
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthetic macromolecular peptide-mimetics with amino acid substructure residues as protein stabilising excipients†
Ruggero
Foralosso
a,
Rafał Jerzy
Kopiasz
ab,
Cameron
Alexander
 *a,
Giuseppe
Mantovani
*a and
Snow
Stolnik
*a
*a,
Giuseppe
Mantovani
*a and
Snow
Stolnik
*a
aUniversity of Nottingham, School of Pharmacy, NG7 2RD, UK. E-mail: giuseppe.mantovani@nottingham.ac.uk; snow.stolnik@nottingham.ac.uk; cameron.alexander@nottingham.ac.uk
bWarsaw University of Technology, Faculty of Chemistry, Noakowskiego 3 St., 00-664, Warsaw, Poland
First published on 29th December 2023
Abstract
The clinical use of protein and peptide biotherapeutics requires fabrication of stable products. This particularly concerns stability towards aggregation of proteins or peptides. Here, we tested a hypothesis that interactions between a synthetic peptide, which is an aggregation-prone region analogue, and its homologous sequence on a protein of interest, could be exploited to design excipients which stabilise the protein against aggregation. A peptide containing the analogue of lysozyme aggregation-prone region (GILQINSRW) was conjugated to a RAFT agent and used to initiate the polymerisation of N-hydroxyethyl acrylamide, generating a GILQINSRW-HEA90 polymer, which profoundly reduced lysozyme aggregation. Substitution of tryptophan in GILQINSRW with glycine, to form GILQINSRG, revealed that tryptophan is a critical amino acid in the protein stabilisation by GILQINSRW-HEA90. Accordingly, polymeric peptide-mimetics of tryptophan, phenylalanine and isoleucine, which are often present in aggregation-prone regions, were synthesized. These were based on synthetic oligomers of acrylamide derivatives of indole-3 acetic acid (IND), phenylacetic acid (PHEN), or 2-methyl butyric acid (MBA), respectively, conjugated with hydrophilic poly(N-hydroxyethyl acrylamide) blocks to form amphiphilic copolymers denoted as INDm-, PHENm- and MTBm-b-HEAn. These materials were tested as protein stabilisers and it was shown that solution properties and the abilities of these materials to stabilise insulin and the peptide IDR 1018 towards aggregation are dependent on the chemical nature of their side groups. These data suggest a structure–activity relationship, whereby the indole-based INDm-b-HEAn peptide-mimetic displays properties of a potential stabilising excipient for protein formulations.
Introduction
The translation of proteins or peptides from biotechnology to biotherapeutic products offers potential treatments to a variety of diseases with unmet clinical need. The rationale for these biomolecules as therapeutics stems from their known mode of action, their inherent degradability as well as typical ‘drug attributes’ such as specificity and potency. This translation from an often complex and delicate macromolecule towards a medicine however requires fabrication of a stable product. This particularly concerns stability towards aggregation of protein or peptide molecules – the process by which unfolded molecules adopt a conformation that causes their clustering into aggregates or/and fibrils.1 Hence, solutions are required to ensure stability to different stress factors during all stages of development and use of peptide or protein formulations. There are currently over 130 protein biotherapeutics in clinical practice, covering disease indications from diabetes, haemophilia, multiple sclerosis, hepatitis C and many cancers.2 These employ a range of stabilising strategies, including: addition of low-molecular weight compounds like amino acids glycine, proline and histidine,3 sugars like trehalose and sucrose, or polyols such as sorbitol. Covalent attachment of polyethylene glycol (PEG) to therapeutic proteins is another option which provides formulation stabilisation, and which can also impact positively on in vivo performance. PEGylated colony-stimulating factors, interferons, erythropoietin and mAbs4–6 are biotherapeutic products that have already reached clinical application. An alternative strategy is a non-covalent PEGylation of a protein, for example as shown by the use of cholanic acid-PEG to increase the half-life of recombinant human growth hormone (rh-GH) and recombinant human granulocyte colony stimulating factor (rh-G-CSF) in vivo,7,8 and tryptophan-PEG was used to reduce significantly the aggregation of salmon calcitonin in vitro.9 In a related context, mPEG2k-poly(glutamic acid) copolymers have been utilised to form reversible protein complexes formulated for application to the lung.10 Synthetic copolymers containing carbohydrate pendant units, especially trehalose-based, have also been used to reduce/modulate protein aggregation.11–14The aggregation of many protein or peptide molecules can be attributed to the existence of 5–15 amino acid long regions that display high hydrophobicity, low net charge, and a high tendency to form β-structures. These are usually termed aggregation-prone regions (APR), and have been shown to be responsible for the aggregation of a range of proteins.15,16 APRs are typically buried within a protein's hydrophobic core and/or positioned close to the active site of enzymes or polypeptide ligands,17,18 and the aggregation process is normally initiated by their exposure to the solvent. Protein unfolding, caused by stress factors (e.g. temperature or pH), or intrinsic factors (e.g. destabilisation of the native structure by genetic mutation) can expose the amino acid sequence of the APR and initiate their self-assembly and consequent protein aggregation. Disruption of APR self-assembly has thus been investigated as an approach to prevent the formation of amyloid aggregates by endogenous proteins, observed to occur in neurodegenerative diseases such as Alzheimer's.19 It has been suggested that synthetic peptides, termed β-sheet breakers, could be developed to be capable of binding the amyloid precursor αβ fragment, but unable to self-assemble into a β-sheet structure.20 Such peptides have been shown to prevent aggregation of the αβ peptide into amyloid fibrils in vitro.21–25
The starting premise of this study was that a stabilizing excipient could be designed comprising a peptide with an amino acid sequence analogous to that of APR, in order to bind strongly to an ‘unfolding protein’ on the path to aggregation, but with a hydrophilic chain conjugated to the peptide in order to provide a steric barrier towards protein aggregation. This approach might be considered analogous to bioconjugation and PEGylation strategies, but with an ‘associative linker’ rather than a conjugate, which interfered in the kinetics of aggregation by blocking access to an intermediate in the protein fragment self-association pathway. The protein lysozyme, and the peptide Gly-Ile-Leu-Gln-Ile-Asn-Ser-Arg-Trp (GILQINSRW) previously identified as the aggregation-prone region in lysozyme,26 were selected as a model system. The GILQINSRW peptide was synthesized, incorporated into a RAFT chain-transfer agent (CTA), which was used to mediate the polymerisation of N-hydroxyethylacrylamide (HEA), forming the required hydrophilic block. Initial experiments identified the crucial role of tryptophan (Trp/W) amino acid of the GILQINSRW sequence in the stabilisation of lysozyme towards aggregation. We then further focused on the synthesis of a tryptophan peptide-mimetic based on indole – a side group of tryptophan, with the aim to test if such a synthetic material could achieve a stabilising effect comparable to that of tryptophan in a peptide or polymer sequence. Accordingly, RAFT CTAs incorporating indole oligomers were synthesised and used to mediate the polymerisation of HEA as a hydrophilic block. For comparison, and to assess if a structure–activity relationship could be established, we also synthesized peptide-mimetics of two other amino acids normally present in APRs, phenylalanine (Phe/F) and isoleucine (Ile/I) – based on phenylacetic acid, and methyl butyric acid structures, respectively. The resulting materials were compared for their ability to moderate the aggregation of insulin and the anti-microbial peptide, IDR 1018,27 as representative therapeutic biomolecules, and promising initial activities of these materials were obtained.
