
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMachine learning empowered next generation DNA sequencing: perspective and prospectus
Sneha
Mittal
 ,
Milan Kumar
Jena
and
Biswarup
Pathak
*
,
Milan Kumar
Jena
and
Biswarup
Pathak
*
Department of Chemistry, Indian Institute of Technology (IIT) Indore, Indore, Madhya Pradesh 453552, India. E-mail: biswarup@iiti.ac.in
First published on 8th July 2024
Abstract
The pursuit of ultra-rapid, cost-effective, and accurate DNA sequencing is a highly sought after aspect of personalized medicine development. With recent advancements, mainstream machine learning (ML) algorithms hold immense promise for high throughput DNA sequencing at the single nucleotide level. While ML has revolutionized multiple domains of nanoscience and nanotechnology, its implementation in DNA sequencing is still in its preliminary stages. ML-aided DNA sequencing is especially appealing, as ML has the potential to decipher complex patterns and extract knowledge from complex datasets. Herein, we present a holistic framework of ML-aided next-generation DNA sequencing with domain knowledge to set directions toward the development of artificially intelligent DNA sequencers. This perspective focuses on the current state-of-the-art ML-aided DNA sequencing, exploring the opportunities as well as the future challenges in this field. In addition, we provide our personal viewpoints on the critical issues that require attention in the context of ML-aided DNA sequencing.
 Sneha Mittal | Sneha Mittal received her M.Sc. degree in chemistry from Ramjas College, University of Delhi (DU). Currently, she is carrying out her doctorate studies under the supervision of Prof. Biswarup Pathak at the Department of Chemistry, Indian Institute of Technology Indore (IITI), India. Her current research work focuses on the application of molecular electronics and machine learning in next-generation DNA and protein sequencing. |
 Milan Kumar Jena | Milan Kumar Jena obtained his UG degree from Ravenshaw University, Cuttack, Odisha. He completed his master's degree from the Indian Institute of Technology Guwahati, Assam. Then, he joined Prof. Biswarup Pathak's group as a graduate student in January 2020 at the Department of Chemistry, Indian Institute of Technology Indore, where his primary research interest is the computational investigation of solid-state nanopores for next-generation DNA sequencing. |
 Biswarup Pathak | Biswarup Pathak is a Professor in the Department of Chemistry at IIT Indore, India. He obtained his PhD from Hyderabad Central University under the supervision of Professor E. D. Jemmis. Soon after his PhD, he completed his four-year postdoctoral study with Prof. Jerzy Leszczynski's group at Jackson State University, USA (January 2008–July 2009) and Prof. Rajeev Ahuja's group at Uppsala University, Sweden (September 2009–May 2012). Prof. Pathak is currently working on developing solid-state materials for clean energy (hydrogen storage, photocatalysis, fuel cells, batteries, and solar cells) and biological (DNA sequencing) applications. |
1. Introduction
Next-generation DNA sequencing (NGS) is presently a powerful paradigm for decoding genetic information encoded within DNA with high resolution and industrial scalability.1–6 This technology has opened new frontiers in genomics research, clinical diagnostics, and personalized medicine, propelling advancements in diverse fields such as cancer research, hereditary diseases, and evolutionary biology.7–9 In NGS, ionic current and transverse tunneling current are two overarching concepts used to identify the individual DNA nucleotides.10–12 The basic principle of electric measurements assisted NGS is that the molecule of interest is translocated or dragged through the device under the influence of an electric field, which causes variations in the electric signals associated with each nucleotide. Based on these variations, sequencing is achieved.In NGS measurements, electric current and translocation time are two key parameters that make biomolecule identification possible.13–16 However, one potential difficulty in determining these parameters is that these signals are associated with unwanted noise, which makes their identification difficult. Moreover, the interpretation of these parameters at the single nucleotide level is very tedious and complex due to the similarity in the histograms plots of electric current and translocation time. In this regard, the time, cost, and complexity of NGS measurements necessitate new potential tools such as machine learning (ML) for cost-effective, fast, and accurate prediction of molecules with spatiotemporal resolution.17
ML has the potential to decipher complex patterns and extract knowledge from large datasets. With its ability to autonomously learn and adapt from data, ML holds tremendous promise in revolutionizing various domains. In the realm of DNA sequencing, ML has already begun to reshape the landscape by enhancing the accuracy, efficiency, and scalability of existing NGS methodologies.18 Integrating ML algorithms into NGS workflows can unlock hidden information, accelerate discoveries, and pave the way for breakthroughs in genomics. A generalized ML workflow illustrating the implementation of data-driven ML algorithms into DNA sequencing across traditional NGS architectures: nanopore, nanogap, and nanochannel is given in Fig. 1.
 | ||
| Fig. 1 (a) Schematic illustration of ML workflow for predicting the signature fingerprints of DNA nucleotides with next-generation sequencing architectures: nanopore, nanogap, and nanochannel. (b) ML workflow for predicting the class of DNA nucleotides exclusively from their signature electronic fingerprints. | ||
Given the importance of ML, in this perspective, we focus on the recent ML developments in the field of NGS, encompassing both the theoretical and experimental aspects. This perspective begins with a discussion on why ML should be integrated with NGS, followed by a summary of pioneering studies in the field. Subsequently, we provide a guide to ML developments with the feature engineering process for both theoretical and experimental measurements. Lastly, we outline a prospectus for the advancement of efficient and robust artificially intelligent DNA sequencers.
2. Why we need machine learning?
The primary motivation behind implementing ML in NGS is rooted in the urgent need to overcome the challenge of similarity in the histogram plot of electric current and translocation time of DNA nucleotides. Traditional sequencing technologies often struggle to keep pace with the massive amounts of information generated from the sequencing of a complete genome, leading to bottlenecks in data analysis and interpretation at the single nucleotide level.19,20 ML algorithms, on the other hand, have the potential to tackle the complexity of genomic data interpretation, ranging from DNA fingerprints (molecular conductance, electric current, and translocation time) recognition and noise detection to nucleotide classification with high precision and accuracy.For more than two decades, researchers have been exploring potential strategies for sequencing DNA through theoretical and experimental measurements. However, ML is especially appealing for a number of reasons. First, the process of finding an electrical signature of each DNA nucleotide is a key step in DNA sequencing. Determining the key signatures of each DNA nucleotide at a significantly reduced time and cost of cumbersome theoretical and experimental studies is at the heart of ML. Second, ML has phenomenal potential to resolve the signal overlap between the current signals of DNA nucleotides through classification tools. There are certain reports providing a glimpse of the classification of DNA nucleotides from complex electric current data of the whole DNA strand.21,22 Third, ML regression and classification algorithms have immense potential to extract the individual current signal from complex data with a high magnitude of noise matrix and decode the genetic code efficiently.
