
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Preparation of thermally curable materials using lignin extracted from sawmill co-product†
Libby J.
Marshall
 a,
Daniel J.
Cheney
b,
Jasmine A.
Keith
b,
Christopher
Kelly
a,
Frédéric
Blanc
bcd,
Andrew J.
West
e and
Dave J.
Adams
*a
a,
Daniel J.
Cheney
b,
Jasmine A.
Keith
b,
Christopher
Kelly
a,
Frédéric
Blanc
bcd,
Andrew J.
West
e and
Dave J.
Adams
*a
aSchool of Chemistry, University of Glasgow, Glasgow, G12 8QQ, UK. E-mail: dave.adams@glasgow.ac.uk
bDepartment of Chemistry, University of Liverpool, Liverpool, L69 7ZD, UK
cStephenson Institute for Renewable Energy, University of Liverpool, Liverpool, L69 7ZF, UK
dLeverhulme Research Centre for Functional Materials Design, Materials Innovation Factory, University of Liverpool, Liverpool, L69 7ZD, UK
eSonichem, Unit C5a, Melton Commercial Park, Melton Mowbray, LE14 3JL, UK
First published on 25th February 2025
Abstract
We have exemplified the chemical reactivity of a new commercially available organosolv-like lignin via reaction with various amines to form benzoxazine groups. The resulting benzoxazine-modified lignin has desirable thermal properties, making it an attractive alternative to non-renewable benzoxazine resins. The thermal properties of the lignin-benzoxazine products can be altered by increasing the number of benzoxazine groups within the lignin. This is achieved by increasing the number of unsubstituted phenol groups in the lignin prior to benzoxazine formation, thereby increasing the number of benzoxazine groups added to the lignin. We were ultimately able to form a curable system from phenolated lignin prepared from lignin extracted from low-value biomass using ultrasonic biorefinery technology via functionalisation with benzoxazine groups. The properties of the new lignin-based benzoxazines are compared to those of Kraft lignin (obtained from pulp) modified by the same chemical reactions.
Sustainability spotlightThe UK generates approximately 4.5 million tonnes of waste wood every year, with around 45% of logs that enter a sawmill ending up as low-value by-products. The lignin within these is a potential valuable building block but is often difficult to work with. Naturally occurring lignin is not immediately transferrable to useful types of products and functionalisation is required to create useful materials. Here, we describe the use of a low molecular weight lignin as a building block for useful polybenzoxazines and show that this lignin provides improvements over traditionally used lignin. |
Introduction
Polybenzoxazines (PBZs) are a relatively new class of thermosetting polymer that possess a range of interesting and useful properties. These properties include high thermal stability,1 processability,2 low water absorption,3 and high char yield.1 PBZs are prepared via a Mannich-type reaction between a polyphenol, formaldehyde and an amine.4 The versatility of this reaction, supplied by the vast number of possible amine starting materials, provides enormous scope for designing PBZs for specific applications.5–10 There is also potential for preparing composite materials by mixing PBZs with epoxides.4Conventional benzoxazine (BZ) synthesis relies on petroleum-derived monomers. Due to their adverse effects on the environment, there is a strong need to reduce reliance on petrochemicals. An attractive alternative is use of bio-derived materials,11–13 such as lignin. Lignin is one of the most abundant natural biopolymers found in biomass. Massive amounts of lignin are generated during the processing of wood, with 98% of this lignin being burned as low-value fuel.14 There is therefore a vast quantity of abundant and cheap lignin available for valorisation. Use of such biopolymers reduces reliance on non-renewable, petrochemical-based materials and allows sustainable, environmentally safe and biodegradable materials to be used in their place.
There are several advantages to preparing BZs directly from a polymer. For example, the chain ends present in PBZs derived from monomeric BZs tend to degrade more easily than the main chain, making BZs prepared from polymeric starting material more thermodynamically stable.15 The presence of a pre-formed polymer also allows the system to make use of the properties already held by the polymer.
Lignin has a complex structure with non-uniform repeating units. The aromatic units in lignin are generally referred to as H, G, and S depending on the number of methoxy groups present on the aromatic ring (Fig. 1a).16 The number and distribution of these units within the lignin structure depends on the lignin source (e.g., species of plant, environmental conditions in which the plant has grown) and the method by which the lignin was extracted from its biomass. The aromatic units are held together by various linkers. These linkers are predominantly aryl-ether linkages (e.g. β-O-4, Fig. 1b).17 The main functional groups present in lignin are phenols, ethers and hydroxyl groups.
 | ||
| Fig. 1 Chemical structures of (a) the aromatic units found in lignin and (b) the main linker group present in lignin, β-O-4 (red). | ||
Owing to its structure, lignin can be used to prepare phenolic resins.18–21 Such resins are widely used due to their high heat resistance,22 electrical insulation,23 dimensional stability,4 low flammability22 and smoke generation.24 However, these resins often suffer from short shelf-life, release of by-products during polymerisation and brittleness. BZ-based phenolic resins are an attractive alternative to traditional phenolic materials as they do not produce by-products during polymerisation and have high resistance to water and heat.25 The thermomechanical properties of BZ resins can be improved by introducing components with low glass transition temperature (Tg).26
Previous work has prepared BZ/lignin mixtures to tune the thermal properties of BZ-based materials27,28 Increasing lignin content was shown to reduce the maximum curing temperature and increase Tg.27 Sonichem lignin (SL) is extracted from the by-products of forestry using Sonichem's ultrasonic biorefinery technology.29,30 Sonichem lignin has relatively low molecular weight (∼1500 Da, Fig. S1, ESI†) and high solubility compared to other currently available lignins.
Lignin from sawmill co-product is usually extracted using the Kraft or sulfite pulping process, with a focus on the production of low-lignin cellulose for pulp and paper manufacture. As such, the lignin is essentially a waste product and is most typically burned for energy recovery, and to allow the isolation and recovery of the inorganic processing chemicals to improve the economics of the processing plant. When lignin is isolated, it often has a very broad molecular weight distribution from a few hundred to tens of thousands of Daltons.31 This lignin also has a high sulfur content as a consequence of the chemicals used in the pulping process, which limits its end uses.32 Organosolv lignin is typically obtained using a milder process than either Kraft or sulfite methods, which allows the isolation of more ‘natural’, less-condensed lignin with no added sulfur.33 The properties of organosolv lignin are highly dependent on both the starting biomass and the isolation method. Sonichem lignin is isolated using a low-temperature, ultrasound-assisted process that reduces lignin condensation and fractionation during extraction, producing a highly soluble, low molecular weight powder with a low polydispersity index (PDI).
The low molecular weight of Sonichem lignin removes the need to depolymerise lignin for preparation of BZs.8,34 Here, we show the reactivity and use of Sonichem lignin by reacting it with various amines (Fig. 2), allowing formation of BZ groups within the lignin structure. The resulting materials exhibit favourable thermal properties. The number of BZ groups introduced can be increased by phenolating the lignin prior to BZ formation.35 Phenolation increases the number of unsubstituted phenols, or H units, present in the lignin. We compare the performance of Sonichem lignin with commercially available Kraft lignin (KL) purchased from Sigma-Aldrich. The use of diamines in BZ synthesis provides potential for cross-linking via further BZ formation using the second amine (Fig. 2).