Results and discussion
Peptide–HEA conjugations synthesis
To investigate the possibility for stabilising lysozyme with peptides analogue of its APR region, short peptide Gly-Ile-Leu-Gln-Ile-Asn-Ser-Arg-Trp (GILQINSRW, residues 54–62)26 was synthesised. Interestingly, Tokunaga et al. have shown that the GILQINSRW fragment of lysozyme APR self-assembles through mutual β-sheet interactions, but a single amino acid substitution of tryptophan with glycine (GILQINSRG) completely suppresses the aggregation.28 Hence, to gain further insight to whether GILQINSRW peptide impacts on the aggregation of lysozyme via an interaction of its GILQINSRW sequences with APR, an analogous peptide Gly-Ile-Leu-Gln-Ile-Asn-Ser-Arg-Gly (GILQINSRG) was also synthesised. Both GILQINSRW and GILQINSRG peptides were prepared by solid-state peptide synthesis on 2-chlorotrityl chloride resin using Fmoc-protected amino acids (Fig. 1(a) and Section S3 in ESI†). Additionally, to (i) circumvent water solubility issues and self-aggregation of the peptide itself,28 and (ii) provide steric stabilisation against aggregation of the protein molecules once GILQINSRW and GILQINSRG were able to interact with lysozyme, the linkage of the peptides with a very hydrophilic polymer chain was carried out. In these cases, poly(N-hydroxyethyl acrylamide) (pHEA) and the peptide–polymer conjugates HEA90-GILQINSRW (1) and HEA95-GILQINSRG (2), were also obtained (Fig. 1(a)). This process involved coupling of a 4-cyano-4-(((ethylthio)carbonothioyl)thio)pentanoic acid, as a chain transfer agent (CTA), to the resin-bound peptides (1a and 2a) and subsequent cleavage from the resin to obtain GILQINSRW- and GILQINSRG-RAFT agents 1b and 2b (Fig. 1(a)). These agents were subsequently used to mediate the RAFT polymerisation of HEA, followed by cleavage of the amino acid protecting groups giving the final materials HEA90-GILQINSRW (1) and HEA95-GILQINSRG (2). The obtained polymer–peptide conjugates, as well as RAFT agents 1b and 2b, were characterised by 1H NMR spectroscopy and HR-MS (Fig. 1(c) and Fig. S1–S11, ESI†), which confirmed their structure and purity. Conjugates 1 and 2 were additionally characterised by size-exclusion chromatography (SEC) (Table 1). Additionally, poly(N-hydroxyethyl acrylamide) without additional functionalities (HEA100) was synthesised as control polymers as shown in Fig. 1(b) (characterisation data in Table 1).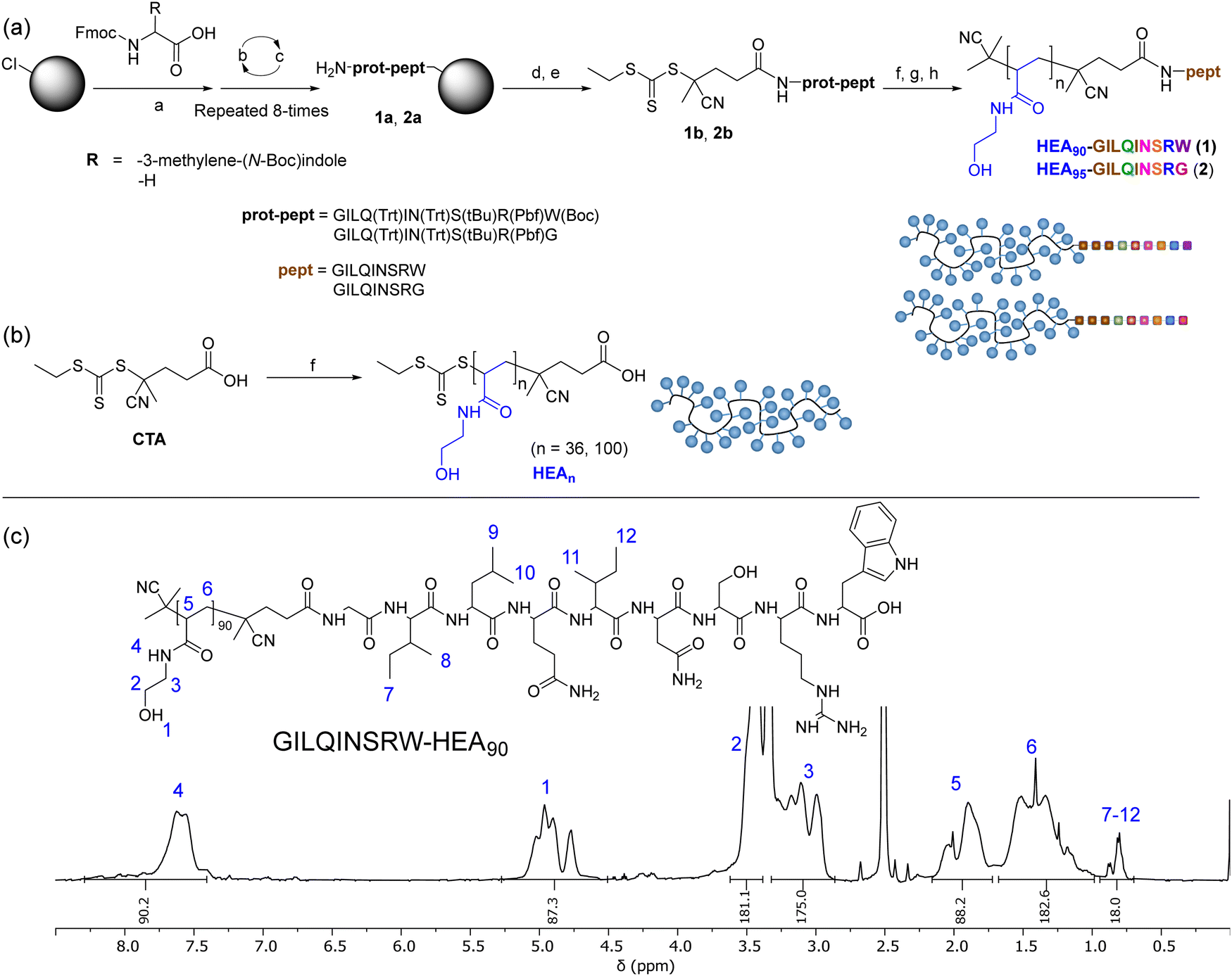 | ||
Fig. 1 (a) Synthesis of HEA90-GILQINSRW, and HEA95-GILQINSRG; (b) synthesis of HEAn as control polymers; (c) 1H NMR (400 MHz, DMSO-d6) spectra of obtained GILQINSRW-HEA90 conjugation. Reagents and conditions: (a) DMF, DIPEA, rt; (b) DMF, piperidine, rt; (c) DMF, Fmoc-amino acid, HATU, DIPEA, rt; (d) CTA, HATU, TEA, DMF, rt; (e) DCM/TFE 8![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 v/v, rt; (f) N-hydroxyethyl acrylamide, V501, DMF, 70 °C; (g) AIBN, DMF, 80 °C; (h) 0.1 N HCl in hexafluoro-2-propanol (HFIP), rt. :2 v/v, rt; (f) N-hydroxyethyl acrylamide, V501, DMF, 70 °C; (g) AIBN, DMF, 80 °C; (h) 0.1 N HCl in hexafluoro-2-propanol (HFIP), rt. | ||
| Composition | M n,THEO (kDa) | M n,NMR (kDa) | M n,SEC (kDa) | Đ |
|---|---|---|---|---|
| Composition is indicated by numbers in subscript.a Calculated from initial [HEA]:[CTA] and final monomer conversion.b Number of repeating units of HEA calculated from 1H NMR spectra by comparing the integral of peptides’ methyl groups with the hydroxyl peak of HEA repeating units.c Obtained from SEC in DMF + 0.1% LiBr (PMMA standards). | ||||
| HEA90-GILQINSRW (1) | 10.5 | 10.8b | 17.0 | 1.09 |
| HEA95-GILQINSRG (2) | 10.3 | 12.1b | 14.8 | 1.13 |
| HEA100 | 7.6 | — | 12.1 | 1.14 |
Moderation of lysozyme aggregation
We tested the potential of HEA90-GILQINSRW to stabilize lysozyme in an alkaline medium which induces its aggregation. While the peptide GILQINSRW was insoluble in water, the polymer–peptide conjugate was freely soluble in both water and in phosphate buffer up to the highest tested concentration of 0.035 mg mL−1. Accordingly, phosphate buffer was chosen as an appropriate medium for the lysozyme aggregation studies, in this case at a pH of 12.3 with assays conducted at room temperature, as these are conditions known to denature lysozyme.29,30 This approach assumes that these conditions promote unfolding of the protein exposing its APR, which makes it more accessible to APR from other lysozyme protein molecules and to the APR-like sequence from HEA90-GILQINSRW.The results of the lysozyme aggregation study are shown in Fig. 2 and confirm the formation of large aggregates in control samples containing only lysozyme. Conversely, in the presence of HEA90-GILQINSRW (1:1 mol with respect to lysozyme) the onset of aggregation was delayed by 10 hours, and within 24 hours of the experiment the aggregation remains relatively low. The profiles further indicated an increased rate of aggregation in the presence of the HEA100 homopolymer, which is analogous to HEA90-GILQINSRW but lacks the GILQINSRW peptide block. This may be ascribed to the macromolecular crowding induced by the simultaneous presence in solution of two macromolecules, the protein and the polymer, at relatively high concentration (10 mg mL−1 lysozyme, 9 mg mL−1 HEA100) and the ‘excluded volume’ occupied by the polymer increasing the chances of protein–protein interaction and precipitation.29,30 The aggregation profile of HEA95-GILQINSRG shows that it does not modulate lysozyme aggregation. On the contrary, it appears that HEA95-GILQINSRG causes an increase in the rate of aggregation, similar to the control polymer HEA100, further indicating the absence of associative interactions with the protein-aggregation region.
