
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEngineering hemin-loaded hyaluronan needle-like microparticles with photoprotective properties against UV-induced tissue damage†
Amir M.
Alsharabasy
 *a,
Amal
Aljaabary
a,
Pau
Farràs
ab and
Abhay
Pandit
*a
*a,
Amal
Aljaabary
a,
Pau
Farràs
ab and
Abhay
Pandit
*a
aCÚRAM, SFI Research Centre for Medical Devices, University of Galway, H91 W2TY, Ireland. E-mail: amir.abdo@universityofgalway.ie; abhay.pandit@universityofgalway.ie
bSchool of Biological and Chemical Sciences, Ryan Institute, University of Galway, H91 TK33, Ireland
First published on 28th August 2024
Abstract
This study aimed to develop hyaluronan (HA)-based hydrogel microparticles (MPs) loaded with hemin to address the limitations of traditional macroscale hydrogels. The objective is to design MPs such that they can modulate their physicochemical properties. Given the widespread use of ultraviolet C (UVC) light in various industries and the need for protective measures against accidental exposure, this study evaluated the potential of hemin-loaded MPs to protect human dermal fibroblasts from oxidative stress and cell death caused by UVC exposure. Multiple MP formulations were developed and analysed for size, surface charge, swelling behaviour, degradation rate, and radical scavenging capabilities, both with and without hemin loading. The most promising formulations were tested against UVC-exposed cells to assess cell viability, intracellular nitric oxide (˙NO) and reactive oxygen species levels, and protein carbonylation. The fabricated particles were in the form of microneedles, and the degree of their crosslinking and the role of hemin in the chemical crosslinking reaction were found to influence the surface charge and hydrodynamic diameter of the MPs. Increased crosslinking resulted in reduced swelling, slower degradation, and decreased hemin release rate. MPs with a higher degree of swelling were capable of releasing hemin into the culture medium, leading to enhanced bilirubin generation in dermal fibroblasts following cellular uptake. Pre-treatment with these MPs protected the cells from UVC-induced cell death, nitrosative stress, and protein carbonylation. These findings highlight the potential of the studied MPs to release hemin and to minimise the harmful effects of UVC on dermal fibroblasts.
1. Introduction
Hydrogels have been employed in various biomedical applications due to their tuneable fabrication conditions with programmable properties, and biocompatibility. Hydrogels are promising devices for the controlled delivery of pharmaceuticals and growth factors, maintaining their bioactivity using minimally invasive techniques.1,2 However, bulk (macroscale) hydrogels are not suitable for certain applications, particularly those requiring injections of specific sizes.3 Additionally, challenges such as adjusting hydrogel composition to achieve certain porosity while maintaining the bioactivity of loaded drugs during storage pose significant issues for some applications.3,4 Some of these drawbacks can be mitigated by fabricating hydrogels as microscale particles. These microsystems retain most of the properties of bulk hydrogels while offering additional advantages. Firstly, their small size allows for injection through small needles, facilitated by their shear-thinning behaviour.5 Secondly, the tailorability of their design, composition, and size enables the creation of microparticles (MPs) with various properties within a multifunctional device.5 Achieving such a multifactorial effect with a single bulk hydrogel structure is challenging. Finally, controlling the porosity between packed MPs by adjusting their size provides an advantage in managing fluid flow and permeability without requiring prior hydrogel degradation.6As the largest organ and the outermost layer of the body, the skin is vulnerable to UV radiation, which damages the DNA in its cells. This can lead to mutagenic lesions, changes in appearance, potential photoaging, and the progression of skin cancer.7,8 Exposure to UVB (280–320 nm) and UVA (320–400 nm) is the primary cause of sunlight-related skin cancer. While UVB, despite being less abundant than UVA, causes more significant DNA damage.9,10 UVC (100–280 nm) is the most harmful form of UV radiation due to its short wavelength; however, most UVC rays are blocked by the Earth's atmosphere, making natural exposure rare.11 Therefore, exposure to UVC typically occurs from artificial sources such as lamps and lasers used in various industries.12,13 To address this, Directive 2006/25/EC was adopted, defining the limits for UVC exposure from artificial sources.14 For example, the exposure limit for effective radiant exposure within the UVC range is 30 J m−2, and it should not exceed 60 J m−2 over an 8-hour workday at 254 nm. Additionally, Directive 2014/35/EU regulates the marketing of UVC lamps in electrical equipment designed for use within specified voltage ranges.
In general, overexposure to UV radiation is responsible for oxidative–nitrosative stress, leading to a series of inflammatory reactions, which cause erythema and chronic damage.15 Oxidative stress is mediated by various reactive oxygen species (ROS). Excessive ROS production results in the oxidation of different cellular biomolecules, altering their functions.16 Various types of antioxidants have demonstrated protective effects against UV-induced oxidative damage.8 Nitrosative stress is mainly mediated by the excessive production of nitric oxide (˙NO) and its toxic congeners, such as nitrogen dioxide (˙NO2), in response to the simultaneous production of oxygen radicals. Additionally, protein nitration plays a vital role in modulating the functionality of different proteins and may contribute to the development of some inflammatory diseases. The overproduction of ˙NO has been observed both immediately after UV exposure17,18 and 8–10 hours later.19 These studies primarily focused on UVA and UVB radiation. While few studies have investigated the influence of UVC on the skin, they have generally found similar effects to those of UVB, including DNA damage induction. However, the dose–response curve for UVC-induced erythema in human skin was less steep than that for UVB.20–22
Hyaluronan (HA), a negatively charged, unbranched glycosaminoglycan, is one of the main components of the extracellular matrix (ECM) in various tissues.23 HA is composed of D-glucuronic acid and β-N-acetylglucosamine (GlcNAc), linked by alternating β-(1,4) and β-(1,3) glycosidic bonds. Its accumulation forms gel-filled spaces with highly hydrated coils in the ECM.24 Consequently, HA has been applied in numerous biomedical fields. For instance, it has been used in developing various drug delivery systems and devices25,26 and creating in vitro 3D cell culture models for drug screening.27,28 Notably, HA-based hydrogel MPs have been developed using different chemistries and fabrication techniques for multiple biological applications.29–32 HA's reactive oxygen and nitrogen scavenging activities have been reported previously,33,34 demonstrating antioxidant activity against UV-induced damage.35,36
Hemin, a coordination complex consisting of iron (Fe(III)) and protoporphyrin IX, exhibits distinctive structural and redox properties that are responsible for its various catalytic functions.37 It has been suggested as a chemopreventive agent due to its potential to disrupt the activity of carcinogens involved in the development of skin cancer.38,39 Additionally, hemin has been reported to elicit cytoprotective effects against UVC radiation by scavenging hydroxyl radicals and peroxynitrite ions,40 and against UVB radiation by inhibiting keratinocyte apoptosis and reducing the maturation and infiltration of bone marrow neutrophils.41 Furthermore, hemin, through the activation of heme oxygenase (HOX-1), reduces intracellular levels of reactive oxygen species (ROS), mitigates oxidative injury, and prevents the apoptosis of dermal fibroblasts.42 Its functionality as a nitric oxide (˙NO) scavenger has also been studied in detail.43,44