3. Pioneering studies of machine learning-aided DNA sequencing
In 2012, Lindsay and co-workers pioneered a concept for the classification of DNA nucleobases through the support vector machine (SVM) algorithm in conjunction with gold nanogap electrodes (Fig. 2a).21 They proposed a recognition tunneling (RT) technique to identify single nucleobases within a DNA oligomer using 4(5)-(2-mercaptoethyl)-1H-imidazole-2-carboxamide (Iz) as an adapter molecule. The adapter molecule helps in reducing the distribution of current signals, enabling better resolution among DNA bases (dAMP, dGMP, dCMP, and dTMP) and epigenetic modification, 5-methyl cytosine (dmeCMP).24 The authors extracted features from the collected recognition tunneling current data, and the optimized multiple parameters fit allowed nucleobase classification from a single peak with an accuracy of 80% and achieved 95% accuracy when analyzing multiple spikes in a signal cluster. The SVM-assisted separation of five bases and water into distinct clusters is displayed in Fig. 2b. To ensure precise DNA sequencing, achieving higher accuracy is paramount. The challenge remains to accurately distinguish the subtle variances in a comprehensive DNA sequence with 1-D conductance measurements. In this context, Kim and co-workers pioneered the approach of two-dimensional molecular electronics spectroscopy. The technique presents a promising avenue to recognize the single molecule signatures of both DNA and cancerous methylated DNA nucleobases at atomic resolution.25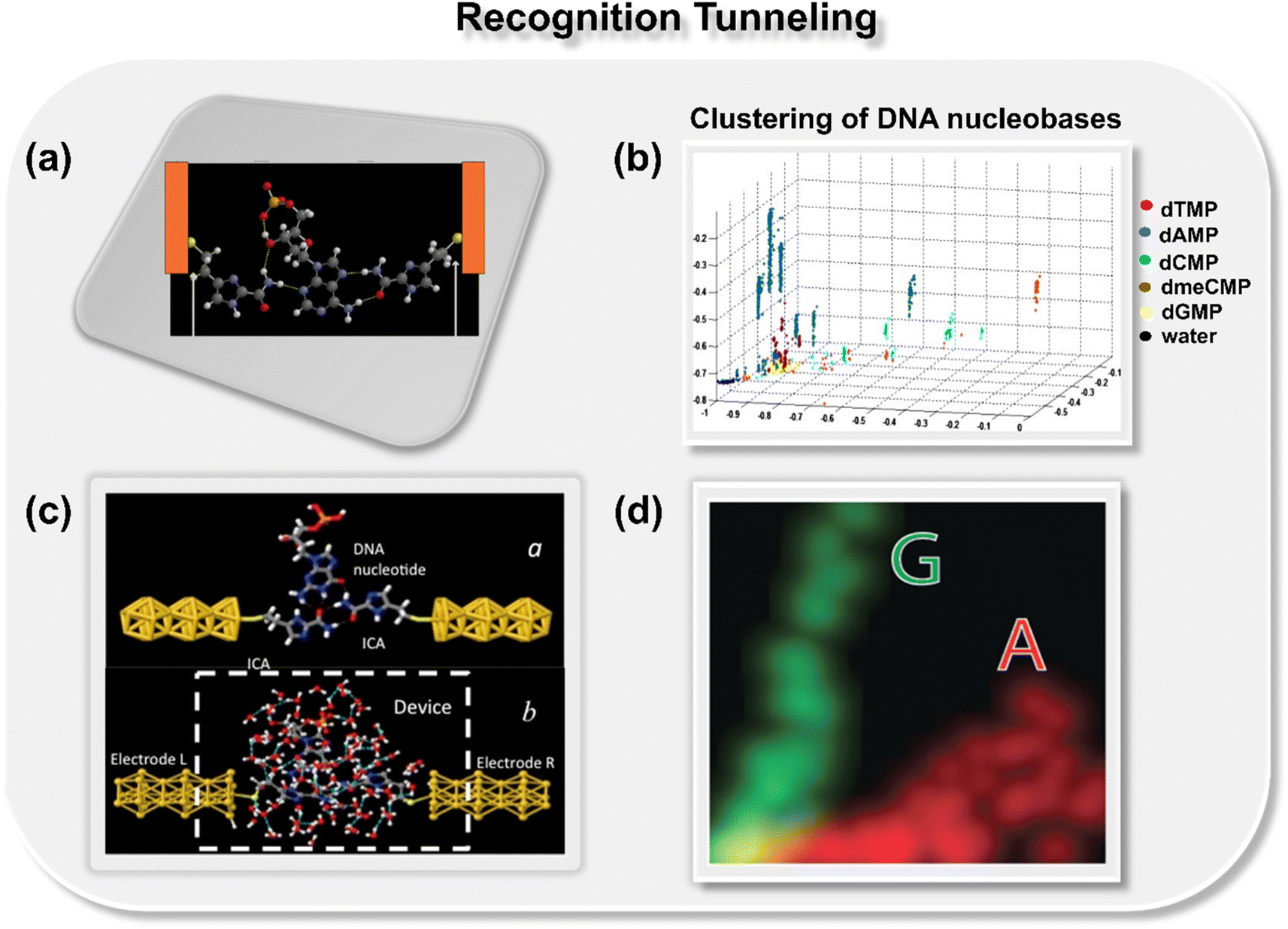 | ||
| Fig. 2 (a) Schematic of 4(5)-(2-mercaptoethyl)-1H-imidazole-2-carboxamide adapter molecule (Iz) trapping the dAMP molecule between gold nanogap electrodes via hydrogen bond.21 Atom color code: C (grey), H (white), O (red), S (yellow), and N (royal purple); (b) 2D projection of a 3D plot displaying the separation of five bases and water into distinct clusters.21 Reprinted with permission from ref. 21. Copyright 2012 IOP science. (c) Schematic of Iz functionalized gold electrodes trapping guanosine molecule with transient hydrogen bonds with and without water molecules23 and (d) two-dimensional distribution of probability densities of two signal features illustrating well-separated data points for A and G nucleobases.23 Reprinted with permission from ref. 23. Copyright 2015 IOP science. | ||
Motivated by the previous work, Biswas et al. next tried to increase the accuracy of the base calling by fine-tuning the chemical structure of adapter molecule Iz, leading to a change in the recognition tunneling current.26 By tuning the chemical structure of Iz, they synthesized three new adapter molecules: 1-(2-mercaptoethyl)-1H-pyrrole-3-carboxamide (Pr), 5-mercapto-1-benzo[d]imidazole-2-carboxamide (Bz), and 3-(2-mercaptoethyl)-1H-1,2,4-triazole-5-carboxamide (Tz). Instead of gold tunneling electrodes, in this work, they utilized palladium (Pd) electrodes as Pd has better conductance and metal-oxide semiconductor compatibility. For SVM classification, recognitional tunneling events were defined by two types of signals, i.e., spikes (individual single peaks) and clusters (a subset of close spikes). The results showed the following order of accuracy for classifying DNA nucleotides: Bz > Iz > Tz > Pr.
While these studies successfully introduced a novel approach for accurately calling nucleobases from stochastic signals, the studies posed an important unanswered question: whether real-time signals in a model system exhibit sufficient variation with the chemical properties of the target molecule in simulations to allow an ML algorithm to identify individual signals with good accuracy. To provide a theoretical underpinning to the proposed approach, Krstić et al. utilized the gold nanogap electrodes functionalized with a reader molecule (Iz) and employed multiscale theory and both all-atom and coarse-grained DNA models (Fig. 2c and d).23 The authors reported that the frequency characteristics of Brownian fluctuations could be leveraged to identify trapped molecules using a SVM algorithm.
Pathak and co-workers theoretically explored the potential of a germanene nanogap toward single-molecule DNA sequencing using the quantum tunneling approach (Fig. 3a).27 Herein, to reduce the overlapping issue of electric conductance signals, the authors combined the quantum transport method with ML classification algorithms to achieve high-throughput DNA sequencing. They report that using a random forest classifier (RFC) algorithm trained with the transmission function dataset, each binary, ternary, and quaternary classification of DNA nucleotides can be achieved with high precision and accuracy (Fig. 3b). Moreover, they also explored the effect of electrode-nucleotide coupling on transmission function and noticed that transmission signatures are sensitive to electrode-nucleotide coupling, and the RFC algorithm is capable of extrapolating that information well during the classification.
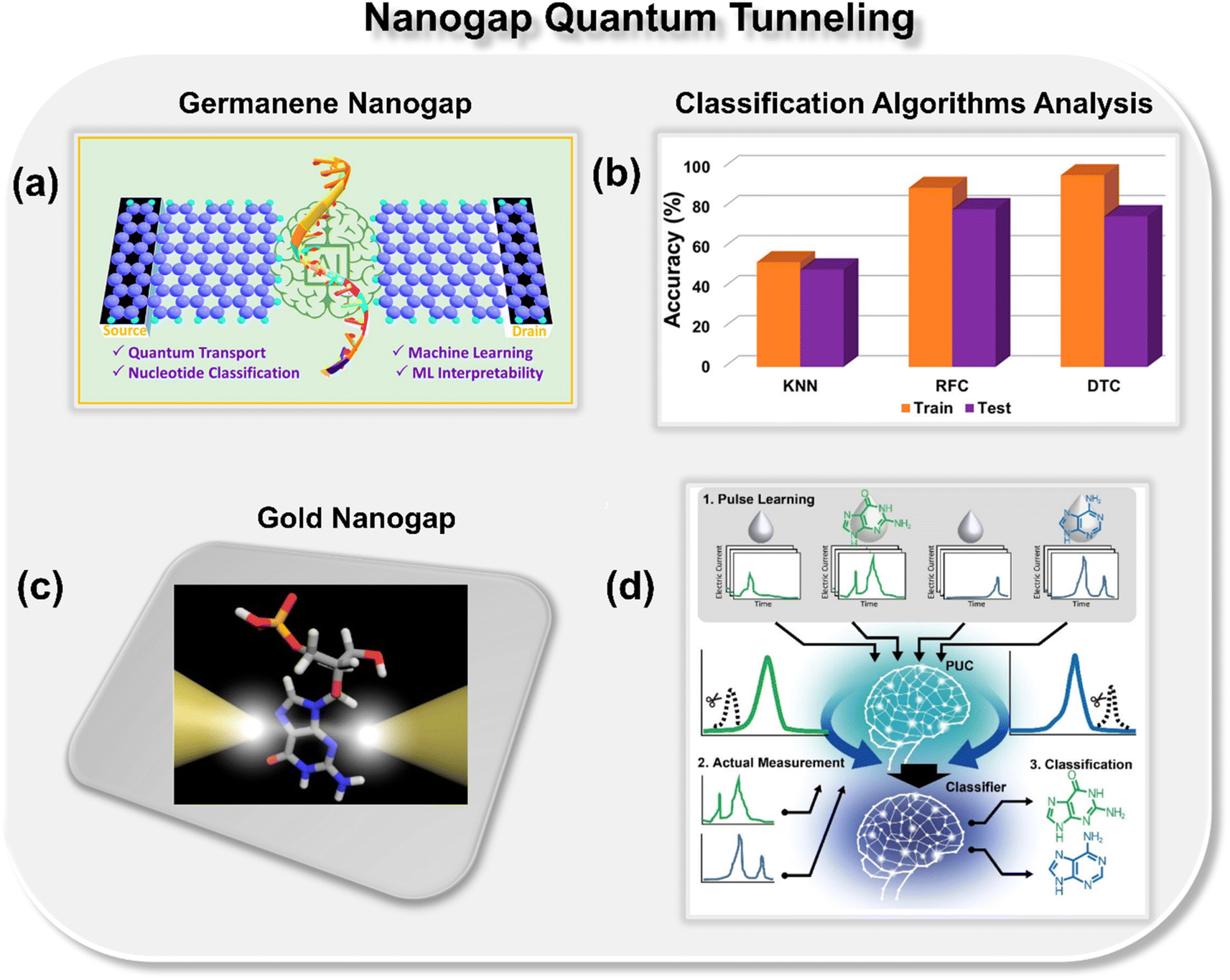 | ||
| Fig. 3 (a) Schematic of germanene nanogap consisting of left and right electrodes which act as the source and drains of electrons, respectively.27 (b) Analysis of ML classification algorithms toward prediction of class of DNA nucleotides.27 Reprinted with permission from ref. 27. Copyright 2023 American Chemical Society. (c) Schematic of single molecule gold nanogap tunneling junction, where the tunneling current passes through the trapped molecule.28 (d) Schematic of pulse learning analysis.28 Reprinted with permission from ref. 28. Copyright 2019 American Chemical Society. | ||
Taniguchi et al. proposed a new method, namely, one electric current pulse method using ML to discern the DNA nucleotides from the background noise through gold nanogap electrodes (Fig. 3c and d).28 The novelty of the approach is that, in contrast to the traditional ionic current and translocation time features, the authors focused on extracting the pulse from the current–time profiles of DNA nucleobases, followed by extraction of features from each pulse. To extract the features, the pulse wave width was divided into ten equal segments for learning and validation. The collected tunneling current data was then averaged for each section, leading to a 10-dimensional feature vector. By using the positive unlabeled classification (PUC), 2-, 3-, and 4-types of DNA nucleotides were identified with a high degree of accuracy. The approach benefits from the fact that by utilizing this method, one can identify the DNA nucleotides within the noise matrix. From the discussion so far, it is evident that nanogap architectures are promising for DNA sequencing, and after integration with ML tools, ultrarapid and accurate identification of DNA nucleotides can be achieved.
