
Balancing the relationship between the activity and stability of anode oxide-based electrocatalysts in acid for PEMWE electrolyzers
Yingying
Xu
ab,
Yingxia
Zhao
bc,
Zihui
Yuan
ab,
Yue
Sun
ab,
Shaomin
Peng
 ab,
Yuanhong
Zhong
abc,
Ming
Sun
*abc and
Lin
Yu
*ab
ab,
Yuanhong
Zhong
abc,
Ming
Sun
*abc and
Lin
Yu
*ab
aJieyang Branch of Chemistry and Chemical Engineering Guangdong Laboratory (Rongjiang Laboratory), Jieyang 515200, P. R. China
bGuangDong Engineering Technology Research Center of Modern Fine Chemical Engineering, School of Chemical Engineering and Light Industry, Guangdong University of Technology, 510006 Guangzhou, P. R. China
cGuangdong Yuntao Hydrogen Energy Technology Co., Ltd, 551040 Guangzhou, China
First published on 4th July 2024
Abstract
Fueled by renewable energy, the paradigm of “green hydrogen” production via water electrolysis stands as an imperative pathway toward the evolution of novel energy frameworks. Proton exchange membrane water electrolysis (PEMWE) emerges as a pivotal route for sustainable power generation, offering substantial advantages. However, the quest to enhance the efficiency of proton exchange membrane water electrolysis (PEMWE) encounters formidable obstacles, prominently centered on attaining a delicate equilibrium between heightened activity and prolonged stability of oxygen evolution reaction (OER) catalysts. The pursuit of OER electrocatalysts that epitomize efficiency, longevity, and cost-effectiveness under high operating potential and corrosive acidic conditions assumes paramount importance in propelling the frontiers of electrochemical water splitting. This review provides a comprehensive overview of recent advancements in both noble and non-noble metal oxides utilized for acidic oxygen evolution reaction (OER). It underscores the significance of analyzing the instability of catalysts based on the OER mechanism, while delving into effective strategies aimed at bolstering the activity and stability of IrO2, RuO2, and transition metal (TM) oxide electrocatalysts. Finally, some insights are provided into the indirect challenges confronting current anode catalysts employed in proton exchange membrane (PEM) electrolyzes, along with perspectives on future research avenues and potential directions.
1. Introduction
With the rapid development of the global economy, the growth of energy demand has become the focus of global attention.1,2 However, the limited reserves of fossil fuels, such as oil, coal and natural gas, and the environmental problems caused by their large-scale burning have become increasingly concerning.3–5 Therefore, there is an urgent need for countries around the world to find environmentally efficient green energy alternatives. In this context, hydrogen energy as a resource-rich, high-energy density, pollution-free green energy attracts much attention. Its wide application is considered to be one of the most promising solutions for energy conversion.6–9 With the development of science and technology, hydrogen production by electrolysis of water, as a sustainable energy mode of production, is receiving more and more attention and research.8,10–15To date, to realize the vision of green hydrogen production, leveraging renewable energy sources such as wind, solar, and tidal energy to drive water electrolysis for hydrogen production has been a promising approach.16,17 However, traditional alkaline electrolysis has limitations in terms of its dynamic response, especially when coupled with fluctuating renewable energy sources. This can lead to reduced overall efficiency and hinder the effective utilization of sustainable energy. Proton Exchange Membrane Water Electrolysis (PEMWE) holds great promise as a method for hydrogen production.18–20 It utilizes an acidic solid polymer electrolyte membrane, which offers several outstanding advantages over traditional electrolysis methods. One key advantage is its high efficiency. The solid polymer electrolyte membrane enables rapid proton transport, facilitating faster electrochemical reactions at the electrodes.21–23 This results in improved energy efficiency and higher hydrogen production rates compared to conventional alkaline electrolysis. Additionally, PEMWE systems offer greater flexibility and scalability. They can operate under a wide range of operating conditions and can be easily scaled up or down to match varying hydrogen production demands.24–26 This makes them suitable for applications ranging from small-scale distributed hydrogen production to large industrial-scale operations. Furthermore, PEMWE technology allows for better integration with renewable energy sources. The rapid response time of PEMWE systems makes them well-suited for coupling with intermittent renewable energy sources such as solar and wind power. This enables more efficient utilization of renewable energy and helps to address the intermittency challenges associated with these energy sources.27–29
In recent years, significant strides have been made in the development of effective metal oxide catalysts for acidic oxygen evolution reaction (OER), with notable examples including IrO2,30,31 RuO2,28,32–34 and various other transition metal oxides.13,35–38 However, a common challenge encountered by most metal oxides is their susceptibility to metal dissolution in acidic electrolytes, often leading to diminished stability. Previous studies39 have revealed an intriguing phenomenon: while the most reactive oxides in acids (Au ≪ Pt < Ir < Ru < Os) exhibit high reactivity, they paradoxically tend to be the most unstable materials (Au ≫ Pt > Ir > Ru ≫ Os). This intricate interplay between activity and stability is governed by the properties of metal cations and the potential conversion of stable metal cations with a valence state of n = +4 to unstable metal cations with a valence state greater than +4. Therefore, achieving the optimal anode material for proton exchange membrane water electrolysis (PEMWE) necessitates striking a delicate balance between stability and activity, ensuring that the dissolution rate remains neither too fast nor too slow.40,41
In this review, we comprehensively explore the latest developments in the fields of IrO2 based catalysts, RuO2 based catalysts, and non-noble metal oxide catalysts related to acidic oxygen evolution reaction (OER). We first introduced the working mechanism of acidic OER, mainly discussing the complex equilibrium dynamics between catalyst activity and stability. Building upon this understanding, the recent advancements in both noble and non-noble metal oxides as anodic catalysts in acidic environments are comprehensively reviewed. Finally, we analysed the indirect challenges faced by existing anode catalysts in proton exchange membrane (PEM) electrolysis cells and shared insights into future research approaches and potential directions (as shown in Fig. 1).
 | ||
| Fig. 1 Schematic diagram of the main content of acidic oxygen evolution reaction in this review. | ||
2. Mainstream OER mechanisms in acid
Understanding the oxygen evolution reaction (OER) mechanism is paramount for guiding the rational design of efficient catalysts. Currently, three main mechanisms are widely accepted for acidic OER: the adsorbate evolution mechanism (AEM), lattice oxygen evolution mechanism (LOM), and oxide pathway mechanism (OPM). A comprehensive comprehension of these mechanisms is instrumental in the development of superior OER electrocatalysts.2.1 Adsorbate evolution mechanism (AEM)
The adsorbate evolution mechanism (AEM) stands as the prevailing traditional OER mechanism, encompassing multiple intermediates and reaction steps. As illustrated in Fig. 2a and b, * denotes a catalytic active site on a specific surface. Initially, the water molecules bind to metal (M) active sites on the surface through a single electron process, resulting in the formation of *OH molecules at the M sites. Subsequently, the *OH molecule undergoes the first step of deprotonation, transitioning into the *O molecule. Following this, the *O molecule combines with a water molecule and undergoes a second oxidation step, resulting in the formation of the intermediate *OOH species. Eventually, *OOH undergoes a third dehydrogenation process, liberating oxygen molecules and reverting the original M site to its initial state. | ||
| Fig. 2 (a) Schematic diagram of the AEM pathway of the OER in acids. (b) The concrete path of the AEM. (c) Contour plot of theoretical overpotential as a function of ΔGO*–ΔGOH* and ΔGOH*.42 Copyright 2023, Wiley-VCH. (d) DEMS measurements of 16O18O and 16O16O signals from the reaction products for NiO in 1 M KOH.43 Copyright 2024, Wiley-VCH. | ||
The OER mechanism dominated by the AEM involves intricate four-electron processes and iterations of intermediates *OH, *O, and *OOH, contributing to overpotential. These steps are intricately dependent on the binding energies involved. According to Sabatier's principle, the ideal oxygen binding energy should be moderate. Excessive binding energy can hinder desorption, while insufficient binding energy leads to inadequate activity. Typically, the step with the largest difference in binding energy is considered the rate-limiting step (RLS) of the reaction. In the current OER, the key overpotential primarily arises from the oxidation of *OH and the formation of *OOH. Optimizing the binding strength of intermediates is pivotal for enhancing OER catalyst performance. To this end, a descriptor for the binding energy difference between *OH and *OOH intermediates, denoted as ΔGOH*–ΔGOOH*, proves valuable for evaluating catalyst performance and predicting OER activity. Theoretical calculations play a crucial role in achieving the optimal binding energy of oxygen intermediates in catalysts, facilitating the precise design of efficient OER catalysts. For instance, Yu et al.42 investigated the binding energy of oxidation intermediates under the interaction between Co doping and cation vacancies using density functional theory calculations. Their findings revealed a scaling relationship between the energy of O* and OOH* provided by metal active sites, expressed as GOOH* = 1.08GOH* + 2.97. Moreover, the overpotential η was found to be related to ΔGO*–ΔGOH*, showing a linear increase. This indicates that Co doping and cationic vacancies can reduce the OER overpotential of NiFe OOH to 0.99 V and 0.82 V, respectively. The identification of AEM pathways relies on monitoring key intermediates, for which effective methods such as in situ Raman spectroscopy, in situ XAS, and isotope-labeled mass spectrometry (DEMs) have been widely employed in recent years. Yan et al.43 labeled the original NiO catalyst with O18 and obtained labeled NiO18 for the OER electrode reaction. In the DEM results, NiO18 exhibited a signal of O,32 indicating that all the generated oxygen originated from water molecules in the electrolyte. This confirmed that the OER performance on the NiO surface is primarily governed by the mechanism of adsorbate evolution.
Addressing the challenges associated with the adsorbate evolution mechanism requires a thorough investigation into the dynamic behavior of adsorbates on catalyst surfaces under acidic electrolyzed water conditions. The complexity and diversity of the reaction, influenced by protons and oxygen in the solution, necessitate a deeper understanding and control of this mechanism. Moreover, the surface structure and composition of catalysts further complicate the evolution of adsorbates. To overcome these challenges, efficient experimental and theoretical methods must be developed to analyze and predict the evolution process of adsorbates accurately.
2.2 Lattice oxygen evolution mechanism (LOM)
In the mechanism of lattice oxygen evolution (LOM) in acidic electrolyzed water, oxygen atoms within the structure of oxide catalysts, known as lattice oxygen, play a pivotal role in the water decomposition process. Interacting with water and protons in solution on the catalyst surface, lattice oxygen becomes involved in the oxygen evolution process. The activation of lattice oxygen is achieved through the evolution of adsorbates, inducing the saturation of active sites on the catalyst surface and facilitating water decomposition. In the schematic diagram shown in Fig. 3a and b, water molecules adsorbed on the metal active site undergo dehydrogenation to form M–O* intermediates, a process that is identical to the adsorption evolution mechanism. Subsequently, *O can bind to oxygen atoms within the lattice of the catalyst, leading to the release of oxygen and the formation of abundant oxygen vacancies (VO) in situ. These vacancies are then replenished by water molecules, forming *OH, which ultimately oxidizes dehydrogenation. In the lattice oxygen evolution mechanism (LOM) pathway, the thermodynamic barrier of the oxygen evolution reaction (OER) is diminished as the formation of the intermediate *OOH is circumvented, and the plentiful oxygen atoms within the catalyst lattice are efficiently utilized. Consequently, catalysts dominated by the LOM pathway exhibit outstanding activity in the OER process. However, the involvement of lattice oxygen in the lattice oxygen evolution mechanism (LOM) may potentially compromise the structural integrity of the crystal and hasten the dissolution of the metal, thereby diminishing operational stability. Consequently, research efforts focusing on the LOM aim to effectively utilize oxygen vacancies while ensuring the stability of the crystal structure. For instance, Sargent et al.44 precisely introduced strontium (Sr) and iridium (Ir) doping into rutile-type RuO2, which modulates the electronic structure of ruthenium (Ru) and optimizes the binding energy of the oxidizing species at the high-valence Ru sites, thus partially inhibiting the LOM. The optimized catalyst exhibits an overpotential of 190 mV and maintains stability for more than 1500 hours (@10 mA cm−2). Xing et al.45 optimized the lattice oxygen evolution mechanism (LOM) by modifying the binding properties of Ru–O in the high entropy oxide (HEO) of ruthenium (Ru) and enhancing the lattice disorder in the HEO. | ||
| Fig. 3 (a) Schematic diagram of the LOM pathway of the OER in acids. (b) The concrete path of the LOM. (c) The electrochemical cell configuration used in DEMS measurements. (d) DEMS signals of 32O2 (16O16O) and 34O2 (16O18O) from the reaction products for 18O-labeled SrRuIr catalysts in H218O aqueous sulfuric acid electrolyte and corresponding CV cycles.44 Copyright 2021, American Chemical Society. | ||
Advancing the understanding of the lattice oxygen mechanism is paramount for overcoming the challenges posed by the stability of lattice oxygen in acidic electrolyzed water. The dissociation and deactivation of lattice oxygen are exacerbated by the harsh conditions of high temperature and high pressure in acidic environments, further underscoring the complexity of this mechanism. To address these challenges, it is imperative to develop stable lattice oxygen structures, elucidate the active sites involved in lattice oxygen reactions, and explore novel reaction conditions. Leveraging advanced characterization techniques and theoretical calculation methods can facilitate in-depth studies of the active sites and reaction mechanisms of lattice oxygen in acidic electrolyzed water. This knowledge can offer valuable theoretical insights for the rational design of catalysts with enhanced activity and stability.