In this study, high-molecular-weight HA MPs were fabricated and the properties of different formulations were evaluated following hemin loading (Scheme 1). The objective of this study was to develop hydrogel-forming MPs with protective functions against UVC-induced pro-oxidant levels and cell death in human dermal fibroblasts. Initially, the morphology, size, and surface charges of both unloaded and hemin-loaded MPs from various formulations were assessed. This was followed by evaluating their swelling behaviour, biodegradation kinetics, and the rate of hemin release (Scheme 2). Subsequently, the properties of candidate formulations, loaded with different concentrations of hemin, were investigated, with assessing the cellular uptake of released hemin and its impact on intracellular bilirubin generation in cells. The study then examined the effects of UVC radiation on cell viability, the generation of intracellular ROS, ˙NO, and the carbonylation of intracellular proteins. These parameters were evaluated both without and with prior cell treatment using the candidate concentrations of unloaded and hemin-loaded MPs. Cell-based experiments also included treatment with hemin alone to gain insights into the mechanism of the intracellular effects of both hemin and the studied MPs.
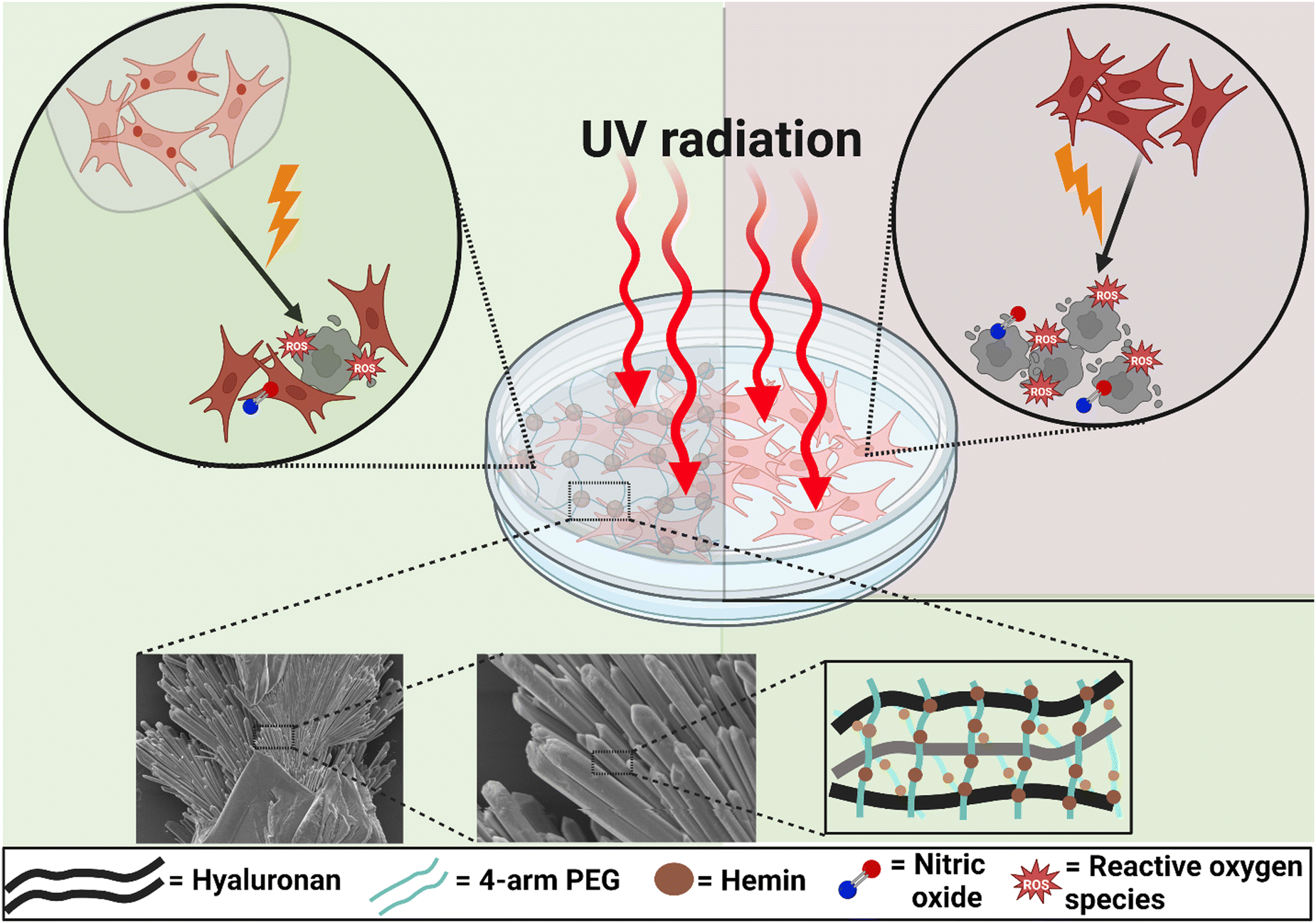 | ||
| Scheme 1 Schematic illustration of the role played by hemin-loaded microparticles in protecting human dermal fibroblasts against the damaging effects of UV radiation and the accompanying oxidative and nitrosative stress, indicated by reactive oxygen species (ROS) and nitric oxide, respectively (A), compared to the MP-free cultures (B). The lower panel shows the needle-like structure of these MPs and a general scheme for the hemin-loaded hyaluronan matrix. Scheme created with BioRender.com. | ||
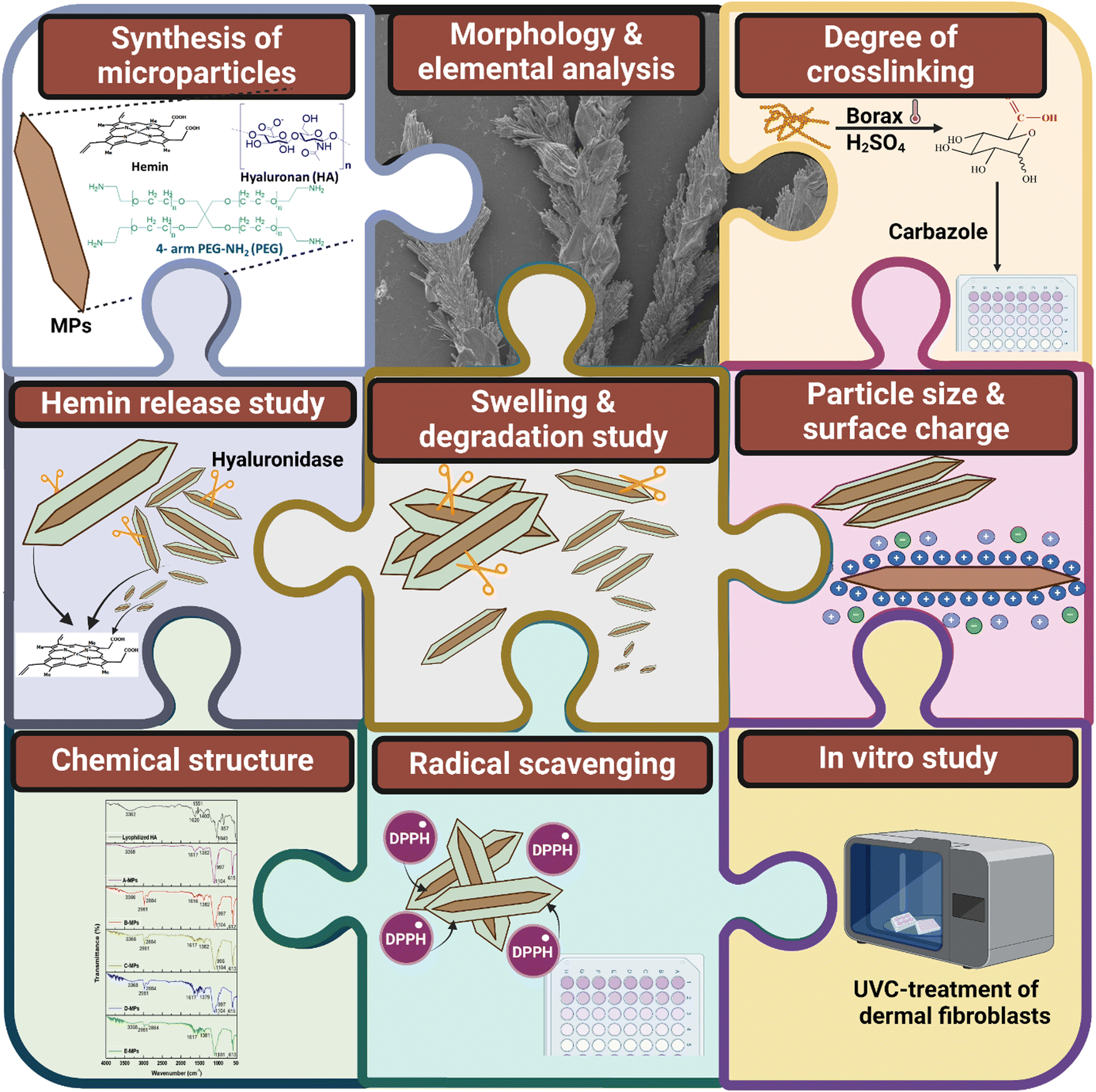 | ||
| Scheme 2 A summary of the microparticle (MPs) synthesis, components and characterization. The assessment involves: morphology and elemental composition examined using SEM/EDX; degree of crosslinking determined through a carbazole assay; particle size and surface charge measurement; swelling and degradation study; hemin release study; analysis of the chemical structure of MPs using FTIR; evaluation of the DPPH radical scavenging efficiency. Finally, an in vitro study was performed where cells were treated with UVC irradiation to assess the microparticles’ effects. | ||
2. Materials and methods
2.1. Materials
Human Primary Dermal Fibroblasts (C-12302™) were from PromoCell. HA (1000 kDa) was provided by Lifecore Biomedical, Chaska, US. 4 arm-polyethylene glycol (PEG)-amine (2 kDa) (4-arm-PEG-NH2) was from JenKem Technology, China. 4-(4,6-Dimethoxy-1,3,5-triazin-2-yl) (DMTMM), 2-(N-morpholino)ethanesulfonic acid (MES), hemin, zinc(II) protoporphyrin IX (Zn-PPIX), anhydrous DMSO, sodium chloride (NaCl), sodium tetraborate decahydrate (Borax), carbazole, 2,2-diphenyl-1-picrylhydrazyl (DPPH), sulfuric acid, ethanol, Dulbecco's Modified Eagle Medium (DMEM), L-glutamine, penicillin, streptomycin, fetal bovine serum (FBS), phosphate-buffered saline (PBS), ethidium homodimer (EthD1), hemin assay kit (MAK036), bilirubin assay kit (MAK126), and transparent 96-well microplates were all purchased from Sigma-Aldrich. AlamarBlue™ cell viability reagent, Pierce™ bicinchoninic acid (BCA) protein assay kit, SuperSignal™ West Pico PLUS chemiluminescent substrate, Invitrogen™ Calcein AM, CellROX® Deep Red Reagent, Invitrogen™ 4-Amino-5-ethylamino-2′,7′-difluorofluorescein diacetate (DAF-FM DA), sodium dodecyl sulfate (SDS), acrylamide/bisacrylamide mixture, and isopropanol were obtained from Fisher Scientific. The protein carbonyl assay Kit (ab178020) was from abcam. The μ-Plate 96 Well Black ibiTreat #1.5 polymer coverslip were provided by IBIDI GMBH, and the black, clear bottom 96-well microplates were from Corning.2.2. Fabrication of HA microparticles
The fabrication of HA MPs followed the methodology outlined by Krishna et al.,45 with minor adjustments. Initially, after dissolving of HA in 0.1 M MES buffer at room temperature, it was mixed with 20% Na2SO4 to facilitate ionization of the HA chains, undergoing continuous stirring for two hours. Subsequently, DMTMM and 4-arm-PEG-NH2 were added to this mixture, which was then stirred overnight at room temperature. The concentration of HA in the final solution was 4 mg mL−1. For fabrication of hemin-loaded MPs, hemin was initially dissolved in DMSO, before being incorporated into the HA/Na2SO4 mixture, along with DMTMM and 4-arm-PEG-NH2. This mixture was stirred overnight at room temperature, protected from light. To precipitate the hydrogel MPs, the solution underwent treatment with twice the volume of an 8% NaCl solution. It was subsequently titrated with four times the volume of absolute ethanol while vigorously stirring for one hour at room temperature, followed by centrifugation at 2000g for 10 minutes. The resultant particle pellets were reconstituted in deionized (DI) water, undergoing repeated titration against ethanol, and subjected to centrifugation twice, before being subjected to freeze-drying of the purified particles. Different formulations of MPs with different ratios between DMTMM and 4 arm-PEG-NH2 were fabricated (Table 1). Following initial precipitation and each round of washing hemin-loaded MPs, the solution was analysed for the presence of hemin by measurement of absorbance at 388 nm using a microplate reader (Varioskan™ Flash, Thermo Scientific™, Finland). The hemin entrapment efficiency was determined using eqn (1):46| Hemin entrapment efficiency (%) = (Molo − Molf/Molo) × 100 | (1) |
| ID | HA | DMTMM | PEG |
|---|---|---|---|
| A | 1 | 0.93 | 0.08 |
| B | 1 | 0.93 | 0.24 |
| C | 1 | 2.78 | 0.08 |
| D | 1 | 1.86 | 0.16 |
| E | 1 | 2.78 | 0.24 |
2.3. Characterization of the synthesized MPs
| Degree of swelling = [(Ms/Md) − 1] × 100 | (2) |
| Remaining weight (%) = (M/Md) × 100 | (3) |
The DPPH radical scavenging efficiency was calculated using eqn (4):
| DPPH radical scavenging efficiency (%) = (AC − Asm) × 100/AC | (4) |
2.4. In vitro studies
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1000) and anti-β-actin (Sigma, A5441, 1:10000), followed by HRP-conjugated goat anti-mouse (Invitrogen, 31430, 1:10000). Protein bands were detected using the SuperSignal™ West Pico PLUS Chemiluminescent Substrate and an Omega Lum™ G Imaging System. Blotting was performed in triplicate with two samples per group.
:1000) and anti-β-actin (Sigma, A5441, 1:10000), followed by HRP-conjugated goat anti-mouse (Invitrogen, 31430, 1:10000). Protein bands were detected using the SuperSignal™ West Pico PLUS Chemiluminescent Substrate and an Omega Lum™ G Imaging System. Blotting was performed in triplicate with two samples per group.
2.5. Statistical analysis
The data were analysed by Microsoft Excel using a student's t-test, and the differences were considered statistically significant at (p < 0.05).3. Results
3.1. UV-Vis spectroscopy
This study evaluated the aggregation of hemin molecules after dissolving in Na2SO4-containing MES buffer. The dilution for 30 minutes resulted in changes in the UV-Vis spectra, the Soret band became broader compared to the original solvent, DMSO, and its intensity slightly decreased after 16 hours of incubation. Additionally, the main bands within the charge transfer region were indistinguishable in the buffer (Fig. S1, ESI†). These results suggest that hemin molecules begin to aggregate in the aqueous solution, but at varying levels.3.2. Morphology of microparticles
Previously, we reported the fabrication conditions and properties of hydrogels made using the same components employed formicroparticle fabrication: HA, 4-arm PEG, and DMTMM.47 In this study, microparticles of different formulations were fabricated, exhibiting needle-like morphologies with similar perimeters after drying (Fig. S2, ESI†). This morphology suites the intended application for hemin delivery considering the reported advantages of non-spherical particles for drug delivery and more effective adhesion to cell surface (Doshi et al., 2010; Howard et al., 2014).48,49However, the length of these needles could not be measured due to their aggregates. The optimal drying period to reach these clear morphologies was determined to be five days at room temperature. Subsequently, both unloaded, and hemin-loaded particles were fabricated for each studied formulation, with a final hemin concentration of 240 μM when mixed with HA. These formulations were designated as A(H), B(H), C(H), D(H) and E(H). Despite varying the ratios of HA, 4-arm PEG, and DMTMM to control the degree of crosslinking, no significant differences were observed in the entrapment efficiency of hemin among the different formulations (Table S1, ESI†).