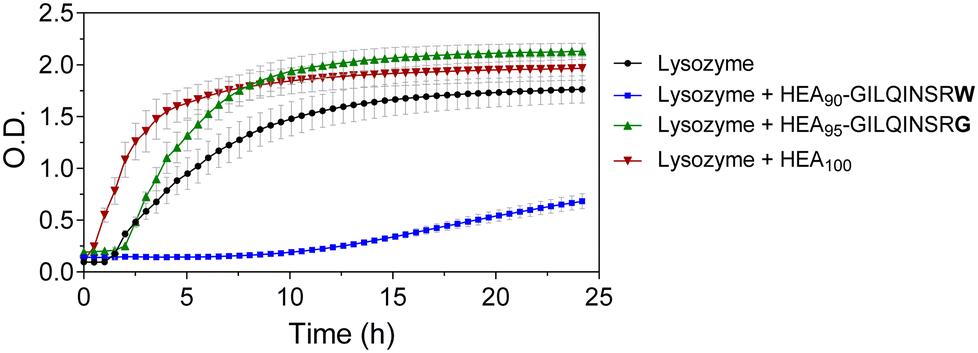 | ||
| Fig. 2 Aggregation assay for lysozyme (10 mg mL−1) in the presence of HEA90-GILQINSRW, HEA95-GILQINSRG, and HEA100 in 100 mM phosphate buffer pH 12.3; molar ratio of peptides and HEA100 to lysozyme was 1:1. Lysozyme aggregation assessed by turbidimetry at λ = 500 nm, readings were taken every 30 min for 24 h. | ||
These data indicate that the modification of a single amino acid in the peptide sequence from GILQINSRW to GILQINSRG suppresses interaction of the corresponding HEA95-GILQINSRG conjugate with lysozyme, in line with the reported loss of self-aggregation of the GILQINSRW peptide when its terminal W is replaced with G residue.28 This suggests that the stabilisation effect of HEA90-GILQINSRW may be attributed to the specific interaction between its GILQINSRW moiety and the homologous aggregation-prone region on lysozyme. The results further highlight the decisive role of tryptophan to maintain anti-aggregation properties of the GILQINSRW sequence.
Synthesis of copolymeric peptide-mimetics
After identifying the key role of tryptophan in the HEA90-GILQINSRW stabilisation of lysozyme (Fig. 2), we synthesised tryptophan peptide-mimetic block copolymers based on indole-3 acetic acid (Fig. 3(a)), acknowledging that the pendant indole group defines many physico-chemical properties of tryptophan.31 As other hydrophobic amino acids, like phenylalanine and isoleucine, play an important role in the protein aggregation process and are abundant in APRs of many proteins, our studies on peptide mimetics were extended to include block copolymers containing phenyl and iso-butyl groups, as mimics of the side chains of these amino acids (Fig. 3(a)).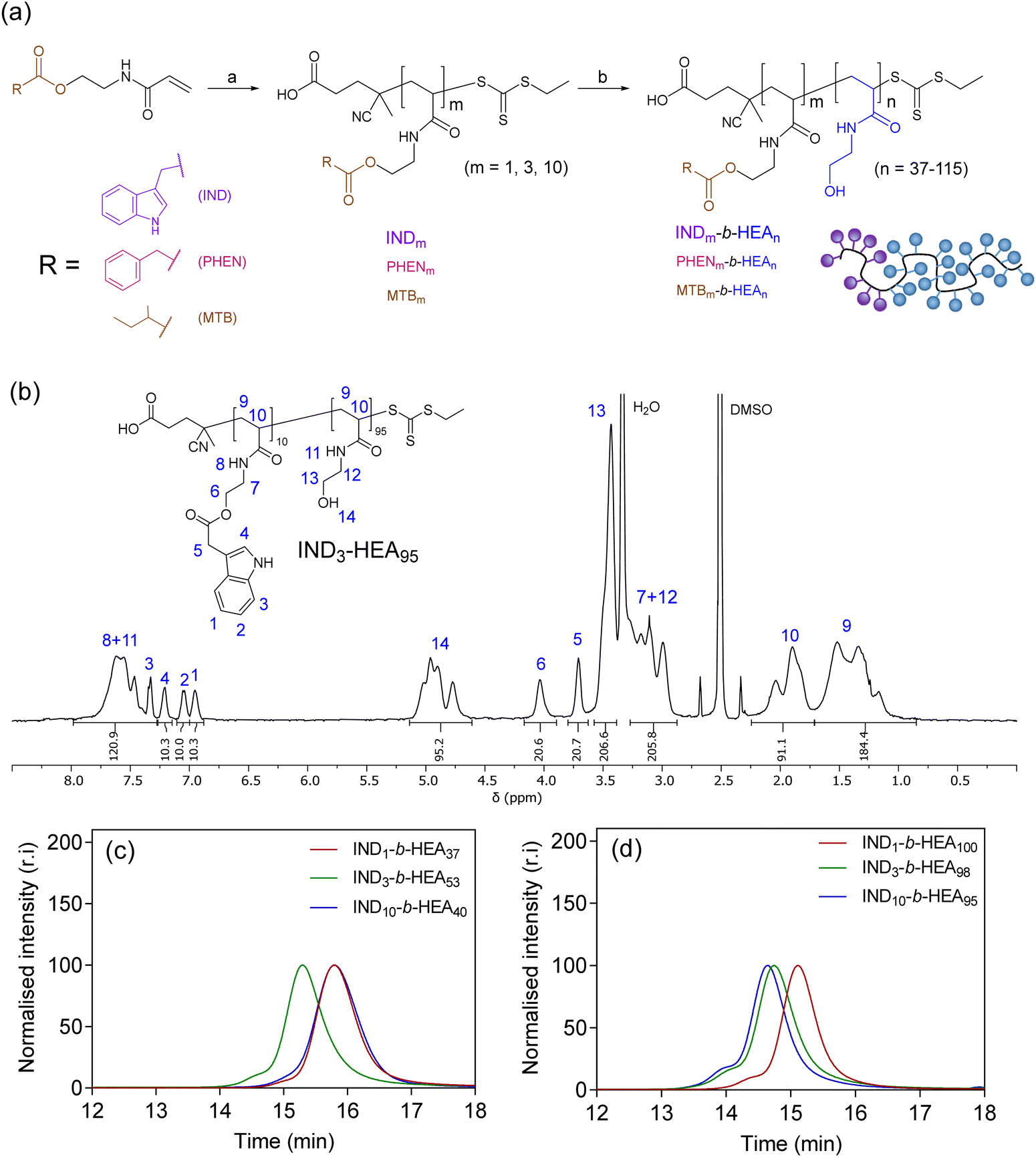 | ||
| Fig. 3 (a) Synthesis of INDm-, PHENm-, MTBm-b-HEAn block copolymers. Reagents and conditions: (a) AIBN, CTA, DMF, 80 °C; (b) N-hydroxyethyl acrylamide, V501, DMF, 70 °C; (b) 1H NMR (DMSO-d6, 400 MHz) spectrum of IND3-HEA95 as a representative spectrum of the synthesised block copolymers; SEC chromatograms of INDm-b-HEAn copolymers with (c) shorter and (d) longer hydrophilic blocks, using DMF with 0.1% LiBr as the mobile phase. Spectra and SEC chromatograms of all polymers are shown in Sections S5.3.1 and S5.3.2 in the ESI.† | ||
As the first step, acrylic monomers IND, PHEN, and MTB were obtained via coupling of N-hydroxyethyl acrylamide with indole-3 acetic acid, phenylacetic acid, and methylbutyric acid, respectively (Scheme S3 and Section S5.1 in the ESI†). Then, these monomers were polymerised by RAFT polymerisation, to synthesise hydrophobic oligomeric chain transfer agents (CTAs) INDm, PHENm, and MTBm (m = 1, 3, 10, Fig. 3(a) and Section S5.2 in ESI†). Purification of the oligomeric CTAs with m = 1 and m = 3 was carried out by flash chromatography, in analogy with the purification of discrete oligomers on silica described by Hawker and co-workers,32 whereas CTAs with average m = 10 were used as obtained from the RAFT reaction, without the additional chromatographic step.