2.3 Oxide path mechanism (OPM)
The recently proposed oxide pathway mechanism (OPM) offers a promising explanation for the exceptional activity observed in acidic OER catalysts, integrating aspects of both the adsorbate evolution mechanism and the lattice oxygen mechanism. Central to the OPM is the direct coupling of oxygen atoms (O–O) at two adjacent metal active sites, following the adsorption and deprotonation of water molecules. This mechanism circumvents the formation of *OOH intermediates and minimizes damage to the catalyst lattice by avoiding the generation of oxygen vacancies. The optimal metal atomic spacing enables efficient oxygen release through direct coupling, making the OPM an attractive mechanism for researchers seeking highly efficient OER catalysts in recent years. In general, acidic OER catalysts governed by the oxide pathway mechanism (OPM) impose stricter demands on the geometric arrangement of metal active sites. Symmetric bimetallic sites with precise atomic distances are anticipated to facilitate the OPM. Mori et al.46 delved into the OER characteristics of perovskite-type oxides hinged on the covalent bond networks of Cu2+ and Fe4+ transition metal ions. Specifically, upon doping Fe4+ and Cu2+ into CaCu3Fe4O12, the Fe–O–Fe bond undergoes significant bending, reducing the oxygen–oxygen distance to 2.6 Å through the highly bent Fe–O–Fe bond (140°), thereby directly fostering the formation of the O–O bond and activating the OPM (see Fig. 4d–f). Conversely, the straightforward SrFeO3 (SFO) struggles to engage directly with oxygen molecules due to its large oxygen–oxygen distance (≈3.9 Å). In this scenario, Cu2+ and Fe4+ doping in CaCu3Fe4O12 circumvents one of the two potential rate-determining steps in the OER process (i.e., deprotonation of oxygen and hydroxide groups to form peroxides), resulting in heightened OER activity. | ||
| Fig. 4 (a) Schematic diagram of the OPM pathway of the OER in acids. (b) The concrete path of the OPM. (c) The schematic diagram of dual-site adsorbate evolution mechanisms for coupled O–O bonding during the OER (blue for Co atoms, red for Fe atoms, and yellow for S atoms).47 Copyright 2024, Springer Nature. (d–f) OH− adsorbed surfaces for SFO and CCFO and the corresponding OER mechanism.46 Copyright 2015, Springer Nature. | ||
Hence, confining the metal active center within a covalent network with symmetric atomic centers (e.g., α-MnO2) stands as one of the effective strategies for designing efficient OER catalysts via the OPM. Lee's team48 utilized a cation exchange method, substituting Ru into MnO2 crystals to form periodic arrangements of Ru atomic chains. XAS and aberration-corrected HAADF-STEM results showed a reduction in the distance between Ru atoms in Ru/MnO2 from the original 3.1 Å to 2.9 Å, facilitating the coupling of O–O radicals. The signal of the key intermediate *O–O* bond directly related to the OPM was demonstrated in electrochemical in situ experiments. Additionally, the authors recorded the changes in the ICP-OES values of Ru and Mn in solution during the OER process. The results indicate the dissolution–redeposition process of Ru, albeit at the expense of Mn dissolution. However, due to in situ self-reconfiguration, the intrinsic OER activity of Ru is more than 600 times higher than that of commercial RuO2. These findings unveil the possibility of triggering the OPM by designing the geometry of the metal active site.
Overall, we have described three typical OER mechanisms in acidic electrolytes: AEM, LOM, and OPM. In the AEM, various oxygen reaction intermediates such as *OH, *O, and *OOH are generated, with their binding energies following a scaling relationship ΔGOOH = ΔGOH + 3.2 ± 0.2 eV, and a theoretical limit of about 370 ± 100 mV. The LOM reduces the theoretical overpotential needed for the OER by avoiding the formation of *OOH intermediates, but it may damage the catalyst lattice structure, posing challenges for long-term stability. New mechanisms like the OPM have emerged in recent years, but specific reaction steps require further research to clarify the intrinsic relationship between activity and stability. It is important to note that these mechanisms are not mutually exclusive, and many new OER mechanisms have evolved from combinations or extensions of the AEM, LOM, and OPM. In Peng's study,49 the structure of Ru-doped Co3O4 was finely tuned at the atomic level, embedding Ru atoms in cation vacancies to activate a proton donor-receptor function (PDAM) mechanism. This optimized mechanism resulted in significantly lower overpotential, with the Ru(anc)–Co3O4 catalyst requiring only 198.5 mV overpotential at 10 mA cm−2 for the OER. In another study by Xu et al.,50 non-catalytic Zn2+ was introduced into CoOOH to alter the OER mechanism from the AEM to the LOM by adjusting the local configuration. They proposed that the OER via the LOM occurs only when two adjacent lattice oxygen ions are oxidized and combined, and two adjacent lattice oxygen vacancies are formed without breaking metal–oxygen bonds. These findings demonstrate how precise control over the catalyst composition and structure can dictate the underlying OER mechanism, leading to enhanced catalytic performance.
3. Noble metal IrO2 OER electrocatalysts
In the current landscape, IrO2-based catalysts stand at the pinnacle under acidic conditions, owing to their dual advantages of high activity and stability. Strasser et al.30 observed that the OER activity in acidic solutions followed the trend of Ru > Ir > Pt, yet only IrO2 nanoparticles retained substantial OER stability along with their high activity. Certainly, Ir-based catalysts pose challenges due to their scarcity and cost. Moreover, the OER properties of IrO2 are influenced by various factors such as particle size, crystallinity, and morphology, among others. Typically, these factors are controlled through different synthesis conditions or methods. To address these challenges, several effective strategies have emerged this year to reduce Ir loading while maintaining high catalytic performance. In this section, we review three such strategies—morphology engineering, perovskite-type oxides, and support effect—that enhance the catalytic activity of IrO2-based catalysts.3.1 Morphology engineering
Morphology engineering of catalysts entails adjusting the morphology or surface structure to enhance their catalytic performance. This approach allows control over the particle size, shape, surface structure, and crystal structure, thereby optimizing catalyst activity, selectivity, stability, and durability. In recent years, many IrO2 catalysts with specific morphologies have been synthesized, such as nanoribbons,51,52 nanowires,53,54 nanoparticles55 and so on. Shen et al.56 utilized metal–organic frameworks (MOFs) as a template to coat IrCo nanoparticles onto high-density carbon nanotubes (IrCo@CNT/CC) through a dicyandiamide-assisted pyrolysis strategy. The resulting IrCo@CNT/CC composite was prepared with a very low Ir loading (0.027 mg cm−2) and exhibited remarkable stability, demonstrating continuous electrolysis in 0.5 M H2SO4 electrolyte for 90 hours. Lee et al.57 synthesized ultrafine IrO2 nanowires with a diameter of 2 nm (Fig. 5c), exhibiting an overpotential of 0.313 V at a current density of 10 mA cm−2. By optimizing the synthesis conditions, the Ir loading was reduced, resulting in a mass activity of 51.6 A g−1. The catalyst demonstrated remarkable stability, operating continuously for 200 hours at a high current density of 2 A cm−2. | ||
| Fig. 5 (a) SEM image showing the uniform distribution of the IrO2NR. (b) A typical long IrO2NR with a length of 22.53 μm.51 Copyright 2023, Springer Nature. (c) TEM image of IrO2 nanoneedles. (d) Initial OER performance of IrO2.57 Copyright 2017, Wiley-VCH. (e) The SEM image of 1T-IrO2, showing its ultrathin morphology. (f) Polarization curves of 1T-IrO2, rutile-IrO2, and commercial catalysts (C-IrO2 and C-Ir/C) in O2-saturated 0.1 M HClO4 electrolyte with iR-correction.58 Copyright 2021, Springer Nature. (g–i) The nanosheet structure of 2D IrIrOx/C-20. (j) Water oxidation polarization curves recorded with a linear scan of potential at 2 mV s−1 for Ir–IrOx/C, commercial crystalline c-IrO2, Ir/C, and carbon black.59 Copyright 2022, American Chemical Society. | ||
Furthermore, extensive studies have been conducted on two-dimensional materials made of IrO2. Shao et al.58 reported the synthesis of 1T-phase-IrO2 nanosheets in strongly alkaline media through a combination of mechanochemical and heat treatment methods, as depicted in Fig. 5e. Atomic force microscopy (AFM) analysis revealed that the thickness of the ultra-thin IrO2 nanosheets was in the range of 3–5 nm. Linear sweep voltammetry (LSV) results (shown in Fig. 5f) indicated that 1T-IrO2 exhibited an ultralow overpotential of 197 mV (at 10 mA cm−2), which was 100 mV lower than that of rutile-IrO2. Further extended X-ray absorption fine structure (EXAFS) analysis suggested that the optimized 1T phase Ir active sites and ultra-thin 2D structure were crucial for achieving high-performance acidic OER catalysis. Zhao et al.59 synthesized ultrathin Ir–IrOx/C nanosheets with ordered interlayer spaces through a nano-self-assembly effect, as illustrated in Fig. 5g and h. On these nanosheets, IrO2 particles were well dispersed, forming a state of Ir/IrO2 coexistence. Due to the abundant pore structure of Ir–IrOx/C and the electronic interaction between Ir and IrO2, the prepared Ir–IrOx/C electrocatalysts exhibited the lowest overpotential (η) of 198 mV at 10 mA cm−2 for the OER in acidic media. This report is of significant importance for the application of two-dimensional materials in acidic OER. In general, apart from nano-self-assembled materials, the morphology of most materials relies on the carrier to prevent sintering. Uniform nucleation points are formed on the carrier with the help of other additives. The interaction between the support and the catalytic metal will be discussed in a later section.
3.2 Perovskite-type oxides
The dissolution of Ir4+ cations and the uncontrolled crystallization of Ir4+ cations are primary factors that affect the stability of catalysts in acidic oxygen evolution reaction (OER). Recent literature has proposed a strategy of incorporating IrO2 into frameworks such as perovskite. Perovskite-type oxides, denoted as ABX3, typically feature cations like alkaline earth metals or rare earth metals in the A site, while the B site commonly hosts transition metals like Co, Cu, Ir, and Ru. The resulting octahedral BO6 units, formed with oxygen atoms, typically exhibit robust stability and corrosion resistance, ensuring sustained performance and longevity across various reaction environments. Hence, Ir-based perovskites have garnered significant attention for acidic OER, where IrO6 or RuO6 octahedra serve as the active sites. Using DFT calculations, Jaramillo et al.60 discovered that under relevant experimental conditions, the thermodynamic driving force for the formation of Sr2+ and rutile-type IrO2 from each strontium (Sr) atom is 3.2 eV. The formation of IrOx sites may be responsible for the high activity of SrIrO3. The subsequent characterization after the reaction further confirmed the dissolution of strontium and the in situ formation of IrOx.Moreover, the unique structure of perovskite-type iridium oxides has also recently garnered widespread attention. Zou et al.61 recently reported a honeycomb layered strontium iridium salt (SrIr2O6) and demonstrated its potential as an efficient OER electrocatalyst with strong structural stability in acids. In stark contrast to conventional iridium-based catalysts, SrIr2O6 distinguishes itself by upholding the crystalline integrity of both its bulk and surface structures throughout the OER process, thus sidestepping the formation of an amorphous active phase. Among them, the short Sr–O bond and strong Sr–O interaction are fundamental factors contributing to the excellent structural stability of SrIr2O6 (Fig. 6a). Impressively, SrIr2O6 outperforms the benchmark catalyst IrO2 by a factor of ten in terms of efficiency, showcasing an exceedingly low total iridium leaching rate of a mere 0.03% during extended open electrolytic reduction tests, while maintaining catalytic activity for over 300 hours. Friedrich62et al. reported the development of an anode catalyst utilizing Sr2CaIrO6, boasting a loading density of merely 0.2 mg cm−2, a striking ten-fold reduction compared to commercially available catalyst-coated membranes (CCMs) laden with iridium. In practical proton exchange membrane water electrolysis (PEMWE) cells integrating this innovative anode, exceptional performance was achieved, with a voltage of 1.78 V recorded at a current density of 2 A cm−2, operating at 80 °C under ambient pressure conditions. Impressively, this remarkable performance was sustained for over 1000 hours, underscoring the catalyst's robust stability. Following this extended operational period, the surface morphology underwent a notable transformation, adopting a sponge-like structure attributed to the rapid leaching of calcium and strontium. Interestingly, this altered morphology featured small nanoscale domains of Ir–O–H, attributed to the small amorphous region of about 2 nm, believed to contribute significantly to the catalyst's superior oxygen evolution reaction (OER) performance. Koper et al.31 introduced a groundbreaking oxygen evolution catalyst featuring iridium double-perovskite. Double perovskites (DPs) are compounds with the generic formula A2BB′O6, with A denoting a large cation and B and B′ smaller cations (Fig. 6d). A wide range of compounds can be prepared by different combinations of A, B and B′ cations, which allows fine tuning of DPs. Remarkably, this catalyst boasts a 32 wt% reduction in iridium content compared to IrO2, yet demonstrates over three-fold higher activity in acidic environments. The 3D network of corner sharing octahedra is a necessary prerequisite for enhancing the activity of Ir DPs and their chemical stability under anode working conditions. Moreover, this double-perovskite strategy holds promise for extension to other noble metal oxide systems, such as ruthenium (Ru), platinum (Pt), and beyond.
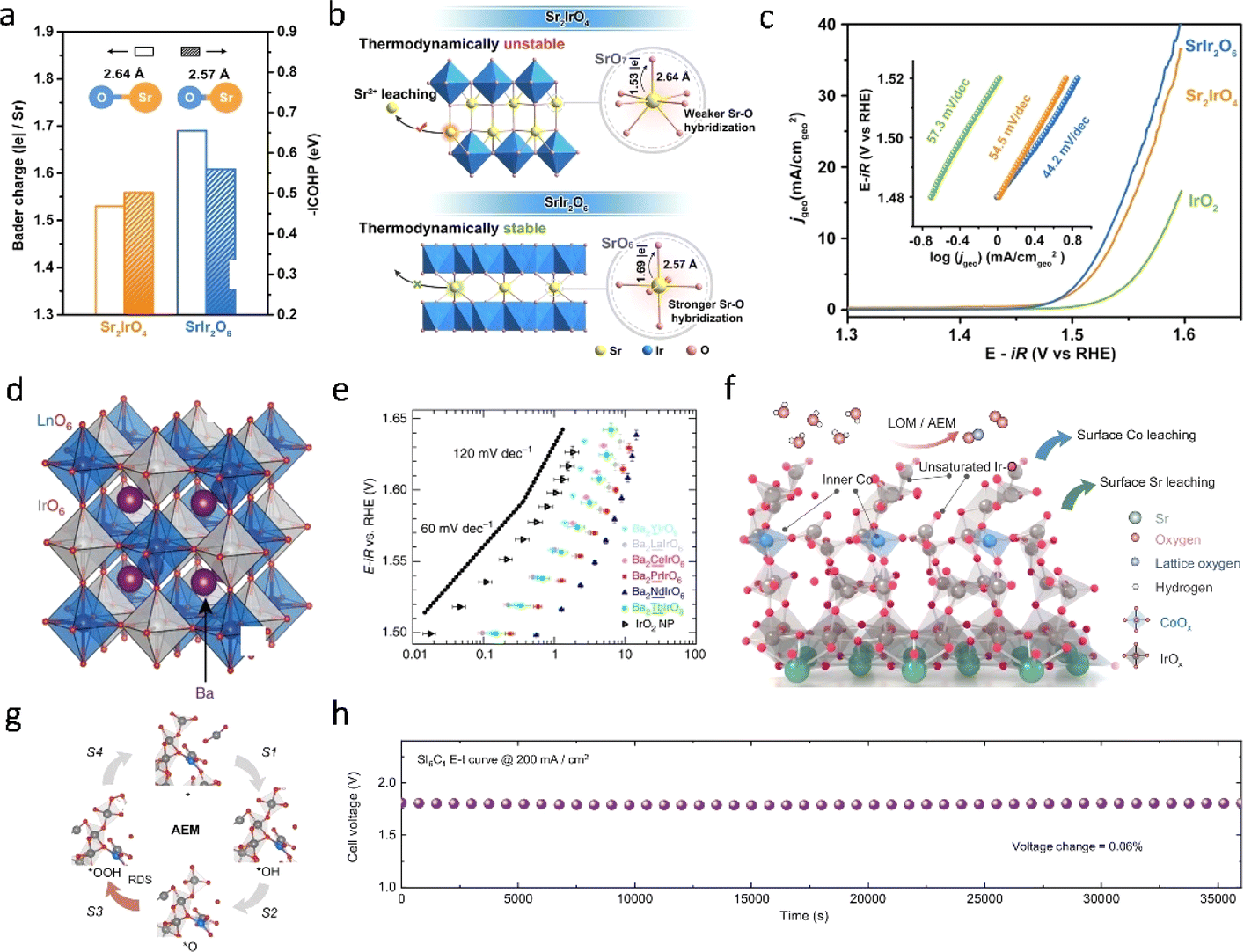 | ||
| Fig. 6 (a) Sr–O bond length, the integrated −ICOHP values of the Sr–O bond, and the Bader charge analysis of SrIr2O6 and Sr2IrO4. (b) Schematic showing the key factors controlling the structural stability of SrIr2O6 and Sr2IrO4. (c) Linear polarization curves for the OER of SrIr2O6, Sr2IrO4, and IrO2 at 1 mV s−1 in a 0.1 M HClO4 electrolyte, with the corresponding Tafel slopes in the inset.61 Copyright 2023, American Chemical Society. (d) Crystal structure of a generic Ba2MIrO6 DP. (e) OER activity in 0.1 M HClO4 of Ba2MIrO6, compared with the benchmark activity of IrO2 nanoparticles; the dotted lines show Tafel slopes of 60 and 120 mV per decade.31 Copyright 2016, Springer Nature. (f) OER catalytic mechanism diagram of the Co doped SrIrO3 catalyst. (g) Adsorbate evolution mechanism (AEM) diagram. (h) OER performance of the samples.63 Copyright 2024, Springer Nature. | ||
In addition to the dissolution of the A site, the evolution of the B site also significantly influences the activity of IrO2, which contributes to understanding the mechanism of acidic OER in perovskite-type Ir oxides. Gao et al.63 elucidated the crucial role of Co dissolution in acidic OER and the impact of surface and bulk Co on IrOx sites. Using in situ experiments and DFT calculations, it was revealed that Co initially dissolves on the catalyst surface under acidic conditions, leading to decreased stability of surface oxygen in SrIrO3 and removal of some bridging lattice oxygen, ultimately resulting in the formation of a low-coordination IrOx structure. Subsequently, adsorbent filling and the LOM reach a dynamic equilibrium, forming a unique mechanism called lattice oxygen promoted acidic evolution mechanism (LOPAEM) that is dominated by the adsorbate evolution mechanism (AEM) and promoted by the LOM.