 | ||
| Fig. 2 BZ synthesis using lignin as the phenol and aniline, amine A (para-phenylene diamine) or amine B (bis-(4-aminophenyl)ether) as the amine. Using diamine compounds, such as amine A or amine B, provides potential for cross-linking via formation of a second BZ group. | ||
Results and discussion
Sonichem lignin is isolated from the by-products of sawmills using a low energy ultrasonic process. This method of extraction results in Sonichem lignin possessing lower molecular weight than most commercially available lignin. Results from solid-state 13C cross polarisation (CP) magic angle spinning (MAS) NMR spectroscopy (Fig. 3), liquid-state 31P NMR spectroscopy of phosphitylated materials (Fig. 4 and 10) and FTIR spectroscopy (Fig. 5) show Sonichem lignin contains similar functional groups to commercially available Kraft lignin, but in different proportions, highlighting the potential for different properties and reactivities. Interestingly, the FTIR spectra of Sonichem lignin is very similar to spectra found in literature corresponding to organosolv lignin, suggesting the two may have similar properties. Unfortunately, organosolv lignin was not readily available for purchase in the UK. Instead, Kraft lignin was selected for comparison. Elemental analysis (Table 1) and XPS data (Table 2) suggest Kraft lignin contains a higher percentage of oxygen than Sonichem lignin. However, these values do not confirm the type of functional group the oxygen atoms are part of.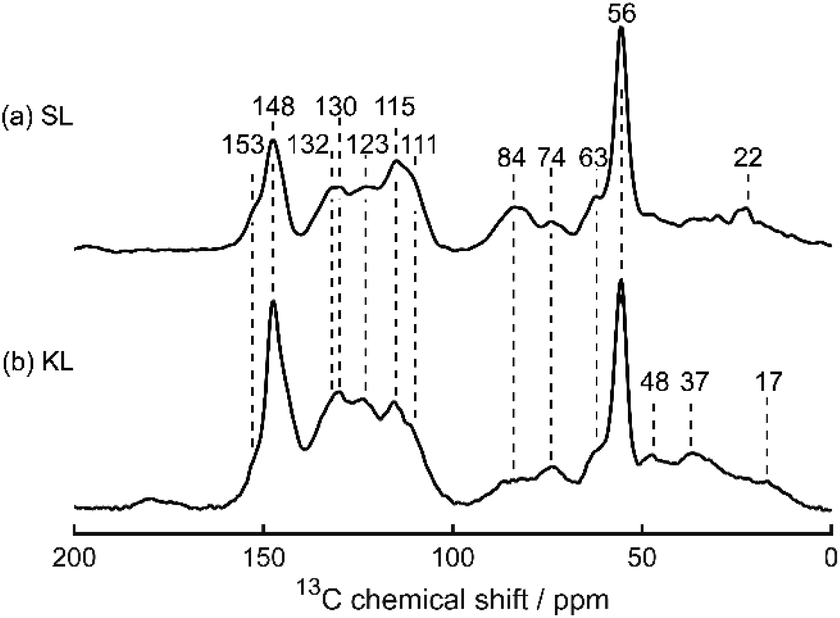 | ||
| Fig. 3 Solid-state 13C CP MAS NMR spectra of (a) Sonichem lignin and (b) Kraft lignin. Chemical shift values (in ppm) of each peak are indicated. | ||
 | ||
| Fig. 4 (a) Schematics showing phosphitylation of the OH groups in lignin using the chosen phosphitylating agent, phosphitylation of the internal standard, and the aromatic units present in SL. (b) Liquid-state 31P NMR spectra recorded of phosphitylated Sonichem lignin. | ||
 | ||
| Fig. 5 FTIR spectra of unmodified (a) SL (black), SL-BZ-aniline (purple), SL-BZ-A (red) and SL-BZ-B (blue) and (b) KL (black), KL-BZ-aniline (purple), KL-BZ-A (red) and KL-BZ-B (blue). | ||
| Sample | C | H | N | Other elements |
|---|---|---|---|---|
| SL | 66.07% | 6.96% | 0.00% | 26.97% |
| SL-BZ-aniline | 68.51% | 6.05% | 3.72% | 21.73% |
| SL-BZ-A | 63.91% | 5.79% | 1.90% | 28.42% |
| SL-BZ-B | 64.05% | 5.96% | 2.15% | 27.85% |
| KL | 61.79% | 5.59% | 0.48% | 32.16% |
| KL-BZ-aniline | 71.26% | 6.01% | 5.74% | 17.01% |
| KL-BZ-A | 60.44% | 5.54% | 1.93% | 32.10% |
| KL-BZ-B | 57.60% | 5.71% | 1.53% | 35.17% |
| Lignin sample | Element | C | O | N | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| C | O | N | O–C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O O |
CO |
C–OH/C–O–C/C–N | C–C sp2 | C–C sp3 | π–π* satellite | C–OH | C–O–C | CO |
N–sp2 C | N–sp3 C | |
| SL | 64.1 | 28.1 | — | 1.22 | 1.94 | 14.4 | 8.28 | 74.2 | — | 68.6 | 31.4 | — | — | |
| SL-BZ-aniline | 57.9 | 38.5 | 3.57 | 3.14 | 19.3 | 24.9 | 52.6 | — | 92.3 | 7.72 | 67.9 | 32.1 | ||
| SL-BZ-A | 51.8 | 41.5 | 2.42 | 0.94 | 41.2 | 57.9 | — | 77.5 | 22.5 | 53.4 | 46.6 | |||
| SL-BZ-B | 59.8 | 29.1 | 7.26 | 1.10 | 35.7 | 61.0 | 1.70 | 63.8 | 19.5 | 16.7 | 59.4 | 40.6 | ||
| KL | 56.2 | 43.8 | — | 2.85 | 39.7 | 55.3 | 2.14 | 88.2 | 11.8 | — | — | |||
| KL-BZ-aniline | 61.1 | 33.3 | 5.62 | 3.91 | 37.1 | 57.4 | 1.67 | 78.5 | 21.5 | 52.1 | 47.9 | |||
| KL-BZ-A | 53.7 | 41.7 | 3.38 | 1.47 | 3.29 | 41.0 | 54.2 | — | 82.0 | 18.0 | 89.3 | 10.7 | ||
| KL-BZ-B | 33.8 | 52.4 | 1.87 | 1.24 | 1.58 | 40.8 | 56.4 | — | 94.4 | 5.57 | 84.4 | 15.6 | ||
Fig. 3 shows the solid-state 13C CP MAS NMR spectra of Sonichem lignin and Kraft lignin prior to functionalisation, with both exhibiting the signals typically expected for lignins,36–42 notably the intense methoxy group signal at 56 ppm, Cγ–OH at 63 ppm, Cα–OH in β-O-4 linker groups at 74 ppm, and Cβ at 84 ppm. Signals arising from aromatic C–H positions appear at 100–127 ppm, as confirmed by the fact that these signals remain if a shorter CP contact time (50 μs) is used (Fig. S17(a) and S18(a), ESI†). Consistent with previous observations, C–C positions appear at 127–140 ppm, and C–O positions appear at 140–160 ppm. Additionally, signals arising from aliphatic CH2 and CH3 groups appear below 56 ppm.
Because of the broad NMR line shapes leading to multiple overlapping peaks, a more detailed assignment for Sonichem lignin is less straightforward. However, peaks arising from the G units can be clearly identified: C3(G) and C4(G) at 148 ppm, C1(G) at 130 ppm, C6(G) at 123 ppm, C5(G) at 115 ppm, and C2(G) at 111 ppm.36 The liquid-state 31P NMR spectrum of phosphitylated lignin, used to quantify the different hydroxyl groups present (Fig. 4b), only has a weak signal at 137.1–138.4 ppm (corresponding to H units) and no signal at 142.0–144.5 ppm (corresponding to S units), which implies that the latter is absent in Sonichem lignin. Both these signals are also weak for Kraft lignin (Fig. 10a), implying that both lignins are primarily composed of G units. The higher concentration of H units in Sonichem lignin shows it lignin has more sites available for BZ formation compared to Kraft lignin.
The signal at 132 ppm in the solid-state 13C CP MAS NMR spectra could be assigned to C1(H), appearing at a slightly higher frequency than C1(G) as it is more deshielded due to the absence of a methoxy group in the ortho position.43 The expected solid-state 13C CP MAS NMR signals for C2(H) and C6(H) at 127 pm, and C3(H) and C5(H) at 117 ppm, are less easily identified, but may be obscured by the more intense signals from the G units nearby.44 However, no signal appears at 159 ppm, as previously reported for C4(H), while a weak signal does appear at 153 ppm, as expected for C4(S).
For Sonichem lignin, this 13C signal appears to be at odds with the liquid-state 31P NMR data, which does not show any signals in the region 142.0–144.5 ppm for the S units (Fig. 4).45 The absence of S–OH signals could be due to the S units not dissolving prior to phosphitylation. If this is assumed then other solid-state 13C CP MAS NMR signals may be tentatively assigned to carbons in the S units: a weak shoulder at 136 ppm may arise from C1(S) and C4(S), and while a signal from C2(S) and C6(S) at 105 ppm is not clearly visible, it may be obscured by the much more intense C2(G) signal. Alternatively, the signal at 153 ppm could arise from C4(H), particularly given the limited published data on the 13C NMR characterisation of H units.44
BZ synthesis
BZs have previously been synthesised from diamines.46–48 Here, we followed synthetic methods previously reported by Podschun et al. for aniline35 and Adjaoud et al. for amines A and B.9 FTIR spectroscopy, TGA, elemental analysis and XPS were used to prove successful introduction of BZ groups into the lignin structure. Solution-state NMR techniques such as two-dimensional 1H–13C Heteronuclear Single Quantum Coherence NMR Spectroscopy (2D-HSQC NMR) and 31P NMR spectroscopy were not possible due to the low solubility of lignin-BZ products in the required solvent systems.The FTIR spectra recorded from Sonichem lignin samples following BZ synthesis clearly show a reduction in the intensity of the broad stretch between 3600 and 3100 cm−1 due to the H units in lignin being used up in BZ formation (Fig. 5a). FTIR spectra from Kraft lignin samples (Fig. 5b) show lower intensity in the phenol region compared to Sonichem lignin, making the expected reduction in intensity in this region less obvious than observed for Sonichem lignin. The peaks at 3000–2842 cm−1 corresponding to methyl and methylene groups49 reduce in intensity following BZ formation. Peaks at 1600 and 1495 cm−1 significantly increase in intensity following BZ synthesis from Kraft lignin. This change is not observed in Sonichem lignin as these peaks are already intense before BZ formation.
The strong peaks at 1510 cm−1 derived from vibrations in the aromatic skeleton50 and at 1030 cm−1 derived from aromatic C–H bonds in G units50 remain largely unchanged following BZ formation. Peaks in the range 1200–1100 cm−1 derived from aromatic skeletal vibrations50 show some subtle changes following BZ formation using each of the different amines. The FTIR spectra collected for Sonichem lignin and Kraft lignin are very similar following BZ formation, particularly when aniline was used as the amine. BZ samples formed using amines A and B show some changes in the fingerprint region.
Peaks corresponding to other expected changes in structure on BZ formation are difficult to distinguish from peaks already present in the spectra. Techniques such as XPS and elemental analysis are therefore useful as these show that N has been added during BZ synthesis since unmodified Sonichem lignin and Kraft lignin do not contain a significant amount of N. Results from elemental analysis show a clear increase in N content following BZ synthesis using both Sonichem lignin and Kraft lignin (Table 1). Kraft lignin and Sonichem lignin show similar increases in N content following BZ formation with each amine. Using aniline as the amine results in the largest increase in N content, perhaps due to steric effects resulting from the larger diamines.
