
Understanding structural isomerism in organoiridium picolinamidate complexes and its consequences on reactivity and biological properties†
Hieu D.
Nguyen
 ,
Croix J.
Laconsay
,
Rahul D.
Jana
,
Tuhin
Ganguly
,
Sally T.
Hoang
,
Kanika
Kaushal
,
Judy I.
Wu
and
Loi H.
Do
*
,
Croix J.
Laconsay
,
Rahul D.
Jana
,
Tuhin
Ganguly
,
Sally T.
Hoang
,
Kanika
Kaushal
,
Judy I.
Wu
and
Loi H.
Do
*
Department of Chemistry, University of Houston, 4800 Calhoun Rd., Houston, Texas 77204, USA. E-mail: loido@uh.edu
First published on 13th September 2024
Abstract
Organoiridium picolinamidate complexes are promising for intracellular applications because of their biocompatibility, activity in living systems, and ease of derivatization. To shield their metal centers from inhibition by biological nucleophiles (e.g., glutathione), attempts were made to increase the steric bulk of the supporting N-(2,6-R2-phenyl)picolinamidate ligand. It was found that when R = H (Ir1) or methyl (Ir2), the ligand adopts N,N′-coordination to iridium, whereas when R = isopropyl (Ir3) or phenyl (Ir4), N,O-coordination was observed. Based on experimental measurements and density functional theory calculations, it was revealed that the carbon chemical shift of the C(O)NR group can be used as a diagnostic handle to distinguish between the N,N′- and N,O-isomers in solution. Computational studies indicate that the former is favored thermodynamically but the latter is preferred when the R group is overly bulky. Complexes Ir1–Ir4 exhibit differences in lipophilicity, cellular uptake, cytotoxicity, and the propensity to generate reactive oxygen species in living cells. Reaction studies showed that Ir1/Ir2 are more efficient than Ir3/Ir4 in promoting the reduction of aldehydes to alcohols via transfer hydrogenation but both isomer types were susceptible to catalyst poisoning by glutathione. This work has led to new insights into structural isomerism in organoiridium picolinamidate complexes and suggests that steric tuning alone is insufficient to protect the Ir center from poisoning by biological nucleophiles.
Introduction
Half-sandwich metal complexes have been used in numerous biological applications, including as therapeutic agents,1–3 intracellular catalysts,4–9 and cofactors for artificial metalloenzymes.10 They typically comprise metal arenes (where M = Ru, Rh, Os, or Ir) coordinated by a multidentate ligand and at least one labile X donor (e.g., chloride). In biological media, the M–X complex can be aquated to yield M–OH2 species or attacked by glutathione (GSH) to generate M–SG species (Chart 1A).11,12 Because the metal–sulfur interaction is strong, binding of GSH to M can interfere with its catalytic abilities by occupying a coordination site.13,14 Given that GSH and other endogenous nucleophiles (e.g., nitrogenous bases) are prevalent in living systems, strategies to prevent their coordination to small-molecule metal catalysts would significantly enhance their intracellular activity.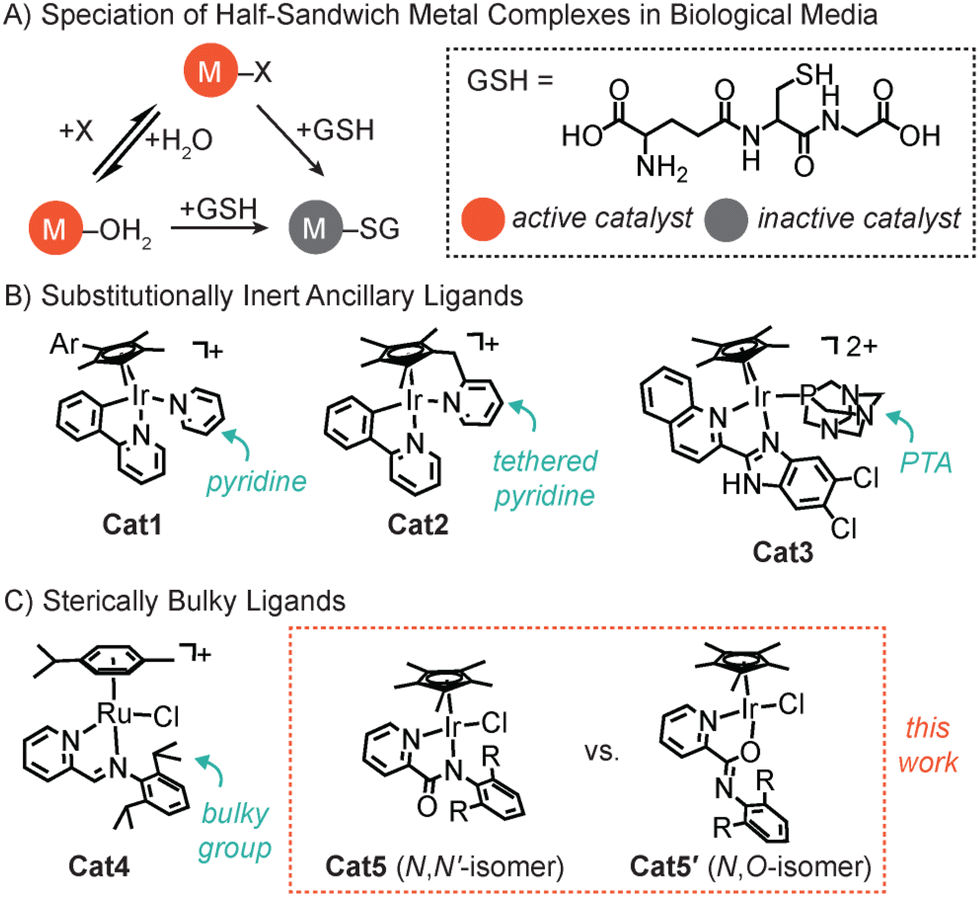 | ||
| Chart 1 Scheme showing the possible speciation of half-sandwich metal complexes in biological media (A) and different strategies used to increase its lifetime/selectivity, such as by using substitutionally inert ancillary ligands (B) or bulky ligands (C). | ||
To prevent the inhibition of half-sandwich organometallic complexes, a variety of methods have been explored. It was reported that the use of substitutionally inert ancillary ligands can protect metal catalysts against GSH attack. For example, Sadler and coworkers demonstrated that replacing chloride with pyridine in a Cp*Ir complex (Cp* = pentamethylcyclopentadienyl anion, Cat1) led to more productive reactive oxygen species (ROS) generation and cancer cell death compared to that of the parent catalyst (Chart 1B).11,15 It was proposed that because Cat1 is slow to react with external donors (e.g., H2O or GSH), it was able to reach its biological targets more effectively. Expanding on this concept, Pizarro and coworkers designed a series of organoiridium catalysts bearing a pyridine ring tethered to Cp* (Cat2, Chart 1B).16–18 The investigators showed that the pyridine-tethered catalysts were substantially more potent against cancer cells than their non-tethered variants. The enhanced effects were attributed to the tethered pyridine being able to shield the Ir center from external nucleophiles and minimize premature catalyst activation. Finally, Das and Paira reported that a Cp*Ir complex (Cat3) with a coordinated 1,3,5-triaza-7-phosphaadamantane (PTA) was capable of achieving high phototoxicity against triple-negative breast cancer cells (Chart 1B).19 Once again, the slow dissociation of the ancillary ligand from Ir was invoked as the reason for its resistance to GSH inhibition.