All in all, limiting IrO2 to perovskite structures can effectively reduce the loading of traditional IrO2-based materials while achieving high-quality activity. The high activity is mainly attributed to the dissolution of the IrOx phase, typically accompanied by the collapse of the perovskite structure. Thus, ensuring the long-term stability of perovskite-type iridium oxides remains a significant challenge. To address this, effective characterization or computational methods are essential to further understanding the catalyst deactivation mechanism. Approaches such as combining Pourbaix plots with in situ data can aid in comprehending the working mechanism of IrO2 and offer crucial insights for the design and synthesis of high-performance perovskite-type iridium oxides.
3.3 Support effect
Introducing a carrier into IrO2 is an effective strategy for reducing Ir loading, enhancing catalyst particle dispersion and optimizing pore structure, thereby maximizing the specific surface area of activity. Beyond offering a suitable surface and mechanical strength, the carrier can optimize the microcrystalline concentration of the active component during preparation, thereby mitigating or preventing sintering and reducing the active component content.64 Additionally, the support can foster strong metal–support interactions (SMSIs) with the active component, such as electron interaction and spillover. These interactions further contribute to enhancing catalytic performance. Jian's group recently unveiled an innovative Ir-based catalyst that capitalizes on metal–carrier interactions to modulate the active species of IrOx in the oxygen evolution reaction (OER). Their Au@MnO2–IrOx core–shell nanoparticles (NPSs) were synthesized via a straightforward method, yielding nanoparticles with a distinctive nanostructure featuring highly dispersed IrOx clusters on an amorphous MnO2 shell. With an overpotential of 230 mV at 10 mA cm−2, these nanoparticles outperformed traditional Ir catalysts. In situ surface-enhanced Raman spectroscopy (SERS) revealed the oxidation of Ir4+ and the adsorption of *O on IrOx, optimizing the electrocatalytic capability of the catalyst. This suggests that pre-activation of the Ir center facilitates superoxide formation. Furthermore, density functional theory (DFT) calculations (as shown in Fig. 7b) demonstrated that the MnO2 substrate optimized the binding energy of intermediates on IrOx, facilitating O–O coupling and enhancing the OER rate. Xiong et al.65 reported on IrO2 particles loaded onto graphene, with morphometric characterization revealing a high dispersion of IrO2 particles on reduced graphene oxide (rGO). The optimized IrO2@rGO-350 exhibited an overpotential of 50 mV at a current density of 10 mA cm−2. The rGO serves as a carrier, preventing the agglomeration of IrO2 nanoparticles and resulting in a high catalytic site surface area. Additionally, the rGO support promotes electron transfer between IrO2 nanoparticles and itself. | ||
| Fig. 7 (a) Schematic illustration of Au@MnO2–IrOx core–shell nanoparticle-enhanced Raman spectroscopy for an in situ electrochemical study. (b) DFT-calculated Gibbs free energies of OER pathways on MnO2–IrO2 and IrO2. (c) LSV polarization curves of Au@MnO2–IrOx, Au@Ir, Au@MnO2 NPs, and commercial IrO2.66 Copyright 2023, Royal Society of Chemistry. (d) Schematic structure of Ir/Nb2O5−x. (e) In situ Ir L3-edge XANES spectra of Ir/Nb2O5−x at different potentials. (f and g) Electrocatalytic performance of Ir/Nb2O5−x.67 Copyright 2022, Wiley-VCH. (h) The illustration of oxygen diffusion in the ZrOx/TaOx interface. (i) Chronopotentiometry curve of the PEM electrolyzer with Ir–ZrTaOx as the anode operated at 1 A cm−2 at 80 °C with a Nafion 117 membrane.68 Copyright 2023, Elsevier. | ||
Indeed, an acidic environment imposes limitations on the use of traditional carbon-based supports due to their solubility in acid. Consequently, inert carriers such as Nb2O5, TiO2,69,70 SnO2,71–73 and TaOx74 have garnered significant attention. These supports remain stable under acidic conditions, providing a robust support platform for catalysts without undergoing dissolution or degradation. As a result, IrO2 catalysts supported on these inert supports exhibit enhanced stability and longevity, making them ideal candidates for various electrochemical applications in acidic media. Xing et al.67 conducted a comprehensive study using Nb2O5−x supported iridium (Ir/Nb2O5−x) as a model catalyst, where they monitored the dynamic evolution of the catalyst under operating conditions and uncovered the robust interaction between the carrier and metal. In the Ir/Nb2O5−x system, oxygen migration from the carrier to the metal Ir nanoparticles facilitated the formation of Ir–O coordination structures. Additionally, excess Ir–O species could be re-incorporated into Nb4+ ions within the Nb2O5−x support. This dynamic migration of interfacial oxygen enabled the prepared catalyst to effectively maintain the oxidation state of Ir–O even at high potentials, thus ensuring the stability of IrOx. In practical performance tests, the catalyst achieved a remarkable current density of 3 A cm−2 at a potential of 1.839 V in proton exchange membrane water electrolysis (PEMWE) cells and demonstrated stability for over 2000 hours at a current density of 2 A cm−2 (Fig. 7d–g).
The continuous triggering of the lattice oxygen mechanism (LOM) by the carrier/metal catalyst inevitably leads to the destruction of the carrier's crystal structure. In addition to selecting carriers with high acid insolubility, modifying the carrier through composite formation is employed to enhance the stability of the carrier. For instance, Zhang et al.68 discovered that the formation of heterogeneous interfaces (Fig. 7h) between ZrOx and TaOx in the support could potentially construct an oxygen diffusion pathway, thereby enhancing the stability. Leveraging this insight, they synthesized a supported catalyst (Ir–ZrTaOx), exhibiting mass-specific activity two orders of magnitude higher than conventional IrO2, capable of sustaining electrolysis for 1000 hours under operating conditions. In a PEMWE single cell, only 1/4 of the conventional Ir dosage was required to operate for over 200 hours (Fig. 7i), with a decay rate of only 4.7 μV h−1. In situ Raman and XAS analyses revealed that oxygen diffusion pathways between ZrOx/TaOx interfaces protected the carriers from over-oxidation and detrimental structural re-modelling, thus enhancing operational stability.
At present, the exploration of supported IrO2-based catalysts remains a forefront area of research, and understanding the interaction between the carrier and metal is crucial for designing OER-friendly IrO2 catalysts. While attributing the support-induced metal–support interaction (SMSI) effect solely to electron interaction may be oversimplified, it poses an important challenge to leverage existing characterization techniques to comprehensively address the promotion of acidic OER by support/metal interaction in IrO2-based catalysts. Building upon current characterization levels involves employing advanced techniques such as in situ spectroscopy, X-ray absorption spectroscopy75 (XAS), X-ray photoelectron spectroscopy (XPS), and transmission electron microscopy (TEM) to elucidate the structural and electronic changes occurring at the support/metal interface during OER operation. Additionally, computational methods like density functional theory (DFT) calculations can provide valuable insights into the nature of support/metal interactions and their influence on OER activity. By integrating experimental and theoretical approaches, researchers can gain a deeper understanding of the mechanisms underlying the enhanced OER performance of supported IrO2 catalysts, paving the way for the rational design of more efficient catalysts for acidic electrolysis applications.
Overall, although the oxygen evolution reaction (OER) activity of IrO2 is lower than that of RuO2, its excellent long-term stability makes it more widely used in acidic OER catalysis. The traditional AEM of IrO2-based catalysts has been reported in a large amount of literature (Table 1); DFT simulations and in situ characterization techniques have been employed to investigate this mechanism, highlighting the formation of OOH* intermediates as the main limiting factor contributing to the high overpotential of IrO2. Consequently, researchers have explored various synthetic methods to reduce the iridium load while preparing catalysts with high acidic OER activity and thermal stability. Future research will be pivotal in balancing the activity and stability of IrO2 catalysts by investigating their reaction mechanisms more thoroughly.
| Catalyst | In a three-electrode system | In a PEMWE | Ref. | |||
|---|---|---|---|---|---|---|
| η 10 (mV) | Stability | Pathway | Stability | T (°C) | ||
| M–RuIrFeCoNiO2 | 189 | 120 h @ 10 mA cm−2 | AEM | 500 h @ 1 A cm−2 | 80 | 76 |
| SI6C1 | 280 | — | AEM/LOM | 10 h @ 200 mA cm−2 | 85 | 63 |
| Mn0.98Ir0.02O2 | 100 | 48 h @ 100 mA cm−2 | AEM | — | — | 77 |
| IrO2@TaB2 | 288 | 120 h @ 10 mA cm−2 | AEM | 100 h @ 1 A cm−2 | 80 | 74 |
| IrCo@CNT/CC | 241 | 90 h @ 10 mA cm−2 | AEM | — | — | 56 |
| SrIr2O6 | 303 | 300 h @ 10 mA cm−2 | AEM | — | — | 61 |
| Sr2CaIrO6 | — | — | — | 100 h @ 1 A cm−2 | 80 | 62 |
| Ir/D-ATO | 293 | 20 h @ 10 mA cm−2 | — | 250 h @ 1 A cm−2 | 80 | 72 |
| M–RuIrFeCoNiO2 | 190 | 120 h @ 10 mA cm−2 | AEM | 500 h @ 1 A cm−2 | 80 | 76 |
| Ir/Nb2O5−x | 218 | 100 h @ 10 mA cm−2 | LOM | 2000 h @ 2 A cm−2 | 80 | 67 |
| 1T-IrO2 | 197 | 50 h @ 50 mA cm | — | — | — | 58 |
4. Noble metal RuO2 OER electrocatalysts
Compared to the expensive Ir-based systems, Ru possesses abundant reserves and is regarded as a promising alternative for OER catalysis due to its lower overpotential.1,2,24,25,78 However, the dissolution of Ru at elevated potentials due to over-oxidation poses a significant challenge for the practical application of RuO2-based catalysts.29,79 Mayrhofer et al.80 provided a comprehensive analysis of the dissolution behaviors of Ru and other noble metals, as illustrated in Fig. 8a–d. It can be observed that, relative to Ir and Pt, Ru displayed a higher dissolution rate across all tested currents, underscoring the metastable nature of Ru. The dissolution of RuO2 in acid primarily arises from the highly covalent nature of the Ru–O bonds within the RuO2 lattice, which triggers the lattice oxygen mechanism (LOM). This mechanism results in the formation of oxygen vacancies and the subsequent dissolution of RuO2 peroxide, as illustrated in Fig. 8.33,81,82 Ling et al.83 pointed out in the calculated RuO2 Pourbaix diagram (Fig. 8e) that RuO2 gradually dissolves to form the highly oxidized RuO4− species within the OER working range, a conclusion supported by in situ XPS data (Fig. 8f). Thus, striking a delicate equilibrium between the stability and activity of RuO2 remains a formidable task. A pivotal challenge lies in mitigating the dissolution of RuO2 at elevated potentials. In this section, we present a review of several effective strategies to optimize the activity and stability of RuO2, encompassing heterometallic doping, heterogeneous structure engineering, and defect engineering (Table 2). | ||
| Fig. 8 (a) Schematic diagram of the structural evolution of RuO2 under acidic conditions.81 Copyright 2024, Springer Nature. (b–d) Dissolution of Ru, Ir, and Pt under steady-state galvanostatic conditions.80 Copyright 2014, Wiley-VCH. (e) Calculated Pourbaix diagrams of RuO2; the potentials are referenced to the standard hydrogen electrode (SHE).83 (f) Ru3+ and Ru4+ content ratios corresponding to the in situ Ru 3d XPS spectra recorded at applied potentials ranging from 1.00 to 2.00 V vs. RHE.83 Copyright 2022, Springer Nature. | ||
| Catalyst | In a three-electrode system | In a PEMWE | Ref. | |||
|---|---|---|---|---|---|---|
| η 10 (mV) | Stability | Pathway | Stability | T (°C) | ||
| Ru/Ti4O7 | 150 | 500 h @ 10 mA cm−2 | — | 200 h @ 200 mA cm−2 | 80 | 84 |
| Mn–RuO2-450 | 168 | 150 h @ 10 mA cm−2 | — | 150 h @ 50 mA cm−2 | 60 | 85 |
| MnxRu1−xO2/MnO2/CFs | 161 | 600 h @ 10 mA cm−2 | — | 24 h @ 500 mA cm−2 | 70 | 86 |
| RuO2–CeO2 | 180 | 1000 h @ 10 mA cm−2 | AEM/OPM | — | — | 87 |
| RuO2/BNNS | 180 | 350 h @ 10 mA cm−2 | AEM | 200 h @ 200 mA cm−2 | 80 | 88 |
| Ru3PbOx | 201 | 72 h @ 10 mA cm−2 | — | 300 h @ 100 mA cm−2 | 25 | 89 |
| RuO2/Ag | 690 (η2A) PEM | 120 h @ 200 mA cm−2 | — | 100 h @ 1 A cm−2 | 90 | |
| SnRuOx | 194 | 250 h @ 100 mA cm−2 | AEM | 1300 h @ 1 A cm−2 | 50 | 91 |
| Nb0.1Ru0.9O2 | 204 | 360 h @ 200 mA cm−2 | — | 100 h @ 300 mA cm−2 | 80 | 92 |
| (Ru–W)Ox | 170 | 300 h @ 10 mA cm−2 | AEM | 500/300/50 h @ 0.1/0.5/1.0 A cm−2 | 80 | 93 |
| Ni–RuO2 | 214 | 200 h @ 10 mA cm−2 | AEM | 1000 h @ 200 mA cm−2 | 25 | 94 |
| SrRuIr | 190 | 1500 h @ 10 mA cm−2 | AEM | 150 h @ 1 A cm−2 | 80 | 44 |
| W0.2Er0.1Ru0.7O2−δ | 168 | 500 h @ 10 mA cm−2 | AEM | 120 h @ 100 mA cm−2 | — | 95 |