Subsequently, the oligomeric CTAs were used to mediate the RAFT polymerisation of N-hydroxyethyl acrylamide, aiming at degrees of polymerisation (DP) of 40 and 100, to investigate the influence of the size of the polyHEA block on protein–copolymer interaction. The resulting block copolymers INDm-b-HEAn, PHENm-b-HEAn, MTBm-b-HEAn were characterised using 1H NMR spectroscopy and SEC (Fig. 3(b)–(d), Table 2, and Section S5.3 in the ESI†). A small shoulder at higher molar masses was in the observed in the SEC traces of INDn-b-HEAm block copolymers, which may be ascribable to a small extent of bimolecular coupling termination, which can occur in RAFT polymerisation.33 The discrepancy between number average molar mass determined by 1H NMR (Mn,NMR) and SEC (Mn,SEC) may result from differences in polymer hydrodynamic volume compared to the standards employed for calibration of SEC.
| Composition | M n,THEO (kDa) | M n,NMR (kDa) | M n,SEC (kDa) | Đ | Composition | M n,THEO (kDa) | M n,NMR (kDa) | M n,SEC (kDa) | Đ |
|---|---|---|---|---|---|---|---|---|---|
| a Calculated from initial [HEA]:[CTA] and final monomer conversion. b Number of repeating units of HEA monomers calculated by 1H NMR, in DMSO-d6, by comparing the integral of indole aromatic. c Number of repeating units of HEA monomers calculated by 1H NMR, in DMSO-d6, by comparing the integral of methylene. d Number of repeating units of HEA monomers calculated by 1H NMR, in DMSO-d6, by comparing the integral of methylbutyril methyl peaks with the hydroxyl peak of HEA repeating units. e Obtained from SEC in DMF + 0.1% LiBr (PMMA standards). | |||||||||
| IND1-b-HEA37 | 5.5 | 5.0b | 8.7 | 1.07 | IND1-b-HEA100 | 9.0 | 8.7b | 13 | 1.08 |
| PHEN1-b-HEA37 | 5.1 | 4.8c | 9.0 | 1.07 | PHEN1-b-HEA100 | 12 | 12.2c | 18.6 | 1.08 |
| MTB1-b-HEA37 | 5.9 | 4.8d | 10.4 | 1.05 | MTB1-b-HEA111 | 13.1 | 13.2d | 19.8 | 1.09 |
| IND3-b-HEA53 | 5.4 | 6.7b | 11.7 | 1.09 | IND3-b-HEA98 | 11.5 | 12b | 17 | 1.11 |
| PHEN3-b-HEA45 | 5.4 | 6.2c | 9.6 | 1.07 | PHEN3-b-HEA99 | 11.7 | 12.5c | 17.5 | 1.06 |
| MTB3-b-HEA42 | 6.4 | 7.3d | 10.9 | 1.05 | MTB3-b-HEA82 | 10.2 | 10.5d | 16.8 | 1.07 |
| IND10-b-HEA40 | 6.1 | 7.7b | 10.5 | 1.15 | IND10-b-HEA95 | 10.7 | 14.1b | 18.4 | 1.11 |
| PHEN10-b-HEA38 | 6.8 | 7.0c | 10.7 | 1.08 | PHEN10-b-HEA88 | 12.6 | 12.9c | 17.4 | 1.07 |
| MTB10-b-HEA43 | 6.7 | 7.2d | 10.0 | 1.07 | MTB10-b-HEA105 | 12.5 | 15.0d | 18.7 | 1.07 |
| HEA36 | 4.5 | 8.5 | 1.04 | HEA100 | 13 | 12.1 | 1.14 | ||
Characterisation of block copolymer self-assemblies
The synthesised INDm-, PHENm-, and MTBm-b-HEAn copolymers are amphiphilic molecules consisting of a hydrophobic and a hydrophilic block. Therefore, to further characterise them, their ability to self-assemble in aqueous environment was investigated by dynamic light scattering (Fig. 4 and Table 3).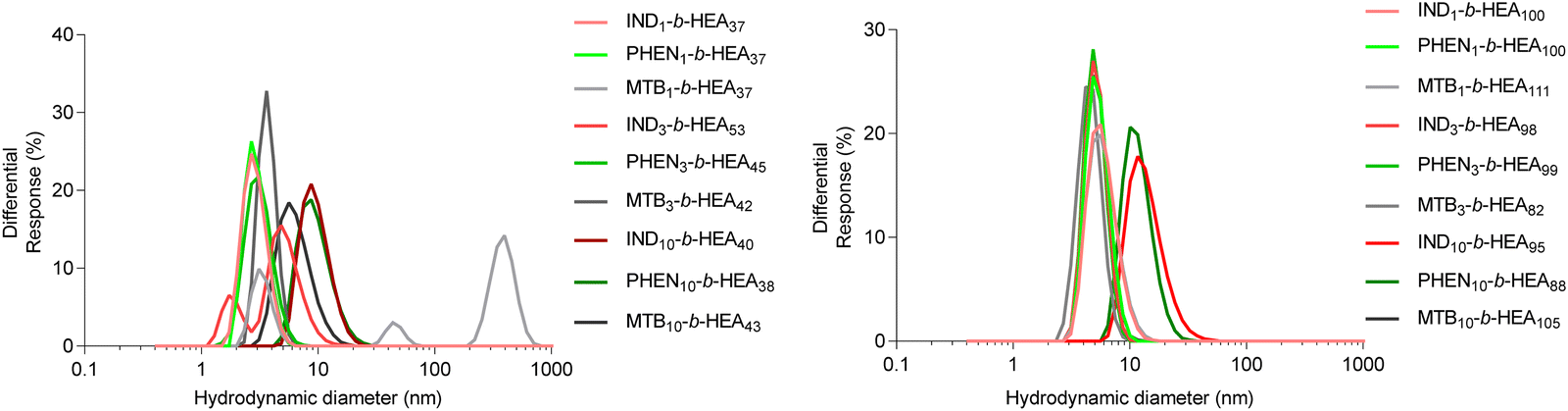 | ||
| Fig. 4 Size distribution (volume%) of copolymers’ self-assemblies (1 mg mL−1) in DI water as determined by DLS. | ||
| Polymer: | IND1-b-HEA37 | PHEN1-b-HEA37 | MTB1-b-HEA37 | IND1-b-HEA100 | PHEN1-b-HEA100 | MTB1-b-HEA111 |
|---|---|---|---|---|---|---|
| Size (PDI) nm | 4.43 (0.26) | 3.28 (0.62) | 3.15 (0.36) | 6.05 (0.11) | 5.69 (0.05) | 6.81 (0.43) |
| Polymer: | IND3-b-HEA53 | MTB3-b-HEA42 | MTB3-b-HEA42 | IND3-b-HEA98 | PHEN3-b-HEA99 | MTB3-b-HEA82 |
| Size (PDI) nm | 5.54 (0.18) | 3.53 (0.07) | 4.73 (1.62) | 5.21 (0.27) | 6.23 (0.86) | 5.40 (1.0) |
| Polymer: | IND10-b-HEA40 | PHEN10-b-HEA38 | MTB10-b-HEA43 | IND10-b-HEA95 | PHEN10-b-HEA88 | MTB10-b-HEA105 |
| Size (PDI) nm | 9.73 (016) | 9.71 (0.18) | 6.54 (0.20) | 14.28 (0.15) | 12.03 (0.16) | 5.38 (0.33) |
Data for INDm-, PHENm- and MTBm-HEAn with m = 1 and 3 show average hydrodynamic particle diameters Dh of around 3-5 nm for shorter HEAn (n ≈ 37), and of around 5–7 nm for longer HEAn (n ≈ 100), with polydispersity indices representative of a relatively narrow particles size distribution. This is consistent with the presence of highly hydrated individual chains of these copolymers in a solvated extended conformation. For instance, the gyration radius of free PEG with molecular mass within the range of 2 kDa to 15 kDa ranging from 1.3 nm to 4 nm.34,35 However, IND10- and PHEN10-b-HEAn show significantly higher hydrodynamic diameters of around 10 nm for shorter HEAn (n ≈ 40), and 12–14 nm for longer HEAn (n ≈ 100). Interestingly, there is no measurable increase in diameter of MTB10-b-HEAn for either n = 43 or 105 in comparison with MTB1- and MTB3-. Taken together, these data indicate minimal or no aggregation in water of copolymers with m = 1 and 3, whilst a prolongation of the hydrophobic block content to 10 repeating units for indole- and phenyl-containing materials induces self-association into species of about 10–14 nm in size, with relatively narrow size distributions, as indicated by their PDI values. A possible explanation for this observation is an intermolecular interaction between aromatic indole and phenyl rings (e.g. π-stacking), which is not present in the case of the branched aliphatic methyl butyryl residues of MTBm-b-HEAn copolymers.