3.3. Degree of crosslinking, zeta potential and size of the microparticles
The degree of crosslinking was estimated using a carbazole assay based on a method developed before.50 This assay works by hydrolysing HA in H2SO4, which breaks it down into its monomeric units. The –COOH groups in the released glucuronic acid then react with carbazole, producing a pink-coloured solution. By measuring the amount of free –COOH groups, the crosslinked groups and the degree of crosslinking can be determined. In this study, A-MPs exhibited the lowest degree of crosslinking, with 37.09% of the –COOH groups being crosslinked (Table 2). Conversely, C- and D-MPs showed the highest degree of crosslinking, with similar percentages of crosslinked –COOH groups. This correlates with an increased amount of crosslinker and DMTMM in the latter formulations, which enhances crosslinking among the HA chains. The B- and E-MPs displayed an intermediate degree of crosslinking, with similar percentages of crosslinked –COOH groups. Despite expectations that E-MPs would exhibit the highest degree of crosslinking, the rapid reaction rate, combined with a relatively high concentration of 4-arm PEG and DMTMM, likely saturated the active sites quickly. This was evidenced by the formation of a stiff mixture after overnight stirring.| Formulation | Free –COOH (%) | Crosslinked –COOH (%) | Zeta potential (mV) | Hydrodynamic diameter (μm) | Formulation | Zeta potential (mV) | Hydrodynamic diameter (μm) |
|---|---|---|---|---|---|---|---|
| a P < 0.05 for the zeta potential and hydrodynamic diameter of hemin-loaded MPs, compared to the respective unloaded MPs of the same formulation, respectively. b P < 0.05 for the hydrodynamic diameter of hemin-loaded B-, C-, D-, E-MPs, compared to the unloaded A-MPs, using a two-tailed unpaired student's t-test. | |||||||
| A | 62.91 | 37.09 | −27.6 ± 0.4 | 1.2 ± 0.1 | A(H) | −32.3 ± 0.3a | 1.9 ± 0.063a |
| B | 51.11 | 48.89 | −27.4 ± 0.9 | 4.8 ± 0.24b | B(H) | −29.9 ± 0.5 | 1.2 ± 0.03a |
| C | 40.58 | 59.42 | −29.3 ± 1 | 1.4 ± 0.05 | C(H) | −29 ± 0.7 | 0.6 ± 0.06a |
| D | 39.07 | 60.93 | −28.3 ± 1 | 1.6 ± 0.1b | D(H) | −27.6 ± 0.3 | 1.2 ± 0.03 |
| E | 51.05 | 48.95 | −24.7 ± 0.4 | 1.6 ± 0.17b | E(H) | −25.2 ± 0.7 | 1.4 ± 0.111 |
The zeta potential of the synthesized particles was measured to assess their stability in aqueous solutions.51 The electrophoretic mobility was used to calculate the surface zeta potential of both unloaded and hemin-loaded MPs. In general, the zeta potential ranged between −24.7 and −29.3 mV, with the highest net negative charge observed on the surface of C-MPs and the lowest on E-MPs (Table 2). These values are influenced by the pH and ionic strength of the solution, which were kept constant among the tested samples, and align with previously reported values for HA-MPs.45 DMTMM is responsible for activating the –COOH groups, which is expected to decrease the zeta potential.52 However, given the threefold concentration of DMTMM in C-MPs compared to A-MPs, with the same 4-arm PEG content, this activation in C-MPs led to an increase in the measured zeta potential. In contrast, the zeta potential decreased significantly in E-MPs due to the consumption of activated HA chains in crosslinking with the increased 4-arm PEG content. Hemin loading generally enhanced the zeta potential values, but significant effects were observed only in hemin-loaded A-MPs. We reported before that the amount of hemin molecules on the surface of hemin-loaded HA hydrogels is inversely proportional to the degree of crosslinking.47 Therefore, more hemin molecules are expected on the surface of A-MPs, which have the lowest degree of crosslinking. This number decreases in other microparticle formulations, with more hemin molecules conjugated to the hydrogel matrix. This explains the significant increase in zeta potential for hemin-loaded A-MPs, with a slight increase in B-MPs.
These results highlight the importance of both DMTMM, as a conjugation agent, and 4-arm PEG in determining the surface charge of the final particles. Moreover, these factors are expected to influence the final size of the formed MPs. Of a note, it should be emphasized here that DLS is a tool mainly used for characterization of the spherical particles based on certain mathematical models, and the hydrodynamic radius measured will not reflect the real geometric particle diameter for non-spherical particles.53,54 However, this technique has been commonly used for size characterization of suspended non-spherical particles, mainly for primary assessment of the aggregation behaviours of particles.55–57 The reported particle size distribution would be an equivalent spherical distribution of the tested particles.
Notably, increasing the concentration of DMTMM has been reported to enhance the activation of HA, leading to more crosslinking and larger HA nanogels.52 This accounts for the increase in measured hydrodynamic diameter with the rise in DMTMM concentration from A- to E-formulations, with sizes ranging between 1.2 and 4.8 μm (Table 2). Additionally, hemin loading increased the size of A-MPs but decreased the size of the other MP formulations. The former effect may be attributed to the accompanying enhancement in zeta potential and increased resistance to aggregation, leading to more hydration and increased size of the MPs. However, considering the reactivity of hemin with DMTMM, less activation of HA occurs in hemin-loaded MPs, resulting in a decreased particle size. Similar effects of hemin on the zeta potential of hemin-loaded nanoparticles have been previously reported.58
3.4. Swelling and degradation behaviour of microparticles
Studying the swelling kinetics and liquid uptake by the different microparticle formulations helps in understanding their pharmaceutical behaviour.59 When the crosslinked polymeric chains absorb water, they generate open diffusion portals for the delivery of the loaded therapeutic molecules.60 This also facilitates the interaction between the hydrogel matrix and degrading enzymes, initiating hydrogel degradation and releasing the conjugated therapeutic molecules into the body. The A-formulation-based MPs showed rapid swelling once mixed with PBS, reaching a maximum after one hour of incubation at room temperature (Fig. 1A). The degree of swelling was highest for A-MPs, reaching 4875 ± 81%, followed by C-MPs, reaching 1324 ± 300%, with B-MPs showing the lowest degree of swelling. E-MPs and F-MPs exhibited intermediate swelling kinetics, with a general maximum swelling observed after one hour of incubation as well. These findings are generally governed by the increased cross-linking degree from A to E formulations, which limits the swelling degree of hydrogel-MPs. Furthermore, these results indicate that the extreme hydrophilicity and water-holding properties of HA and HA-hydrogels, as previously reported,61,62 are responsible for the swelling of the unloaded MPs. Additionally, the interactions of 4-arm PEG with PBS also contribute to the overall swelling kinetics. Moreover, the partial disintegration of the particles after one hour of incubation highlights the importance of the crosslinking bonds and their stability against prolonged water uptake. In contrast, hemin loading introduces a hydrophobic domain to the hydrogels, which suppresses their swelling compared to the unloaded counterparts (Fig. 1B).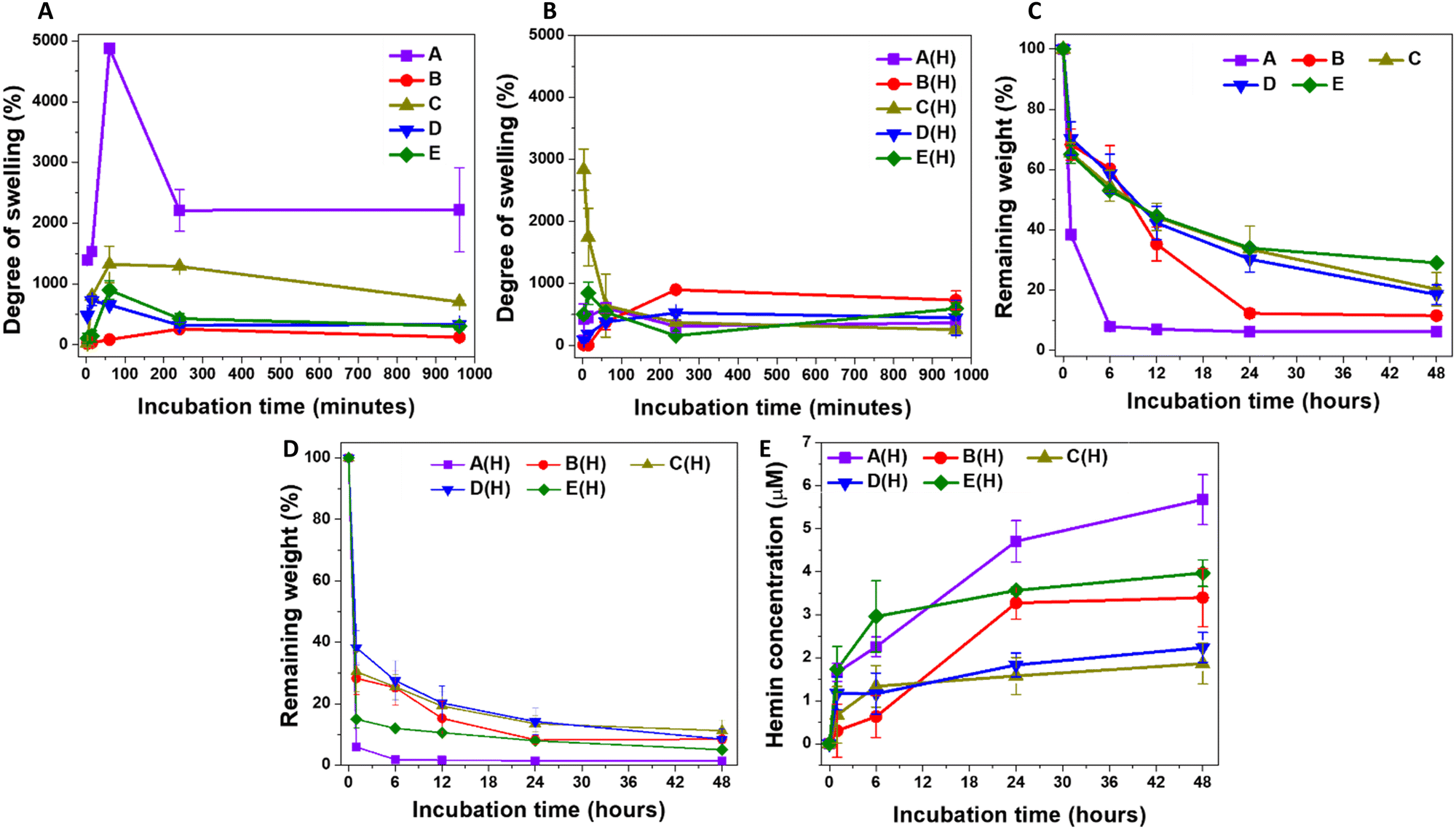 | ||
| Fig. 1 A comparison of the swelling kinetics of: (A) the unloaded A-, B-, C-, D-, E-MPs and (B) their respective hemin-loaded MPs, indicated as A(H), B(H), C(H), D(H), and E(H) in PBS at room temperature, and the degradation, in response to HAase (10 U mL−1), of the unloaded (C) and hemin-loaded MPs (D). (E) The concentration of hemin following degradation of the MPs. Results are presented as mean ± S.D, n = 3. | ||
Considering the influence of particle size on water uptake, smaller particle sizes have a larger surface area, which facilitates the internalization of water molecules and creates more pores.63 This explains the highest rate of swelling observed in A-MPs, followed by a lower swelling degree in C-MPs, and an even lower degree in the largest particles, B-MPs, while D-MPs and E-MPs exhibited intermediate swelling kinetics. However, this phenomenon does not explain the observed behaviour of hemin-loaded MPs, which showed variations in swelling kinetics that did not correlate directly with the decrease in microparticle size (Fig. 1B). It is worth noting that there is still controversy regarding the influence of particle size on swelling. For instance, an inverse relationship between particle size and the degree of swelling was reported.63 In contrast, Soundaranathan et al. found improved swelling in larger particles compared to smaller ones.64 Moreover, Schulte et al. found that the swelling rate was independent of microparticle size.65
The swelling of the hydrogel allows interaction between its matrix and the hyaluronidase enzyme, inducing its degradation. Accordingly, it would be expected that the degradation rate of hemin-loaded MPs would be higher than that of unloaded MPs of the same formulation. However, the degradation kinetics were assessed here by comparing the weights of lyophilized degraded hydrogels to those of the lyophilized particles before swelling. Therefore, the most accurate comparison should be among the formulations of either unloaded or loaded MPs. For the hemin-free MPs, the highest degradation rate was observed in A-MPs, followed by B-MPs, with C-, D- and E-MPs showing the lowest rates (Fig. 1C). These results indicate the lower stability of A-MPs, likely due to their lowest degree of crosslinking, compared to C-, D- and E-MPs. However, enzymatic treatment of hemin-loaded MPs accelerated their degradation (Fig. 1D), exhibiting different hemin release kinetics (Fig. 1E). The highest release rate was generally observed in A-MPs, corresponding to their highest degradation rate. This was followed by a lower rate for E-MPs, then B-MPs, with C- and D-MPs demonstrating the lowest degradation and hemin release kinetics.