| InRuO2/G | 187 | — | — | 350 h @ 100 mA cm−2 | — | 96 |
| Si–RuO2-0.1 | 224 | 800 h @ 10 mA cm−2 | AEM | — | — | 81 |
4.1 Heterometallic doping
In recent years, various strategies have emerged to enhance the acidic OER performance of RuO2 by doping hetero-metals (e.g., Mn,32 Co,49 Mo,97 Pb,89etc.). These approaches have been extensively documented, demonstrating that the stability and activity of metal–RuO2 hybrids surpass those of commercial RuO2 by enhancing the intrinsic electrochemical properties of RuO2 or optimizing active sites through electronic effects.98 A common heterometallic doping strategy is to delay or prevent the dissolution of RuO2 at the expense of the added metal.93,99 This strategy usually combines RuO2 with more corrosion-resistant metals during the synthesis process and controls the dispersion of RuO2 to avoid direct contact with the electrolyte. Sun et al.92 introduced an innovative strategy by incorporating insoluble high-valent Nb into RuO2, aiming to overcome the stability constraints associated with conventional RuO2. In their electrochemical evaluations (Fig. 9a–c), the synthesized Nb0.1Ru0.9O2 displayed remarkably low overpotential (204 mV @ 10 mA cm−2) and excellent stability, exhibiting an ultra-low degradation rate (25 mV h−1) during a 360-hour durability test at 200 mA cm−2. In contrast, pure RuO2 started to deteriorate within the initial 5 hours during the durability test. Further detailed characterization and theoretical calculations (Fig. 9d and e) indicate that Nb doping not only lowers the OER overpotential but also prevents the over-oxidation of Ru sites at high current densities by stabilizing the lattice oxygen of RuO2 and modulating the electron density of Ru sites. | ||
| Fig. 9 (a–c) OER performance of the prepared NbxRu1−xO2 in a three-electrode system and PEM electrolyzer. (d and e) EXAFS and XPS characterization of Nb0.1Ru0.9O2.92 Copyright 2023, Elsevier. (f) RuO6 octahedron before and after lithium intercalation. (g) The charge density distribution of the O* absorbed on the (110) surface of RuO2 (up) and Li0.5RuO2 (down) and the charge density distribution of the OOH* absorbed on the (110) surface of RuO2 (up) and Li0.5RuO2 (down).98 Copyright 2022, Springer Nature. | ||
However, most of the doping strategies activate the lattice oxygen mechanism (LOM) of RuO2, which inherently increases the covalency of Ru–O bonds. As previously discussed, this tendency poses challenges for the long-term stability of RuO2. Therefore, it is imperative to stabilize the RuO2 lattice bonds both before and after the reaction while optimizing the reaction pathway. Wang94 reported a nickel-stabilized RuO2 (Ni–RuO2) catalyst exhibiting high activity and durability in acidic oxygen evolution reaction (OER) for proton exchange membrane (PEM) water electrolysis. The original RuO2 demonstrated poor stability in acid OER and degraded rapidly, whereas the RuO2 lattice was significantly stabilized by the addition of nickel, extending its durability by more than an order of magnitude. When utilized as the anode of a PEM water electrolyzer, the prepared Ni–RuO2 catalyst exhibited stability exceeding 1000 hours at a water decomposition current of 200 mA cm−2, indicating its potential for practical applications. Wei et al.81 introduced a novel gap silicon doping strategy aimed at stabilizing the RuO2 lattice and inhibiting the direct involvement of lattice oxygen in the acidic oxygen evolution reaction (OER) process. The resulting Si–RuO2 electrode exhibited remarkable activity and stability in acid. As shown in Fig. 10a and b, the overpotential of the catalyst at 10 mA cm−2 was 226 mV. Moreover, after continuous oxygen evolution for up to 800 hours, the degradation rate of the catalyst does not exceed 52 μV per hour. Furthermore, metallized silicon demonstrates excellent acid resistance and possesses a suitable ionic radius for insertion into the interstitial positions of the RuO2 lattice. Additionally, the bond dissociation energy of the Si–O bond (798 kJ mol−1) surpasses that of the Ru–O bond (481 kJ mol−1), effectively reducing the covalency of the Ru–O bond (Fig. 10d–f). These strong Si–O bonds favor the inhibition of the lattice oxygen mechanism (LOM) pathway and enhance the associative elementary mechanism (AEM) pathway (Fig. 10g). The effective doping strategy proposed in this study, which attenuates the covalency of Ru–O bonds while preserving the quantity of Ru–O structures, carries profound implications. It emphasizes the necessity of evaluating the covalent characteristics of Ru–O bonds and the precise delineation of oxygen evolution reaction (OER) mechanisms as pivotal considerations in the design of acidic OER catalysts. This highlights the need for more sophisticated characterization methodologies like synchrotron radiation, along with the adoption of online in situ techniques such as in situ electrochemical mass spectrometry.
 | ||
| Fig. 10 (a and b) OER performance of the prepared Si–RuO2 in acidic electrolyte. (c) Comparison of the number of leached Ru ions during the OER process observed for Si–RuO2. (d) Normalized Ru K-edge XANES spectra of Si–RuO2−0.1 and com-RuO2. (e) Fourier-transform EXAFS spectra of Si–RuO2−0.1 and com-RuO2. (f) Schematic diagram of the band structures of Si–RuO2 and RuO2 based on Ru and O atoms on the (110) plane. (g) Schematic diagram of the enhanced stability caused by interstitial Si doping.81 Copyright 2024, Springer Nature. | ||
In addition to introducing insoluble doped metals to stabilize the RuO2 lattice and reduce the covalency of Ru–O, Hao et al.95 reduced/avoided the dissolution of RuO2 by decreasing the formation energy (ΔG) of oxygen vacancies (VO). They co-doped tungsten (W) and erbium (Er) into RuO2 to adjust its electronic structure. The introduction of W and Er significantly increased the formation energy of oxygen vacancies (VO) compared to the redox reaction of H2O/O2, which prevented the dissolution of Ru. As a result, the overpotential of W and Er co-doped RuO2 under acidic conditions was reduced to 168 mV at 10 mA cm−2, and it remained stable for at least 500 hours without apparent decay even at a high current density of 100 mA cm−2.
Additionally, heterometallic doping also benefits the asymmetric structure of metal–O–Ru, thereby disrupting the traditional adsorption or lattice oxygen mechanism. For instance, Wu et al.96 prepared In–RuO2 catalysts (In–RuO2/G) on graphene to enhance the oxidation of acidic water. By breaking the Ru–O–Ru symmetry in conventional RuO2, the electronic microenvironment and the local RuO bond strength of the catalyst were optimized, resulting in a significant improvement in acid OER performance. The overpotential was reduced to 187 mV at 10 mA cm−2, and persistent stability reached 350 hours at 100 mA cm−2. Further theoretical calculations revealed that the asymmetric structure of Ru–O–In disrupts the thermodynamic activity limit of the traditional adsorption evolution mechanism and notably weakens the formation energy barrier of OOH*, thus leading to a new rate-determining step for OH* adsorption. Similarly, in the work of Peng et al.93 asymmetric Ru–O–W active units were created around the interface of (Ru–W)Ox heterostructures, inducing valence oscillations at the Ru site. This cyclic transformation of the oxidation state of the active metals promoted the continuous operation of the active sites. The prepared catalyst demonstrated excellent stability for 300 hours in acidic electrolyte and showed potential for practical application in proton exchange membrane water electrolysis (PEMWE), maintaining stability for 300 hours at 0.5 A cm−2. In summary, the heterometallic doping strategy effectively adjusts the electronic structure of RuO2, stabilizes the crystal lattice, reduces the covalency of Ru–O bonds, enhances the formation energy of oxygen vacancies, and breaks the symmetry of Ru–O–Ru. These adjustments enable unconventional OER mechanisms and reduce barriers, ultimately improving the OER activity and stability of RuO2.
4.2 Heterogeneous structure engineering
Heterogeneous interface engineering represents a method to tailor the properties of catalysts through precise manipulation of the interface or interfacial structure. Within the structure of heterojunction catalysts, the heterojunction interface not only provides additional active sites to promote catalytic reactions but also regulates the electronic structure of the catalyst, optimizing adsorption energies and thereby adjusting catalytic activity. Currently, researchers employ synthetic approaches to design heterogeneous interfacial catalysts with tailored structures and compositions to achieve efficient OER electrocatalytic activity in acidic environments. Additionally, theoretical simulations and computational methods are widely utilized to elucidate the mechanisms of heterogeneous interfaces and provide guidance for the design of heterogeneous interface catalysts. Liu et al.100 synthesized a unique heterojunction catalyst (Ru-MOF-400) by pyrolyzing a ruthenium metal–organic framework (Ru-MOF) and introducing Co3O4 into the rutile lattice of RuO2 (Fig. 11a–c). In their electrochemical evaluation, the catalyst exhibited an overpotential of 190 mV at a current density of 10 mA cm−2 and a mass activity of 2557 μA gRu−1. The formation of Ru–Co bonds, facilitated by the addition of Co to the rutile lattice of RuO2, effectively inhibited the dissolution of Ru. Furthermore, in situ electrochemical and density functional theory studies revealed that the oxidation pathway of Ru-MOF-400 involves asymmetric bi-active sites during acidic oxygen evolution. This asymmetry is primarily mediated by the presence of Ru–Co bi-activity sites at the interface between Co3O4 and CoRuOx. Xu et al.101 fabricated a RuO2/(Co, Mn)3O4 heterostructure on carbon cloth, altering the electronic properties of RuO2 and generating electron-rich Ru species. In contrast to commercial RuO2 catalysts, RuO2/(Co, Mn)3O4 demonstrated enhanced acidic OER activity (η = 270 mV, 10 mA cm−2) and long-term stability. Mu et al.102 adeptly engineered a layered heterostructure of Ru/RuS2, exhibiting remarkable catalytic activity for both the OER (201 mV @ 10 mA cm−2) and the HER (45 mV @ 10 mA cm−2) in acidic media, facilitated by interface charge redistribution and enhanced electron conduction (Fig. 11d and e). This phenomenon serves to optimize the adsorption of crucial intermediates on Ru atoms with surface electron deficiency, thereby mitigating reaction barriers.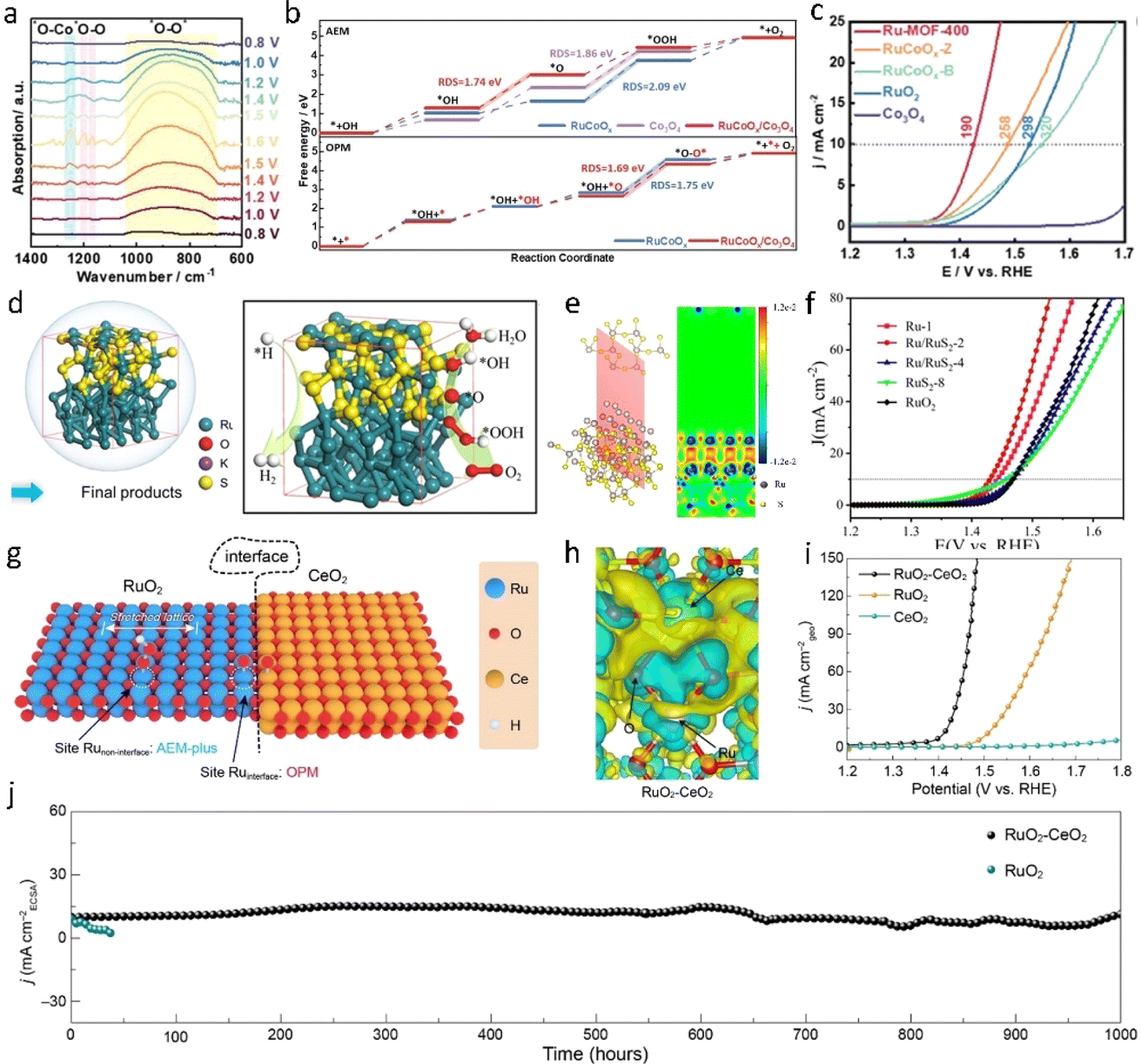 | ||
| Fig. 11 (a) Operando synchrotron FTIR spectra were recorded in a cyclic potential range from 0.8 to 1.6 V versus RHE for Ru-MOF-400. (b) The free energy diagrams of the OER process at VRHE = 0 V (via the AEM) for RuCoOx, Co3O4, and RuCoOx/Co3O4 (upper pattern) and the calculated free energy diagrams of the OER process (via the OPM) for RuCoOx and RuCoOx/Co3O4 (lower pattern). (c) LSV curves of Ru-MOF-400, RuCoOx-Z, RuCoOx-B, RuO2, and Co3O4.100 Copyright 2023, Wiley-VCH. (d and e) Charge density difference of a two-dimensional slice on the RuS2–Ru model. (f) LSV curves with iR correction.102 Copyright 2021, Wiley-VCH. (g and h) Differential charge density analysis of RuO2–CeO2. (i and j) OER performance in 0.5 M H2SO4.87 Copyright 2024, American Chemical Society. | ||
Indeed, beyond the electronic effect, the lattice effect of a heterogeneous interface can induce lattice distortion or the formation of new lattice structures, both of which can significantly alter the surface activity of the catalyst. Lu et al.87 recently constructed RuO2–CeO2 heterojunction electrocatalysts using a lattice matching strategy. At the interface of the Ru–O–Ce heterostructure, a pathway for electron transfer between Ru and Ce is established (Fig. 11h), inducing lattice stress and distorting the local structure of RuO2. This resulted in a catalyst that demonstrated remarkable stability, maintaining OER activity for at least 1000 hours in 0.5 M H2SO4 with negligible degradation and an overpotential of merely 180 mV at 10 mA cm−2 (Fig. 11i and j). In situ ATR-SEIRAS and DEMS analyses revealed that both the interface and non-interface sites of RuO2–CeO2 underwent the oxide path mechanism (OPM) and adsorbate evolution mechanism (AEM) during the OER. In essence, the introduction of CeO2 initially distorts the RuO2 lattice, facilitating the deprotonation of *OH at the Ru site, thereby promoting stable *OOH adsorption. Consequently, the energy barrier of the AEM reaction pathway is lowered. Additionally, the unique electronic structure at the interface further promotes the direct coupling of hydrogen peroxide radicals in the OER process. Overall, Ru-based heterointerfaces serve as pivotal components in acidic electrolyzed water, substantially enhancing the performance of the electrocatalytic oxygen evolution reaction through diverse pathways such as component effects, electronic effects, band modulation effects, and lattice effects.