Further analysis of self-assembly behaviour of IND3- and IND10-b-HEAn molecules in water by surface tension (γ) measurements indicates that the former copolymer is a weak whereas the latter a strong surfactant, reducing γ to 66 mN m−1 and 58 mN m−1, respectively (Fig. 5). The γ vs. concentration plot for IND10-b-HEAn presents a formation of supramolecular aggregates at a concentration between 1.0 and 2.0 mg mL−1 (critical aggregation concentration, CAC). In the case of IND3-b-HEA98, the γ vs. concentration plot has no clear discontinuity or plateau at higher concentrations. This result agrees with our DLS analysis results (Table 3) that indicate an absence of self-assembled species for IND3-b-HEA98.
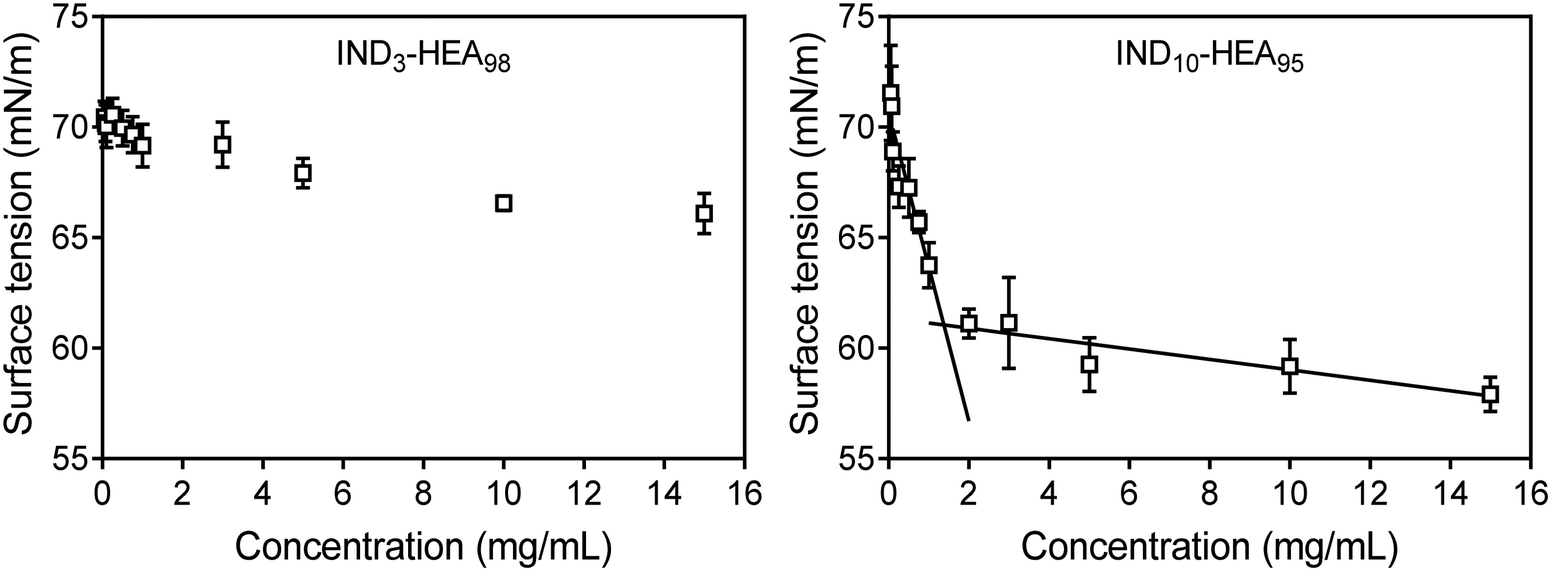 | ||
| Fig. 5 Surface tension measurements of representative copolymers IND3-b-HEA98 and IND10-b-HEA95 carried out in water by pendant drop method.36 | ||
Modulation of insulin aggregation by block copolymers
After identifying the key role of tryptophan in the HEA90-GILQINSRW stabilisation of lysozyme, we studied tryptophan peptide-mimetic oligomers based on indole-3 acetic acid (INDm-b-HEAn), on the basis that the indole ring contributes the defining physicochemical properties of tryptophan.31 Unfortunately, under the alkaline conditions that induce lysozyme aggregation, INDm-, PHENm- and MTBm-b-HEAn materials failed to show any stabilizing effect (Fig. S32 and S33, Section S6 in ESI†). Further analysis revealed that these aggregation favouring conditions (pH 12.3) induce hydrolysis of the ester groups linking the pendant indole group to the polymer backbone; the 1H NMR spectrum of IND10-b-HEA after incubation under such conditions displayed no indole aromatic protons peaks between 6.9–7.3 ppm (Spectrum S34, Section S6 in the ESI†).Accordingly, we selected insulin as a model protein to replace lysozyme in the aggregations studies as it is well-established that the aggregation of insulin is initiated at pH around 5.3, corresponding to the protein isoelectric point.37 The residues of insulin B-chain including GB8, SB9, VB12, EB13, YB16, GB23, FB24, FB25, YB26, and TB27 are believed to play an important role in insulin self-assembly.38 The impact of the peptide-mimetic materials synthesized in this work on the aggregation of insulin is shown in Fig. 6. Only 17% of insulin (0.34 mg mL−1 out of 2 mg mL−1) remained in a solution after incubation at conditions inducing its aggregation. However, the addition of IND3- and IND10-b-HEAm polymers at polymer:peptide molar ratio of 10 prevented aggregation very efficiently – almost 100% of insulin remained in its non-aggregated state after 20 h. Conversely, this effect was not observed for IND1-b-HEAn which possesses only one indole group. For PHENm-based copolymers, only the longest copolymer, PHEN10-b-HEA88, reduces aggregation (61% non-aggregated insulin retained), when compared to HEA100 control polymers (only 24% of residual insulin in solution). No measurable impact against insulin aggregation is seen for copolymers containing isoleucine-mimicking groups – MTBm-b-HEAn.