13C NMR signals for BZ have previously been reported at 48 ppm for Ar–CH2–N, and 75–82 ppm for N–CH2–O.9,35 In the solid-state 13C CP MAS NMR spectra of our BZ-functionalised lignins, however, the former of these is obscured by the Cα and Cβ linker signals, while the latter only appears for SL-BZ-aniline (Fig. 6). It is possible that, given the relatively small number of BZ groups introduced, these signals are below the detection limit. When aniline is used as the amine, a prominent signal appears at 130 ppm, which is easily distinguished from the lignin signals in that region as it still appears with a CP contact time of 50 μs (Fig. S17b, ESI†). This may be assigned to aniline's meta carbons.51 SL-BZ-A and SL-BZ-B both show a new signal at 67 ppm. Because this signal only appears when BZ is formed using diamines, we tentatively assign it to Cβ–OH, suggesting that the formation of cross-links using amines A and B result in the breaking of the β-O-4 linkers. Similar effects are seen for Kraft Lignin (Fig. S18, ESI†), although an intense signal is observed at 88 ppm for KL-BZ-B. This signal likely originates from paraformaldehyde, a key reagent in the synthesis of BZ.52 2D HETeronuclear CORrelation (HETCOR) measurements were performed to further elucidate the structure of the benzoxazine-functionalised lignins (Fig. S19 to S22, ESI†), although due to the low sensitivity and resolution, these did not provide any further information.
 | ||
| Fig. 6 Solid-state 13C CP MAS NMR spectra of (a) SL, (b) SL-BZ-aniline, (c) SL-BZ-A, and (d) SL-BZ-B. Chemical shift values (in ppm) of each peak are indicated. | ||
XPS was used to determine the chemical composition of the lignin samples.53 The results from XPS agree reasonably well with the results from elemental analysis. Before BZ synthesis, no peaks were observed in the N region. Peaks corresponding to Si at 96 and 139 eV were observed in some samples. Hence, not all values from the survey scan sum to 100%. These are ignored for clarity of discussion here. As expected for lignin, the carbon content of Sonichem lignin is made up of mostly sp2 carbon atoms within the aromatic skeleton. sp3 carbon atoms correspond to the aliphatic linkers that connect the aromatic units. Peaks corresponding to ester, carbonyl, ether and hydroxyl groups are observed in the O region.
Before modification, Sonichem lignin and Kraft lignin exhibit different C/O ratios and notably different proportions of each type of C and O atom (Table 2), showing they have different structures and therefore different properties. On BZ formation, the characteristic peak corresponding to N 1s emerged at 400 eV,53 in the survey spectra. The peak at 286 eV in the C region corresponds to C–N bonding energy.53 In most cases, it is not possible to distinguish the C–N peak from the C–OH/C–O–C peak and the C–OH/C–O–C/C–N peak increases on BZ formation as OH groups are used up and C–N and C–O–C groups introduced. This peak tends to increase more significantly when using diamines. The proportion of sp2 and sp3 carbons tends to decrease as BZ groups are introduced owing to the addition of further O- and N- containing functional groups.
We were able to resolve two peaks in the N region of the XPS spectra of all N-containing samples. We expect these two peaks to arise from N directly bonded to either a sp2 C atom or a sp3 carbon atom.54 The ratio of N–sp2 C to N–sp3 C changes on going from aniline to diamines A and B. This could be related to the diamines ability to undergo cross-linking via BZ formation. Since a third peak relating to N–H electron binding energy is not observed in the diamine samples, we can conclude that cross-linking has occurred at each BZ site, leaving no unreacted amine groups.
Following BZ formation, the N and C content of Sonichem lignin increased while the O content decreased (Table 2) matching the expected changes as both N and sp2 carbon atoms are introduced during BZ formation using aromatic amines. The same trend is seen in KL-BZ-aniline while the opposite trend is observed when reacting Kraft lignin with diamines A and B. The C–OH/C–O–C/C–N peak in the C region increases in all SL-BZ samples. A far less significant change is observed in the Kraft lignin samples with KL-BZ-aniline showing a small decrease and KL-BZ-A and KL-BZ-B showing a small increase in this peak. In the O region, all SL-BZ samples show an overall increase in the C–OH/C–O–C region and a decrease in the CO region. Since C–OH is expected to be used up during BZ formation, this increase must be due to increased C–O–C content. This agrees with results from the SL-BZ-B XPS spectra where we were able to resolve the C–OH and C–O–C peaks during fitting. Here, the C–OH content decreases compared to the overall C–OH/C–O–C content measured in SL. Unfortunately, we were unable to resolve these peaks in the other samples. Again, Kraft lignin shows inconsistent trends with C–OH/C–O–C increasing in KL-BZ-aniline and KL-BZ-A and decreasing in KL-BZ-B.
The results from acetone solubility studies also indicate that cross-linking has occurred in lignin-BZ products prepared from diamines as these are far less soluble in acetone than both unmodified lignin and lignin-BZ-aniline (Fig. 7). BZ synthesis using aniline as the amine should not cross-link through further BZ formation since aniline contains only one amine. Kraft lignin clearly has lower solubility in acetone than Sonichem lignin. Solubility decreases on functionalisation with BZ groups in all cases, with KL-derivatives showing lower solubility than SL-derivatives.
 | ||
| Fig. 7 Photographs of (a(i)) unmodified SL, (a(ii)) SL-BZ-aniline, (a(iii)) SL-BZ-A a, (a(iv)) SL-BZ-B, (b(i)) unmodified KL, (b(ii)) KL-BZ-aniline, (b(iii)) KL-BZ-A and (b(iv)) KL-BZ-B dissolved in acetone (10 mg mL−1) by stirring at room temperature for 10 minutes before leaving any undissolved solid to settle. | ||
We expect the increased solubility of Sonichem lignin compared to Kraft lignin contributes to its increased reactivity and thereby greater degree of functionalisation. It could also be the case that Sonichem lignin contains a greater number of phenolic OH groups with unsubstituted ortho positions, despite containing a smaller proportion of oxygen atoms. This results in greater propensity for BZ functionalisation.
Phenolation
The method followed for lignin phenolation was the same as previously reported by Podschun et al.35 Phenolation prior to BZ synthesis increases the number of H units available for BZ formation. This will result in more BZ groups available for cross-linking and therefore a more thermomechanically stable material. The properties of lignin-BZ materials can therefore be controlled using this additional phenolation step prior to BZ formation. Successful phenolation was verified using liquid-state 31P NMR and FTIR spectroscopy.Results from liquid-state 31P NMR spectroscopy show that ∼77% of aliphatic hydroxyl groups in Sonichem lignin are converted to phenols during the phenolation reaction (Fig. 8). As a result, the number of unsubstituted phenol groups (H units) present in the lignin increases from 0.06 μmol mg−1 to 2.73 μmol mg−1 (Fig. 8). There is also a decrease in the signal attributed to G-OH. This suggests that these are also involved in phenolation.
 | ||
| Fig. 8 (a) Liquid-state 31P NMR spectra recorded of SL-Ph. (b) Graph summarising the amounts of each type of OH group in SL before (black) and after (red) phenolation of SL, according to comparison with the internal standard. | ||
FTIR spectroscopy also provides some insights into lignin functionalisation. The stretch in the region 3600–3100 cm−1 is clearly broadened in the spectrum collected from phenolated Sonichem lignin (SL-Ph), indicating an increased number of phenol groups (Fig. 9a). The FTIR spectrum of SL-Ph also shows two new peaks at 819 and 750 cm−1 (Fig. 9a). This shows that during the phenolation reaction, phenol forms C–C bonds to lignin both ortho and para to the hydroxyl group on phenol (Fig. 9b). This observation was previously reported by Abarro et al.35 FTIR spectra recorded from SL-Ph and KL-Ph are almost identical.
 | ||
| Fig. 9 (a) FTIR spectra of unmodified SL (black) and SL-Ph (red). (b) Scheme showing phenolation of lignin, resulting in phenol groups in both ortho and para positions. | ||
Results from liquid-state 31P NMR and FTIR spectroscopy clearly show that phenolation was successful using Kraft lignin (Fig. 10). According to liquid-state 31P NMR, ∼81% of aliphatic OH groups in Kraft lignin reacted with phenol resulting in an increase in H units from 0.42 μmol mg−1 to 2.30 μmol mg−1.
 | ||
| Fig. 10 (a) Liquid-state 31P NMR collected of phosphitylated (i) KL and (ii) and KL-Ph. (b) Graph summarising the amounts of each different OH group present in KL before (black) and after (red) phenolation. (c) FTIR spectra of KL (black) and KL-Ph (red). | ||
Solid-state 13C CP MAS NMR spectra (Fig. 11) provide further evidence for successful phenolation: new peaks appear at 155 ppm, corresponding to the C1 carbons (as shown in Fig. 9b) and 129 ppm, corresponding to C2/3/5 of ortho-substituted phenol and C3–5 of para-substituted phenol.55 Furthermore, the linker signals have been shifted to a lower chemical shift than the methoxyl peak, due to the replacement of the hydroxyl groups with the less electron withdrawing phenol groups.
 | ||
| Fig. 11 Solid-state 13C CP MAS NMR spectra of (a) Sonichem lignin and (b) phenolated Sonichem lignin. Chemical shift values (in ppm) of each peak are indicated. | ||
BZ synthesis using SL-Ph results in a massive reduction in intensity of the O–H stretch between 3600 and 3100 cm−1 during FTIR spectroscopy, confirming the use of lignin phenol groups in BZ formation (Fig. 12a). More obvious changes are observed in the peaks within 1200–1100 cm−1 derived from aromatic skeletal vibrations following BZ formation using SL-Ph compared to non-phenolated Sonichem lignin. These more obvious changes may result from more BZ groups being introduced. As with the unmodified lignin, more subtle changes to the FTIR spectra are not clearly visible.
 | ||
| Fig. 12 FTIR spectra of unmodified (a) SL-Ph (black), SL-PhBZ-aniline (purple), SL-PhBZ-A (red) and SL-PhBZ-B (blue) and (b) KL-Ph (black), KL-PhBZ-aniline (purple), KL-PhBZ-A (red) and KL-PhBZ-B (blue). | ||
Similar changes in FTIR spectra of Kraft lignin samples following BZ synthesis suggest the reactions were successful (Fig. 12b.) As with Sonichem lignin, a clear increase in intensity of the broad O–H peak in FTIR spectra is observed following phenolation. After BZ synthesis, this O–H peak reduced in intensity as the O–H groups on H units in lignin are replaced with BZ rings.