Although metalloenzymes can restrict access to their active sites using protein channels,20–22 small-molecule metal catalysts lack this ability. However, there has been some success in using bulky ligands to protect the metal centers from reaction with undesired species. For example, Mukherjee and coworkers found that having sterically encumbering 2,6-diisopropyl groups in the ligand framework of the Ru complex Cat4 was key to preventing its deactivation by GSH (Chart 1C).23 Inspired by these studies, we proceeded to explore the application of steric tuning to enhance the GSH tolerance of Cp*Ir picolinamidate complexes (Cat5).24–26 There is growing interest in these catalysts because they are active inside living cells and are amenable to further functionalization for specialized applications.27–29 Attempts to use 2,2′-bipyridine, which is electronically similar to the iminopyridine ligand in Cat4, with Cp*Ir did not afford active transfer hydrogenation catalysts under mild conditions.30,31 In the present work, we have systematically varied the 2,6-substituents on the N-phenyl ring in Cat5. Our results showed that increasing the steric bulk in the picolinamidate ligand led to a preference for N,O- rather than N,N′-coordination to iridium.32–34 The two isomers were distinguishable by NMR spectroscopy and the chemical basis for their existence was studied by density functional theory (DFT) calculations. Although GSH tolerance was not achieved, these investigations offered new insights into the design criteria for constructing Cp*Ir picolinamidate-based systems with high activity.
Results and discussion
Synthesis and characterization of the Ir complexes
To study the effects of steric hindrance on the reactivity of Cp*Ir picolinamidate complexes, we prepared a series of N-phenylpicolinamides (4a–4f) with different R substituents at the 2,6-positions of the phenyl ring (Fig. 1A). The ligands were obtained by combining the desired aniline with 2-picolinic acid using either 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide hydrochloride and hydroxybenzotriazole (for R = H (4a), Me (4b), and iPr (4c)) or propylphosphonic anhydride (for R = Ph (4e) or 3,5-Me2Ph (4f)) as amide coupling reagents (Scheme S1†). Attempts to synthesize 4d with R = t-Bu were unsuccessful using either amide coupling reagents. We found that metalation of the picolinamides required different methods depending on the steric bulk of the R group. For example, 4a and 4b were treated with [Cp*IrCl2]2 and NH4PF6 in EtOH to furnish Ir1 and Ir2 with ≥70% yield, respectively (Method A). The preparation of Ir1 was reported previously.24 In contrast, the bulkier ligands 4c and 4e were metalated by reaction with [Cp*IrCl2]2 and NEt3 in CH2Cl2 to provide Ir3 and Ir4, respectively (Method B). The yields were modest (≤41%), potentially due to the slower rates of reactions as a result of increased steric hindrance. The bulkiest ligand, 4f, could not be metalated using either Methods A or B (Scheme S1†). | ||
| Fig. 1 Synthesis of Ir1–Ir4 (part A) and their UV-vis absorption (CH2Cl2; part B), infrared (ATR-IR; part B), and 1H NMR (CDCl3; part C) spectra. | ||
Next, the iridium complexes were characterized using a variety of spectroscopic methods. We observed that Ir1–Ir4 displayed strong absorbances below 450 nm, similar to other organoiridium picolinamidate complexes reported in the literature (Fig. 1B, left).31 They also exhibit multiple vibrational features around 1560–1630 cm−1, most likely due to the presence of the C(O)NR group (Fig. 1B, right).35 These spectroscopic data, however, do not provide information about the structure or atom connectivity of the Ir species.
To obtain further insights into the nature of the Ir complexes in solution, we used NMR spectroscopy. The 1H NMR spectra of Ir1 (Fig. S33†) and Ir4 (Fig. S39†) showed sharp resonances in the aliphatic and aromatic regions, indicating that the Ir center is coordinated to Cp* and the picolinamidate ligand, respectively. We observed that in the 1H NMR spectrum of Ir2, there are two signals centered at 2.1 and 2.3 ppm (Fig. 1C, top), which were assigned to the methyl substituents of the picolinamidate ligand. Because these peaks are magnetically inequivalent, it suggests that the 2,6-dimethylphenyl group has restricted rotational freedom. In contrast, the 1H NMR spectrum of Ir3 exhibits two sharp signals (∼1.1 and 1.2 ppm) corresponding to the –CH3 of the isopropyl groups (Fig. 1C, bottom). If the 2,6-diisopropylphenyl ring was conformationally rigid, four signals originating from the –CH3 units would be expected. Thus, these results suggest that the 2,6-diisopropylphenyl moiety in Ir3 has greater rotational freedom than the 2,6-dimethylphenyl moiety in Ir2, which is counterintuitive based on their relative steric volumes.
The surprising observation above led us to compare the 13C NMR spectra of the iridium complexes more closely. We found that the carbon chemical shifts of the C(O)NR group appeared at 168.4, 169.9, 163.0, and 163.8 ppm in Ir1, Ir2, Ir3, and Ir4, respectively (Fig. 2). A survey of the literature revealed that all reported Cp*Ir complexes with confirmed or presumed N,N′-coordination of the picolinamidate ligand to Ir exhibit C(O)NR peaks at ≥167 ppm, regardless of electronic differences in their ligands (e.g., complexes I–VI in Fig. 2A; additional examples in Fig. S55†).24,25,28,31,36–42 Given that the C(O)NR resonances in Ir3 and Ir4 clearly fall outside of the expected range, we suspected that they are structurally distinct from the N,N′-coordinated Cp*Ir picolinamidate species.