In addition to nanogap architectures, researchers have also delved into utilizing ML-integrated solid-state nanopores for DNA sequencing. In 2019, Fyta and co-workers introduced a new approach to identify distinct DNA molecular events through ionic current measurements of the 2D MoS2 nanopore.22 Different from the previous studies, in this work, the authors introduced an unsupervised ML model for clustering nanopore ionic traces into different classes, remarkably avoiding the need for labels. Within this approach, the authors introduced four key features: dwell time, ionic current blockade height, ionic blockade mean current, and levels, denoting the quantity of putative distinct DNA configurations during nanopore translocation events. To provide insight into the molecular features inherent in the nanopore data, k-means clustering is employed, as implemented in the scikit-learn library.29 The feature space analysis with respect to ionic blockade mean current and ionic blockade height for all four DNA nucleotides is shown in Fig. 4a. To find the optimum number of clusters further, Silhouette (S) and Calinski-Harabasz (CH) scores are calculated for each nucleotide and the results demonstrate that the dwell time is not as efficient as the newly introduced feature ionic blockade height for classification (Fig. 4b).
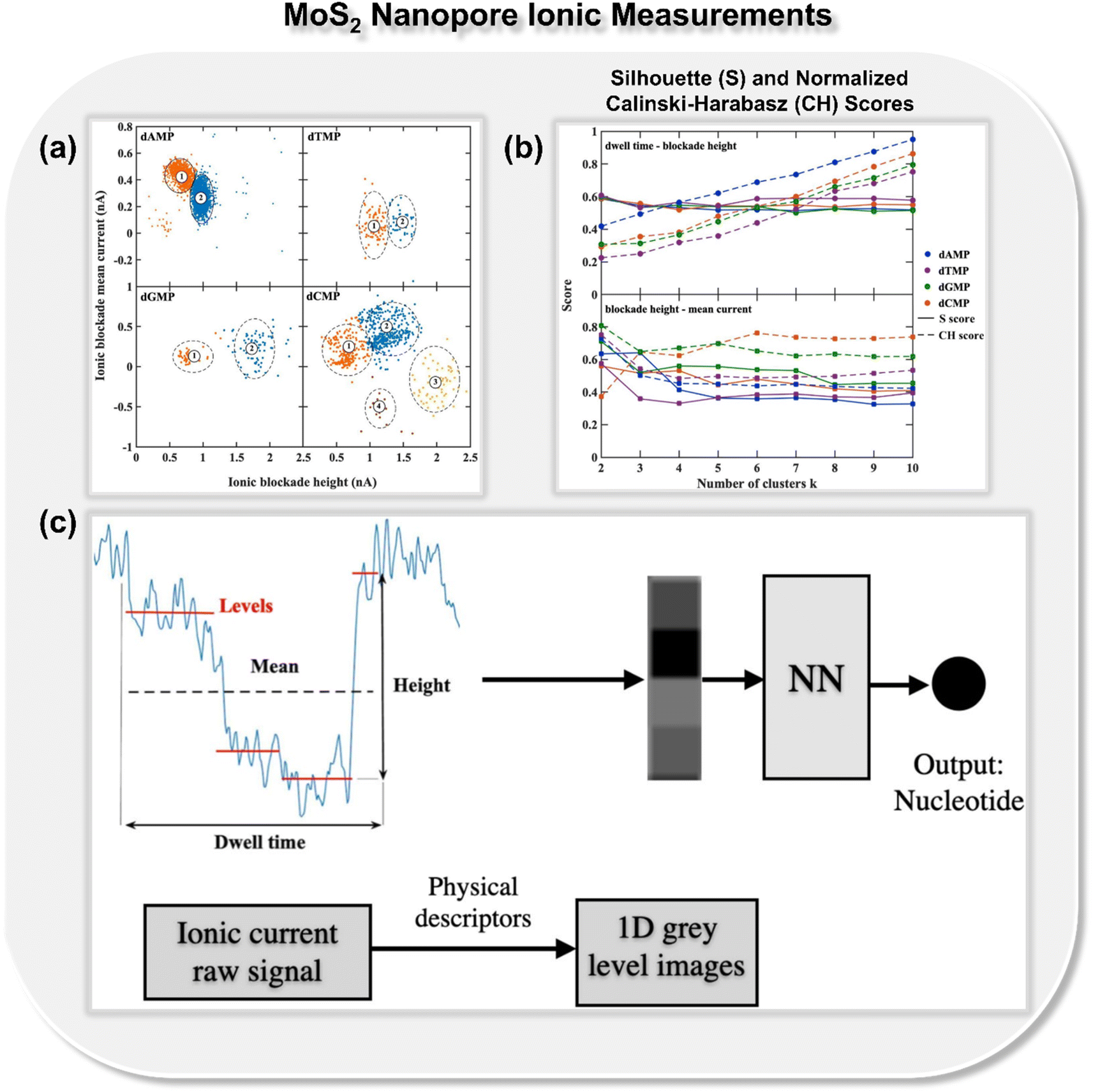 | ||
| Fig. 4 (a) Feature space analysis for the translocation events of all four DNA nucleotides (dAMP, dGMP, dCMP, and dTMP),22 (b) Silhouette (S) and normalized Calinski-Harabasz (CH) scores for all single-nucleotide translocation measurements.22 Reprinted with permission from ref. 22. Copyright 2019 IOP science. (c) Schematic of mapping ionic current traces by means of features, dwell time, ionic blockade height, ionic blockade mean current, and levels into grey-level scale images, which ultimately leads to the prediction of class of DNA nucleotides.30 Reprinted with permission from ref. 30. Copyright 2021 IOP science. | ||
Building on the importance of the newly introduced feature—i.e., ionic blockade current height—the authors next reported the deep neural network to enhance the read-out-efficiency of DNA nucleotide identification (Fig. 4c).30 In place of using the models trained with full ionic current traces, a new method was introduced, which reduced the current traces to a few descriptors and thus reduced the data dimensionality. Leveraging convolutional networks and deep neural networks trained on lower dimensional data, an average accuracy of 94% is achieved. It was noticed that the feature, ‘ionic blockade height’ in combination with other features, leads to the well-separated and dense data clustering in the feature space, significantly enhancing the applicability of MoS2 nanopore toward DNA sequencing.
Recently, Pathak and co-workers proposed a concept for sequencing DNA nucleotides using the ML-integrated gold nanopore with a transverse current approach (Fig. 5a).31 In earlier reported methods, ML algorithms were used only for the classification of DNA nucleotides. Here, for the very first time, the authors explored the potential of ML in sequencing DNA nucleotides. To achieve this goal, the features were extracted directly from the chemical and electrical properties of the peripheral atoms of the nucleotides and the transmission function was considered as output. It is observed that by utilizing the extreme gradient boosting regression (XGBR) algorithm, it is possible to identify the DNA nucleotides with a low root mean square error (∼0.12), provided the model is trained with a transmission function dataset of only one nucleotide. The scatter plot for DFT calculated transmission and XGBR predicted transmission for dGMP is shown in Fig. 5b.
 | ||
| Fig. 5 (a) Schematic of gold nanopore consisting of left and right electrode and a central scattering region.31 (b) Scatter plot of DFT calculated transmission vs. predicted transmission.31 Reprinted with permission from ref. 31. Copyright 2022 American Chemical Society. (c) Schematic of graphene nanopore32 and (d) confusion matrix for classification of DNA nucleotides (dAMP, dGMP, dCMP, and dTMP).32 Reprinted with permission from ref. 32. Copyright 2023 American Chemical Society. | ||
To further explore how well a solid-state nanopore architecture can be integrated with ML tools and utilized for high-throughput DNA sequencing, the authors delved deeper and studied other solid-state materials such as graphene (Fig. 5c and d) and C3N (Fig. 6a and b) nanopores in the realm of ML-aided DNA sequencing.32,33 They observed that nanopore architectures are promising toward both writing (prediction of electronic signatures) and reading (identification of the class of nucleotides) of DNA nucleotides. In integration with artificial intelligence, solid-state nanopores allow the identification and classification of each DNA nucleotide with good precision and accuracy. Training upon only single nucleotide datasets prediction of other nucleotides with configurational variations is possible by using an extreme boosting gradient regressor (XGBR). Moreover, using ML classification tools such as SVM, RFC, and decision tree classifier (DTC) models trained with only four transmission features, each binary, ternary, and quaternary classification of unlabeled DNA nucleotides can be achieved with good accuracy.32,33
 | ||
| Fig. 6 (a) Schematic of C3N nanopore33 and (b) ML predicted sensitivity plot for DNA nucleotides dTMP, dGMP, and dCMP.33 Reprinted with permission from ref. 33. Copyright 2023 American Chemical Society. (c) Schematic illustration of neural network-assisted MspA biological nanopore readout processing for different combinations and ordered sequences of natural and chemically modified nucleotides34 and (d) confusion matrix with lowest and highest accuracy scores.34 Reprinted with permission from ref. 34. Copyright 2022 American Chemical Society. | ||
Other than solid-state nanopores, biological nanopores are also integrated with ML classification tools and utilized for high-throughput DNA sequencing. Tabatabaei et al. reported an extended molecular alphabet for DNA data storage combining natural and chemically modified DNA nucleotides (Fig. 6c and d).34 It has been noticed that using the Mycobacterium smegmatis porin A (MspA) biological nanopore, it is possible to detect different combinations and ordered sequences of natural and modified nucleotides in oligomers. In addition, a neural network architecture is demonstrated for sequencing the extended alphabet with increased accuracy by 60%.
