatov, I. L. Moudrakovski and J. A. Ripmeester, Angew. Chem., Int. Ed. Engl., 2004, 43, 6308–6311 CrossRef CAS PubMed.
- I. Moudrakovski, D. V. Soldatov, J. A. Ripmeester, D. N. Sears and C. J. Jameson, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 17924–17929 CrossRef CAS PubMed.
- R. Anedda, D. V. Soldatov, I. L. Moudrakovski, M. Casu and J. A. Ripmeester, Chem. Mater., 2008, 20, 2908–2920 CrossRef CAS.
- M. Reches and E. Gazit, Nano Lett., 2004, 4, 581–585 CrossRef CAS.
- M. Reches and E. Gazit, Phys. Biol., 2006, 3, S10–S19 CrossRef CAS PubMed.
- O. Carny, D. E. Shalev and E. Gazit, Nano Lett., 2006, 6, 1594–1597 CrossRef CAS PubMed.
- M. Yemini, M. Reches, J. Rishpon and E. Gazit, Nano Lett., 2005, 5, 183–186 CrossRef CAS PubMed.
- M. Yemini, M. Reches, E. Gazit and J. Rishpon, Anal. Chem., 2005, 77, 5155–5159 CrossRef CAS PubMed.
- N. Amdursky, I. Koren, E. Gazit and G. Rosenman, J. Nanosci. Nanotechnol., 2012, 11, 9282–9286 CrossRef PubMed.
- N. Amdursky, M. Molotskii, D. Aronov, L. Adler-Abramovich, E. Gazit and G. Rosenman, Nano Lett., 2009, 9, 3111–3115 CrossRef CAS PubMed.
- L. Adler-Abramovich, M. Badihi-Mossberg, E. Gazit and J. Rishpon, Small, 2010, 6, 825–831 CrossRef CAS PubMed.
- A. Comotti, S. Bracco, G. Distefano and P. Sozzani, Chem. Commun., 2009, 284–286 RSC.
- N. S. de Groot, T. Parella, F. X. Aviles, J. Vendrell and S. Ventura, Biophys. J., 2007, 92, 1732–1741 CrossRef PubMed.
- R. Orbach, L. Adler-Abramovich, S. Zigerson, I. Mironi-Harpaz, D. Seliktar and E. Gazit, Biomacromolecules, 2009, 10, 2646–2651 CrossRef CAS PubMed.
- A. Mahler, M. Reches, M. Rechter, S. Cohen and E. Gazit, Adv. Mater., 2006, 18, 1365–1370 CrossRef CAS.
- S. Guha, M. G. B. Drew and A. Banerjee, Chem. Mater., 2008, 20, 2282–2290 CrossRef CAS.
- S. Guha, M. G. Drew and A. Banerjee, Small, 2008, 4, 1993–2005 CrossRef CAS PubMed.
- S. Guha and A. Banerjee, Adv. Funct. Mater., 2009, 19, 1949–1961 CrossRef CAS.
- S. Guha, T. Chakraborty and A. Banerjee, Green Chem., 2009, 11, 1139–1145 RSC.
- M. Reches and E. Gazit, Isr. J. Chem., 2005, 45, 363–371 CrossRef CAS.
- M. Zhou, A. M. Smith, A. K. Das, N. W. Hodson, R. F. Collins, R. V. Ulijn and J. E. Gough, Biomaterials, 2009, 30, 2523–2530 CrossRef CAS PubMed.
- T. Liebmann, S. Rydholm, V. Akpe and H. Brismar, BMC Biotechnol., 2007, 7, 88 CrossRef PubMed.
- V. Jayawarna, M. Ali, T. A. Jowitt, A. F. Miller, A. Saiani, J. E. Gough and R. V. Ulijn, Adv. Mater., 2006, 18, 611–614 CrossRef CAS.
- V. Jayawarna, A. Smith, J. E. Gough and R. V. Ulijn, Biochem. Soc. Trans., 2007, 35, 535–537 CrossRef CAS PubMed.
- Z. Yang, G. Liang and B. Xu, Chem. Commun., 2006, 738–740 RSC.
- G. Liang, Z. Yang, R. Zhang, L. Li, Y. Fan, Y. Kuang, Y. Gao, T. Wang, W. W. Lu and B. Xu, Langmuir, 2009, 25, 8419–8422 CrossRef CAS.
- B. Xing, C. W. Yu, K. H. Chow, P. L. Ho, D. Fu and B. Xu, J. Am. Chem. Soc., 2002, 124, 14846–14847 CrossRef CAS PubMed.