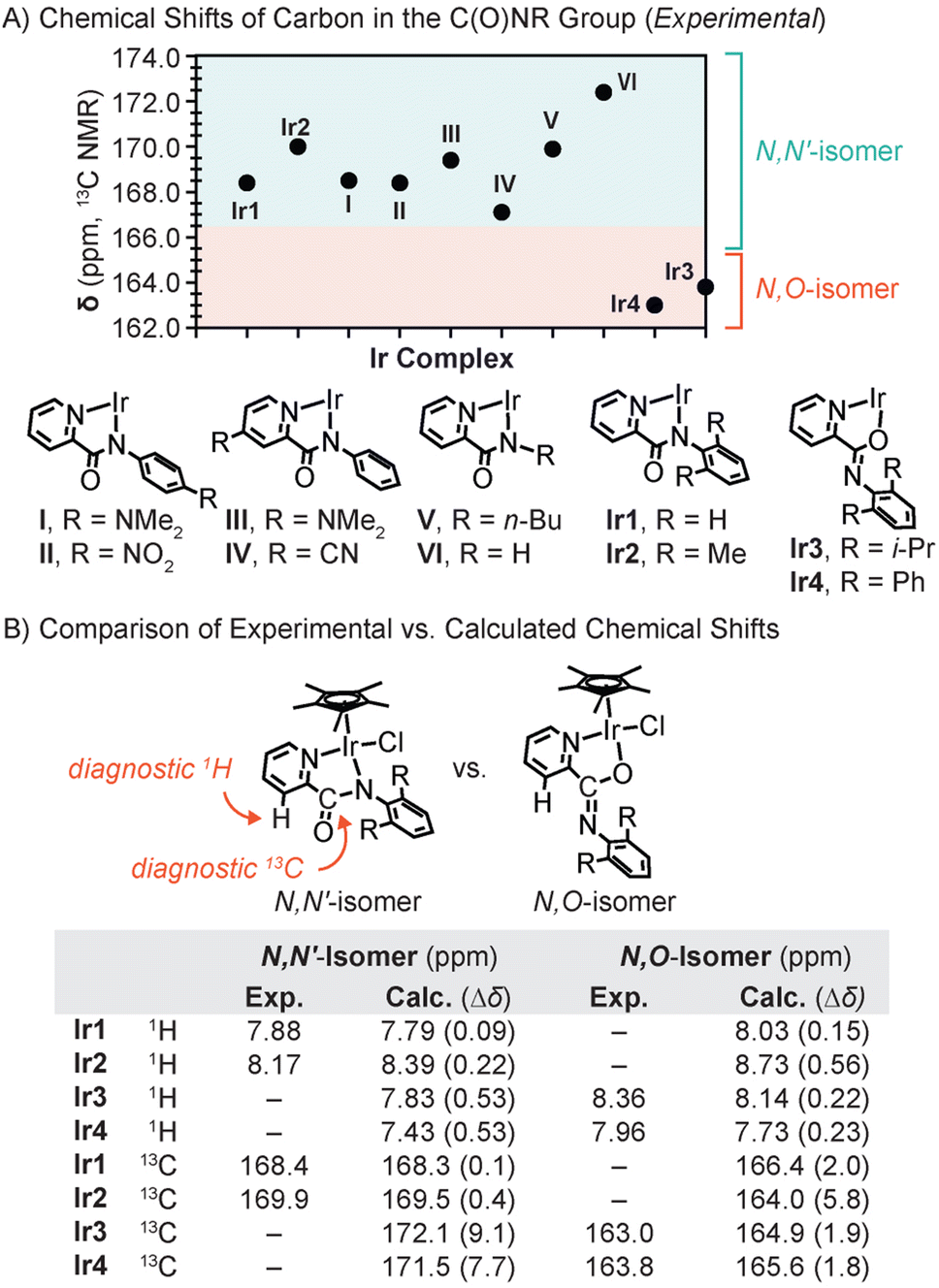 | ||
| Fig. 2 (A) The 13C NMR chemical shifts of the C(O)NR group in various Cp*Ir picolinamidate complexes. The Cp* and Cl− were omitted from the Ir complexes for clarity. (B) Comparison of the experimental vs. calculated chemical shifts of the diagnostic proton and carbon atoms in the N,N′- vs. N,O-isomers of Ir1–Ir4. | ||
To elucidate the structures of the iridium complexes, we grew single crystals of Ir1–Ir4 and analyzed them by X-ray crystallography (Table S9†). We found that all four complexes have the formula [Cp*Ir(picolinamidate)Cl] but they differ in the binding mode of their supporting ligands. Complexes Ir1 (Fig. S47†) and Ir2 (Fig. 3A) feature N,N′-coordination whereas Ir3 (Fig. 3A) and Ir4 (Fig. S50†) feature N,O-coordination to the metal. Based on the bond distances (Fig. 3B), the C(O)NR group in Ir1 and Ir2 is best described as an amidate with C–O and C–N bond distances of ∼1.24 and ∼1.33 Å, respectively.43 In contrast, the C(O)NR group in Ir3 and Ir4 has bond metrics that are consistent with an iminolate group (C–O = 1.30 Å and C–N = 1.29 Å).43
 | ||
| Fig. 3 (A) Molecular structures of Ir2 (left) and Ir3 (right) determined by X-ray crystallography. The displacement ellipsoids are drawn at 50% probability level and the hydrogen atoms have been omitted for clarity. Color code: orange = iridium, blue = nitrogen, red = oxygen, black = carbon, green = chlorine. (B) Comparison of the bond distances (Å) in Ir1–Ir4 focusing on the C(O)NR unit to differentiate between the N,N′- vs. N,O-isomers. | ||
In light of these results, our NMR data above can be further clarified. Because Ir2 exists as the N,N′-isomer, its 2,6-dimethylphenyl group has limited rotational freedom due to possible steric clash with the Cp* ring. However, the 2,6-diisopropylphenyl group in Ir3 has free rotation because it is positioned further away from Cp* in the N,O-isomer structure. The percent buried volumes (%Vbur) of the Ir complexes,44 which measure the steric bulk of the picolinamidate ligands, corroborate the finding that N,O-binding results in reduced steric encumbrance relative to N,N′-binding (Fig. S46 and Table S8†).
Density functional theory calculations
To verify the binding mode of the structural isomers, we turned to density functional theory (DFT) to compute the NMR chemical shifts (δ) for the N,N′- and N,O-forms of the iridium complexes. The chemical shifts were computed at the SMD-(CH2Cl2)-B97-2//M11/def2-TZVPP level,45 and the results for select diagnostic peaks (proton at the 3-position of pyridine and carbon in the C(O)NR unit) are provided in Fig. 2B. Our studies revealed that the computed 1H chemical shifts matched the experimental chemical shifts better for the N,N′-isomer of Ir1/Ir2 and the N,O-isomer of Ir3/Ir4. For example, the absolute deviation (Δδ) was 0.09 and 0.22 ppm for the N,N′-isomers of Ir1 and Ir2, respectively, which is smaller than that for their corresponding N,O-isomers. In contrast, the Δδ for Ir3 and Ir4 was 0.53 ppm for the N,N′-isomer and ≤0.23 ppm for the N,O-isomer. Given that the tolerable error for the computed δ is <0.3 ppm, the N,O-isomer was clearly a closer match than the N,N′-isomer for Ir3 and Ir4.Comparison of the computed vs. experimentally measured carbon chemical shifts showed similar trends (Fig. 2B). We found that the Δδ for the diagnostic carbon in the N,N′-isomer of Ir1 and Ir2 was ≤0.4 ppm whereas that in the corresponding N,O-isomer was ≥2.0 ppm. For Ir3 and Ir4, the opposite was observed, in which the Δδ for the N,N′-isomer was larger (≥7.7 ppm) than that for the N,O-isomer (≤1.9 ppm). These results suggest that Ir1 and Ir2 exists exclusively in the N,N′-form whereas Ir3 and Ir4 exist exclusively in the N,O-form, which is consistent with our crystallographic data. Studies of Ir3 using variable temperature NMR spectroscopy in CD3OD did not show any changes from −35 to 25 °C, indicating that it cannot interconvert between different structural isomers in solution under the conditions tested.