So far, using ML, the potential of nanopore and nanogap architectures in single-molecule DNA sequencing has been thoroughly investigated from both theoretical and experimental aspects. It has been demonstrated that both architectures are promising to identify DNA nucleotides with high precision and accuracy. Further, to check the potential of nanochannel architecture, Pathak and co-workers thoroughly investigated the potential of MXene and MoS2 nanochannels toward ML-aided DNA sequencing. MXene nanochannels are demonstrated to be promising for both genome and epigenome sequencing.35 Using Ti2NS2 MXene nanochannel, the transmission of both DNA and methylated DNA nucleobases is predicted with low root mean square error (∼0.16) (Fig. 7a and b). Notably, if the ML algorithm is trained with the data of methylated DNA bases, selective identification of all four DNA nucleobases is possible.
 | ||
| Fig. 7 (a) Schematic of Ti2NS2 MXene-based nanochannel device leveraging ML regression tools for identification of both DNA and methylated DNA.35 (b) Scatter plot of DFT calculated transmission vs. ML predicted transmission for 5-methyl cytosine (5 mC) nucleobase.35 Reprinted with permission from ref. 35. Copyright 2023 American Chemical Society. (c) Schematic of MoS2 nanochannel device leveraging artificial intelligence (AI) for prediction of transmission fingerprints and class of DNA nucleotides36 and (d) principal component analysis plot reducing the dimensionality of the dataset to three principal components.36 Reprinted with permission from ref. 36. Copyright 2023 Royal Society of Chemistry. | ||
Currently, transition metal dichalcogenides are emerging as a potential material for molecule DNA sequencing. It has been previously demonstrated that using the MoS2 nanopore, the DNA nucleobase can be identified with a high signal-to-noise ratio (SNR ∼15).37 On account of the importance of MoS2, Pathak and co-workers proposed an artificially intelligent MoS2 nanochannel in high throughput recognition and classification of DNA nucleotides (Fig. 7c).36 Different from the previous works, here they introduced RDKit fingerprints as important features toward DNA recognition. To reduce the dimensionality of features, principal component analysis has been employed, which reduces the 2048 RDKit features to 3 principal components (Fig. 7d). RDKit fingerprints, in combination with previously reported elemental features, led to a noteworthy reduction of 16% in the mean absolute error values. Moreover, herein, for AI calling of DNA nucleotides exclusively from their transmission readouts, the authors leveraged the supervised ML classification tools and predicted the class of DNA nucleotides with a perfect accuracy of 100%. To check real-time viability of the proposed approach, they try to predict the DNA nucleotides in different rotated configurations. For each rotated configuration, binary, ternary, and quaternary classification of DNA nucleotides, a perfect classification accuracy of 100% is achieved. To provide insight into the machine decision making process, the feature importance plots and SHAP summary bar plots are evaluated and the feature ‘MIN’ is found to be the most dominant toward DNA classification. For better understanding, year-wise documentation of experimental and theoretical studies reported on ML aided DNA sequencing studies is tabulated in Table 1.
| S. No. | Material | Nanodevice | Molecular carrier | Study | Comments | Year | Ref. |
|---|---|---|---|---|---|---|---|
| 1 | Gold electrodes | Functionalized nanogap | Nucleoside 5′-monophosphates and DNA oligomer | Experimental + SVM | 80% accuracy of base calling from a single peak and the accuracy increased to 95% when multiple spikes in a signal cluster were analyzed | 2012 | 21 |
| 2 | Gold electrodes | Functionalized nanogap | DNA bases and DNA homopolymer | Experimental + SVM | Theoretical underpinning to machine learning-driven identification of single molecule signals | 2015 | 23 |
| 3 | Palladium electrodes | Functionalized nanogap | DNA nucleotides | Experimental + SVM | Both benzimidazole carboxamide and triazole-carboxamide are noted to function much better than imidazole carboxamide toward DNA nucleotide classification | 2016 | 26 |
| 4 | 2D-MoS2 | Nanopores of different diameters | DNA nucleotides | Experimental + unsupervised learning | Ionic blockade height is reported as a superior feature to the traditional feature dwell time toward well-defined clustering of DNA events | 2019 | 22 |
| 5 | Gold electrodes | Nanogap | DNA nucleotides | Experimental + supervised machine learning | High-precision single molecule identification is achieved via a one-electric current pulse method using a random forest classifier | 2019 | 28 |
| 6 | 2D-MoS2 | Solid-state nanopore | DNA nucleotides | Experimental + deep neural networks | Leveraging ionic blockade height in combination with other features and deep neural network led to an increased accuracy up to 94% | 2021 | 30 |
| 7 | Mycobacterium smegmatis porin A (MspA) | Biological nanopore | ssDNA | Experimental + neural network | Single-molecule sequencing of both natural and modified chemical nucleotides is achieved using neural networks | 2022 | 34 |
| 8 | Gold | Nanopore | DNA nucleotides | DFT + NEGF + ML | Using interpretable XGBR, transmission prediction of DNA nucleotides and their rotated configurations is reported with good accuracy | 2022 | 31 |
| 9 | Graphene | Nanopore | DNA nucleotides | DFT + NEGF + ML | Using XGBR and SVC, high throughput DNA sequencing is achieved with high accuracy | 2023 | 32 |
| 10 | Ti2NS2 MXene | Nanochannel | DNA nucleobases | DFT + NEGF + ML | Using random forest regression identification of both DNA and methylated DNA nucleotides is reported with root mean square error as low as 0.16 | 2023 | 35 |
| 11 | Germanene | Nanogap | DNA nucleotides | DFT + NEGF + ML | Random forest classifier identified DNA nucleotides and performed high precision classification with accuracy as high as 97% | 2023 | 27 |
| 12 | MoS2 | Nanochannel | DNA nucleotides | DFT + NEGF + ML | New SMILES features are introduced and accuracy for each binary, ternary, and quaternary classification of DNA nucleotides is achieved with 100% accuracy | 2023 | 36 |
| 13 | C3N | Nanopore | DNA nucleotides | DFT + NEGF + ML | Transmission prediction of each unknown nucleotide and their rotation dynamics is reported with interpretable XGBR model | 2023 | 33 |
4. Guide to ML developments
Feature designing is a crucial step in the machine learning (ML) pipeline for making accurate predictions, as the quality and relevance of features directly impact the model's ability to understand patterns and make accurate predictions. The process of feature designing involves creating, selecting, or transforming the input feature variables to enhance a model's predictive capabilities. In the ML framework of sequencing DNA, depending on the experimental and theoretical measurements, different input features have been designed and utilized to decode the complex genetic code exclusively from their electric readouts. The key parameters considered in designing features in both theoretical and experimental studies are as follows.4.1. Experimental feature engineering
Earlier experimental reports utilizing ML are based on recognition tunneling (RT) measurements.21,23,26 They mainly leveraged ML classification tools such as SVM and RFC to identify the class of DNA nucleotides. To extract features, RT events are first divided into two parts: spikes and clusters. The spike is an individual single RT peak and the cluster is a subset of close spikes. Considering the importance of spikes and clusters, different features have been extracted and utilized to identify DNA patterns. Later, new features like ionic blockade current height and dwell time were introduced, and ionic blockade height was reported to be a superior method for the precise identification of DNA nucleobases.22,30 Recently, a new electric current pulse method has been introduced and features have been extracted directly from the pulse.28 A detailed description of feature engineering in experimental measurements is listed in Table 2.| S. No. | Measurement | Device | Features | Description | ML algorithm | Ref. |
|---|---|---|---|---|---|---|
| 1 | Recognition tunneling | Au nanogap | Spike amplitude | Average peak amplitude | SVM | 21 and 23 |
| 2 | Recognition tunneling | Au nanogap | Spike width | Full width of the peak at half of the average peak height | SVM | 21 and 23 |
| 3 | Recognition tunneling | Au nanogap | Spike Fourier component N | Obtained Fourier components | SVM | 21 and 23 |
| 4 | Recognition tunneling | Au nanogap | Spike phase component N | Phase averaged over four frequency intervals | SVM | 21 and 23 |
| 5 | Recognition tunneling | Au nanogap | Spike wavelet component N | Decomposition of the spike into Haar wavelet components | SVM | 21 and 23 |
| 6 | Recognition tunneling | Au nanogap | Number of peaks in a cluster | Parameters assigned to each peak in the cluster | SVM | 21 and 23 |
| 7 | Recognition tunneling | Au nanogap | Cluster on-time | The ratio of the sum of the full widths of all peaks in a cluster to the total duration of the cluster | SVM | 21 and 23 |
| 8 | Recognition tunneling | Au nanogap | Spike frequency | Number of peaks found within ±2000 0.02 ms sample points of the center of a given peak | SVM | 21 and 23 |
| 9 | Recognition tunneling | Au nanogap | Cluster frequency N | Each cluster is loaded into an array of 4096 points, and the FFT is calculated for the entire cluster as described above for spikes. It is resolved into nine bins covering the frequency range up to the Nyquist limit | SVM | 21 and 23 |
| 10 | Recognition tunneling | Au nanogap | Cluster phase N | Calculated analogous to spike phase but for the whole cluster | SVM | 21 and 23 |
| 11 | Recognition tunneling | Pd nanogap | Primary features | P_max amplitude, P_average amplitude, P_top average, P_peak width, P_roughness, P_frequency, C_peaks in cluster, C_frequency, C_average amplitude, C_top average, C_cluster width, C_roughness, C_max amplitude | SVM | 26 |
| 12 | Recognition tunneling | Pd nanogap | Secondary features | P_peakFFT_Whole1 ∼ 51, C_totalPower, C_iFFTLow, C_iFFTMedium, C_iFFTHigh, C_clusterFFT1 ∼ 61, C_highLow, C_freq_Maximum_Peak1 ∼ 4, C_clusterCepstrum1 ∼ 61, C_clusterFFT_Whole1 ∼ 51 | SVM | 26 |
| 13 | Ionic current | MoS2 nanopore | Translocation time | Maps the duration of the event | K-means clustering, DNN, CNN, LSTM network, XGBoost | 22 and 30 |
| 14 | Ionic current | MoS2 nanopore | Ionic current blockade height | Difference between the maximum and minimum values of a single current blockade peak | K-means clustering, DNN, CNN, LSTM network, XGBoost | 22 and 30 |
| 15 | Ionic current | MoS2 nanopore | Ionic blockade mean current | Average current value | K-means clustering, DNN, CNN, LSTM network, XGBoost | 22 and 30 |
| 16 | Ionic current | MoS2 nanopore | Levels | Number of the presumably different DNA configurations through the nanopore | K-means clustering, DNN, CNN, LSTM network, XGBoost | 22 and 30 |
| 15 | Transverse tunneling | Au nanogap | Pulse wave width | Pulse wave width is divided into ten equal sections and generated electric current data were averaged for each section to be a 10-dimensional feature vector | Random forest classification and positive unlabeled classification (PUC) methods | 28 |
| 16 | Ionic current | MspA nanopore | Oligo data corresponding to different combinations and orderings of chemically modified nucleotides | 1D convolution layers (conv) serve as feature extractors | 1D-residual neural network model | 34 |