4.3 Defect engineering
Defect engineering, encompassing the doping of anionic and cationic vacancies, stands as a potent strategy for enhancing the properties of nanomaterials.103 Through the doping of foreign atoms, the relative position between the center of the d-band and the Fermi energy level can be finely adjusted,104,105 consequently enabling the fine-tuning of adsorption energy and efficiency for intermediates. Vacancy defects, such as oxygen vacancies in oxides, serve to lower the energy band of O 2p and Fermi levels106 (Fig. 12a and b). This action augments the covalency of metal–oxygen bonds, thereby activating lattice oxygen and amplifying the activity of the Oxygen Evolution Reaction (OER). At present, the presence vacancies is mainly proven by means of atomic-level characterization such as STEM. Moreover, the correlation between vacancies and the active mechanism is extensively explored through theoretical density functional calculations, providing insights into their influence on catalytic performance (Fig. 12c and d). | ||
| Fig. 12 (a) Illustration of the strategies for enhancing the performance of electrocatalytic water splitting via vacancy engineering.103 Copyright 2020, Royal Society of Chemistry. (b) A schematic illustration of the role of defects in the modulation of electrocatalytic performance.8 Copyright 2021, Royal Society of Chemistry. (c) STEM image of a CVD-grown monolayer MoS2 flake film after etching. The yellow dotted circles represent the S-vacancies. (d) The projected electronic density of states of the d-band for the Mo atoms of P-MoS2, MoS2-FGJK, and MoS2-EGMO.104 Copyright 2020, American Chemical Society. (e and f) Two-dimensional volcano plot constructed using the descriptors of ΔGO*–ΔGOH* and ΔGOOH*–ΔGO* and a schematic showing the origin of OER activity improvement upon introducing a Ru vacancy. Insets show the configurations of O-terminated RuO2(110) and RuO2(100) surfaces.28 Copyright 2020, Royal Society of Chemistry. | ||
Firstly, metal vacancies have garnered widespread attention and experienced rapid development in recent years, primarily due to their unique electronic structure and high surface activity density. The presence of metal vacancies typically results in the formation of additional active sites on the catalyst surface, thereby enhancing the adsorption and activation of reactants. Furthermore, the change in the electronic structure of the catalyst induced by metal vacancies, particularly the shift in the position of the d-band center, can optimize the adsorption energy of reaction intermediates. The introduction of a metal vacancy can alter the reaction pathway and mechanism of the catalyst, thereby accelerating the chemical kinetics. Metal vacancies can lower the energy barrier of the reaction, facilitating easier reaction processes. Although the incorporation of metal vacancies can introduce structural defects, careful design and optimization can significantly enhance the stability of the catalyst. Overall, the role of metal vacancies in electrocatalysis is profound, demonstrating substantial potential by modulating the electronic structure, enhancing catalytic activity, promoting chemical kinetics, and improving catalyst stability.
Li et al.28 employed a molten salt method to fabricate ultra-thin RuO2 nanosheets abundant in Ru vacancies, yielding numerous multi-scale defects including dislocations and grain boundaries. The presence of Ru vacancy phases on the surface substantially diminishes the binding energy of O*, consequently reducing the energy barrier for the conversion of O* to OOH* and significantly enhancing the performance of acidic OER. Remarkably, at a current density of 10 mA cmgeo−2 and 125 μg cmgeo−2 loading, an exceptionally low overpotential of 199 mV was achieved along with the demonstrated stability (Fig. 12e and f). Similarly, Li et al.107 reported defect-rich amorphous/crystalline RuO2 porous particles, showcasing outstanding OER performance under acidic conditions with a low overpotential of 165 mV at a current density of 10 mA cm−2.
On the one hand, the presence of oxygen vacancies can activate neighboring metal atoms, inducing high reactivity, by shifting the density of states (DOS) closer to the Fermi level, thereby facilitating electron transfer. On the other hand, electrons residing on the vacancies can be excited into the conduction band (CB), introducing fresh sites conducive to hydrogen adsorption within the CB. Cho et al.108 introduced a novel modification to amorphous/crystalline RuO2 (a/c-RuO2) by incorporating oxygen-enriched vacancy sodium. Through DFT calculations, they demonstrated that the introduction of oxygen vacancies leads to an increased separation between the d-band center of RuO2 and the Fermi energy level. This effect weakens the chemical bond between oxygen intermediates and the RuO2 surface, consequently enhancing both the activity and stability of RuO2 in electrocatalytic applications. Li et al.109 devised a novel oxygen-enriched RuO2 nanoparticle–carbon coupled hybrid aerogel (VO-RuO2@CA), demonstrating that oxygen vacancies (VO) can notably diminish the binding energy of O* relative to OOH*, thereby enhancing the oxidation activity and anti-peroxidation capability of RuO2 in natural water. Notably, the representative VO-RuO2@CA achieved an ultra-low overpotential of 207 mV under acidic water oxidation conditions at 10 mA cm−2. Additionally, the emergence of metal-induced oxygen vacancies has garnered significant attention. For instance, Qi et al.110 devised a distinctive array of Mo-doped ruthenium–cobalt oxide (Mo–RuCoOx) nanoplates. During surface reconstruction in the oxygen evolution reaction (OER), these nanoplates generate oxygen vacancies, optimizing the adsorption energy of hydrogen/oxygen intermediates. As a result, the kinetics of both the hydrogen evolution reaction (HER) and OER in acidic conditions are greatly accelerated.
While vacancy engineering has showcased remarkable activity in electrocatalysis, the unsaturated coordination and heightened reactivity of the defective catalyst make it unable to withstand prolonged continuous operation under harsh electrolytic conditions.34 Consequently, the catalyst is prone to restructuring during the reaction. In situ characterization becomes imperative to monitor the catalyst's state under operational conditions, necessitating techniques such as in situ Raman spectroscopy, in situ infrared spectroscopy, in situ XPS, and in situ synchrotron radiation. Moreover, structural evolution may lead to mechanistic shifts, warranting further characterization, such as in situ electrochemical mass spectrometry, and computational methods to elucidate the mechanism of the defective catalyst. This aids in gaining a deeper understanding of the relationship between defects and activity and stability.
In conclusion, the dominant AEM restricts the further enhancement of RuO2 catalytic activity. At the same time, the traditional LOM related to lattice oxygen, although helpful to enhance the catalytic activity, initiated the structural variation and dissolution of RuO2, which significantly reduced its long-term stability. It is necessary to carry out the effective coordination and management between the AEM and LOM mechanisms, therefore balance the activity and stability of RuO2 electrocatalysts. This optimization not only helps to improve the application of RuO2 in acidic electrolyzed water, but also provides an important design idea and method for developing new efficient and stable electrocatalysts.
5. Non-noble metal-oxide OER electrocatalysts
Previous studies have elucidated the irreplaceable status of noble metal oxide catalysts in the case of acidic OER;18 however non-precious metals, because of their low price and abundant availability on Earth, can align well with the concept of green water electrolysis. Consequently, there has been a surge in research focused on the development of novel non-noble metal-based catalysts capable of efficiently catalyzing the electrolysis of water under acidic conditions. The acid-stable periodic table, as proposed by Nørskov et al.111 serves as a valuable roadmap for identifying potential non-noble metal catalysts capable of showing both stability and activity in acidic OER. Within this framework, particular attention is given to spinel Co3O4 and Mn-based oxides as promising candidates for acidic OER. These materials have garnered significant interest due to their inherent stability in acidic environments and their demonstrated catalytic activity for oxygen evolution. In the following section, we will delve into the specific characteristics and performance of spinel Co3O4 and Mn-based oxides as electrocatalysts for acidic OER, shedding light on their potential role in catalytic OER performance in acidic electrolyte.5.1 Co-oxide catalysts
Researchers are drawn to spinel Co3O4-based OER electrocatalysts due to their distinctive electronic structure and remarkable resistance to corrosion. Within the spinel Co3O4 lattice, cobalt ions predominantly occupy tetrahedral positions in the Co(II) oxidation state, while octahedral positions are predominantly occupied by Co(III) ions. It is generally accepted that these Co(III) ions in the octahedral positions serve as active sites driving the oxygen evolution reaction in acidic environments. Consequently, considerable research effort has been devoted to modifying these octahedral sites, including strategies such as heteroatomic substitution and the regulation of the coordination environment surrounding the octahedral cobalt ions. These approaches aim to enhance the catalytic activity and stability of spinel Co3O4-based catalysts for acidic OER. Liu et al.112 introduced a novel spinel-type Co3O4 co-doped with lanthanum (La) and manganese (Mn), denoted as LMCF. In this material, La-doped Co3O4 contributed to stabilizing the corrosion resistance across multiple facets of Co3O4, while Mn3+ ions replaced Co3+ sites within the octahedral structure, resulting in a mean oxidation state of cobalt less than 2.67. This substitution induced the formation of defect states occupying two distinct regions within the intermediate band gap. Consequently, LMCF exhibited an overpotential of 353 ± 30 mV at 10 mA cm−2 and demonstrated stability for 300 hours under operating conditions at 10 mA cm−2. This innovative approach offers promising prospects for advancing the performance and durability of spinel Co3O4-based electrocatalysts for acidic oxygen evolution reaction. Liang et al.113 devised a method to enhance the acidic oxygen evolution reaction (OER) activity and stability of Co3O4 spinel by strategically doping atomic ruthenium (Ru) onto octahedral cobalt (Co) sites. This selective doping resulted in highly symmetric octahedral Ru–O–Co synergistic coordination and strong electron coupling effects, facilitating a direct oxygen free radical coupling OER pathway. Moreover, they observed electron transfer behaviour on the tetrahedral site of the Co atom, thus inhibiting the excessive oxidation and dissolution of active Ru and Co species and enhancing catalyst stability. With a low Ru loading, the catalyst achieved an overpotential of 200 mV at 10 mA cm−2 and exhibited a potential decay rate not exceeding 0.45 mV h−1. This innovative approach offers promising avenues for improving the performance and durability of Co3O4-based spinel catalysts in acidic OER applications.In addition to noble metal doping, the incorporation of non-noble metals into Co3O4 has emerged as a promising avenue. Lou et al.114 explored the introduction of 13 different metals as isolated metal sites within ultrathin Co3O4 nanosheets. Their investigation revealed that Fe-doped Co3O4 significantly enhanced the electrocatalytic activity for the oxygen evolution reaction (OER). Leveraging their advantageous structure and composition, the Fe-doped Co3O4 laminated nanosheets demonstrated remarkable electrocatalytic performance in the OER. At an overpotential of 262 mV at 10 mA cm−2, these nanosheets exhibited excellent stability even under high current density conditions of 100 mA cm−2, maintaining their performance for 50 hours.