It should also be noted that the upfield shifted carbon peaks assigned to the C(O)NR group in Ir3 and Ir4, relative to that in Ir1 and Ir2, were reproduced computationally at 164.9 (Δδ = 1.9) and 165.6 ppm (Δδ = 1.8), respectively. Thus, it appears that the carbon chemical shift of the C(O)NR unit can be used as a diagnostic handle to differentiate between N,N′- and N,O-coordination in these systems (Fig. 2A).
To understand the chemical basis for the N,N′- vs. N,O-binding preference, the relative Gibbs free energies of the iridium complexes were determined by DFT. In all cases, the N,N′-isomers were energetically more favorable than the N,O-isomers by 8.9, 6.7, 5.4, and 1.7 kcal mol−1 for Ir1, Ir2, Ir3, and Ir4, respectively (Fig. 4A). Although these results would suggest that only the N,N′-isomer should exist, further DFT studies revealed that steric hindrance may be responsible for the isomer preference. Our results showed that the N,N′-isomer of Ir1 has minimal repulsive noncovalent interactions (NCIs) whereas the N,N′-isomers of Ir3 and Ir4 have several regions of significant steric clash (Fig. 4B and S51–S54†).46 Specifically, the R groups (i.e., iPr in Ir3 and Ph in Ir4) have unfavorable NCIs with the Cp* ring and the amidate moiety in the case of Ir4. In contrast, the N,O-isomers of Ir3 and Ir4 exhibit mostly attractive van der Waals forces (Fig. S53 and S54†). Thus, our results revealed that although the N,N′-isomer is more thermodynamically stable than the N,O-isomer, the presence of bulky R groups leads to a preference for the latter to minimize steric repulsion.
 | ||
| Fig. 4 The relative Gibbs free energy calculated for the N,N′- and N,O-isomers using DFT (SMD-(CH2Cl2)-B97-2//M11/def2-TZVPP) (A) and their key non-covalent interactions (B). Color code: orange = iridium, blue = nitrogen, red = oxygen, black = carbon, green = chlorine, light gray = hydrogen. | ||
Transfer hydrogenation studies
Our new iridium complexes offered an opportunity to compare the reactivity of the N,N′- vs. N,O-isomers. Using our standard conditions for transfer hydrogenation reported in previous work,30,31 we evaluated the ability of Ir1–Ir4 to catalyze the conversion of aldehydes to alcohols using HCOONa in DMSO/H2O (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :9) under air at 37 °C (Table 1). After 24 h, Ir1 and Ir2 gave quantitative yields of benzyl alcohol starting from benzaldehyde but Ir3 produced only 32% yield and Ir4 was inactive (entry 1). A time-dependence study using Ir1 and Ir2 showed that the former is more active than the latter (Table S2†), suggesting that when R = Me, the increased steric bulk may lead to slower catalysis. The lower activity of the N,O-isomers (Ir3/Ir4) compared to the N,N′-isomers (Ir1/Ir2) is likely due to their more electron poor nature, which has been shown to reduce the efficiency of hydride transfer from the Ir–H intermediate to aldehyde acceptors.31 In the case of Ir4, we found that it does not react readily with HCOONa, which is the first step in the transfer hydrogenation process. We hypothesize that the excessive steric bulk in Ir4 due to the presence of the phenyl substituents is most likely responsible for its inefficient reactions with both formate and substrates.
:9) under air at 37 °C (Table 1). After 24 h, Ir1 and Ir2 gave quantitative yields of benzyl alcohol starting from benzaldehyde but Ir3 produced only 32% yield and Ir4 was inactive (entry 1). A time-dependence study using Ir1 and Ir2 showed that the former is more active than the latter (Table S2†), suggesting that when R = Me, the increased steric bulk may lead to slower catalysis. The lower activity of the N,O-isomers (Ir3/Ir4) compared to the N,N′-isomers (Ir1/Ir2) is likely due to their more electron poor nature, which has been shown to reduce the efficiency of hydride transfer from the Ir–H intermediate to aldehyde acceptors.31 In the case of Ir4, we found that it does not react readily with HCOONa, which is the first step in the transfer hydrogenation process. We hypothesize that the excessive steric bulk in Ir4 due to the presence of the phenyl substituents is most likely responsible for its inefficient reactions with both formate and substrates.
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Aldehyde | GSH (mM) | Alcohol yieldb (%) | |||
| Ir1 | Ir2 | Ir3 | Ir4 | |||
|
a Reaction conditions: aldehyde (15 μmol), HCOONa (135 μmol), Ir complex (0.15 μmol), DMSO:H2O or DMSO-d6:D2O (1:9), 37 °C, 24 h.
b Yields of benzyl alcohol were determined by GC-MS whereas yields of butanol and hexanol were determined by 1H NMR spectroscopy. The reported yields are the average values from duplicate runs. See ESI† for details.
c The partially reduced products butanal and but-2-en-1-ol were obtained; no butanol was observed.
|
||||||
| 1 | Benzaldehyde | 0 | 99 | 99 | 32 | 0 |
| 2 | Benzaldehyde | 1.0 | 18 | 27 | 0 | 0 |
| 3 | Crotonaldehyde | 0 | 99 | 99 | 95 | 0c |
| 4 | Hexanal | 0 | 99 | 99 | 99 | 9 |

We observed that the efficiency of benzaldehyde reduction using the Ir catalysts and HCOONa is dependent on the reaction medium. For example, reactions in DMSO/Dulbecco's Modified Eagle Medium (DMEM) (1:9) gave similar results as those in DMSO/H2O (1:9) (Table S1†). However, the addition of 10% fetal bovine serum (FBS) to the DMSO/DMEM mixture afforded lower yields of benzyl alcohol (90% for Ir1, 95% for Ir2, 22% for Ir3, and 0% for Ir4; Table S1†). The use of DMSO/Roswell Park Memorial Institute-1640 (RPMI-1640) also lowered the transfer hydrogenation efficiency. Consistent with our previous findings,47 the presence of biological components and nucleophiles can coordinatively inhibit the iridium catalysts.
When the reactions were performed in the presence of 1.0 mM GSH (Table 1, entry 2), the yields of benzyl alcohol for Ir1 and Ir2 dropped substantially compared to that in the absence of GSH and Ir3 was completely inhibited. Although Ir2 was slightly more tolerant of GSH than Ir1 (27 vs. 18% yield, respectively), these results suggest that using bulky ligands to protect the Ir centers in Cp*Ir picolinamidate catalysts from coordination inhibition is challenging because the less active N,O-isomer is generated if the R group is too bulky.
The iridium complexes were next tested for their transfer hydrogenation activity toward biologically relevant aldehydes.48 Under our standard conditions using HCOONa as the hydride source, Ir1, Ir2, and Ir3 fully reduced crotonaldehyde to 1-butanol with ≥95% yield (Table 1, entry 3). In contrast, Ir4 afforded the partially reduced species butanal and but-2-en-1-ol in 9% and 7% yield, respectively (Table S3†). When hexanal was employed as the substrate, Ir1, Ir2, and Ir3 gave quantitative amounts of hexane-1-ol whereas Ir4 gave only a 9% yield (Table 1, entry 4). In general, these studies demonstrated that the N,N′-isomers are more active than the N,O-isomers for transfer hydrogenation and that having sterically-encumbering R groups in the picolinamidate ligand decrease the catalytic efficiency.