4.2. Theoretical feature engineering
Different from the experimental reports, in theoretical studies, both ML regression and classification algorithms have been wisely utilized for high throughput DNA sequencing. ML regression algorithms leverage the elemental and molecular properties of individual DNA nucleotides as input features to predict the transmission fingerprints of each individual molecule.32,36 While ML classification algorithms extract features directly from the transmission profiles and train on these features, the ML classifiers predict the class of each nucleotide with high precision and accuracy. A detailed description of designed features in theoretical studies is listed in Table 3.| S. No. | Device | Features | Best-fitted ML algorithms | Ref. |
|---|---|---|---|---|
| 1 | Au nanopore | Average Mendeleev number, average Vander walls radii, average polarizability, average valence electrons, average effective nuclear charge, average ionization energy, average electron affinity, average molecular weight, electronegativity, the (E–EF) energy scale for the transmission spectra | eXtreme gradient boosting regression (XGBR) model | 31 |
| 2 | Graphene nanopore | Average valence electrons, average molecular weight, average electronegativity, minimum distance between nucleotide H-atom and pore edge H-atom, minimum distance between nucleotide H-atom and pore edge C-atom, the minimum distance between nucleotide H-atom and pore edge N-atom, the minimum distance between nucleotide H-atom and pore edge O-atom, average electron affinity, average Vander walls radii, average dipole polarizability, average ionic radii, average covalent radii, average ionization energy, average effective nuclear charge, the energy range of transmission spectra; for classification transmission, maxima normalized transmission, minima normalized transmission, average normalized transmission | XGBR regression model and radial basis function support vector classification model (SVCrbf) | 32 |
| 3 | Ti2NS2 MXene nanochannel | Energy, sum Pauling electronegativity, sum atomic radius, sum valence electrons, sum polarizability, LUMO, HOMO, sum atomic charge nitrogen atoms, and sum atomic charge oxygen atoms | Random forest regressor (RFR) | 35 |
| 4 | Germanene nanogap | Transmission, maxima normalized transmission, minima normalized transmission, average normalized transmission | Random forest classification (RFC) | 27 |
| 5 | MoS2 nanochannel | Average atomic radius, average ionic radius, average covalent radius, average Pauling electronegativity, average number of valance electrons, and average polarizability, HOMO, LUMO, and RDkit features; for classification transmission, maxima normalized transmission, minima normalized transmission, average normalized transmission | XGBR and logistic regression (LR) | 36 |
| 6 | C3N nanopore | Mean valence electrons, mean molecular weight, mean electronegativity, Minimum distance between nucleotide H-atom and pore edge H atom, Minimum distance between nucleotide H-atom and pore edge C atom, Minimum distance between nucleotide H-atom and pore edge O atom, mean electron affinity, mean van der Waals radii, mean dipole polarizability, mean ionic radii, mean covalent radii, mean ionization energy, mean effective nuclear charge, energy range of transmission; for classification transmission, maxima normalized transmission, minima normalized transmission, average normalized transmission | XGBR, RFC, and decision tree classification (DTC) | 33 |
5. Data regimes and ML methodologies
As discussed above, ML tools have been successfully integrated with NGS datasets, including both theoretical and experimental datasets, to accelerate the process of high-throughput DNA sequencing. Here, it should be noted that ML models are inherently data-driven, and the size of NGS datasets can significantly influence their performance. Understanding the distinction between experimental and theoretical studies in terms of dataset size, as well as the suitability of different ML methods in various data regimes, is crucial for advancing DNA sequencing technologies.5.1. Experimental big data regime
In the real NGS measurements, sequencing of native DNA and RNA molecules is achieved through devices from Oxford Nanopore Technologies such as MinION, GridION, and PromethION, resulting in a massive amount of data in FAST5 format (∼1.3 TB FAST5 files for ∼30× human genome).38–40 The primary challenge associated with FAST5 files is signal processing. Generally, signal processing of ionic current data obtained through nanopore sequencing consists of four steps: denoising raw data, spike recognition, feature extraction, and analysis.41 For each step of signal processing, different ML algorithms were utilized. The primary step is the denoising of raw data, i.e., the accurate interpretation of the noisy and complex ionic current signals that occur as the analyte passes through the nanopore. ML algorithms, especially deep learning models, are adept at handling this complexity.42,43 They are used to enhance signal processing by identifying patterns and distinguishing between the current disruptions caused by different nucleotides, significantly improving the accuracy of base calling. The second step involves spike recognition, i.e., identification and extraction of translocation events, for which the most suitable algorithm is the Hidden Markov Model (HMM).44,45 To reduce the overlapping issue of residual current and duration time of DNA nucleobases obtained from the biological nanopore, the AdaBoost-based ML model is utilized, which is trained on feature vectors obtained from HMM, enabling the identification of each nucleobase.46,47 For feature extraction, the most common ML algorithm is Bi-path Network (B-net), which is based on the residual neural network (ResNet).48 Other than that, a different method, namely, shapelet, has also been proposed for feature extraction.49 The Learning Time-Series Shapelets (LTS) algorithm successfully differentiates between various DNA oligomers that have nearly identical blockade current amplitudes and durations. The last and most important step involves the analyte classification. Major attempts have been directed toward the utilization of three algorithms: SVM, decision trees (DTs), and random forests (RFs).41 Using SVM trained on features extracted from characterization of DNA translocating event including relative intensity, surface area, dwell time, and both the left and right slope, both short ssDNA (10-mers) and dsDNA (40-mers) are classified with good accuracy.50 For better understanding, the commonly used ML algorithms for processing of experimental NGS nanopore signals are depicted in Table 4.| S. No. | Algorithm | Function | Ref. |
|---|---|---|---|
| 1 | Deep neural network (DNN) | Data denoising | 42 and 43 |
| 2 | Hidden Markov model (HMM) | Spike recognition | 44 and 45 |
| 3 | Fuzzy-c means clustering (FCM) | Spike recognition | 51 |
| 4 | DBSCAN | Spike recognition | 52 |
| 5 | Bi-path network (B-net) | Feature extraction | 48 |
| 6 | Learning time-series shapelets (LTS) | Feature extraction | 49 |
| 7 | Support vector machines (SVM) | Analyte classification | 26 |
| 8 | Decision trees (DTs) | Analyte classification | 41 |
| 9 | Random forests (RFs) | Analyte classification | 53 and 54 |
5.2. Theoretical low data regime
Compared to experimental studies, which deal with full DNA strands (∼3.2 billion nucleobase pairs) or DNA oligomers, theoretical studies often rely on simulated or smaller datasets of single nucleobase, highlighting specific aspects of DNA sequencing.55 In experimental NGS measurements, conductance and ionic blockade current signals are the major identifying parameters, while ML-coupled theoretical studies mainly deal with transmission function (∼500 data points per nucleotide) as targeted regression and classification parameter.35,36,56,57 ML regression algorithms are generally used for the prediction of transmission profiles of DNA nucleotides.31,36 ML classification algorithms are also utilized for the classification of DNA nucleotides based on features extracted from their transmission profiles.32,36 As shown in Table 3, for the identification of transmission profiles of DNA nucleotides, the best-fitted regression algorithms are noted to be XGBR and RFR and the algorithms LR, RFC, DTC, and SVCrbf are found to be well-suited to DNA nucleotide classification.6. Signal denoising
A major stumbling block in achieving high throughput DNA sequencing is reducing unwanted noise signals.55 Noise in sequencing data can arise from various sources, including sequencing platform errors (such as base calling and phasing errors), instrumental noise (signal intensity variations and background noise), sample quality issues (DNA degradation and contamination), amplification biases (PCR errors and uneven coverage), genomic complexities (repetitive regions and structural variations), and procedural inconsistencies (library preparation errors and data processing mistakes). This noise can obscure true genetic variations and lead to incorrect conclusions, making it critical to employ effective denoising strategies. In this regard, various algorithms, including both ML-based and non-ML-based, have been developed to isolate the target analyte signals from the background noise.41 Traditional methods of signal denoising include low-pass filters, which restrict the bandwidth of the signal and filter out the noisy background.58 The low-pass filters rely on the hard frequency threshold, which may lead to unwanted filtration of high-frequency signal components, which are crucial for efficient analyte characterization. To mitigate this effect, a potential alternative is the Kalman filter, which can efficiently isolate a signal from severely overlapped background noise.59 Other than that, wavelet transform-based filtering technology60,61 and consensus filter62 have also been utilized for denoising of nanopore signals.Compared to traditional non-ML-based algorithms, the utilization of ML algorithms for signal denoising is critically less explored. One notable approach is the use of convolutional neural networks (CNNs) and recurrent neural networks (RNNs), which are well-suited for handling sequential data and can effectively model the dependencies in DNA sequences to detect and correct errors.63 For instance, ‘DeepVariant,’ developed by Google, uses deep learning techniques to identify variants in sequencing data with high accuracy, outperforming traditional methods such as GATK, Hidden Markov Model (HMM), naive bayes classification, and the gaussian mixture model in several benchmarks.64 Similarly, the algorithm ‘Clairvoyante’ leverages deep neural networks to improve the calling of genetic variants from nanopore sequencing data, showing significant improvements in accuracy compared to existing tools.65 Moreover, unsupervised learning methods, such as clustering algorithms, have been used to distinguish between true genetic variants and sequencing errors. A study by Liu et al. demonstrated the use of a Gaussian mixture model to reduce noise in single-cell RNA sequencing data, leading to more accurate downstream analysis.66 Despite nanopore signal denoising, an attempt toward denoising of nanogap signals has also been made.28 By using the positive unlabeled classification (PUC) method, single DNA molecule information has been precisely discerned from the background of electrical noises generated through gold nanogap tunneling measurements. For better understanding, a summary of non-ML-based and ML-based algorithms used in denoising the nanopore/nanogap sequencing data is provided in Table 5.