Collectively, based on these results, A-MPs were selected for further investigation and in vitro testing to determine their potential in protecting dermal fibroblasts against UV-induced oxidative injury. Their higher degree of swelling and faster degradation rate, which facilitates quicker hemin release compared to other formulations, make them suitable candidates as protective hydrogel-forming particles. These properties align with the need for a relatively rapid effect in such applications.
3.5. Chemical composition
The chemical composition of unloaded and hemin-loaded MPs was investigated using FTIR. The peak assignments for HA (Fig. S3A, ESI†) have been previously discussed.34 The broad peak at around 3362 cm−1, corresponding to the stretching vibrations of –OH and –NH functional groups in lyophilized HA, was retained in the spectra of both unloaded (Fig. S3B–F, ESI†) and hemin-loaded MPs (A and B formulations) (Fig. S3H and I, ESI†), though with reduced intensities in the latter cases. Additionally, peaks corresponding to alkyl groups (–CH) at 2980 cm−1 were detected in all microparticle spectra except for A-MPs. The remaining peaks in the HA spectrum were also observed in the spectra of all tested MPs. However, the conjugation of hemin to HA in the hemin-loaded MPs could not be confirmed using FTIR (Fig. S3H–L, ESI†).3.6. DPPH-radical scavenging efficiency
The efficiency of HA to scavenge DPPH radicals has been reported previously.34 Moreover, our study further revealed varying affinities of crosslinked HA hydrogels towards the scavenging of these radicals (data not shown). However, all MPs studied exhibited similar reducing effects on DPPH radical levels, with C-MPs demonstrating the highest, albeit not statistically significant, DPPH radical scavenging efficiency. Additionally, the hemin-loaded MPs did not show any significant differences in scavenging efficiency compared to their unloaded counterparts. An example for these results after one hour of incubation is shown in Fig. S4 (ESI†).3.7. Characterization of A-MPs
A-MPs, loaded with different concentrations of hemin were next fabricated, with their details shown in Table S2 (ESI†). It shows the calculations for the final amount of hemin loaded onto particles, prepared with different initial concentrations of hemin mixed with HA. The maximum entrapment efficiency was observed in MPs prepared with an initial hemin concentration of 480 μM, which was slightly higher than those with initial hemin concentration of 240 μM. Moreover, the lowest entrapment efficiency was seen at an initial hemin concentration of 2 μM. Collectively, these results confirm the stability of hemin conjugation to the HA hydrogel after particle synthesis and repeated washing.Both the synthesized unloaded and hemin-loaded MPs exhibited a needle-like morphology (Fig. 2A and D), with particle perimeters generally ranging from 0.3 to 3.5 μm. An example for these morphologies in case of unloaded, and MPs loaded with 0.01, 0.61 and 2.57 μmol hemin is shown in Fig. S5 (ESI†). These were referred to as A1, A5 and A7-MPs, respectively. Fig. 2B and E compare the perimeter distribution of the microneedles of both unloaded and hemin-loaded A7-MPs of A-formulation. 23% of the unloaded microneedles had perimeters in the range of 0.8–1 μm. In contrast, 25% and 22% of the A7-MPs microneedles had perimeters in the ranges of 0.5–0.8 μm and 0.8–1 μm, respectively. Therefore, hemin loading did not drastically change the average perimeter of the synthesized microparticles.
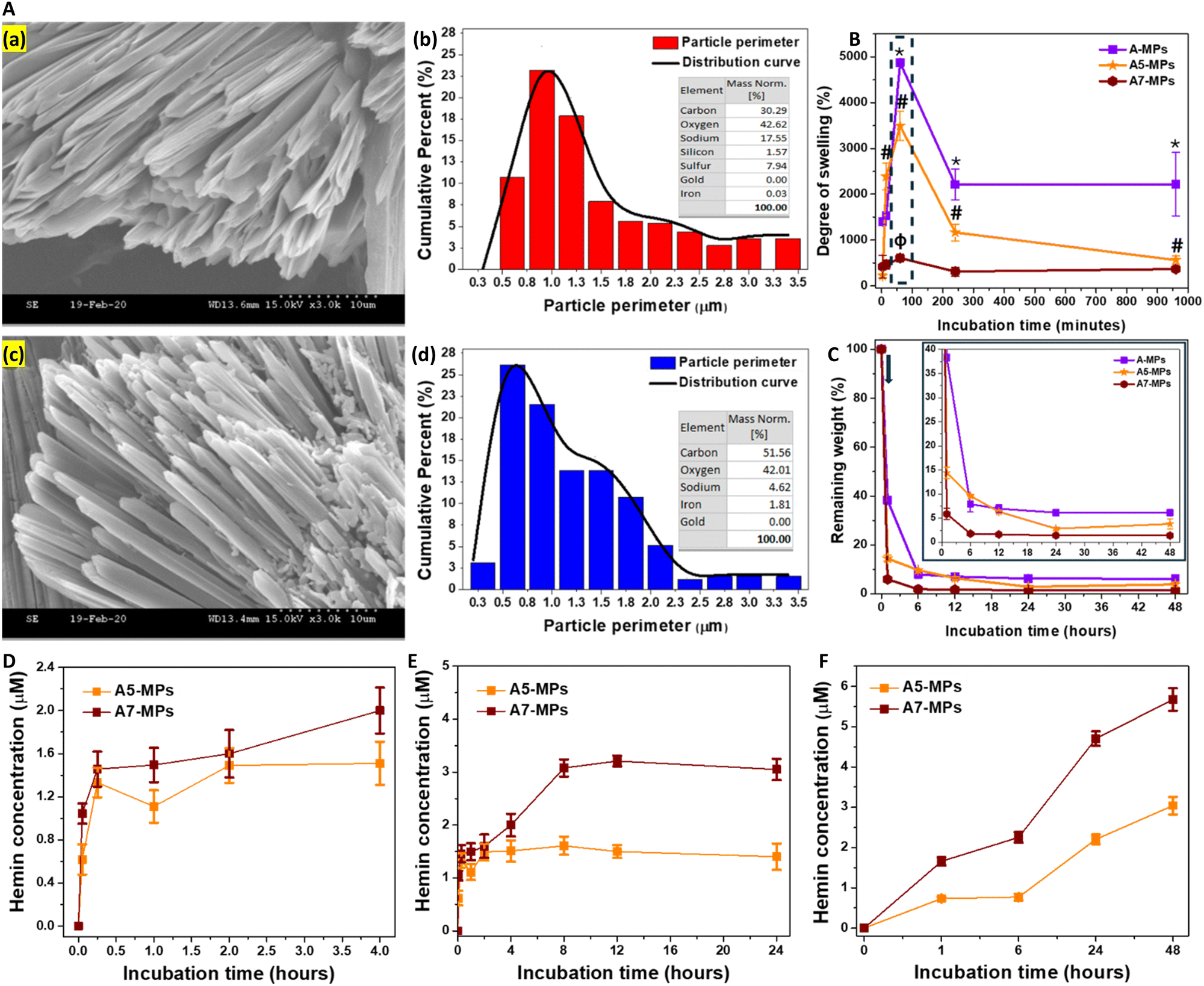 | ||
| Fig. 2 Characterization of A-MPs. (A) Morphology of MPs: (a) and (c) SEM images of unloaded and hemin-loaded MPs (A7), respectively. Scale bar: 10 μm, indicated by dotted line. (b) and (d) The calculated particle perimeter, represented as cumulative percentage, with the respective weight percentage of the major elements. (B) and (C) A comparison of the swelling kinetics and enzymatic degradation of the unloaded A-MPs, A5, and A7-MPs. The arrow in figure (C) indicates the time point of the treatment with HAase. (D) and (E) The concentration of hemin released following swelling of the MPs over the first four hours in PBS (D), and after 24 hours (E). (F) the concentration of hemin following degradation of the MPs. Results are presented as mean ± SD, n = 3. *,#,ϕp < 0.05 for the degree of swelling at each time point compared to the swelling after three minutes of mixing with PBS for A-MPs, A5, and A7-MPs, respectively using a two-tailed unpaired student's t-test. | ||
Additionally, the EDX diagram for unloaded particles showed only 0.03% detected iron, whereas the hemin-loaded MPs exhibited significantly higher levels, reaching 1.81% (Fig. 2C and F). These were comparable to the elements detected in case of the other hemin-loaded microparticle formulations (Fig. S6B, S7B, S8B and S9F, ESI†).
While the weight increase due to swelling was the highest in unloaded A-MPs, the swelling rate was relatively lower in hemin-loaded MPs. Specifically, a comparison of the swelling kinetics of unloaded MPs (A) and hemin-loaded MPs (A5 and A7) was performed. The maximum degree of swelling reached 4875 ± 81% for A-MPs, 3494 ± 319% for A5-MPs, and 604 ± 86% for A7-MPs, respectively (Fig. 2G). However, after one hour, the MPs began to decompose, leading to a drop in the swelling degree, particularly in unloaded and A5-MPs. Additionally, the higher the concentration of hemin loaded, the lower the degree of swelling.
HAase was utilized for the degradation of the MPs and testing hemin release following swelling for one hour. Like the results shown in Fig. 1C and D, the swollen masses decreased after one hour of incubation with the enzyme, and the hemin-loaded A-formulation MPs showed a higher rate of degradation than the unloaded MPs, with slight changes at later time points (Fig. 2H). For instance, while the unloaded A-MPs lost around 62% of their mass, the A5 and A7-MPs lost nearly 86% and 94% of their masses, respectively, after one hour of degradation. These findings may relate to possible enhancing effects of hemin on enzyme activity. However, studying these effects was beyond the scope of the current work. These fast biodegradation kinetics are suitable for the dermal application of these MPs and protection against the UV irradiation.
A slight release of hemin was detected in PBS during the first few hours of swelling of hemin-loaded MPs (Fig. 2D). However, prolonged incubation at 37 °C without hyaluronidase led to significant hemin release due to particle fragmentation (Fig. 2E), although at a lower rate compared to incubation with hyaluronidase (Fig. 2F). This release pattern mirrors that observed in previous studies with other drugs loaded into HA-based nano- and microparticle formulations. The PBS release pattern includes: (1) an initial phase within 15 minutes, associated with the release of physically adsorbed molecules within the hydrogel matrix or on the surface of particles, and (2) a later phase, after 15 minutes, characterized by controlled hemin release due to partial particle fragmentation. These phases have been detailed previously.66–68 However, the fragmentation was enhanced in the presence of hyaluronidase, which degrades HA and consequently facilitates greater hemin release.