The addition of Mn into the spinel lattice of Co3O4 represents a significant advancement in improving the stability of non-noble metal-based OER catalysts under acidic conditions. Xiao et al.'s work115 demonstrated a remarkable increase in the catalyst's lifetime, extending from just a few hours to hundreds or even thousands of hours. Theoretical calculations revealed that the stability of the Mn–O bond played a crucial role in preventing the dissolution of lattice oxygen, thereby enhancing the catalyst's stability. This breakthrough represents a promising development in the field of Co3O4-based OER catalysts, showing their potential for practical applications in acidic electrolysis systems. By addressing the stability limitations at high current densities, this research opens up new possibilities for the design of robust and efficient non-noble metal-based catalysts for water electrolysis. Zhai et al.116 applied a coating of Mn-doped Co3O4 with N (Mn–CO3O4@CN) layers. Through in situ isotopic labeling spectroscopy and electron localization function (ELF) evaluation, they unveiled the presence of hydrogen bond interactions between CN units and H2O molecules, forming a continuous hydrogen bond network at the catalyst–electrolyte interface (Fig. 13h and i). Additionally, strong charge regulation between Mn/CN and Co sites was observed to reduce the OER barrier. The results demonstrated that the Co3O4-based electrode exhibited a low overpotential of 395 mV (at 10 mA cm−2) in acidic media, surpassing the activity of RuO2 even at high current densities (100 mA cm−2 @ ∼538 mV). In addition, the acidic OER properties of Co3O4-based catalysts in recent years are listed in Table 3.
 | ||
| Fig. 13 (a) Frequency at which each element appears in acid-stable oxides. Elements with zero frequency are shaded in gray. Lanthanoids and actinoids are omitted for clarity because no oxide containing these elements was predicted to be stable.111 Copyright 2020, American Chemical Society. (b) Calculated Pourbaix diagram of CoOx.83 Copyright 2022, Springer Nature. (c) Activity trends towards oxygen evolution for rutile, anatase, Co3O4, and MnxOy oxides.117 Copyright 2011, Wiley-VCH. (d and e) Identification of the Ruoct–O–Cooct coordination environment in RuCoOx. (f) Simulative structural model of a Ruoct–O–Cooct unit with two adsorbed oxygen radicals. (g) Geometric LSV curves of RuO2, Co3O4 and RuCoOx.113 Copyright 2023, American Chemical Society. (h) Schematic illustration of Mn–Co3O4@CN with a hydrophilous CN layer to tailor the local chemical environment at the interface for optimum OER in acidic electrolyte. (i) In situ Raman spectra of interfacial water. (j) Tafel curves obtained from the polarization curves.116 Copyright 2024, Wiley-VCH. | ||
| Catalyst | In a three-electrode system | In a PEMWE | Ref. | |||
|---|---|---|---|---|---|---|
| η 10 (mV) | Stability | Pathway | Stability | T (°C) | ||
| Mn–Co3O4@CN | 395 | — | — | 50 h @ 200 mA cm−2 | 80 | 116 |
| RuCoOx | 200 | 100 h @ 10 mA cm−2 | DOM | 10 h @ 100 mA cm−2 | 85 | 113 |
| Co3−xBaxO4 | 278 | 110 h @ 10 mA cm−2 | OPM | — | — | 118 |
| Co9S8/Co3O4 | 275 | 40 h @ 10 mA cm−2 | OPM | 50 h @ 50 mA cm−2 | 80 | 119 |
| Ir–Co3O4 | 236 | 30 h @ 10 mA cm−2 | — | — | — | 120 |
| Co3O4@C/GPO | 360 | 40 h @ 10 mA cm−2 | — | — | — | 121 |
| Co2MnO4 | — | 1600 h @ 200 mA cm−2 | LOM | — | — | 115 |
| γ-MnO2 | 428 | 8000 h @ 10 mA cm−2 | — | 400 h @ 10 mA cm−2 | 25 | 122 |
| LiMnP2O7 | 680 | — | — | — | — | 123 |
5.2 Mn-oxide catalysts
The stability of MnO2, particularly γ-MnO2, was demonstrated in a previous study where it underwent electrolysis at a pH of 2 for 8000 hours without showing any signs of deactivation.122 Despite this remarkable stability, the development of Mn-based oxides faces challenges primarily due to their high overpotential.115 Kumta et al.124 first reported the F-doping of Cu1.5Mn1.5O4 with a method of combining theory with experiment. They conducted first-principles calculations to determine the total energy and electronic structure of the Cu1.5Mn1.5O4 electrocatalyst system, leading to the identification of the appropriate composition. The system exhibited outstanding oxygen evolution reaction (OER) activity on par with IrO2, coupled with remarkable long-term stability validated through 6000 cycles in acidic media, thus confirming the theoretical predictions. Stephens125et al. introduced a novel approach to enhance the catalytic performance of manganese dioxide (MnO2) under acidic conditions by modifying disparate sites on its surface with stable TiO2. The theoretical prediction suggesting that titanium can stabilize MnO2-based catalysts under oxygen evolution reaction (OER) conditions was validated experimentally. This finding offers valuable insights for the utilization of MnO2-based materials in proton exchange membrane water electrolysis (PEMWE) applications.Certainly, as with all transition metal oxides in acidic environments, catalysts featuring MnO2 as the anode unavoidably confront dissolution in acidic electrolytes. Previous studies123,126,127 have suggested that the higher overpotential observed for MnO2 catalysts could be attributed to the instability arising from charge disproportionation caused by Mn3+ intermediates. Recently, several effective strategies have been proposed to mitigate the dissolution of Mn under acidic conditions.
For instance, Nakamura128 investigated the oxygen evolution reaction (OER) of rutile-type MnO2 with (101) and (110) crystal faces. Through in situ monitoring of Mn3+ intermediates, a linear relationship between the quantity and activity of Mn3+ intermediates was revealed. It was found that the presence of Mn3+ intermediates could enhance stability, a characteristic determined by the surface electronic structure of MnO2. While much of the development of MnO2 catalysts still occurs at low current densities, recent work by Xiao et al.129 achieved a remarkable improvement in stability. By increasing the proportion of planar oxygen in the MnO2 lattice, they enhanced stability by 40 times. Additionally, they achieved a current density of 2 amperes at 2 volts, with catalytic activity comparable to that of iridium. Overall, research on MnO2-based acidic OER catalysts remains in its nascent stages. One pressing issue to address is the alteration of MnO2 valence states at high potentials, which can result in instability in MnO2 performance. Additionally, the intrinsic activity of MnO2 requires further enhancement through strategies such as doping, introducing defects, and inducing strain, among others. The rational utilization of Pourbaix diagrams and other theoretical verification methods can offer guidance for screening non-noble metal elements stable in acids.
Overall, the high activity and stability of noble metal-based catalysts in acidic oxygen evolution reaction (OER) currently make them the most effective choice. However, their high cost and scarcity limit their large-scale application. Non-noble metal-based catalysts have significant potential for application due to their low cost and abundant resources, but their activity and stability require further enhancement. Future research should prioritize improving the performance of non-noble metal-based catalysts through nanostructure design, doping, and alloying, and developing new catalytic materials that balance high efficiency and cost control. These efforts are essential to achieving comprehensive advances in acid water electrolysis technology.
6. Conclusion and prospects
In summary, we systematically considered the origins and strategies to address the stability challenges encountered in acidic OER. It provides a comprehensive review of the research progress pertaining to noble metal oxide-based catalysts such as IrO2 and RuO2, as well as non-noble metal oxides, for acidic OER. In acidic environments, OER catalysts are initially confronted with the issues of oxidation or even dissolution at high potentials, leading to diminished catalytic activity and stability. As a result, IrO2-based catalysts have garnered significant attention in the PEMWE industry and have found practical applications. However, the high cost of Ir necessitates further exploration into achieving low loading while maintaining high activity and stability in IrO2-based catalysts. Ru-based catalysts, particularly RuO2, are recognized as among the most active electrocatalysts for anodic oxygen evolution reaction (OER) in water electrolysis. However, the thermodynamic instability of RuO2 throughout the pH range during the OER has been well-documented. Extensive theoretical and experimental investigations have revealed that the OER process involves the transition from stable Ru4+ to unstable Run+ (n > 4), leading to gradual catalyst dissolution and deactivation. Hence, to enhance the stability and activity of ruthenium-based catalysts, novel strategies are imperative. Current research efforts on RuO2-based catalysts predominantly concentrate on enhancing their stability, necessitating meticulous exploration of their material structure and mechanisms. Despite these challenges, RuO2 has demonstrated practical applications across various industries. Under acidic conditions, the oxidation of most transition metals (TMs) at high potentials poses a challenge, leading to reduced catalytic activity and stability. However, a select few TMs, such as Co and Mn, have exhibited notable activity against acidic OER. However, their stability and activity still require a lot of development.The advancement of earth-abundant OER catalysts with high activity and stability in acidic environments remains a formidable challenge. Progress in this direction requires a deeper comprehension of these catalysts' operational states and the characteristics of their active sites to enable precise control and adjustment of their properties. The integration of theoretical insights, computational modelling, and sophisticated in situ/operational characterization techniques will be instrumental in addressing these pivotal challenges. From the foregoing discussion, several key points emerge. Firstly, regarding IrO2-based catalysts, current research focuses on achieving low loading, high stability, and intrinsic activity. Strategies such as leveraging the structure effect, support effect, interface effect, defect effect, and molecular self-assembly effect show promise in enhancing the stability of IrO2 catalysts. Secondly, a significant challenge lies in reducing the dissolution rate of RuO2 at high potential. Effective approaches such as incorporating sacrificial components have been widely explored. Additionally, RuO2-based catalysts are emerging as the preferred alternative to Ir-based catalysts. Furthermore, research on non-noble metal acidic OER catalysts is still in its nascent stages. While practical application remains challenging at present, the field holds considerable potential for development. Lastly, it is crucial to underscore the importance of in situ characterization techniques such as in situ XPS, in situ synchrotron radiation X-ray absorption spectrometry (XAS), operando differential electrochemical mass spectrometry (DEMS), and in situ Raman analysis. These methods play a vital role in tracing catalyst evolution during OER processes. When combined with mechanism elucidation and catalyst design strategies, they enable more efficient catalyst design, leading to high-performance catalytic acidic OER under high current densities. Overall, the high activity and stability of noble metal-based catalysts in acidic oxygen evolution reaction (OER) currently make them the most effective choice. However, their high cost and scarcity limit their large-scale application. Non-noble metal-based catalysts, due to their low cost and abundant resources, hold significant potential for application, but their activity and stability require further enhancement. Future research should prioritize improving the performance of non-noble metal-based catalysts through nanostructure design, doping, and alloying, as well as developing new catalytic materials that balance high efficiency and cost control. These efforts are essential to achieving comprehensive advancements in acidic water electrolysis technology. The further development of advanced anode oxide-based electrocatalysts for proton exchange membrane water electrolyzers (PEMWEs) in acidic environments needs to address several key areas.
6.1 A comprehensive understanding of the catalytic mechanism
Although oxide-based anodic catalysts have been widely reported, the true active sites remain controversial. In this context, we emphasize the importance of further mechanism-based understanding of how catalysts work. A more detailed analysis of the OER mechanisms will not only elucidate the deactivation pathways of catalysts but also reveal the specific properties and behaviours of the active sites, thereby providing a theoretical foundation for optimizing catalytic performance. This comprehension serves as the linchpin for overcoming the constraints associated with balancing catalyst activity and stability, thereby fostering the transition of catalyst design from an empirical to a more scientific and systematic approach. It lays the groundwork for the development of new, highly efficient, and stable catalysts. In this case, the application of advanced characterization techniques is paramount for elucidating the catalytic mechanism. For instance, in situ differential electrochemical mass spectrometry combined with isotopic labelling (DEMS) offers unparalleled insights. This technology accurately tracks intermediates and final products in electrochemical reactions by utilizing isotopically labeled reactants and products, thereby revealing the reaction pathways and mechanisms. Such detailed analysis not only elucidates the actual behaviour of the catalyst during reactions but also identifies key factors influencing catalyst efficiency and stability. This approach provides invaluable data support and a theoretical foundation for the optimization and design of high-performance catalysts.6.2 Exploring the reconstruction of the catalysts under operational conditions
At the operational potential of the oxygen evolution reaction (OER), it is crucial to closely study the evolution of the catalyst structure in order to understand the OER mechanism. Studying these structural changes can uncover the surface and bulk phase reconstructions of the catalyst during electrocatalysis, which directly affect its activity and stability. Recently, various in situ characterization techniques have been widely reported. For example, high-resolution microscopy and in situ spectroscopy can observe the morphology, lattice distortion, and phase transitions of the catalyst under OER conditions. This information is invaluable in identifying the formation and deactivation mechanisms of active sites. In addition, theoretical calculations and simulations, combined with experimental data, offer a deeper understanding of electronic structure changes and reaction pathways. This multi-dimensional approach comprehensively reveals the dynamic behaviour of catalysts during the OER and provides a scientific basis and guidance for the design of efficient and stable oxide-based catalysts.6.3 Satisfying the criteria for industrial applications
Although significant advances have been made in anodic catalysts for acidic oxygen evolution reaction (OER), such as noble metal oxide-based catalysts with overpotentials of 100–250 mV at 10 mA cm−2, the overpotential of non-noble metal oxides is between 300 and 500 mV. Most catalysts, however, are still confined to laboratory evaluation stages. In general, acid OER catalysts should not only exhibit high catalytic activity and long-term stability but also meet more stringent requirements for mass transfer and mechanical stability. Achieving these goals necessitates more in-depth and systematic research in the design, synthesis, and structural and performance optimization of catalysts. Such efforts will be crucial for advancing the transition of these catalysts from laboratory settings to industrial applications. In practical proton exchange membrane water electrolysis (PEMWE), the preparation conditions of the catalyst electrodes, such as hot-pressing temperature and pressure, significantly impact the efficiency of the monolithic membrane electrode assembly (MEA). Optimizing these parameters is crucial for enhancing electrolysis efficiency and prolonging equipment lifespan. To understand and optimize PEMWE operations, finite element simulation techniques can be employed to simulate the multi-physical fields within the cell, including electric fields, temperature fields, concentration fields, and flow fields. This approach allows for an in-depth analysis of how different process parameters and operating conditions affect cell performance, thereby guiding experimental design and improving the preparation process of the membrane electrode module. Ultimately, this methodology enables the realization of high-efficiency and stable hydrogen production through water electrolysis.Data availability
No primary research results, software or code has been included and no new data were generated or analysed as part of this review.Author contributions
Yingying Xu: conceptualization, investigation, methodology and writing – original draft. Yingxia Zhao: conceptualization and formal analysis. Zihui Yuan: investigation. Yue Sun: investigation. Shaomin Peng: methodology and writing – review & editing. Yuanhong Zhong: methodology, conceptualization and writing – review & editing. Ming Sun: validation, supervision, writing – review & editing and methodology. Lin Yu: funding acquisition, validation, resources, supervision and project administration.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the Foundation of Basic and Applied Basic Research of Guangdong Province (2023B1515120043), the Foundation of the Smart Medical Innovation Technology Center in Guangdong University of Technology (ZYZX22034), and the Yangfan Project of Maoming City (MMGCIRI-2022YFJH-Y-014).References
- H. Over, Chem. Rev., 2012, 112, 3356–3426 CrossRef CAS PubMed.
- Z. W. Seh, J. Kibsgaard, C. F. Dickens, I. Chorkendorff, J. K. Nørskov and T. F. Jaramillo, Science, 2017, 355, 146 CrossRef PubMed.
- S. M. El-Refaei, P. A. Russo and N. Pinna, ACS Appl. Mater. Interfaces, 2021, 13, 22077–22097 CrossRef CAS PubMed.
- Y. Zhou and H. J. Fan, ACS Mater. Lett., 2020, 3, 136–147 CrossRef.
- T. Kou, S. Wang and Y. Li, ACS Mater. Lett., 2021, 3, 224–234 CrossRef CAS.
- Z. Y. Yu, Y. Duan, X. Y. Feng, X. Yu, M. R. Gao and S. H. Yu, Adv. Mater., 2021, 33, e2007100 CrossRef PubMed.
- J. Zhu, L. Hu, P. Zhao, L. Y. S. Lee and K. Y. Wong, Chem. Rev., 2020, 120, 851–918 CrossRef CAS PubMed.
- S. Jiao, X. Fu, S. Wang and Y. Zhao, Energy Environ. Sci., 2021, 14, 1722–1770 RSC.
- J. Ying and H. Wang, Front. Chem., 2021, 9, 700020 CrossRef CAS PubMed.