The solid-state 13C CP MAS NMR spectra of the phenolated samples (Fig. 13) show similar changes upon benzoxazine formation to those shown in Fig. 6. However, the peak at 67 ppm for the SL-PhBZ-A and SL-PhBZ-B is much more intense than for their non-phenolated counterparts, potentially suggesting extensive severance of the β-O-4 linkers. Interestingly, this signal does not appear for the Kraft lignin samples (Fig. S18, ESI†), in line with the lower BZ yields implied from elemental analysis (Table 3). When a CP contact time of 50 μs is used, the phenol signal at 129 ppm disappears in SL-PhBZ-A and SL-PhBZ-B, indicating that the carbon atoms in these positions are used to form benzoxazine and are therefore deprotonated (Fig. S17, ESI†). In addition, we observe signals at 73 ppm and 48 ppm, which are likely to correspond to N–CH2–O and N–CH2–Ar carbons, respectively.9,35 For SL-PhBZ-A, a new signal appears at 144 ppm, which could tentatively be assigned to the aromatic N–C carbon in the p-phenylene diamine linker.
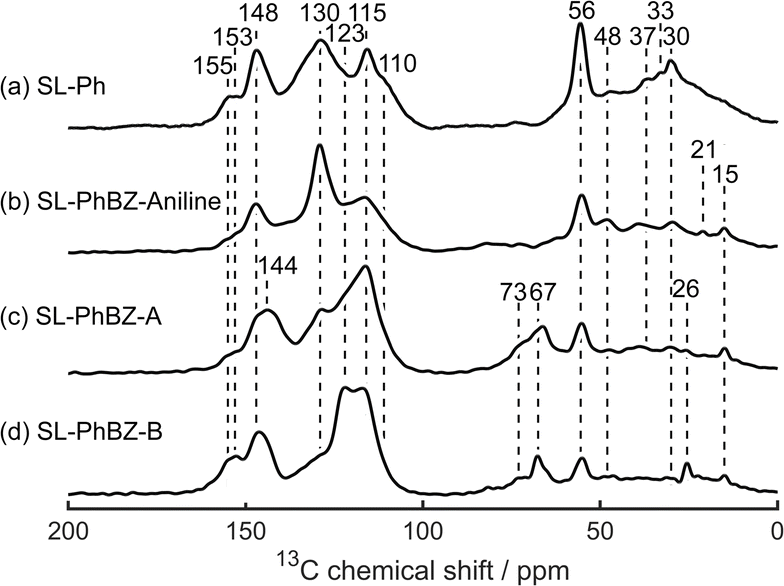 | ||
| Fig. 13 Solid-state 13C CP MAS NMR spectra of (a) SL-Ph, (b) SL-PhBZ-aniline, (c) SL-PhBZ-A, and (d) SL-PhBZ-B. Chemical shift values (in ppm) of each peak are indicated. | ||
| Sample | C | H | N | Other elements |
|---|---|---|---|---|
| SL-Ph | 67.76% | 6.15% | 0.00% | 26.10% |
| SL-PhBZ-aniline | 69.33% | 5.60% | 3.57% | 21.51% |
| SL-PhBZ-A | 66.79% | 5.67% | 10.43% | 17.13% |
| SL-PhBZ-B | 68.78% | 5.39% | 7.18% | 18.67% |
| KL-Ph | 61.34% | 4.92% | 0.02% | 35.17% |
| KL-PhBZ-aniline | 67.22% | 5.18% | 2.91% | 24.70% |
| KL-PhBZ-A | 63.00% | 4.94% | 2.86% | 29.20% |
| KL-PhBZ-B | 63.33% | 4.77% | 2.28% | 29.63% |
Again, we used elemental analysis and XPS to show the introduction of N into the lignin. N content increased massively following BZ formation using SL-Ph (Table 3). The increase in N content following BZ synthesis using phenolated Sonichem lignin is much greater than that observed using phenolated Kraft lignin (KL-Ph).
XPS data further confirms successful phenolation of Sonichem lignin, showing an increase in the C–OH/C–O–C peak (Table 4). The C–OH/C–O–C peak recorded for KL-Ph decreases compared to unmodified Kraft lignin. This may be due to surface gases, such as CO2 causing an increase in the CO peak. N was also observed in the KL-Ph sample, despite no N-containing groups being introduced during phenolation. Again, this could be due to N2 gas being present on the surface of the sample, even after etching. The C/O ratio of Sonichem lignin flipped following phenolation. The C/O ratio of Kraft lignin did not change significantly after phenolation. This may be due to the differences in structure between the two lignin types.
| Lignin sample | Element | C | O | N | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| C | O | N | O–CO |
CO |
C–OH/C–O–C/C–N | C–C sp2 | C–C sp3 | π–π* satellite | C–OH | C–O–C | CO |
N–sp2 C | N–sp3 C | |
| SL-Ph | 67.9 | 25.9 | — | 0.71 | 1.23 | 8.16 | 81.6 | 8.34 | — | 80.4 | 19.6 | — | — | |
| SL-PhBZ-aniline | 47.0 | 40.6 | 3.37 | 1.13 | 1.99 | 32.2 | 63.4 | 1.37 | 87.6 | 12.4 | 32.4 | 67.6 | ||
| SL-PhBZ-A | 57.7 | 34.7 | 3.75 | 1.62 | — | 33.1 | 65.4 | — | 76.4 | 11.9 | 11.8 | 76.3 | 23.7 | |
| SL-PhBZ-B | 54.2 | 38.9 | 1.62 | 2.00 | — | 37.8 | 60.2 | — | 57.3 | 42.7 | 57.3 | 42.7 | ||
| KL-Ph | 55.0 | 41.2 | 0.69 | 0.74 | 1.86 | 35.8 | 59.8 | 1.98 | 81.2 | 18.8 | 15.7 | 84.3 | ||
| KL-PhBZ-aniline | 36.4 | 48.7 | 1.95 | 2.27 | — | 32.2 | 63.5 | 2.10 | 93.0 | 7.01 | 29.8 | 70.2 | ||
| KL-PhBZ-A | 34.9 | 51.5 | 1.55 | 2.84 | 2.11 | 31.5 | 62.0 | 1.52 | 92.1 | 7.87 | 14.4 | 85.6 | ||
| KL-PhBZ-B | 28.5 | 55.2 | 1.32 | 0.47 | 0.75 | 35.6 | 62.1 | 1.14 | 93.8 | 6.20 | 6.66 | 93.3 | ||
XPS spectra collected from phenolated samples show different trends following BZ formation compared to non-phenolated samples. This likely arises from the different C and O content of the phenolated lignins, owing to the considerable increase in H units following phenolation. For example, all BZ samples show decreased C content and increased O and N content (Table 4).
Interestingly, all SL-Ph samples show a decrease in C–C content while all KL-Ph samples show an increase in C–C content. The C–OH/C–O–C/C–N peak in the C region of SL-Ph shows a massive increase following BZ formation with all three amines. This suggests enhanced BZ formation using phenolated Sonichem lignin and agrees with elemental analysis data. Phenolated Kraft lignin shows a small decrease in the C–OH/C–O–C/C–N region. This may be due to surface gases.
Results from fitting the XPS data show that C–OH groups persist after BZ formation. While some of these C–OH groups will be aliphatic and therefore unable to participate in BZ formation, there may still be unreacted OH groups available for further BZ formation under optimised conditions. Increases in N and C–O–C content confirm successful BZ formation using phenolated Kraft lignin.
Phenolation of lignin prior to BZ synthesis appears to further decrease solubility in acetone (Fig. 14). This agrees with the expectation that increasing the number of phenol groups present in lignin will increase the number of sites available for BZ formation and thereby increase cross-linking.
 | ||
| Fig. 14 Photographs of (a(i)) SL-Ph, (a(ii)) SL-PhBZ-aniline, (a(iii)) SL-PhBZ-A, (a(iv)) SL-PhBZ-B, (b(i)) KL-Ph, (b(ii)) KL-PhBZ-aniline, (b(iii)) KL-PhBZ-A and (b(iv)) KL-PhBZ-B dissolved in acetone (10 mg mL−1) by stirring at room temperature for 10 minutes before leaving any undissolved solid to settle. | ||
Thermal behaviour of lignin-BZ products
BZ rings are stable at low temperature. Increasing temperature causes the BZ ring to open, resulting in formation of a phenolic hydroxyl group and a tertiary amine.56 This ring-opening allows polymerisation of the BZ monomers. PBZs have desirable properties for many applications, including aerospace, packaging, composite material manufacturing and powder coating.15 Their unusual properties arise from the complex hydrogen bonding of BZs.15 For example, PBZs can form a six-membered ring that provides the polymer with hydrophobicity, low dielectric constant, high char yield and high modulus.15The complex, aromatic structure of lignin causes it to decompose over a wide range of temperatures.57 Thermal decomposition of lignin tends to be divided into three stages: dehydration at temperatures below 100 °C, cleavage of α-aryl ether and β-aryl ether linkages at temperatures between 150 and 260 °C and cleavage of C–C bonds at temperatures between 260 and 480 °C. The thermal decomposition of the lignin derivatives was studied using thermogravimetric analysis (TGA).