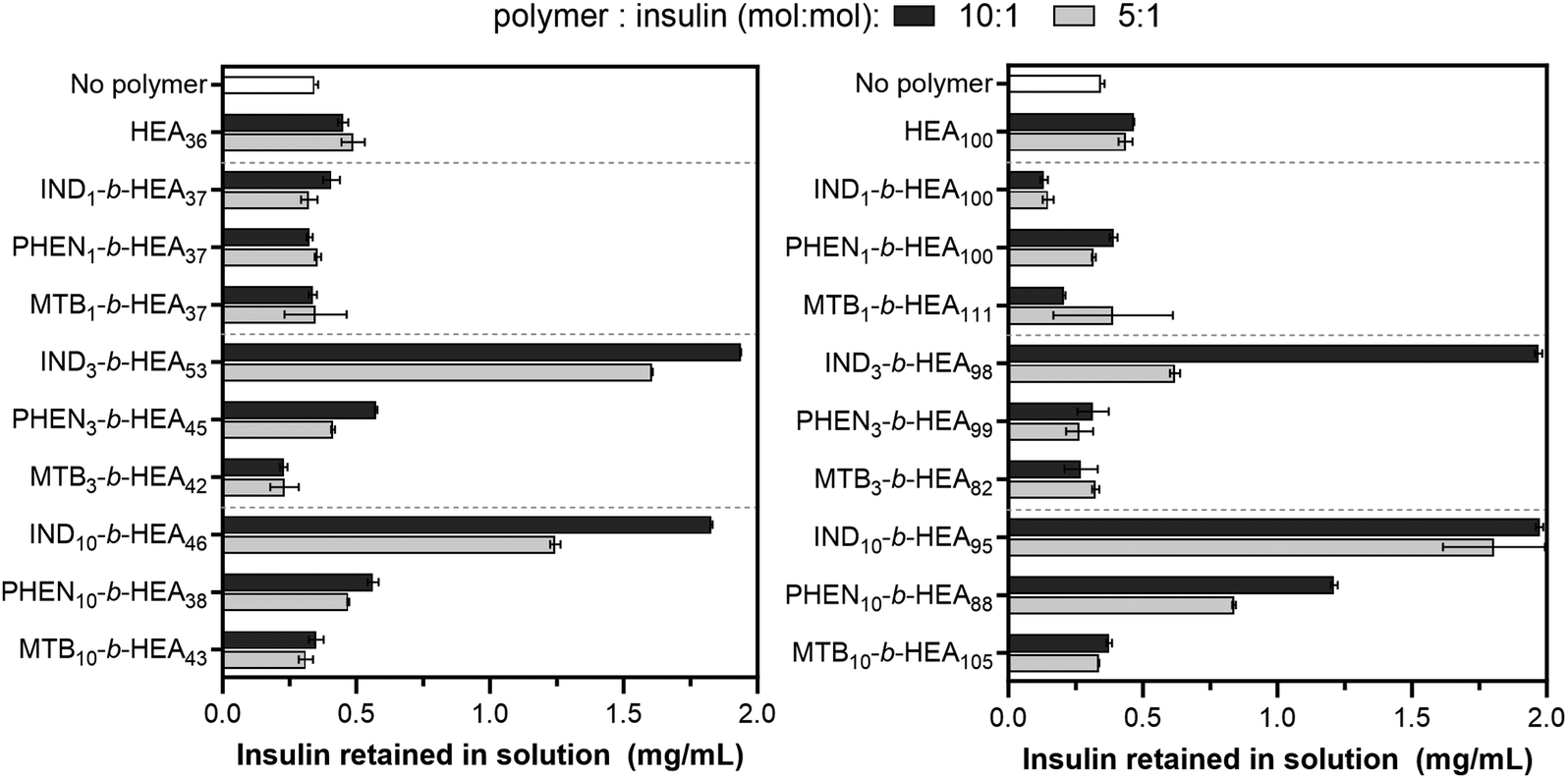 | ||
| Fig. 6 Insulin aggregation in the presence of INDm-, PHENm- and MTBm-b-HEAn materials. Concentration of insulin remaining in a solution after incubation in the aggregation inducing conditions (pH 5.2) for 20 h without and with synthesised copolymers at different molar ratio; the initial concentration of insulin was 2 mg mL−1. Higher values in the graph correspond to better protein stabilisation and hence reduced aggregation. | ||
Interaction of peptide-mimetic oligomers with insulin.
In the next step potential effects of the IND10-based oligomers on the secondary structure of insulin were assessed by circular dichroism (CD). The analysis of samples in the presence of IND10-b-HEA95 produced poor signal-to-noise ratio, likely due to the presence of indole groups, as reported previously.39 The analysis hence measured the CD profile after separation of insulin and IND10-b-HEA95. Whilst this experiment cannot inform directly on secondary structure alterations that may be occurring during insulin interactions with stabilizing IND10-b-HEA95, it however provides an insight to whether these interactions induce irreversible changes of the protein secondary structure (Fig. S31, ESI†). The profile obtained from the ‘native’ insulin, i.e. before any manipulation, exhibits two negative peaks, at 208 nm and 222 nm, as previously described for bovine insulin, and is typical of an α-helix secondary structure.40 A similar profile is seen for the sample subjected to HPLC purification. The two samples taken from insulin aggregation experiments in the presence of IND10-b-HEA95 at 10:1 and 5:1 insulin molar ratios also show [θ] vs. wavelength profiles similar to that of native insulin. The experiment hence indicates that the stabilizing IND10-b-HEA95 peptide-mimetic does not cause irreversible changes in the protein secondary structure.
Moderation of IDR 1018 aggregation by block copolymers
To exemplify the potential of our synthetic peptide-mimetic materials to moderate aggregation of peptides, the IDR 1018 peptide, consisting of the amino acid sequence VRLIVAVRIWRR was selected. This peptide has been reported to show antimicrobial and immunomodulatory activities,41–43 but clinical applications have been hampered by its tendency to aggregation under aqueous conditions. This aggregation has been attributed to the 5 consecutive hydrophobic residues between L3 and V7, which are believed to form the peptide's aggregation-prone region. The effect of INDm-, PHENm-, and MTBm-b-HEAn materials on aggregation of the peptide was investigated in 100 mM pH 7.2 phosphate buffer, where IDR 1018 is known to aggregate,44 and the results are presented in Fig. 7.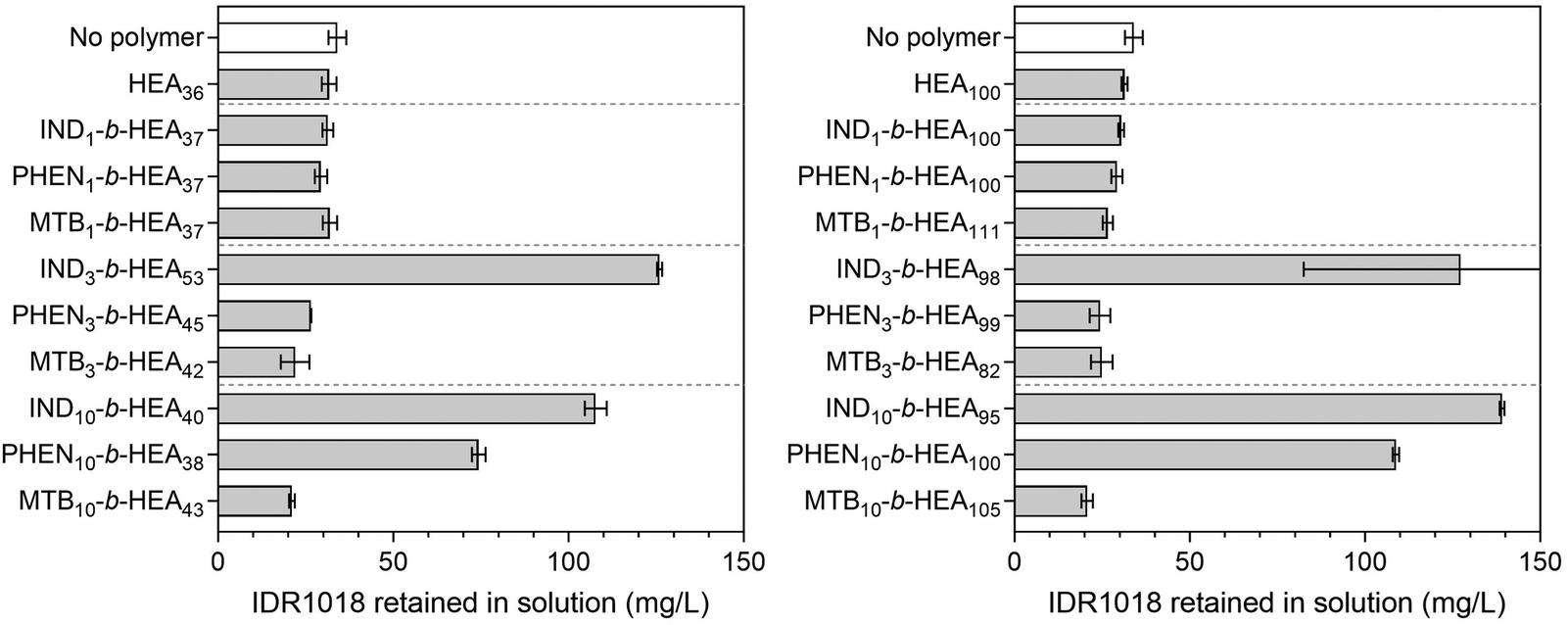 | ||
| Fig. 7 The concentration of IDR 1018 remaining in a solution after incubation in aggregation conditions (100 mM phosphate buffer, pH 7.2) for 5 h without and with synthesised copolymers at 1:1 molar ratio; the initial concentration of the peptide was 0.153 mg mL−1. Higher values in the graph correspond to better protein stabilisation and hence reduced aggregation. | ||
Incubation of IDR 1018 in 100 mM phosphate buffer, pH 7.2 for 5 hours resulted in 75% peptide aggregation – 38.3 mg mL−1 final concentration from an initial 153 mg L−1. However, in the presence of indole-based materials (IND3-b-HEA53, IND10-b-HEA40, IND3-b-HEA98 and IND10-b-HEA95) in a 1:1 (co)polymer:IDR 1018 molar ratio, as much as 70–80% of the peptide remained in solution under identical conditions, whereas for phenyl-based materials (PHEN10-b-HEA38 and PHEN10-b-HEA100) it was around 50–75%. The other copolymers tested did not show any stabilization effect on IDR 1018 (Fig. 7).