- Z. Yang, H. Gu, Y. Zhang, L. Wang and B. Xu, Chem. Commun., 2004, 208–209 RSC.
- E. K. Johnson, D. J. Adams and P. J. Cameron, J. Am. Chem. Soc., 132, 5130–5136 CrossRef CAS PubMed.
- S. Gilead and E. Gazit, Supramol. Chem., 2005, 17, 87 CrossRef CAS.
- Y. Song, S. R. Challa, C. J. Medforth, Y. Qiu, R. K. Watt, D. Pena, J. E. Miller, F. van Swol and J. A. Shelnutt, Chem. Commun., 2004, 1044–1045 RSC.
- X. Yan, Q. He, K. Wang, L. Duan, Y. Cui and J. Li, Angew. Chem., Int. Ed. Engl., 2007, 46, 2431–2434 CrossRef CAS PubMed.
- X. Yan, Y. Cui, Q. He, K. Wang, J. Li, W. Mu, B. Wang and Z. C. Ou-Yang, Chemistry, 2008, 14, 5974–5980 CrossRef CAS PubMed.
- J. Naskar and A. Banerjee, Chem. – Asian J., 2009, 4, 1817–1823 CrossRef CAS PubMed.
- A. Mishra, J. J. Panda, A. Basu and V. S. Chauhan, Langmuir, 2008, 24, 4571–4576 CrossRef CAS PubMed.
- S. Alam, J. J. Panda and V. S. Chauhan, Int. J. Nanomed., 2012, 7, 4207–4221 CAS.
- D. M. Ryan, T. M. Doran, S. B. Anderson and B. L. Nilsson, Langmuir, 2011, 27, 4029–4039 CrossRef CAS PubMed.
- Z. Yang, H. Gu, D. Fu, P. Gao, J. K. Lam and B. Xu, Adv. Mater., 2004, 16, 1440–1444 CrossRef CAS.
- Z. Yang and B. Xu, Chem. Commun., 2004, 2424–2425 RSC.
- Z. Yang, G. Liang and B. Xu, Acc. Chem. Res., 2008, 41, 315–326 CrossRef CAS PubMed.
- L. Adler-Abramovich, L. Vaks, O. Carny, D. Trudler, A. Magno, A. Caflisch, D. Frenkel and E. Gazit, Nat. Chem. Biol., 2012, 8, 701–706 CrossRef CAS PubMed.
- S. Tsonchev, G. C. Schatz and M. A. Ratner, Nano Lett., 2003, 3, 623–626 CrossRef CAS.
- M. McCullagh, T. Prytkova, S. Tonzani, N. D. Winter and G. C. Schatz, J. Phys. Chem. B, 2008, 112, 10388–10398 CrossRef CAS PubMed.
- S. Tsonchev, A. Troisi, G. C. Schatz and M. A. Ratner, J. Phys. Chem. B, 2004, 108, 15278 CrossRef CAS.
- O.-S. Lee, S. I. Stupp and G. C. Schatz, J. Am. Chem. Soc., 2011, 133, 3677–3683 CrossRef CAS PubMed.
- O.-S. Lee, V. Cho and G. C. Schatz, Nano Lett., 2012, 12, 4907–4913 CrossRef CAS PubMed.
- Y. S. Velichko, S. I. Stupp and M. O. de la Cruz, J. Phys. Chem. B, 2008, 112, 2326 CrossRef CAS PubMed.
- E. T. Pashuck, H. G. Cui and S. I. Stupp, J. Am. Chem. Soc., 2010, 132, 6041 CrossRef CAS PubMed.
- J. C. Stendahl, M. S. Rao, M. O. Guler and S. I. Stupp, Adv. Funct. Mater., 2006, 16, 499 CrossRef CAS.
- H. Z. Jiang, M. O. Guler and S. I. Stupp, Soft Matter., 2007, 3, 454 RSC.
- D. Flock, G. Rossetti, I. Daidone, A. Amadei and A. Di Nola, Proteins, 2006, 65, 914–921 CrossRef CAS PubMed.
- P. Tamamis, L. Adler-Abramovich, M. Reches, K. Marshall, P. Sikorski, L. Serpell, E. Gazit and G. Archontis, Biophys. J., 2009, 96, 5020–5029 CrossRef CAS PubMed.
- A. Villa, N. F. A. van der Vegt and C. Peter, Phys. Chem. Chem. Phys., 2009, 11, 2068–2076 RSC.
- A. Villa, C. Peter and N. F. A. van der Vegt, Self-Assembling Dipeptides: Conformational Sampling in Solvent-Free Coarse-Grained Simulation, Phys. Chem. Chem. Phys., 2009, 11, 2077–2086 RSC.