The minimum in the derivative thermogram observed in lignin samples at ∼200 °C results from breaking of the β-O-4 linkages in the lignin resulting in release of terminal phenolic groups.58 The larger minimum at ∼320 °C most likely results from the breakage of C–C bonds.58 A new minimum between 120 and 140 °C appears in the Sonichem lignin-BZ samples (Fig. 15). This peak is not observed in SL-PhBZ-aniline or in SL-PhBZ-A. A peak at ∼100 °C is observed in SL-PhBZ-B. Additional peaks are observed in SL-BZ-aniline at 175 °C, 250 °C and 395 °C, and in SL-BZ-A and SL-BZ-B at 120 °C, 230 °C and 400 °C. The presence of peaks after the β-O-4 peak show further reactions can occur after cleavage of β-O-4 bonds.58 Previous work has identified the monomeric phenols released from lignin during thermal decomposition.58
 | ||
| Fig. 15 TGA thermograms collected for (a(i)) SL-BZ-aniline, (a(ii)) SL-BZ-A, (a(iii)) SL-BZ-B, (b(i)) SL-PhBZ-aniline, (b(ii)) SL-PhBZ-A and SL-PhBZ-B (dark red and grey). Thermograms for (a) SL and (b) SL-Ph are shown in light red and black in each graph for comparison. | ||
Following phenolation, the C–C peak appears to shift to 400 °C (Fig. 15b). Following BZ synthesis using aniline, the β-O-4 peak shifts from 215 °C in SL-Ph to 230 °C while the C–C peak remains at 400 °C (Fig. 15b(i)). In SL-PhBZ-A, the β-O-4 breakage temperature decreases slightly to 210 °C while the C–C breakage temperature increases to 395 °C (Fig. 15b(ii)). As with its non-phenolated counterpart, SL-PhBZ-B shows an additional peak at ∼100 °C (Fig. 15(iii)). The other three peaks in SL-PhBZ-B are observed at 275 °C (β-O-4), 370 °C (C–C) and 500 °C. KL-PhBZ-A and KL-PhBZ-B also show a new peak in their derivative thermograms at ∼550 °C (Fig. 16b). Confirmation of the source of these peaks would require direct injection of released volatiles into a gas chromatograph.58
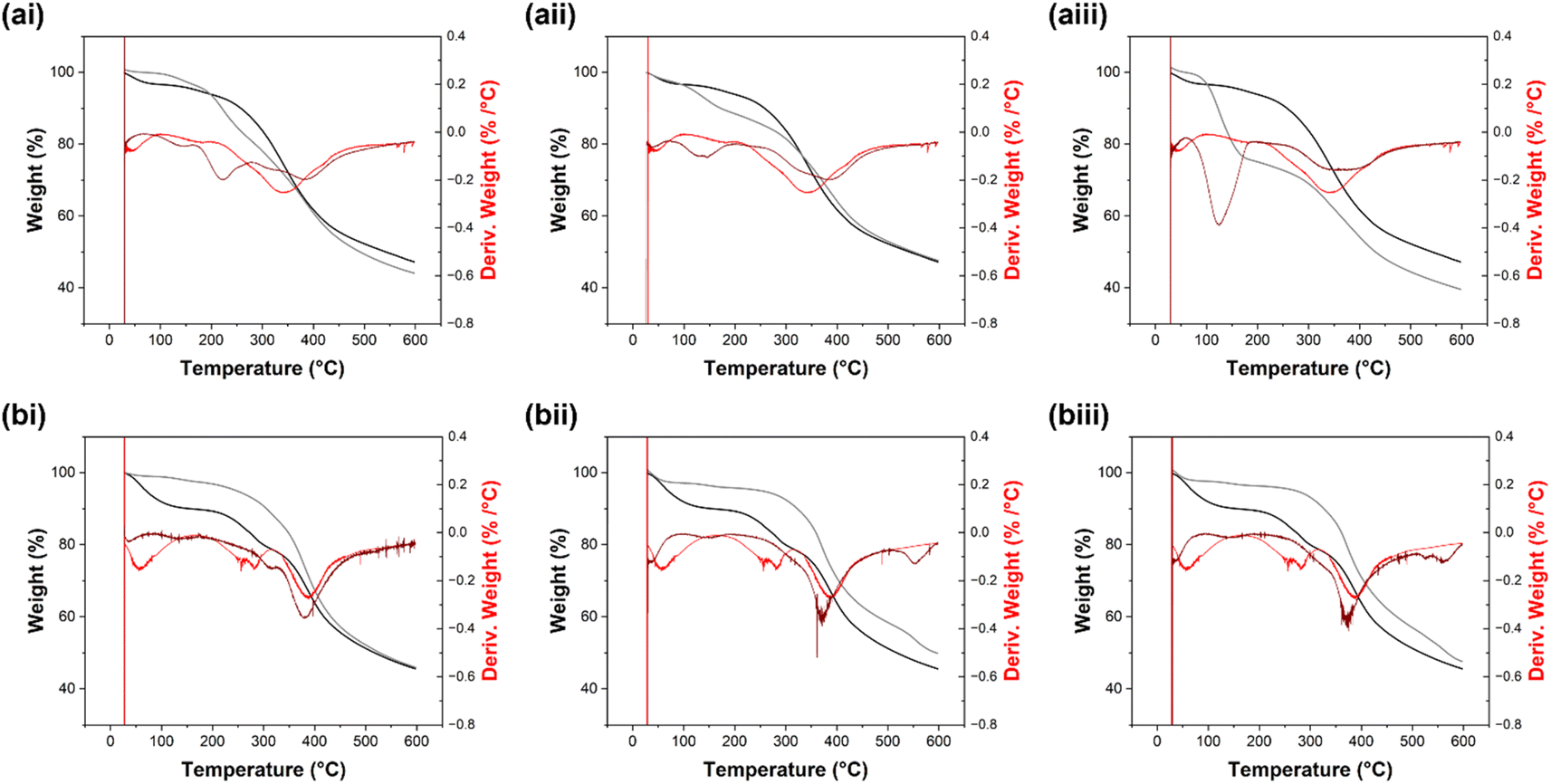 | ||
| Fig. 16 TGA thermograms collected for (a(i)) KL-BZ-aniline, (a(ii)) KL-BZ-A, (a(iii)) KL-BZ-B, (b(i)) KL-PhBZ-aniline, (b(ii)) Kl-PhBZ-A and (b(iii)) KL-PhBZ-B. Thermograms for (a) KL and (b) KL-Ph are shown in light red and black in each graph for comparison. | ||
5% degradation temperature is regularly used to measure the thermal stability of lignin-based materials, as this shows the temperature at which the material begins to degrade.59,60 The 5% degradation temperature of Sonichem lignin decreases in most cases following BZ modification, with only SL-PhBZ-A and SL-PhBZ-B showing an increase (Fig. 15). This could be related to phenolation increasing the number of BZ groups introduced, or to the potential for amines A and B to form cross-links.
While KL-BZ-aniline showed an increase in 5% degradation temperature, KL-BZ-A and KL-BZ-B did not (Fig. 16). Following phenolation, all KL-PhBZ derivatives showed significantly increased 5% degradation temperatures. For both Sonichem lignin and Kraft lignin, phenolation significantly reduces the 5% degradation temperature. Cases where BZ formation does not increase 5% degradation temperature could be due to not all H units being used up in the BZ formation reaction. It is noted that the 5% degradation temperature is lower than values previously reported for other lignin-based benzoxazines9 and considerably lower than those recorded for benzoxazines prepared from petrochemical-derived compounds.61,62 However, we expect such properties can be tuned by varying the amine precursors used for benzoxazine formation.
The char yield of Sonichem lignin clearly increases following BZ synthesis with all three amines (Fig. 15). High char yield is desirable for fire-retardant applications.63–66 The char yield of Kraft lignin does not change significantly on modification and in some cases decreases (Fig. 16). Kraft lignin shows similar changes in TGA to Sonichem lignin following modification with some peaks slightly shifted. There is no obvious trend in how the peaks shift. The char yield values calculated for the lignin-BZ materials prepared here are similar to those previously recorded for lignin/benzoxazine monomer mixtures.27,28,67 The char yields observed here were also comparable to those recorded from petrochemical-derived benzoxazines61,62 or from other renewable resources.68
Differential scanning calorimetry (DSC) curves generally indicate energy consumption and release with negative heat flow showing endothermic processes and positive heat flow showing exothermic processes. Introduction of BZ rings causes the endothermic peak observed at ∼100 °C in the DSC curves to shift to higher temperatures (Fig. 17). Introduction of BZ groups into the lignin structure therefore increases the thermal stability of the lignin. Increasing the number of BZ groups in the lignin via phenolation of lignin prior to BZ formation results in an even more dramatic shift in this endothermic peak. The SL-PhBZ-aniline products also exhibit an exothermic peak at 210 °C for SL-PhBZ-A and 186 °C for SL-PhBZ-B. This exothermic peak results from polymerisation of the BZ rings.46,48 A slight exothermic inflection is observed in the same region for the non-phenolated products, suggesting polymerisation may be taking place but to a much smaller degree. It may be the case that there is not sufficient density of BZ groups in the non-phenolated samples for effective polymerisation to take place owing to its low molecular weight and relatively low number of H units.35 It is therefore necessary to first phenolate Sonichem lignin for effective use as a BZ resin. It is unlikely that the SL-BZ products had already undergone curing during their synthesis as they were prepared at relatively low temperatures (70–80 °C).