We noticed that in the reactions above, the amount of HCOONa consumed was consistently greater than the amount of substrate reduced. Because it has been shown in previous studies that Cp*Ir catalysts are capable of converting O2 to H2O2 in the presence of HCOONa,11,28 we measured the hydrogen peroxide levels under various reaction conditions using semi-quantitative color test strips. When the Ir complexes and excess HCOONa were stirred in DMSO/H2O (1:9) under air at 37 °C, formation of H2O2 was detected in all reactions (Fig. 5, left). Complex Ir2 generated significantly more H2O2 than the other Ir catalysts (up to ∼300 μM) and the H2O2 concentration fluctuated over the course of 72 h. The changing amounts of H2O2 over time may be due to its disproportionation.49,50 These results indicate that formate dehydrogenation to generate Ir–H species can occur in all four Ir catalysts, albeit with different rates, but the subsequent hydride transfer from Ir–H to O2 is more facile than to aldehyde substrates. When the reactions were repeated in the presence of benzaldehyde, similar results were obtained (Fig. 5, right). The most noticeable difference was that the maximum concentration of H2O2 generated by Ir2 was lowered to ∼200 μM, which is likely due to competitive reaction of the Ir–H intermediate with both O2 and the organic electrophile.28,51,52 The higher activity of Ir2 relative to that of Ir1 for H2O2 production is the opposite of what was observed for the reduction of benzaldehyde (Table S2†). These results can be rationalized by the fact that Ir1 is a less bulky catalyst than Ir2 so it can react more readily with larger substrates, leading to greater reaction efficiency with small-molecule aldehydes (and vice versa). These studies suggest that the iridium catalysts may be capable of producing ROS inside living cells since endogenous reduced nicotinamide adenine dinucleotide (NADH) can serve as a hydride source.11,53
 | ||
| Fig. 5 Determination of hydrogen peroxide concentration under transfer hydrogenation conditions without (left) and with (right) benzaldehyde substrate. Measurements using Quantofix Peroxide 25 test strips are semi-quantitative. | ||
Biological properties
Because we are interested in using the iridium complexes for applications in living systems,4,5,7,54 we proceeded to evaluate their reactions with biologically-relevant species. To probe their tendency to be aquated, the Ir complexes were dissolved in DMSO-d6 and then their 1H NMR spectra were recorded before and after the addition of water. We observed that in the presence of up to 0.5 equiv. of H2O, relative to Ir, no significant changes in the chemical shifts of Ir2 had occurred (Fig. S41†), indicating that it remained in the Ir–Cl form. In contrast, when Ir4 was combined with 0.5 equiv. of H2O, two distinct sets of peaks appeared in the NMR spectra (Fig. S42 and S43†). The Cp* peak at 1.4 ppm was assigned to the starting Ir–Cl species whereas the Cp* peak at 1.2 ppm was assigned to the newly formed Ir–OH2 species. These results indicate that N,N′- and N,O-isomers have different propensities toward aquation.Next, the interactions of Ir1, Ir2, and Ir3 with endogenous nucleophiles in DMSO/H2O (1:9) were investigated by UV-vis absorption spectroscopy. We found that the addition of up to 10 equiv. of 2-acetamido-6-hydroxypurine (Fig. S4–S6†), cysteine (Fig. S7–S9†), or glutathione (Fig. S10–S12†), relative to Ir, led to distinct spectral changes. Although the nature of these interactions was not investigated further, coordination of biological nucleophiles to Cp*Ir complexes is well documented.13,14
To estimate the lipophilicity of the iridium complexes, their octanol/water partition coefficients (logP) were measured.55 Sodium chloride (0.25 M) was added to the aqueous solutions to mimic the high salt concentrations of biological media. Our data revealed that the Ir complexes had logP values in the order Ir4 (1.32) > Ir3 (0.88) > Ir2 (0.78) > Ir1 (0.48) (Table 2, column 2). As expected, when the R group of the picolinamidate ligand is alkyl or Ph, the Ir complexes’ overall hydrophobicity increases relative to that of the parent complex Ir1.
| Complex | LogP |
[Ir]cellb (ng per 106 cells) | IC50c (μM) | ROS (RFU) |
|---|---|---|---|---|
| a Biological properties (cell uptake, cytotoxicity, and ROS generation) were measured in NIH-3T3 mouse fibroblast cells. Reported values are the average of experiments performed in at least triplicate. See ESI for details. b Cellular uptake was measured using ICP-MS. c Cell viability was measured using colorimetric SRB assays. | ||||
| Ir1 | 0.48 | 46 | 71 | 4.5 |
| Ir2 | 0.78 | 105 | 45 | 11.2 |
| Ir3 | 0.88 | 45 | >200 | 3.7 |
| Ir4 | 1.32 | 163 | 60 | 1.3 |
Next, the cellular uptake of the iridium complexes in NIH-3T3 mouse fibroblast cells was determined. This cell line was chosen due to its robustness and common use in mammalian cell studies.56–58 The cells were incubated with 10 μM of Ir1, Ir2, Ir3, or Ir4 for 24 h, washed with fresh media, and then lysed so that the iridium content of the cells could be analyzed by inductively coupled plasma mass spectrometry (ICP-MS). According to our data, the following cell uptake efficiency was observed: Ir4 > Ir2 > Ir1 ∼ Ir3, with intracellular Ir concentrations ranging from 45–163 ng per 106 cells (Table 2, column 3). Although the cell uptake trend does not correlate with the logP values, differences in the speciation or cell uptake mechanism of the iridium complexes may account for this discrepancy.
Given that the iridium complexes are readily internalized in the NIH-3T3 cells, we next evaluated their cytotoxicity. For these experiments, the cells were exposed to various concentrations of the iridium complexes for 24 h and then their cell viability was measured using a sulforhodamine B (SRB) assay. The 50% inhibition concentrations (IC50) were found to be 71, 45, >200, and 60 μM for Ir1, Ir2, Ir3, and Ir4, respectively (Table 2, column 4). The cytotoxicity of the iridium complexes does not appear to correlate with either their logP values or cell uptake efficiency.
To assess whether the cytotoxicity of the Ir catalysts is cell line dependent, we also screened them using A549 human lung cancer cells and BEAS-2B human lung non-cancer cells (Table S15†). Our results showed that in A549, the IC50 values ranged from ∼45 to 129 μM in the order Ir2 < Ir4 < Ir1 < Ir3, which is consistent with the cytotoxicity trend observed in NIH-3T3 cells. However, in the BEAS-2B cell line, the IC50 values were observed in the order Ir4 < Ir1 < Ir3 ∼ Ir2 (Table S15†). For comparison, the IC50 of Cat1 was reported to be ∼4 μM in A549 cells.15 Because there are many factors that contribute to cytotoxicity, further studies are needed to understand the origin of these different effects.
Although the N,N′- and N,O-isomers exhibit clear differences in their reactivity, as demonstrated in the transfer hydrogenation section above, their biological properties are not dependent on their picolinamidate ligand binding mode. Thus, the characteristics of each iridium complex inside cells must be measured individually rather than relying on their molecular structures to predict their biological behavior.