3.8. Cell viability
While evaluating the biological response to particles and hydrogels is crucial for their biomedical applications,69,70 this study specifically aimed to determine whether our tested MPs could protect against the cytotoxic effects of UVC on the tested cells. The in vitro study started with testing the viability of dermal fibroblasts in response to UVC radiation for 10 and 60 seconds as well as the treatment with hemin or MPs. This was followed by evaluating the influence of UV radiation cell viability, treated initially with candidate concentrations of hemin, unloaded and hemin-loaded MPs belonging to the A-formulation. This was performed by measuring the metabolic activity and cell viability after certain periods of treatment. It should be noted that the UVC doses tested in this study were higher than those typically used in industry, representing scenarios of accidental exposure.Generally, HDFs exhibited reduced metabolic activity following 10 and 60 seconds of UV irradiation, with more pronounced effects observed after the longer exposure period (Fig. 3A). The corresponding cell viability results are shown in Fig. S10A and B (ESI†). Notably, hemin demonstrated a more substantial protective effect against UVC-induced cell damage following a 10-second treatment (Fig. 3B and Fig. S6C, F, I, L, ESI†). Moreover, exposure to hemin maintained the metabolic activity at three- and 24-hours post-irradiation, although a decline was noted after 48 hours of culture (Fig. 3B). Conversely, under the 60-second treatment, sustained metabolic activity was observed only after three-hours of culture, declining with extended culture periods. The effects of different concentrations of hemin on the metabolic activity and viability of cells without UV exposure are shown in Fig S11A and B (ESI†), respectively, while Fig. S12A–D (ESI†) show the corresponding calcein AM/EthD-1-stained cells. The cell viability decreased generally after 48 hours of culture due to the use of FBS-free medium. Hemin has been reported to exhibit antioxidant properties by inducing HOX expression.42 These effects have been shown to improve the viability of dermal fibroblasts and reduce fibroblast functional disorders.71 Further details on HOX-1 expression will be discussed in the next section. Additionally, real-time measurement of mitochondrial function in response to 24-hour hemin treatment revealed a significant increase in basal and maximal respiration, along with ATP production (Fig. S13, ESI†). The influence of hemin on these parameters and its correlation with HOX-1 expression has been explained in detail earlier.72,73 Collectively, these results explain the enhanced metabolic activity and maintained cell viability observed with hemin treatment for 24 hours at concentrations up to 8 μM. However, the decline in metabolic activity and viability after 48 hours is likely due to excessive hemin accumulation, nutrient depletion, and oxidative stress, as we previously described.72
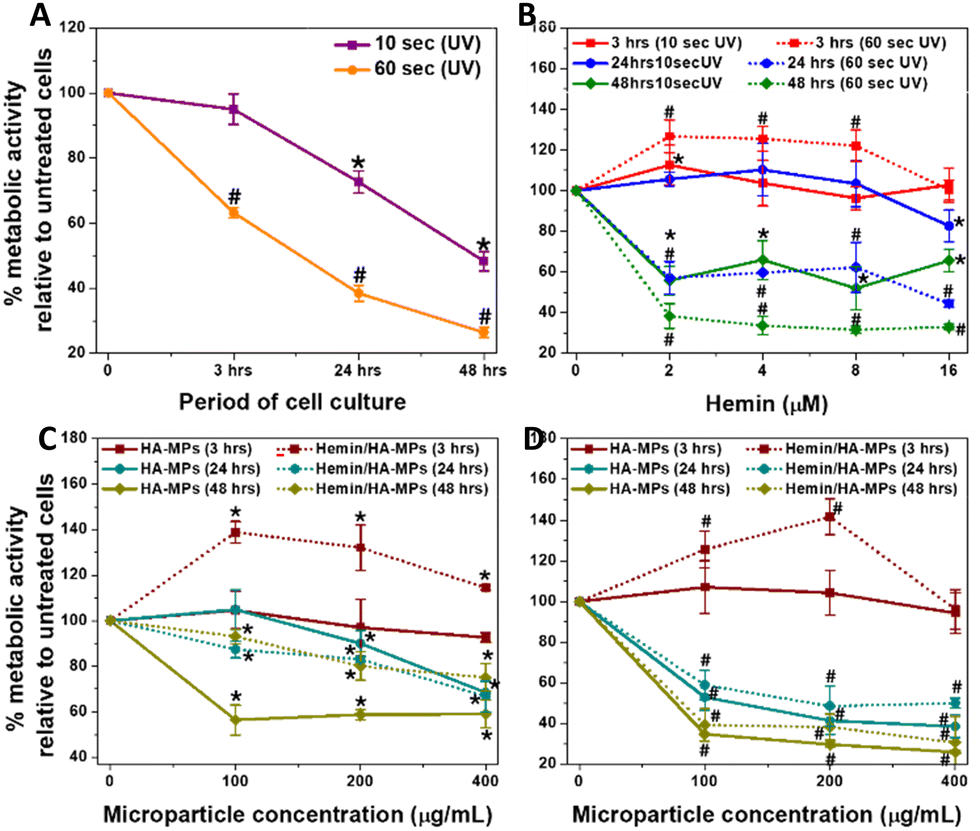 | ||
| Fig. 3 Metabolic activity of HDFs. (A) The influence of UVC irradiation for 10 and 60 seconds, followed by culturing for 3, 24, and 48 hours. (B) The influence of UVC irradiation on HDFs pre-treated with hemin, followed by culturing for 3, 24, and 48 hours. (C) and (D) The effects of UV C irradiation for 10 and 60 seconds, respectively, on HDFs pre-treated with both unloaded (HA) and hemin-loaded (Hemin/HA) MPs, followed by culturing for 3, 24, and 48 hours. Results are presented as mean values ± SD, n = 3. *,#, P < 0.05 for cells, treated for 10 and 60 seconds, respectively compared to the untreated cells, using a two-tailed unpaired student's t-test. | ||
When HDFs were treated with MPs, significant protective effects were observed after three hours of culture following both 10 and 60 seconds of irradiation (Fig. 3C and D). Furthermore, the metabolic activity notably increased, particularly with hemin-loaded MPs compared to the unloaded counterparts. However, a general decrease in the metabolic activity and cell viability (Fig. S10D, G, J, E, H and K, ESI†) were observed at longer periods of culture, with a severe drop in the metabolic activity (Fig. 3D) in case of 60 second-UV irradiation. Fig. S14A and B (ESI†) show the changes in cell viability for HDFs pre-treated with hemin and different MPs, then irradiated for 10 seconds before being cultured for 24 hours. While the changes in cell viability with hemin treatment were consistent with the metabolic activity measurements, there were differences with microparticle treatment, indicating that membrane integrity was maintained in response to MPs or their degradation products. As a result, the cell viability was preserved in MPs-treated cells, unlike metabolic activity results, which rely on a different mechanism to assess cell viability.59
The effects of different concentrations of MPs on the metabolic activity and viability of cells without UV exposure are shown in Fig S15A and B (ESI†), while Fig. S12E–J (ESI†) show the corresponding calcein AM/EthD-1-stained cells. It should be noted that cell treatment with MPs did not include hyaluronidase, so the hemin release patterns in the medium follow the kinetics shown in Fig. 2E and F. Moreover, measuring the enzyme secreted by cells was outside the scope of this study.
3.9. Hemin uptake and bilirubin generation
Intracellular hemin concentration, indicative of hemin uptake, was assessed following HDFs treatment, both in FBS-free and FBS-containing media. Notably, hemin concentration was higher in cultures treated in FBS-free medium compared to those with FBS, owing to hemin chelation by FBS, which reduces its availability to cells (Fig. 4A). These measured concentrations correlated with the diluted hemin concentrations in the media, a phenomenon previously observed in hemin-treated cancer cells.44 Consequently, all experiments involving primary HDFs treatment with hemin and MPs were conducted in FBS-free medium. Similarly, the treatment with hemin-loaded MPs led to elevated intracellular hemin levels, directly proportional to the particle concentration (Fig. 4B). Moreover, the unloaded MPs failed to induce any increase in intracellular hemin levels, displaying no difference from the untreated HDFs.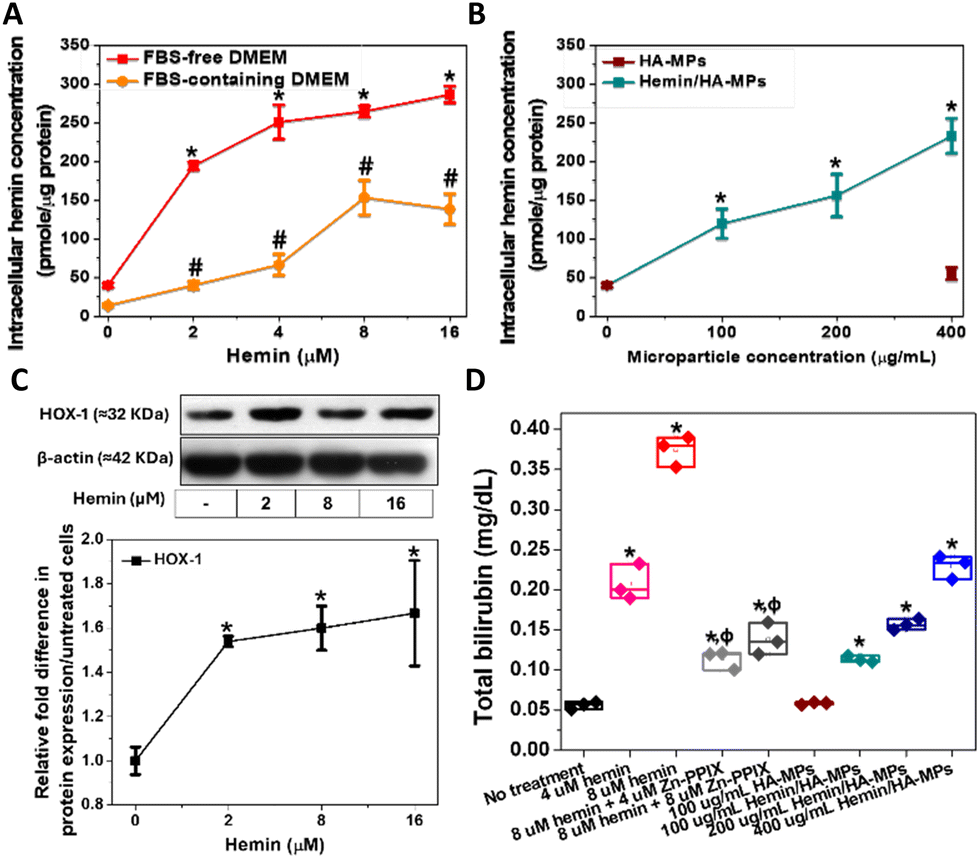 | ||
| Fig. 4 Concentration of cellular hemin isolated from HDFs after: (A) treatment with hemin in FBS-free and FBS-containing medium, and (B) treatment with hemin-loaded and unloaded MPs in FBS-free medium. (C) Immunoblots after cell treatment with different concentrations of hemin utilizing 5 μg proteins per well, with the relative quantification of HOX-1. (D) Bilirubin extracted from cells upon treatment with hemin with/without Zn-PPIX as well as HA- and Hemin/HA-MPs. Results are presented as mean values ± SD, n = 3. *,#, P < 0.05 for cells treated with hemin or MPs in FBS-free and FBS-containing medium, respectively compared to the untreated cells. ϕ, P < 0.05 for cells, treated with hemin/Zn-PPIX mixtures compared to the hemin alone-treated cells, using a two-tailed unpaired student's t-test. | ||
HOX catalyses the oxidative cleavage of the protoporphyrin IX ring of heme and hemin, yielding biliverdin, carbon monoxide (CO), and labile iron.74,75 Therefore, HOX-1 protein expression was measured following the treatment of HDFs with hemin. Increasing concentrations of hemin led to the upregulation of HOX-1 (Fig. 4C). Biliverdin is subsequently reduced to bilirubin, which accounts for the antioxidant activity of hemin. Therefore, measuring bilirubin serves as an indicator of heme oxygenase activation in HDFs and the degradation of hemin. The concentrations of extracted bilirubin were found to be proportional to the hemin concentration, confirming the hemin-mediated enzyme activation (Fig. 4C). These results also confirm the antioxidative properties of hemin, which are responsible for maintaining cell viability and metabolic activity.
Additionally, HDFs were treated with hemin in combination with Zn-PPIX, known as an inhibitor of HOX.76,77 This resulted in a significant reduction in generated bilirubin compared to cells treated with hemin alone, confirming the inducible effects of hemin on HOX. Additionally, similar results were observed in HDFs treated with hemin-loaded MPs, showing a significant increase in bilirubin concentration proportional to the loaded hemin concentration (Fig. 4C). No significant changes were noted in cells treated with unloaded MPs.