 | ||
| Fig. 17 DSC curves collected for (a) SL (black), SL-Ph (grey), SL-BZ-aniline (purple), SL-PhBZ-aniline (pale purple), SL-BZ-A (red), SL-PhBZ-A (pale red), SL-BZ-B (blue), SL-PhBZ-B (pale blue); (b) KL (black), KL-Ph (grey), KL-BZ-aniline (purple), KL-PhBZ-aniline (pale purple), KL-BZ-A (red), KL-PhBZ-A (pale red), KL-BZ-B (blue), KL-PhBZ-B (pale blue). | ||
Phenols and a free ortho-position in lignin catalyse the ring-opening polymerisation of BZ, resulting in reduced polymerisation temperature compared to BZ in the absence of lignin.27 Previously studied non-covalent BZ/lignin mixtures exhibited increased Tg and char yield with increasing amounts of lignin.27 This change in thermal behaviour resulted from the increased cross-linking density and more compact structure within the lignin/BZ composites.27,28
As seen in previous studies,27 introduction of BZ groups into the lignin increases the glass transition temperature (Tg) of the lignin, showing increased thermal stability (Table 5). Interestingly, phenolation also increased the Tg of the lignin. While introduction of BZ using aniline as the amine further enhanced the Tg of the lignin, introduction of cross-linkable BZ groups reduced Tg of phenolated lignin. Kraft lignin shows notably higher Tg than Sonichem lignin. As with Sonichem lignin, Tg increases on phenolation and following BZ formation. The Tg values recorded for the benzoxazine-modified lignin samples prepared here are comparable to those reported for benzoxazines prepared from non-lignin sources.62,67–69
| Sample name | 5% degradation temperature (°C) | T g (°C) | Char yield at 600 °C (%) |
|---|---|---|---|
| Sonichem lignin | 202 | 95 | 36 |
| SL-BZ-aniline | 145 | 89 | 47 |
| SL-BZ-A | 136 | 117 | 47 |
| SL-BZ-B | 200 | 115 | 51 |
| SL-Ph | 152 | 121 | 36 |
| SL-PhBZ-aniline | 132 | 128 | 48 |
| SL-PhBZ-A | 183 | 112 | 42 |
| SL-PhBZ-B | 161 | 111 | 40 |
| Kraft lignin | 172 | 121 | 47 |
| KL-BZ-aniline | 188 | 141 | 44 |
| KL-BZ-A | 118 | 148 | 48 |
| KL-BZ-B | 108 | 146 | 40 |
| KL-Ph | 69 | 154 | 46 |
| KL-PhBZ-aniline | 247 | 146 | 46 |
| KL-PhBZ-A | 251 | 140 | 50 |
| KL-PhBZ-B | 271 | 137 | 47 |
The large gap between the melting temperature and curing temperature of SL-PhBZ-A provides a large processing window, making it useful for industrial applications.46 Steric hindrance in the ring-opened product caused by smaller linker in SL-PhBZ-A compared to SL-PhBZ-B may be the reason for this.32 Of the Kraft lignin samples, only KL-BZ-aniline shows a clear endothermic peak suggesting curing has taken place. This suggests Sonichem lignin provides a more versatile starting material for preparation of curable materials than Kraft lignin. Use of cross-linkable amines A and B results in a larger increase in Tg for both Sonichem and Kraft lignin. However, this is not the case when using phenolated lignins.
Previous work has investigated the thermal properties of benzoxazine/lignin mixtures.27 The DSC curves of pure benzoxazine monomers exhibited narrow exothermic peaks, showing curing temperatures of around 260 °C. Lignin alone does not exhibit an exotherm transaction within the tested temperature ranges, showing that without benzoxazine groups, no cross-linking takes place. By mixing benzoxazine monomers with varying amounts of lignin, Nour-Eddine et al.27 observed a reduction in the curing temperature. The curing temperature decreased further with increasing amounts of lignin, with 30% lignin exhibiting curing temperatures as low as 203 °C. The curing temperature of SL-PhBz-B at 186 °C was even lower than those reported by Nour-Eddine et al., exemplifying the ability of Sonichem lignin to activate benzoxazine polymerisation. The extent of this activation can be tuned by changing the structure of the benzoxazine by changing the identity of the amine used for benzoxazine formation within the lignin, as exemplified by the higher curing temperature of SL-PhBZ-B.
Conclusions
Successful lignin-BZ formation was achieved using both Sonichem lignin and Kraft lignin. A wide range of thermal properties were obtained depending on the identity of the amine used for BZ synthesis, the type of lignin and whether the lignin had been phenolated prior to BZ formation. We were able to achieve curing behaviour from all BZ-modified Sonichem lignin samples that had previously been phenolated. Curing behaviour was only observed in one of the phenolated Kraft lignin samples.Sonichem lignin has clear advantages over Kraft lignin in its solubility and thus ease of handling and characterisation. Sonichem lignin consistently reacted with various amines to form BZs, with the number of BZ groups introduced clearly increasing when phenolated lignin was used as the starting material. This allows easy tuning of BZ content and thereby thermal properties. No consistent increase in BZ content was observed when using phenolated Kraft lignin. Sonichem lignin also consistently exhibited increased char yield, providing an advantage in flame-retardant applications. The thermal properties of the lignin-based benzoxazines studied here are comparable to those observed in benzoxazines prepared from other renewable and non-renewable sources.
The environmentally friendly extraction process, low molecular weight, relatively high solubility and high and versatile reactivity of Sonichem lignin makes it attractive compared to other types of lignin. The differences in thermal behaviour between SL-BZ samples prepared using aniline, amine A and amine B highlight the possibility for tuning the properties of lignin-BZ materials at the molecular design phase.
Experimental
Sonichem lignin was extracted from Sitka Spruce from a single sawmill in Scotland (James Jones and Sons). The lignin is then extracted using Sonichem's ultrasound-assisted fractionation process. The Kraft lignin used for comparison was purchased from Sigma-Aldrich (source material used was not stated in the product information). Reagents used for synthesis and for liquid-state 31P NMR spectroscopy were purchased from Merck (paraformaldehyde; aniline, p-phenylene diamine; bis-(4-aminophenyl)ether; phenol; pyridine; 2-chloro-4,4,5,5-tetramethyl-1,3,2-dioxaphospholane, Cr(acac)) or from Fluorochem (N-hydroxy-5-norborene-2,3-dicarboxoimide) and used as received.Dialysis
Sonichem lignin was dry loaded into dialysis sacs from Merck and dialysed in water for 7 days. The resulting solid was dried in a vacuum oven at 50 °C for 16 hours to ensure the product was completely dry before use.BZ synthesis
For BZ preparation using aniline as the amine, we used a method adapted from work by Podschun et al.55 Paraformaldehyde (516 mg, 17.2 mmol) was dissolved in 5 mL of toluene![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :ethanol (1:1 v/v) in a Schlenk flask or round-bottomed flask equipped with a condenser. Aniline (786 μL, 8.6 mmol) was added to the paraformaldehyde solution and allowed to react at 95 °C for 15 minutes. Lignin or phenolated lignin (1 g) was dissolved in 5 mL of toluene:ethanol (1:1 v/v) using sonication. The lignin solution was added to the reaction mixture and reacted at 95 °C for 20 hours. The reaction mixture was allowed to cool to room temperature. The product was precipitated in excess diethyl ether (∼50 mL). The resulting precipitate was filtered and washed with excess diethyl ether (>200 mL).
:ethanol (1:1 v/v) in a Schlenk flask or round-bottomed flask equipped with a condenser. Aniline (786 μL, 8.6 mmol) was added to the paraformaldehyde solution and allowed to react at 95 °C for 15 minutes. Lignin or phenolated lignin (1 g) was dissolved in 5 mL of toluene:ethanol (1:1 v/v) using sonication. The lignin solution was added to the reaction mixture and reacted at 95 °C for 20 hours. The reaction mixture was allowed to cool to room temperature. The product was precipitated in excess diethyl ether (∼50 mL). The resulting precipitate was filtered and washed with excess diethyl ether (>200 mL).
For BZ synthesis using amine A (p-phenylene diamine) and amine B (bis-(4-aminophenyl)ether), we used a method adapted from work by Adjoud et al.9 Lignin, amine (1 equivalent for unmodified lignin and 7 equivalents for phenolated lignin) and paraformaldehyde (2 equivalents with respect to the amine) were dissolved in a minimum amount of THF. The temperature was increased to 80 °C and reacted for 6–20 hours. The reaction was allowed to cool to room temperature before being concentrated under vacuum. The product was then precipitated in excess diethyl ether. The resulting fine precipitate was filtered under gravity and washed with excess diethyl ether. The final product was then dried in a vacuum oven at 50 °C. The mass balances for each reaction were as follows: 100% (SL-BZ-aniline), 69% (SL-BZ-A), 89% (SL-BZ-B), 100% (SL-PhBZ-aniline), 100% (SL-PhBZ-A), 100% (SL-PhBZ-B), 100% (KL-BZ-aniline), 89% (KL-BZ-A), 100% (KL-BZ-B), 87% (KL-PhBZ-aniline), 83% (KL-PhBZ-A), 100% (KL-PhBZ-B).