Data on behaviour of IND10- and PHEN10-b-HEAn copolymers in aqueous solutions (Table 3 and Fig. 4 and 5), together with protein aggregation results (Fig. 6 and 7), could be explained with the existence of an equilibrium between polymer self-assemblies and their interactions with the proteins and peptides tested, leading to their stabilization towards aggregation. This is apparent for IND10-HEAn and PHEN10-b-HEAn materials. Interestingly, similar protein stabilization induced by IND3-b-HEAn and IND10-b-HEAn may suggest that polymer self-assembly does not hamper interactions with proteins, as the former one does not self-assemble (Fig. 5).
Collectively, the data indicate different impacts of copolymers bearing indole, phenyl, or methyl butyric side groups mimicking W, F, and I, respectively, on protein stabilisation. This observation may be ascribed to the physicochemical properties of tryptophan, strongly affected by its indole ring, observed via the influence of tryptophan on protein folding, as well as its positioning in lipid bilayers and in the membrane-spanning regions of transmembrane proteins. Tryptophan has a number of unique molecular features:45 the largest nonpolar (hydrophobic) area of all amino acids due to the two-sided π-electron face of its indole side chain that is also polarizable and highly accessible because of its planar topology, the strongest electrostatic potential for cation–π interactions, and an indole hydrogen-bond donor N–H moiety. The single phenyl ring, present in phenylalanine and PHENm-b-HEAn copolymers, nor the short branched alkyl group present in isoleucine and MTBm-b-HEAn, do not exhibit the full range of these properties, hence their interactions with protein regions involved with the aggregation of the proteins and peptide tested may be limited. Furthermore, the presented data indicate to exert a protein stabilizing activity INDm-b-HEAn copolymers must comprise more than one indole-containing repeating unit (IND), as a tryptophan mimetic unit. Intriguingly, a PEG2k-tryptophan conjugate reported by Mueller et al.46 has shown a significant stabilization effect on salmon calcitonin despite bearing only a single tryptophan unit, thus indicating that that the structure of tryptophan-containing stabilisers may need fine-tuning to achieve the desired protein/peptide stabilisation effect. Importantly, the proteins and peptides tested to date with tryptophan-containing stabilisers – lysozyme, insulin, IDR 1018, and salmon calcitonin – possess chemically distinct aggregation-prone regions (APRs), which underlines the versatility of indole moiety in protein stabilization.
Conclusions
This work started with a hypothesis that interactions could be exploited between a synthetic peptide, which is an aggregation-prone region analogue, and its homologous sequence on a protein, to design excipients to stabilise the protein against aggregation. The hypothesis further suggested that attachment of a hydrophilic polymer to such a peptide will provide a stabilisation effect, such that the associative interactions at the aggregation-prone region of the peptide mimetic with the protein would prevent full aggregation by the steric demands of the attached hydrophilic polymer. The synthesized GILQINSRW-HEA90 polymer containing the lysozyme aggregation-prone region analogue indeed shows a dramatic reduction in lysozyme aggregation. Substitution of tryptophan in GILQINSRW with glycine to form GILQINSRG showed, via the lack of activity of the mutated peptide, that tryptophan was a decisive factor in the protein stabilisation by GILQINSRW-b-HEA90. This led to the successful synthesis of peptide-mimetics from tryptophan, phenylalanine and isoleucine amino acid analogues, based on synthetic oligomers of indole-3 acetic acid, phenylacetic acid, or methylbutyric acid, and with a block of poly(HEA) to provide aqueous solubility and steric stabilisation: INDm-, PHENm- and MTBm-b-HEAn. The study then demonstrates that the solution properties and their ability of these materials to stabilise insulin and IDR 1018 peptide towards aggregation are dependent on the chemical nature of their side groups – indole vs. phenyl vs. methylbutyrate – with indole based INDm-b-HEAn peptide-mimetic material displaying properties of a potential stabilising excipient to protein formulations.Experimental section
For materials, synthetic procedures and analytical methods see the ESI.†Author contributions
Ruggero Foralosso – investigation, methodology, formal analysis. Rafal Kopiasz – formal analysis, writing – original draft, writing – review and editing. Giuseppe Mantovani – supervision, writing – review and editing. Cameron Alexander – funding acquisition, supervision, writing – review and editing. Snow Stolnik – funding acquisition, supervision, writing – review and editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was funded by the Engineering and Physical Science Research Council [EP/L01646X/1] and the Royal Society [Wolfson Research Merit Award WM150086 to CA]. We thank Dr Marco di Antonio (University of Cambridge) for conducting the circular dichroism experiments. We also thank Tom Hyde, Esme Ireson and Paul Cooling for expert technical support.References
- M. Stefani, Biochim. Biophys. Acta, Mol. Basis Dis., 2004, 1739, 5–25 CrossRef CAS PubMed.
- M. Evans, P. M. Schumm-Draeger, J. Vora and A. B. King, Diabetes, Obes. Metab., 2011, 13, 677–684 CrossRef CAS PubMed.
- S. Saurabh, C. Kalonia, Z. Li, P. Hollowell, T. Waigh, P. Li, J. Webster, J. M. Seddon, J. R. Lu and F. Bresme, Mol. Pharmaceutics, 2022, 19, 3288–3303 CrossRef CAS PubMed.
- M. J. Roberts, M. D. Bentley and J. M. Harris, Adv. Drug Delivery Rev., 2002, 54, 459–476 CrossRef CAS PubMed.
- P. L. Turecek, M. J. Bossard, F. Schoetens and I. A. Ivens, J. Pharm. Sci., 2016, 105, 460–475 CrossRef CAS PubMed.
- F. M. Veronese and G. Pasut, Drug Discovery Today, 2005, 10, 1451–1458 CrossRef CAS PubMed.