Finally, we wanted to assess whether the ROS-generating ability of the Ir complexes could be predicted based on their reactivity in solution.11 In these studies, NIH-3T3 cells were incubated with 5 μM of an iridium complex and 2 mM of HCOONa (if any) for 24 h prior to measuring the ROS levels using a fluorescein-based assay (Fig. S17†). We observed that cells containing Ir1, Ir2, and Ir3, with or without HCOONa, displayed higher amounts of ROS compared to those in the untreated control. No statistically significant increase in ROS was found in cells exposed to Ir4. It should be noted that Ir2 produced the largest amounts of ROS in cells (Table 2, column 5), which is consistent with our studies showing that it was also the most active for H2O2 production in the reaction flask (Fig. 5). Thus, the reactivity of the iridium complexes with O2 in solution appears to roughly mirror their relative reaction rates in cells. However, this observation is likely true only under limited circumstances since the efficiency of intracellular reactions is dependent on numerous factors (e.g., local pH, GSH concentrations, catalyst localization, etc.).7
Conclusions
We have prepared and characterized a series of Cp*Ir picolinamidate complexes bearing different R groups at the 2,6-positions of the N-phenyl moiety. We observed that the picolinamidate ligand binds to iridium via N,N′-coordination when R = H or Me but via N,O-coordination when R = iPr or Ph. The carbon chemical shift of the C(O)NR group is a useful handle for differentiating between the structural isomers in solution, in which peaks at ≥167 ppm are typical for N,N′-isomers and peaks at ∼164 ppm are typical for N,O-isomers. DFT studies suggest that the N,N′-isomer is energetically more favorable than the N,O-isomer but the presence of bulky R groups in the picolinamidate ligand leads to a preference for the latter. In terms of their transfer hydrogenation activity for the reduction of benzaldehyde to benzyl alcohol, Ir1 and Ir2 (N,N′-isomers) exhibited higher activity than Ir3 and Ir4 (N,O-isomers). Unfortunately, all the iridium catalysts showed reduced or no activity when GSH was added. The biological properties of the iridium complexes, such as lipophilicity, cell uptake, and cytotoxicity, are not dependent on their picolinamidate ligand binding mode. Thus, it is necessary to evaluate each complex individually to assess their behavior in living systems. Complex Ir2 was found to produce the highest amounts of ROS in cells, which is consistent with our solution studies showing that it was the most active in generating H2O2 from reaction with HCOONa and O2 in air.In conclusion, this work has led to a better understanding of structural isomerism in Cp*Ir picolinamidate complexes and offers additional design rules for creating new variants. These studies also suggest that steric tuning alone is insufficient to protect the Ir center from poisoning by biological nucleophiles. Thus, additional strategies are needed to prevent the deactivation of small-molecule organometallic catalysts and maximize their catalytic potential inside living systems.
Author contributions
H. D. N.: conceptualization, investigation, writing; C. J. L.: investigation, writing; R. D. J.: investigation; T. G.: investigation; S. H.: investigation; K. K.: investigation; J. I. W.: supervision, funding acquisition; L. H. D.: conceptualization, supervision, writing, funding acquisition.Data availability
The data supporting this article have been included as part of the ESI.† Crystallographic data for Ir1–Ir4 have been deposited in the CCDC under #2365892, 2365893, 2665894, and 2635985.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the National Institute of General Medicine Sciences of the National Institutes of Health (R01GM129276 to L. H. D. and R35GM133548 to J. I. W.), and the Alfred P. Sloan Research Foundation (FG-2020-12811 to J. I. W.) for supporting this research.References
- S. Swaminathan, J. Haribabu, N. Balakrishnan, P. Vasanthakumar and R. Karvembu, Piano stool Ru(II)-arene complexes having three monodentate legs: A comprehensive review on their development as anticancer therapeutics over the past decade, Coord. Chem. Rev., 2022, 459, 214403 CrossRef CAS
.
- G. Gasser, I. Ott and N. Metzler-Nolte, Organometallic Anticancer Compounds, J. Med. Chem., 2011, 54, 3–25 CrossRef CAS PubMed
- W. D. J. Tremlett, D. M. Goodman, T. R. Steel, S. Kumar, A. Wieczorek-Błauż, F. P. Walsh, M. P. Sullivan, M. Hanif and C. G. Hartinger, Design concepts of half-sandwich organoruthenium anticancer agents based on bidentate bioactive ligands, Coord. Chem. Rev., 2021, 445, 213950 CrossRef CAS
- J. J. Soldevila-Barreda and N. Metzler-Nolte, Intracellular Catalysis with Selected Metal Complexes and Metallic Nanoparticles: Advances toward the Development of Catalytic Metallodrugs, Chem. Rev., 2019, 119, 829–869 CrossRef CAS PubMed
- S. Banerjee and P. J. Sadler, Transfer Hydrogenation Catalysis in Cells, RSC Chem. Biol., 2021, 21, 12–29 RSC
- Z. Liu and P. J. Sadler, Organoiridium Complexes: Anticancer Agents and Catalysts, Acc. Chem. Res., 2014, 47, 1174–1185 CrossRef CAS PubMed
- A. H. Ngo, S. Bose and L. H. Do, Intracellular Chemistry: Integrating Molecular Inorganic Catalysts with Living Systems, Chem. – Eur. J., 2018, 24, 10584–10594 CrossRef CAS PubMed
- D. Chauhan, P. Prasad and P. K. Sasmal, Organoiridium-catalyzed bioorthogonal chemistry, Coord. Chem. Rev., 2024, 520, 216139 CrossRef CAS
- S. W. M. Crossley, L. Tenney, V. N. Pham, X. Xie, M. W. Zhao and C. J. Chang, A Transfer Hydrogenation Approach to Activity-Based Sensing of Formate in Living Cells, J. Am. Chem. Soc., 2024, 146, 8865–8876 CrossRef CAS PubMed