Phenolation
We used a method adapted from work by Podschun et al.55 Lignin (1 g), phenol (2 g) and H2SO4 (6.7 wt% with respect to lignin and phenol, 0.368 mL) were added to a Schlenk flask or round-bottomed flask equipped with a condenser and reacted at 110 °C for 20 minutes. The reaction was quenched with and dissolved in a 5-fold amount of acetone:water (9:1, 5 mL). The product was precipitated in a 4-fold amount of pH1 H2SO4 (20 mL). The precipitate was filtered under vacuum and washed with a large excess of deionised water. To ensure all phenol had been removed, the dried solid was added to a large excess of deionised water and stirred for a few hours before filtering under vacuum and drying in a vacuum oven at 50 °C. The mass balance of the phenolation reaction was 80% for both Sonichem lignin and Kraft lignin.
FTIR spectroscopy
FTIR spectra were recorded using an Agilent Technologies Cary 630 FTIR spectrometer in the range 4000–650 cm−1.Solid-state 13C CP MAS NMR spectroscopy
Solid-state NMR data were collected using a Bruker 400 MHz (9.4 T) Avance III HD NMR spectrometer equipped with a 4 mm triple-resonance HXY MAS probe in double resonance mode tuned to 1H at ν0(1H) = 400.1 MHz and 13C at ν0(13C) = 100.6 MHz. The samples were packed into HR-MAS inserts, which were themselves inserted into 4 mm ZrO2 rotors and spun at a MAS rate of νr = 10 kHz. The 1H radio frequency (rf) field amplitude was set to 69 kHz for the 90° pulse in the 1H → 13C cross polarisation (CP). This CP experiment was carried out using Hartman-Hahn matched rf amplitudes of 50 kHz (with a 70 to 100% linear amplitude ramp) and 42 kHz for 1H and 13C, respectively. CP contact times of 50 μs and 1 ms were used. SPINAL-64 decoupling was applied during 13C NMR signal acquisition with a 1H rf pulse amplitude of 69 kHz.70 A recycle delay of 3 s was used, which in all cases exceeded 1.3 times the 1H spin-lattice relaxation time (T1), as measured using saturation recovery. The 13C chemical shifts were externally referenced to the CH carbons of adamantane at 29.45 ppm.712D 1H 13C HETCOR NMR spectra were collected on a Bruker 400 MHz Avance III NMR spectrometer equipped with a 4 mm double-resonance HX MAS probe. The 1H rf field amplitude was set to 69 kHz for the 90° pulse, and CP was carried out using Hartman-Hahn matched rf amplitudes of 50 kHz (with a 70 to 100% linear amplitude ramp) and 61 kHz for 1H and 13C, respectively. SPINAL-64 decoupling was applied during 13C NMR signal acquisition with a 1H rf pulse amplitude of 69 kHz. Frequency-Switched Lee-Goldberg (FSLG) homonuclear decoupling at a rf amplitude of 69 kHz and a LG offset of 0 Hz was applied during the 1H evolution time.72 This resulted in a 1H chemical shift scaling factor, λexp, of 0.81, which was obtained by comparing the 1H spectrum of L-alanine to its HETCOR projection.73 The 1H chemical shift range was then scaled accordingly to recover the true 1H chemical shifts.
Elemental analysis
Elemental analysis was performed using an Exeter CE-440 elemental analyser. C, H and N contents were measured, and the content of any other elements present in the samples was calculated by difference.X-ray photoelectron spectroscopy (XPS)
Samples were prepared for XPS by first preparing a solution of each sample at a concentration of 10 mg mL−1 in THF. These samples were left to stir for 16 hours at room temperature before filtering through 0.45 μm PTFE syringe filters. 100 μL of the filtered solution was dropped onto a circular glass slide (diameter = 12 mm) and spun at 1000 rpm for 60 seconds using a spin coater. The samples were left to dry for 1 hour in a vacuum oven at 50 °C before analysis by XPS. XPS data were collected on a Kratos AXIS Supra+ X-ray photoelectron spectrometer with an Al Kα X-ray source.TGA
TGA was performed using a TA Instruments TGA 5500 with a temperature ramp of 10 °C per minute.DSC
DSC was performed using a TA Instruments DSC 25 with a temperature ramp of 25 °C per minute.Liquid-state 31P NMR spectroscopy
40 mg of lignin was dissolved in 0.4 mL of pyridine:CDCl3 (1.6 mL:1 mL) and sonicated until completely dissolved. 50 μL of Cr(acac)3 relaxation agent (11.4 mg mL−1, dissolved in pyridine:CDCl3 solvent system), 100 μL of internal standard (∼0.12 M, dissolved in pyridine:CDCl3 solvent system) N-hydroxy-5-norborene-2,3-dicarboxoimide (CAS no.: 21715-90-2) were added to the lignin. The ratio of internal standard to lignin should be ∼0.3 μmol mg−1 100 μL of phosphitylating agent, 2-chloro-4,4,5,5-tetramethyl-1,3,2-dioxaphospholane (CAS no.: 14812-59-0) was added to the sample. The sample was vortexed for ∼30 seconds to ensure the contents were thoroughly mixed. The sample was then transferred to an NMR tube. NMR experiments were performed using a Bruker AVIII HD 400 MHz NMR spectrometer. Liquid-state 31P NMR spectra were collected without proton decoupling experiment using an acquisition time of 1 second, a relaxation delay of 5 seconds and 500 scans.74
Acetone solubility study
Each sample was dissolved in acetone (10 mg mL−1) and allowed to stir at room temperature for 10 minutes before photographs were taken.Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
Sample preparation, characterisation and functionalisation of lignin samples and data analysis were performed by Libby J. Marshall. Solid-state 13C CP MAS NMR data were collected and analysed by Daniel J. Cheney, Jasmine A. Keith and Frédéric Blanc. XPS data were collected and processed by Christopher Kelly. Libby J. Marshall wrote the initial draft with subsequent text addition and editing by all authors.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the EPSRC for funding via the University of Glasgow's 2022–2025 EPSRC Impact Acceleration Account (EP/X525716/1) and for enabling the use of the X-ray photoelectron spectrometer within the School of Chemistry at the University of Glasgow through the ESPRC Strategic Equipment Award (EP/W02134X/1). We thank the IBioIC for funding (FF-2023-02). Daniel J. Cheney was supported by the Leverhulme Trust under Research Project grant RPG-2020-066. We thank the University of Liverpool for funding Jasmine A. Keith. We also thank Sonichem for providing the lignin samples, Connor Fleming (University of Glasgow) for carrying out the elemental analysis measurements, and Andy Monaghan (University of Glasgow) for carrying out the TGA and DSC analysis.Notes and references
- S. Ohashi, J. Kilbane, T. Heyl and H. Ishida, Macromolecules, 2015, 48, 8412–8417 CrossRef CAS.
- G. Lligadas, A. Tüzün, J. C. Ronda, M. Galià and V. Cádiz, Polym. Chem., 2014, 5, 6636–6644 RSC.
- H. Ishida and H. Y. Low, Macromolecules, 1997, 30, 1099–1106 CrossRef CAS.
- S. Rimdusit, S. Pirstpindvong, W. Tanthapanichakoon and S. Damrongsakkul, Polym. Eng. Sci., 2005, 45, 288–296 CrossRef CAS.
- P. Froimowicz, C. R. Arza, L. Han and H. Ishida, ChemSusChem, 2016, 9, 1921–1928 CrossRef CAS PubMed.
- L. Puchot, P. Verge, T. Fouquet, C. Vancaeyzeele, F. Vidal and Y. Habibi, Green Chem., 2016, 18, 3346–3353 RSC.
- A. Martos, R. M. Sebastián and J. Marquet, Eur. Polym. J., 2018, 108, 20–27 CrossRef CAS.
- A. Trejo-Machin, A. Adjaoud, L. Puchot, R. Dieden and P. Verge, Eur. Polym. J., 2020, 124, 109468 CrossRef CAS.
- A. Adjaoud, L. Puchot, C. E. Federico, R. Das and P. Verge, Chem. Eng. J., 2023, 453, 10–16 CrossRef.
- N. N. Ghosh, B. Kiskan and Y. Yagci, Prog. Polym. Sci., 2007, 32, 1344–1391 CrossRef CAS.
- M. L. Salum, D. Iguchi, C. R. Arza, L. Han, H. Ishida and P. Froimowicz, ACS Sustain. Chem. Eng., 2018, 6, 13096–13106 CrossRef CAS.
- L. R. V. Kotzebue, J. R. de Oliveira, J. B. da Silva, S. E. Mazzetto, H. Ishida and D. Lomonaco, ACS Sustain. Chem. Eng., 2018, 6, 5485–5494 CrossRef CAS.
- M. Comí, G. Lligadas, J. C. Ronda, M. Galià and V. Cádiz, J. Polym. Sci., Part A: Polym. Chem., 2013, 51, 4894–4903 CrossRef.
- M. Margarida Martins, F. Carvalheiro and F. Gírio, Biomass Convers. Biorefin., 2022, 14, 3183–3207 CrossRef.
- H. Ishida, in Handbook of Benzoxazine Resins, ed. H. Ishida and T. Agag, Elsevier, 2011, pp. 3–81 Search PubMed.
- T. Rashid, F. Sher, T. Rasheed, F. Zafar, S. Zhang and T. Murugesan, J. Mol. Liq., 2021, 321, 114577 CrossRef CAS.
- A. Moreno and M. H. Sipponen, Mater. Horiz., 2020, 7, 2237–2257 RSC.