| S. No. | Algorithm | Algorithm type | Comments | Ref. |
|---|---|---|---|---|
| 1 | Low-pass filter | Non-ML-based | Efficiently restricts the bandwidth and filters out noise signals | 58 |
| 2 | Kalman filter | Non-ML-based | Signal isolation from severely overlapped background noise | 59 |
| 3 | Wavelet-transform based technology | Non-ML-based | Signal and background noise can be separated in the wavelet domain rather than overlapped frequency domain | 60 and 61 |
| 4 | Consensus filter | Non-ML-based | Removes the uncorrelated events as noise from the channels | 62 |
| 5 | DeepVariant | ML-based | Identifies variants in sequencing data | 64 |
| 6 | Clairvoyante | ML-based | Sample agnostic and finds variants in less than 2 hours on a standard server | 65 |
| 7 | Gaussian mixture model | ML-based | Effective tool for accurately clustering and identifying cell types from single-cell RNA-Seq data | 66 |
| 8 | Positive unlabeled classification (PUC) | ML-based | Identification of 2-, 3-, and 4-type nucleotides is achieved at a single-molecule resolution | 28 |
7. Opportunities and challenges
7.1. ML-guided ionic current techniques
As discussed above, ML-guided ionic current techniques offer substantial opportunities to enhance DNA sequencing but also present serious challenges. On the opportunities front, ML models, especially deep learning algorithms like convolutional neural networks (CNNs) and recurrent neural networks (RNNs), significantly improve sequencing accuracy by distinguishing true genetic signals from noise, thereby enhancing base calling and variant detection.63 These models enable real-time analysis, providing immediate feedback during sequencing, which optimizes throughput and data quality. Furthermore, ML techniques excel in recognizing complex patterns in ionic current data, allowing for the detection of subtle genetic variations with high sensitivity and specificity.65 Their ability to continuously learn and adapt to new data further refines their predictive capabilities. Additionally, ML-guided methods are versatile, applicable across various sequencing platforms, and capable of integrating multiomics data for comprehensive biological insights.41 However, several challenges need to be addressed. High-quality, annotated datasets are essential for training ML models, but acquiring such data can be resource-intensive. Sequencing data often contains noise and systematic biases, complicating model training and requiring robust error-handling mechanisms. The black-box nature of many advanced ML models poses interpretability issues, hindering their acceptance in clinical and research settings where understanding the basis of predictions is critical.67 Moreover, the computational demands of training and deploying these models necessitate substantial processing power and efficient algorithms, posing scalability issues as data volumes increase. Addressing these challenges through collaborative efforts, improved data quality, and advancements in computational techniques will be crucial to fully realize the potential of ML in ionic current-based DNA sequencing.7.2. ML-guided transverse current techniques
In transverse current techniques for DNA sequencing, ML is essential for overcoming the technical complexities and enhancing the accuracy of base identification. These techniques involve measuring the electronic transport properties of nucleotides as a DNA strand passes near a nanoscale gap between electrodes. Through unsupervised learning and other advanced ML approaches, these algorithms can classify and interpret the nuanced electronic signatures of different nucleotides with high precision, improving the reliability of sequencing data.18 Furthermore, ML techniques are crucial for noise reduction, filtering out extraneous signals to achieve a higher signal-to-noise ratio.28 Besides, certain challenges must still be addressed, such as the measurement of electronic transport properties in transverse current techniques, which is technically complex and requires sophisticated pattern recognition capabilities that can be challenging to develop and optimize. Like ionic current techniques, transverse current methods also require high-quality, annotated datasets for effective ML training. The resource-intensive nature of producing these datasets poses a significant challenge. Integrating ML-guided transverse current techniques with other sequencing methods and multi-omics data can be complex, requiring sophisticated algorithms and comprehensive data management strategies. Overcoming these challenges will involve the development of more advanced computational techniques, enhanced data quality and annotation methods, and collaborative efforts across the scientific community.8. Perspective and prospectus
So far, we have discussed how ML regression and classification algorithms can propel biosensors toward rapid and accurate DNA sequencing without prior knowledge of sequence information. This is primarily achieved by the astute feature engineering and rational selection of prediction models for rapid and accurate prediction of key features of DNA nucleotides. Importantly, we shed light on the indispensable role of ML in the identification of DNA nucleotides through the elucidation of complex ionic current and transverse-tunneling current data. Moving forward, the following aspects could be helpful in the efficient development of artificially intelligent DNA sequencers: (1) building a generalized ML approach for efficient DNA sequencing, (2) strengthening the ML prediction with knowledge of the structure–property relationship, (3) leveraging advanced ML algorithms for key signatures determination of DNA nucleotides, (4) theory-driven ML approaches accelerating experimental efforts, and (5) meeting the demands for active collaboration.8.1. Building a generalized ML approach for efficient DNA sequencing
The advent of ML algorithms in NGS technology has made a drastic reduction in the time and cost of tedious theoretical and experimental demonstration of DNA sequencing.18,32 To date, however, the research efforts are mainly focusing on ML algorithms trained with certain features. To improve the efficiency of ML predictions, possible conformations of DNA nucleotides or the bonding configurations of DNA nucleotides with each other could also be a promising aspect of feature extraction. To streamline the feature extraction, device-DNA interactions are of particular importance. In this regard, the inclusion of well-sampled data with features extracted directly from the inherent molecular and geometrical properties of individual DNA nucleotides can accelerate the development of a generalized ML model for ultrafast DNA sequencing.We need more precise and easily available features, extracted from the molecular and chemical properties of a DNA strand, close to the realistic environment for a generalized ML model promising efficient and accurate DNA sequencing. To account for realistic conditions, features extracted from the characteristic properties of DNA-solvent interactions can also be of particular importance. Theoretical tools such as the molecular dynamics (MD) simulations and quantum mechanics-molecular mechanics (QM-MM) approach can be helpful in the generation of more efficient features with improved information on dynamic interactions of DNA nucleotides within the solvent environment.
Moreover, the need for various targeted properties used in ML-aided sequencing is of growing importance. In addition, the full potential of ML algorithms can be harnessed by training the algorithms with a well-sampled dataset. To make sure the model performance is maximized, a large number of training datasets is needed. For a generalized ML approach of nucleotide determination, it becomes crucial to extract features from the easily available data without the need for information on a complete DNA strand. For example, one can establish a standardized library of molecular features of DNA nucleotides (both geometrical and molecular properties are important) to sequence the complete DNA strand without the need for additional information on environmental conditions. We postulate that the integration of ML algorithms with complex transverse-tunneling current and ionic current data could be synergistically concatenated to create a biosensor capable of single nucleotide resolution. A standardized data library of DNA nucleotides would be key in accelerating the growth of ML-integrated next-generation DNA sequencing. Using the best-fitted algorithm, each DNA nucleotide can concurrently be identified with pooled computational and experimental data. Such an ML-integrated next-generation DNA sequencing approach is expected to guide the robust development of biosensors with industrial scalability, which is essential for personalized medicine development.
8.2. Strengthening the ML prediction with knowledge of the structure–property relationship
The complexity of data resulting from the enormous number of DNA nucleotides undergoing orientational variations demands more robust and sophisticated ML algorithms. One noteworthy trend in ML-aided DNA sequencing is the interpretation and explanation of utilized ML algorithms, enabling a better understanding of the structure–property relationship. It is of particular importance in designing an efficient biosensor because, through this, one can determine why certain predictions are made. In addition, interpretable ML algorithms can provide valuable mechanical insight and can help in improving the model's performance. The interpretability can also help in the identification and mitigation of biases and discrimination in ML algorithms which in turn can help in enhancing the ability of ML algorithms toward accurate prediction.In our reports, we tried to introduce transparency into the ML model predictions by performing the cooperative game theory-based SHapley Additive exPlanations (SHAP) analysis.27,33,35 The SHAP analysis, in combination with the feature importance plot helps to anchor the ML prediction toward the establishment of device-DNA interaction with base knowledge of scientific reasoning. We postulate that interpretable ML models would help in guiding feature selection techniques, through which more robust prediction of output with domain knowledge of ML decision making process can be made.
Despite significant attempts made to explore the potential key signatures (descriptors) elucidated from signal variations, it still remains difficult to determine how a rationale DNA sequencing would be manifested through such descriptors in a future outbreak. The current ML-aided DNA sequencing strategies revolve around only ionic current and translocation time as the potential targeted output. Well, this process can be further hastened if we consider more potential output variables encoding key signatures of individual DNA nucleotides. Reviewing the existing ML-aided DNA sequencing reports (both experimental and theoretical), we postulate that apart from the used output variables (ionic current height, ionic current magnitude, translocation time, and transmission function), more output variables determining the key characteristics of DNA nucleotides while translocating through the NGS device could be incorporated to enhance the signal to noise ratio characteristic.