- C.-J. Tsai, J. Zheng and R. Nussinov, PLoS Comput. Biol., 2006, 2, e42 CrossRef PubMed.
- C. H. Gorbitz, Chem. Commun., 2006, 2332 RSC.
- C. Guo, Y. Luo, R. Zhou and G. Wei, ACS Nano, 2012, 6, 3907–3918 CrossRef CAS PubMed.
- J. Jeon, C. E. Mills and M. S. Shell, J. Phys. Chem. B, 2013, 117, 3935–3943 CrossRef CAS PubMed.
- D. Peer, J. M. Karp, S. Hong, O. C. Farokhzad, R. Margalit and R. Langer, Nat. Nanotechnol., 2007, 2, 751–760 CrossRef CAS PubMed.
- W. Wiwanitkit, A. Sereemaspun and R. Rojanathanes, Fertil. Steril., 2009, 91, e7–e8 Search PubMed.
- W. H. De Jong and P. J. A. Borm, Int. J. Nanomed., 2008, 3, 133–149 CAS.
- D. B. Warheit, B. R. Laurence, K. L. Reed, D. H. Roach, G. A. Reynolds and T. R. Webb, Toxicol. Sci., 2004, 77, 117–125 CrossRef CAS PubMed.
- K. R. Vega-Villa, J. K. Takemoto, J. A. Yáñez, C. M. Remsburg, M. L. Forrest and N. M. Davies, Adv. Drug Delivery Rev., 2008, 60, 929–938 CrossRef CAS PubMed.
- S. I. Wada and R. Tanaka, Bioorg. Med. Chem. Lett., 2004, 14, 2563–2566 CrossRef CAS PubMed.
- L. Liu, K. Xu, H. Wang, P. K. Tan, W. Fan, S. S. Venkatraman, L. Li and Y. Y. Yang, Nat. Nanotechnol., 2009, 4, 457–463 CrossRef CAS PubMed.
- M. R. Ghadiri, J. R. Granja and L. K. Buehler, Nature, 1994, 369, 301–304 CrossRef CAS PubMed.
- V. Dartois, J. Sanchez-Quesada, E. Cabezas, E. Chi, C. Dubbelde, C. Dunn, J. Granja, C. Gritzen, D. Weinberger, M. R. Ghadiri and T. R. Parr, Jr, Antimicrob. Agents Chemother., 2005, 49, 3302–3310 CrossRef CAS PubMed.
- A. Montero, P. Gastaminza, M. Law, G. Cheng, F. V. Chisari and M. R. Ghadiri, Chem. Biol., 2011, 18, 1453–1462 CrossRef CAS PubMed.
- M. C. Morris, P. Vidal, L. Chaloin, F. Heitz and G. Divita, Nucleic Acids Res., 1997, 25 Search PubMed.
- F. Simeoni, M. C. Morris, F. Heitz and G. Divita, Nucleic Acids Res., 2003, 31, 2717–2724 CrossRef CAS PubMed.
- L. Crombez, M. C. Morris, S. Deshayes, F. Heitz and G. Divita, Curr. Pharm. Des., 2008, 14, 3656–3665 CrossRef CAS.
- D. Bhadra, S. Bhadra and N. K. Jain, Pharm. Res., 2006, 23, 623–633 CrossRef CAS PubMed.
- N. Wiradharma, M. Khan, Y. W. Tong, S. Wang and Y.-Y. Yang, Adv. Funct. Mater., 2008, 18, 943–951 CrossRef CAS.
- N. Wiradharma, Y. W. Tong and Y. Y. Yang, Biomaterials, 2009, 30, 3100–3109 CrossRef CAS PubMed.
- G. von Maltzahn, S. Vauthey, S. Santoso and S. U. Zhang, Langmuir, 2003, 19, 4332–4337 CrossRef CAS.
- X. Zhao and S. Zhang, Trends Biotechnol., 2004, 22, 470–476 CrossRef CAS PubMed.
- J. Gorman, Sci. News, 2003, 163, 43–44 CrossRef.
- Y. B. Lim, E. Lee, Y. R. Yoon, M. S. Lee and M. Lee, Angew. Chem., Int. Ed. Engl., 2008, 47, 4525–4528 CrossRef CAS PubMed.
- X. Li, J. Li, Y. Gao, Y. Kuang, J. Shi and B. Xu, J. Am. Chem. Soc., 2010, 132, 17707–17709 CrossRef CAS PubMed.
- J. Naskar, S. Roy, A. Joardar, S. Das and A. Banerjee, Org. Biomol. Chem., 2011, 9, 6610–6615 CAS.