- X. Yu, Z. Y. Yu, X. L. Zhang, Y. R. Zheng, Y. Duan, Q. Gao, R. Wu, B. Sun, M. R. Gao, G. Wang and S. H. Yu, J. Am. Chem. Soc., 2019, 141, 7537–7543 CrossRef CAS PubMed.
- Y. Zhao, M. Sun, Q. Wen, S. Wang, S. Han, L. Huang, G. Cheng, Y. Liu and L. Yu, J. Mater. Chem. A, 2022, 10, 10209–10218 RSC.
- Y. Liang, J. Wang, D. Liu, L. Wu, T. Li, S. Yan, Q. Fan, K. Zhu and Z. Zou, J. Mater. Chem. A, 2021, 9, 21785–21791 RSC.
- M. Gong, W. Zhou, M. C. Tsai, J. Zhou, M. Guan, M. C. Lin, B. Zhang, Y. Hu, D. Y. Wang, J. Yang, S. J. Pennycook, B. J. Hwang and H. Dai, Nat. Commun., 2014, 5, 4695 CrossRef CAS PubMed.
- S. S. Shinde, J. Y. Jung, N. K. Wagh, C. H. Lee, D.-H. Kim, S.-H. Kim, S. U. Lee and J.-H. Lee, Nat. Energy, 2021, 6, 592–604 CrossRef CAS.
- C.-T. Dinh, A. Jain, F. P. G. de Arquer, P. De Luna, J. Li, N. Wang, X. Zheng, J. Cai, B. Z. Gregory, O. Voznyy, B. Zhang, M. Liu, D. Sinton, E. J. Crumlin and E. H. Sargent, Nat. Energy, 2018, 4, 107–114 CrossRef.
- M. N. Islam, A. B. Mansoor Basha, V. O. Kollath, A. P. Soleymani, J. Jankovic and K. Karan, Nat. Commun., 2022, 13, 6157 CrossRef CAS PubMed.
- T. W. Song, C. Xu, Z. T. Sheng, H. K. Yan, L. Tong, J. Liu, W. J. Zeng, L. J. Zuo, P. Yin, M. Zuo, S. Q. Chu, P. Chen and H. W. Liang, Nat. Commun., 2022, 13, 6521 CrossRef CAS PubMed.
- C. Wei, Z. Wang, K. Otani, D. Hochfilzer, K. Zhang, R. Nielsen, I. Chorkendorff and J. Kibsgaard, ACS Catal., 2023, 13, 14058–14069 CrossRef CAS.
- Q. Qian, Y. Zhu, N. Ahmad, Y. Feng, H. Zhang, M. Cheng, H. Liu, C. Xiao, G. Zhang and Y. Xie, Adv. Mater., 2023, e2306108 Search PubMed.
- Z. Zhong, J. Fang, K. Hu, D. Huang, X. Ai, X. Yang, J. Wen, Y. Pan and S. Cheng, CSEE J. Power Energy Syst., 2023, 9, 1266–1283 Search PubMed.
- R. Deng, M. Guo, C. Wang and Q. Zhang, Nano Mater. Sci., 2024, 2, 139–173 CrossRef.
- K. Zhang, X. Liang, L. Wang, K. Sun, Y. Wang, Z. Xie, Q. Wu, X. Bai, M. S. Hamdy, H. Chen and X. Zou, Nano Research Energy, 2022, 1, e9120032 CrossRef.
- N. T. T. Thao, J. U. Jang, A. K. Nayak and H. Han, Small Sci., 2023, 2300109 Search PubMed.
- C. Rong, K. Dastafkan, Y. Wang and C. Zhao, Adv. Mater., 2023, 35, e2211884 CrossRef PubMed.
- I. Katsounaros, S. Cherevko, A. R. Zeradjanin and K. J. Mayrhofer, Angew Chem. Int. Ed. Engl., 2014, 53, 102–121 CrossRef CAS PubMed.
- X. Wang, Y. Xu, Y. Xi, X. Yang, J. Wang, X. Huang, W. Li, K. Xu, K. Zhang, R. Duan, D. Liu, N. Hou, Z. Yang, H. Wang and X. Li, J. Mater. Chem. A, 2024, 12, 9268–9295 RSC.
- M. Sohail, W. Lv and Z. Mei, ACS Sustain. Chem. Eng., 2023, 11, 17564–17594 CrossRef CAS.
- Z. L. Zhao, Q. Wang, X. Huang, Q. Feng, S. Gu, Z. Zhang, H. Xu, L. Zeng, M. Gu and H. Li, Energy Environ. Sci., 2020, 13, 5143–5151 RSC.
- N. Hodnik, P. Jovanovič, A. Pavlišič, B. Jozinović, M. Zorko, M. Bele, V. S. Šelih, M. Šala, S. Hočevar and M. Gaberšček, J. Phys. Chem. C, 2015, 119, 10140–10147 CrossRef CAS.
- T. Reier, M. Oezaslan and P. Strasser, ACS Catal., 2012, 2, 1765–1772 CrossRef CAS.
- O. Diaz-Morales, S. Raaijman, R. Kortlever, P. J. Kooyman, T. Wezendonk, J. Gascon, W. T. Fu and M. T. Koper, Nat. Commun., 2016, 7, 12363 CrossRef CAS PubMed.
- S. Chen, H. Huang, P. Jiang, K. Yang, J. Diao, S. Gong, S. Liu, M. Huang, H. Wang and Q. Chen, ACS Catal., 2019, 10, 1152–1160 CrossRef.
- A. Grimaud, O. Diaz-Morales, B. Han, W. T. Hong, Y. L. Lee, L. Giordano, K. A. Stoerzinger, M. T. M. Koper and Y. Shao-Horn, Nat. Chem., 2017, 9, 457–465 CrossRef CAS PubMed.
- M. Lu, Y. Zheng, Y. Hu, B. Huang, D. Ji, M. Sun, J. Li, Y. Peng, R. Si, P. Xi and C.-H. Yan, Sci. Adv., 2022, 8, eabq3563 CrossRef CAS PubMed.
- A. Yang, J. Wang, K. Su, W. Lei, X. Qiu and Y. Tang, Chem.–Eur. J., 2021, 27, 4869–4875 CrossRef CAS PubMed.
- C. Wei, Y. Sun, G. G. Scherer, A. C. Fisher, M. Sherburne, J. W. Ager and Z. J. Xu, J. Am. Chem. Soc., 2020, 142, 7765–7775 CrossRef CAS PubMed.
- K. Xu, P. Chen, X. Li, Y. Tong, H. Ding, X. Wu, W. Chu, Z. Peng, C. Wu and Y. Xie, J. Am. Chem. Soc., 2015, 137, 4119–4125 CrossRef CAS PubMed.
- H. Li, S. Chen, Y. Zhang, Q. Zhang, X. Jia, Q. Zhang, L. Gu, X. Sun, L. Song and X. Wang, Nat. Commun., 2018, 9, 2452 CrossRef PubMed.
- N. Danilovic, R. Subbaraman, K. C. Chang, S. H. Chang, Y. J. Kang, J. Snyder, A. P. Paulikas, D. Strmcnik, Y. T. Kim, D. Myers, V. R. Stamenkovic and N. M. Markovic, J. Phys. Chem. Lett., 2014, 5, 2474–2478 CrossRef CAS PubMed.
- C. Wang, R. Deng, M. Guo and Q. Zhang, Int. J. Hydrogen Energy, 2023, 48, 31920–31942 CrossRef CAS.
- Z. Yan, S. Guo, Z. Tan, L. Wang, G. Li, M. Tang, Z. Feng, X. Yuan, Y. Wang and B. Cao, Materials, 2024, 17, 1637 CrossRef CAS PubMed.
- Y. Zhao, Q. Wen, D. Huang, C. Jiao, Y. Liu, Y. Liu, J. Fang, M. Sun and L. Yu, Adv. Energy Mater., 2023, 13, 2203595 CrossRef CAS.
- J. Jin, J. Yin, Y. Hu, Y. Zheng, H. Liu, X. Wang, P. Xi and C. H. Yan, Angew Chem. Int. Ed. Engl., 2023, e202313185 Search PubMed.
- Y. Wen, P. Chen, L. Wang, S. Li, Z. Wang, J. Abed, X. Mao, Y. Min, C. T. Dinh, P. Luna, R. Huang, L. Zhang, L. Wang, L. Wang, R. J. Nielsen, H. Li, T. Zhuang, C. Ke, O. Voznyy, Y. Hu, Y. Li, W. A. Goddard, III, B. Zhang, H. Peng and E. H. Sargent, J. Am. Chem. Soc., 2021, 143, 6482–6490 CrossRef CAS PubMed.
- J. Ni, Z. Shi, Y. Wang, J. Yang, H. Wu, P. Wang, K. Li, M. Xiao, C. Liu and W. Xing, Nano Res., 2023, 17, 1107–1113 CrossRef.
- S. Yagi, I. Yamada, H. Tsukasaki, A. Seno, M. Murakami, H. Fujii, H. Chen, N. Umezawa, H. Abe, N. Nishiyama and S. Mori, Nat. Commun., 2015, 6, 8249 CrossRef PubMed.
- S. Xu, S. Feng, Y. Yu, D. Xue, M. Liu, C. Wang, K. Zhao, B. Xu and J. N. Zhang, Nat. Commun., 2024, 15, 1720 CrossRef CAS PubMed.
- C. Lin, J.-L. Li, X. Li, S. Yang, W. Luo, Y. Zhang, S.-H. Kim, D.-H. Kim, S. S. Shinde, Y.-F. Li, Z.-P. Liu, Z. Jiang and J.-H. Lee, Nat. Catal., 2021, 4, 1012–1023 CrossRef CAS.
- Y. Hao, S. F. Hung, W. J. Zeng, Y. Wang, C. Zhang, C. H. Kuo, L. Wang, S. Zhao, Y. Zhang, H. Y. Chen and S. Peng, J. Am. Chem. Soc., 2023, 145, 23659–23669 CrossRef CAS PubMed.
- Z.-F. Huang, J. Song, Y. Du, S. Xi, S. Dou, J. M. V. Nsanzimana, C. Wang, Z. J. Xu and X. Wang, Nat. Energy, 2019, 4, 329–338 CrossRef CAS.
- F. Liao, K. Yin, Y. Ji, W. Zhu, Z. Fan, Y. Li, J. Zhong, M. Shao, Z. Kang and Q. Shao, Nat. Commun., 2023, 14, 1248 CrossRef CAS PubMed.
- S. D. Ghadge, O. I. Velikokhatnyi, M. K. Datta, P. M. Shanthi, S. Tan, K. Damodaran and P. N. Kumta, ACS Catal., 2019, 9, 2134–2157 CrossRef CAS.
- L. Li, P. Wang, Z. Cheng, Q. Shao and X. Huang, Nano Res., 2021, 15, 1087–1093 CrossRef.
- D. Cao, J. Wang, H. Zhang, H. Xu and D. Cheng, Chem. Eng. J., 2021, 416, 129128 CrossRef CAS.
- H. N. Nong, L. Gan, E. Willinger, D. Teschner and P. Strasser, Chem. Sci., 2014, 5, 2955–2963 RSC.
- X. Wang, Z. Qin, J. Qian, L. Chen and K. Shen, ACS Catal., 2023, 13, 10672–10682 CrossRef CAS.
- J. Lim, D. Park, S. S. Jeon, C. W. Roh, J. Choi, D. Yoon, M. Park, H. Jung and H. Lee, Adv. Funct. Mater., 2017, 28, 1704796 CrossRef.
- Q. Dang, H. Lin, Z. Fan, L. Ma, Q. Shao, Y. Ji, F. Zheng, S. Geng, S. Z. Yang, N. Kong, W. Zhu, Y. Li, F. Liao, X. Huang and M. Shao, Nat. Commun., 2021, 12, 6007 CrossRef CAS PubMed.
- L. Zu, X. Qian, S. Zhao, Q. Liang, Y. E. Chen, M. Liu, B. J. Su, K. H. Wu, L. Qu, L. Duan, H. Zhan, J. Y. Zhang, C. Li, W. Li, J. Y. Juang, J. Zhu, D. Li, A. Yu and D. Zhao, J. Am. Chem. Soc., 2022, 144, 2208–2217 CrossRef CAS PubMed.
- L. C. Seitz, C. F. Dickens, K. Nishio, Y. Hikita, J. Montoya, A. Doyle, C. Kirk, A. Vojvodic, H. Y. Hwang, J. K. Norskov and T. F. Jaramillo, Science, 2016, 353, 1011–1014 CrossRef CAS PubMed.
- L. Wang, L. Shi, Q. Liu, Y. Huang, W. Yan, X. Liang, X. Zhao, H. Chen and X. Zou, ACS Catal., 2023, 13, 7322–7330 CrossRef CAS.
- J. Torrero, T. Morawietz, D. García Sanchez, D. Galyamin, M. Retuerto, V. Martin-Diaconescu, S. Rojas, J. A. Alonso, A. S. Gago and K. A. Friedrich, Adv. Energy Mater., 2023, 13, 2204169 CrossRef CAS.
- J. W. Zhao, K. Yue, H. Zhang, S. Y. Wei, J. Zhu, D. Wang, J. Chen, V. Y. Fominski and G. R. Li, Nat. Commun., 2024, 15, 2928 CrossRef CAS PubMed.
- V. Muravev, A. Parastaev, Y. van den Bosch, B. Ligt, N. Claes, S. Bals, N. Kosinov and E. J. M. Hensen, Science, 2023, 380, 1174–1179 CrossRef CAS PubMed.
- J. Chu, J. Gong, Y. Cheng and S. Xiong, Ionics, 2023, 29, 2417–2425 CrossRef CAS.
- G.-Y. Xu, M.-F. Yue, Z.-X. Qian, Z.-Y. Du, X.-Q. Xie, W.-P. Chen, Y.-J. Zhang and J.-F. Li, J. Mater. Chem. A, 2023, 11, 15204–15210 RSC.
- Z. Shi, J. Li, J. Jiang, Y. Wang, X. Wang, Y. Li, L. Yang, Y. Chu, J. Bai, J. Yang, J. Ni, Y. Wang, L. Zhang, Z. Jiang, C. Liu, J. Ge and W. Xing, Angew Chem. Int. Ed. Engl., 2022, 61, e202212341 CrossRef CAS PubMed.
- R. Huang, Y. Wen, P. Miao, W. Shi, W. Niu, K. Sun, Y. Li, Y. Ji and B. Zhang, Chem Catal., 2023, 3, 100667 CrossRef CAS.
- E. Oakton, D. Lebedev, M. Povia, D. F. Abbott, E. Fabbri, A. Fedorov, M. Nachtegaal, C. Copéret and T. J. Schmidt, ACS Catal., 2017, 7, 2346–2352 CrossRef CAS.
- P. Mazúr, J. Polonský, M. Paidar and K. Bouzek, Int. J. Hydrogen Energy, 2012, 37, 12081–12088 CrossRef.
- G. Shi, T. Tano, D. A. Tryk, T. Uchiyama, A. Iiyama, M. Uchida, K. Terao, M. Yamaguchi, K. Tamoto, Y. Uchimoto and K. Kakinuma, ACS Catal., 2023, 13, 12299–12309 CrossRef CAS.