3.10. ˙NO & ROS detection
Imaging of the intracellular NO was performed using our previously outlined method using DAF-FM-DA.34 The probe, upon hydrolysis inside the cells by the intracellular esterases, produces DAF-FM, which becomes nitrosated in response to N2O3, resulting from the autoxidation of ˙NO,78 generating a highly fluorescent triazole derivative.79 Moreover, DAF-FM probe undergoes one-electron oxidation, producing the radical intermediate, DAF˙, which interacts directly with ˙NO giving the fluorescence triazole.80,81 The UV treatment of cells resulted in significant increased levels of intracellular ˙NO (Fig. 5A), accompanied with initial high fluorescence intensity after 30 minutes of UV-irradiation, which dropped gradually, reaching nearly steady levels after six hours. These levels were generally higher than those of untreated cells. The comparison of the fluorescence intensity in response to UV radiation, with/without cell treatment with DAF-FM confirms its sensitivity to the ˙NO generated intracellularly (Fig. S16A, ESI†). Two main mechanisms have been proposed to explain UV-induced nitrosative stress, characterized by the excessive production of ˙NO and its toxic congeners. The first mechanism involves the increased expression of inducible nitric oxide synthase in the skin, occurring within 8 hours of UV exposure.19 The second mechanism is non-enzymatic and involves the direct release of ˙NO and oxygen radicals because of UV-induced nitrite decomposition, which ultimately produces the toxic ˙NO2 radicals.17 However, this decomposition has been studied only in response to UVA exposure at 365 nm. The levels of intracellular ˙NO in response to UV radiation were reported to correlate with the radiation dose,18 but with general higher levels in response to UVA, than UVB radiation. Hence, our results complement these findings, focusing on the effects of UVC radiation.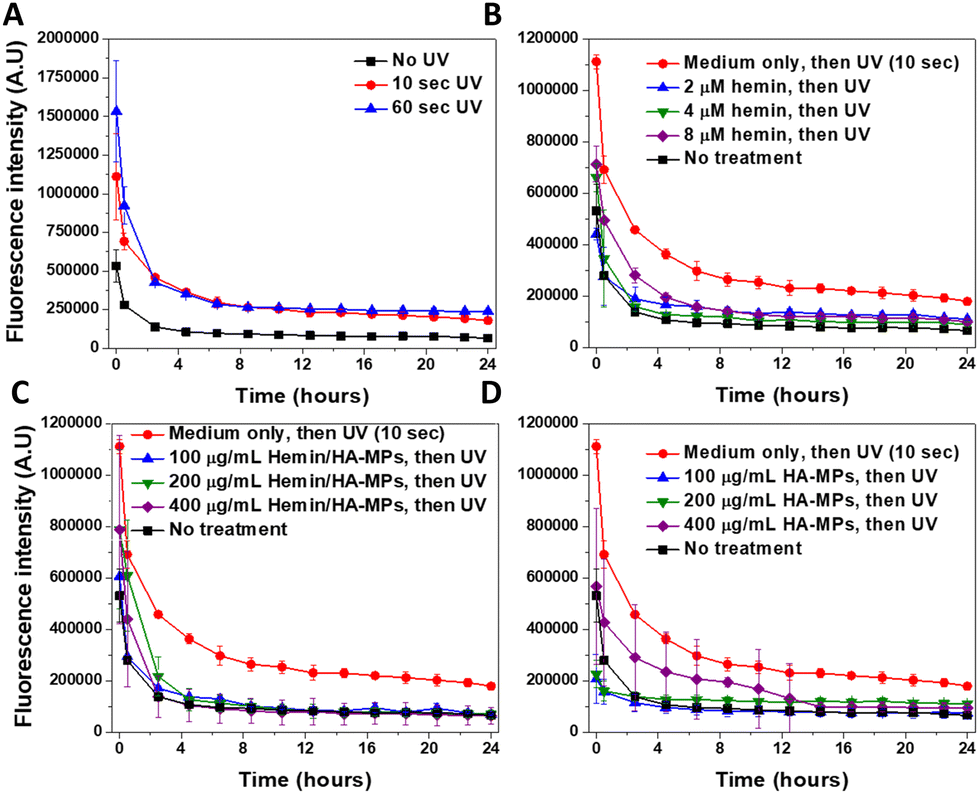 | ||
| Fig. 5 Kinetics of changes in intracellular ˙NO levels in HDFs as revealed by the ˙NO-specific indicator DAF-FM. (A) Fluorescence levels following cell treatment with UVC radiation for 10 and 60 seconds. (B)–(D) Fluorescence levels following pre-treatment with different concentrations of hemin, A-MPs, or A7-MPs, respectively for 24 hours, followed by UVC treatment for 10 seconds. Generally, after different treatments, cells were incubated with DAF-FM-DA for 1 hour before UVC exposure and subsequently imaged using the real-time Incucyte® imaging system. Results are presented as mean fluorescence intensity ± SD, n = 3. | ||
The prior treatment with hemin reduced the elevated levels of ˙NO in case of 10 (Fig. 5B) and 60 seconds of UV radiation (Fig. S17A, ESI†), with more significant effects observed under the former treatment. These effects relate to the ˙NO-scavenging effects of hemin, as we reported before,44,73 referring to the potential role of hemin in protection against the nitrosative stress, induced by the UV treatment of cells. Similar effects were observed in HDFs, with prior treatment with the hemin/HA-MPs (Fig. 5C), which showed higher potential in reducing the levels of ˙NO, compared to that of unloaded HA-MPs (Fig. 5D). These findings confirm the potency of released hemin molecules to maintain their binding affinity towards ˙NO, besides the photoprotective roles of HA36 and its ˙NO-binding efficiency.34 However, after UV-treatment for 60 seconds, these protecting effects were lower in case of the hemin/HA-MPs (Fig. S17B, ESI†), which relates to the excessive production of ˙NO, which cannot be tolerated by the released hemin. However, the unloaded MPs maintained the affinity towards ˙NO (Fig. S17C, ESI†), which can relate to partial depolymerization of HA.82 Of a note, we found that the fragmentation of HA enhances its ˙NO-binding affinity (data not shown).
CellROX® Deep Red Reagent was used for general detection of the intracellular ROS, without targeting specific type of these reactive species. Like its effects on ˙NO, UV radiation caused a dose-dependent increase in the generated ROS, whose levels gradually dropped over time (Fig. S18A, ESI†). This phenomenon has been extensively discussed.83–85 The comparison of the fluorescence intensity in response to UV radiation, with/without cell treatment with CellROX® reagent confirms its sensitivity to the ROS generated intracellularly (Fig. S16B, ESI†).
The effects of hemin and hemin/HA-MPs on the levels of intracellular ROS cannot be studied under the current experimental conditions, due to the affinity of hemin molecules to be oxidized intracellularly. This induces cycles of oxidation/reduction reactions, leading to generation of various radicals, particularly hydroperoxyl and hydroxyl radicals via Haber–Weiss reaction.86,87 These reactions lead to enhanced generation of ROS in case of both 10 (Fig. S18B and C, ESI†) and 60 seconds of UV treatment (Fig. S19A and B, ESI†) in response to hemin and hemin/HA-MPs, respectively. Fig. S20 (ESI†) shows the increased ROS levels, due to different concentrations of hemin without UV application, which confirm these reactions. However, the unloaded HA-MPs showed protection against the UV-induced ROS generation with lower fluorescence intensities in case of both 10 (Fig. S18D, ESI†) and 60 seconds of UV treatment (Fig. S19C, ESI†). These effects relate to the antioxidant activity of HA and ability to scavenge different ROS.34,36 However, this activity could not reverse the intracellular redox reactions in response to hemin.
3.11. Protein carbonylation
Protein carbonylation is a marker of oxidative stress induced by UV radiation. The excessive production of reactive oxygen species (ROS) leads directly to the oxidative modification of various proteins, commonly occurring at the side chains of arginine, lysine, threonine, and proline residues.88 Moreover, protein carbonylation can occur indirectly through the Michael addition of lipid peroxidation products as well as the formation of advanced glycation end products.89 These modifications result in alterations/abnormalities in protein functions, impaired clearance, and potential cancer progression.90–92 In this study, total protein carbonylation in HDFs was measured following exposure to UV radiation for 10 and 60 seconds, with/without prior exposure to hemin, HA, or hemin-loaded MPs. The testing of carbonylation was performed after three hours of UV treatment due to the reported decline in protein carbonylation within four hours of irradiation.22 Moreover, the conjugation of extracted proteins to 2,4-dinitrophenylhydrazone (DNPH) was performed immediately after cell homogenization and protein quantification to prevent the formation of Schiff bases between carbonyl groups and lysine residues on proteins during storage.89 The carbonyl levels were proportional to the duration of UV exposure (Fig. S21, ESI†), with distinct effects on proteins of different molecular weights, especially following treatment with hemin or the tested MPs. For instance, hemin did not significantly alter carbonylation levels in the 60 kDa protein band in the absence of UV radiation or after 10 seconds of irradiation (Fig. S21A, B and D, ESI†). However, it significantly protected against carbonylation after 60 seconds of UV treatment. Significant inhibitory effects of hemin on protein carbonylation were observed at the 40 kDa protein band (Fig. S21E, ESI†). For comparison, the negative controls without derivatization with DNPH is shown in (Fig. S21C, ESI†). It should be mentioned here that the blots shown were developed after 10 seconds (Fig. S21A, ESI†) and 30 minutes (Fig. S21C, ESI†) of exposure. Hemin has been reported as a protective compound against UVB-induced skin damage, inhibiting the expression of inflammation-associated cytokines in UVB-treated keratinocytes, such as IL-1α, IL-1β, IL-6, IL-8, and TNF-α.41 Despite these effects, hemin did not provide complete protection against UV radiation, as overall protein carbonyl levels were still higher compared to untreated cells.When cells were pre-treated with microparticles, the levels of protein carbonyls at both studied molecular weight bands generally decreased in the absence of UV irradiation (Fig. 6A–C). The reduction was more significant with hemin/HA-MPs compared to HA-MPs alone (Fig. 6D and E), exhibiting similar protecting effects against the 10-second UV-induced carbonylation. HA-MPs restored protein carbonyl levels to those seen in untreated cells, while hemin/HA-MPs led to even lower carbonyl levels. The protective effects of HA against UVB radiation for keratinocytes and its modulatory role in cell response have been reported previously.35,36 These effects are attributed to HA's ability to suppress the UVB-induced secretion of pro-inflammatory cytokines (IL-6, IL-8, TGF-β) and reduce cell viability,35 as well as its radical-scavenging capacity.36 These findings support the observed protective roles of the unloaded MPs against UV-induced protein oxidation. However, UVC irradiation was used in this study instead of UVB. Collectively, these results highlight the complementary roles of both HA and loaded hemin in protecting against UVC-induced protein oxidation.