- M. C. Branco, D. J. Pochan, N. J. Wagner and J. P. Schneider, Biomaterials, 2009, 30, 1339–1347 CrossRef CAS PubMed.
- J. Naskar, G. Palui and A. Banerjee, J. Phys. Chem. B, 2009, 113, 11787–11792 CrossRef CAS PubMed.
- J. H. Kim, S. Y. Lim, D. H. Nam, J. Ryu, S. H. Ku and C. B. Park, Biosens. Bioelectron., 2011, 26, 1860 CrossRef CAS PubMed.
- J. Nanda and A. Banerjee, Soft Matter, 2012, 8, 3380–3386 RSC.
- J. J. Panda, A. Kaul, S. Alam, A. K. Babbar, A. K. Mishra and V. S. Chauhan, Ther. Delivery, 2011, 2, 193–204 CrossRef CAS.
- J. J. Panda, A. Kaul, S. Kumar, S. Alam, A. K. Mishra, G. C. Kundu and V. S. Chauhan, Nanomedicine, 2013, 1927–1942 CrossRef CAS PubMed.
- J. J. Panda, S. Yandrapu, R. S. Kadam, V. S. Chauhan and U. B. Kompella, J. Controlled Release, 2013, 172, 1151–1160 CrossRef CAS PubMed.
- J. J. Panda, A. Varshney and V. S. Chauhan, J. Nanobiotechno., 2013, 11, 18 CAS.
- D. Mendel, J. A. Ellman, Z. Chang, D. L. Veenstra, P. A. Kollman and P. G. Schultz, Science, 1992, 256, 1798–1802 CAS.
- M. L. English and C. H. Stammer, Biochem. Biophys. Res. Commun., 1978, 83, 1464 CrossRef CAS.
- A. C. Jen, M. C. Wake and A. G. Mikos, Biotechnol. Bioeng., 1996, 50, 357–364 CrossRef CAS.
- M. J. Lysaght and P. Aebischer, Sci. Am., 1999, 280, 76–82 CrossRef CAS PubMed.
- K. Y. Lee and D. J. Mooney, Chem. Rev., 2001, 101, 1869–1879 CrossRef CAS PubMed.
- S. Kyle, A. Aggeli, E. Ingham and M. J. McPherson, Trends Biotechnol., 2009, 27, 423–433 CrossRef CAS PubMed.
- E. Genove, C. Shen, S. Zhang and C. E. Semino, Biomaterials, 2005, 26, 3341–3351 CrossRef CAS PubMed.
- A. Schneider, J. A. Garlick and C. Egles, PLoS One, 2008, 3, e1410 Search PubMed.
- F. Gelain, D. Bottai, A. Vescovi and S. Zhang, PLoS One, 2006, 1, e119 Search PubMed.
- A. Horii, X. Wang, F. Gelain and S. Zhang, PLoS One, 2007, 2, e190 Search PubMed.
- M. E. Davis, J. P. Motion, D. A. Narmoneva, T. Takahashi, D. Hakuno, R. D. Kamm, S. Zhang and R. T. Lee, Circulation, 2005, 111, 442–450 CrossRef CAS PubMed.
- J. D. Hartgerink, E. Beniash and S. I. Stupp, Science, 2001, 294, 1684–1688 CrossRef CAS PubMed.
- M. O. Guler, L. Hsu, S. Soukasene, D. A. Harrington, J. F. Hulvat and S. I. Stupp, Biomacromolecules, 2006, 7, 1855–1863 CrossRef CAS PubMed.
- M. O. Guler, S. Soukasene, J. F. Hulvat and S. I. Stupp, Nano Lett., 2005, 5, 249–252 CrossRef CAS PubMed.
- H. Storrie, M. O. Guler, S. N. Abu-Amara, T. Volberg, M. Rao, B. Geiger and S. I. Stupp, Biomaterials, 2007, 28, 4608–4618 CrossRef CAS PubMed.
- Z. Huang, T. D. Sargeant, J. F. Hulvat, A. Mata, P. Bringas Jr, C. Y. Koh, S. I. Stupp and M. L. Snead, J. Bone Miner. Res., 2008, 23, 1995–2006 CrossRef CAS PubMed.
- D. A. Harrington, E. Y. Cheng, M. O. Guler, L. K. Lee, J. L. Donovan, R. C. Claussen and S. I. Stupp, J. Biomed. Mater. Res., Part A, 2006, 78, 157–167 CrossRef PubMed.