- G. R. Lee, J. Kim, D. Hong, Y. J. Kim, H. Jang, H. J. Han, C. K. Hwang, D. Kim, J. Y. Kim and Y. S. Jung, Nat. Commun., 2023, 14, 5402 CrossRef CAS PubMed.
- R. A. Krivina, M. Zlatar, T. N. Stovall, G. A. Lindquist, D. Escalera-López, A. K. Cook, J. E. Hutchison, S. Cherevko and S. W. Boettcher, ACS Catal., 2022, 13, 902–915 CrossRef.
- Y. Wang, M. Zhang, Z. Kang, L. Shi, Y. Shen, B. Tian, Y. Zou, H. Chen and X. Zou, Nat. Commun., 2023, 14, 5119 CrossRef CAS PubMed.
- S. Chen, S. Zhang, L. Guo, L. Pan, C. Shi, X. Zhang, Z. F. Huang, G. Yang and J. J. Zou, Nat. Commun., 2023, 14, 4127 CrossRef CAS PubMed.
- H. Chun, Y. Kaihang, H. Jiajia, L. Lijia, L. Qiunan, K. Qingyu, P. Chih-Wen, H. Zhiwei, S. Kazu, S. Dong, Z. Qiaobao, W. Xianying, T. Yuanzhi and H. Xiaoqing, Sci. Adv., 2023, 9, eadf9144 CrossRef PubMed.
- W. Zhao, F. Xu, L. Liu, M. Liu and B. Weng, Adv. Mater., 2023, 35, e2308060 CrossRef PubMed.
- Y. Yao, S. Hu, W. Chen, Z.-Q. Huang, W. Wei, T. Yao, R. Liu, K. Zang, X. Wang, G. Wu, W. Yuan, T. Yuan, B. Zhu, W. Liu, Z. Li, D. He, Z. Xue, Y. Wang, X. Zheng, J. Dong, C.-R. Chang, Y. Chen, X. Hong, J. Luo, S. Wei, W.-X. Li, P. Strasser, Y. Wu and Y. Li, Nat. Catal., 2019, 2, 304–313 CrossRef CAS.
- W. Zhao, Y. Liu, X. Fu and W. Wang, Renewables, 2023, 1, 638–667 CrossRef.
- S. Cherevko, A. R. Zeradjanin, A. A. Topalov, N. Kulyk, I. Katsounaros and K. J. J. Mayrhofer, ChemCatChem, 2014, 6, 2219–2223 CrossRef CAS.
- X. Ping, Y. Liu, L. Zheng, Y. Song, L. Guo, S. Chen and Z. Wei, Nat. Commun., 2024, 15, 2501 CrossRef CAS PubMed.
- L. Cao, Q. Luo, J. Chen, L. Wang, Y. Lin, H. Wang, X. Liu, X. Shen, W. Zhang, W. Liu, Z. Qi, Z. Jiang, J. Yang and T. Yao, Nat. Commun., 2019, 10, 4849 CrossRef PubMed.
- K. Du, L. Zhang, J. Shan, J. Guo, J. Mao, C.-C. Yang, C.-H. Wang, Z. Hu and T. Ling, Nat. Commun., 2022, 13, 5448 CrossRef CAS PubMed.
- S. Zhao, S. F. Hung, L. Deng, W. J. Zeng, T. Xiao, S. Li, C. H. Kuo, H. Y. Chen, F. Hu and S. Peng, Nat. Commun., 2024, 15, 2728 CrossRef CAS PubMed.
- M. Xiao, J. Liu, R. Li, Y. Sun, F. Liu, J. Gan and S. Gao, Small, 2024, e2400754 CrossRef PubMed.
- X. Wu, C. Lin, W. Hu, C. Fu, L. Tan, H. Wang, F. Meharban, X. Pan, P. Fu, H. D. Um, Q. Xiao, X. Li, M. Yamauchi and W. Luo, Small Struct., 2024, 2300518, DOI:10.1002/sstr.202300518.
- H. Song, X. Yong, G. I. N. Waterhouse, J. Yu, H. Wang, J. Cai, Z. Tang, B. Yang, J. Chang and S. Lu, ACS Catal., 2024, 14, 3298–3307 CrossRef CAS.
- Y. Hao, S. F. Hung, C. Tian, L. Wang, Y. Y. Chen, S. Zhao, K. S. Peng, C. Zhang, Y. Zhang, C. H. Kuo, H. Y. Chen and S. Peng, Angew Chem. Int. Ed. Engl., 2024, e202402018 CAS.
- F.-Y. Chen, C. Qiu, Z.-Y. Wu, T.-U. Wi, Y. Z. Finfrock and H. Wang, Nano Res., 2024, 1–7 Search PubMed.
- J. Tang, Y. Zhong, C. Su and Z. Shao, Small Sci., 2023, 3, 2300055 CrossRef CAS.
- Z. Shi, J. Li, Y. Wang, S. Liu, J. Zhu, J. Yang, X. Wang, J. Ni, Z. Jiang, L. Zhang, Y. Wang, C. Liu, W. Xing and J. Ge, Nat. Commun., 2023, 14, 843 CrossRef CAS PubMed.
- H. Liu, Z. Zhang, J. Fang, M. Li, M. G. Sendeku, X. Wang, H. Wu, Y. Li, J. Ge, Z. Zhuang, D. Zhou, Y. Kuang and X. Sun, Joule, 2023, 7, 558–573 CrossRef CAS.
- L. Deng, S. F. Hung, Z. Y. Lin, Y. Zhang, C. Zhang, Y. Hao, S. Liu, C. H. Kuo, H. Y. Chen, J. Peng, J. Wang and S. Peng, Adv. Mater., 2023, 35, e2305939 CrossRef PubMed.
- Z. Y. Wu, F. Y. Chen, B. Li, S. W. Yu, Y. Z. Finfrock, D. M. Meira, Q. Q. Yan, P. Zhu, M. X. Chen, T. W. Song, Z. Yin, H. W. Liang, S. Zhang, G. Wang and H. Wang, Nat. Mater., 2022, 100–108 Search PubMed.
- S. Hao, M. Liu, J. Pan, X. Liu, X. Tan, N. Xu, Y. He, L. Lei and X. Zhang, Nat. Commun., 2020, 11, 5368 CrossRef CAS PubMed.
- Y. Wang, X. Lei, B. Zhang, B. Bai, P. Das, T. Azam, J. Xiao and Z. S. Wu, Angew Chem. Int. Ed. Engl., 2024, 63, e202316903 CrossRef CAS PubMed.
- Y. Zhang, J. Dong, T. Sun, X. Zhang, J. Chen and L. Xu, Small, 2024, 20, e2305889 CrossRef PubMed.
- Y. Qin, T. Yu, S. Deng, X. Y. Zhou, D. Lin, Q. Zhang, Z. Jin, D. Zhang, Y. B. He, H. J. Qiu, L. He, F. Kang, K. Li and T. Y. Zhang, Nat. Commun., 2022, 13, 3784 CrossRef CAS PubMed.
- X. Wang, H. Jang, S. Liu, Z. Li, X. Zhao, Y. Chen, M. G. Kim, Q. Qin and X. Liu, Adv. Energy Mater., 2023, 13, 2301673 CrossRef CAS.
- J. Liang, X. Gao, K. Xu, J. Lu, D. Liu, Z. Zhao, E. C. M. Tse, Z. Peng, W. Zhang and J. Liu, Small, 2023, 2304889 CrossRef CAS PubMed.
- S. Niu, X.-P. Kong, S. Li, Y. Zhang, J. Wu, W. Zhao and P. Xu, Appl. Catal., B, 2021, 297, 120442 CrossRef CAS.
- J. Zhu, Y. Guo, F. Liu, H. Xu, L. Gong, W. Shi, D. Chen, P. Wang, Y. Yang, C. Zhang, J. Wu, J. Luo and S. Mu, Angew Chem. Int. Ed. Engl., 2021, 60, 12328–12334 CrossRef CAS PubMed.
- S. Choi, Y. Park, H. Yang, H. Jin, G. M. Tomboc and K. Lee, CrystEngComm, 2020, 22, 1500–1513 RSC.
- X. Wang, Y. Zhang, H. Si, Q. Zhang, J. Wu, L. Gao, X. Wei, Y. Sun, Q. Liao, Z. Zhang, K. Ammarah, L. Gu, Z. Kang and Y. Zhang, J. Am. Chem. Soc., 2020, 142, 4298–4308 CrossRef CAS PubMed.
- R. Zhang, Y.-C. Zhang, L. Pan, G.-Q. Shen, N. Mahmood, Y.-H. Ma, Y. Shi, W. Jia, L. Wang, X. Zhang, W. Xu and J.-J. Zou, ACS Catal., 2018, 8, 3803–3811 CrossRef CAS.
- M. Bi, Y. Zhang, X. Jiang, J. Sun, X. Wang, J. Zhu and Y. Fu, Adv. Funct. Mater., 2023, 34, 2309330 CrossRef.
- C. Wang, Q. Geng, L. Fan, J.-X. Li, L. Ma and C. Li, Nano Research Energy, 2023, 2, e9120070 CrossRef.
- L. Zhang, H. Jang, H. Liu, M. G. Kim, D. Yang, S. Liu, X. Liu and J. Cho, Angew Chem. Int. Ed. Engl., 2021, 60, 18821–18829 CrossRef CAS PubMed.
- X. Zhang, Y. Qiu, X. Guo, J. Chang, Y. Zhang, J. Cao, Y. Jiang, J. Bai, W. Wang, J. Tian and X. Li, Mol. Catal., 2024, 558, 114041 CrossRef CAS.
- Y. Zhang, R. Lu, C. Wang, Y. Zhao and L. Qi, Adv. Funct. Mater., 2023, 33, 2303073 CrossRef CAS.
- Z. Wang, Y.-R. Zheng, I. Chorkendorff and J. K. Nørskov, ACS Energy Lett., 2020, 5, 2905–2908 CrossRef CAS.
- L. Chong, G. Gao, J. Wen, H. Li, H. Xu, Z. Green, J. D. Sugar, A. J. Kropf, W. Xu, X.-M. Lin, H. Xu, L.-W. Wang and D.-J. Liu, Science, 2023, 380, 609–616 CrossRef CAS PubMed.
- W. Zhu, F. Yao, K. Cheng, M. Zhao, C.-J. Yang, C.-L. Dong, Q. Hong, Q. Jiang, Z. Wang and H. Liang, J. Am. Chem. Soc., 2023, 145, 17995–18006 CrossRef CAS PubMed.
- S. L. Zhang, B. Y. Guan, X. F. Lu, S. Xi, Y. Du and X. W. D. Lou, Adv. Mater., 2020, 32, e2002235 CrossRef PubMed.
- A. Li, S. Kong, C. Guo, H. Ooka, K. Adachi, D. Hashizume, Q. Jiang, H. Han, J. Xiao and R. Nakamura, Nat. Catal., 2022, 5, 109–118 CrossRef CAS.
- S. Zhu, R. Yang, H. Li, S. Huang, H. Wang, Y. Liu, H. Li and T. Zhai, Angew. Chem., Int. Ed., 2024, e202319462 CAS.
- I. C. Man, H. Y. Su, F. Calle-Vallejo, H. A. Hansen, J. I. Martínez, N. G. Inoglu, J. Kitchin, T. F. Jaramillo, J. K. Nørskov and J. Rossmeisl, ChemCatChem, 2011, 3, 1159–1165 CrossRef CAS.
- N. Wang, P. Ou, R. K. Miao, Y. Chang, Z. Wang, S. F. Hung, J. Abed, A. Ozden, H. Y. Chen, H. L. Wu, J. E. Huang, D. Zhou, W. Ni, L. Fan, Y. Yan, T. Peng, D. Sinton, Y. Liu, H. Liang and E. H. Sargent, J. Am. Chem. Soc., 2023, 145, 7829–7836 CrossRef CAS PubMed.
- L. Wang, H. Su, Z. Zhang, J. Xin, H. Liu, X. Wang, C. Yang, X. Liang, S. Wang, H. Liu, Y. Yin, T. Zhang, Y. Tian, Y. Li, Q. Liu, X. Sun, J. Sun, D. Wang and Y. Li, Angew Chem. Int. Ed. Engl., 2023, 62, e202314185 CrossRef CAS PubMed.
- Y. Zhu, J. Wang, T. Koketsu, M. Kroschel, J.-M. Chen, S.-Y. Hsu, G. Henkelman, Z. Hu, P. Strasser and J. Ma, Nat. Commun., 2022, 13, 7754 CrossRef CAS PubMed.
- J. Yu, F. A. Garces-Pineda, J. Gonzalez-Cobos, M. Pena-Diaz, C. Rogero, S. Gimenez, M. C. Spadaro, J. Arbiol, S. Barja and J. R. Galan-Mascaros, Nat. Commun., 2022, 13, 4341 CrossRef CAS PubMed.
- A. Li, H. Ooka, N. Bonnet, T. Hayashi, Y. Sun, Q. Jiang, C. Li, H. Han and R. Nakamura, Angew Chem. Int. Ed. Engl., 2019, 58, 5054–5058 CrossRef CAS PubMed.
- J. Park, H. Kim, K. Jin, B. J. Lee, Y. S. Park, H. Kim, I. Park, K. D. Yang, H. Y. Jeong, J. Kim, K. T. Hong, H. W. Jang, K. Kang and K. T. Nam, J. Am. Chem. Soc., 2014, 136, 4201–4211 CrossRef CAS PubMed.
- P. P. Patel, M. K. Datta, O. I. Velikokhatnyi, R. Kuruba, K. Damodaran, P. Jampani, B. Gattu, P. M. Shanthi, S. S. Damle and P. N. Kumta, Sci. Rep., 2016, 6, 28367 CrossRef CAS PubMed.
- R. Frydendal, E. A. Paoli, I. Chorkendorff, J. Rossmeisl and I. E. L. Stephens, Adv. Energy Mater., 2015, 5, 1500991 CrossRef.
- D. M. Robinson, Y. B. Go, M. Mui, G. Gardner, Z. Zhang, D. Mastrogiovanni, E. Garfunkel, J. Li, M. Greenblatt and G. C. Dismukes, J. Am. Chem. Soc., 2013, 135, 3494–3501 CrossRef CAS PubMed.
- T. Takashima, K. Hashimoto and R. Nakamura, J. Am. Chem. Soc., 2012, 134, 18153–18156 CrossRef CAS PubMed.
- H. Kakizaki, H. Ooka, T. Hayashi, A. Yamaguchi, N. Bonnet-Mercier, K. Hashimoto and R. Nakamura, Adv. Funct. Mater., 2018, 28, 1706319 CrossRef.
- S. Kong, A. Li, J. Long, K. Adachi, D. Hashizume, Q. Jiang, K. Fushimi, H. Ooka, J. Xiao and R. Nakamura, Nat. Catal., 2024, 7, 252–261 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2024 |