8.3. Leveraging advanced ML algorithms for accurate DNA sequencing
The full suite of ML algorithms in long-read DNA sequencing is yet to be harnessed. For complex data mining, potential alternatives are principal component analysis (PCA), isometric mapping (ISOMAP), and uniform manifold approximation (UMAP), among others.68–71 Moreover, through the utilization of more advanced ML tools, such as direct neural networks, graph-neutral networks could be helpful in reducing prediction errors.30,65 For improved algorithm interpretation, Shapley additive explanations (SHAP), local interpretable model-agnostic explanations (LIME), and contrastive explanation methods (CEM) can be of particular importance in avoiding the pitfalls of ML, such as underfitting and overfitting.72,73 For real time data, we envision that leveraging the advanced ML tools could spark a paradigm shift in the area of single-molecule DNA sequencing, enabling the identification of individual nucleotides without the need for sequence information of complete DNA strand.8.4. Theory-driven ML approaches accelerating experimental efforts
There is also growing interest in integrating theory-driven ML approaches with experimental data to accelerate the process of precision base calling from real-time data. Theory-driven ML involves incorporating domain knowledge and theoretical models into ML algorithms to enhance their performance and interpretability. In this direction, Fyta and co-workers have made pioneering efforts. The authors utilized the experimental data of MoS2 nanopores and by using unsupervised and deep learning tools, the ionic blockade current height was demonstrated as an efficient feature for clustering of DNA translocation events.22,30 As the field is in its nascent stage, other potential alternatives such as physics-informed neural networks (PINNs),74 hybrid models,75 and transfer learning76 can also be of significant importance in accelerating the theory-driven ML experimental efforts. PINNs integrate physical laws and constraints into neural networks, allowing them to learn more effectively from data that adheres to known theoretical principles. Hybrid models, combining mechanistic models with data-driven approaches, can improve the robustness and generalizability of ML algorithms. For example, the integration of mechanistic understanding of gene regulatory networks with data-driven methods can enhance the prediction accuracy of sequencing outcomes.75 Through transfer learning (applying models trained on large, annotated datasets to new, smaller datasets), one can leverage existing knowledge and improve performance in low-data regimes. This approach could be more useful in genomic studies where annotated data is often scarce.8.5. Meeting the demand for active collaboration
In the current landscape, the application of machine learning (ML) in DNA sequencing is in its early stages with only a limited number of reports, primarily theoretical, leveraging ML regression and classification algorithms. However, the realm of real-time DNA sequencing generates vast and intricate genomic data. The main hindrance is caused by a lack of standardized protocols and testing and validation of new data. To harness the full potential of ML, collaborative efforts are imperative, bringing together bioinformaticians, geneticists, and data scientists to seamlessly integrate ML tools into existing sequencing workflows and optimize their performance for real-time analysis. Implementation of ML in the field involves several key steps. Firstly, collaboration facilitates the integration of ML techniques into experimental design, allowing researchers to optimize data collection processes for subsequent analysis. Secondly, ML can aid in the preprocessing and cleaning of raw genomic data, handling noise, and extracting meaningful features. Thirdly, collaborative efforts empower the development of predictive models that can uncover hidden associations, predict outcomes, and contribute to the identification of potential genetic markers or targets. We believe that meeting the demand for collaboration will fortify the applicability of ML in achieving ultra-rapid, accurate, and high-throughput capabilities in DNA sequencing.8.6. Conclusions and outlook
In this perspective, a summary of the research progress of ML-guided next-generation DNA sequencing is provided. The existing NGS nanoarchitectures include field-effect-transistor-based nanopore, nanogap, and nanochannel devices utilizing ionic current and transverse-tunneling current approaches. ML has tremendous potential for extensive integration into DNA sequencing devices. Resolving the current signal overlapping issue of DNA nucleotides with classification algorithms and identification of key signatures of DNA nucleotides through regression algorithms are two major breakthroughs in ML-aided DNA sequencing applications. Despite the exciting breakthroughs, the usage of ML in DNA sequencing is still in its preliminary stage. For widespread adoption of ML in real DNA sequencing, a significant number of challenges must be addressed, such as efficient training of machines, real-time efficient data, fast processing of electronic data, etc. So far, various alternatives have been proposed to achieve single-nucleotides resolution, such as two-dimensional molecular electronics spectroscopy,25 different edge functionalization,77,78 labeling of DNA nucleotides,79,80 different device architecture,81,82 nucleobase analogs,82,83 heterostructure,84,85 hybrid nanopore,55,86 and so on. However, the potential of integrating ML with these methods to accelerate the DNA sequencing process has not yet been fully explored. By combining ML algorithms with these advanced techniques, one can potentially improve the accuracy, speed, and efficiency of DNA sequencing, paving the way for significant advancements in genomic research and medical diagnostics. The significant ongoing efforts from various science disciplines, e.g., material science, artificial intelligence, biology, and chemistry, among others, can revolutionize the field of DNA sequencing. The overlapping of electric signals is a major bottleneck in decoding the genetic code and we foresee that a viable solution lies in seamlessly integrating next-generation DNA sequencing with artificial intelligence.Data availability
No primary research results, software or code have been included and no new data were generated or analyzed as part of this perspective.Author contributions
S. M. contributed to the outline and wrote the original draft of the manuscript; M. K. J. contributed to the review of the manuscript; and B. P. contributed to the outline, reviewed and edited the manuscript.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
This work is acknowledged by grants DST-SERB (project number: CRG/2022/000836), BRNS (project number: 2023-BRNS/12356), and CSIR (project number: 01(3046)/21/EMR-II). S. M. and M. K. J. acknowledge UGC and MHRD for their research fellowships, respectively.References
- J. Shendure and H. Ji, Nat. Biotechnol., 2008, 26, 1135–1145 CrossRef CAS PubMed.
- T. P. Niedringhaus, D. Milanova, M. B. Kerby, M. P. Snyder and A. E. Barron, Anal. Chem., 2011, 83, 4327–4341 CrossRef CAS PubMed.
- S. Behjati and P. S. Tarpey, Arch. Dis. Child., 2013, 98, 236–238 Search PubMed.
- W. J. Ansorge, New Biotechnol., 2009, 25, 195–203 CrossRef CAS.
- S. Goodwin, J. D. McPherson and W. R. McCombie, Nat. Rev. Genet., 2016, 17, 333–351 CrossRef CAS PubMed.
- X. Chen, Y. Kang, J. Luo, K. Pang, X. Xu, J. Wu, X. Li and S. Jin, Front. Cell. Infect. Microbiol., 2021, 632490 CrossRef CAS.
- T. A. Manolio, L. D. Brooks and F. S. Collins, J. Clin. Invest., 2008, 118, 1590–1605 CrossRef CAS PubMed.
- S. Feng, S. E. Jacobsen and W. Reik, Science, 2010, 330, 622–627 CrossRef CAS.
- J. Chen, Q. Huang, D. Gao, J. Wang, Y. Lang, T. Liu, B. Li, Z. Bai, J. Luis Goicoechea, C. Liang, C. Chen, W. Zhang, S. Sun, Y. Liao, X. Zhang, L. Yang, C. Song, M. Wang, J. Shi, G. Liu, J. Liu, H. Zhou, W. Zhou, Q. Yu, N. An, Y. Chen, Q. Cai, B. Wang, B. Liu, J. Min, Y. Huang, H. Wu, Z. Li, Y. Zhang, Y. Yin, W. Song, J. Jiang, S. A. Jackson, R. A. Wing, J. Wang and M. Chen, Nat. Commun., 2013, 4, 1595 CrossRef.
- M. Zwolak and M. Di Ventra, Rev. Mod. Phys., 2008, 80, 141–165 CrossRef.
- H. S. Kim and Y.-H. Kim, Biosens. Bioelectron., 2015, 69, 186–198 CrossRef CAS PubMed.
- L. Gasparyan, I. Mazo, V. Simonyan and F. Gasparyan, Open J. Biophys., 2019, 9, 169–197 CrossRef CAS.
- A. Meller, L. Nivon and D. Branton, Phys. Rev. Lett., 2001, 86, 3435–3438 CrossRef CAS.
- S. Benner, R. J. A. Chen, N. A. Wilson, R. Abu-Shumays, N. Hurt, K. R. Lieberman, D. W. Deamer, W. B. Dunbar and M. Akeson, Nat. Nanotechnol., 2007, 2, 718–724 CrossRef CAS PubMed.
- A. Meller, L. Nivon, E. Brandin, J. Golovchenko and D. Branton, Proc. Natl. Acad. Sci. U.S.A., 2000, 97, 1079–1084 CrossRef CAS.
- J. Li, M. Gershow, D. Stein, E. Brandin and J. A. Golovchenko, Nat. Mater., 2003, 2, 611–615 CrossRef CAS PubMed.
- F. J. Rang, W. P. Kloosterman and J. de Ridder, Genome Biol., 2018, 19, 90 CrossRef.
- M. Taniguchi, ACS Omega, 2020, 5, 959–964 CrossRef CAS.
- D. Branton, D. W. Deamer, A. Marziali, H. Bayley, S. A. Benner, T. Butler, M. Di Ventra, S. Garaj, A. Hibbs, X. Huang, S. B. Jovanovich, P. S. Krstic, S. Lindsay, X. S. Ling, C. H. Mastrangelo, A. Meller, J. S. Oliver, Y. V. Pershin, J. M. Ramsey, R. Riehn, G. V. Soni, V. Tabard-Cossa, M. Wanunu, M. Wiggin and J. A. Schloss, Nat. Biotechnol., 2008, 26, 1146–1153 CrossRef CAS PubMed.
- J. J. Gooding and K. Gaus, Angew Chem. Int. Ed. Engl., 2016, 55, 11354–11366 CrossRef CAS PubMed.
- S. Chang, S. Huang, H. Liu, P. Zhang, F. Liang, R. Akahori, S. Li, B. Gyarfas, J. Shumway, B. Ashcroft, J. He and S. Lindsay, Nanotechnology, 2012, 23, 235101 CrossRef PubMed.
- A. D. Carral, C. S. Sarap, K. Liu, A. Radenovic and M. Fyta, 2D Mater., 2019, 6, 045011 CrossRef CAS.
- P. Krstić, B. Ashcroft and S. Lindsay, Nanotechnology, 2015, 26, 084001 CrossRef.
- S. Chang, S. Huang, J. He, F. Liang, P. Zhang, S. Li, X. Chen, O. Sankey and S. Lindsay, Nano Lett., 2010, 10, 1070–1075 CrossRef CAS PubMed.
- A. C. Rajan, M. R. Rezapour, J. Yun, Y. Cho, W. J. Cho, S. K. Min, G. Lee and K. S. Kim, ACS Nano, 2014, 8, 1827–1833 CrossRef CAS PubMed.
- S. Biswas, S. Sen, J. Im, S. Biswas, P. Krstic, B. Ashcroft, C. Borges, Y. Zhao, S. Lindsay and P. Zhang, ACS Nano, 2016, 10, 11304–11316 CrossRef CAS.
- M. K. Jena, D. Roy, S. Mittal and B. Pathak, ACS Mater. Lett., 2023, 5, 2488–2498 CrossRef CAS.
- M. Taniguchi, T. Ohshiro, Y. Komoto, T. Takaai, T. Yoshida and T. Washio, J. Phys. Chem. C, 2019, 123, 15867–15873 CrossRef CAS.
- F. Pedregosa, G. Varoquaux, A. Gramfort, V. Michel, B. Thirion, O. Grisel, M. Blondel, P. Prettenhofer, R. Weiss, V. Dubourg, J. Vanderplas, A. Passos, D. Cournapeau, M. Brucher, M. Perrot and É. Duchesnay, J. Mach. Learn. Res., 2011, 12, 2825–2830 Search PubMed.
- Á. Díaz Carral, M. Ostertag and M. Fyta, J. Chem. Phys., 2021, 154, 044111 CrossRef.
- M. K. Jena, D. Roy and B. Pathak, J. Phys. Chem. Lett., 2022, 13, 11818–11830 CrossRef CAS.
- M. K. Jena and B. Pathak, Nano Lett., 2023, 23, 2511–2521 CrossRef CAS.
- M. K. Jena, S. Mittal, S. S. Manna and B. Pathak, Nanoscale, 2023, 15, 18080–18092 RSC.