- Z. Gao, X. Lang, S. Chen and C. Zhao, Energy Fuels, 2021, 35, 18385–18395 CrossRef CAS.
- Q. Liu, Y. Xu, F. Kong, H. Ren and H. Zhai, Wood Sci. Technol., 2022, 56, 1527–1549 CrossRef CAS.
- Z. Peng, X. Jiang, C. Si, A. Joao Cardenas-Oscanoa and C. Huang, ChemSusChem, 2023, 16, e202300174 CrossRef CAS PubMed.
- W. Li, H. Sun, G. Wang, W. Sui, L. Dai and C. Si, Green Chem., 2023, 25, 2241–2261 RSC.
- P. L. de Hoyos-Martínez, H. Issaoui, R. Herrera, J. Labidi and F. Charrier-El Bouhtoury, ACS Sustain. Chem. Eng., 2021, 9, 1729–1740 CrossRef.
- A. Y. Kharade and D. D. Kale, Eur. Polym. J., 1998, 34, 201–205 CrossRef CAS.
- N. Zhang, Z. Li, Y. Xiao, Z. Pan, P. Jia, G. Feng, C. Bao, Y. Zhou and L. Hua, J. Bioresour. Bioprod., 2020, 5, 67–77 Search PubMed.
- X. Ning and H. Ishida, J. Polym. Sci., Part A: Polym. Chem., 1994, 32, 1121–1129 CrossRef CAS.
- R. Jain, A. K. Narula and V. Choudhary, J. Appl. Polym. Sci., 2007, 106, 3327–3334 CrossRef CAS.
- E. M. Nour-Eddine, Q. Yuan and F. Huang, J. Appl. Polym. Sci., 2012, 125, 1773–1781 CrossRef CAS.
- H. M. E. Haque, Z. Islam, T. Kawauchi and T. Takeichi, Appl. Mech. Mater., 2012, 217, 571–577 Search PubMed.
- S. Allies, Sonichem, accessed 2nd December, 2024.
- G. Barclay, Scottish startup Sonichem turns sawdust into sustainable cosmetics, accessed 2nd December, 2024.
- O. Sevastyanova, M. Helander, S. Chowdhury, H. Lange, H. Wedin, L. Zhang, M. Ek, J. Kadla, C. Crestini and M. Lindström, J. Appl. Polym. Sci., 2014, 131, 40799 CrossRef.
- A. Evdokimov, A. Kurzin, O. Fedorova, P. Lukanin, V. Kazakov and A. Trifonova, Wood Sci. Technol., 2018, 52, 1165–1174 CrossRef CAS.
- M. Karlsson, J. Romson, T. Elder, A. Emmer and M. Lawoko, Biomacromolecules, 2023, 24, 2314–2326 CrossRef CAS PubMed.
- K. Chiou and H. Ishida, Curr. Org. Chem., 2013, 17, 913–925 CrossRef CAS.
- G. J. Abarro, J. Podschun, L. J. Diaz, S. Ohashi, B. Saake, R. Lehnen and H. Ishida, RSC Adv., 2016, 6, 107689–107698 RSC.
- A. T. Martínez, G. Almendros, F. J. González-Vila and R. Fründ, Solid State Nucl. Magn. Reson., 1999, 15, 41–48 CrossRef PubMed.
- Y. Feng, J. Lan, P. Ma, X. Dong, J. Qu and H. He, Wood Sci. Technol., 2016, 51, 135–150 Search PubMed.
- S. Wang, B. Ru, H. Lin, W. Sun and Z. Luo, Bioresour. Technol., 2015, 182, 120–127 CrossRef CAS PubMed.
- R. El Hage, N. Brosse, P. Sannigrahi and A. Ragauskas, Polym. Degrad. Stab., 2010, 95, 997–1003 Search PubMed.
- F. Huang, P. M. Singh and A. J. Ragauskas, J. Agric. Food Chem., 2011, 59, 12910–12916 Search PubMed.
- O. Gordobil, R. Moriana, L. Zhang, J. Labidi and O. Sevastyanova, Ind. Crop. Prod., 2016, 83, 155–165 CrossRef CAS.
- J. Bortoluz, A. Cemin, L. R. Bonetto, F. Ferrarini, V. I. Esteves and M. Giovanela, Cellulose, 2019, 26, 4895–4908 Search PubMed.
- R. M. Silverstein, F. X. Webster and D. J. Kiemle, Spectrometric Identification of Organic Compounds, Wiley, New York, 7th edn, 2005 Search PubMed.
- X. Kang, A. Kirui, M. C. Dickwella Widanage, F. Mentink-Vigier, D. J. Cosgrove and T. Wang, Nat. Commun., 2019, 10, 347 CrossRef CAS PubMed.
- R. C. Sun, ChemSusChem, 2020, 13, 4385–4393 CrossRef CAS PubMed.
- C. H. Lin, S. L. Chang, C. W. Hsieh and H. H. Lee, Polymer, 2008, 49, 1220–1229 CrossRef CAS.
- S. Li and L. Wang, J. Appl. Polym. Sci., 2005, 99, 1359–1366 CrossRef.
- C. Andronescu, S. A. Gârea, C. Deleanu and H. Iovu, Thermochim. Acta, 2012, 530, 42–51 Search PubMed.
- J. Y. Kim, E. J. Shin, I. Y. Eom, K. Won, Y. H. Kim, D. Choi, I. G. Choi and J. W. Choi, Bioresour. Technol., 2011, 102, 9020–9025 Search PubMed.
- Z. J. Berkson, S. Bjorgvinsdottir, A. Yakimov, D. Gioffre, M. D. Korzynski, A. B. Barnes and C. Coperet, JACS Au, 2022, 2, 2460–2465 CrossRef CAS PubMed.
- G. Vijaykumar and S. K. Mandal, Dalton Trans., 2016, 45, 7421–7426 Search PubMed.
- K. Albert, B. Peters and E. Bayer, Z. Naturforsch., 1986, 41b, 351–358 CrossRef CAS.
- H. Pan, G. Sun and T. Zhao, Int. J. Biol. Macromol., 2013, 59, 221–226 CrossRef CAS PubMed.
- O. Auciello, J.-F. Veyan and M. J. Arellano-Jimenez, Front. Carbon, 2023, 2, 1279356 Search PubMed.
- J. Podschun, B. Saake and R. Lehnen, Eur. Polym. J., 2015, 67, 1–11 CrossRef CAS.
- G. Riess, J. M. Schwob, G. Guth, M. Roche and B. Lande, in Advances in Polymer Synthesis, ed. B. M. Culbertson and J. E. McGrath, Plenum Press, New York, 1986, pp. 27–50 Search PubMed.
- C. Wang, J. Pan, J. Li and Z. Yang, Bioresour. Technol., 2008, 99, 2778–2786 Search PubMed.
- J. Y. Kim, H. Hwang, S. Oh, Y. S. Kim, U. J. Kim and J. W. Choi, Int. J. Biol. Macromol., 2014, 66, 57–65 CrossRef CAS PubMed.
- K. G. Hanson, C.-H. Lin and M. M. Abu-Omar, Polymer, 2021, 233, 124202 Search PubMed.
- J. Sun, C. Wang, J. C. C. Yeo, D. Yuan, H. Li, L. P. Stubbs and C. He, Macromol. Mater. Eng., 2015, 301, 328–336 CrossRef.
- C. Lin, S. Cai, T. Leu, T. Hwang and H. Lee, J. Polym. Sci. Polym. Chem., 2006, 44, 3454–3468 CrossRef CAS.
- G. Cao, W. Chen and X. Liu, Polym. Degrad. Stabil., 2008, 93, 739–744 Search PubMed.
- S. Chen, S. Lin, Y. Hu, M. Ma, Y. Shi, J. Liu, F. Zhu and X. Wang, Polym. Adv. Technol., 2018, 29, 3142–3150 Search PubMed.
- L. Liu, B. Shi, A. Zhang, Y. Xue, J. Zhang, J. Dai, M. Hassanpour, L.-C. Tang, Y. Shi and P. Song, Compos. – A: Appl. Sci. Manuf., 2022, 160, 107028 Search PubMed.
- Y. Liu, Q. Ran and Y. Gu, Polym. Degrad. Stab., 2019, 163, 15–24 CrossRef CAS.
- L. Zhao, C. Zhao, C. Guo, Y. Li, S. Li, L. Sun, H. Li and D. Xiang, ACS Omega, 2019, 4, 20275–20284 CrossRef CAS PubMed.
- H. Ding, X. Wang, L. Song and Y. Hu, J. Renewable Mater., 2021, 10, 871 Search PubMed.
- Y. Liu and C. Chou, J. Polym. Sci. Polym. Chem., 2005, 43, 5267–5282 CrossRef CAS.
- Y. Liu, W. Zhang, Y. Chen and S. Zheng, J. Appl. Polym. Sci., 2006, 99, 927–936 CrossRef CAS.
- B. M. Fung, A. K. Khitrin and K. Ermolaev, J. Magn. Reson., 2000, 142, 97–101 CrossRef CAS PubMed.
- C. R. Morcombe and K. W. Zilm, J. Magn. Reson., 2003, 162, 479–486 CrossRef CAS PubMed.
- B.-J. van Rossum, H. Förster and H. J. M. de Groot, J. Magn. Reson., 1997, 124, 516–519 CrossRef CAS.
- F. M. Paruzzo and L. Emsley, J. Magn. Reson., 2019, 309, 106598 Search PubMed.
- M. Balakshin and E. Capanema, J. Wood Chem. Technol., 2015, 35, 220–237 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4su00767k |
| This journal is © The Royal Society of Chemistry 2025 |