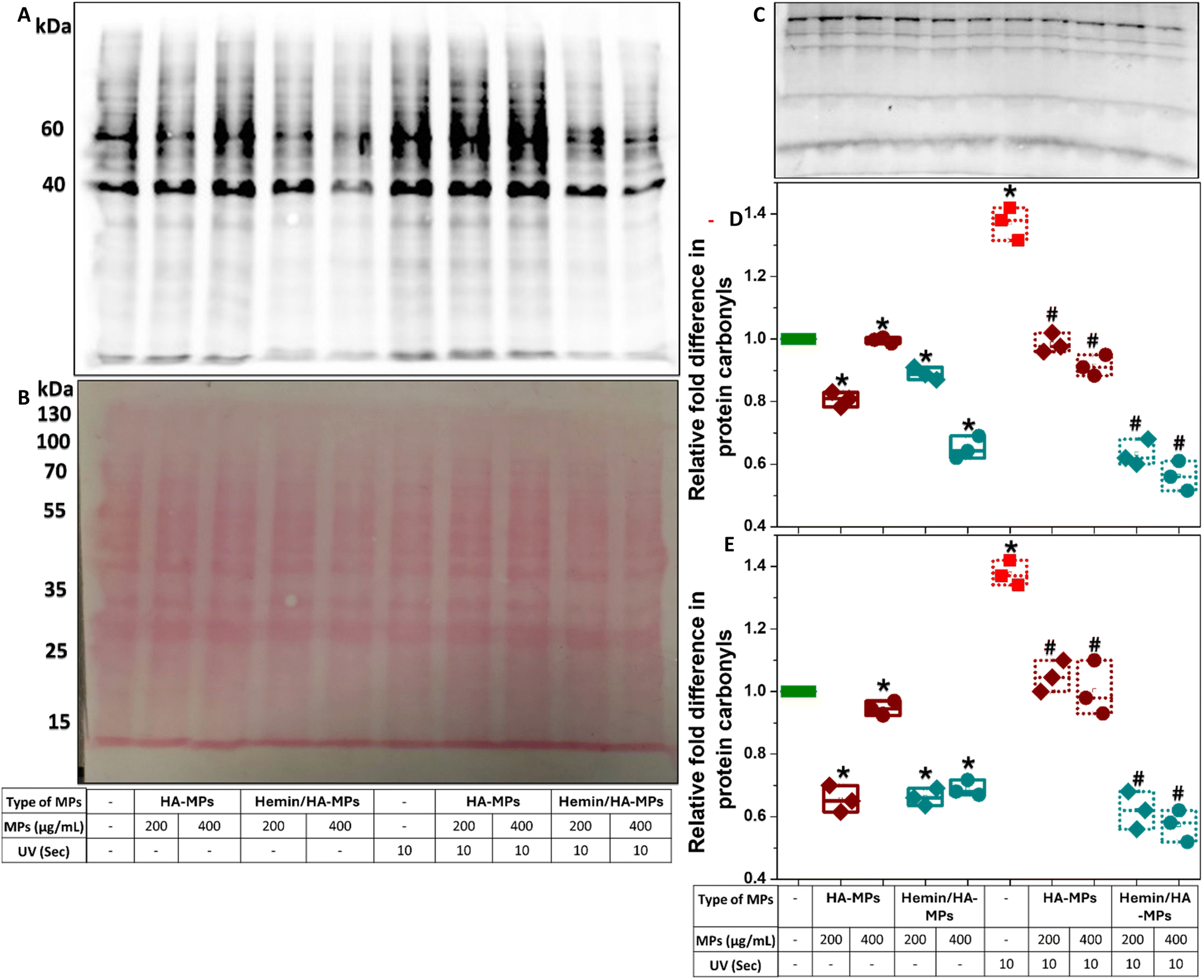 | ||
| Fig. 6 Protein carbonylation following HDFs treatment with HA-MPs and hemin/HA-MPs and UV irradiation. (A) Representative oxyblot of total cell proteins (DNPH) after cell treatment with 200 and 400 μg mL−1 HA-MPs and hemin/HA-MPs without/with subsequent UV treatment for 10 seconds, with the corresponding membranes stained with Ponceau S (B). (C) The blot of negative controls. (D) and (E) Relative quantification of protein carbonyl levels at the detected MWs of 60 and 40 kDa, respectively, obtained from triplicate samples, measured by scanning densitometry, and normalized to the corresponding densities from the Ponceau S-stained membranes. Following MPs treatment for 24 hours, the cells were cultured in FBS-containing medium for three hours, with/without prior treatment with UV for 10 seconds, followed by extraction of the intracellular proteins. After quantification using BCA assay, the proteins were derivatized to DNPH, subjected to SDS-PAGE using 12% polyacrylamide gel, followed by membrane transfer, staining with Ponceau S, and probing with primary antibody, specific to the DNP moiety of the proteins. Results are presented as mean values ± SD, n = 3. *P < 0.05 compared to untreated cells. #P < 0.05 compared to UV-only treated cells, using a two-tailed unpaired student's t-test. | ||
In case of 60 second UV-induced carbonylation, the protecting roles of the unloaded MPs were more significant than those of hemin/HA-MPs (Fig. S22A–D, ESI†). These results confirm the higher efficiency of unloaded MPs in reducing ROS levels compared to hemin/HA-MPs as presented in (Fig. S18 and S19, ESI†). Hemin may interfere with protein carbonylation and contribute to ROS generation, particularly under 60 seconds of UV exposure. However, studying these effects was beyond the scope of the current work.
4. Conclusions
The development of hydrogels as microscale particles has garnered attention to overcome limitations associated with traditional macroscale structures. In this study, we reported the creation of HA needle-like MPs loaded with hemin and examined how their physicochemical properties can be modulated. Given the widespread use of UVC radiation across various industries and the need for protective measures against accidental exposure, we evaluated the protective roles of these hemin-loaded MPs against UVC-induced oxidative stress and cell death in human dermal fibroblasts, as an application strategy of the developed MPs. The degree of crosslinking, influenced by the ratio of crosslinker to DMTMM, determined the final properties of the MPs and the rate of hemin release. The formulations exhibiting the highest degree of swelling were selected for in vitro testing. Rapid swelling facilitated the degradation of MPs and the release of hemin, which was evidenced by increased intracellular hemin levels and bilirubin production. Pre-treatment with hemin and the candidate MPs effectively inhibited the cytotoxic effects of UVC radiation during a 10-second exposure of HDFs. This offered protection against UVC-induced nitrosative stress, as indicated by reduced intracellular ˙NO levels, and against oxidative stress, as indicated by reduced protein carbonylation. However, under the current experimental conditions, the actual effect of hemin and hemin-loaded MPs on intracellular ROS levels could not be distinguished, despite the ROS quenching effects of the unloaded MPs. These findings suggest that the studied MPs have the potential to release hemin and mitigate the harmful effects of UVC on dermal fibroblasts. For further applications, particularly when localized drug release is required, incorporating these MPs into a secondary hydrogel would be essential to achieve more controlled and localized drug delivery through a two-phase approach.Author contributions
A. M. Alsharabasy: conducting experiments, collecting data, administration, analysis and writing original draft. A. Aljaabary: assisting with experimental operations and data curation. Pau Farràs: administration, data visualization and revising manuscript. A. Pandit: project administration, funding acquisition, data verification, and revising manuscript. All authors have read and agreed to the submitted version of the manuscript.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
The research presented in this publication was jointly funded by a research grant from Science Foundation Ireland (SFI), co-funded under the European Regional Development Fund (Grant number 13/RC/2073_P2), the Irish Research Council (Grant number GOIPD/2023/1640), and the College of Engineering and Informatics Scholarship Scheme at the University of Galway, Ireland. The authors acknowledge the facilities of the Anatomy Imaging and Microscopy Facility at the University of Galway and the technical assistance of Dr Éadaoin Timmins (https://imaging.universityofgalway.ie/imaging/).References
- M. Guvendiren, H. D. Lu and J. A. Burdick, Soft Matter, 2012, 8, 260–272 RSC.
- R. Dimatteo, N. J. Darling and T. Segura, Adv. Drug Delivery Rev., 2018, 127, 167–184 CrossRef CAS PubMed.
- A. C. Daly, L. Riley, T. Segura and J. A. Burdick, Nat. Rev. Mater., 2019, 5, 20–43 CrossRef PubMed.
- J. Elisseeff, Nat. Mater., 2008, 7, 271–273 CrossRef CAS PubMed.
- J. E. Mealy, J. J. Chung, H. Jeong, D. Issadore, D. Lee, P. Atluri and J. A. Burdick, Adv. Mater., 2018, 30, 1705912 CrossRef PubMed.
- L. Riley, L. Schirmer and T. Segura, Curr. Opin. Biotechnol, 2019, 60, 1–8 CrossRef CAS PubMed.
- M. Berneburg, H. Plettenberg and J. Krutmann, Photodermatol. Photoimmunol. Photomed., 2000, 16, 239–244 CrossRef CAS PubMed.
- R. Pandel, B. Poljšak, A. Godic and R. Dahmane, ISRN Dermatol., 2013, 1–11 Search PubMed.
- L. B. Alexandrov, S. Nik-Zainal, D. C. Wedge, S. A. J. R. Aparicio, S. Behjati, A. V. Biankin, G. R. Bignell, N. Bolli, A. Borg, A.-L. Børresen-Dale, S. Boyault, B. Burkhardt, A. P. Butler, C. Caldas, H. R. Davies, C. Desmedt, R. Eils, J. E. Eyfjörd, J. A. Foekens, M. Greaves, F. Hosoda, B. Hutter, T. Ilicic, S. Imbeaud, M. Imielinski, N. Jäger, D. T. W. Jones, D. Jones, S. Knappskog, M. Kool, S. R. Lakhani, C. López-Otín, S. Martin, N. C. Munshi, H. Nakamura, P. A. Northcott, M. Pajic, E. Papaemmanuil, A. Paradiso, J. V. Pearson, X. S. Puente, K. Raine, M. Ramakrishna, A. L. Richardson, J. Richter, P. Rosenstiel, M. Schlesner, T. N. Schumacher, P. N. Span, J. W. Teague, Y. Totoki, A. N. J. Tutt, R. Valdés-Mas, M. M. van Buuren, L. van’t Veer, A. Vincent-Salomon, N. Waddell, L. R. Yates, J. Zucman-Rossi, P. Andrew Futreal, U. McDermott, P. Lichter, M. Meyerson, S. M. Grimmond, R. Siebert, E. Campo, T. Shibata, S. M. Pfister, P. J. Campbell and M. R. Stratton, Nature, 2013, 500, 415–421 CrossRef CAS PubMed.
- J. Cadet, T. Douki and J. Ravanat, Photochem. Photobiol., 2015, 91, 140–155 CrossRef CAS PubMed.
- M. Norval, A. P. Cullen, F. R. de Gruijl, J. Longstreth, Y. Takizawa, R. M. Lucas, F. P. Noonan and J. C. van der Leun, Photochem. Photobiol. Sci., 2007, 6, 232–251 CrossRef CAS PubMed.
- P. Elsner and S. Hassam, Contact Dermatitis, 1996, 35, 180–181 CrossRef CAS PubMed.
- A. Dixon, Aust. Fam. Physician, 2007, 36, 255–256 Search PubMed.
- SCHEER, 2016, 1–43.
- C. Oresajo, M. Yatskayer, A. Galdi, P. Foltis and S. Pillai, J. Cosmet. Laser Ther., 2010, 12, 157–162 CrossRef PubMed.
- C. S. Sander, H. Chang, S. Salzmann, C. S. L. Müller, S. Ekanayake-Mudiyanselage, P. Elsner and J. J. Thiele, J. Invest. Dermatol., 2002, 118, 618–625 CrossRef CAS PubMed.
- C. Opländer, M. M. Cortese, H.-G. Korth, M. Kirsch, C. Mahotka, W. Wetzel, N. Pallua and C. V. Suschek, Free Radic. Biol. Med., 2007, 43, 818–829 CrossRef PubMed.
- G. Holliman, D. Lowe, H. Cohen, S. Felton and K. Raj, Sci. Rep., 2017, 7, 11105 CrossRef PubMed.
- M. Mowbray, S. McLintock, R. Weerakoon, N. Lomatschinsky, S. Jones, A. G. Rossi and R. B. Weller, J. Invest. Dermatol., 2009, 129, 834–842 CrossRef CAS PubMed.
- P. M. Farr, J. E. Besag and B. L. Diffey, J. Invest. Dermatol., 1988, 91, 454–457 CrossRef CAS PubMed.
- B. L. Diffey and P. M. Farr, Clin. Phys. Physiol. Meas., 1991, 12, 311–325 CrossRef CAS PubMed.
- R. Brem, P. Macpherson, M. Guven and P. Karran, Sci. Rep., 2017, 7, 4310 CrossRef PubMed.
- T. C. Laurent and J. R. E. Fraser, FASEB J., 1992, 6, 2397–2404 CrossRef CAS PubMed.
- M. Dovedytis, Z. J. Liu and S. Bartlett, Eng. Regen, 2020, 1, 102–113 Search PubMed.
- I. L. Mohd Isa, S. A. Abbah, M. Kilcoyne, D. Sakai, P. Dockery, D. P. Finn and A. Pandit, Sci. Adv., 2018, 4, eaaq0597 CrossRef PubMed.
- N. G. Kotla, S. R. Bonam, S. Rasala, J. Wankar, R. A. Bohara, J. Bayry, Y. Rochev and A. Pandit, J. Controlled Release, 2021, 336, 598–620 CrossRef CAS PubMed.
- A. F. Jeffery, M. A. Churchward, V. K. Mushahwar, K. G. Todd and A. L. Elias, Biomacromolecules, 2014, 15, 2157–2165 CrossRef CAS PubMed.
- B. J. Engel, P. E. Constantinou, L. K. Sablatura, N. J. Doty, D. D. Carson, M. C. Farach-Carson, D. A. Harrington and T. I. Zarembinski, Adv. Healthcare Mater., 2015, 4, 1664–1674 CrossRef CAS PubMed.
- A. Khademhosseini, G. Eng, J. Yeh, J. Fukuda, J. Blumling, R. Langer and J. A. Burdick, J. Biomed. Mater. Res., Part A, 2006, 79A, 522–532 CrossRef CAS PubMed.