- F. Schwizer, Y. Okamoto, T. Heinisch, Y. Gu, M. M. Pellizzoni, V. Lebrun, R. Reuter, V. Köhler, J. C. Lewis and T. R. Ward, Artificial Metalloenzymes: Reaction Scope and Optimization Strategies, Chem. Rev., 2018, 118, 142–231 CrossRef CAS PubMed
- Z. Liu, I. Romero-Canelón, B. Qamar, J. M. Hearn, A. Habtemariam, N. P. E. Barry, A. M. Pizarro, G. J. Clarkson and P. J. Sadler, The Potent Oxidant Anticancer Activity of Organoiridium Catalysts, Angew. Chem., Int. Ed., 2014, 53, 3941–3946 CrossRef CAS PubMed
- J. M. Poljarević, G. Tamás Gál, N. V. May, G. Spengler, O. Dömötör, A. R. Savić, S. Grgurić-Šipka and É. A. Enyedy, Comparative solution equilibrium and structural studies of half-sandwich ruthenium(II)(η6-toluene) complexes of picolinate derivatives, J. Inorg. Biochem., 2018, 181, 74–85 CrossRef PubMed
- Y. M. Wilson, M. Dürrenberger, E. S. Nogueira and T. R. Ward, Neutralizing the Detrimental Effect of Glutathione on Precious Metal Catalysts, J. Am. Chem. Soc., 2014, 136, 8928–8932 CrossRef CAS PubMed
- H. D. Nguyen and L. H. Do, Taming glutathione potentiates metallodrug action, Curr. Opin. Chem. Biol., 2022, 71, 102213 CrossRef CAS PubMed
- Z. Liu, I. Romero-Canelón, A. Habtemariam, G. J. Clarkson and P. J. Sadler, Potent Half-Sandwich Iridium(III) Anticancer Complexes Containing C^N-Chelated and Pyridine Ligands, Organometallics, 2014, 33, 5324–5333 CrossRef CAS PubMed
- A. C. Carrasco, V. Rodríguez-Fanjul, A. Habtemariam and A. M. Pizarro, Structurally Strained Half-Sandwich Iridium(III) Complexes As Highly Potent Anticancer Agents, J. Med. Chem., 2020, 63, 4005–4021 CrossRef CAS PubMed
- A. C. Carrasco, V. Rodríguez-Fanjul and A. M. Pizarro, Activation of the Ir–N(pyridine) Bond in Half-Sandwich Tethered Iridium(III) Complexes, Inorg. Chem., 2020, 59, 16454–16466 CrossRef CAS PubMed
- S. Infante-Tadeo, V. Rodríguez-Fanjul, A. Habtemariam and A. M. Pizarro, Osmium(II) tethered half-sandwich complexes: pH-dependent aqueous speciation and transfer hydrogenation in cells, Chem. Sci., 2021, 12, 9287–9297 RSC
- U. Das and P. Paira, Exploring the phototoxicity of GSH-resistant 2-(5,6-dichloro-1H-benzo[d]imidazol-2-yl)quinoline-based Ir(III)-PTA complexes in MDA-MB-231 cancer cells, Dalton Trans., 2024, 53, 6459–6471 RSC
- R. Banerjee and J. D. Lipscomb, Small-Molecule Tunnels in Metalloenzymes Viewed as Extensions of the Active Site, Acc. Chem. Res., 2021, 54, 2185–2195 CrossRef CAS PubMed
- S. Zacarias, A. Temporão, M. d. Barrio, V. Fourmond, C. Léger, P. M. Matias and I. A. C. Pereira, A Hydrophilic Channel Is Involved in Oxidative Inactivation of a [NiFeSe] Hydrogenase, ACS Catal., 2019, 9, 8509–8519 CrossRef CAS
- M. L. Zastrow and V. L. Pecoraro, Influence of Active Site Location on Catalytic Activity in de Novo-Designed Zinc Metalloenzymes, J. Am. Chem. Soc., 2013, 135, 5895–5903 CrossRef CAS PubMed
- K. Purkait, S. Karmakar, S. Bhattacharyya, S. Chatterjee, S. K. Dey and A. Mukherjee, A hypoxia efficient imidazole-based Ru(II) arene anticancer agent resistant to deactivation by glutathione, Dalton Trans., 2015, 44, 5969–5973 RSC
- Z. Almodares, S. J. Lucas, B. D. Crossley, A. M. Basri, C. M. Pask, A. J. Hebden, R. M. Phillips and P. C. McGowan, Rhodium, Iridium, and Ruthenium Half-Sandwich Picolinamide Complexes as Anticancer Agents, Inorg. Chem., 2014, 53, 727–736 CrossRef CAS PubMed
- L. Tensi, A. Dall’Anese, A. Annunziata, S. Mearini, V. Nofrini, G. M. Rodriguez, A. Carotti, R. Sardella, F. Ruffo and A. Macchioni, Synthesis and Characterization of Chiral Iridium Complexes Bearing Carbohydrate Functionalized Pyridincarboxamide Ligands and Their Application as Catalysts in the Asymmetric Transfer Hydrogenation of α-Ketoacids in Water, Organometallics, 2023, 42, 157–166 CrossRef CAS
- R. Kanega, N. Onishi, L. Wang, K. Murata, J. T. Muckerman, E. Fujita and Y. Himeda, Picolinamide-Based Iridium Catalysts for Dehydrogenation of Formic Acid in Water: Effect of Amide N Substituent on Activity and Stability, Chem. – Eur. J., 2018, 24, 18389–18392 CrossRef CAS PubMed
- S. Bose, A. H. Ngo and L. H. Do, Intracellular Transfer Hydrogenation Mediated by Unprotected Organoiridium Catalysts, J. Am. Chem. Soc., 2017, 139, 8792–8795 CrossRef CAS PubMed
- H. T. H. Nguyen and L. H. Do, Organoiridium–quinone conjugates for facile hydrogen peroxide generation, Chem. Commun., 2020, 56, 13381–13384 RSC
- S. Bose, H. D. Nguyen, A. H. Ngo and L. H. Do, Fluorescent half-sandwich iridium picolinamidate complexes for in-cell visualization, J. Inorg. Biochem., 2022, 234, 111877 CrossRef CAS PubMed
- A. H. Ngo, M. Ibañez and L. H. Do, Catalytic Hydrogenation of Cytotoxic Aldehydes Using Nicotinamide Adenine Dinucleotide (NADH) in Cell Growth Media, ACS Catal., 2016, 6, 2637–2641 CrossRef CAS
- A. H. Ngo and L. H. Do, Structure–Activity Relationship Study of Half-Sandwich Metal Complexes in Aqueous Transfer Hydrogenation Catalysis, Inorg. Chem. Front., 2020, 7, 583–591 RSC
- C. Jiang and T. S. Teets, Impacts of ancillary ligand coordination modes on red-emitting cyclometalated iridium complexes, Inorg. Chem. Front., 2024, 11, 1501–1510 RSC
- R. M. Lord, M. Zegke, A. M. Basri, C. M. Pask and P. C. McGowan, Rhodium(III) Dihalido Complexes: The Effect of Ligand Substitution and Halido Coordination on Increasing Cancer Cell Potency, Inorg. Chem., 2021, 60, 2076–2086 CrossRef CAS PubMed
- A. M. Basri, R. M. Lord, S. J. Allison, A. Rodríguez-Bárzano, S. J. Lucas, F. D. Janeway, H. J. Shepherd, C. M. Pask, R. M. Phillips and P. C. McGowan, Bis-picolinamide Ruthenium(III) Dihalide Complexes: Dichloride-to-Diiodide Exchange Generates Single trans Isomers with High Potency and Cancer Cell Selectivity, Chem. – Eur. J., 2017, 23, 6341–6356 CrossRef CAS PubMed
- Y. Ji, X. Yang, Z. Ji, L. Zhu, N. Ma, D. Chen, X. Jia, J. Tang and Y. Cao, DFT-Calculated IR Spectrum Amide I, II, and III Band Contributions of N-Methylacetamide Fine Components, ACS Omega, 2020, 5, 8572–8578 CrossRef CAS PubMed