- S. K. Tabatabaei, B. Pham, C. Pan, J. Liu, S. Chandak, S. A. Shorkey, A. G. Hernandez, A. Aksimentiev, M. Chen, C. M. Schroeder and O. Milenkovic, Nano Lett., 2022, 22, 1905–1914 CrossRef CAS PubMed.
- S. Mittal, S. Manna, M. K. Jena and B. Pathak, ACS Mater. Lett., 2023, 1570–1580 CrossRef CAS.
- S. Mittal, S. Manna, M. K. Jena and B. Pathak, Digital Discovery, 2023, 2, 1589–1600 RSC.
- A. B. Farimani, K. Min and N. R. Aluru, ACS Nano, 2014, 8, 7914–7922 CrossRef CAS PubMed.
- D. Deamer, M. Akeson and D. Branton, Nat. Biotechnol., 2016, 34, 518–524 CrossRef CAS PubMed.
- H. Gamaarachchi, H. Samarakoon, S. P. Jenner, J. M. Ferguson, T. G. Amos, J. M. Hammond, H. Saadat, M. A. Smith, S. Parameswaran and I. W. Deveson, Nat. Biotechnol., 2022, 40, 1026–1029 CrossRef CAS PubMed.
- K. Shafin, T. Pesout, R. Lorig-Roach, M. Haukness, H. E. Olsen, C. Bosworth, J. Armstrong, K. Tigyi, N. Maurer, S. Koren, F. J. Sedlazeck, T. Marschall, S. Mayes, V. Costa, J. M. Zook, K. J. Liu, D. Kilburn, M. Sorensen, K. M. Munson, M. R. Vollger, J. Monlong, E. Garrison, E. E. Eichler, S. Salama, D. Haussler, R. E. Green, M. Akeson, A. Phillippy, K. H. Miga, P. Carnevali, M. Jain and B. Paten, Nat. Biotechnol., 2020, 38, 1044–1053 CrossRef CAS.
- C. Wen, D. Dematties and S.-L. Zhang, ACS Sens., 2021, 6, 3536–3555 CrossRef CAS.
- M. U. Ahsan, A. Gouru, J. Chan, W. Zhou and K. Wang, Nat. Commun., 2024, 15, 1448 CrossRef CAS PubMed.
- K. Misiunas, N. Ermann and U. F. Keyser, Nano Lett., 2018, 18, 4040–4045 CrossRef CAS.
- J. Schreiber and K. Karplus, Bioinformatics, 2015, 31, 1897–1903 CrossRef CAS PubMed.
- M. Landry and S. Winters-Hilt, BMC Bioinf., 2007, 8, S12 CrossRef PubMed.
- X.-J. Sui, M.-Y. Li, Y.-L. Ying, B.-Y. Yan, H.-F. Wang, J.-L. Zhou, Z. Gu and Y.-T. Long, J. Anal. Test., 2019, 3, 134–139 CrossRef.
- A. Churbanov, C. Baribault and S. Winters-Hilt, BMC Bioinf., 2007, 8, S14 CrossRef.
- D. Dematties, C. Wen, M. D. Pérez, D. Zhou and S.-L. Zhang, ACS Nano, 2021, 15, 14419–14429 CrossRef CAS.
- Z.-X. Wei, Y.-L. Ying, M.-Y. Li, J. Yang, J.-L. Zhou, H.-F. Wang, B.-Y. Yan and Y.-T. Long, Anal. Chem., 2019, 91, 10033–10039 CrossRef CAS PubMed.
- N. Meyer, J.-M. Janot, M. Lepoitevin, M. Smietana, J.-J. Vasseur, J. Torrent and S. Balme, Biosensors, 2020, 10, 140 CrossRef CAS.
- J. Zhang, X. Liu, Y.-L. Ying, Z. Gu, F.-N. Meng and Y.-T. Long, Nanoscale, 2017, 9, 3458–3465 RSC.
- J.-H. Zhang, X.-L. Liu, Z.-L. Hu, Y.-L. Ying and Y.-T. Long, Chem. Commun., 2017, 53, 10176–10179 RSC.
- M. Kolmogorov, E. Kennedy, Z. Dong, G. Timp and P. A. Pevzner, PLoS Comput. Biol., 2017, 13, e1005356 CrossRef.
- N. Cardozo, K. Zhang, K. Doroschak, A. Nguyen, Z. Siddiqui, N. Bogard, K. Strauss, L. Ceze and J. Nivala, Nat. Biotechnol., 2022, 40, 42–46 CrossRef CAS PubMed.
- R. L. Kumawat, M. K. Jena, S. Mittal and B. Pathak, Small, 2024, 2401112 CrossRef PubMed.
- S. Mittal, S. Manna and B. Pathak, ACS Appl. Mater. Interfaces, 2022, 14, 51645–51655 CrossRef CAS.
- S. Mittal, M. K. Jena and B. Pathak, Chem.–A Euro. J., 2023, 29, e202301667 CrossRef CAS.
- D. Pedone, M. Firnkes and U. Rant, Anal. Chem., 2009, 81, 9689–9694 CrossRef CAS.
- C. R. O'Donnell, D. M. Wiberg and W. B. Dunbar, in 2012 IEEE 51st IEEE Conference on Decision and Control (CDC), 2012, pp. 2304–2309 Search PubMed.
- S. Shekar, C.-C. Chien, A. Hartel, P. Ong, O. B. Clarke, A. Marks, M. Drndic and K. L. Shepard, Nano Lett., 2019, 19, 1090–1097 CrossRef CAS PubMed.
- A. V. Jagtiani, R. Sawant, J. Carletta and J. Zhe, Meas. Sci. Technol., 2008, 19, 065102 CrossRef.
- B. Yan, H. Cui, J. Zhou and H. Wang, Quim. Nova, 2020, 43, 837–843 CAS.
- S. Min, B. Lee and S. Yoon, Briefings Bioinf., 2017, 18, 851–869 Search PubMed.
- R. Poplin, P.-C. Chang, D. Alexander, S. Schwartz, T. Colthurst, A. Ku, D. Newburger, J. Dijamco, N. Nguyen, P. T. Afshar, S. S. Gross, L. Dorfman, C. Y. McLean and M. A. DePristo, Nat. Biotechnol., 2018, 36, 983–987 CrossRef CAS PubMed.
- R. Luo, F. J. Sedlazeck, T.-W. Lam and M. C. Schatz, Nat. Commun., 2019, 10, 998 CrossRef PubMed.
- B. Yu, C. Chen, R. Qi, R. Zheng, P. J. Skillman-Lawrence, X. Wang, A. Ma and H. Gu, Briefings Bioinf., 2021, 22, bbaa316 CrossRef.
- T. Albrecht, G. Slabaugh, E. Alonso and S. M. R. Al-Arif, Nanotechnology, 2017, 28, 423001 CrossRef PubMed.
- I. T. Jolliffe and J. Cadima, Philos. Trans. R. Soc., A, 2016, 374, 20150202 CrossRef.
- R. W. Epps, M. S. Bowen, A. A. Volk, K. Abdel-Latif, S. Han, K. G. Reyes, A. Amassian and M. Abolhasani, Adv. Mater., 2020, 32, 2001626 CrossRef CAS.
- E. Becht, L. McInnes, J. Healy, C.-A. Dutertre, I. W. H. Kwok, L. G. Ng, F. Ginhoux and E. W. Newell, Nat. Biotechnol., 2019, 37, 38–44 CrossRef CAS.
- N. Wu, X.-Y. Zhang, J. Xia, X. Li, T. Yang and J.-H. Wang, ACS Nano, 2021, 15, 19522–19534 CrossRef CAS PubMed.
- A. K. Chew, J. A. Pedersen and R. C. Van Lehn, ACS Nano, 2022, 16, 6282–6292 CrossRef CAS PubMed.
- G. Pilania, Comput. Mater. Sci., 2021, 193, 110360 CrossRef CAS.
- S. Cuomo, V. S. Di Cola, F. Giampaolo, G. Rozza, M. Raissi and F. Piccialli, J. Sci. Comput., 2022, 92, 88 CrossRef.
- A. Greenfield, C. Hafemeister and R. Bonneau, Bioinformatics, 2013, 29, 1060–1067 CrossRef CAS PubMed.
- X. Huang, J. Xu, M. Sun and Y. Liu, in Proceedings of the 59th Annual Meeting of the Association for Computational Linguistics and the 11th International Joint Conference on Natural Language Processing (Volume 1: Long Papers), ed. C. Zong, F. Xia, W. Li and R. Navigli, Association for Computational Linguistics, 2021, pp. 5738–5750 Search PubMed.
- R. G. Amorim, A. R. Rocha and R. H. Scheicher, J. Phys. Chem. C, 2016, 120, 19384–19388 CrossRef CAS.
- J. Prasongkit, A. Grigoriev, B. Pathak, R. Ahuja and R. H. Scheicher, J. Phys. Chem. C, 2013, 117, 15421–15428 CrossRef CAS.
- S. Mittal, M. K. Jena and B. Pathak, ACS Appl. Nano Mater., 2022, 5, 9356–9366 CrossRef CAS.
- S. Mittal and B. Pathak, ACS Appl. Bio Mater., 2023, 6, 218–227 CrossRef CAS.
- A. Choudhary, H. Joshi, H.-Y. Chou, K. Sarthak, J. Wilson, C. Maffeo and A. Aksimentiev, ACS Nano, 2020, 14, 15566–15576 CrossRef CAS.
- S. Mittal and B. Pathak, Nanoscale, 2023, 15, 757–767 RSC.
- T. Furuhata, T. Ohshiro, G. Akimoto, R. Ueki, M. Taniguchi and S. Sando, ACS Nano, 2019, 13, 5028–5035 CrossRef CAS.
- R. Balasubramanian, S. Pal, A. Rao, A. Naik, B. Chakraborty, P. K. Maiti and M. M. Varma, ACS Appl. Bio Mater., 2021, 4, 451–461 CrossRef CAS PubMed.
- B. Luan and M. A. Kuroda, ACS Nano, 2020, 14, 13137–13145 CrossRef CAS.
- R. Balasubramanian, S. Pal, H. Joshi, A. Rao, A. Naik, M. Varma, B. Chakraborty and P. K. Maiti, J. Phys. Chem. C, 2019, 123, 11908–11916 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2024 |