- X.-H. Qin, P. Gruber, M. Markovic, B. Plochberger, E. Klotzsch, J. Stampfl, A. Ovsianikov and R. Liska, Polym. Chem., 2014, 5, 6523–6533 RSC.
- J. Chen, K. Huang, Q. Chen, C. Deng, J. Zhang and Z. Zhong, ACS Appl. Mater. Interfaces, 2018, 10, 3929–3937 CrossRef CAS PubMed.
- D. Nikjoo, I. van der Zwaan, M. Brülls, U. Tehler and G. Frenning, Pharmaceutics, 2021, 13, 1878 CrossRef CAS PubMed.
- C. Ke, L. Sun, D. Qiao, D. Wang and X. Zeng, Food Chem. Toxicol., 2011, 49, 2670–2675 CrossRef CAS PubMed.
- A. M. Alsharabasy, S. Glynn, P. Farràs and A. Pandit, Biomacromolecules, 2022, 23, 3621–3647 CrossRef CAS PubMed.
- M. Hašová, T. Crhák, B. Šafránková, J. Dvořáková, T. Muthný, V. Velebný and L. Kubala, Arch. Dermatol. Res., 2011, 303, 277–284 CrossRef PubMed.
- A. E. Shaheen, H. M. Gebreel, L. A. Moussa, A. E. Zakaria and W. A. Nemr, Curr. Microbiol., 2023, 80, 262 CrossRef CAS PubMed.
- E. Tomat, Comments Inorg. Chem., 2016, 36, 327–342 CrossRef CAS.
- W.-Y. Chung, J.-M. Lee, W.-Y. Lee, Y.-J. Surh and K.-K. Park, Mutat. Res. Toxicol. Environ. Mutagen., 2000, 472, 139–145 CrossRef CAS PubMed.
- J. H. Park, C. K. Lee, Y. S. Hwang, K.-K. Park and W.-Y. Chung, Mutat. Res. Mol. Mech. Mutagen., 2008, 642, 68–73 CrossRef CAS PubMed.
- S. Fotiou, D. Fotiou and G. Deliconstantinos, In Vivo, 2009, 23, 281–286 CAS.
- F. Bai, C. Fan, X. Lin, H. Y. Wang, B. Wu, C. L. Feng, R. Zhou, Y. W. Wu and W. Tang, J. Photochem. Photobiol., B, 2023, 238, 112604 CrossRef CAS PubMed.
- Q. Li, R. Guo, K. Zhao, D. Lin, X. Ye and L. Chen, J. Wound Care, 2018, 27, 780–789 CrossRef PubMed.
- C. E. Cooper, Biochim. Biophys. Acta, Bioenerg., 1999, 1411, 290–309 CrossRef CAS PubMed.
- A. M. Alsharabasy, S. Glynn, P. Farràs and A. Pandit, Nitric Oxide, 2022, 124, 49–67 CrossRef CAS PubMed.
- K. V. Krishna, A. Benito, J. Alkorta, C. Gleyzes, D. Dupin, I. Loinaz and A. Pandit, Nano Sel., 2020, 1, 353–371 CrossRef.
- Y. Shi, A. Wan, Y. Shi, Y. Zhang and Y. Chen, Biomed Res. Int., 2014, 1–8 Search PubMed.
- A. M. Alsharabasy, M. Sankar, M. Biggs, P. Farràs and A. Pandit, Talanta, 2024, 126522 CrossRef CAS PubMed.
- N. Doshi, B. Prabhakarpandian, A. Rea-Ramsey, K. Pant, S. Sundaram and S. Mitragotri, J. Controlled Release, 2010, 146, 196–200 CrossRef CAS PubMed.
- M. Howard, B. J. Zern, A. C. Anselmo, V. V. Shuvaev, S. Mitragotri and V. Muzykantov, ACS Nano, 2014, 8, 4100–4132 CrossRef CAS PubMed.
- T. Bitter and H. M. Muir, Anal. Biochem., 1962, 4, 330–334 CrossRef CAS PubMed.
- V. Selvamani, Characterization and Biology of Nanomaterials for Drug Delivery, Elsevier, 2019, pp. 425–444 Search PubMed.
- Y. R. Choi, H.-J. Kim, G. Y. Ahn, M. J. Lee, J. R. Park, D.-R. Jun, T.-K. Ryu, J. W. Park, E. Shin and S.-W. Choi, Colloids Surf., B, 2018, 171, 690–697 CrossRef CAS PubMed.
- R. Pecora, J. Nanopart. Res., 2000, 2, 123–131 CrossRef CAS.
- M. Hassellöv, J. W. Readman, J. F. Ranville and K. Tiede, Ecotoxicology, 2008, 17, 344–361 CrossRef PubMed.
- M. Lotya, A. Rakovich, J. F. Donegan and J. N. Coleman, Nanotechnology, 2013, 24, 265703 CrossRef PubMed.
- Y. Boluk and C. Danumah, J. Nanopart. Res., 2014, 16, 2174 CrossRef.
- M. Zhang, S. A. Bradford, J. Šimůnek, H. Vereecken and E. Klumpp, Water Res., 2017, 109, 358–366 CrossRef CAS PubMed.
- R. Geng, R. Chang, Q. Zou, G. Shen, T. Jiao and X. Yan, Small, 2021, 17, 2008114 CrossRef CAS PubMed.
- S. Martin-Saldaña, M. Al Waeel, A. M. Alsharabasy, A. Daly and A. Pandit, Matter, 2022, 5, 3659–3705 CrossRef.
- R. F. Donnelly, D. I. J. Morrow, M. T. C. McCrudden, A. Z. Alkilani, E. M. Vicente-Pérez, C. O’Mahony, P. González-Vázquez, P. A. McCarron and A. D. Woolfson, Photochem. Photobiol., 2014, 90, 641–647 CrossRef CAS PubMed.
- A. Albersdörfer and E. Sackmann, Eur. Phys. J. B, 1999, 10, 663–672 CrossRef.
- Y. Zhao, B. Yi, J. Hu, D. Zhang, G. Li, Y. Lu and Q. Zhou, Adv. Funct. Mater., 2023, 33, 2300710 CrossRef CAS.
- G. Golomb, P. Fisher and E. Rahamim, J. Controlled Release, 1990, 12, 121–132 CrossRef CAS.
- M. Soundaranathan, P. Vivattanaseth, E. Walsh, K. Pitt, B. Johnston and D. Markl, Int. J. Pharm., 2020, 590, 119903 CrossRef CAS PubMed.
- J. Schulte, T. Pütz and R. Gebhardt, Food Hydrocolloids Health, 2021, 1, 100014 CrossRef CAS.
- S. T. Lim, G. P. Martin, D. J. Berry and M. B. Brown, J. Controlled Release, 2000, 66, 281–292 CrossRef CAS PubMed.
- A. M. Al-Ghananeem, A. H. Malkawi, Y. M. Muammer, J. M. Balko, E. P. Black, W. Mourad and E. Romond, AAPS PharmSciTech, 2009, 10, 410–417 CrossRef CAS PubMed.
- O. Galvin, A. Srivastava, O. Carroll, R. Kulkarni, S. Dykes, S. Vickers, K. Dickinson, A. L. Reynolds, C. Kilty, G. Redmond, R. Jones, S. Cheetham, A. Pandit and B. N. Kennedy, J. Controlled Release, 2016, 233, 198–207 CrossRef CAS PubMed.
- D. F. Williams, Biomaterials, 2008, 29, 2941–2953 CrossRef CAS PubMed.
- International Organization for Standardization, Biological evaluation of medical devices (ISO Standard No. 10993-1:2018), 2018, https://www.iso.org/standard/68936.html.
- Q. Li, S. Liang, Q. Lai, L. Shen, Y. Zhang and R. Guo, Exp. Ther. Med., 2021, 22, 1212 CrossRef CAS PubMed.
- A. M. Alsharabasy, P. I. Lagarias, K. D. Papavasileiou, A. Afantitis, P. Farràs, S. Glynn and A. Pandit, J. Med. Chem., 2024 DOI:10.1021/acs.jmedchem.4c00989.
- A. M. Alsharabasy, A. Aljaabary, R. Bohara, P. Farràs, S. A. Glynn and A. Pandit, ACS Pharmacol. Transl. Sci., 2023, 6, 1416–1432 CrossRef CAS PubMed.
- R. Tenhunen, H. S. Marver and R. Schmid, Proc. Natl. Acad. Sci. U. S. A., 1968, 61, 748–755 CrossRef CAS PubMed.
- R. Larsen, Z. Gouveia, M. P. Soares and R. Gozzelino, Front. Pharmacol., 2012, 3, 77 Search PubMed.
- D. Nowis, M. Bugajski, M. Winiarska, J. Bil, A. Szokalska, P. Salwa, T. Issat, H. Was, A. Jozkowicz, J. Dulak, T. Stoklosa and J. Golab, BMC Cancer, 2008, 8, 197 CrossRef PubMed.
- C. Zhou, J. Zhou, F. Sheng, H. Zhu, X. Deng, B. Xia and J. Lin, Acta Biochim. Biophys. Sin., 2012, 44, 815–822 CrossRef CAS PubMed.
- H. Kojima, N. Nakatsubo, K. Kikuchi, S. Kawahara, Y. Kirino, H. Nagoshi, Y. Hirata and T. Nagano, Anal. Chem., 1998, 70, 2446–2453 CrossRef CAS PubMed.
- M. M. Tarpey, D. A. Wink and M. B. Grisham, Am. J. Physiol. Integr. Comp. Physiol., 2004, 286, R431–R444 CrossRef CAS PubMed.
- M. M. Cortese-Krott, A. Rodriguez-Mateos, G. G. C. Kuhnle, G. Brown, M. Feelisch and M. Kelm, Free Radic. Biol. Med., 2012, 53, 2146–2158 CrossRef CAS PubMed.
- J. Li, A. LoBue, S. K. Heuser, F. Leo and M. M. Cortese-Krott, Nitric Oxide, 2021, 115, 44–54 CrossRef CAS PubMed.
- E. Hvidberg, S. A. Kvorning, A. Schmidt and J. Schou, Acta Pharmacol. Toxicol., 1959, 15, 356–364 CrossRef CAS PubMed.
- J.-L. Ravanat, T. Douki and J. Cadet, J. Photochem. Photobiol., B, 2001, 63, 88–102 CrossRef CAS PubMed.
- J. Cadet, E. Sage and T. Douki, Mutat. Res. Mol. Mech. Mutagen., 2005, 571, 3–17 CrossRef CAS PubMed.
- T. L. de Jager, A. E. Cockrell and S. S. Du Plessis, Adv. Exp. Med. Biol., 2017, 996, 15–23 CrossRef CAS PubMed.
- A. Bhattacharyya, R. Chattopadhyay, S. Mitra and S. E. Crowe, Physiol. Rev., 2014, 94, 329–354 CrossRef CAS PubMed.
- P. Kord Forooshani, R. Pinnaratip, E. Polega, A. G. Tyo, E. Pearson, B. Liu, T. O. Folayan, L. Pan, R. M. Rajachar, C. L. Heldt and B. P. Lee, Chem. Mater., 2020, 32, 8182–8194 CrossRef CAS PubMed.
- E. R. Stadtman and R. L. Levine, Ann. N. Y. Acad. Sci., 2000, 899, 191–208 CrossRef CAS PubMed.
- A. G. Madian and F. E. Regnier, J. Proteome Res., 2010, 9, 3766–3780 CrossRef CAS PubMed.
- A. Castegna, M. Aksenov, V. Thongboonkerd, J. B. Klein, W. M. Pierce, R. Booze, W. R. Markesbery and D. A. Butterfield, J. Neurochem., 2002, 82, 1524–1532 CrossRef CAS PubMed.
- R. Thanan, S. Oikawa, P. Yongvanit, Y. Hiraku, N. Ma, S. Pinlaor, C. Pairojkul, C. Wongkham, B. Sripa, N. Khuntikeo, S. Kawanishi and M. Murata, Free Radic. Biol. Med., 2012, 52, 1465–1472 CrossRef CAS PubMed.
- B. Aryal and V. A. Rao, PLoS One, 2018, 13, e0194164 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4tb01529k |
| This journal is © The Royal Society of Chemistry 2024 |