- S. R. Bull, M. O. Guler, R. E. Bras, T. J. Meade and S. I. Stupp, Nano Lett., 2005, 5, 1–4 CrossRef CAS PubMed.
- T. D. Sargeant, M. O. Guler, S. M. Oppenheimer, A. Mata, R. L. Satcher, D. C. Dunand and S. I. Stupp, Biomaterials, 2008, 29, 161–171 CrossRef CAS PubMed.
- J. Guo, H. Su, Y. Zeng, Y. X. Liang, W. M. Wong, R. G. Ellis-Behnke, K. F. So and W. Wu, Nanomedicine, 2007, 3, 311–321 CrossRef CAS PubMed.
- K. M. Galler, A. Cavender, V. Yuwono, H. Dong, S. Shi, G. Schmalz, J. D. Hartgerink and R. N. D'Souza, Tissue Eng., Part A, 2008, 14, 2051–2058 CrossRef CAS PubMed.
- E. Beniash, J. D. Hartgerink, H. Storrie, J. C. Stendahl and S. I. Stupp, Acta Biomater., 2005, 1, 387–397 CrossRef PubMed.
- K. Rajangam, H. A. Behanna, M. J. Hui, X. Han, J. F. Hulvat, J. W. Lomasney and S. I. Stupp, Nano Lett., 2006, 6, 2086–2090 CrossRef CAS PubMed.
- J. Kisiday, M. Jin, B. Kurz, H. Hung, C. Semino, S. Zhang and A. J. Grodzinsky, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 9996–10001 CrossRef CAS PubMed.
- T. C. Holmes, Trends Biotechnol., 2002, 20, 16–21 CrossRef CAS.
- G. A. Silva, C. Czeisler, K. L. Niece, E. Beniash, D. A. Harrington, J. A. Kessler and S. I. Stupp, Science, 2004, 303, 1352–1355 CrossRef CAS PubMed.
- J. K. Kretsinger, L. A. Haines, B. Ozbas, D. J. Pochan and J. P. Schneider, Biomaterials, 2005, 26, 5177–5186 CrossRef CAS PubMed.
- Z. Wang, H. Wang, W. Zheng, J. Zhang, Q. Zhao, S. Wang, Z. Yang and D. Kong, Chem. Commun., 2011, 47, 8901–8903 RSC.
- H. Misawa, N. Kobayashi, A. Soto-Gutierrez, Y. Chen, A. Yoshida, J. D. Rivas-Carrillo, N. Navarro-Alvarez, K. Tanaka, A. Miki, J. Takei, T. Ueda, M. Tanaka, H. Endo, N. Tanaka and T. Ozaki, Cell Transplant., 2006, 15, 903–910 Search PubMed.
- R. G. Ellis-Behnke, Y. X. Liang, S. W. You, D. K. Tay, S. Zhang, K. F. So and G. E. Schneider, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 5054–5059 CrossRef CAS PubMed.
- M. E. Davis, P. C. Hsieh, T. Takahashi, Q. Song, S. Zhang, R. D. Kamm, A. J. Grodzinsky, P. Anversa and R. T. Lee, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 8155–8160 CrossRef CAS PubMed.
- V. M. Tysseling-Mattiace, V. Sahni, K. L. Niece, D. Birch, C. Czeisler, M. G. Fehlings, S. I. Stupp and J. A. Kessler, J. Neurosci., 2008, 28, 3814–3823 CrossRef CAS PubMed.
- R. F. Service, Science, 2005, 308, 44 CAS.
- J. Kirkham, A. Firth, D. Vernals, N. Boden, C. Robinson, R. C. Shore, S. J. Brookes and A. Aggeli, J. Dent. Res., 2007, 86, 426–430 CrossRef CAS PubMed.
- A. Firth, A. Aggeli, J. L. Burke, X. Yang and J. Kirkham, Nanomedicine, 2006, 12, 189–199 CrossRef PubMed.
- C. J. Bell, L. M. Carrick, J. Katta, Z. Jin, E. Ingham, A. Aggeli, N. Boden, T. A. Waigh and J. Fisher, J. Biomed. Mater. Res., Part A, 2006, 78, 236–246 CrossRef PubMed.
- L. Sasso, I. Vedarethinam, J. Emnéus, W. E. Svendsen and J. Castillo-Leóon, J. Nanosci. Nanotechnol., 2012, 12, 3077–3083 CrossRef CAS PubMed.
- J. J. Panda, R. Dua, A. Mishra, B. Mittra and V. S. Chauhan, ACS Appl. Mater. Interfaces, 2010, 2, 2839–2848 CAS.
| This journal is © The Royal Society of Chemistry 2014 |