- S. Salmaso, S. Bersani, F. Mastrotto, G. Tonon, R. Schrepfer, S. Genovese and P. Caliceti, J. Controlled Release, 2012, 162, 176–184 CrossRef CAS PubMed.
- S. Salmaso, S. Bersani, A. Scomparin, A. Balasso, C. Brazzale, M. Barattin and P. Caliceti, J. Controlled Release, 2014, 194, 168–177 CrossRef CAS PubMed.
- C. Mueller, M. A. H. Capelle, E. Seyrek, S. Martel, P.-A. Carrupt, T. Arvinte and G. Borchard, J. Pharm. Sci., 2012, 101, 1995–2008 CrossRef CAS PubMed.
- A. Nieto-Orellana, M. Di Antonio, C. Conte, F. H. Falcone, C. Bosquillon, N. Childerhouse, G. Mantovani and S. Stolnik, Polym. Chem., 2017, 8, 2210–2220 RSC.
- R. J. Mancini, J. Lee and H. D. Maynard, J. Am. Chem. Soc., 2012, 134, 8474–8479 CrossRef CAS PubMed.
- J. Madeira do O, F. Mastrotto, N. Francini, S. Allen, C. F. van der Walle, S. Stolnik and G. Mantovani, J. Mater. Chem. B, 2018, 6, 1044–1054 RSC.
- M. Milewska, A. Milewski, I. Wandzik and M. H. Stenzel, Polym. Chem., 2022, 13, 1831–1843 RSC.
- A. N. Bristol, J. Saha, H. E. George, P. K. Das, L. K. Kemp, W. L. Jarrett, V. Rangachari and S. E. Morgan, Biomacromolecules, 2020, 21, 4280–4293 CrossRef CAS PubMed.
- J. Beerten, J. Schymkowitz and F. Rousseau, Curr. Top. Med. Chem., 2013, 12, 2470–2478 CrossRef PubMed.
- Aggregation of Therapeutic Proteins, ed. W. Wang and C. J. Roberts, John Willey & Sons, Inc., New Jersey, 2010 Search PubMed.
- G. De Baets, J. Schymkowitz and F. Rousseau, Essays Biochem., 2014, 56, 41–52 CrossRef PubMed.
- A.-M. Fernandez-Escamilla, F. Rousseau, J. Schymkowitz and L. Serrano, Nat. Biotechnol., 2004, 22, 1302–1306 CrossRef CAS PubMed.
- A. Aguzzi and T. O’Connor, Nat. Rev. Drug Discovery, 2010, 9, 237–248 CrossRef CAS PubMed.
- S. C. Li, N. K. Goto, K. A. Williams and C. M. Deber, Proc. Natl. Acad. Sci. U. S. A., 1996, 93, 6676–6681 CrossRef CAS PubMed.
- C. Soto, M. S. Kindy, M. Baumann and B. Frangione, Biochem. Biophys. Res. Commun., 1996, 226, 672–680 CrossRef CAS PubMed.
- C. Soto, E. M. Sigurdsson, L. Morelli, R. Asok Kumar, E. M. Castaño and B. Frangione, Nat. Med., 1998, 4, 822–826 CrossRef CAS PubMed.
- C. Adessi and C. Soto, Drug Dev. Res., 2002, 56, 184–193 CrossRef CAS.
- L. O. Tjernberg, D. J. E. Callaway, A. Tjernberg, S. Hahne, C. Lilliehöök, L. Terenius, J. Thyberg and C. Nordstedt, J. Biol. Chem., 1999, 274, 12619–12625 CrossRef CAS PubMed.
- W. P. Esler, E. R. Stimson, J. R. Ghilardi, Y.-A. Lu, A. M. Felix, H. V. Vinters, P. W. Mantyh, J. P. Lee and J. E. Maggio, Biochemistry, 1996, 35, 13914–13921 CrossRef CAS PubMed.
- Y. Sugimoto, Y. Kamada, Y. Tokunaga, H. Shinohara, M. Matsumoto, T. Kusakabe, T. Ohkuri and T. Ueda, Biochem. Cell Biol., 2011, 89, 533–544 CrossRef CAS PubMed.
- S. C. Mansour, C. de la Fuente-Núñez and R. E. W. Hancock, J. Pept. Sci., 2015, 21, 323–329 CrossRef CAS PubMed.
- Y. Tokunaga, Y. Sakakibara, Y. Kamada, K. Watanabe and Y. Sugimoto, Int. J. Biol. Sci., 2013, 9, 219–227 CrossRef CAS PubMed.
- I. Kuznetsova, K. Turoverov and V. Uversky, Int. J. Mol. Sci., 2014, 15, 23090–23140 CrossRef PubMed.
- I. Kuznetsova, B. Zaslavsky, L. Breydo, K. Turoverov and V. Uversky, Molecules, 2015, 20, 1377–1409 CrossRef PubMed.
- T. Bar and Y. Okon, Can. J. Microbiol., 2011, 39, 81–86, DOI:10.1139/m93-011.
- J. Lawrence, S.-H. Lee, A. Abdilla, M. D. Nothling, J. M. Ren, A. S. Knight, C. Fleischmann, Y. Li, A. S. Abrams, B. V. K. J. Schmidt, M. C. Hawker, L. A. Connal, A. J. McGrath, P. G. Clark, W. R. Gutekunst and C. J. Hawker, J. Am. Chem. Soc., 2016, 138, 6306–6310 CrossRef CAS PubMed.
- S. Perrier, Macromolecules, 2017, 50, 7433–7447 CrossRef CAS.
- F. Piguet, H. Ouldali, F. Discala, M. F. Breton, J. C. Behrends, J. Pelta and A. Oukhaled, Sci. Rep., 2016, 6, 38675, DOI:10.1038/srep38675.
- K. Devanand and J. C. Selser, Asymptotic Behavior and Long-Range Interactions in Aqueous Solutions of Polyethylene oxide, 1991, vol. 24 Search PubMed.
- C. E. Stauffer, J. Phys. Chem., 1965, 69, 1933–1938 CrossRef CAS.
- F. Fischel-Ghodsian, L. Brown, E. Mathiowitz, D. Brandenburg and R. Langer, Proc. Natl. Acad. Sci. U. S. A., 1988, 85, 2403–2406 CrossRef CAS PubMed.
- A. Das, M. Shah and I. Saraogi, ACS Bio Med Chem Au, 2022, 2, 205–221 CrossRef CAS PubMed.
- D. Andersson, U. Carlsson and P.-O. Freskgård, Eur. J. Biochem., 2001, 268, 1118–1128 CrossRef CAS PubMed.
- J. Goldman and F. H. Carpenter, Biochemistry, 1974, 13, 4566–4574 CrossRef CAS PubMed.
- O. M. Pena, N. Afacan, J. Pistolic, C. Chen, L. Madera, R. Falsafi, C. D. Fjell and R. E. W. Hancock, PLoS One, 2013, 8, e52449 CrossRef CAS PubMed.
- H. Choe, A. S. Narayanan, D. A. Gandhi, A. Weinberg, R. E. Marcus, Z. Lee, R. A. Bonomo and E. M. Greenfield, Clin. Orthop. Relat. Res., 2015, 473, 2898–2907 CrossRef PubMed.
- M. Wieczorek, H. Jenssen, J. Kindrachuk, W. R. P. Scott, M. Elliott, K. Hilpert, J. T. J. Cheng, R. E. W. Hancock and S. K. Straus, Chem. Biol., 2010, 17, 970–980 CrossRef CAS PubMed.
- E. F. Haney, B. (Catherine) Wu, K. Lee, A. L. Hilchie and R. E. W. Hancock, Cell. Chem. Biol., 2017, 24, 969–980.e4 CrossRef CAS PubMed.
- D. E. Schlamadinger, J. E. Gable and J. E. Kim, J. Phys. Chem. B, 2009, 113, 14769–14778 CrossRef CAS PubMed.
- C. Mueller, M. A. H. Capelle, T. Arvinte, E. Seyrek and G. Borchard, Eur. J. Pharm. Biopharm., 2011, 79, 646–657 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3tb02102e |
| This journal is © The Royal Society of Chemistry 2024 |