- A. Stein, D. Chen, N. V. Igareta, Y. Cotelle, J. G. Rebelein and T. R. Ward, A Dual Anchoring Strategy for the Directed Evolution of Improved Artificial Transfer Hydrogenases Based on Carbonic Anhydrase, ACS Cent. Sci., 2021, 7, 1874–1884 CrossRef CAS PubMed
- Y.-A. Young, H. T. H. Nguyen, H. D. Nguyen, T. Ganguly, Y. H. Nguyen and L. H. Do, A ratiometric substrate for rapid evaluation of transfer hydrogenation efficiency in solution, Dalton Trans., 2024, 53, 8887–8892 RSC
- J. G. Rebelein, Y. Cotelle, B. Garabedian and T. R. Ward, Chemical Optimization of Whole-Cell Transfer Hydrogenation Using Carbonic Anhydrase as Host Protein, ACS Catal., 2019, 9, 4173–4178 CrossRef CAS PubMed
- X. Liu, W.-Z. Dong, Y. Li, X. Yu, W.-H. Wang, Y. Himeda and M. Bao, Efficient β-alkylation of secondary alcohols to α-substituted ketones catalyzed by functionalized Ir complexes via borrowing hydrogen in water, Org. Chem. Front., 2023, 10, 355–362 RSC
- W.-H. Wang, W.-Y. Shao, J.-Y. Sang, X. Li, X. Yu, Y. Yamamoto and M. Bao,
N,N-Dialkylation of Acyl Hydrazides with Alcohols Catalyzed by Amidato Iridium Complexes via Borrowing Hydrogen, Organometallics, 2023, 42, 2623–2631 CrossRef CAS
- K. Tanaka, T. Miki, K. Murata, A. Yamaguchi, Y. Kayaki, S. Kuwata, T. Ikariya and M. Watanabe, Reductive Amination of Ketonic Compounds Catalyzed by Cp*Ir(III) Complexes Bearing a Picolinamidato Ligand, J. Org. Chem., 2019, 84, 10962–10977 CrossRef CAS PubMed
- L. Tensi, A. V. Yakimov, C. Trotta, C. Domestici, J. De Jesus Silva, S. R. Docherty, C. Zuccaccia, C. Copéret and A. Macchioni, Single-Site Iridium Picolinamide Catalyst Immobilized onto Silica for the Hydrogenation of CO2 and the Dehydrogenation of Formic Acid, Inorg. Chem., 2022, 61, 10575–10586 CrossRef CAS PubMed
-
A. G. Orpen, L. Brammer, F. H. Allen, O. Kennard, D. G. Watson and R. Taylor, Appendix A: Typical Interatomic Distances in Organic Compounds and Organometallic Compounds and Coordination Complexes of the d- and f-block metals, in Structure Correlation, 1994, pp. 752–858 Search PubMed
- L. Falivene, R. Credendino, A. Poater, A. Petta, L. Serra, R. Oliva, V. Scarano and L. Cavallo, SambVca 2. A Web Tool for Analyzing Catalytic Pockets with Topographic Steric Maps, Organometallics, 2016, 35, 2286–2293 CrossRef CAS
-
A. T. Merrill, W. Guo and D. J. Tantillo, Chapter 8: NMR Prediction with Computational Chemistry, in Enabling Tools and Techniques for Organic Synthesis: A Practical Guide to Experimentation, Automation, and Computation, ed. S. G. Newman, John Wiley & Sons, 2023 Search PubMed
- E. R. Johnson, S. Keinan, P. Mori-Sánchez, J. Contreras-García, A. J. Cohen and W. Yang, Revealing Noncovalent Interactions, J. Am. Chem. Soc., 2010, 132, 6498–6506 CrossRef CAS PubMed
- R. D. Jana, A. H. Ngo, S. Bose and L. H. Do, Organoiridium Complexes Enhance Cellular Defense Against Reactive Aldehydes Species, Chem. – Eur. J., 2023, 29, e202300842 CrossRef CAS PubMed
- R. M. LoPachin and T. Gavin, Molecular Mechanisms of Aldehyde Toxicity: A Chemical Perspective, Chem. Res. Toxicol., 2014, 27, 1081–1091 Search PubMed
- K. Sengupta, S. Chatterjee and A. Dey, Catalytic H2O2 Disproportionation and Electrocatalytic O2 Reduction by a Functional Mimic of Heme Catalase: Direct Observation of Compound 0 and Compound I in Situ, ACS Catal., 2016, 6, 1382–1388 Search PubMed
- D. M. Stanbury, The principle of detailed balancing, the iron-catalyzed disproportionation of hydrogen peroxide, and the Fenton reaction, Dalton Trans., 2022, 51, 2135–2157 RSC
- D. Bridget Williams, W. Kaminsky, J. M. Mayer and K. I. Goldberg, Reactions of iridium hydride pincer complexes with dioxygen: new dioxygen complexes and reversible O2 binding, Chem. Commun., 2008, 4195–4197 RSC
- L. Boisvert and K. I. Goldberg, Reactions of Late Transition Metal Complexes with Molecular Oxygen, Acc. Chem. Res., 2012, 45, 899–910 CrossRef CAS PubMed
- J. J. Soldevila-Barreda, I. Romero-Canelón, A. Habtemariam and P. J. Sadler, Transfer Hydrogenation Catalysis in Cells as a New Approach to Anticancer Drug Design, Nat. Commun., 2015, 6, 6582 CrossRef CAS PubMed
- N. Singh, A. Gupta, P. Prasad, P. Mahawar, S. Gupta and P. K. Sasmal, Iridium-Triggered Allylcarbamate Uncaging in Living Cells, Inorg. Chem., 2021, 60, 12644–12650 CrossRef CAS PubMed
- G. Hodges, C. Eadsforth, B. Bossuyt, A. Bouvy, M.-H. Enrici, M. Geurts, M. Kotthoff, E. Michie, D. Miller, J. Müller, G. Oetter, J. Roberts, D. Schowanek, P. Sun and J. Venzmer, A comparison of log Kow (n-octanol–water partition coefficient) values for non-ionic, anionic, cationic and amphoteric surfactants determined using predictions and experimental methods, Environ. Sci. Eur., 2019, 31, 1 CrossRef CAS
- P. Krishnamoorthy, P. Sathyadevi, A. H. Cowley, R. R. Butorac and N. Dharmaraj, Evaluation of DNA binding, DNA cleavage, protein binding and in vitro cytotoxic activities of bivalent transition metal hydrazone complexes, Eur. J. Med. Chem., 2011, 46, 3376–3387 CrossRef CAS PubMed
- R. Mailhot, T. Traviss-Pollard, R. Pal and S. J. Butler, Cationic Europium Complexes for Visualizing Fluctuations in Mitochondrial ATP Levels in Living Cells, Chem. – Eur. J., 2018, 24, 10745–10755 CrossRef CAS PubMed
- A. Foucault-Collet, K. A. Gogick, K. A. White, S. Villette, A. Pallier, G. Collet, C. Kieda, T. Li, S. J. Geib, N. L. Rosi and S. Petoud, Lanthanide near infrared imaging in living cells with Yb3+ nano metal organic frameworks, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 17199–17204 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2365892–2365895. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4qi01955e |
| This journal is © the Partner Organisations 2024 |